Co-reporter:Jesus F. Barajas;Gaurav Shakya;Gabriel Moreno;David R. Jackson;Caitlyn L. Topper;Heriberto Rivera, Jr.;Shiou-Chuan Tsai;Anna L. Vagstad;Michael D. Burkart;James J. La Clair
PNAS 2017 Volume 114 (Issue 21 ) pp:E4142-E4148
Publication Date(Web):2017-05-23
DOI:10.1073/pnas.1609001114
Product template (PT) domains from fungal nonreducing polyketide synthases (NR-PKSs) are responsible for controlling the aldol
cyclizations of poly-β-ketone intermediates assembled during the catalytic cycle. Our ability to understand the high regioselective
control that PT domains exert is hindered by the inaccessibility of intrinsically unstable poly-β-ketones for in vitro studies.
We describe here the crystallographic application of “atom replacement” mimetics in which isoxazole rings linked by thioethers
mimic the alternating sites of carbonyls in the poly-β-ketone intermediates. We report the 1.8-Å cocrystal structure of the
PksA PT domain from aflatoxin biosynthesis with a heptaketide mimetic tethered to a stably modified 4′-phosphopantetheine,
which provides important empirical evidence for a previously proposed mechanism of PT-catalyzed cyclization. Key observations
support the proposed deprotonation at C4 of the nascent polyketide by the catalytic His1345 and the role of a protein-coordinated
water network to selectively activate the C9 carbonyl for nucleophilic addition. The importance of the 4′-phosphate at the
distal end of the pantetheine arm is demonstrated to both facilitate delivery of the heptaketide mimetic deep into the PT
active site and anchor one end of this linear array to precisely meter C4 into close proximity to the catalytic His1345. Additional
structural features, docking simulations, and mutational experiments characterize protein–substrate mimic interactions, which
likely play roles in orienting and stabilizing interactions during the native multistep catalytic cycle. These findings afford
a view of a polyketide “atom-replaced” mimetic in a NR-PKS active site that could prove general for other PKS domains.
Co-reporter:Rongfeng Li, Ryan A. Oliver, Craig A. Townsend
Cell Chemical Biology 2017 Volume 24, Issue 1(Volume 24, Issue 1) pp:
Publication Date(Web):19 January 2017
DOI:10.1016/j.chembiol.2016.11.010
•The first monobactam biosynthetic gene cluster is isolated and characterized•Two non-ribosomal peptide synthetases play essential biosynthetic roles•A new mechanism of β-lactam formation is catalyzed in an aberrant thioesterase domainThe monobactams, exemplified by the natural product sulfazecin, are the only class of β-lactam antibiotics not inactivated by metallo-β-lactamases, which confer bacteria with extended-spectrum β-lactam resistance. We screened a transposon mutagenesis library from Pseudomonas acidophila ATCC 31363 and isolated a sulfazecin-deficient mutant that revealed a gene cluster encoding two non-ribosomal peptide synthetases (NRPSs), a methyltransferase, a sulfotransferase, and a dioxygenase. Three modules and an aberrant C-terminal thioesterase (TE) domain are distributed across the two NRPSs. Biochemical examination of the adenylation (A) domains provided evidence that L-2,3-diaminopropionate, not L-serine as previously thought, is the direct source of the β-lactam ring of sulfazecin. ATP/PPi exchange assay also revealed an unusual substrate selectivity shift of one A domain when expressed with or without the immediately upstream condensation domain. Gene inactivation analysis defined a cluster of 13 open reading frames sufficient for sulfazecin production, precursor synthesis, self-resistance, and regulation. The identification of a key intermediate supported a proposed NRPS-mediated mechanism of sulfazecin biosynthesis and β-lactam ring formation distinct from the nocardicins, another NRPS-derived subclass of monocyclic β-lactam. These findings will serve as the basis for further biosynthetic research and potential engineering of these important antibiotics.Download high-res image (139KB)Download full-size image
Co-reporter:Adam G. Newman
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:4219-4228
Publication Date(Web):March 3, 2016
DOI:10.1021/jacs.6b00633
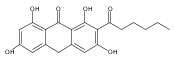
Perylenequinones are a class of photoactivated polyketide mycotoxins produced by fungal plant pathogens that notably produce reactive oxygen species with visible light. The best-studied perylenequinone is cercosporin—a product of the Cercospora species. While the cercosporin biosynthetic gene cluster has been described in the tobacco pathogen Cercospora nicotianae, little is known of the metabolite’s biosynthesis. Furthermore, in vitro investigations of the polyketide synthase central to cercosporin biosynthesis identified the naphthopyrone nor-toralactone as its direct product—an observation in conflict with published biosynthetic proposals. Here, we present an alternative biosynthetic pathway to cercosporin based on metabolites characterized from a series of biosynthetic gene knockouts. We show that nor-toralactone is the key polyketide intermediate and the substrate for the unusual didomain protein CTB3. We demonstrate the unique oxidative cleavage activity of the CTB3 monooxygenase domain in vitro. These data advance our understanding of perylenequinone biosynthesis and expand the biochemical repertoire of flavin-dependent monooxygenases.
Co-reporter:Victor K. Outlaw, Jiawang Zhou, Arthur E. Bragg and Craig A. Townsend
RSC Advances 2016 vol. 6(Issue 66) pp:61249-61253
Publication Date(Web):03 Jun 2016
DOI:10.1039/C6RA10605F
6-Amino-8-cyanobenzo[1,2-b]indolizines, a new class of photoluminescent materials, exhibit reversible pH-dependent optical properties characterized by an uncommon and dramatic blue shift in fluorescence emission when protonated. Acid titration and NMR spectroscopy experiments reveal that, rather than the anticipated N-protonation, C-protonation and loss of aromaticity is responsible for the observed photophysical changes. Efficient synthesis from indole-2-carboxaldehydes makes variously substituted versions of this nucleus readily available to tune optical and pH effects.
Co-reporter:Victor K. Outlaw, Felipe B. d’Andrea, and Craig A. Townsend
Organic Letters 2015 Volume 17(Issue 8) pp:1822-1825
Publication Date(Web):March 27, 2015
DOI:10.1021/ol5036936
An efficient route to substituted N-fused aromatic heterocycles, including indolizines, imidazo[1,2-a]pyridines, and imidazo[1,5-a]pyridines from azole aldehydes, is reported. Wittig olefination of the aldehydes with fumaronitrile and triethylphosphine affords predominantly E-alkenes that undergo rapid cyclization upon treatment with a mild base. Substituent control of the 1-, 2-, and 3-positions of the resulting heteroaromatic bicycles is shown. Alternatively, the isolable E-alkene undergoes selective alkylation with electrophiles, followed by in situ annulation to indolizines additionally substituted at the 6-position.
Co-reporter:Callie R. Huitt-Roehl, Eric A. Hill, Martina M. Adams, Anna L. Vagstad, Jesse W. Li, and Craig A. Townsend
ACS Chemical Biology 2015 Volume 10(Issue 6) pp:1443
Publication Date(Web):February 25, 2015
DOI:10.1021/acschembio.5b00005
Nonreducing polyketide synthases (NR-PKSs) are unique among PKSs in their domain structure, notably including a starter unit:acyl-carrier protein (ACP) transacylase (SAT) domain that selects an acyl group as the primer for biosynthesis, most commonly acetyl-CoA from central metabolism. This clan of mega-enzymes resembles fatty acid synthases (FASs) by sharing both their central chain elongation steps and their capacity for iterative catalysis. In this mode of synthesis, catalytic domains involved in chain extension exhibit substrate plasticity to accommodate growing chains as small as two carbons to 20 or more. PksA is the NR-PKS central to the biosynthesis of the mycotoxin aflatoxin B1 whose SAT domain accepts an unusual hexanoyl starter from a dedicated yeast-like FAS. Explored in this paper is the ability of PksA to utilize a selection of potential starter units as substrates to initiate and sustain extension and cyclization to on-target, programmed polyketide synthesis. Most of these starter units were successfully accepted and properly processed by PksA to achieve biosynthesis of the predicted naphthopyrone product. Analysis of the on-target and derailment products revealed trends of tolerance by individual PksA domains to alternative starter units. In addition, natural and un-natural variants of the active site cysteine were examined and found to be capable of biosynthesis, suggesting possible direct loading of starter units onto the β-ketoacyl synthase (KS) domain. In light of the data assembled here, the predictable synthesis of unnatural products by NR-PKSs is more fully defined.
Co-reporter:Daniel R. Marous;Evan P. Lloyd;Anthony J. Blaszczyk;Squire J. Booker;Tyler L. Grove;Kristos A. Moshos;Andrew R. Buller
PNAS 2015 Volume 112 (Issue 33 ) pp:10354-10358
Publication Date(Web):2015-08-18
DOI:10.1073/pnas.1508615112
Despite their broad anti-infective utility, the biosynthesis of the paradigm carbapenem antibiotic, thienamycin, remains largely
unknown. Apart from the first two steps shared with a simple carbapenem, the pathway sharply diverges to the more structurally
complex members of this class of β-lactam antibiotics, such as thienamycin. Existing evidence points to three putative cobalamin-dependent
radical S-adenosylmethionine (RS) enzymes, ThnK, ThnL, and ThnP, as potentially being responsible for assembly of the ethyl side chain
at C6, bridgehead epimerization at C5, installation of the C2-thioether side chain, and C2/3 desaturation. The C2 substituent
has been demonstrated to be derived by stepwise truncation of CoA, but the timing of these events with respect to C2–S bond
formation is not known. We show that ThnK of the three apparent cobalamin-dependent RS enzymes performs sequential methylations
to build out the C6-ethyl side chain in a stereocontrolled manner. This enzymatic reaction was found to produce expected RS
methylase coproducts S-adenosylhomocysteine and 5′-deoxyadenosine, and to require cobalamin. For double methylation to occur, the carbapenam substrate
must bear a CoA-derived C2-thioether side chain, implying the activity of a previous sulfur insertion by an as-yet unidentified
enzyme. These insights allow refinement of the central steps in complex carbapenem biosynthesis.
Co-reporter:Adam G. Newman ; Anna L. Vagstad ; Philip A. Storm
Journal of the American Chemical Society 2014 Volume 136(Issue 20) pp:7348-7362
Publication Date(Web):April 25, 2014
DOI:10.1021/ja5007299
Iterative, nonreducing polyketide synthases (NR-PKSs) are multidomain enzymes responsible for the construction of the core architecture of aromatic polyketide natural products in fungi. Engineering these enzymes for the production of non-native metabolites has been a long-standing goal. We conducted a systematic survey of in vitro “domain swapped” NR-PKSs using an enzyme deconstruction approach. The NR-PKSs were dissected into mono- to multidomain fragments and recombined as noncognate pairs in vitro, reconstituting enzymatic activity. The enzymes used in this study produce aromatic polyketides that are representative of the four main chemical features set by the individual NR-PKS: starter unit selection, chain-length control, cyclization register control, and product release mechanism. We found that boundary conditions limit successful chemistry, which are dependent on a set of underlying enzymatic mechanisms. Crucial for successful redirection of catalysis, the rate of productive chemistry must outpace the rate of spontaneous derailment and thioesterase-mediated editing. Additionally, all of the domains in a noncognate system must interact efficiently if chemical redirection is to proceed. These observations refine and further substantiate current understanding of the mechanisms governing NR-PKS catalysis.
Co-reporter:Craig A. Townsend
Natural Product Reports 2014 vol. 31(Issue 10) pp:1260-1265
Publication Date(Web):31 Jul 2014
DOI:10.1039/C4NP00092G
In this viewpoint highlights are drawn from a deep analysis of the multifaceted problem of aflatoxin biosynthesis, one of the most highly rearranged polyketide natural products known. Fundamental chemical insights have emerged into how cytochrome P450-mediated skeletal rearrangements occur through probable cationic intermediates and oxidative dearomatizations, which are applicable more widely in natural product catabolism. So to where current experimental methods have failed in our hands, bioinformatic tools and fresh experimental strategies have been developed to identify linker regions in large, polydomain proteins and guide the dissection and reassembly of their component parts. It has been possible to deduce individual catalytic roles, how overall synthesis is coordinated and how these enzymes can be re-engineered in a rational manner to prepare non-natural products. These insights and innovations were often not planned or anticipated, but sprung from the inability to answer fundamental questions. Advances in science can take place by chance favoring the prepared mind, other times by refusing to give up and devising new solutions to address hard questions. Both ways forward played important roles in the investigation of aflatoxin biosynthesis. For these contributions I am pleased to share this special issue of NPR with John Vederas and Tom Simpson, who have been leaders in this field for the last third of a century.
