Co-reporter:Andrea Gini;Tobias Brandhofer;Olga García Mancheño
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 6) pp:1294-1312
Publication Date(Web):2017/02/07
DOI:10.1039/C6OB02474B
Over the past few years, the development of oxidative methodologies towards efficient and selective direct Csp3–H bond functionalization processes has attracted tremendous attention from synthetic chemists. However, only a little attention has been given to the key role of the nature of the oxidant. This review aims at providing a brief summary of the recent advances in mild and more benign oxidative Csp3–H bond functionalization reactions, which are classified according to the type of oxidation system employed.
Co-reporter:Sebastian Stockerl, Daniel Gutiérrez, Olga García Mancheño
Journal of Molecular Catalysis A: Chemical 2017 Volume 426, Part B(Volume 426, Part B) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.molcata.2016.09.006
•Synthesis of a new family of C2-symmetric triazole-phosphoric acid catalysts.•Intramolecular asymmetric CH bond functionalization of N-aryl substituted tetrahydroisoquinolines.•Important effects of the triazoles and the regiomeric structures for optimal enantioselectivity.•π–π vs. cooperative π–π and triazole-substrate interactions.Counteranion-catalysis represents an appealing but challenging approach for the development of enantioselective oxidative CH bond functionalization reactions. In this work, a new family of 3,3′-triazolyl BINOL-derived phosphoric acids was synthesized and employed in the intramolecular asymmetric CH bond functionalization of N-aryl substituted tetrahydroisoquinolines. As previously reported with related structures, the presence of the triazole groups on the catalysts was key to attain enantioselectivity. Our study also shows the importance of choosing the appropriate regioisomeric triazole groups at the BINOL backbone to achieve a more efficient chirality transfer. Moderate enantiomeric ratios were obtained with the N-benzamide substrates, whereas the change of the nature of the nucleophile fragment was translated to a dramatic loss of the enantioselectivity. Therefore, it can be foreseen that there is a need for designing further superior catalyst structures to develop future counter-anion organocatalyzed asymmetric CH bond functionalization reactions.Download high-res image (82KB)Download full-size image
Co-reporter:Mercedes Zurro;Dr. Sören Asmus;Julia Bamberger;Dr. Stephan Beckendorf;Dr. Olga GarcíaMancheño
Chemistry - A European Journal 2016 Volume 22( Issue 11) pp:3785-3793
Publication Date(Web):
DOI:10.1002/chem.201504094
Abstract
Easily accessible and tunable chiral triazoles have been introduced as a novel class of C−H bond-based H-donors for anion-binding organocatalysis. They have proven to be effective catalysts for the dearomatization reaction of different N-heteroarenes. Although this dearomatization approach represents a powerful strategy to build chiral heterocycles, to date only a few catalytic methods to this end exist. In this work, the organocatalyzed enantioselective Reissert-type dearomatization of isoquinoline derivatives employing a number of structurally diverse chiral triazoles as anion-binding catalysts was realized. The here presented method was employed to synthesize a number of chiral 1,2-dihydroisoquinoline substrates with an enantioselectivity up to 86:14 e.r. Moreover, a thorough study of the determining parameters affecting the activity of this type of anion- binding catalysts was carried out.
Co-reporter:Dr. Olga GarcíaMancheño;Sören Asmus;Mercedes Zurro;Theresa Fischer
Angewandte Chemie International Edition 2015 Volume 54( Issue 30) pp:8823-8827
Publication Date(Web):
DOI:10.1002/anie.201502708
Abstract
The asymmetric dearomatization of N-heterocycles is an important synthetic method to gain bioactive and synthetically valuable chiral heterocycles. However, the catalytic enantio- and regioselective dearomatization of the simplest six-membered-ring N-heteroarenes, the pyridines, is still very challenging. The first anion-binding-catalyzed, highly enantioselective nucleophilic dearomatization of pyridines with triazole-based H-bond donor catalysts is presented. Contrary to other more common NH-based H-bond donors, this type of organocatalyst shows a prominent higher C2-regioselectivity and is able to promote high enantioinductions via formation of a close chiral anion-pair complex with a preformed N-acyl pyridinium ionic intermediate. This method offers a straightforward and useful synthetic approach to chiral N-heterocycles from abundant and readily available pyridines.
