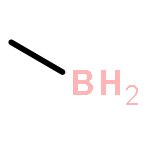
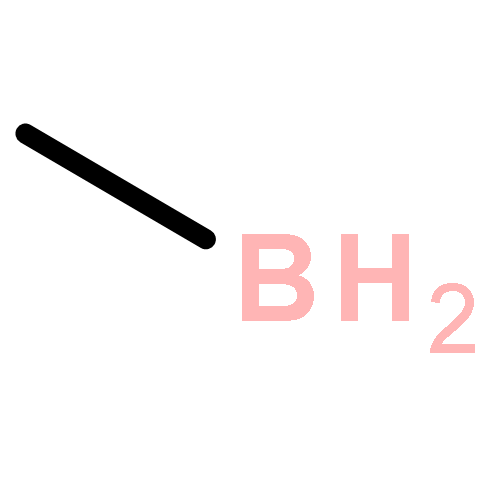
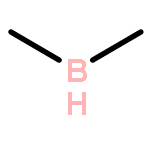
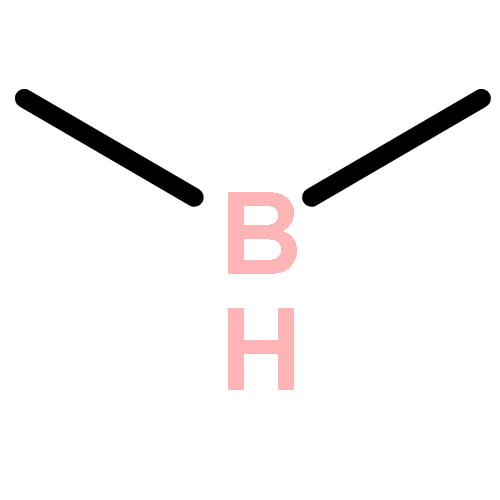
Co-reporter:Ying-ying Xue, Jing-jing Sui, Jing Xu, and Yi-hong Ding
ACS Omega September 2017? Volume 2(Issue 9) pp:5407-5407
Publication Date(Web):September 5, 2017
DOI:10.1021/acsomega.7b00487
It is well-known that the chemistry of aluminum is dominated by Al(III) in the +3 oxidation state. Only during the past 2 decades has the chemistry of Al(I) and Al(II) been rapidly developed. However, if Al(I) and Al(III) are combined, the inherently high reactivities of Al(I) and Al(III) mostly result in their coupling with each other or interacting with surrounding elements, which easily results in significant deactivation or quenching of the desired oxidation states, as in the case of reported mixed valent Al-compounds. In this article, we report an unprecedented type of organoaluminum system, C2Al4R4 (R = H, SiH3, Si(C6H5)3, SiiPrDis2, SiMe(SitBu3)2), whose lowest-energy structure, C2Al4R4-01, contains two Al(I) and two Al(III) atoms. The global nature and bonding motif of the parent C2Al4R4-01 (R = H) were supported by an extensive global isomeric search, CBS-QB3 energy calculations, adaptive natural density partitioning, and bond order analysis. Interestingly and in sharp contrast to most organoaluminum species, C2Al4R4-01 is associated with little multicenter bonding. C2Al4R4-01 has a high feasibility of being observed either in the gas or condensed phases (with suitable substitutents). With well-separated Al(I) and Al(III), C2Al4R4-01 (with suitable substitutents) could serve as the first Al/Al frustrated Lewis pair.Topics: Electronic structure; Enthalpy; Enthalpy; Frustrated Lewis pairs; Molecular structure; Molecular structure-property relationship; Organometallic chemistry; Organometallic compounds; Thermodynamic properties;
Co-reporter:Nan-nan LiuYing-ying Xue, Yi-hong Ding
The Journal of Physical Chemistry A February 9, 2017 Volume 121(Issue 5) pp:
Publication Date(Web):January 12, 2017
DOI:10.1021/acs.jpca.6b11066
[5]Radialene, the missing link for synthesis of radialene family, has been finally obtained via the preparation and decomplexation of the [5]radialene–bis-Fe(CO)3 complex. The stability of [5]radialene complex benefits from the coordination with Fe(CO)3 by losing free 1,3-butadiene structures to avoid polymerization. In light of the similar coordination ability of half-sandwiches CpM(Cp = η5-C5H5; M = Fe, Co, Ni), there is a great possibility that the sandwiched complexes of [5]radialene with CpM are available. Herein, we present the first theoretical prediction on the geometry, spin states and bonding of (CpM)(C10H10) and (CpM)2(C10H10). For M = Fe, Co, Ni, the ground states of (CpM)(C10H10) and (CpM)2(C10H10) are doublet and triplet, singlet and singlet, and doublet and triplet states, where each Fe, Co, and Ni adopts 17, 18, and 19 electron-configuration, respectively. In particular, (CpFe)2(C10H10) and (CpNi)2(C10H10) have considerable open-shell singlet features. Generally the trans isomers of (CpM)2(C10H10) with two CpM fragments on the opposite sides of the [5]radialene plane are apparently more stable than the cis ones with CpM fragments on the same side. However, for the singlet and triplet isomers of (CpNi)2(C10H10) (both cis and trans isomers), the energy differences are relatively small, indicating that these isomers all have the opportunity to exist. Besides, the easy Diels–Alder (DA) dimerization between the [3]dendralene-like fragments of (CpM)(C10H10) suggests the great difficulty in isolating the (CpM)(C10H10) monomer.