Co-reporter:Victor K. Outlaw and Craig A. Townsend
Organic Letters 2014 Volume 16(Issue 24) pp:6334-6337
Publication Date(Web):December 5, 2014
DOI:10.1021/ol503078h
Among privileged structures, indoles occupy a central place in medicinal chemistry and alkaloid research. Here we report a flexible and efficient conversion of pyrrole-3-carboxaldehydes to substituted 7-amino-5-cyanoindoles. Phosphine addition to fumaronitrile proceeds with prototropic rearrangement of the initially formed zwitterion to the thermodynamically favored phosphonium ylide, which is poised for in situ Wittig olefination. The predominantly E-alkene product positions the allylic nitrile for facile intramolecular Hoeben–Hoesch reaction in the presence of BF3·OEt2. Syntheses of 2,5- and 3,5-disubstituted 7-aminoindoles are illustrated. Additionally, dianion alkylation of the allylic nitrile is demonstrated to furnish, after cyclization, 5,6-disubstituted 7-aminoindoles to further exemplify this scalable and high-yielding method.
Co-reporter:Andrew R. Buller, Michael F. Freeman, Joel F. Schildbach, and Craig A. Townsend
Biochemistry 2014 Volume 53(Issue 26) pp:4273-4281
Publication Date(Web):June 16, 2014
DOI:10.1021/bi500385d
In the past decade, there have been major achievements in understanding the relationship between enzyme catalysis and protein structural plasticity. In autoprocessing systems, however, there is a sparsity of direct evidence of the role of conformational dynamics, which are complicated by their intrinsic chemical reactivity. ThnT is an autoproteolytically activated enzyme involved in the biosynthesis of the β-lactam antibiotic thienamycin. Conservative mutation of ThnT results in multiple conformational states that can be observed via X-ray crystallography, establishing ThnT as a representative and revealing system for studing how conformational dynamics control autoactivation at a molecular level. Removal of the nucleophile by mutation to Ala disrupts the population of a reactive state and causes widespread structural changes from a conformation that promotes autoproteolysis to one associated with substrate catalysis. Finer probing of the active site polysterism was achieved by EtHg derivatization of the nucleophile, which indicates the active site and a neighboring loop have coupled dynamics. Disruption of these interactions by mutagenesis precludes the ability to observe a reactive state through X-ray crystallography, and application of this insight to other autoproteolytically activated enzymes offers an explanation for the widespread crystallization of inactive states. We suggest that the N → O(S) acyl shift in cis-autoproteolysis might occur through a si-face attack, thereby unifying the fundamental chemistry of these enzymes through a common mechanism.
Co-reporter:Victor K. Outlaw, Edward A. Wydysh, Aravinda Vadlamudi, Susan M. Medghalchi and Craig A. Townsend
MedChemComm 2014 vol. 5(Issue 6) pp:826-830
Publication Date(Web):06 May 2014
DOI:10.1039/C4MD00126E
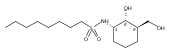
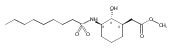
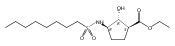
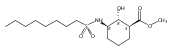
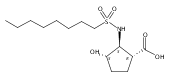
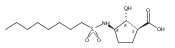
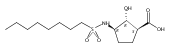
Despite a rising demand for anti-obesity therapeutics, few effective pharmacological options are clinically available that target the synthesis and accumulation of body fat. Moderate inhibition of mammalian glycerol-3-phosphate acyltransferase (GPAT) with 2-(alkanesulfonamido)benzoic acids has recently been described in vitro, accompanied by promising weight loss in vivo. In silico docking studies with 2-(octanesulfonamido)benzoic acid modeled into the active site of squash GPAT revealed an unoccupied volume lined with hydrophobic residues proximal to C-4 and C-5 of the benzoic acid ring. In an effort to produce more potent GPAT inhibitors, a series of 4- and 5-substituted analogs were designed, synthesized, and evaluated for inhibitory activity. In general, compounds containing hydrophobic substituents at the 4- and 5-positions, such as biphenyl and alkylphenyl hydrocarbons, exhibited an improved inhibitory activity against GPAT in vitro. The most active compound, 4-([1,1′-biphenyl]-4-carbonyl)-2-(octanesulfonamido)benzoic acid, demonstrated an IC50 of 8.5 μM and represents the best GPAT inhibitor discovered to date. Conversely, further substitution with hydroxyl or fluoro groups, led to a 3-fold decrease in activity. These results are consistent with the presence of a hydrophobic pocket and may support the binding model as a potential tool for developing more potent inhibitors.
Co-reporter:Dr. Rongfeng Li;Evan P. Lloyd;Dr. Kristos A. Moshos; Craig A. Townsend
ChemBioChem 2014 Volume 15( Issue 2) pp:320-331
Publication Date(Web):
DOI:10.1002/cbic.201300319
Abstract
Nearly 50 naturally occurring carbapenem β-lactam antibiotics, most produced by Streptomyces, have been identified. The structural diversity of these compounds is limited to variance of the C-2 and C-6 side chains as well as the stereochemistry at C-5/C-6. These structural motifs are of interest both for their antibiotic effects and their biosynthesis. Although the thienamycin gene cluster is the only active gene cluster publically available in this group, more comparative information is needed to understand the genetic basis of these structural differences. We report here the identification of MM 4550, a member of the olivanic acids, as the major carbapenem produced by Streptomyces argenteolus ATCC 11009. Its gene cluster was also identified by degenerate PCR and targeted gene inactivation. Sequence analysis revealed that the genes encoding the biosynthesis of the bicyclic core and the C-6 and C-2 side chains are well conserved in the MM 4550 and thienamycin gene clusters. Three new genes, cmmSu, cmm17 and cmmPah were found in the new cluster, and their putative functions in the sulfonation and epimerization of MM 4550 are proposed. Gene inactivation showed that, in addition to cmmI, two new genes, cmm22 and -23, encode a two-component response system thought to regulate the production of MM 4550. Overexpression of cmmI, cmm22 and cmm23 promoted MM 4550 production in an engineered strain. Finally, the involvement and putative roles of all genes in the MM 4550 cluster are proposed based on the results of bioinformatics analysis, gene inactivation, and analysis of disruption mutants. Overall, the differences between the thienamycin and MM 4550 gene clusters are reflected in characteristic structural elements and provide new insights into the biosynthesis of the complex carbapenems.
Co-reporter:Jason W. Labonte and Craig A. Townsend
Chemical Reviews 2013 Volume 113(Issue 3) pp:2182
Publication Date(Web):December 3, 2012
DOI:10.1021/cr300169a
Co-reporter:Jeanne M. Davidsen ; David M. Bartley
Journal of the American Chemical Society 2013 Volume 135(Issue 5) pp:1749-1759
Publication Date(Web):January 18, 2013
DOI:10.1021/ja307710d
Nocardicin A is a monocyclic β-lactam isolated from the actinomycete Nocardia uniformis that shows moderate antibiotic activity against a broad spectrum of Gram-negative bacteria. The monobactams are of renewed interest due to emerging Gram-negative strains resistant to clinically available penicillins and cephalosporins. Like isopenicillin N, nocardicin A has a tripeptide core of non-ribosomal origin. Paradoxically, the nocardicin A gene cluster encodes two non-ribosomal peptide synthetases (NRPSs), NocA and NocB, predicted to encode five modules pointing to a pentapeptide precursor in nocardicin A biosynthesis, unless module skipping or other nonlinear reactions are occurring. Previous radiochemical incorporation experiments and bioinformatic analyses predict the incorporation of p-hydroxy-l-phenylglycine (l-pHPG) into positions 1, 3, and 5 and l-serine into position 4. No prediction could be made for position 2. Multidomain constructs of each module were heterologous expressed in Escherichia coli for determination of the adenylation domain (A-domain) substrate specificity using the ATP/PPi exchange assay. Three of the five A-domains, from modules 1, 2, and 4, required the addition of stoichiometric amounts of MbtH family protein NocI to detect exchange activity. On the basis of these analyses, the predicted product of the NocA and NocB NRPSs is l-pHPG–l-Arg–d-pHPG–l-Ser–l-pHPG, a pentapeptide. Despite being flanked by non-proteinogenic amino acids, proteolysis of this pentapeptide by trypsin yields two fragments from cleavage at the C terminus of the l-Arg residue. Thus, a proteolytic step is likely involved in the biosynthesis of nocardicin A, a rare but precedented editing event in the formation of non-ribosomal natural products that is supported by the identification of trypsin-encoding genes in N. uniformis.
Co-reporter:Ryan M. Phelan
Journal of the American Chemical Society 2013 Volume 135(Issue 20) pp:7496-7502
Publication Date(Web):April 23, 2013
DOI:10.1021/ja311078s
The carbapenem class of β-lactam antibiotics is known for its remarkable potency, antibacterial spectrum, and resistance to β-lactamase-mediated inactivation. While the biosynthesis of structurally “complex” carbapenems, such as thienamycin, share initial biochemical steps with carbapenem-3-carboxylate (“simple” carbapenem), the requisite inversion at C5 and formation of the characteristic α,β-unsaturated carboxylate are different in origin between the two groups. Here, we consider carbapenem synthase, a mechanistically distinct bifunctional non-heme iron α-ketoglutarate-dependent enzyme responsible for the terminal reactions, C5 epimerization and desaturation, in simple carbapenem production. Interestingly, this enzyme accepts two stereoisomeric substrates and transforms each to a common active antibiotic. Owing both to enzyme and product instability, resorting to saturation mutagenesis of active site and selected second-sphere residues gave clearly differing profiles of CarC tolerance to structural modification. Guided by a crystal structure and the mutational data, in silico docking was used to suggest the positioning of each disastereomeric substrate in the active site. The two orientations relative to the reactive iron-oxo center are manifest in the two distinct reactions, C5-epimerization and C2/3-desaturation. These observations favor a two-step reaction scheme involving two complete oxidative cycles as opposed to a single catalytic cycle in which an active site tyrosine, Tyr67, after hydrogen donation to achieve bicyclic ring inversion, is further hypothesized to serve as a radical carrier.
Co-reporter:Katherine Belecki
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14339-14348
Publication Date(Web):September 17, 2013
DOI:10.1021/ja406697t
Despite considerable interest in the enediyne family of antitumor antibiotics, assembly of their polyketide core structures in nature remains poorly understood. Discriminating methods to access enzyme-bound intermediates are critical for elucidating unresolved polyketide and nonribosomal peptide biosynthetic pathways. Here, we describe the development of broadly applicable techniques for the mild chemical release and analysis of intermediates bound to carrier proteins (CPs), providing access to these species even in sensitive systems. These techniques were applied to CalE8, the polyketide synthase (PKS) involved in calicheamicin biosynthesis, facilitating the unambiguous identification of enzyme-bound polyketides on an enediyne PKS. Moreover, these methods enabled the preparation of fully unloaded CalE8, providing a “clean slate” for reconstituted activity and allowing us to demonstrate the preferential accumulation of a PKS-bound octaketide with evidence of programmed processing control by CalE8. This intermediate, which has the expected chain length for enediyne core construction, could previously only be indirectly inferred. These studies prove that this polyketide is an authentic product of CalE8 and may be a key precursor to the enediyne core of calicheamicin, as it is the only programmed, enzyme-bound species observed for any enediyne system to date. Our experimental advances into a generally inaccessible system illustrate the utility of these techniques for investigating CP-based biosynthetic pathways.
Co-reporter:Nicole M. Gaudelli and Craig A. Townsend
The Journal of Organic Chemistry 2013 Volume 78(Issue 13) pp:6412-6426
Publication Date(Web):June 12, 2013
DOI:10.1021/jo4007893
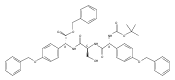
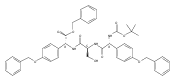
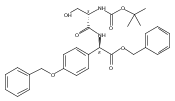
