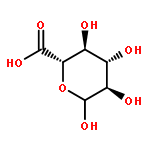
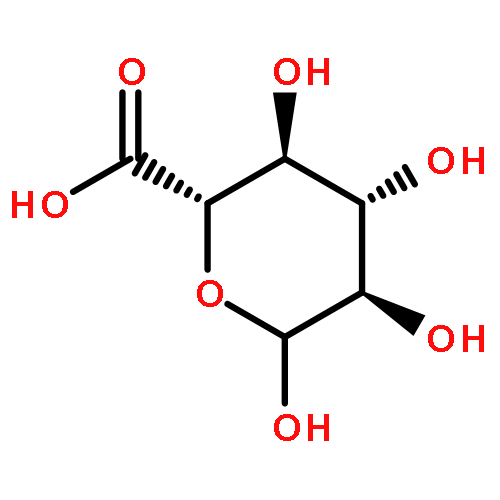
Co-reporter:Julie L. Sutcliffe;Lina Y. Hu;Kimberly A. Kelly
Molecular Imaging and Biology 2017 Volume 19( Issue 2) pp:
Publication Date(Web):2017/04/01
DOI:10.1007/s11307-016-1016-z
Molecular imaging allows for the visualization of changes at the cellular level in diseases such as cancer. A successful molecular imaging agent must rely on disease-selective targets and ligands that specifically interact with those targets. Unfortunately, the translation of novel target-specific ligands into the clinic has been frustratingly slow with limitations including the complex design and screening approaches for ligand identification, as well as their subsequent optimization into useful imaging agents. This review focuses on combinatorial library approaches towards addressing these two challenges, with particular focus on phage display and one-bead one-compound (OBOC) libraries. Both of these peptide-based techniques have proven successful in identifying new ligands for cancer-specific targets and some of the success stories will be highlighted. New developments in screening methodology and sequencing technology have pushed the bounds of phage display and OBOC even further, allowing for even faster and more robust discovery of novel ligands. The combination of multiple high-throughput technologies will not only allow for more accurate identification, but also faster affinity maturation, while overall streamlining the process of translating novel ligands into clinical imaging agents.
Co-reporter:Robin C. Cumming, Dag Erlend Olberg and Julie L. Sutcliffe
RSC Advances 2014 vol. 4(Issue 90) pp:49529-49534
Publication Date(Web):25 Sep 2014
DOI:10.1039/C4RA10520F
To date the majority of 18F-peptide radiolabeling approaches are multi-step, low yielding and time-consuming processes. Given the short half-life of 18F (109.8 min), it is critical that methods are developed to increase the efficiency of this process with simpler, higher yielding and faster reactions that can be rapidly translated into clinical use. Here, we demonstrate the first microfluidic synthesis of the [18F]F-Py-TFP prosthetic group with radiochemical yields of up to 97% and a synthesis time of 3 min. In addition, we utilized a single microfluidics device to prepare the [18F]F-Py-YGGFL peptide using [18F]F-Py-TFP, from [18F]fluoride in a two-step, fully automated approach. The model peptide NH2-YGGFL was radiolabeled with [18F]F-Py-TFP in up to 28% overall radiochemical yield within 8 minutes starting from anhydrous [18F]fluoride.
Co-reporter:Dag Erlend Olberg, Kjetil Wessel Andressen, Finn Olav Levy, Jo Klaveness, Ira Haraldsen, Julie L. Sutcliffe
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 7) pp:1846-1850
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmcl.2014.02.002
Two novel small molecule gonadotropin-releasing hormone (GnRH) receptor antagonists (12 and 13) of the furamide-class were synthesized and evaluated in vitro for their receptor binding affinities for the rat GnRH receptor. Radiolabeling with no carrier added fluorine-18 of the appropriate precursors was investigated in a one-step reaction. Log P (Octanol/PBS pH 7.4) and serum stability of the compounds were investigated. The antagonists showed low nM affinity for the rat GnRH receptor. 18F-radiolabled compounds were obtained in high radiochemical purity (>95%) and specific activity (>75 GBq/μmol). These findings suggest this class of compounds holds promise as potential probes for PET targeting of GnRH-receptor expression.
