Co-reporter:Hongguang Ma, Victoria N. Stone, Huiqun Wang, Glen E. Kellogg, Ping Xu, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.06.056
Two diastereomeric analogs (1 and 2) of diaminopimelic acid (DAP) bearing an isoxazoline moiety were synthesized and evaluated for their inhibitory activities against meso-diaminopimelate dehydrogenase (m-Ddh) from the periodontal pathogen, Porphyromonas gingivalis. Compound 2 showed promising inhibitory activity against m-Ddh with an IC50 value of 14.9 µM at pH 7.8. The two compounds were further tested for their antibacterial activities against a panel of periodontal pathogens, and compound 2 was shown to be selectively potent to P. gingivalis strains W83 and ATCC 33277 with minimum inhibitory concentration (MIC) values of 773 µM and 1.875 mM, respectively. Molecular modeling studies revealed that the inversion of chirality at the C-5 position of these compounds was the primary reason for their different biological profiles. Based on these preliminary results, we believe that compound 2 has properties consistent with it being a lead compound for developing novel pathogen selective antibiotics to treat periodontal diseases.Download high-res image (135KB)Download full-size image
Co-reporter:Huiqun Wang, Saheem A. Zaidi, Yan Zhang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 8(Issue 8) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bmc.2017.03.005
Mu opioid receptor selective antagonists are highly desirable because of their utility as pharmacological probes for receptor characterization and functional studies. Furthermore, the mu opioid receptors act as an important target in drug abuse and addiction treatment. Previously, we reported NAP as a novel lead compound with high selectivity and affinity towards the mu opioid receptor. Based on NAP, we have synthesized its derivatives and further characterized their binding affinities and selectivity towards the receptor. NMP and NGP were identified as the two most selective MOR ligands among NAP derivatives. In the present study, molecular modeling methods were applied to assess the dual binding modes of NAP derivatives, particularly on NMP and NGP, in three opioid receptors, in order to analyze the effects of structural modifications on the pyridyl ring of NAP on the binding affinity and selectivity. The results indicated that the steric hindrance, electrostatic, and hydrophobic effects caused by the substituents on the pyridyl ring of NAP contributed complimentarily on the binding affinity and selectivity of NAP derivatives to three opioid receptors. Analyses of these contributions provided insights on future design of more potent and selective mu opioid receptor ligands.Binding modes NMP in the site A of the MOR (a) and NGP in the site B of the MOR (b) with key residues shown after 10 ns MD simulations in a membrane bilayer system.Download high-res image (255KB)Download full-size image
Co-reporter:Guoyan G. Xu, Olga Yu. Zolotarskaya, Dwight A. Williams, Yunyun Yuan, Dana E. Selley, William L. Dewey, Hamid I. Akbarali, Hu Yang, and Yan Zhang
ACS Medicinal Chemistry Letters 2017 Volume 8(Issue 1) pp:
Publication Date(Web):November 21, 2016
DOI:10.1021/acsmedchemlett.6b00382
Opioids are the mainstay for cancer and noncancer pain management. However, their use is often associated with multiple adverse effects. Among them, the most common and persistent one is probably opioid-induced constipation (OIC). Periphery selective opioid antagonists may alleviate the symptoms of OIC without compromising the analgesic effects of opioids. Recently our laboratories have identified one novel lead compound, 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)acetamido]morphinan (NAP), as a peripherally selective mu opioid receptor ligand carrying subnanomolar affinity to the mu opioid receptor and over 100-folds of selectivity over both the delta and kappa opioid receptors, with reasonable oral availability and half-life, and potential to treat OIC. Nanoparticle-based drug delivery systems are now widely considered due to their technological advantages such as good stability, high carrier capacity, low therapeutic side effects, etc. Herein we report nanoparticle supported NAP as a potential candidate for OIC treatment with improved peripheral selectivity over the original lead compound NAP.Keywords: Mu opioid receptor antagonist; nanoconjugate; NAP; Opioid-induced constipation; Periphery selective;
Co-reporter:Piyusha P. Pagare, Saheem A. Zaidi, Xiaomei Zhang, Xia Li, Xiaofei Yu, Xiang-Yang Wang, Yan Zhang
Journal of Molecular Graphics and Modelling 2017 Volume 77(Volume 77) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jmgm.2017.08.013
•Monomer and dimer homology models of SRA was constructed.•Docking study and molecular dynamics simulations was done to identify putative binding domain(s) of the inhibitors.•The dimer model provided more consistent information regarding to the binding pocket composition and ligand binding mode.•Treatment of SRA with rhein led to significant dissociation of SRA oligomers to its trimer and dimer form.•Above information is helpful to design more potent inhibitor to modulate SRA function for therapeutic application.Scavenger receptor A (SRA), as an immune regulator, has been shown to play important roles in lipid metabolism, cardiovascular diseases, and pathogen recognition. Several natural product inhibitors of SRA have been studied for their potential application in modulating SRA functions. To understand the binding mode of these inhibitors on SRA, we conducted systematic molecular modeling studies in order to identify putative binding domain(s) that may be responsible for their recognition to the receptor as well as their inhibitory activity. Treatment of SRA with one of the natural product inhibitors, rhein, led to significant dissociation of SRA oligomers to its trimer and dimer forms, which further supported our hypothesis on their putative mechanism of action. Such information is believed to shed light on design of more potent inhibitors for the receptor in order to develop potential therapeutics through immune system modulation.Dimeric SRA model with natural product ligand rhein shown in sticks at the most preferred docking mode.Download high-res image (144KB)Download full-size image
Co-reporter:Dwight A. Williams, Yi Zheng, Bethany G. David, Yunyun Yuan, Saheem A. Zaidi, David L. Stevens, Krista L. Scoggins, Dana E. Selley, William L. Dewey, Hamid I. Akbarali, and Yan Zhang
ACS Chemical Neuroscience 2016 Volume 7(Issue 8) pp:1120
Publication Date(Web):June 6, 2016
DOI:10.1021/acschemneuro.6b00075
The 6β-N-heterocyclic naltrexamine derivative, NAP, has been demonstrated to be a peripherally selective mu opioid receptor modulator. To further improve peripheral selectivity of this highly potent ligand, its pyridal ring was quaterinized with benzyl bromide to produce BNAP. In radioligand binding assay, the Ki of BNAP for MOR was 0.76 ± 0.09 nM and was >900-fold more selective for MOR than DOR. The Ki for KOR was 3.46 ± 0.05 nM. In [35S]GTPγS ligand stimulated assay, BNAP showed low agonist efficacy with 14.6% of the maximum response of DAMGO with an EC50 of 4.84 ± 0.6 nM. However, unlike its parent compound NAP, BNAP displayed partial agonist activity at KOR with % maximum response at 45.9 ± 1.7% of U50,488H. BNAP did not reverse morphine-induced antinociception when administered subcutaneously but did antagonize when administered intracerebroventricularly. BNAP antagonized morphine-induced contractions of the circular muscle in mice colon. BNAP inhibition of field-stimulated contractions in longitudinal muscle strips for the guinea-pig ileum were also blocked by nor-BNI, a kappa opioid receptor antagonist. BNAP induced inhibition of acetic acid induced abdominal stretching in chronic morphine treated mice. These findings suggest that BNAP is a dual MOR antagonist/KOR agonist and may have functional use in irritable bowel patients.Keywords: irritable bowel syndrome; mixed opioid ligand; opioid induced constipation; Periphery selectivity; visceral pain