Methods have been developed to synthesize tri- and pentapeptide thioesters containing one or more p-(hydroxyphenyl)glycine (pHPG) residues and l-serine, some where the latter is O-phosphorylated, O-acetylated, or exists as a β-lactam. Selection of orthogonal protection strategies and development of conditions to achieve seryl O-phosphorylation without β-elimination and to maintain stereochemical control, especially simultaneously at exceptionally base-labile pHPG α-carbons, are described. Intramolecular closure of a seryl peptide to a β-lactam-containing peptide and the syntheses of corresponding thioester analogues are also reported. Modification of classical Mitsunobu conditions is described in the synthesis of the β-lactam-containing products, and in a broadly useful observation, it was found that simple exclusion of light from the P(OEt)3-mediated Mitsunobu ring closure afforded yields of >95%, presumably owing to reduced photodegradation of the azodicarboxylate used. These sensitive potential substrates and products will be used in mechanistic studies of the two nonribosomal peptide synthetases NocA and NocB that lie at the heart of nocardicin biosynthesis, a family of monocyclic β-lactam antibiotics.
Co-reporter:Andrew R. Buller
PNAS 2013 Volume 110 (Issue 8 ) pp:E653-E661
Publication Date(Web):2013-02-19
DOI:10.1073/pnas.1221050110
The study of proteolysis lies at the heart of our understanding of biocatalysis, enzyme evolution, and drug development. To
understand the degree of natural variation in protease active sites, we systematically evaluated simple active site features
from all serine, cysteine and threonine proteases of independent lineage. This convergent evolutionary analysis revealed several
interrelated and previously unrecognized relationships. The reactive rotamer of the nucleophile determines which neighboring
amide can be used in the local oxyanion hole. Each rotamer–oxyanion hole combination limits the location of the moiety facilitating
proton transfer and, combined together, fixes the stereochemistry of catalysis. All proteases that use an acyl-enzyme mechanism
naturally divide into two classes according to which face of the peptide substrate is attacked during catalysis. We show that
each class is subject to unique structural constraints that have governed the convergent evolution of enzyme structure. Using
this framework, we show that the γ-methyl of Thr causes an intrinsic steric clash that precludes its use as the nucleophile
in the traditional catalytic triad. This constraint is released upon autoproteolysis and we propose a molecular basis for
the increased enzymatic efficiency introduced by the γ-methyl of Thr. Finally, we identify several classes of natural products
whose mode of action is sensitive to the division according to the face of attack identified here. This analysis of protease
structure and function unifies 50 y of biocatalysis research, providing a framework for the continued study of enzyme evolution
and the development of inhibitors with increased selectivity.
Co-reporter:Dr. Anna L. Vagstad;Adam G. Newman;Philip A. Storm;Katherine Belecki; Jason M. Crawford; Craig A. Townsend
Angewandte Chemie 2013 Volume 125( Issue 6) pp:1762-1765
Publication Date(Web):
DOI:10.1002/ange.201208550
Co-reporter:Dr. Anna L. Vagstad;Adam G. Newman;Philip A. Storm;Katherine Belecki; Jason M. Crawford; Craig A. Townsend
Angewandte Chemie International Edition 2013 Volume 52( Issue 6) pp:1718-1721
Publication Date(Web):
DOI:10.1002/anie.201208550
Co-reporter:Anna L. Vagstad ; Stefanie B. Bumpus ; Katherine Belecki ; Neil L. Kelleher
Journal of the American Chemical Society 2012 Volume 134(Issue 15) pp:6865-6877
Publication Date(Web):March 27, 2012
DOI:10.1021/ja3016389
Nonreducing iterative polyketide synthases (NR-PKSs) are responsible for assembling the core of fungal aromatic natural products with diverse biological properties. Despite recent advances in the field, many mechanistic details of polyketide assembly by these megasynthases remain unknown. To expand our understanding of substrate loading, polyketide elongation, cyclization, and product release, active site occupancy and product output were explored by Fourier transform mass spectrometry using the norsolorinic acid anthrone-producing polyketide synthase, PksA, from the aflatoxin biosynthetic pathway in Aspergillus parasiticus. Here we report the simultaneous observation of covalent intermediates from all catalytic domains of PksA from in vitro reconstitution reactions. The data provide snapshots of iterative catalysis and reveal an underappreciated editing function for the C-terminal thioesterase domain beyond its recently established synthetic role in Claisen/Dieckmann cyclization and product release. The specificity of thioesterase catalyzed hydrolysis was explored using biosynthetically relevant protein-bound and small molecule acyl substrates and demonstrated activity against hexanoyl and acetyl, but not malonyl. Processivity of polyketide extension was supported by the inability of a nonhydrolyzable malonyl analog to trap products of intermediate chain lengths and by the detection of only fully extended species observed covalently bound to, and as the predominant products released by, PksA. High occupancy of the malonyl transacylase domain and fast relative rate of malonyl transfer compared to starter unit transfer indicate that rapid loading of extension units onto the carrier domain facilitates efficient chain extension in a manner kinetically favorable to ultimate product formation.
Co-reporter:Adam G. Newman, Anna L. Vagstad, Katherine Belecki, Jonathan R. Scheerer and Craig A. Townsend
Chemical Communications 2012 vol. 48(Issue 96) pp:11772-11774
Publication Date(Web):29 Oct 2012
DOI:10.1039/C2CC36010A
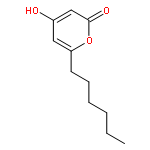
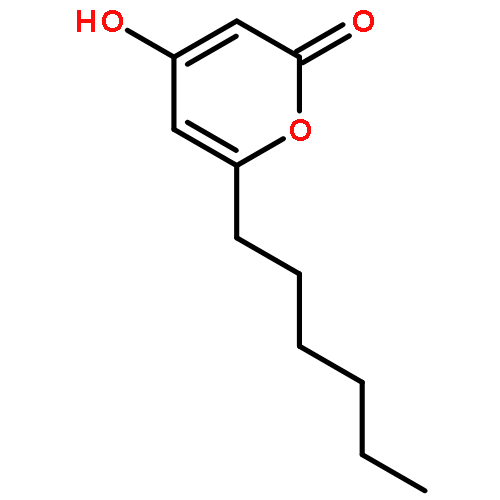
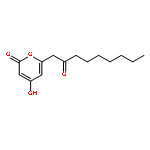
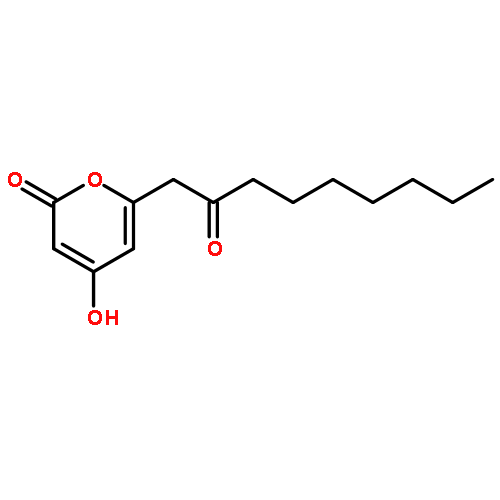
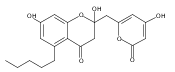
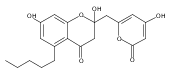
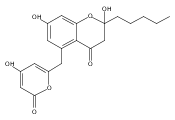
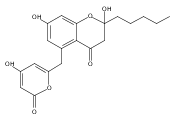
The polyketide synthase CTB1 is demonstrated to catalyze pyrone formation thereby expanding the known biosynthetic repertoire of thioesterase domains in iterative, non-reducing polyketide synthases.
Co-reporter:Jeanne M. Davidsen, Craig A. Townsend
Chemistry & Biology 2012 Volume 19(Issue 2) pp:297-306
Publication Date(Web):24 February 2012
DOI:10.1016/j.chembiol.2011.10.020
Two nonribosomal peptide synthetases (NRPS), NocA and NocB, together comprising five modules, are essential for the biosynthesis of the D,L,D configured tripeptide backbone of the monocyclic β-lactam nocardicin A. We report a double replacement gene strategy in which point mutations were engineered in the two encoding NRPS genes without disruption of the nocABC operon by placing selective markers in adjacent genes. A series of mutants was constructed to inactivate the thiolation (T) domain of each module and to evaluate an HHxxxDR catalytic motif in NocA and an atypical extended histidine motif in NocB. The loss of nocardicin A production in each of the T domain mutants indicates that all five modules are essential for its biosynthesis. Conversely, production of nocardicin A was not affected by mutation of the NocB histidine motif or the R828G mutation in NocA.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (302 K)Download as PowerPoint slideHighlights► In vivo double replacement gene strategy for preparation of markerless mutants ► Inactivation of T domain of each module and in vivo evaluation of atypical motifs ► All five NRPS modules required for biosynthesis of tripeptide core of nocardicin A
Co-reporter:Anna L. Vagstad, Eric A. Hill, Jason W. Labonte, Craig A. Townsend
Chemistry & Biology 2012 Volume 19(Issue 12) pp:1525-1534
Publication Date(Web):21 December 2012
DOI:10.1016/j.chembiol.2012.10.002
Melanins are a broad class of darkly pigmented macromolecules formed by oxidative polymerization of phenolic monomers. In fungi, melanins are known virulence factors that contribute to pathogenicity. Their biosynthesis generally involves polymerization of 1,8-dihydroxynaphthalene via a 1,3,6,8-tetrahydroxynaphthalene (THN) precursor assembled by multidomain, nonreducing polyketide synthases. Convergent routes to THN have evolved in fungi. Parallel heptaketide and hexaketide pathways exist that utilize conventional C-terminal thioesterase/Claisen cyclase domains and separate side-chain deacylases. Here, in vitro characterization of Pks1 from Colletotrichum lagenarium establishes a true THN synthase with a bifunctional thioesterase (TE) catalyzing both cyclization and deacetylation of an enzyme-bound hexaketide substrate. Chimeric TE domains were generated by swapping lid regions of active sites between classes of melanin TEs to gain insight into this unprecedented catalysis of carbon–carbon bond making and breaking by an α/β-hydrolase fold enzyme.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (211 K)Download as PowerPoint slideHighlights► Revised C. lagenarium Pks1 coding sequence enabling in vitro analysis ► Clarified third fungal route to dihydroxynaphthalene melanin ► Revealed thioesterase-catalyzed tandem Claisen cyclization and deacetylation ► Lid swap chimera illuminate features necessary for C-C bond making and breaking
Co-reporter:Ryan M. Phelan, Benjamin J. DiPardo, and Craig A. Townsend
ACS Chemical Biology 2012 Volume 7(Issue 5) pp:835
Publication Date(Web):March 19, 2012
DOI:10.1021/cb200504g
High-throughput screens and selections have had profound impact on our ability to engineer proteins possessing new, desired properties. These methods are especially useful when applied to the modification of existing enzymes to create natural and unnatural products. In an advance upon existing methods we developed a high-throughput, genetically regulated screen for the in vivo production of β-lactam antibiotics using a green fluorescent protein (gfp) reporter. This assay proved reliable and sensitive and presents a dynamic range under which a wide array of β-lactam architectural subclasses can be detected. Moreover, the graded response elicited in this assay can be used to rank mutant activity. The utility of this development was demonstrated in vivo and then applied to the first experimental investigation of a putative catalytic residue in carbapenem synthase (CarC). Information gained about the mutability of this residue defines one parameter for enzymatic activity and sets boundaries for future mechanistic and engineering efforts.
Co-reporter:Jason W. Labonte, Fumitaka Kudo, Michael F. Freeman, Mary L. Raber and Craig A. Townsend
MedChemComm 2012 vol. 3(Issue 8) pp:960-966
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2MD00305H
The 2-azetidinone ring of the Class A and D β-lactamase inhibitor clavulanic acid (1) is synthesized by the ATP-utilizing enzyme β-lactam synthetase (βLS). A hydroxyethyl group attached to C-6 of 1 in the (S) configuration markedly enhances the efficacy of this compound against Class C β-lactamases. Guided by a series of X-ray structures of βLS, we have engineered this enzyme to act upon a methylated substrate analogue to give selectively the (3S)-methyl β-lactam core, which, upon closure of the second ring of the bicyclic system of 1, would lead to the (6S)-methylated clavulanic acid derivative.
Co-reporter:Dr. Jennifer Foulke-Abel ; Dr. Craig A. Townsend
ChemBioChem 2012 Volume 13( Issue 13) pp:1880-1884
Publication Date(Web):
DOI:10.1002/cbic.201200267
Co-reporter:Michael F. Freeman;Andrew R. Buller;Joel F. Schildbach;Nathan T. Wright
PNAS 2012 Volume 109 (Issue 7 ) pp:
Publication Date(Web):2012-02-14
DOI:10.1073/pnas.1113633109
ThnT is a pantetheine hydrolase from the DmpA/OAT superfamily involved in the biosynthesis of the β-lactam antibiotic thienamycin.
We performed a structural and mechanistic investigation into the cis-autoproteolytic activation of ThnT, a process that has
not previously been subject to analysis within this superfamily of enzymes. Removal of the γ-methyl of the threonine nucleophile
resulted in a rate deceleration that we attribute to a reduction in the population of the reactive rotamer. This phenomenon
is broadly applicable and constitutes a rationale for the evolutionary selection of threonine nucleophiles in autoproteolytic
systems. Conservative substitution of the nucleophile (T282C) allowed determination of a 1.6-Å proenzyme ThnT crystal structure,
which revealed a level of structural flexibility not previously observed within an autoprocessing active site. We assigned
the major conformer as a nonreactive state that is unable to populate a reactive rotamer. Our analysis shows the system is
activated by a structural rearrangement that places the scissile amide into an oxyanion hole and forces the nucleophilic residue
into a forbidden region of Ramachandran space. We propose that conformational strain may drive autoprocessing through the
destabilization of nonproductive states. Comparison of our data with previous reports uncovered evidence that many inactivated
structures display nonreactive conformations. For penicillin and cephalosporin acylases, this discrepancy between structure
and function may be resolved by invoking the presence of a hidden conformational state, similar to that reported here for
ThnT.
Co-reporter:Katherine Belecki ;Dr. Craig A. Townsend
Angewandte Chemie International Edition 2012 Volume 51( Issue 45) pp:11316-11319