Co-reporter:Nannan Liu, Shuang Yu, Yihong Ding
Journal of Organometallic Chemistry 2017 Volume 828() pp:75-82
Publication Date(Web):1 January 2017
DOI:10.1016/j.jorganchem.2016.11.023
•[3]radialene might be Y-aromatic instead of traditionally cyclic aromatic.•The LUMO of radialene reshapes to be cyclic aromatic when forming a dative bond.•The metal-aromatic interaction may exist between metal and radialene in CpM(C2nH2n).The coordination and electron configuration of radialene complexes CpM(C2nH2n) (n = 3, 4) are investigated. For M = Sc, Ti, Fe, Co, Ni, the ground states of CpM(C2nH2n) are low-spin states (singlet or doublet), while for M = V, Cr, Mn, the ground states are high-spin states (triplet or quartet). For M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, [3]radialene in the ground states of CpM(C6H6) are ŋ6-, ŋ6-, ŋ6-, ŋ4-, ŋ4-, ŋ4-, ŋ4-, and ŋ2-coordinated; and [4]radialene in the ground states of CpM(C8H8) are ŋ8-, ŋ8-, ŋ6-, ŋ4-, ŋ4-, ŋ4-, ŋ4-, and ŋ2-coordinated, respectively. Between metal atom and radialene in CpM(C2nH2n), the metal-alkene interactions, which are caused by the exocyclic CC π-bonds that donate the lone pair electrons to the empty d-orbitals of metal atom, are dominant and fundamental. Besides, the special π-backbonding metal-aromatic interactions, which is overlapped by the lone pair on metal atom and the empty 2π-aromatic orbital on the cyclic C3 or C4 ring of [n]radialenes, may also exist in some ground states of CpM(C2nH2n) complexes.The coordination and electron configuration of radialene complexes CpM(C2nH2n) are investigated. In spite of the [3]- and [4]radialene and their dianions generally being non-aromatic, the special π-backbonding metal-aromatic interactions may exist in CpM(C2nH2n) complexes.
Co-reporter:Jing-fan Xin;Fei-fei He
RSC Advances (2011-Present) 2017 vol. 7(Issue 14) pp:8533-8541
Publication Date(Web):2017/01/23
DOI:10.1039/C6RA28358F
Seeking high-energy-density materials (HEDMs) with balanced huge energy release and good stability has remained quite a tough task for both experimentalists and theoreticians. The current HEDM design mostly concentrates on the chemical modification of either the skeletons or ligands. To increase the number of HEDM candidates, a novel design strategy is highly desired. In this paper, we computationally proposed a bottom-up strategy, i.e., a suitable HEDM seed (e.g., cyc-N2CO) can form novel HEDMs while retaining good stability and good performance. Starting from the experimentally known diazirinone (cyc-N2CO) as a “seed” and by considering various bond-addition channels (2 + 2/2 + 3/3 + 3 cyclo-addition at the NN/CO/C–N bonds), we found that the cyc-N2CO dimer isomer 1 (i.e., (N2CO)2 containing a COCO ring with an exocyclic side-N2 at each C-atom) possess the rate-determining barrier of 29.9 kcal mol−1 and exothermicity of 168.7 kcal mol−1 into 2N2 + 2CO at the composite CBS-QB3 level. Moreover, the trimer and tetramer of cyc-N2CO each possess high rate-determining barriers of 25.8 and 30.3 kcal mol−1, respectively, at the CBS-QB3 level. Even higher oligomers with n = 5–8 have rate-determining barriers around 25 and 34 kcal mol−1. The spiral skeletons were shown to have a contribution to their good inherent kinetic stability. By comparing the detonation properties with some known HEDM compounds, the oligomers of cyc-N2CO may well deserve future synthetic trials for novel HEDMs. Our designed (N2CO)n with all the untouched NN bonds differed sharply from the recently reported high-pressure polymerized forms, in which all the double bonds have been transformed into single bonds. The present bottom-up strategy from an HEDM seed (i.e., cyc-N2CO) to novel oligomeric HEDMs confirmed by the CBS-QB3 calculations seems to be quite promising and may open a new way of designing in the HEDM realm.
Co-reporter:Qingyun Wang;Yihong Ding
Journal of Molecular Modeling 2017 Volume 23( Issue 2) pp:
Publication Date(Web):2017 February
DOI:10.1007/s00894-017-3249-4
Understanding the bond-cleavage ability of metal clusters is very important in various fields, such as catalysis and surface science. In this work, we performed density functional theory calculations on the first dehydrogenation process (also the key step) of methanol on Ptnq (n = 1–3, q = 0, +1, −1) clusters in varied charge states using quantum chemical calculations. It is shown that methanol is adsorbed much more easily to the cationic Ptn+ than to the neutral and anionic Ptn0/−. By contrast, the intrinsic bond cleavage barriers of both C–H and O–H on the cationic Ptn+ are significantly higher than on Ptn0/− (the only exception is the C–H bond cleavage on Pt+). Promisingly, injecting an electron to the neutral Ptn0 to give Ptn− can greatly reduce the C–H/O–H bond scission barrier while maintaining appreciable adsorption energy. The charging effect can be nicely interpreted by the nature of the frontier orbitals of Ptnq.
Co-reporter:Zhong-hua Cui;Valentin Vassilev-Galindo;José Luis Cabellos;Edison Osorio;Mesías Orozco;Sudip Pan;Gabriel Merino
Chemical Communications 2017 vol. 53(Issue 1) pp:138-141
Publication Date(Web):2016/12/20
DOI:10.1039/C6CC08273D
Viable planar pentacoordinate carbon (ppC) systems with a ppC bonded to a transition metal and embedded in a metallocene framework are reported. Our detailed global minima search shows that CAl4MX2 (M = Zr and Hf; X = F–I and C5H5) clusters with ppCs are appropriate candidates for experimental realization in the gas phase. The fulfillment of the 18 electron rule and electron delocalization is found to be crucial for the stabilization of these ppC arrangements.