Co-reporter:Andrea Gini;Marwin Segler;Dominik Kellner;Dr. Olga García Mancheño
Chemistry - A European Journal 2015 Volume 21( Issue 34) pp:12053-12060
Publication Date(Web):
DOI:10.1002/chem.201501314
Abstract
N-carbamoyl nitrones represent an important class of reagents for the synthesis of a variety of natural and biologically active compounds. These compounds are generally converted into valuable 4-isoxazolines upon cyclization reaction with dipolarophiles. However, these types of N-protected nitrones are highly unstable, which limits their synthesis, storage and practical use, enforcing alternative lengthy or elaborated synthetic routes. In this work, a 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO)-mediated formal “dehydrogenation” of N-protected benzyl-, allyl- and alkyl-substituted hydroxylamines followed by in situ trapping of the generated unstable nitrones into N-carbamoyl 4-isoxazolines is presented. A plausible mechanism is also proposed, in which the dipolarophile shows an important assistant role in the generation of the active nitrone intermediate. This simple protocol avoids the problematic isolation of N-carbamoyl protected nitrones, providing new synthetic possibilities in isoxazoline chemistry.
Co-reporter:Dr. Olga GarcíaMancheño;Sören Asmus;Mercedes Zurro;Theresa Fischer
Angewandte Chemie 2015 Volume 127( Issue 30) pp:8947-8951
Publication Date(Web):
DOI:10.1002/ange.201502708
Abstract
Die asymmetrische Dearomatisierung von N-Heterocyclen ist ein wichtiges Verfahren zur Synthese bioaktiver und präparativ wertvoller chiraler Heterocyclen. Allerdings stellt die katalytische, enantio- und regioselektive Dearomatisierung der einfachsten sechsgliedrigen N-Heteroarene, der Pyridine, immer noch eine Herausforderung dar. In dieser Arbeit präsentieren wir die erste hoch enantioselektive, nukleophile Dearomatisierung von Pyridinen durch Anionenbindungskatalyse mit Triazol-basierten H-Donor-Katalysatoren. Im Vergleich zu den geläufigen NH-basierten H-Bindungsdonoren zeigt dieser Organokatalysator eine sehr viel höhere C2-Regioselektivität. Er ist in der Lage, hohe Enantioselektivitäten zu induzieren, indem er einen chiralen Ionenpaar-Komplex mit einem ionischen N-Acylpyridinium-Intermediat bildet. Die Methode bietet eine direkte und nützliche Synthese von chiralen N-Heterocyclen ausgehend von kommerziell erhältlichen Pyridinen.
Co-reporter:Mercedes Zurro ; Sören Asmus ; Stephan Beckendorf ; Christian Mück-Lichtenfeld ;Olga García Mancheño
Journal of the American Chemical Society 2014 Volume 136(Issue 40) pp:13999-14002
Publication Date(Web):September 11, 2014
DOI:10.1021/ja507940k
Helical chirality and selective anion-binding processes are key strategies used in nature to promote highly enantioselective chemical reactions. Although enormous efforts have been made to develop simple helical chiral systems and thus open new possibilities in asymmetric catalysis and synthesis, the efficient use of synthetic oligo- and polymeric helical chiral catalysts is still very challenging and rather unusual. In this work, structural unique chiral oligotriazoles have been developed as C–H bond-based anion-binding catalysts for the asymmetric dearomatization of N-heteroarenes. These rotational flexible catalysts adopt a reinforced chiral helical conformation upon binding to a chloride anion, allowing high levels of chirality transfer via a close chiral anion-pair complex with a preformed ionic substrate. This methodology offers a straightforward and potent entry to the synthesis of chiral (bioactive)heterocycles with added synthetic value from simple and abundant heteroarenes.
Co-reporter: Dr. O. Garcia Mancheno
Chemie Ingenieur Technik 2014 Volume 86( Issue 9) pp:1487-1488
Publication Date(Web):
DOI:10.1002/cite.201450657
No abstract is available for this article.
Co-reporter:Andrea Gini, Tobias Brandhofer and Olga García Mancheño
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 6) pp:NaN1312-1312
Publication Date(Web):2017/01/05
DOI:10.1039/C6OB02474B
Over the past few years, the development of oxidative methodologies towards efficient and selective direct Csp3–H bond functionalization processes has attracted tremendous attention from synthetic chemists. However, only a little attention has been given to the key role of the nature of the oxidant. This review aims at providing a brief summary of the recent advances in mild and more benign oxidative Csp3–H bond functionalization reactions, which are classified according to the type of oxidation system employed.