Co-reporter:Drishty Satpati;Sven H. Hausner;Nadine Bauer;Julie L. Sutcliffe
Journal of Labelled Compounds and Radiopharmaceuticals 2014 Volume 57( Issue 9) pp:558-565
Publication Date(Web):
DOI:10.1002/jlcr.3215
Cerenkov luminescence imaging (CLI) is an emerging preclinical molecular imaging modality that tracks the radiation emitted in the visible spectrum by fast moving charged decay products of radionuclides. The aim of this study was in vitro and in vivo evaluation of the two radiotracers, 90Y-DOTA-PEG28-A20FMDV2 (90Y-1) and 90Y-DOTA-Ahx-A20FMDV2 (90Y-2) (>99% radiochemical purity, 3.7 GBq/µmol specific activity) for noninvasive assessment of tumors expressing the integrin αvβ6 and their future use in tumor targeted radiotherapy. Cell binding and internalization in αvβ6-positive cells was 90Y-1: 10.1 ± 0.8%, 50.3 ± 2.1%; 90Y-2: 22.4 ± 1.7%, 44.7 ± 1.5% with <5% binding to αvβ6-negative control cells. Biodistribution studies showed maximum αvβ6-positive tumor uptake of the radiotracers at 1-h post injection (p.i.) (90Y-1: 0.64 ± 0.15% ID/g; 90Y-2: 0.34 ± 0.11% ID/g) with high renal uptake (>25% ID/g at 24 h). Because of the lower tumor uptake and high radioactivity accumulation in kidneys (that could not be reduced by pre-administration of either lysine or furosemide), the luminescence signal from the αvβ6-positive tumor was not clearly detectable in CLI images. The studies suggest that CLI is useful for indicating major organ uptake for both radiotracers; however, it reaches its limitation when there is low signal-to-noise ratio.
Co-reporter:Zibo Li;Lina Y. Hu;Leah M. Knight;Shuanglong Liu;Peter S. Conti;Carolyn J. Anderson;Julie L. Sutcliffe;Nadine Bauer
Molecular Imaging and Biology 2014 Volume 16( Issue 4) pp:
Publication Date(Web):2014/08/01
DOI:10.1007/s11307-013-0717-9
The integrin αvβ6 is overexpressed in a variety of aggressive cancers and serves as a prognosis marker. This study describes the conjugation, radiolabeling, and in vitro and in vivo evaluation of four chelators to determine the best candidate for 64Cu radiolabeling of A20FMDV2, an αvβ6 targeting peptide.Four chelators were conjugated onto PEG28-A20FMDV2 (1): 11-carboxymethyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane-4-methanephosphonic acid (CB-TE1A1P), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA), and 4,4′-((3,6,10,13,16,19-hexazazbicyclo[6.6.6]ico-sane-1,8-diylbis(aza-nediyl))bis(methylene)dibenzoic acid (BaBaSar). All peptides were radiolabeled with 64Cu in ammonium acetate buffer at pH 6 and formulated to pH 7.2 in PBS for use. The radiotracers were evaluated using in vitro cell binding and internalization assays and serum stability assays. In vivo studies conducted include blocking, biodistribution, and small animal PET imaging. Autoradiography and histology were also conducted.All radiotracers were radiolabeled in good radiochemical purity (>95 %) under mild conditions (37–50 °C for 15 min) with high specific activity (0.58–0.60 Ci/μmol). All radiotracers demonstrated αvβ6-directed cell binding (>46 %) with similar internalization levels (>23 %). The radiotracers 64Cu-CB-TE1A1P-1 and 64Cu-BaBaSar-1 showed improved specificity for the αvβ6 positive tumor in vivo over 64Cu-DOTA-1 and 64Cu-NOTA-1 (+/− tumor uptake ratios—3.82 +/- 0.44, 3.82 ± 0.41, 2.58 ± 0.58, and 1.29 ± 0.14, respectively). Of the four radiotracers, 64Cu-NOTA-1 exhibited the highest liver uptake (10.83 ± 0.1 % ID/g at 4 h).We have successfully conjugated, radiolabeled, and assessed the four chelates CB-TE1A1P, DOTA, NOTA, and BaBaSar both in vitro and in vivo. However, the data suggests no clear “best candidate” for the 64Cu-radiolabeling of A20FMDV2, but instead a trade-off between the different properties (e.g., stability, selectivity, pharmacokinetics, etc.) with no obvious effects of the individual chelators.