Co-reporter:Qing-Yun Wang, Yi-Hong Ding
Electrochimica Acta 2016 Volume 216() pp:140-146
Publication Date(Web):20 October 2016
DOI:10.1016/j.electacta.2016.08.052
Graphene has been increasingly used as an effective support for Pt-electrocatalyst in the direct methanol fuel cells (DMFCs). Very recently, it has been found that introducing the defects in graphenes can not only stabilize the Pt-particles on graphene, but also greatly increase the tolerance to CO-poisoning. Here we explored for the first time the influence of graphene-defects on the Pt-electrocatalytic behavior. At the B3LYP and M06 (single-point) levels, the adsorption and the first dehydrogenation (C-H/O-H) of methanol on Pt supported by three types of graphenes, i.e., pristine, in Stone-Wales (SW) defect and in single-vacancy (SV) defect. The calculations revealed that the SV-defect has the most stabilization and CO-tolerance for Pt, compared to SW- and pristine graphenes. However, meanwhile, the SV-defect significantly increases the intrinsic C-H/O-H cleavage barrier, which would deteriorate the electrocatalytic activity of Pt.
Co-reporter:Jing-jing Sui, Jing Xu and Yi-hong Ding
Dalton Transactions 2016 vol. 45(Issue 1) pp:56-60
Publication Date(Web):16 Nov 2015
DOI:10.1039/C5DT03989D
Through a global isomeric study, we computationally identified the first structural template C2Si2X that could encompass a planar tetracoordinate X for all the heavier group 14 elements X in the 0, +1 or −1 charge state. We thus significantly expanded the traditional 16/17/18ve rules to 19/20/21ve for ptX.
Co-reporter:Fei-fei He and Yi-hong Ding
RSC Advances 2016 vol. 6(Issue 31) pp:26441-26450
Publication Date(Web):01 Mar 2016
DOI:10.1039/C5RA27576H
Molecules containing carbon (C), nitrogen (N) and oxygen (O) atoms have received considerable attention due to their great relevance in astrophysical, atmospheric and high-energy-density material (HEDM) realms. Greatly differing from most studies that mainly center on the low-lying isomers, study of C, N and O systems appeals for the additional consideration of high-energy species. Thus, understanding the thermodynamic and kinetic stability of diversified isomers is of vital importance to assess their role in these processes. In this work, we investigated a pentatomic CN2O2 system, the isomeric study of which was initiated 20 years ago and up to now its three isomers OCNNO, CNNO2 and NCNO2 have been experimentally characterized. Based on our global search strategies for both isomers and transition states, we constructed hitherto the most comprehensive potential energy surface of CN2O2, covering 15 new isomers and 29 new transition states. The ring-containing isomers, i.e., 14, 22 and 29, were shown to possess considerable rate-determining Gibbs free energy barriers with respect to the radical–radical (P3 NCO + NO, P6 3NCN + 3O2 or P10 3NNC + 3O2) and lowest-energy product (P1 CO2 + N2) at the (U)CCSD(T)/CBS level. Thus, they are expected to be experimentally observable. After the experimentally known OCNNO, CNNO2 and NCNO2, the presently found three isomers 14, 22 and 29 warmly welcome future laboratory investigations. In addition, for CNNO2 09, we located a previously unreported transition state, which provides a new viewpoint on its kinetic stability.
Co-reporter:Nannan Liu;Yihong Ding
Science China Chemistry 2016 Volume 59( Issue 6) pp:760-764
Publication Date(Web):2016 June
DOI:10.1007/s11426-016-5590-3
The bridging Re–Xe–Re bond with a remarkable stability is firstly predicted. The average binding energies for Re–Xe bond in Re2Cp2(PF3)4Xe with bridging Xe are calculated to be higher than that in ReCp(CO)2Xe, ReCp(CO)(PF3)Xe and ReCp(PF3)2Xe with terminal Xe. The interaction between two ReCp(PF3)2 fragments provides an additional contribution for the stability of bridging Re–Xe–Re bond. Besides, the Re2Cp2(PF3)4Xe isomers with bridging Xe are also stable in energy than the isomers with bridging PF3. As the terminal Re–Xe bond was found to exist in experiments, the more stable bridging Re–Xe–Re bond might be existent under similar or even milder condition.
Co-reporter:Fei-fei He, Si-meng Gao, Giulia de Petris, Marzio Rosi, and Yi-hong Ding
The Journal of Physical Chemistry A 2016 Volume 120(Issue 27) pp:4812-4817
Publication Date(Web):February 11, 2016
DOI:10.1021/acs.jpca.5b12275
Fulminates containing the CNO– ion have been widely utilized as high-energy density materials (HEDMs) for more than 120 years. Yet no purely covalently bound CNO molecule, i.e., nitrile oxide, is known to behave as an HEDM. In this study, we performed a thorough investigation of the potential energy surface of nitrile oxide ONCNO and related isomers, applying various sophisticated methods including G4, CBS-QB3, W1BD, CCSD(T)/CBS, and CASPT2/CBS. The Gibbs free energy calculations showed that the decomposition of ONCNO to the considerably endothermic products CNO + NO is favored compared to that into the highly exothermic products CO2 + N2. Thus, ONCNO fails to be the long expected nitrile oxide HEDM. However, with the rate-determining barrier of 23.3 kcal mol–1 at the W1BD level, ONCNO should be experimentally accessible.
Co-reporter:Nan-nan Liu, Si-meng Gao and Yi-hong Ding
Dalton Transactions 2015 vol. 44(Issue 1) pp:345-350
Publication Date(Web):23 Sep 2014
DOI:10.1039/C4DT02282C
The inverse sandwich Ca–C8H8–Ca is predicted to be an open-shell singlet state. Since the C8H8 ligand prevents the spin-up and spin-down electrons of different calcium atoms from forming Ca–Ca bonds, the spin-coupling electrons lead to a singlet diradical character. The metal–ligand interaction contributes to the stability of Ca–C8H8–Ca against dissociation and isomerization. For the coordination complex (DME)3Ca–C8H8–Ca(DME)3, the open-shell singlet state is unavailable, while the closed-shell singlet state with direct Ca–Ca bonds is more favorable, because dimethyl ether molecules could push the spin-paired electrons of different calcium atoms to migrate towards the direction of Ca–Ca bonding. For Ca–C4H4–Ca, the ground state is an open-shell singlet state, of which the diradical character is very similar to that of Ca–C8H8–Ca. For (DME)3Ca–C4H4–Ca(DME)3, the lowest energy is the triplet state.