Publication Date(Web):
DOI:10.1002/anie.201206462
Co-reporter:Katherine Belecki ;Dr. Craig A. Townsend
Angewandte Chemie 2012 Volume 124( Issue 45) pp:11478-11481
Publication Date(Web):
DOI:10.1002/ange.201206462
Co-reporter:Dr. Micah J. Bodner;Dr. Rongfeng Li;Ryan M. Phelan;Dr. Michael F. Freeman;Dr. Kristos A. Moshos;Evan P. Lloyd ; Dr. Craig A. Townsend
ChemBioChem 2011 Volume 12( Issue 14) pp:2159-2165
Publication Date(Web):
DOI:10.1002/cbic.201100366
Abstract
Approximately 50 naturally occurring carbapenem β-lactam antibiotics are known. All but one of these have been isolated from Streptomyces species and are disubstituted structural variants of a simple core that is synthesized by Pectobacterium carotovorum (Erwinia carotovora), a phylogenetically distant plant pathogen. While the biosynthesis of the simple carbapenem, (5R)-carbapen-2-em-3-carboxylic acid, is impressively efficient requiring only three enzymes, CarA, CarB and CarC, the formation of thienamycin, one of the former group of metabolites from Streptomyces, is markedly more complex. Despite their phylogenetic separation, bioinformatic analysis of the encoding gene clusters suggests that the two pathways could be related. Here we demonstrate with gene swapping, stereochemical and kinetics experiments that CarB and CarA and their S. cattleya orthologues, ThnE and ThnM, respectively, are functionally and stereochemically equivalent, although their catalytic efficiencies differ. The biosynthetic pathways, therefore, to thienamycin, and likely to the other disubstituted carbapenems, and to the simplest carbapenem, (5R)-carbapen-2-em-3-carboxylic acid, are initiated in the same manner, but share only two common steps before diverging.
Co-reporter: Dr. Craig A. Townsend
ChemBioChem 2011 Volume 12( Issue 15) pp:2267-2269
Publication Date(Web):
DOI:10.1002/cbic.201100431
Co-reporter:Jason M. Crawford
&
Craig A. Townsend
Nature Reviews Microbiology 2010 8(12) pp:879
Publication Date(Web):2010-12-16
DOI:10.1038/nrmicro2465
Fungal aromatic polyketides constitute a large family of bioactive natural products and are synthesized by the non-reducing group of iterative polyketide synthases (PKSs). Their diverse structures arise from selective enzymatic modifications of reactive, enzyme-bound poly-β-keto intermediates. How iterative PKSs control starter unit selection, polyketide chain initiation and elongation, intermediate folding and cyclization, selective redox or modification reactions during assembly, and product release are central mechanistic questions underlying iterative catalysis. This Review highlights recent insights into these questions, with a particular focus on the biosynthetic programming of fungal aromatic polyketides, and draws comparisons with the allied biosynthetic processes in bacteria.
Co-reporter:Edward A. Wydysh, Aravinda Vadlamudi, Susan M. Medghalchi, Craig A. Townsend
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 17) pp:6470-6479
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmc.2010.06.091
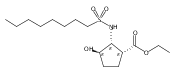
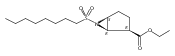
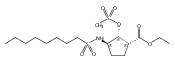
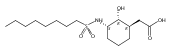
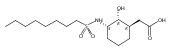
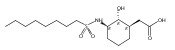
Glycerol 3-phosphate acyltransferase (GPAT) isozymes are central control points for fat synthesis in mammals. Development of inhibitors of these membrane-bound enzymes could lead to an effective treatment for obesity, but is thwarted by an absence of direct structural information. Based on a highly successful study involving conformationally constrained glycerol 3-phosphate analogs functioning as potent glycerol 3-phosphate dehydrogenase inhibitors, several series of cyclic bisubstrate and transition state analogs were designed, synthesized, and tested as GPAT inhibitors. The weaker in vitro inhibitory activity of these compounds compared to a previously described benzoic acid series was then examined in docking experiments with the soluble squash chloroplast GPAT crystal structure. These in silico experiments indicate that cyclopentyl and cyclohexyl scaffolds prepared in this study may be occluded from the enzyme active site by two protein loops that sterically guard the phosphate binding region. In view of these findings, future GPAT inhibitor design will be driven toward compounds based on planar frameworks able to slide between these loops and enter the active site, resulting in improved inhibitory activity.
Co-reporter:Tyler Paz Korman;Jason M. Crawford;Jason W. Labonte;Adam G. Newman;Justin Wong;Shiou-Chuan Tsai;
Proceedings of the National Academy of Sciences 2010 107(14) pp:6246-6251
Publication Date(Web):March 23, 2010
DOI:10.1073/pnas.0913531107
Polyketide natural products possess diverse architectures and biological functions and share a subset of biosynthetic steps
with fatty acid synthesis. The final transformation catalyzed by both polyketide synthases (PKSs) and fatty acid synthases
is most often carried out by a thioesterase (TE). The synthetic versatility of TE domains in fungal nonreducing, iterative
PKSs (NR-PKSs) has been shown to extend to Claisen cyclase (CLC) chemistry by catalyzing C–C ring closure reactions as opposed
to thioester hydrolysis or O–C/N–C macrocyclization observed in previously reported TE structures. Catalysis of C–C bond formation
as a product release mechanism dramatically expands the synthetic potential of PKSs, but how this activity was acquired has
remained a mystery. We report the biochemical and structural analyses of the TE/CLC domain in polyketide synthase A, the multidomain
PKS central to the biosynthesis of aflatoxin B1, a potent environmental carcinogen. Mutagenesis experiments confirm the predicted identity of the catalytic triad and its
role in catalyzing the final Claisen-type cyclization to the aflatoxin precursor, norsolorinic acid anthrone. The 1.7 Å crystal
structure displays an α/β-hydrolase fold in the catalytic closed form with a distinct hydrophobic substrate-binding chamber.
We propose that a key rotation of the substrate side chain coupled to a protein conformational change from the open to closed
form spatially governs substrate positioning and C–C cyclization. The biochemical studies, the 1.7 Å crystal structure of
the TE/CLC domain, and intermediate modeling afford the first mechanistic insights into this widely distributed C–C bond-forming
class of TEs.
Co-reporter:Katherine Belecki ; Jason M. Crawford
Journal of the American Chemical Society 2009 Volume 131(Issue 35) pp:12564-12566
Publication Date(Web):August 18, 2009
DOI:10.1021/ja904391r
Enediyne antibiotics are categorized according to the presence of either a 9- or 10-membered ring within their polyketide-derived core structures. Recent literature reports have favored the notion that biosynthetic divergence of the two structural families is determined by the enediyne polyketide synthases (PKSs) alone. We now disclose the simultaneous in vitro production of three octaketide polyenes by biosynthetic enzymes for the 10-membered enediyne calicheamicin γ1I, including the elusive β-keto acid precursor to a previously described C15 methyl hexaenone. Alongside these two polyene products, we have additionally detected a hydrocarbon heptaene previously isolated only from 9-membered enediyne systems. The discovery of the heptaene in the calicheamicin system promotes a more convergent model for the early steps of enediyne biosynthesis. Furthermore, the synthesis of this set of octaketides by the enediyne PKS CalE8 and thioesterase CalE7 suggests, in contrast to recent biosynthetic proposals, that accessory enzymes may be necessary to initiate differentiation to 9- or 10-membered enediyne precursors, either by modulation of enediyne PKS activity or by interception and modification of polyketide chain-extension intermediates.
Co-reporter:Micah J. Bodner ; Ryan M. Phelan ; Michael F. Freeman ; Rongfeng Li
Journal of the American Chemical Society 2009 Volume 132(Issue 1) pp:12-13
Publication Date(Web):December 17, 2009
DOI:10.1021/ja907320n
Carbapenems are a clinically important antibiotic family. More than 50 naturally occurring carbapenam/ems are known and are distinguished primarily by their C-2/C-6 side chains where many are only differentiated by the oxidation states of these substituents. With a limited palette of variations the carbapenem family comprises a natural combinatorial library, and C-2/C-6 oxidation is associated with increased efficacy. We demonstrate that ThnG and ThnQ encoded by the thienamycin gene cluster in Streptomyces cattleya oxidize the C-2 and C-6 moieties of carbapenems, respectively. ThnQ stereospecifically hydroxylates PS-5 (5) giving N-acetyl thienamycin (2). ThnG catalyzes sequential desaturation and sulfoxidation of PS-5 (5), giving PS-7 (7) and its sulfoxide (9). The enzymes are relatively substrate selective but are proposed to give rise to the oxidative diversity of carbapenems produced by S. cattleya, and orthologues likely function similarly in allied streptomyces. Elucidating the roles of ThnG and ThnQ will focus further investigations of carbapenem antibiotic biosynthesis.
Co-reporter:Micah J. Bodner, Ryan M. Phelan and Craig A. Townsend
Organic Letters 2009 Volume 11(Issue 16) pp:3606-3609
Publication Date(Web):July 17, 2009
DOI:10.1021/ol901269d
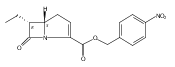
Efficient syntheses of N-acetyl thienamycin and epithienamycin A in their readily deprotected form are reported where three contiguous stereocenters are established in a single catalytic asymmetric azetidinone-forming reaction. These examples are a template for synthesizing C-5/C-6 cis or trans carbapenems with independent control of the C-8 stereocenter. A library of oxidatively and sterochemically defined azetidinone precursors to a variety of naturally occurring carbapenems and potential biosynthetic intermediates has been prepared to facilitate studies of carbapenem antibiotic biosynthesis.
Co-reporter:Edward A. Wydysh ; Susan M. Medghalchi ; Aravinda Vadlamudi
Journal of Medicinal Chemistry 2009 Volume 52(Issue 10) pp:3317-3327
Publication Date(Web):April 23, 2009
DOI:10.1021/jm900251a
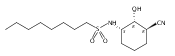
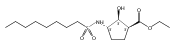
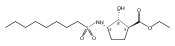
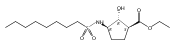
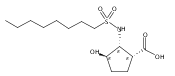
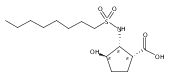
The incidence of obesity and other diseases associated with an increased triacylglycerol mass is growing rapidly, particularly in the United States. Glycerol 3-phosphate acyltransferase (GPAT) catalyzes the rate-limiting step of glycerolipid biosynthesis, the acylation of glycerol 3-phosphate with saturated long-chain acyl-CoAs. In an effort to produce small molecule inhibitors of this enzyme, a series of benzoic and phosphonic acids was designed and synthesized. In vitro testing of this series has led to the identification of several compounds, in particular 2-(nonylsulfonamido)benzoic acid (15g), possessing moderate GPAT inhibitory activity in an intact mitochondrial assay.
Co-reporter:Mary L. Raber, Samantha O. Arnett and Craig A. Townsend
Biochemistry 2009 Volume 48(Issue 22) pp:
Publication Date(Web):April 16, 2009
DOI:10.1021/bi900432n
β-Lactam-synthesizing enzymes carbapenam synthetase (CPS) and β-lactam synthetase (β-LS) are evolutionarily linked to a common ancestor, asparagine synthetase B (AS-B). These three relatives catalyze substrate acyl-adenylation and nucleophilic acyl substitution by either an external (AS-B) or internal (CPS, β-LS) nitrogen source. Unlike AS-B, crystal structures of CPS and β-LS revealed a putative Tyr-Glu dyad (CPS, Y345/E380; β-LS, Y348/E382) proposed to deprotonate the respective internal nucleophile. CPS and β-LS site-directed mutagenesis (Y345/8A, Y345/8F, E380/2D, E380/2Q, E380A) resulted in the reduction of their catalytic efficiency, with Y345A, E380A, and E382Q producing undetectable amounts of β-lactam product. However, [32P]PPi−ATP exchange assays demonstrated Y345A and E380A undergo the first half-reaction, with the remaining active mutants showing decreased forward commitment to β-lactam cyclization. pH−rate profiles of CPS and β-LS supported the importance of a Tyr-Glu dyad in β-lactam formation and suggested its reverse protonation in β-LS. The kinetics of CPS double-site mutants reinforced the synergism of Tyr-Glu in catalysis. Furthermore, significant solvent isotope effects on kcat (Dkcat) for Y345F (1.9) and Y348F (1.7) maintained the assignment of Y345/8 in proton transfer. A proton inventory on Y348F determined its D(kcat/Km) = 0.2 to arise from multiple reactant-state fractionation factors, presumably from water molecule(s) replacing the missing Tyr hydroxyl. The role of a CPS and β-LS Tyr-Glu catalytic dyad was solidified by a significant decrease in mutant kcat viscosity dependence with respect to the wild-type enzymes. The evolutionary relation and potential for engineered biosynthesis were demonstrated by β-LS acting as a carbapenam synthetase.
Co-reporter:Ryan M. Phelan, Marc Ostermeier, Craig A. Townsend
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 4) pp:1261-1263
Publication Date(Web):15 February 2009