Co-reporter:Drishty Satpati;Nadine Bauer
Journal of Radioanalytical and Nuclear Chemistry 2014 Volume 302( Issue 2) pp:765-771
Publication Date(Web):2014 November
DOI:10.1007/s10967-014-3197-8
Catalyst-free click reactions are effective chemical tools for synthesis of radiometal-based radiopharmaceuticals offering advantages towards preparation of non-toxic agents with high specific activity. In the present study the radiotracer [64Cu]DOTA-ADIBON3-Ala-PEG28-A20FMDV2, [64Cu]3, was synthesized for positron emission tomography imaging of integrin αvβ6 expressing tumors via a strain-promoted click reaction using both a “pre-click” and “post-click” approaches. The radiotracer, prepared in >99 % radiochemical yield, was evaluated in vitro (64.6 ± 2.8 % binding to αvβ6-positive cells vs. <5 % to αvβ6-negative cells) and in vivo (αvβ6-positive tumor uptake: 1.52 ± 0.16 % ID/g, 24 h p.i.). While the high initial renal uptake (76.2 ± 10.7 % ID/g at 1 h p.i.) was comparable to a previously reported radiotracer, [64Cu]DOTA-PEG28-A20FMDV2, [64Cu]3 showed notably improved renal clearance (11.3 ± 2.5 % ID/g at 24 h p.i.). Thus, the introduction of a chelator-strained alkyne system resulted in improved pharmacokinetics for the present radiotracer, highlighting the attractive prospects of strain-promoted click-based preparations in the construction of radiometalated bioconjugates for targeted molecular imaging and therapy.
Co-reporter:Richard D. Carpenter, Sven H. Hausner, and Julie L. Sutcliffe
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 12) pp:885
Publication Date(Web):October 7, 2011
DOI:10.1021/ml200187j
The strain-promoted click 1,3-dipolar cycloaddition reactions involving azides and cyclooctynes for the synthesis of triazoles offer the advantage of being able to be performed in biological settings via copper-free chemistries. While strained reagents conjugated to optical dyes and radiometal conjugates have been reported, cyclooctyne reagents labeled with fluorine-18 (18F) and radiochemically evaluated in a copper-free click reaction have yet to be explored. This report describes the conversion of a bifunctional azadibenzocyclooctyne (ADIBO) amine to the 18F-labeled cyclooctyne 4, the subsequent fast copper-free 1,3-dipolar cycloaddition reaction with alkyl azides at 37 °C (>70% radiochemical conversion in 30 min), and biological evaluations (serum stability of >95% at 2 h). These findings demonstrate the excellent reactivity of the 18F-labeled cyclooctyne 4 with readily available azides that will allow future work focusing on rapid copper-free in vitro and in vivo click chemistries for PET imaging using 18F-labeled cyclooctyne derivatives of ADIBO.Keywords: Copper-free click chemistry; cycloaddition; cyclooctyne; fluorine-18; positron emission tomography
Co-reporter:M. Karen J. Gagnon;Sven H. Hausner;Jan Marik;Craig K. Abbey;John F. Marshall;Julie L. Sutcliffe
PNAS 2009 Volume 106 (Issue 42 ) pp:17904-17909
Publication Date(Web):2009-10-20
DOI:10.1073/pnas.0906925106
The rapid development and translation of targeted molecular imaging agents from bench to bedside is currently a slow process,
with a clear bottleneck between the discovery of new compounds and the development of an appropriate molecular imaging agent.