Co-reporter:Zhong-hua Cui, Jing-jing Sui and Yi-hong Ding
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 47) pp:32016-32022
Publication Date(Web):30 Oct 2015
DOI:10.1039/C5CP04776E
With the high preference in forming multi-center bonding, boron has been a miracle ligand in constructing diverse planar multi-coordinate (pM) (tetra/hyper) species. Unfortunately, the boron ligand usually dislikes encompassing a pM carbon (pMC) due to the high competition with pM boron (pMB), which makes the realization of boron-based pMC very difficult and quite challenging. Herein, we propose a strategy that by means of cooperative doping and charge-compensation, we can successfully improve and tune the stability of pMC relative to pMB for CB42−. In the free CBxEy2− (E = Al/Ga) species, ptC is thermodynamically less stable than the global ptB in mono- and di-substituted systems, in agreement with the results of Boldyrev and Wang. However, the thermodynamic preference of pMC increases along with the Al/Ga-doping. The pMC species can be further stabilized by the introduction of the alkaline-earth counterion (Mg2+). CB2E2Mg (E = Al, Ga) designed in the present study represents the first successful design of a boron-based planar penta-coordinate carbon (ppC) structures as the global minima. The strategy proposed in this study should be useful in the manipulation of competition between exotic pMC and pMB in B-based systems.
Co-reporter:Nan-nan Liu and Yi-hong Ding
New Journal of Chemistry 2015 vol. 39(Issue 3) pp:1558-1562
Publication Date(Web):10 Dec 2014
DOI:10.1039/C4NJ01832J
The Sc–Sc bond through the hole of C8H8 in X[Sc–C8H8–Sc]X (X = F∼Br) and the uninterrupted Sc–Sc–Sc–Sc bond-chain with both the Sc–Sc bond through C8H8 and the Sc–Sc bond between two [Sc–C8H8–Sc] units in X[Sc–C8H8–Sc]2X are predicted. The molecules are roughly estimated as potentially electro-conductive by the low HOMO–LUMO gaps.
Co-reporter:Jing-jing Sui, Jing Xu and Yi-hong Ding
RSC Advances 2015 vol. 5(Issue 122) pp:101193-101199
Publication Date(Web):17 Nov 2015
DOI:10.1039/C5RA22310E
The composition and valence electrons of molecules usually have a great impact on the eventual topology. With a high tendency to form sp/sp2-hybridized multiple bonding of C2, the main-group dicarbides (C2Xn) usually adopt a non-closo (or open) topology. By contrast, naked Zintl ions (e.g., Si52−) usually feature deltahedral structures. In this paper, we report an unexpected example of a pentatomic carbon-silicon cluster C2Si32− which has the global minimum C2Si32−-01 featuring a closo-structure with all deltahedras. The global minimum nature of C2Si32−-01 was confirmed by various sophisticated methods including G3B3, G4, CBS-QB3 and W1BD as well as CCSD(T) extrapolated up to the complete basis set limit based on the CCSD(T)/aug-cc-pVTZ, CCSD(T)/aug-cc-pVQZ and CCSD(T)/aug-cc-pV5Z calculations. The AdNDP analysis revealed that C2Si32−-01 possesses a lone pair of electrons on each of the three silicon atoms, four 2c–2e bonds and four 3c–2e bonds, among which the two 2c–2e bonds between the two carbons indicate the existence of a multiply bonded C2 (1.320 Å) that carries the most negative charges. With a total of 22 valence electrons, C2Si32−-01 formally resembles the known Wade–Mingos clusters with (n + 1) polyhedral skeletal electron pairs (PSEPs). Replacement of Si2 by the highly electron-withdrawing C2 does not break the deltahedral topology of the Zintl ion Si52−. To the best of our knowledge, C2Si32− represents the smallest deltahedral main-group dicarbide and also the first deltahedral main-group dicarbide with (n + 1) PSEPs. To direct its organometallic applications, we designed the hetero-deckered sandwich compounds CpMg(C2Si32−)MgCp, in which the C2Si32−-01 unit can be nicely maintained.
Co-reporter:Fei-fei He, Si-meng Gao, Giulia de Petris, Marzio Rosi and Yi-hong Ding
RSC Advances 2015 vol. 5(Issue 111) pp:91581-91586
Publication Date(Web):20 Oct 2015
DOI:10.1039/C5RA19895J
Seeking promising molecular species with huge energy release and significant kinetic stability continues to be a hot topic and a great challenge in the field of high-energy density materials (HEDMs). CO4 is the first high-order carboxide that has the potential as an energetic molecule. However, the intrinsic kinetic stability of its two most studied energy-rich isomers, i.e., 11 (monocyclic) and 12 (bicyclic), has remained quite unclear in spite of numerous studies. This has greatly hindered the quantitative stability assessment of 11 and 12 under various conditions as well as the justification of their prospect as energetic candidates. In this work, for the first time we report the rate-determining transition states associated with the CO2-elimination from 11 and 12. The thermodynamics of 11 and 12 was described using G3B3, CBS-QB3, G4, W1BD, CCSD(T)/CBS and CASPT2/CBS, while the kinetic stability was analyzed based on broken-symmetry UCCSD(T)/CBS and CASPT2/CBS single-point energy calculations on UB3LYP geometries. The rate-determining barriers for the dissociation of 11 and 12 into CO2 + 1O2 at 298 K were found to amount to 28.7 and 14.7 kcal mol−1 at the CASPT2(18e,12o)/CBS level of theory, and 23.5 and 21.1 kcal mol−1 at the UCCSD(T)/CBS level of theory, respectively. 11 is a kinetically stable energetic molecule, which releases 45.2 kcal mol−1 upon dissociation into CO2 + 1O2 at the CASPT2(18e,12o)/CBS level and 38.9 kcal mol−1 at the UCCSD(T)/CBS level, and could serve as a rigid energetic building block for larger oxocarbons. The bicyclic 12 releases much higher energy, 79.3 kcal mol−1 at the CASPT2(18e,12o)/CBS level and 73.4 kcal mol−1 at the CASPT2-corrected UCCSD(T)/CBS level whereas the barrier for dissociation is lower than that of monocyclic 11.