DOI:10.1016/j.bmcl.2008.12.057
An efficient synthesis of a 5-fluorouracil-cephalosporin prodrug is described for use against colorectal and other cancers in antibody and gene-directed therapies. The compound shows stability in aqueous media until specifically activated by β-lactamase (βL). The kinetic parameters of the 5-fluorouracil-cephalosporin conjugate were determined in the presence of Enterobacter cloacae P99 βL (ECl βL) revealing a Km = 95.4 μM and Vmax = 3.21 μMol min−1 mg−1. The data compare favorably to related systems that have been reported and enable testing of this prodrug against cancer cell lines in vitro and in vivo.
Co-reporter:Mary L. Raber Dr.;Alvaro Castillo;Alexer Greer Dr. Dr.
ChemBioChem 2009 Volume 10( Issue 18) pp:2904-2912
Publication Date(Web):
DOI:10.1002/cbic.200900389
Abstract
β-Lactam synthetase (β-LS) is the paradigm of a growing class of enzymes that form the critical β-lactam ring in the clavam and carbapenem antibiotics. β-LS catalyzes a two-stage reaction in which N2-(2-carboxyethyl)-L-arginine is first adenylated, and then undergoes intramolecular ring closure. It was previously shown that the forward kinetic commitment to β-lactam formation is high, and that the overall rate of reaction is partially limited to a protein conformational change rather than to the chemical step alone of closing the strained ring. β-Lactam formation was evaluated on the basis of X-ray crystal structures, site-specific mutation, and kinetic and computational studies. The combined evidence clearly points to a reaction coordinate involving the formation of a tetrahedral transition state/intermediate stabilized by a conserved Lys. The combination of substrate preorganization, a well-stabilized transition state and an excellent leaving group facilitates this acyl substitution to account for the strong forward commitment to catalysis and to lower the barrier of four-membered ring formation to the magnitude of a protein conformational change.
Co-reporter:Jason M. Crawford,
Tyler P. Korman,
Jason W. Labonte,
Anna L. Vagstad,
Eric A. Hill,
Oliver Kamari-Bidkorpeh,
Shiou-Chuan Tsai
&
Craig A. Townsend
Nature 2009 461(7267) pp:1139
Publication Date(Web):2009-10-22
DOI:10.1038/nature08475
Regiospecific cyclizations of reactive poly--keto intermediates are known to lead to the structural variability of aromatic products of fungal nonreducing, multidomain iterative polyketide synthases (NR-PKS group of IPKSs), but questions about the process remain. The crystal structure and mutational studies of a dissected product template monodomain from PksA, the NR-PKS that initiates the biosynethesis of the hepatocarcinogen aflatoxin B1, are now presented.
Co-reporter:Jason M. Crawford, Anna L. Vagstad, Kenneth C. Ehrlich, Craig A. Townsend
Bioorganic Chemistry 2008 Volume 36(Issue 1) pp:16-22
Publication Date(Web):February 2008
DOI:10.1016/j.bioorg.2007.11.002
Search of the protein database with the aflatoxin pathway polyketide synthase (PKS) revealed putative PKSs in the pathogenic fungi Coccidioides immitis and Coccidioides posadasii that could require partnerships with a pair of fatty acid synthase (FAS) subunits for the biosynthesis of fatty acid–polyketide hybrid metabolites. A starter unit:acyl-carrier protein transacylase (SAT) domain was discovered in the nonreducing PKS. This domain is thought to accept the fatty acid product from the FAS to initiate polyketide synthesis. We expressed the C. immitis SAT domain in Escherichia coli and showed that this domain, unlike that from the aflatoxin pathway PKS, transferred octanoyl-CoA four times faster than hexanoyl-CoA. The SAT domain also formed a covalent octanoyl intermediate and transferred this group to a free-standing ACP domain. Our results suggest that C. immitis/posadasii, both human fungal pathogens, contain a FAS/PKS cluster with functional similarity to the aflatoxin cluster found in Aspergillus species. Dissection of the PKS and determination of in vitro SAT domain specificity provides a tool to uncover the growing number of similar sequenced pathways in fungi, and to guide elucidation of the fatty acid–polyketide hybrid metabolites that they produce.
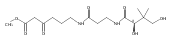
Co-reporter:Michael F. Freeman;Kristos A. Moshos;Rongfeng Li;Micah J. Bodner
PNAS 2008 Volume 105 (Issue 32 ) pp:11128-11133
Publication Date(Web):2008-08-12
DOI:10.1073/pnas.0804500105
The enzymatic activities of three proteins encoded by the thienamycin gene cluster of Streptomyces cattleya (ThnR, ThnH, and ThnT) have been shown to incrementally cleave CoA to afford the active side-chain component of the β-lactam
antibiotic thienamycin. These results supersede proposals based on earlier radiochemical incorporation experiments. For 20
years it has been thought that cysteine was directly incorporated into the antibiotic. Specific, stepwise truncation of CoA
to 4-phosphopantetheine, pantetheine, and finally cysteamine was observed with ThnR, ThnH, and ThnT, respectively, in a series
of coupled enzymatic assays. Pantetheinylated carbapenams were synthesized to address possible thienamycin biosynthetic intermediates
and were shown to be effective substrates for the pantetheine-cleaving enzyme ThnT. Finally, a fourth gene, thnF, was shown to encode a protein capable of N-acetylating a model compound containing cysteamine in the presence of acetyl-CoA, consistent with the production of the S. cattleya cometabolite, N-acetylthienamycin. Taken together, these four enzymes are proposed to siphon CoA from primary metabolism to create the side
chains for the predominant S. cattleya carbapenems, thienamycin and N-acetylthienamycin, in a process likely to be general for the broader class of these antibiotics.
Co-reporter:Jonathan R. Scheerer;Neil L. Kelleher;Jason M. Crawford;Anna L. Vagstad;Paul M. Thomas
Science 2008 Volume 320(Issue 5873) pp:243-246
Publication Date(Web):11 Apr 2008
DOI:10.1126/science.1154711
Abstract
PksA, which initiates biosynthesis of the environmental carcinogen aflatoxin B1, is one of the multidomain iterative polyketide synthases (IPKSs), a large, poorly understood family of biosynthetic enzymes. We found that dissection of PksA and its reconstitution from selected sets of domains allows the accumulation and characterization of advanced octaketide intermediates bound to the enzyme, permitting the reactions controlled by individual catalytic domains to be identified. A product template (PT) domain unites with the ketosynthase and thioesterase in this IPKS system to assemble precisely seven malonyl-derived building blocks to a hexanoyl starter unit and mediate a specific cyclization cascade. Because the PT domain is common among nonreducing IPKSs, these mechanistic features should prove to be general for IPKS-catalyzed production of aromatic polyketides.
Co-reporter:Jason M. Crawford Dr.;Anna L. Vagstad;Kenneth C. Ehrlich Dr.;Daniel W. Udwary Dr. Dr.
ChemBioChem 2008 Volume 9( Issue 10) pp:1559-1563
Publication Date(Web):
DOI:10.1002/cbic.200700659
Co-reporter:Jason M. Crawford Dr.;Anna L. Vagstad;Karen P. Whitworth;Kenneth C. Ehrlich Dr. Dr.
ChemBioChem 2008 Volume 9( Issue 7) pp:1019-1023
Publication Date(Web):
DOI:10.1002/cbic.200700702
Co-reporter:Craig A. Townsend, Jason M. Crawford, Tsion Bililign
Chemistry & Biology 2006 Volume 13(Issue 4) pp:349-351
Publication Date(Web):April 2006
DOI:10.1016/j.chembiol.2006.04.001
Two recent papers in Science reported the X-ray structures of the large, organizationally distinct animal and fungal fatty acid synthases at 5 Å. These new structural insights have unexpected implications for enzyme function for the other “iterative” and “assembly line” megasynthases.
Co-reporter:Jason M. Crawford;Blair C. R. Dancy;Eric A. Hill;Daniel W. Udwary;
Proceedings of the National Academy of Sciences 2006 103(45) pp:16728-16733
Publication Date(Web):October 27, 2006
DOI:10.1073/pnas.0604112103
Polyketides are a class of natural products that exhibit a wide range of functional and structural diversity. They include
antibiotics, immunosuppressants, antifungals, antihypercholesterolemics, and cytotoxins. Polyketide synthases (PKSs) use chemistry
similar to fatty acid synthases (FASs), although building block variation and differing extents of reduction of the growing
polyketide chain underlie their biosynthetic versatility. In contrast to the well studied sequential modular type I PKSs,
less is known about how the iterative type I PKSs carry out and control chain initiation, elongation, folding, and cyclization
during polyketide processing. Domain structure analysis of a group of related fungal, nonreducing PKSs has revealed well defined
N-terminal domains longer than commonly seen for FASs and modular PKSs. Predicted structure of this domain disclosed a region
similar to malonyl-CoA:acyl-carrier protein (ACP) transacylases (MATs). MATs play a key role transferring precursor CoA thioesters
from solution onto FASs and PKSs for chain elongation. On the basis of site-directed mutagenesis, radiolabeling, and kinetics
experiments carried out with individual domains of the norsolorinic acid PKS, we propose that the N-terminal domain is a starter
unit:ACP transacylase (SAT domain) that selects a C6 fatty acid from a dedicated yeast-like FAS and transfers this unit onto the PKS ACP, leading to the production of the aflatoxin
precursor, norsolorinic acid. These findings could indicate a much broader role for SAT domains in starter unit selection
among nonreducing iterative, fungal PKSs, and they provide a biochemical rationale for the classical acetyl “starter unit
effect.”
Co-reporter:Craig A Townsend
Current Opinion in Chemical Biology (December 2016) Volume 35() pp:97-108
Publication Date(Web):December 2016
DOI:10.1016/j.cbpa.2016.09.013
Co-reporter:Andrew R. Buller, Jason W. Labonte, Michael F. Freeman, Nathan T. Wright, ... Craig A. Townsend
Journal of Molecular Biology (28 September 2012) Volume 422(Issue 4) pp:508-518
Publication Date(Web):28 September 2012
DOI:10.1016/j.jmb.2012.06.012
cis-Autoproteolysis is a post-translational modification necessary for the function of ThnT, an enzyme involved in the biosynthesis of the β-lactam antibiotic thienamycin. This modification generates an N-terminal threonine nucleophile that is used to hydrolyze the pantetheinyl moiety of its natural substrate. We determined the crystal structure of autoactivated ThnT to 1.8 Å through X-ray crystallography. Comparison to a mutationally inactivated precursor structure revealed several large conformational rearrangements near the active site. To probe the relevance of these transitions, we designed a pantetheine-like chloromethyl ketone inactivator and co-crystallized it with ThnT. Although this class of inhibitor has been in use for several decades, the mode of inactivation had not been determined for an enzyme that uses an N-terminal nucleophile. The co-crystal structure revealed the chloromethyl ketone bound to the N-terminal nucleophile of ThnT through an ether linkage, and analysis suggests inactivation through a direct displacement mechanism. More importantly, this inactivated complex shows that three regions of ThnT that are critical to the formation of the substrate binding pocket undergo rearrangement upon autoproteolysis. Comparison of ThnT with other autoproteolytic enzymes of disparate evolutionary lineage revealed a high degree of similarity within the proenzyme active site, reflecting shared chemical constraints. However, after autoproteolysis, many enzymes, like ThnT, are observed to rearrange in order to accommodate their specific substrate. We propose that this is a general phenomenon, whereby autoprocessing systems with shared chemistry may possess similar structural features that dissipate upon rearrangement into a mature state.Download high-res image (202KB)Download full-size imageHighlights► A 1.8-Å X-ray structure shows that peptide bond isomerization accompanies autoproteolysis. ► The threonine γ-methyl facilitates formation of a reactive N-terminal nucleophile. ► A halomethyl ketone inhibitor is shown to bind through an ether linkage. ► Computational modeling suggests inactivation through a direct displacement mechanism. ► Comparison with the proteasome β-subunit reveals general features of autoactivation.
Co-reporter:Adam G. Newman, Anna L. Vagstad, Katherine Belecki, Jonathan R. Scheerer and Craig A. Townsend
Chemical Communications 2012 - vol. 48(Issue 96) pp:NaN11774-11774
Publication Date(Web):2012/10/29
DOI:10.1039/C2CC36010A
The polyketide synthase CTB1 is demonstrated to catalyze pyrone formation thereby expanding the known biosynthetic repertoire of thioesterase domains in iterative, non-reducing polyketide synthases.
.jpg)
![PROPANETHIOIC ACID, 3-(METHYLTHIO)-, S-[2-(ACETYLAMINO)ETHYL] ESTER](http://img.cochemist.com/ccimg/866500/866465-15-8.png)
![PROPANETHIOIC ACID, 3-(METHYLTHIO)-, S-[2-(ACETYLAMINO)ETHYL] ESTER](http://img.cochemist.com/ccimg/866500/866465-15-8_b.png)
![Ethanethioic acid, (methylthio)-, S-[2-(acetylamino)ethyl] ester](http://img.cochemist.com/ccimg/866500/866465-14-7.png)
![Ethanethioic acid, (methylthio)-, S-[2-(acetylamino)ethyl] ester](http://img.cochemist.com/ccimg/866500/866465-14-7_b.png)