The ability to identify promising new molecular imaging agents, as well as failures, much earlier in the development process
using high-throughput screening techniques could save significant time and money. This work combines the advantages of combinatorial
chemistry, site-specific solid-phase radiolabeling, and in vivo imaging for the rapid screening of molecular imaging agents.
A one-bead-one-compound library was prepared and evaluated in vitro, leading to the identification of 42 promising lead peptides.
Over 11 consecutive days, these peptides, along with a control peptide, were successfully radiolabeled with 4-[18F]fluorobenzoic acid and evaluated in vivo using microPET. Four peptides were radiolabeled per day, followed by simultaneous
injection of each individual peptide into 2 animals. As a result, 4 promising new molecular imaging agents were identified
that otherwise would not have been selected based solely on in vitro data. This study is the first example of the practical
application of a high-throughput screening approach using microPET imaging of [18F]-labeled peptides for the rapid in vivo identification of potential new molecular imaging agents.
Co-reporter:Sven H. Hausner ; Jan Marik ; M. Karen J. Gagnon ;Julie L. Sutcliffe
Journal of Medicinal Chemistry 2008 Volume 51(Issue 19) pp:5901-5904
Publication Date(Web):September 12, 2008
DOI:10.1021/jm800608s
Numerous radiolabeled peptides have been utilized for in vivo imaging of a variety of cell surface receptors. For applications in PET using [18F]fluorine, peptides are radiolabeled via a prosthetic group approach. We previously developed solution-phase 18F-“click” radiolabeling and solid-phase radiolabeling using 4-[18F]fluorobenzoic and 2-[18F]fluoropropionic acids. Here we compare the three different radiolabeling approaches and report the effects on PET imaging and pharmacokinetics. The prosthetic groups did have an effect; metabolites with significantly different polarities were observed.
Co-reporter:Sven H. Hausner, Nadine Bauer, Julie L. Sutcliffe
Nuclear Medicine and Biology (January 2014) Volume 41(Issue 1) pp:43-50
Publication Date(Web):1 January 2014
DOI:10.1016/j.nucmedbio.2013.09.009
IntroductionIncorporation of fluorine-18 (18F) into radiotracers by capturing ionic [18F]-species can greatly accelerate and simplify radiolabeling for this important positron emission tomography (PET) radioisotope. Among the different strategies, the incorporation of aluminum [18F]fluoride (Al[18F]2 +) into NOTA chelators has recently emerged as a robust approach to peptide radiolabeling. This study presents Al[18F]2 +-radiolabeling of an αvβ6 integrin-targeted peptide (NOTA-PEG28-A20FMDV2) and its in vitro and in vivo evaluation.MethodsAluminum [18F]fluoride was prepared at r.t. from [18F]fluoride (40 MBq–11 GBq) and introduced into NOTA-PEG28-A20FMDV2 (1) in sodium acetate (pH 4.1; 100°C, 15 min). The radiotracer Al[18F] NOTA-PEG28-A20FMDV2 (2) was purified by HPLC, formulated in PBS and evaluated in vitro (stability; binding and internalization in αvβ6(+) and αvβ6(−) cells) and in vivo (paired αvβ6(+) and αvβ6(−) xenograft mice: PET/CT, biodistribution, tumor autoradiography and metabolites).ResultsThe radiotracer 2 was prepared in 90 ± 6 min (incl. formulation; n = 3) in 19.3 ± 5.4% decay corrected radiochemical yield (radiochemical purity: > 99%; specific activity: 158 ± 36 GBq/μmol) and was stable in PBS and serum (2 h). During in vitro cell binding studies, 2 showed high, αvβ6-targeted binding (αvβ6(+): 42.4 ± 1.2% of total radioactivity, ratio (+)/(−) = 8.4/1) and internalization (αvβ6(+): 28.3 ± 