Co-reporter:Yu Tian, Yue-jie Liu, Jing-xiang Zhao and Yi-hong Ding
RSC Advances 2015 vol. 5(Issue 43) pp:34070-34077
Publication Date(Web):08 Apr 2015
DOI:10.1039/C5RA02585K
We investigated the structural and electronic properties of Pt13 nanoparticles on various nitrogen (N)-doped graphene and their interaction with O by density functional theory (DFT) calculations. The results revealed that the N-doping can greatly enhance the binding strength of Pt13 nanoparticles on the graphene surface, thus ensuring their high stability. For NC doping (N atoms directly substituting for C atoms), the enhanced binding strength of the Pt13 cluster is attributed to the activation of the carbon atoms around the N-dopant, while the strong hybridization of the d states of the Pt13 cluster with the sp2 dangling bonds of the N atoms in defective N-doped graphenes contributes to the strong adsorption. Moreover, a certain amount of electrons are transferred from Pt13 to the substrate accompanied by a substantial downshift of the Pt13 d-band center, thus greatly weakening the interaction of O on these composites: the adsorption energy of O is reduced from −3.700 eV on freestanding Pt13 nanoparticles to −1.762, −1.723, and −1.507 eV on deposited Pt13 ones on NC, 3NV, and 4ND structures, respectively. Hence, it is expected that N-doped graphene supported Pt nanoparticles exhibit super catalytic reactivity in the ORR.
Co-reporter:Xiao-yong Zhang and Yi-hong Ding
RSC Advances 2015 vol. 5(Issue 34) pp:27134-27139
Publication Date(Web):10 Mar 2015
DOI:10.1039/C5RA02178B
Due to their fantastic structures and reactivity, carbon–boron mixed cluster hydrides have aroused increasing attention both experimentally and theoretically. Generally, n-vertex borane and carborane containing equal to or more than (n + 1) polyhedral skeletal electron pairs (PSEPs) present similar structural characteristics. However, for the n-PSEP boranes and carboranes, whether their structural pattern is similar or not is still uncertain. In this work, we report a particular example, C2B4H4, the global minimum of which does not conform to the well-known polyhedral skeletal electron pair theory (PSEPT). Through extensive isomeric searching, the global minimum structure of C2B4H4 is addressed to exhibit a ribbon-like structure at the aug-cc-pVTZ-CCSD(T)//B3LYP level. However, the hypercloso structure predicted by PSEPT lies 16.0 kcal mol−1 higher. Our results are also different from a recent theoretical study. These findings are believed to enrich the understanding of hypercloso chemistry of carbon-substituted borane derivatives.
Co-reporter:Xiao-yong Zhang and Yi-hong Ding
RSC Advances 2014 vol. 4(Issue 83) pp:44214-44222
Publication Date(Web):05 Sep 2014
DOI:10.1039/C4RA05523C
Structures and transport behaviors around the ionomer–catalyst interface in polymer electrolyte membrane fuel cells (PEMFCs) have aroused great research interests in recent years. Herein, classical molecular dynamics simulation method is used to investigate the interfacial self-assembly phenomena of three fully hydrated (λ = 23) Nafion films with thicknesses of 2.4, 5.0 and 7.3 nm on the platinum surface. Interestingly, it is found that in the vicinity of the platinum surface, there is an ultra-dense adhesive ionomer layer with a thickness of 0.5 nm, whose compositions are not affected by the hydration levels and film thickness. Due to the lack of sulfonate groups, the Nafion ionomer in regions away from the Pt slab are reorganized in different patterns for films with different thicknesses. Besides this, we have found a thickness-dependence of the wetability of the surfaces exposed to the air in these fully hydrated films. It is also shown that the transport properties of hydronium ions and water molecules in the interfacial films are closely related to film morphologies. Water molecules in the 5.0 nm film are found to possess the lowest mobility as a result of the weakest connectivity of the hydrophilic channels, while in the 7.3 nm film, water diffusion is the fastest since the water channels are most ideally connected throughout this film. Notably, though water molecules cannot be retained inside the ultrathin 2.4 nm film, they could mostly develop into linear hydrophilic channels over the ionomer matrix, which can also provide transport pathways for hydrophilic species without interruption.
Co-reporter:Bo Xiao, Xuefang Yu, Hong Hu, Yihong Ding
Chemical Physics Letters 2014 Volume 608() pp:277-283
Publication Date(Web):21 July 2014
DOI:10.1016/j.cplett.2014.05.095
Highlights
- •
A new kind of ptN structure is obtained via Be-decorated armchair BNNR.
- •
The high thermal stability of such ptN system is confirmed.
- •
The transport properties of such ptN system are significantly enhanced.