![Pentanethioic acid, S-[2-(acetylamino)ethyl] ester](http://img.cochemist.com/ccimg/174600/174509-73-0.png)
![Pentanethioic acid, S-[2-(acetylamino)ethyl] ester](http://img.cochemist.com/ccimg/174600/174509-73-0_b.png)


![6-(4-CHLOROBENZYL)-2,3-DIHYDRO-5H-[1,3]THIAZOLO[3,2-A]PYRIMIDINE-<WBR />5,7(6H)-DIONE](http://img.cochemist.com/ccimg/78000/77912-79-9.png)
![6-(4-CHLOROBENZYL)-2,3-DIHYDRO-5H-[1,3]THIAZOLO[3,2-A]PYRIMIDINE-<WBR />5,7(6H)-DIONE](http://img.cochemist.com/ccimg/78000/77912-79-9_b.png)
![(R)-p-Nitrobenzyl ester7-Oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/75500/75468-46-1.png)
![(R)-p-Nitrobenzyl ester7-Oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/75500/75468-46-1_b.png)
![1-[TERT-BUTYL(DIMETHYL)SILYL]-4-PROP-2-ENYLAZETIDIN-2-ONE](http://img.cochemist.com/ccimg/74900/74819-64-0.png)
![1-[TERT-BUTYL(DIMETHYL)SILYL]-4-PROP-2-ENYLAZETIDIN-2-ONE](http://img.cochemist.com/ccimg/74900/74819-64-0_b.png)
![Butanethioic acid, S-[2-(acetylamino)ethyl] ester](http://img.cochemist.com/ccimg/68300/68231-66-3.png)
![Butanethioic acid, S-[2-(acetylamino)ethyl] ester](http://img.cochemist.com/ccimg/68300/68231-66-3_b.png)
![N5,N5,N10,N10-tetramethyl-5,10-dihydrodipyrrolo[1,2-a:1',2'-d]pyrazine-5,10-diamine](http://img.cochemist.com/ccimg/64500/64435-30-9.png)
![N5,N5,N10,N10-tetramethyl-5,10-dihydrodipyrrolo[1,2-a:1',2'-d]pyrazine-5,10-diamine](http://img.cochemist.com/ccimg/64500/64435-30-9_b.png)