0.5% of total radioactivity, (+)/(−) = 11.7/1). In vivo, 2 maintained αvβ6-targeted binding (biodistribution; 1 h: αvβ6(+): 1.74 ± 0.38% ID/g, (+)/(−) = 2.72/1; 4 h: αvβ6(+): 1.21 ± 0.56% ID/g, (+)/(−) = 4.0/1; 11% intact 2 in tumor at 1 h), with highest uptake around the tumor edge (autoradiography). Most of the radioactivity cleared rapidly in the urine within one hour, but a significant fraction remained trapped in the kidneys (4 h: 229 ± 44% ID/g).ConclusionThe Al[18F]/NOTA-based radiolabeling was rapid and efficient, and the radiotracer 2 showed good αvβ6-selectivity in vitro and in vivo. However, in contrast to A20FMDV2 labeled with covalently bound [18F]-prosthetic groups (e.g., [18F]fluorobenzoic acid), 2 demonstrated significant trapping in kidneys, similar to radiometal-labeled chelator-analogs of 2.
Co-reporter:Jason B. White, David L. Boucher, Kirstin A. Zettlitz, Anna M. Wu, Julie L. Sutcliffe
Nuclear Medicine and Biology (December 2015) Volume 42(Issue 12) pp:945-957
Publication Date(Web):1 December 2015
DOI:10.1016/j.nucmedbio.2015.07.014
IntroductionThis work describes the development and characterization of two antibody fragments that specifically target the αvβ6 integrin, a non-covalent diabody and a disulfide-stabilized cys-diabody. The diabodies were analyzed for their ability to bind both immobilized and cell surface-bound αvβ6. Radiolabeling was done using non-site-specific and site-specific conjugation approaches with N-succinimidyl 4-[18F]fluorobenzoate ([18F]-SFB) and the bifunctional chelator 1,4,7-triazacyclononane-triacetic acid maleimide (NOTA-maleimide) and copper-64 ([64Cu]), respectively. The affects of each radiolabeling method on RCY, RCP, and immunoreactivity were analyzed for the [18F]-FB-αvβ6 diabody, [18F]-FB-αvβ6 cys-diabody, and the [64Cu]-NOTA-αvβ6 cys-diabody.MethodsDiabodies were constructed from the variable domains of the humanized 6.3G9 anti-αvβ6 intact antibody. The anti-αvβ6 cys-diabody was engineered with C-terminal cysteines to enable covalent dimerization and site-specific modification. Biochemical characterization included SDS-PAGE, Western blot, and electrospray ionization to confirm MW, and flow cytometry and ELISA experiments were used to determine binding affinity and specificity to αvβ6. The diabodies were radiolabeled with [18F]-SFB and in addition, the anti-αvβ6 cys-diabody was also radiolabeled site-specifically using NOTA-maleimide and [64Cu]. Immunoreactivities were confirmed using in vitro cell binding to DX3Puroβ6 (αvβ6+) and DX3Puro (αvβ6−) cell lines.ResultsThe diabodies were purified from cell culture supernatants with purities >98%. Subnanomolar binding affinity towards αvβ6 was confirmed by ELISA (diabody IC50 = 0.8 nM, cys-diabody IC50 = 0.6 nM) and flow cytometry revealed high specificity only to the DX3Puroβ6 cell line for both diabodies. RCYs were 22.6% ± 3.6% for the [18F]-FB-αvβ6 diabody, 8.3% ± 1.7% for the [18F]-FB-αvβ6 cys-diabody and 43.5% ± 5.5% for the [64Cu]-NOTA-αvβ6 cys-diabody. In vitro cell binding assays revealed excellent specificity and retention of immunoreactivity ([18F]-FB-αvβ6 diabody = 58.7% ± 6.7%, [18F]-FB-αvβ6 cys-diabody = 80.4% ± 4.4%, [64Cu]-NOTA-αvβ6 cys-diabody = 59.4% ± 0.6%) regardless of the radiolabeling method used.ConclusionsTwo novel diabodies with excellent binding affinity and specificity for the αvβ6 integrin in vitro were developed. Radiolabeling of the diabodies with fluorine-18 ([18F]) and [64Cu] revealed advantages and disadvantages with regards to methodologies and RCYs, however immunoreactivities were well preserved regardless of radiolabeling approach.