Co-reporter:Nan-nan Liu, Yi-hong Ding
Computational and Theoretical Chemistry 2014 Volume 1045() pp:29-34
Publication Date(Web):1 October 2014
DOI:10.1016/j.comptc.2014.06.019
•The Cr–Xe bond energy in bridging Cr–Xe–Cr is slightly lower than terminal Cr–Xe.•The bridging Xe has the possibility to exist at low temperature like terminal Xe.•Xe atom could donor more than one pair electrons.Bridging Xe atom between two Lewis acidic transition metal centers is theoretically proposed. The structures, stabilities and bonding of Cr2(CO)10Xe, Cr2(CO)8Xe2, as well as Cr2(ŋ4-C4H4)4(PF3)2Xe and Cr2(ŋ4-C4H4)4Xe2 containing bridging Xe are detailed studied. Xe atom acts as an electron donor in above complexes. The predicted bond energies for Cr–Xe in these complexes with bridging Xe are lower compared with the terminal Cr(CO)5Xe. However, the dissociation energies of Cr2(ŋ4-C4H4)4(PF3)2Xe and the intermediate product Cr2(ŋ4-C4H4)4Xe, are nearly the same to the existing terminal Cr(CO)5Xe. As being relatively stable against dissociation, once generate, the bridging Xe atom might exist at low temperature like the terminal Xe.Graphical abstract
Co-reporter:Dr. Guosheng Shi;Jinrong Yang; Yihong Ding; Haiping Fang
ChemPhysChem 2014 Volume 15( Issue 12) pp:2588-2594
Publication Date(Web):
DOI:10.1002/cphc.201402075
Abstract
Anion–π interactions generally exist between an anion and an electron-deficient π-ring because of the electron-accepting character of the ring. In this paper, we report orbital effect-induced anomalous binding between electron-rich π systems and F− through anion–π interactions calculated at the MP2/6-31+G(d,p) and ωB97X-D/6-31+G(d,p) levels of theory. We find that anion–π interactions between F− and electron-rich π rings increase markedly with increasing number of π electrons and size of the π rings. This is contrary to intuition because anion–π interactions would be expected to gradually decrease because of gradually increasing Coulombic repulsion between the negative charge of the anions and gradually increasing number of π electrons of the aromatic surfaces. Energy decomposition analysis showed that the key to this anomalous effect is the more effective delocalization of negative charge to the unoccupied π* orbitals of larger π rings, which is termed an “orbital effect”.
Co-reporter:Li-yan Feng ; Yue-jie Liu ; Jing-xiang Zhao
The Journal of Physical Chemistry C 2014 Volume 118(Issue 51) pp:30325-30332
Publication Date(Web):December 1, 2014
DOI:10.1021/jp5109043
Recently, the encapsulation of various species inside boron nitride nanotubes (BNNTs) was identified as an effective method for modifying the physical and chemical properties of boron nitride nanotubes (BNNTs), thus greatly widening their potential applications. In the present work, we have performed comprehensive density functional theory (DFT) calculations to study the effects of the encapsulation of various numbers of Li atoms on the electronic and magnetic properties of a BNNT. The results show that two, three, and four Li atoms can be stably encapsulated inside a BNNT (Eint = −1.198, −2.081, and −2.378 eV, respectively), giving interaction energies that are much larger than that of one Li atom (Eint = −0.052). Because a certain number of electrons are transferred from these encapsulated Li atoms to the BNNT, some impurity levels are induced within the band structures of the BNNT, thus reducing its band gap. Encapsulation of three and four Li atoms renders the BNNT a metallic material, whereas 2Li@BNNT has a semiconducting nature with a small band gap (∼0.70 eV). Furthermore, we explored the catalytic activities of these composites for the oxygen reduction reaction (ORR). The chemisorption of ORR species O2, OOH, and O and the downhill energy landscape of the ORR on the surface of Li-encapsulated BNNTs indicate that Li encapsulation significantly enhances the BNNT chemical reactivity, rendering BNNTs active for the complete four-electron reduction of O2 to 2H2O.
Co-reporter:Dr. Jing Xu;Dr. Yi-Hong Ding;Dr. Diego M. Andrada;Dr. Gernot Frenking
Chemistry - A European Journal 2014 Volume 20( Issue 30) pp:9216-9220
Publication Date(Web):
DOI:10.1002/chem.201403252
Abstract
Quantum chemical calculations show that the N-heterocyclic carbene (NHC)-stabilized silavinylidene, NHCtBuCSiR2 is a strongly bonded complex, which has a linear arrangement of the donor and acceptor moieties. The molecule is the energetically lowest lying isomer of the NHC-stabilized R2CSi isomers and it is stable towards dimerization when R is a bulky substituent. The silavinylidene complex is the only species of the silylidene homologues NHCtBuEE′R2 (E, E′=C–Pb) which possesses a linear arrangement. The unusual bonding situation is explained in terms of donor–acceptor interactions between NHCtBu as σ donor and CSiR2 in the doubly excited singlet state 3a12b2 which leads to a significantly shorter CSiR2 bond compared with free CSiR2.
Co-reporter:Jing Xu
Structural Chemistry 2014 Volume 25( Issue 4) pp:1133-1139
Publication Date(Web):2014 August
DOI:10.1007/s11224-013-0385-z
Understanding the structure, bonding, and stability of the PI compounds has continued to be one of the very important research areas of low-valent main group chemistry. Up to now, many intramolecularly stabilized PI in cyclic form and several intermolecularly stabilized PI in acyclic form have been reported. In this paper, we report the computational design of two novel cyclic intermolecularly stabilized PI types, i.e., RP(μ-R)AlR21 and P(μ-R)2AlR22. For most of the 26 kinds of the studied substituents, both types of the PI structures are more stable than the classical PIII-one R2P-AlR23. The PI feature is supported by the structural, natural bond orbital analysis, and reactivity analysis. Formally, 1 and 2 are cooperatively stabilized by two sets of intermolecular donor–acceptor interactions between AlR3 and PR, i.e., μ-R → P and P → Al in 1 and μ-R1 → P and μ-R2 → Al in 2. These novel PI compounds strongly welcome the future laboratory studies.
Co-reporter:Chen Guo;Chong Wang
Structural Chemistry 2014 Volume 25( Issue 4) pp:1023-1031
Publication Date(Web):2014 August
DOI:10.1007/s11224-013-0372-4
Compounds containing the sulfur triple bonding have continued to attract chemists’ attention. In this article, with an attempt to predict intrinsically stable species with sulfur-related triple bonding, we report a thorough computational study of two charged systems [B,C,O,S]+ and [B,C,O,S]− at the CCSD(T)/aug-cc-pVTZ//B3LYP/6-311+G(3df,2p)+ZPVE level for singlet and triplet potential energy surfaces, aug-cc-pVTZ-B3LYP, M06-2X, and CCSD(T) levels for critical structures, as well as the CCSD(T)/aug-cc-pVQZ and G4 levels for adiabatic bond dissociation energy (ABDE). A total of 26 isomers and 25 transition states were located. The cationic and anionic [B,C,O,S] have the singlet and triplet ground states, respectively. For both systems, the former low-lying isomers are the linear SCBO+/−01 and SBCO+/−02, both of which contain the S≡X (X = C, B) bonding and are kinetically very stable against interconversion and fragmentation. With the increased valence electron number in the order of SCBO/SBCO+ < SCBO/SBCO < SCBO/SBCO−, the SX bond distance elongates as 1.5653 < 1.6126 < 1.6924 Å for X = B and 1.4715 < 1.5319 < 1.6100 Å for X = C at the M06-2X/aug-cc-pVTZ level. Notably, SBCO+ bears the shortest S≡B bond known to date, while SCBO+ bears the shortest S≡C bond among the known classical and non-protonated compounds (the well-known F3SCCF3 and F3SCSF5 have been termed as “nonclassical” because of their unusually low ABDE of SC bond). Future mass spectroscopic studies are greatly appealed for the characterization of the cationic and anionic SCBO+/−01 as well as SBCO+/−02.