Co-reporter:Yan Zhang, Dwight A. Williams, Saheem A. Zaidi, Yunyun Yuan, Amanda Braithwaite, Edward J. Bilsky, William L. Dewey, Hamid I. Akbarali, John M. Streicher, and Dana E. Selley
ACS Chemical Neuroscience 2016 Volume 7(Issue 3) pp:297
Publication Date(Web):December 30, 2015
DOI:10.1021/acschemneuro.5b00245
Mounting evidence has suggested that G protein-coupled receptors can be stabilized in multiple conformations in response to distinct ligands, which exert discrete functions through selective activation of various downstream signaling events. In accordance with this concept, we report biased signaling of one C6-heterocyclic substituted naltrexamine derivative, namely, 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(4′-pyridylcarboxamido)morphinan (NAP) at the mu opioid receptor (MOR). NAP acted as a low efficacy MOR partial agonist in the G protein-mediated [35S]GTPγS binding assay, whereas it did not significantly induce calcium flux or β-arrestin2 recruitment. In contrast, it potently blocked MOR full agonist-induced β-arrestin2 recruitment and translocation. Additionally, NAP dose-dependently antagonized MOR full agonist-induced intracellular calcium flux and β-arrestin2 recruitment. Further results in an isolated organ bath preparation confirmed that NAP reversed the morphine-induced reduction in colon motility. Ligand docking and dynamics simulation studies of NAP at the MOR provided more supporting evidence for biased signaling of NAP at an atomic level. Due to the fact that NAP is MOR selective and preferentially distributed peripherally upon systemic administration while β-arrestin2 is reportedly required for impairment of intestinal motility by morphine, biased antagonism of β-arrestin2 recruitment by NAP further supports its utility as a treatment for opioid-induced constipation.Keywords: biased signaling; functional selectivity; Mu opioid receptor; nalbuphine; NAP; β-arrestin2
Co-reporter:Christopher K. Arnatt, Bethany A. Falls, Yunyun Yuan, Thomas J. Raborg, Ruturaj R. Masvekar, Nazira El-Hage, Dana E. Selley, Anthony V. Nicola, Pamela E. Knapp, Kurt F. Hauser, Yan Zhang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 22) pp:5969-5987
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmc.2016.09.059
•The first SAR report on bivalent ligands targeting the MOR and CCR5 dimerization.•Comprehensive coverage on the chemical synthesis of these novel bivalent ligands.•Systematic biological and pharmacological characterization of these bivalent ligands.•Molecular modeling effort to understand possible mechanism of action of the lead.•Shed insight on future molecular design and development.Modern antiretroviral therapies have provided HIV-1 infected patients longer lifespans and better quality of life. However, several neurological complications are now being seen in these patients due to HIV-1 associated injury of neurons by infected microglia and astrocytes. In addition, these effects can be further exacerbated with opiate use and abuse. One possible mechanism for such potentiation effects of opiates is the interaction of the mu opioid receptor (MOR) with the chemokine receptor CCR5 (CCR5), a known HIV-1 co-receptor, to form MOR–CCR5 heterodimer. In an attempt to understand this putative interaction and its relevance to neuroAIDS, we designed and synthesized a series of bivalent ligands targeting the putative CCR5–MOR heterodimer. To understand how these bivalent ligands may interact with the heterodimer, biological studies including calcium mobilization inhibition, binding affinity, HIV-1 invasion, and cell fusion assays were applied. In particular, HIV-1 infection assays using human peripheral blood mononuclear cells, macrophages, and astrocytes revealed a notable synergy in activity for one particular bivalent ligand. Further, a molecular model of the putative CCR5–MOR heterodimer was constructed, docked with the bivalent ligand, and molecular dynamics simulations of the complex was performed in a membrane-water system to help understand the biological observation.Compound 1b, Ki (MOR) = 3.24 nM; Ki (CCR5) = 239 nM; more effective in inhibiting HIV-1 entry in astrocytes compared to maraviroc alone or mixture of maraviroc and naltrexone with presence of morphine.
Co-reporter:Dwight A. Williams, Saheem A. Zaidi, and Yan Zhang
Journal of Natural Products 2015 Volume 78(Issue 8) pp:1859-1867
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.jnatprod.5b00118
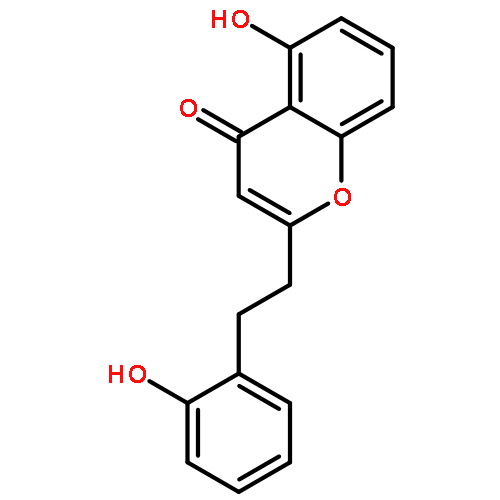
The involvement of the neurotransmitter serotonin (5-HT) in numerous physiological functions is often attributed to the diversity of receptors with which it interacts. Ligands targeting serotonin receptor 2B (5-HT2B) have received renewed interest for their potential to help understand the role of 5-HT2B in migraines, drug abuse, neurodegenerative diseases, and irritable bowel syndrome. To date, most of the ligands targeting 5-HT2B have been nitrogen-containing compounds. The natural product 5-hydroxy-2-(2-phenylethyl)chromone (5-HPEC, 5) has been shown previously to act as a non-nitrogenous antagonist for the 5-HT2B receptor (pKi = 5.6). This report describes further progress on the study of the structure–activity relationship of both naturally occurring and synthetic compounds bearing the 2-(2-phenylethyl)chromone scaffold at the 5-HT2B receptor. The inhibitory activity of the newly synthesized compounds (at 10 μM) was tested against each of the 5-HT2 receptors. Following this assay, the binding affinity and antagonism of the most promising compounds were then evaluated at 5-HT2B. Among all the analogues, 5-hydroxy-2-(2-phenylpropyl)chromone (5-HPPC, 22h) emerged as a new lead compound, showing a 10-fold improvement in affinity (pKi = 6.6) over 5-HPEC with reasonable antagonist properties at 5-HT2B. Additionally, ligand docking studies have identified a putative binding pocket for 5-HPPC and have helped understand its improved affinity.
Co-reporter:Guoyan G. Xu, Saheem A. Zaidi, Feng Zhang, Shilpa Singh, Thomas J. Raborg, Yunyun Yuan, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3721-3725
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.06.029
Prostate cancer is one of the leading causes of death among males in the world. Prostate cancer cells have been shown to express upregulated chemokine receptor CCR5, a G protein-coupled receptor (GPCR) that relates to the inflammation process. Anibamine, a natural product containing a pyridine ring and two aliphatic side chains, was shown to carry a binding affinity of 1 μM at CCR5 as an antagonist with potential anti-cancer activity. However, it is not drug-like according to the Lipinski’s rule of five mainly due to its two long aliphatic side chains. In our effort to improve its drug-like property, a series of anibamine derivatives were designed and synthesized by placement of aromatic side chains through an amide linkage to the pyridine ring. The newly synthesized compounds were tested for their CCR5 affinity and antagonism, and potential anti-proliferation activity against prostate cancer cell lines. Basal cytotoxicity was finally studied for compounds showing potent anti-proliferation activity. It was found that compounds with hydrophobic substitutions on the aromatic systems seemed to carry more promising CCR5 binding and prostate cancer cell proliferation inhibition activities.