Co-reporter:Jason B. White, Sven H. Hausner, Richard D. Carpenter, Julie L. Sutcliffe
Applied Radiation and Isotopes (December 2012) Volume 70(Issue 12) pp:2720-2729
Publication Date(Web):December 2012
DOI:10.1016/j.apradiso.2012.08.003
Co-reporter:Ryan A. Davis, Kevin Lau, Sven H. Hausner and Julie L. Sutcliffe
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 37) pp:NaN8663-8663
Publication Date(Web):2016/08/25
DOI:10.1039/C6OB01636G
Solid-phase peptide synthesis, head-to-tail cyclization, and subsequent radiolabeling provided a reproducible, simple, rapid synthetic method to generate the cyclic peptide radiotracer cRGDyK([18F]FBA). Herein is reported the first on-resin cyclization and 18F-radiolabeling of a cyclic peptide (cRGDyK) in an overall peptide synthesis yield of 88% (cRGDyK(NH2)) and subsequent radiolabeling yield of 14 ± 2% (decay corrected, n = 4). This approach is generally applicable to the development of an automated process for the synthesis of cyclic radiolabeled peptides for positron emission tomography (PET).

![1,4,7,10-Tetraazacyclododecane-1,4,7-triacetic acid,10-[2-[(2,5-dioxo-1-pyrrolidinyl)oxy]-2-oxoethyl]-](http://img.cochemist.com/ccimg/171000/170908-81-3.png)
![1,4,7,10-Tetraazacyclododecane-1,4,7-triacetic acid,10-[2-[(2,5-dioxo-1-pyrrolidinyl)oxy]-2-oxoethyl]-](http://img.cochemist.com/ccimg/171000/170908-81-3_b.png)
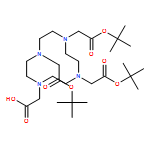
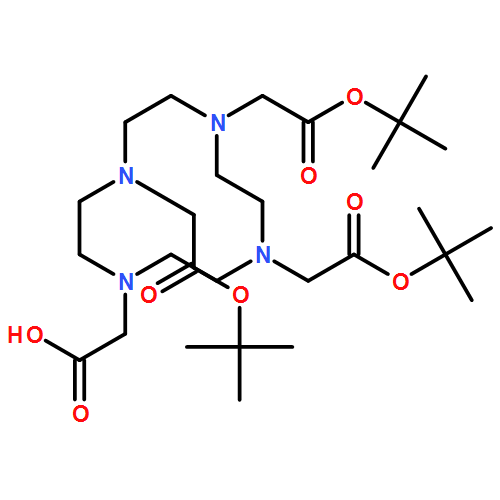







![2-[4,7-BIS(CARBOXYMETHYL)-1,4,7-TRIAZONAN-1-YL]ACETIC ACID](http://img.cochemist.com/ccimg/56500/56491-86-2.png)
![2-[4,7-BIS(CARBOXYMETHYL)-1,4,7-TRIAZONAN-1-YL]ACETIC ACID](http://img.cochemist.com/ccimg/56500/56491-86-2_b.png)