Co-reporter:Chong Wang and Yi-hong Ding
Journal of Materials Chemistry A 2013 vol. 1(Issue 5) pp:1885-1891
Publication Date(Web):03 Dec 2012
DOI:10.1039/C2TA00736C
Graphene with a perfect hexagonal network structure is desirable for various reasons, e.g., mechanical, thermal conductivity and transport properties. Yet, the embedded defects generated either in synthesis or usage stages have posed obstacles for graphene applications. Therefore, removal of the structural defects in graphene has remained an important task. Stone–Wales (SW) defects are one typical topological structure in the carbon nanomaterials. Unfortunately, the SW defects in graphene have to overcome a very high restoration barrier (ca. 6 eV). Very recent theoretical work has shown the promise to reduce the restoration barrier by the adsorbed transition metal atoms down to 2.86 eV (for W) (yet this is still too high). In the present density functional theory (DFT) study, we find that through a mechanically different process, the adsorption of carbon atoms can dramatically reduce the restoration barrier to hitherto the lowest value, i.e., 20.0 kcal mol−1 (0.87 eV), which could make the SW-healing experimentally accessible. Subsequently, the C-adatom can migrate very easily on the graphene surface. As a result, one carbon adatom could principally catalyze the healing of all the SW defects in a cascade mode if no termination steps exist. During the graphene growth, the presently proposed carbon-adatom catalytic mechanism could have played a role in healing the SW defect. Moreover, we propose that in the post-treatment of graphene, adsorption of the carbon adatom could be used as an effective catalyst for the SW-healing. The catalytic role of carbon atoms on the SW defect should be included in the modeling of graphene growth.
Co-reporter:Ying Chen, Yue-jie Liu, Hong-xia Wang, Jing-xiang Zhao, Qing-hai Cai, Xuan-zhang Wang, and Yi-hong Ding
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 13) pp:5994
Publication Date(Web):June 12, 2013
DOI:10.1021/am400563g
Density functional theory (DFT) calculations were performed on the NO reduction on the silicon (Si)-doped graphene. The results showed that monomeric NO dissociation is subject to a high barrier and large endothermicity and thus is unlikely to occur. In contrast, it was found that NO can easily be converted into N2O through a dimer mechanism. In this process, a two-step mechanism was identified: (i) the coupling of two NO molecules into a (NO)2 dimer, followed by (ii) the dissociation of (NO)2 dimer into N2O + Oad. In the energetically most favorable pathway, the trans-(NO)2 dimer was shown to be a necessary intermediate with a total energy barrier of 0.464 eV. The catalytic reactivity of Si-doped graphene to NO reduction was interpreted on the basis of the projected density of states and charge transfer.Keywords: density functional theory; dimer mechanism; direct dissociation mechanism; metal-free catalyst; NO reduction; Si-doped graphene;
Co-reporter:Ying Chen, Hongmei Wang, Hongxia Wang, Jing-xiang Zhao, Qing-hai Cai, Xiao-Guang Wang, Yi-hong Ding
Applied Surface Science 2013 Volume 273() pp:293-301
Publication Date(Web):15 May 2013
DOI:10.1016/j.apsusc.2013.02.034
Abstract
We have performed first-principles calculations to study the chemical functionalization of the BN graphene with divacancy (DV) defect by 12 different transition metal (TM) atoms, including Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Pt, and Au. The results indicate that the DV defect can assist the adsorption of TM atoms on BN graphene. Moreover, some impurity bands are induced within the band gap of DV-BN graphene, leading to the modification of its electronic properties in various ways. Interestingly, Ti- and Co-adsorbed DV-BN graphenes are found to possess ferromagnetic characteristic, while antiferromagnetic state is preferred for V-, Mn-, and Fe-functionalized DV-BN graphenes, and the paramagnetic state is the ground state for Sc-, Cr-, Ni-, Cu, Zn-, Pt-, and Au-decorated DV-BN graphenes. Finally, aiming at evaluating the potential of these functionalized BN graphenes in hydrogen storage, we study their interaction with H2 molecules. It is found that the dispersed Sc, V, and Cr on DV-BN graphene are able to adsorb up to three H2 molecules as strongly as 0.25–0.58 eV/H2, suggesting that the three nanomaterials may be suitable candidates for hydrogen storage.