Co-reporter:Yunyun Yuan, Xia Li, Saheem A. Zaidi, Christopher K. Arnatt, Xiaofei Yu, Chunqing Guo, Xiang-Yang Wang, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 16) pp:3179-3183
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmcl.2015.05.090
Scavenger receptor A (SRA) has been implicated in the processes of tumor invasion and acts as an immunosuppressor during therapeutic cancer vaccination. Pharmacological inhibition of SRA function thus holds a great potential to improve treatment outcome of cancer therapy. Macromolecular natural product sennoside B was recently shown to block SRA function. Here we report the identification and characterization of a small molecule SRA inhibitor rhein. Rhein, a deconstructed analog of sennoside B, reversed the suppressive activity of SRA in dendritic cell-primed T cell activation, indicated by transcription activation of il2 gene and production of IL-2. Rhein also inhibited SRA ligand polyinosinic:polycytidylic acid (poly(I:C)) induced activation of transcriptional factors, including interferon regulatory factor 3 (IRF3) and signal transducer and activator of transcription 1 (STAT1). Additionally, this newly identified lead compound was docked into the homology models of the SRA cysteine rich domain to gain insights into its interaction with the receptor. It was then found that rhein can favorably interact with SRA cysteine rich domain. Collectively, rhein, being the first identified small molecule inhibitors for SRA, warrants further structure–activity relationship studies, which may lead to development of novel pharmacological intervention for cancer therapy.
Co-reporter:Yunyun Yuan, Saheem A. Zaidi, David L. Stevens, Krista L. Scoggins, Philip D. Mosier, Glen E. Kellogg, William L. Dewey, Dana E. Selley, Yan Zhang
Bioorganic & Medicinal Chemistry 2015 23(8) pp: 1701-1715
Publication Date(Web):
DOI:10.1016/j.bmc.2015.02.055
Co-reporter:Christopher K. Arnatt, Joanna L. Adams, Zhu Zhang, Kendra M. Haney, Guo Li, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 10) pp:2319-2323
Publication Date(Web):15 May 2014
DOI:10.1016/j.bmcl.2014.03.073
Chemokine receptor CCR5 plays an important role in the pro-inflammatory environment that aids in the proliferation of prostate cancer cells. Previously, a series of CCR5 antagonists containing a piperidine ring core skeleton were designed based upon the proposed CCR5 antagonist pharmacophore from molecular modeling studies. The developed CCR5 antagonists were able to antagonize CCR5 at a micromolar level and inhibit the proliferation of metastatic prostate cancer cell lines. In order to further explore the structure–activity-relationship of the pharmacophore identified, the molecular scaffold was expanded to contain a piperazine ring as the core. A number of compounds that were synthesized showed promising anti prostate cancer activity and reasonable cytotoxicity profiles based on the biological characterization.
Co-reporter:Mostafa H. Ahmed, Glen E. Kellogg, Dana E. Selley, Martin K. Safo, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1158-1165
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2013.12.119
Cannabinoid receptors are a family of G-protein coupled receptors that are involved in a wide variety of physiological processes and diseases. One of the key regulators that are unique to cannabinoid receptors is the cannabinoid receptor interacting proteins (CRIPs). Among them CRIP1a was found to decrease the constitutive activity of the cannabinoid type-1 receptor (CB1R). The aim of this study is to gain an understanding of the interaction between CRIP1a and CB1R through using different computational techniques. The generated model demonstrated several key putative interactions between CRIP1a and CB1R, including the critical involvement of Lys130 in CRIP1a.
Co-reporter:Dwight A. Williams, Saheem A. Zaidi, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 6) pp:1489-1492
Publication Date(Web):15 March 2014
DOI:10.1016/j.bmcl.2014.02.029
Chromones are a class of natural products found in almost every known terrestrial plant with over 4000 naturally occurring derivatives having been isolated and structurally elucidated. Recently, 5-hydroxy-2-(2-phenylethyl)chromone (5-HPEC), isolated from Imperata cylindrical, showed neuroprotective activity against glutamate induced excitotoxicity in primary cultures of rat cortical cells. In comparison to other naturally occurring neuroprotective chromones, 5-HPEC contains fewer hydroxyl groups. Here we report our most recent characterization on this interesting natural product against a number of CNS receptors for the purpose to identify the potential molecular targets that may be related to its biological activity. Based on our studies, including radiobinding assays, calcium flux functional assays and molecular modeling studies, 5-HPEC may represent a type of novel nonnitrogenous ligands to the 5-HT2B receptor.
Co-reporter:Yunyun Yuan ; Saheem A. Zaidi ; Orgil Elbegdorj ; Lindsey C. K. Aschenbach ; Guo Li ; David L. Stevens ; Krista L. Scoggins ; William L. Dewey ; Dana E. Selley
Journal of Medicinal Chemistry 2013 Volume 56(Issue 22) pp:9156-9169
Publication Date(Web):October 21, 2013
DOI:10.1021/jm4012214
On the basis of a mu opioid receptor (MOR) homology model and the isosterism concept, three generations of 14-heteroaromatically substituted naltrexone derivatives were designed, synthesized, and evaluated as potential MOR-selective ligands. The first-generation ligands appeared to be MOR-selective, whereas the second and the third generation ones showed MOR/kappa opioid receptor (KOR) dual selectivity. Docking of ligands 2 (MOR selective) and 10 (MOR/KOR dual selective) to the three opioid receptor crystal structures revealed a nonconserved-residue-facilitated hydrogen-bonding network that could be responsible for their distinctive selectivity profiles. The MOR/KOR dual-selective ligand 10 showed no agonism and acted as a potent antagonist in the tail-flick assay. It also produced less severe opioid withdrawal symptoms than naloxone in morphine-dependent mice. In conclusion, ligand 10 may serve as a novel lead compound to develop MOR/KOR dual-selective ligands, which might possess unique therapeutic value for opioid addiction treatment.
Co-reporter:Christopher K. Arnatt, Saheem A. Zaidi, Zhu Zhang, Guo Li, Amanda C. Richardson, Joy L. Ware, Yan Zhang
European Journal of Medicinal Chemistry 2013 Volume 69() pp:647-658
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.09.004
•Design and syntheses of a series of novel chemokine receptor CCR5 antagonist.•Application of pharmacophore analysis and molecular modeling in molecular design.•Multi-tier in vitro biological screenings to characterize the synthesized analogs.•Further in vivo study to identify the next generation lead.Accumulating evidence has shown multiple roles that chemokine receptor CCR5 may play to promote the progression of several types of cancer. The mechanism of such promotion is believed to involve chronic inflammation that creates a microenvironment which enhances tumor survival. Therefore, blocking CCR5 function with an antagonist may provide a novel treatment of cancers such as prostate cancer. Currently, several CCR5 antagonists are available, but all have been optimized for their inhibitory activity on HIV-1 cellular membrane invasion process rather than inhibition on cytoplasmic signaling pathways. Thus, there is need to develop CCR5 antagonists focusing on blockage of CCR5 downstream signaling and inhibition of CCR5 related prostate cancer proliferation and progression. In this report, a pharmacophore analysis was conducted based on docking studies of several known CCR5 antagonists in a CCR5 homology model. A unique structural skeleton for CCR5 antagonist was constructed and functionalized, resulting in a new series of small molecules to be synthesized and characterized. A combination of CCR5 calcium flux inhibition, anti prostate cancer cell proliferation, basal cytotoxicity, and in vivo animal model studies were applied to screen the newly synthesized compounds. Results from this study provided a potential lead compound for future CCR5 antagonist development focusing on prostate cancer therapy.