![8-Chloro-1-methyl-2,3,4,5-tetrahydro-1H-benzo[d]azepine hydrochloride](http://img.cochemist.com/ccimg/1431700/1431697-94-7.png)
![8-Chloro-1-methyl-2,3,4,5-tetrahydro-1H-benzo[d]azepine hydrochloride](http://img.cochemist.com/ccimg/1431700/1431697-94-7_b.png)
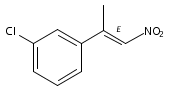
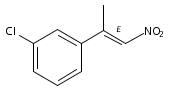














![Silane, (1,1-dimethylethyl)[[(1E)-1-ethoxy-1-propenyl]oxy]dimethyl-](http://img.cochemist.com/ccimg/89100/89043-55-0.png)
![Silane, (1,1-dimethylethyl)[[(1E)-1-ethoxy-1-propenyl]oxy]dimethyl-](http://img.cochemist.com/ccimg/89100/89043-55-0_b.png)


![5H-DIBENZ[B,F]AZEPINE, 5-PHENYL-](http://img.cochemist.com/ccimg/79000/78943-59-6.png)
![5H-DIBENZ[B,F]AZEPINE, 5-PHENYL-](http://img.cochemist.com/ccimg/79000/78943-59-6_b.png)
![5H-Dibenz[b,f]azepine, 5-(phenylmethyl)-](http://img.cochemist.com/ccimg/79000/78943-58-5.png)
![5H-Dibenz[b,f]azepine, 5-(phenylmethyl)-](http://img.cochemist.com/ccimg/79000/78943-58-5_b.png)
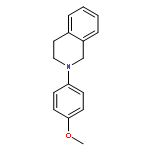
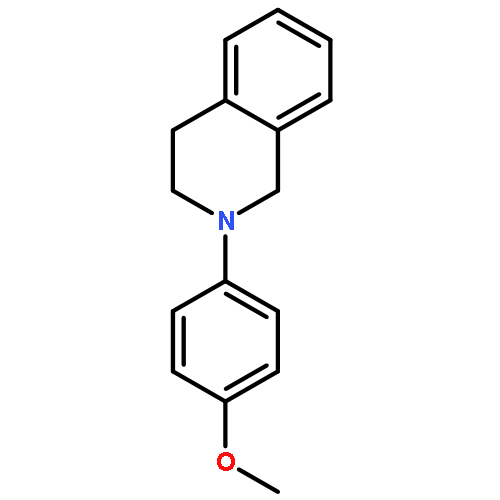


![TERT-BUTYL-DIMETHYL-[1-[(2-METHYLPROPAN-2-YL)OXY]ETHENOXY]SILANE](http://img.cochemist.com/ccimg/74800/74786-02-0.png)
![TERT-BUTYL-DIMETHYL-[1-[(2-METHYLPROPAN-2-YL)OXY]ETHENOXY]SILANE](http://img.cochemist.com/ccimg/74800/74786-02-0_b.png)
![SILANE, (1,1-DIMETHYLETHYL)[[(1Z)-1-ETHOXY-1-PROPENYL]OXY]DIMETHYL-](http://img.cochemist.com/ccimg/74000/73967-98-3.png)
![SILANE, (1,1-DIMETHYLETHYL)[[(1Z)-1-ETHOXY-1-PROPENYL]OXY]DIMETHYL-](http://img.cochemist.com/ccimg/74000/73967-98-3_b.png)


![5-methyl-5H-dibenzo[b,f]azepine](http://img.cochemist.com/ccimg/52300/52249-32-8.png)
![5-methyl-5H-dibenzo[b,f]azepine](http://img.cochemist.com/ccimg/52300/52249-32-8_b.png)
![3-BROMO-N-[(4-CHLOROPHENYL)METHYL]PROPAN-1-AMINE;HYDROBROMIDE](/data/chemimg/59400/40584-09-6.png)
![3-BROMO-N-[(4-CHLOROPHENYL)METHYL]PROPAN-1-AMINE;HYDROBROMIDE](/data/chemimg/59400/40584-09-6_b.png)









![Dibenzo[b,f]thiepin](http://img.cochemist.com/ccimg/300/257-13-6.png)
![Dibenzo[b,f]thiepin](http://img.cochemist.com/ccimg/300/257-13-6_b.png)
![Dibenz[b,f]oxepin](http://img.cochemist.com/ccimg/300/257-05-6.png)
![Dibenz[b,f]oxepin](http://img.cochemist.com/ccimg/300/257-05-6_b.png)