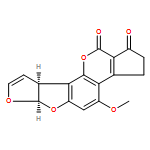
![(3aS,12aR)-4,6,8-trihydroxy-3a,12a-dihydroanthra[2,3-b]furo[3,2-d]furan-5,10-dione](http://img.cochemist.com/ccimg/6900/6807-96-1.png)
![(3aS,12aR)-4,6,8-trihydroxy-3a,12a-dihydroanthra[2,3-b]furo[3,2-d]furan-5,10-dione](http://img.cochemist.com/ccimg/6900/6807-96-1_b.png)



![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)


![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[(2,6-dimethoxybenzoyl)amino]-3,3-dimethyl-7-oxo-, (2S,5R,6R)-](http://img.cochemist.com/ccimg/100/61-32-5_b.png)


















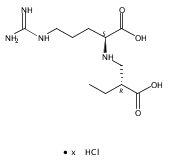
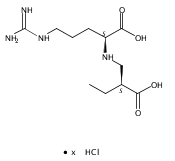


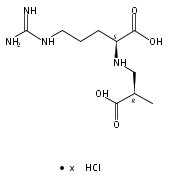
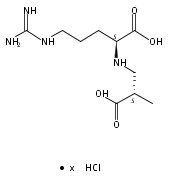
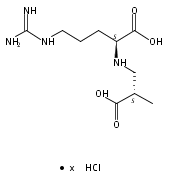
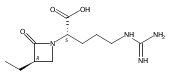
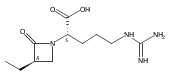
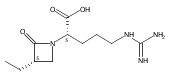
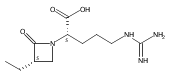
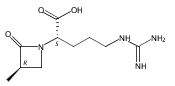






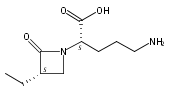


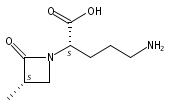



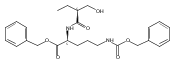

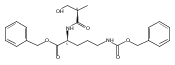
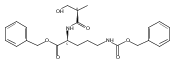


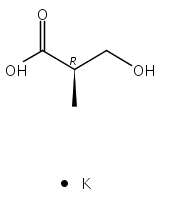

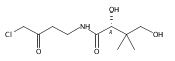
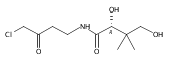



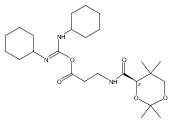
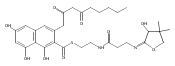

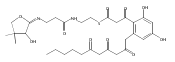
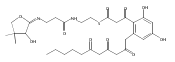
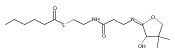

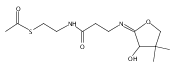






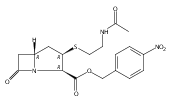


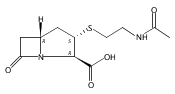
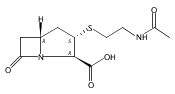


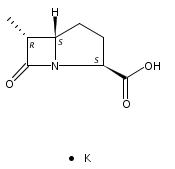
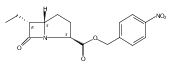



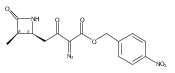



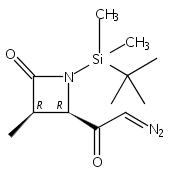
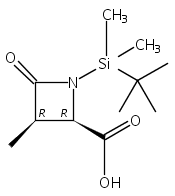


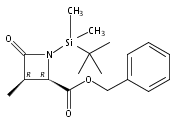
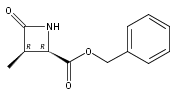
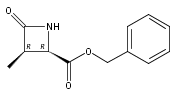




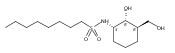





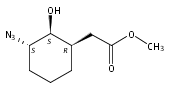

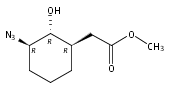

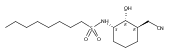



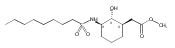







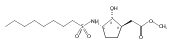
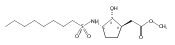
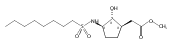
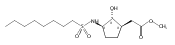
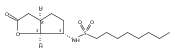

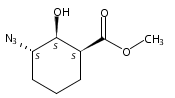

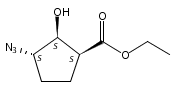

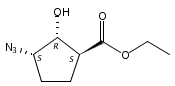
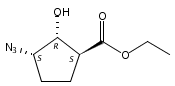
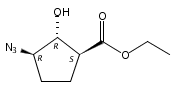

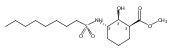

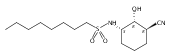












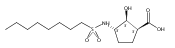



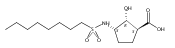
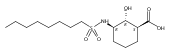
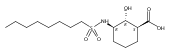






















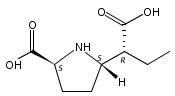
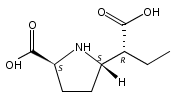

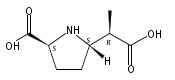
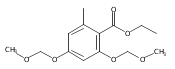
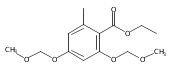





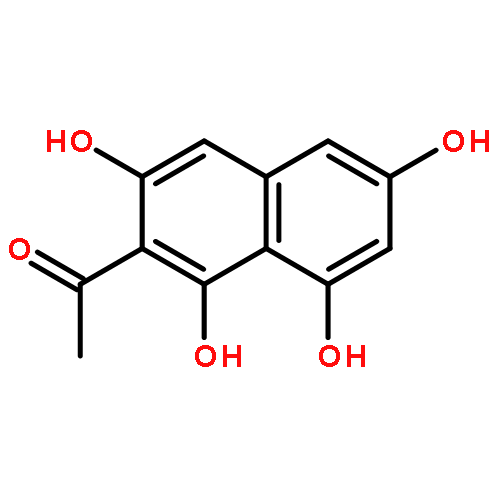















![b-Alanine, N-[(2,2,5,5-tetramethyl-1,3-dioxan-4-yl)carbonyl]-, (R)-](/data/chemimg/115400/167308-62-5.png)
![b-Alanine, N-[(2,2,5,5-tetramethyl-1,3-dioxan-4-yl)carbonyl]-, (R)-](/data/chemimg/115400/167308-62-5_b.png)












![Propanedioic acid, mono[(4-nitrophenyl)methyl] ester, magnesium salt](/data/chemimg/120900/105995-50-4.png)
![Propanedioic acid, mono[(4-nitrophenyl)methyl] ester, magnesium salt](/data/chemimg/120900/105995-50-4_b.png)








![p-Nitrobenzyl ester-(S)-7-Oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/75500/75468-47-2.png)
![p-Nitrobenzyl ester-(S)-7-Oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/75500/75468-47-2_b.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-ethyl-7-oxo-, (5R,6R)-](http://img.cochemist.com/ccimg/74900/74806-75-0.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-ethyl-7-oxo-, (5R,6R)-](http://img.cochemist.com/ccimg/74900/74806-75-0_b.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[[(1E)-2-(acetylamino)ethenyl]thio]-6-ethyl-7-oxo-, (5R,6R)-](http://img.cochemist.com/ccimg/72700/72615-18-0.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[[(1E)-2-(acetylamino)ethenyl]thio]-6-ethyl-7-oxo-, (5R,6R)-](http://img.cochemist.com/ccimg/72700/72615-18-0_b.png)

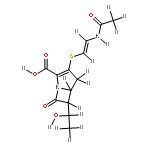
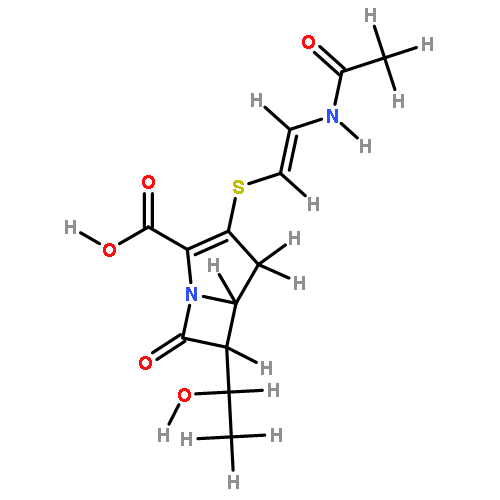
![trans-3-[2-(Acetamido)ethylsulfanyl]-6-ethyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/67100/67007-79-8.png)
![trans-3-[2-(Acetamido)ethylsulfanyl]-6-ethyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/67100/67007-79-8_b.png)
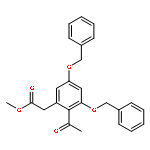
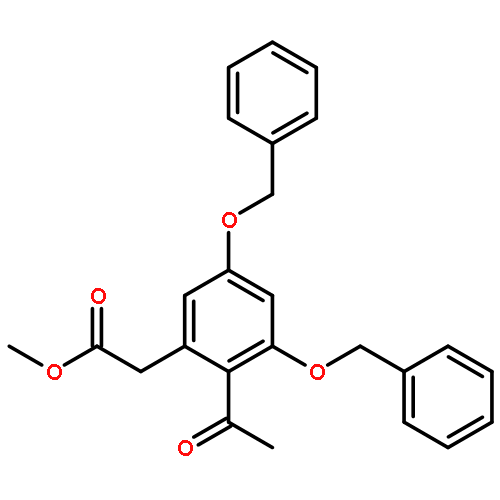
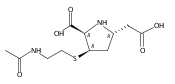
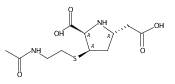
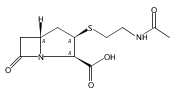
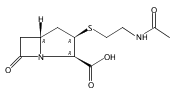
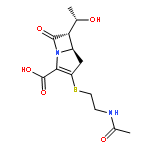
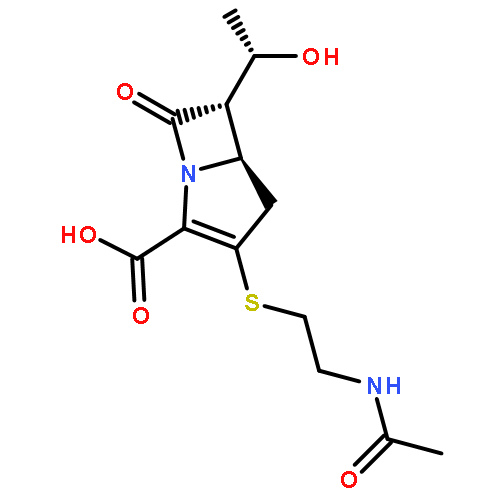
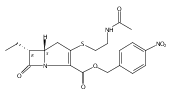
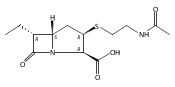
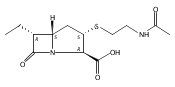
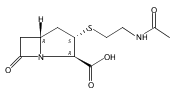
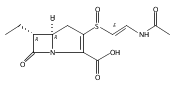
![(5R,6R)-3-{[2-(acetylamino)ethyl]sulfanyl}-6-[(1R)-1-hydroxyethyl]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/63800/63701-32-6.png)
![(5R,6R)-3-{[2-(acetylamino)ethyl]sulfanyl}-6-[(1R)-1-hydroxyethyl]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/63800/63701-32-6_b.png)
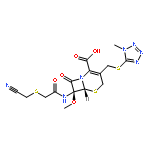
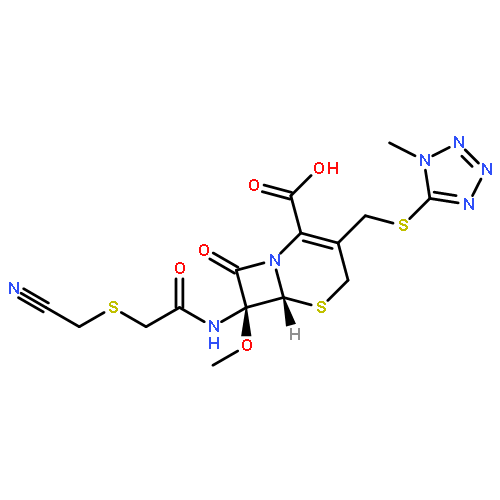