Co-reporter:Saheem A. Zaidi, Christopher K. Arnatt, Hengjun He, Dana E. Selley, Philip D. Mosier, Glen E. Kellogg, Yan Zhang
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6405-6413
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.08.042
Highly selective opioid receptor antagonists are essential pharmacological probes in opioid receptor structural characterization and opioid agonist functional studies. Currently, there is no highly selective, nonpeptidyl and reversible mu opioid receptor antagonist available. Among a series of naltrexamine derivatives that have been designed and synthesized, two compounds, NAP and NAQ, were previously identified as novel leads for this purpose based on their in vitro and in vivo pharmacological profiles. Both compounds displayed high binding affinity and selectivity to the mu opioid receptor. To further study the interaction of these two ligands with the three opioid receptors, the recently released opioid receptor crystal structures were employed in docking studies to further test our original hypothesis that the ligands recognize a unique ‘address’ domain in the mu opioid receptor involving Trp318 that facilitates their selectivity. These modeling results were supported by site-directed mutagenesis studies on the mu opioid receptor, where the mutants Y210A and W318A confirmed the role of the latter in binding. Such work not only enriched the ‘message–address’ concept, also facilitated our next generation ligand design and development.NAP adopted a favorable binding mode in the mu opioid receptor to interact with W318 to achieve its selectivity to the receptor.
Co-reporter:Yan Zhang, Orgil Elbegdorj, Yunyun Yuan, Irina O. Beletskaya, Dana E. Selley
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3719-3722
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.05.027
Isosterism is commonly used in drug discovery and development to address stability, selectivity, toxicity, pharmacokinetics, and efficacy issues. A series of 14-O-substituted naltrexone derivatives were identified as potent mu opioid receptor (MOR) antagonists with improved selectivity over the kappa opioid receptor (KOR) and the delta opioid receptor (DOR), compared to naltrexone. Since esters are not metabolically very stable under typical physiological conditions, their corresponding amide analogs were thus synthesized and biologically evaluated. Unlike their isosteres, most of these novel ligands seem to be dually selective for the MOR and the KOR over the DOR. The restricted flexibility of the amide bond linkage might be responsible for their altered selectivity profile. However, the majority of the 14-N-substituted naltrexone derivatives produced marginal or no MOR stimulation in the 35S-GTP[γS] assay, which resembled their ester analogs. The current study thus indicated that the 14-substituted naltrexone isosteres are not bioisosteres since they have distinctive pharmacological profile with the regard to their opioid receptor binding affinity and selectivity.
Co-reporter:Yunyun Yuan, Orgil Elbegdorj, Irina O. Beletskaya, Dana E. Selley, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 18) pp:5045-5048
Publication Date(Web):15 September 2013
DOI:10.1016/j.bmcl.2013.07.043
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3′-carboxamido)morphinan (NAQ) was previously designed following the ‘message-address’ concept and was identified as a potent and highly selective mu opioid receptor (MOR) ligand based on its pharmacological profile. We here report the preliminary structure activity relationship (SAR) studies of this novel lead compound. For the new ligands synthesized as NAQ analogues, their binding assay results showed that a longer spacer and a saturated ring system of the side chain were unfavorable for their MOR selectivity over the kappa and delta opioid receptors. In contrast, substitutions with different electronic properties at either 1′- or 4′-position of the isoquinoline ring of the side chain were generally acceptable for reasonable MOR selectivity. The majority of NAQ analogues retained low efficacy at the MOR compared to NAQ in the 35S-GTP[γS] binding assays while electron-withdrawing groups at 1′-position of the isoquinoline ring induced higher MOR stimulation than electron-donating groups did. In summary, the electronic characteristics of substituents at 1′- or 4′-position of the isoquinoline ring in NAQ seem to be critical and need to be further tuned up to achieve higher MOR selectivity and lower MOR stimulation.NAQ analogues.
Co-reporter:Dwight A. Williams, Cameron Smith, Yan Zhang
Tetrahedron Letters 2013 Volume 54(Issue 32) pp:4292-4295
Publication Date(Web):7 August 2013
DOI:10.1016/j.tetlet.2013.06.006
Several 2-(2-phenylethyl)chromones have been shown to possess neuroprotective activity. However, limited synthetic methods have been disclosed to construct the 2-(2-phenylethyl)chromone skeleton. Herein, we report a straightforward 3-step preparation of five naturally occurring 2-(2-phenylethyl)chromones utilizing the Claisen condensation as the key step.
Co-reporter:Yunyun Yuan, Christopher K. Arnatt, Nazira El-Hage, Seth M. Dever, Joanna C. Jacob, Dana E. Selley, Kurt F. Hauser and Yan Zhang
MedChemComm 2013 vol. 4(Issue 5) pp:847-851
Publication Date(Web):21 Mar 2013
DOI:10.1039/C3MD00080J
Opioid substitution and antiretroviral therapies have steadily increased the life spans of AIDS patients with opioid addiction, while the adverse drug–drug interactions and persistence of HIV-associated neurocognitive disorders still require new strategies to target opioid abuse and HIV-1 comorbidities. A bivalent ligand 1 with a 21-atom spacer was thus synthesized and explicitly characterized as a novel pharmacological probe to study the underlying mechanism of opioid-enhanced NeuroAIDS. The steric hindrance generated from the spacer affected the binding affinity and Ca2+ flux inhibition functional activity of bivalent ligand 1 at the chemokine receptor CCR5 more profoundly than it did at the mu opioid receptor (MOR). However, the CCR5 radioligand binding affinity and the Ca2+ flux inhibition function of the ligand seemed not necessarily to correlate with its antiviral activity given that it was at least two times more potent than maraviroc alone in reducing Tat expression upon HIV-1 infection in human astrocytes. Furthermore, the ligand was also about two times more potent than the simple mixture of maraviroc and naltrexone in the same viral entry inhibition assay. Therefore bivalent ligand 1 seemed to function more effectively by targeting specifically the putative MOR–CCR5 heterodimer in the viral invasion process. The results reported here suggest that a properly designed bivalent ligand may serve as a useful chemical probe to study the potential MOR–CCR5 interaction during the progression of NeuroAIDS.
Co-reporter:Orgil Elbegdorj, Richard B. Westkaemper, Yan Zhang
Journal of Molecular Graphics and Modelling 2013 Volume 39() pp:50-60
Publication Date(Web):February 2013
DOI:10.1016/j.jmgm.2012.10.005
The orphan G-protein coupled receptor GPR55 was shown to bind to certain cannabinoid compounds which led to its initial classification as the third type of cannabinoid receptor. Later studies showed that lysophosphatidylinositol (LPI) also activated GPR55, in particular 2-arachidonoyl-LPI was proposed to be its endogenous ligand. However, the results of pharmacological studies regarding GPR55 have been quite inconsistent. Despite its contradictory pharmacological profile, GPR55 has been implicated in various disease states including inflammatory and neuropathic pain, metabolic bone diseases, and cancer. Herein, we report the ligand binding properties of GPR55 by applying homology modeling and automated docking algorithms in order to understand its pharmacological profile. The 3D homology model of GPR55 was built based on the adenosine A2A receptor crystal structure. Docking studies of several types of reported ligands were carried out afterwards. The results indicated that both hydrogen bonding and hydrophobic interactions contributed significantly for its ligand binding and the amino acid residue Lys80 seemed to be the anchor residue for receptor recognition. In addition, its putative agonist and antagonist appeared to recognize different domains of the receptor corresponding to their reported pharmacological activities.Graphical abstractGPR55 antagonist CBD binding mode in the GPR55 homology model.Highlight► We constructed the homology model of the orphan G-protein coupled receptor GPR55. ► A number of reported ligands were docked into the homology model of the receptor. ► Ligand binding sites were characterized and analyzed. ► Critical anchor amino acid residue was identified. ► Putative agonist and antagonist binding pockets were discussed.
Co-reporter:Yunyun Yuan ; Orgil Elbegdorj ; Jianyang Chen ; Shashidhar K. Akubathini ; Feng Zhang ; David L. Stevens ; Irina O. Beletskaya ; Krista L. Scoggins ; Zhenxian Zhang ; Phillip M. Gerk ; Dana E. Selley ; Hamid I. Akbarali ; William L. Dewey
Journal of Medicinal Chemistry 2012 Volume 55(Issue 22) pp:10118-10129
Publication Date(Web):November 1, 2012
DOI:10.1021/jm301247n
Peripheral selective μ opioid receptor (MOR) antagonists could alleviate the symptoms of opioid-induced constipation (OIC) without compromising the analgesic effect of opioids. However, a variety of adverse effects were associated with them, partially due to their relatively low MOR selectivity. NAP, a 6β-N-4′-pyridyl substituted naltrexamine derivative, was identified previously as a potent and highly selective MOR antagonist mainly acting within the peripheral nervous system. The noticeable diarrhea associated with it prompted the design and synthesis of its analogues in order to study its structure–activity relationship. Among them, compound 8 showed improved pharmacological profiles compared to the original lead, acting mainly at peripheral while increasing the intestinal motility in morphine-pelleted mice (ED50 = 0.03 mg/kg). The slight decrease of the ED50 compared to the original lead was well compensated by the unobserved adverse effect. Hence, this compound seems to be a more promising lead to develop novel therapeutic agents toward OIC.
Co-reporter:Yunyun Yuan, Christopher K. Arnatt, Guo Li, Kendra M. Haney, Derong Ding, Joanna C. Jacob, Dana E. Selley and Yan Zhang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 13) pp:2633-2646
Publication Date(Web):12 Jan 2012
DOI:10.1039/C2OB06801J
The bivalent ligand approach has been utilized not only to study the underlying mechanism of G protein-coupled receptors dimerization and/or oligomerization, but also to enhance ligand affinity and/or selectivity for potential treatment of a variety of diseases by targeting this process. Substance abuse and addiction have made both the prevention and the treatment of human immunodeficiency virus (HIV) infection more difficult to tackle. Morphine, a mu opioid receptor (MOR) agonist, can accelerate HIV infection through up-regulating the expression of the chemokine receptor CCR5, a well-known co-receptor for HIV invasion to the host cells and this has been extensively studied. Meanwhile, two research groups have described the putative MOR–CCR5 heterodimers in their independent studies. The purpose of this paper is to report the design and synthesis of a bivalent ligand to explore the biological and pharmacological process of the putative MOR–CCR5 dimerization phenomenon. The developed bivalent ligand thus contains two distinct pharmacophores linked through a spacer; ideally one of which will interact with the MOR and the other with the CCR5. Naltrexone and Maraviroc were selected as the pharmacophores to generate such a bivalent probe. The overall reaction route to prepare this bivalent ligand was convergent and efficient, and involved sixteen steps with moderate to good yields. The preliminary biological characterization showed that the bivalent compound 1 retained the pharmacological characteristics of both pharmacophores towards the MOR and the CCR5 respectively with relatively lower binding affinity, which tentatively validated our original molecular design.
Co-reporter:Feng Zhang, Christopher K. Arnatt, Kendra M. Haney, Harrison C. Fang, John E. Bajacan, Amanda C. Richardson, Joy L. Ware, Yan Zhang
European Journal of Medicinal Chemistry 2012 Volume 55() pp:395-408
Publication Date(Web):September 2012
DOI:10.1016/j.ejmech.2012.07.049
Recent studies have indicated that the CCR5 chemokine receptor may be a potential target for treating prostate cancer. Thus, development of CCR5 antagonists may provide novel prostate cancer therapy. Anibamine, a novel pyridine quaternary alkaloid isolated from Aniba sp., was found to effectively compete with 125I-gp120 in binding to the chemokine receptor CCR5, with an IC50 = 1 μM. Anibamine is the first natural product reported as a CCR5 antagonist, and thus provides a novel structural skeleton unique from other lead compounds that have generally been identified from high-throughput screening efforts. In order to refine the lead compound's structure and improve the therapeutic index of anibamine derivatives as potential anti prostate cancer agents, the approach of “deconstruction–reconstruction–elaboration” was applied in the structure–activity relationship studies of this work. Here, we report the design, syntheses and anti prostate cancer activities of anibamine and 17 analogues. The results from the in vitro and in vivo studies described here show that this class of compounds has potential to provide novel leads as anti prostate cancer agents.Graphical abstractAnibamine reduced the subcutaneous growth of M12 tumors in athymic nude mice by nearly 50% at 0.3 mg/kg dose, while the new lead by over 70% at the same dose.Highlights► Design and syntheses of a series of novel analogues of natural product chemokine receptor CCR5 antagonist, anibamine. ► Application of “deconstruction–reconstruction–elaboration” concept in molecular design. ► The adoption of total synthesis route of the parent natural product. ► Multi-tier in vitro biological screenings to characterize the synthesized analogues. ► Further in vivo study of the original and the second generation leads.
Co-reporter:Christopher K. Arnatt, Yan Zhang
Tetrahedron Letters 2012 Volume 53(Issue 13) pp:1592-1594
Publication Date(Web):28 March 2012
DOI:10.1016/j.tetlet.2012.01.066
Nitrocylcohexadienones have been applied as nitration reagents for mild, mono-nitrating reactions. The original synthesis of 2,3,5,6-tetrabromo-4-methyl-4-nitrocylcohexa-2,5-dien-1-one appeared to be difficult to pursue due to both the solvent system and reaction conditions. Therefore, we applied a modified solvent system and optimized the reaction conditions to prepare the dienone at 0 °C, to eventually overcome the difficulties.
Co-reporter:Yan Zhang, Christopher K. Arnatt, Feng Zhang, Jiannan Wang, Kendra M. Haney, Xianjun Fang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 15) pp:5093-5097
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmcl.2012.05.127
Chemokines and their receptors play important roles in the development of primary tumors and their metastases. Particularly CC chemokine receptor 5 (CCR5) and its ligand CC chemokine ligand 5 (CCL5/RANTES) seem to be critical in proliferation and invasion of ovarian cancer, the leading cause of death from gynecological malignancies in the United States. Anibamine, the first natural product CCR5 antagonist, and its analogues were examined for their effects on proliferation of the OVCAR-3 ovarian cancer cells in order to validate their candidacy as leads to develop novel anti-ovarian cancer agents. Acting as CCR5 antagonists, anibamine and its analogues significantly suppressed CCL5-induced intracellular Ca2+ flux. The compounds also inhibited the proliferation of OVCAR-3 at micromolar to submicromolar range. Moreover, anibamine and several analogues did not show significant cytotoxicity in NIH 3T3 cells at concentrations up to 20 μM. Based on these results, anibamine and one of its synthetic analogues were defined as potential leads to develop novel agents against ovarian cancer.Structure–activity relationship of natural product CCR5 antagonist anibamine was studied and all its derivatives were tested for their inhibition of ovarian cancer cell proliferation activity and basal cytotoxicity.
Co-reporter:Yunyun Yuan, David L. Stevens, Amanda Braithwaite, Krista L. Scoggins, Edward J. Bilsky, Hamid I. Akbarali, William L. Dewey, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4731-4734
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.075
A 6β-N-heterocyclic substituted naltrexamine derivative, NAP, was proposed as a peripheral mu opioid receptor (MOR) selective antagonist based on the in vitro and in vivo pharmacological and pharmacokinetic studies. To further validate this notion, several functional assays were carried out to fully characterize this compound. In the charcoal gavage and intestinal motility assay in morphine-pelleted mice, when administered 0.3 mg/kg or higher doses up to 3 mg/kg subcutaneously, NAP significantly increased the intestinal motility compared to the saline treatment. The comparative opioid withdrawal precipitation study and the lower locomotor assay demonstrated that NAP showed only marginal intrinsic effect in the central nervous system either given subcutaneously or intravenously: no jumps were witnessed for the tested animals even given up to a dose of 50 mg/kg, while similar noticeable wet-dog shakes only occurred at the dose 50 times of those for naloxone or naltrexone, and significant reduction of the hyper-locomotion only happened at the dose as high as 32 mg/kg. Collectively, these results suggested that NAP may serve as a novel lead to develop peripheral MOR selective antagonist which might possess therapeutic potential for opioid-induced bowel dysfunction (OBD), such as opioid-induced constipation (OIC).