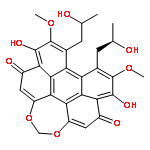
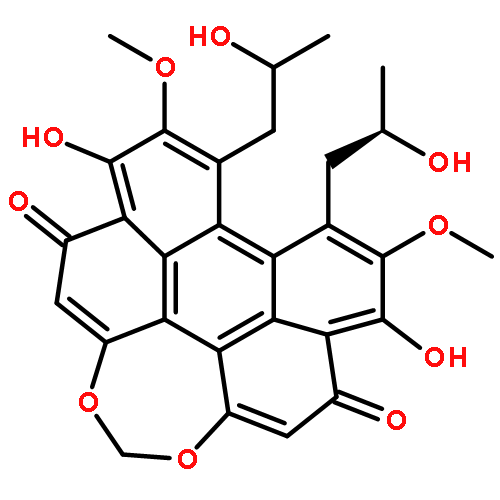
![Perylo[1,12-def]-1,3-dioxepin-5,11-dione,6,12-dihydroxy-8,9-bis[(2R)-2-hydroxypropyl]-7,10-dimethoxy-, (13bR)-](http://img.cochemist.com/ccimg/35100/35082-49-6.png)
![Perylo[1,12-def]-1,3-dioxepin-5,11-dione,6,12-dihydroxy-8,9-bis[(2R)-2-hydroxypropyl]-7,10-dimethoxy-, (13bR)-](http://img.cochemist.com/ccimg/35100/35082-49-6_b.png)
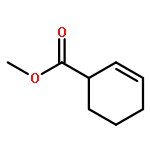
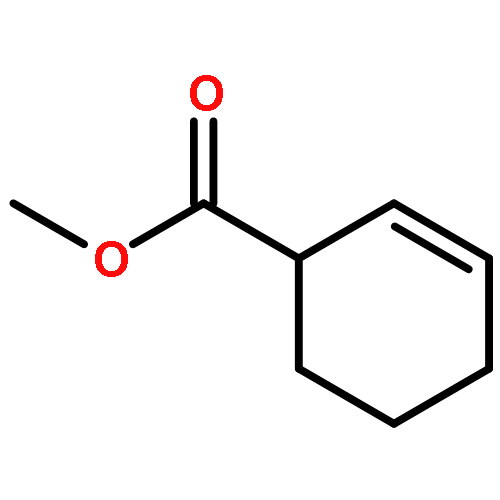

![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
























































































![1,3-Dioxane-4-carboxamide,N-[3-[(2-mercaptoethyl)amino]-3-oxopropyl]-2,2,5,5-tetramethyl-, (4R)-](http://img.cochemist.com/ccimg/167400/167308-64-7.png)
![1,3-Dioxane-4-carboxamide,N-[3-[(2-mercaptoethyl)amino]-3-oxopropyl]-2,2,5,5-tetramethyl-, (4R)-](http://img.cochemist.com/ccimg/167400/167308-64-7_b.png)
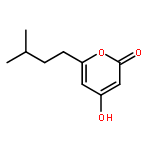
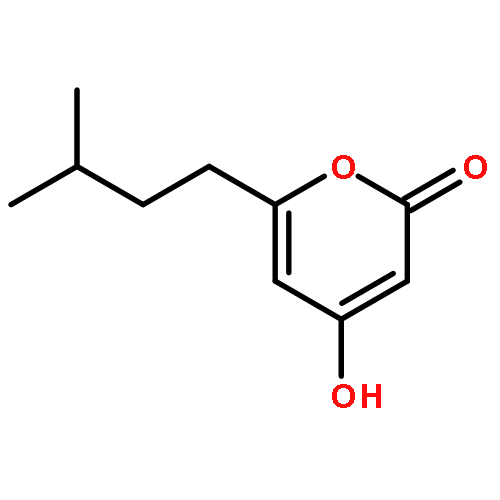
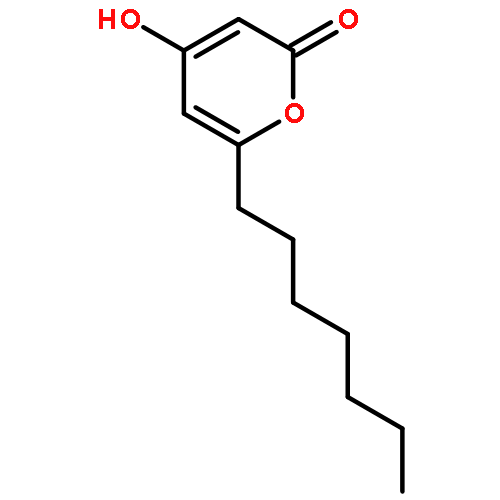
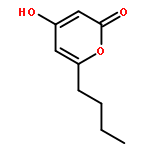
![2H-Naphtho[1,2-b]pyran-2-one,4,8,10-trihydroxy-5-methyl-](http://img.cochemist.com/ccimg/137100/137023-81-5.png)
![2H-Naphtho[1,2-b]pyran-2-one,4,8,10-trihydroxy-5-methyl-](http://img.cochemist.com/ccimg/137100/137023-81-5_b.png)








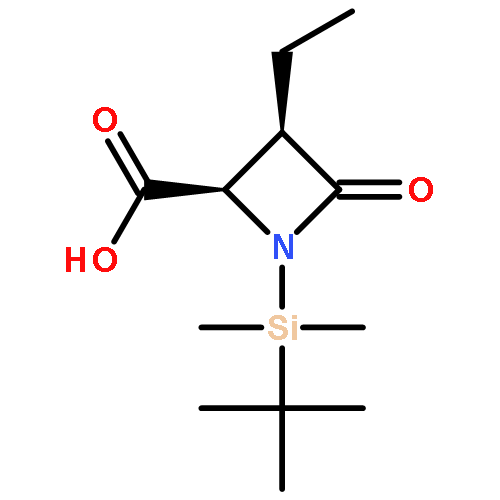
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 7-oxo-, (5R)-](http://img.cochemist.com/ccimg/78900/78854-41-8.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 7-oxo-, (5R)-](http://img.cochemist.com/ccimg/78900/78854-41-8_b.png)
![(2S)-2-(4-hydroxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid](http://img.cochemist.com/ccimg/69700/69651-48-5.png)
![(2S)-2-(4-hydroxyphenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid](http://img.cochemist.com/ccimg/69700/69651-48-5_b.png)
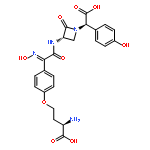
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-[(1R)-1-hydroxyethyl]-7-oxo-, (5R,6S)-](http://img.cochemist.com/ccimg/60000/59995-64-1.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-[(1R)-1-hydroxyethyl]-7-oxo-, (5R,6S)-](http://img.cochemist.com/ccimg/60000/59995-64-1_b.png)
![(2r,3z,5r)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]heptane-2-carboxylic Acid](http://img.cochemist.com/ccimg/58100/58001-44-8.png)
![(2r,3z,5r)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]heptane-2-carboxylic Acid](http://img.cochemist.com/ccimg/58100/58001-44-8_b.png)


![1-Azabicyclo[3.2.0]heptane-2-carboxylicacid, 7-oxo-, (2S,5S)-](http://img.cochemist.com/ccimg/123500/123409-86-9.png)
![1-Azabicyclo[3.2.0]heptane-2-carboxylicacid, 7-oxo-, (2S,5S)-](http://img.cochemist.com/ccimg/123500/123409-86-9_b.png)
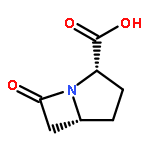


![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-(1-hydroxyethyl)-7-oxo-](http://img.cochemist.com/ccimg/64100/64090-99-9.png)
![1-Azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid, 3-[(2-aminoethyl)thio]-6-(1-hydroxyethyl)-7-oxo-](http://img.cochemist.com/ccimg/64100/64090-99-9_b.png)
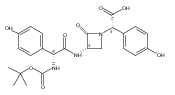
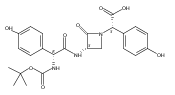
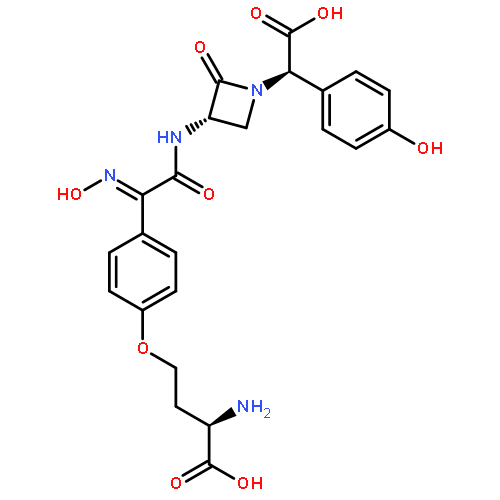
![(3S,αR)-3-[[[4-[(R)-3-Amino-3-carboxypropoxy]phenyl]-[(Z)-hydroxyimino]acetyl]amino]-α-(4-hydroxyphenyl)-2-oxo-1-azetidineacetic acid](http://img.cochemist.com/ccimg/39400/39391-39-4.png)
![(3S,αR)-3-[[[4-[(R)-3-Amino-3-carboxypropoxy]phenyl]-[(Z)-hydroxyimino]acetyl]amino]-α-(4-hydroxyphenyl)-2-oxo-1-azetidineacetic acid](http://img.cochemist.com/ccimg/39400/39391-39-4_b.png)