Co-reporter:Yunyun Yuan, Guo Li, Hengjun He, David L. Stevens, Patrick Kozak, Krista L. Scoggins, Pallabi Mitra, Phillip M. Gerk, Dana E. Selley, William L. Dewey, and Yan Zhang
ACS Chemical Neuroscience 2011 Volume 2(Issue 7) pp:346
Publication Date(Web):May 6, 2011
DOI:10.1021/cn2000348
As important pharmacological probes, highly selective opioid receptor antagonists are essential in opioid receptor structural characterization and opioid agonist functional studies. At present, a nonpeptidyl, highly selective, and reversible mu opioid receptor antagonist is still not available. Among a series of novel naltrexamine derivatives that have been designed and synthesized following molecular modeling studies, two compounds, NAP and NAQ, were identified as leads based on the results of in vitro and in vivo pharmacological assays. Both of them displayed high binding affinity and selectivity to the mu opioid receptor. Further pharmacokinetic and functional characterization revealed that NAP seems to be a peripheral nervous system agent while NAQ seems to be a central one. Such characteristics provide two distinguished potential application routes for these two agents and their derivatives. These results also supported our hypothesis that they may serve as leads to develop more potent and selective antagonists for the mu opioid receptor.Keywords: Mu opioid receptor; NAP; NAQ; selective antagonist
Co-reporter:Kendra M. Haney, Feng Zhang, Christopher K. Arnatt, Yunyun Yuan, Guo Li, Joy L. Ware, David A. Gewirtz, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 18) pp:5159-5163
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmcl.2011.07.058
Prostate cancer is a leading cause of death among males in the United States. As the chemokine receptor CCR5 is over-expressed in more aggressive forms of prostate cancer, and is also a critical receptor in inflammation, chemokine receptor CCR5 antagonists could potentially act as anti-prostate cancer agents. Anibamine, a natural product CCR5 antagonist, provides a unique molecular scaffold for the generation of novel analogs with possible anti-prostate cancer activity. A series of analogs of anibamine were designed, synthesized and tested against several prostate cancer cell lines. The analogs all acted as CCR5 antagonists at micromolar range affinity to the receptor while their anti-proliferative activity varied depending on the cell line type and their chemical structural properties. Further basal cytotoxicity characterization on these compounds indicated some of them may be suitable for in vivo studies.A series of the natural product CCR5 antagonist anibamine analogs have been synthesized and tested for their inhibition of prostate cancer cell proliferation activity and cytotoxicity.
Co-reporter:Giang T. Ha;Ryan K. Wong
Chemistry & Biodiversity 2011 Volume 8( Issue 7) pp:1189-1204
Publication Date(Web):
DOI:10.1002/cbdv.201000269
Abstract
Alzheimer's disease (AD) is the fourth leading cause of death in adults, characterized by hallmark neuritic plaques and neurofibrillary tangles. Current treatments focus only on symptom relief. As a possible new treatment option for AD, huperzine A's chemistry, pharmacology, and clinical effectiveness are assessed. The chemical synthesis of huperzine A has been optimized, while an in vitro technique has provided a renewable plant source. Pharmacological studies showed that the drug inhibits the enzyme acetylcholinesterase reversibly and selectively. Huperzine A also displayed good pharmacokinetics with a rapid absorption and a wide distribution in the body at a low to moderate rate of elimination. Presently, inadequate toxicity data in human have been reported, yet animal studies demonstrated mild to moderate cholinergic side effects at therapeutic doses. Previous clinical trials have shown improvement in memory function using MMSE, MQ, ADAS-COG, and ADL tests. In an unpublished phase II clinical trial, the ADAS-COG and MMSE tests indicated cognitive enhancement at a dose of 0.4 mg, yet no improvement was observed at a dose of 0.2 mg. The MMSE scores indicated cognitive enhancement at 0.4 mg. Promising data suggested that huperzine A is well tolerated at doses up to 0.4 mg for 24 weeks. Therefore, huperzine A seems to be a potential treatment option for AD.
Co-reporter:Feng Zhang, Saheem Zaidi, Kendra M. Haney, Glen E. Kellogg, and Yan Zhang
The Journal of Organic Chemistry 2011 Volume 76(Issue 19) pp:7945-7952
Publication Date(Web):August 29, 2011
DOI:10.1021/jo2013669
The syntheses of the natural product anibamine and its three olefin isomers have been achieved concisely and efficiently via highly regio- and stereoselective reactions. The crucial steps included a regioselective palladium-catalyzed alkynylation by Sonogashira coupling and a stereoselective Suzuki coupling. Further conformation analyses and in vitro calcium mobilization studies were carried out to characterize the compounds’ biological properties.
Co-reporter:Xueping Zhang, Kendra M. Haney, Amanda C. Richardson, Eden Wilson, David A. Gewirtz, Joy L. Ware, Zendra E. Zehner, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 15) pp:4627-4630
Publication Date(Web):1 August 2010
DOI:10.1016/j.bmcl.2010.06.003
Accumulating evidence indicates that the chemokine receptor CCR5 and the chemokine CCL5 may be involved in the proliferation and metastasis of prostate cancer. Consequently, chemokine receptor CCR5 antagonists could potentially act as anti-prostate cancer agents. As the first natural product CCR5 antagonist, anibamine provides a novel chemical structural skeleton compared with other known antagonists identified through high-throughput screening. Our studies demonstrate that anibamine produces significant inhibition of prostate cancer cell proliferation at micromolar to submicromolar concentrations as well as suppressing adhesion and invasion of the highly metastatic M12 prostate cancer cell line. Preliminary in vivo studies indicate that anibamine also inhibits prostate tumor growth in mice. These findings indicate that anibamine may prove to be a novel lead compound for the development of prostate cancer therapeutic agents.Anibamine produces significant inhibition of prostate cancer cell proliferation at micromolar to submicromolar concentrations and inhibits prostate tumor growth in mice.
Co-reporter:Guo Li ; Lindsey C. Aschenbach ; Jianyang Chen ; Michael P. Cassidy ; David L. Stevens ; Bichoy H. Gabra ; Dana E. Selley ; William L. Dewey ; Richard B. Westkaemper
Journal of Medicinal Chemistry 2009 Volume 52(Issue 5) pp:1416-1427
Publication Date(Web):February 6, 2009
DOI:10.1021/jm801272c
Opioid receptor selective antagonists are important pharmacological probes in opioid receptor structural characterization and opioid agonist functional study. Thus far, a nonpeptidyl, highly selective and reversible μ opioid receptor (MOR) antagonist is unavailable. On the basis of our modeling studies, a series of novel naltrexamine derivatives have been designed and synthesized. Among them, two compounds were identified as leads based on the results of in vitro and in vivo assays. Both of them displayed high binding affinity for the MOR (Ki = 0.37 and 0.55 nM). Compound 6 (NAP) showed over 700-fold selectivity for the MOR over the δ receptor (DOR) and more than 150-fold selectivity over the κ receptor (KOR). Compound 9 (NAQ) showed over 200-fold selectivity for the MOR over the DOR and approximately 50-fold selectivity over the KOR. Thus these two novel ligands will serve as leads to further develop more potent and selective antagonists for the MOR.
Co-reporter:Guo Li, Kendra M. Haney, Glen E. Kellogg and Yan Zhang
Journal of Chemical Information and Modeling 2009 Volume 49(Issue 1) pp:120-132
Publication Date(Web):January 5, 2009
DOI:10.1021/ci800356a
Anibamine, a novel pyridine quaternary alkaloid recently isolated from Aniba sp., has been found to effectively bind to the chemokine receptor CCR5 with an IC50 at 1 μM in competition with 125I-gp120, an HIV viral envelope protein binding to CCR5 with high affinity. Since CCR5, a G-protein-coupled receptor, is an essential coreceptor for the human immunodeficiency virus type I (HIV-1) entry to host cells, a CCR5 antagonist that inhibits the cellular entry of HIV-1 provides a new therapy choice for the treatment of HIV. Anibamine provides a novel structural skeleton that is remarkably different from all lead compounds previously identified as CCR5 antagonists. Here, we report comparative docking studies of anibamine with several other known CCR5 antagonists in two CCR5 homology models built based on the crystal structures of bovine rhodopsin and human β2-adrenergic receptor. The binding pocket of anibamine has some common features shared with other high affinity CCR5 antagonists, suggesting that they may bind in similar binding sites and/or modes. At the same time, several unique binding features of anibamine were identified, and it will likely prove beneficial in future molecular design of novel CCR5 antagonists based on the anibamine scaffold.
Co-reporter:Guo Li, Lindsey C.K. Aschenbach, Hengjun He, Dana E. Selley, Yan Zhang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1825-1829
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2008.12.093
Mu opioid receptor antagonists have clinical utility and are important research tools. To develop non-peptide and highly selective mu opioid receptor antagonist, a series of 14-O-heterocyclic-substituted naltrexone derivatives were designed, synthesized, and evaluated. These compounds showed subnanomolar-to-nanomolar binding affinity for the mu opioid receptor. Among them, compound 1 exhibited the highest selectivity for the mu opioid receptor over the delta and kappa receptors. These results implicated an alternative ‘address’ domain in the extracellular loops of the mu opioid receptor.A series of 14-O-heterocyclic-substituted naltrexone derivatives were designed, synthesized, and evaluated. Among them, compound 1 showed binding affinity at subnanomolar level and highest selectivity for the mu opioid receptor.
Co-reporter:Guo Li, Philip D. Mosier, Xianjun Fang, Yan Zhang
Journal of Molecular Graphics and Modelling 2009 Volume 28(Issue 1) pp:70-79
Publication Date(Web):August 2009
DOI:10.1016/j.jmgm.2009.04.004
Lysophosphatidic acid (LPA) is a naturally occurring phospholipid that initiates a broad array of biological processes, including those involved in cell proliferation, survival and migration via activation of specific G protein-coupled receptors located on the cell surface. To date, at least five receptor subtypes (LPA1–5) have been identified. The LPA1–3 receptors are members of the endothelial cell differentiation gene (Edg) family. LPA4, a member of the purinergic receptor family, and the recently identified LPA5 are structurally distant from the canonical Edg LPA1–3 receptors. LPA4 and LPA5 are linked to Gq, G12/13 and Gs but not Gi, while LPA1–3 all couple to Gi in addition to Gq and G12/13. There is also evidence that LPA4 and LPA5 are functionally different from the Edg LPA receptors. Computational modeling has provided useful information on the structure–activity relationship (SAR) of the Edg LPA receptors. In this work, we focus on the initial analysis of the structural and ligand-binding properties of LPA4, a prototype non-Edg LPA receptor. Three homology models of the LPA4 receptor were developed based on the X-ray crystal structures of the ground state and photoactivated bovine rhodopsin and the recently determined human β2-adrenergic receptor. Docking studies of LPA in the homology models were then conducted, and plausible LPA binding loci were explored. Based on these analyses, LPA is predicted to bind to LPA4 in an orientation similar to that reported for LPA1–3, but through a different network of hydrogen bonds. In LPA1–3, the ligand polar head group is reported to interact with residues at positions 3.28, 3.29 and 7.36, whereas three non-conserved amino acid residues, S114(3.28), T187(EL2) and Y265(6.51), are predicted to interact with the polar head group in the LPA4 receptor models.
Co-reporter:Yan Zhang Dr.;Yuk Y. Sham Dr.;Ramkumar Rajamani Dr.;Jiali Gao ;Philip S. Portoghese
ChemBioChem 2005 Volume 6(Issue 5) pp:
Publication Date(Web):18 MAR 2005
DOI:10.1002/cbic.200400207
Three types of opioid receptors—mu, delta, and kappa—belong to the rhodopsin subfamily in the G protein-coupled receptor superfamily. With the recent characterization of the high-resolution X-ray crystal structure of bovine rhodopsin, considerable attention has been focused on molecular modeling of these transmembrane proteins. In this study, a homology model of the mu opioid receptor was constructed based on the X-ray crystal structure of bovine rhodopsin. A phospholipid bilayer was built around the receptor, and two water layers were placed on both surfaces of the lipid bilayer. Molecular-dynamics simulations were carried out by using CHARMM for the entire system, which consisted of 316 amino acid residues, 92 phospholipid molecules, 8327 water molecules, and 11 chloride counter ions—40 931 atoms altogether. The whole system was equilibrated for 250 ps followed by another 2 ns dynamic simulation. The opioid ligand naltrexone was docked into the optimized model, and the critical amino acid residues for binding were identified. The mu opioid receptor homology model optimized in a complete membrane–aqueous system should provide a good starting point for further characterization of the binding modes for opioid ligands. Furthermore, the method developed herein will be applicable to molecular model building to other opioid receptors as well as other GPCRs.
Co-reporter:Guo Li, Yan Zhang
Steroids (June 2007) Volume 72(Issues 6–7) pp:569-572
Publication Date(Web):1 June 2007
DOI:10.1016/j.steroids.2007.03.011
7α-Methylthio-3β-hydroxy-17α-pregn-4-ene-21,17-carbolactone and 7α-methylthio-3α-hydroxy-17α-pregn-4-ene-21,17-carbolactone, the two major metabolites of spironolactone, were prepared stereoselectively by exploring different types of reduction reagents. For the 3β-hydroxyl isomer, the application of NaBH4/CeCl3·7H2O gave the best result while for the 3α-hydroxyl isomer, K-selectride seemed to provide the highest stereoselectivity.
Co-reporter:Yi Zheng, Xia Li, Piyusha P. Pagare, Yunyun Yuan, Xiang-Yang Wang, Yan Zhang
Bioorganic & Medicinal Chemistry Letters (1 January 2017) Volume 27(Issue 1) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.bmcl.2016.11.029
Scavenger receptor A (SRA) has been known as an immunosuppressive factor and therefore therapeutic inhibition of SRA may be potentially exploited for cancer immunotherapy. Our previously work suggested that rhein may act as an inhibitor of SRA in reversing immunosuppression of SRA during T cells activation. Herein, three deconstruction analogs of rhein, compound 1, 2, and 3, were further studied as inhibitors of SRA. These three compounds, particularly compound 1, also known as a natural product danthron, enhanced T cells activation, indicated by increased transcriptional activation of interleukin 2 (Il2) gene, production of IL-2 protein, and proliferation of T cells. Additionally, the interaction between these compounds and SRA was studied by molecular modeling. Compound 1 showed a favorable binding mode with the cysteine rich domain of SRA protein compared to compound 2 and 3. Collectively, those results would provide insight for future design and development of next generation rhein derivatives as SRA inhibitors.
Co-reporter:Yunyun Yuan, Christopher K. Arnatt, Guo Li, Kendra M. Haney, Derong Ding, Joanna C. Jacob, Dana E. Selley and Yan Zhang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 13) pp:NaN2646-2646
Publication Date(Web):2012/01/12
DOI:10.1039/C2OB06801J
The bivalent ligand approach has been utilized not only to study the underlying mechanism of G protein-coupled receptors dimerization and/or oligomerization, but also to enhance ligand affinity and/or selectivity for potential treatment of a variety of diseases by targeting this process. Substance abuse and addiction have made both the prevention and the treatment of human immunodeficiency virus (HIV) infection more difficult to tackle. Morphine, a mu opioid receptor (MOR) agonist, can accelerate HIV infection through up-regulating the expression of the chemokine receptor CCR5, a well-known co-receptor for HIV invasion to the host cells and this has been extensively studied. Meanwhile, two research groups have described the putative MOR–CCR5 heterodimers in their independent studies. The purpose of this paper is to report the design and synthesis of a bivalent ligand to explore the biological and pharmacological process of the putative MOR–CCR5 dimerization phenomenon. The developed bivalent ligand thus contains two distinct pharmacophores linked through a spacer; ideally one of which will interact with the MOR and the other with the CCR5. Naltrexone and Maraviroc were selected as the pharmacophores to generate such a bivalent probe. The overall reaction route to prepare this bivalent ligand was convergent and efficient, and involved sixteen steps with moderate to good yields. The preliminary biological characterization showed that the bivalent compound 1 retained the pharmacological characteristics of both pharmacophores towards the MOR and the CCR5 respectively with relatively lower binding affinity, which tentatively validated our original molecular design.

![2H-Pyran-4-aminium,N-[[4-[[[6,7-dihydro-2-(4-methylphenyl)-5H-benzocyclohepten-8-yl]carbonyl]amino]phenyl]methyl]tetrahydro-N,N-dimethyl-,chloride (1:1)](/data/chemimg/22200/229005-80-5.png)
![2H-Pyran-4-aminium,N-[[4-[[[6,7-dihydro-2-(4-methylphenyl)-5H-benzocyclohepten-8-yl]carbonyl]amino]phenyl]methyl]tetrahydro-N,N-dimethyl-,chloride (1:1)](/data/chemimg/22200/229005-80-5_b.png)
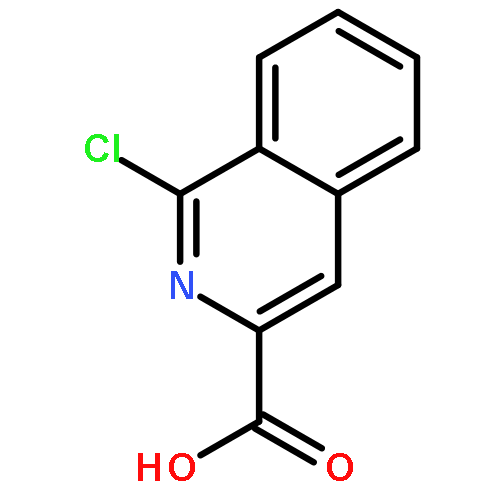
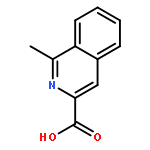
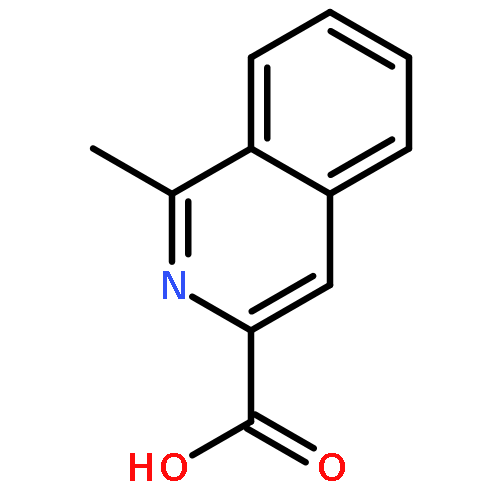
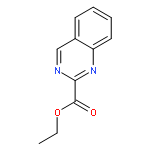
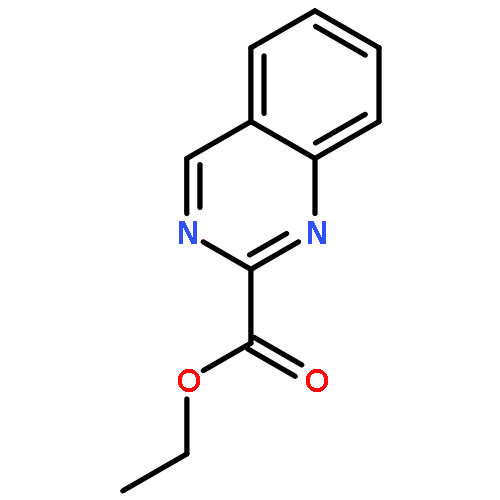
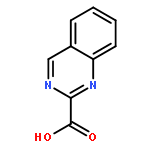
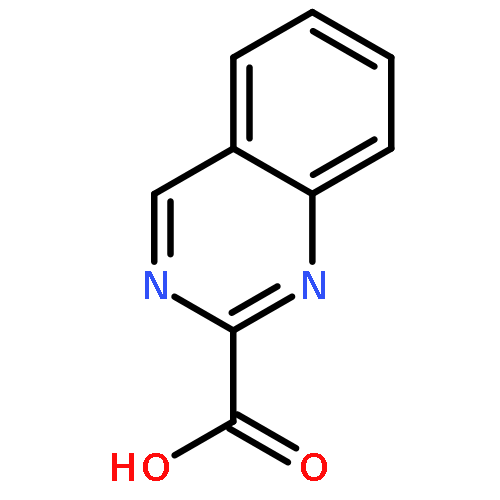
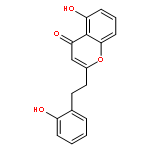
![3-[(1R)-1-AMINOPROPYL]BENZONITRILE HYDROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/349600/349552-70-1.png)
![3-[(1R)-1-AMINOPROPYL]BENZONITRILE HYDROCHLORIDE (1:1)](http://img.cochemist.com/ccimg/349600/349552-70-1_b.png)

![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6_b.png)


![acetic acid; (2S)-2-amino-N-[(1R)-1-[[[(1S)-1-(2-hydroxyethylcarbamoyl)-2-phenyl-ethyl]-methyl-carbamoyl]methylcarbamoyl]ethyl]-3-(4-hydroxyphenyl)propanamide](http://img.cochemist.com/ccimg/101000/100929-53-1.png)
![acetic acid; (2S)-2-amino-N-[(1R)-1-[[[(1S)-1-(2-hydroxyethylcarbamoyl)-2-phenyl-ethyl]-methyl-carbamoyl]methylcarbamoyl]ethyl]-3-(4-hydroxyphenyl)propanamide](http://img.cochemist.com/ccimg/101000/100929-53-1_b.png)


![DAMGO;[D-ALA2,NME-PHE4,GLY-OL5]-ENKEPHALIN](http://img.cochemist.com/ccimg/78200/78123-71-4.png)
![DAMGO;[D-ALA2,NME-PHE4,GLY-OL5]-ENKEPHALIN](http://img.cochemist.com/ccimg/78200/78123-71-4_b.png)
![2-[2-(1H-BENZIMIDAZOL-2-YL)ETHYL]-N-ETHYLHYDRAZINECARBOTHIOAMIDE](http://img.cochemist.com/ccimg/72100/72033-13-7.png)
![2-[2-(1H-BENZIMIDAZOL-2-YL)ETHYL]-N-ETHYLHYDRAZINECARBOTHIOAMIDE](http://img.cochemist.com/ccimg/72100/72033-13-7_b.png)




![Ethanone, 1-[2-hydroxy-4-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-08-6.png)
![Ethanone, 1-[2-hydroxy-4-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-08-6_b.png)









![1-[2-HYDROXY-6-(METHOXYMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/78700/78646-28-3.png)
![1-[2-HYDROXY-6-(METHOXYMETHOXY)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/78700/78646-28-3_b.png)