Co-reporter:Si-meng Gao and Yi-hong Ding
RSC Advances 2012 vol. 2(Issue 31) pp:11764-11776
Publication Date(Web):17 Sep 2012
DOI:10.1039/C2RA21250A
Hetero-doped nitrogen-rich compounds are of great interest for potential use in high energy density materials (HEDM) fields. In this paper, we report in detail the structures and stabilities of a tetra-atomic cluster CN3−, which is isoelectronic to the well-known N4. A series of higher-level energetic calculations were carried out at the CCSD(T)/6-311+G(d)//B3LYP/6-311+G(d), CASPT2(12,12)/6-311+G(d)//CASSCF (12,12)/6-311+G(d) and CASPT2(12,12)/aug-cc-pVTZ//CASSCF(12,12)/aug-cc-pVTZ levels. The kinetic stability of CN3− was evaluated for the first time by studying the dissociation, isomerization and intersystem crossing barriers, as well as the Born–Oppenheimer molecular dynamic (BOMD) simulations. We found that the ground state isomer, i.e., chain-like triplet NCNN−(Cs), and a higher-energy isomer, i.e., tetrahedral-like singlet CN3−(C3v), are both kinetically stable, even when the intersystem crossing (ISC) is included. Therefore, the two isomers, 3NCNN− and 1CN3− (C3v), could be observable in future laboratory studies. By comparison with the isoelectronic N4 system, we conclude that the carbon-doping for XN3 (X = N to C−) greatly enhances the kinetic stability of the chain-like triplet and the tetrahedral-like singlet structures. Finally, for more practical use, our studies on the related salts M+[CN3]− (M = Li, Na, K) showed that both the chain-like 3NCNN− and tetrahedral-like 1CN3− could act as promising building blocks for novel HEDMs.
Co-reporter:Dr. Chong Wang;Dr. Bo Xiao ; Dr. Yi-hong Ding
ChemPhysChem 2012 Volume 13( Issue 3) pp:774-779
Publication Date(Web):
DOI:10.1002/cphc.201100864
Abstract
Many outstanding properties of graphene are blocked by the existence of structural defects. Herein, we propose an important healing mechanism for the growth of graphene, which is produced via plasma-enhanced chemical vapor decomposition (PECVD), that is, the healing of graphene with single vacancies by decomposed CH4 (hydrocarbon radical CHx, x=1, 2, 3). The healing processes undergo three evolutionary steps: 1) the chemisorption of the hydrocarbon radicals, 2) the incorporation of the C atom of the hydrocarbon radicals into the defective graphene, accompanied by the adsorption of the leaving H atom on the graphene surface, 3) the removal of the adsorbed H atom and H2 molecule to generate the perfect graphene. The overall healing processes are barrierless, with a huge released heat of 530.79, 290.67, and 159.04 kcal mol−1, respectively, indicative of the easy healing of graphene with single vacancies by hydrocarbon radicals. Therefore, the good performance of the PECVD method for the generation of graphene might be ascribed to the dual role of the CHx (x=1, 2, 3) species, acting both as carbon source and as defect healer.
Co-reporter:Ying Chen;Hong-xia Wang;Jing-xiang Zhao
Journal of Nanoparticle Research 2012 Volume 14( Issue 1) pp:
Publication Date(Web):2012 January
DOI:10.1007/s11051-011-0675-6
The anchoring of small organic molecules onto the semiconductor surface has a great application for developing various molecular devices, such as novel solar cells, fuel cells, hybrid systems, sensors, and so on. In the present work, by carrying out detailed density-functional theory calculations, we have investigated the adsorption of the formic acid (HCOOH) molecule on planar and various curved silicon carbide (SiC) nanotubes. By considering both the molecular and dissociative adsorptions of HCOOH on these SiC nanomaterials, we found that the HCOOH molecule prefers to dissociate into HCOO and H group. Interestingly, different adsorption modes were found for HCOOH on SiC nanotubes, i.e. dissociative monodentate or bidentate adsorption, which depends on the tube diameter and helicity. For (n, 0) SiC nanotube, the monodentate adsorption mode is energetically favorable when n is less than 10. However, HCOOH prefers to be adsorbed on other (n, 0) SiC nanotubes in a bridged bidentate mode, which is similar to those of on (n, n) SiC nanotubes or planar SiC sheet. Moreover, upon HCOOH adsorption, these SiC nanomaterials remain to be of the semiconducting nature and their band gaps are decreased to different degrees. In addition, we also explored the effects of HCOOH coverage on its adsorption on SiC nanotube.
Co-reporter:Zhong-hua Cui ; Maryel Contreras ; Yi-hong Ding ;Gabriel Merino
Journal of the American Chemical Society 2011 Volume 133(Issue 34) pp:13228-13231
Publication Date(Web):July 28, 2011
DOI:10.1021/ja203682a
In this study, we analyzed CB4 and its cation, CB4+. Using CCSD(T)/aug-cc-pVQZ//CCSD(T)/aug-cc-pVTZ quantum-chemical calculations, we found that the neutral molecule is in accord with the results of Boldyrev and Wang, having a Cs global minimum with a planar tricoordinate carbon structure, contradicting previous studies. In contrast, CB4+, which was reported by an early mass spectroscopic study, has a planar tetracoordinate carbon (ptC) atom, demonstrating that a modification of the charge can promote the stabilization of a ptC structure.
Co-reporter:Ning He, Yi-hong Ding
Microporous and Mesoporous Materials 2011 Volume 144(1–3) pp:67-73
Publication Date(Web):October 2011
DOI:10.1016/j.micromeso.2011.02.016
The catalytic activity of zeolite is well known to be associated with proton-donating ability at the Brønsted acid sites, i.e., high acidity versus high activity. In this paper, we firstly show that for modified zeolites, this simple acidity–activity correlation might be biased. We found that the proton hopping barrier (21.42 kcal/mol) within CpNa-modified HMCM-22 (Cp: cyclopentadienyl) is significantly higher than that of HMCM-22 (13.46 kcal/mol), indicating a clear lowering of acidity upon CpNa-modification. Surprisingly, the activation energy of ethene protonation is also significantly reduced from 30.43 kcal/mol in HMCM-22 to 24.74 kcal/mol in CpNa-modified HMCM-22, suggesting an enhanced catalytic activity upon CpNa-modification. The cause of violation from traditional “acidity–activity” correlation can be ascribed to the dual factor of CpNa, namely, Na+ can “activate” the proton due to its influence on the charges of proton and oxygen atoms, whereas Cp− can “deactivate” the proton due to its effective interaction with proton.Graphical abstractFor the ethylene protonation in HMCM-22, the CpNa-modification can cause the violation of the general acidity-activity correlation.Research highlights► CpNa-modified HMCM-22 possesses high catalytic activity, but low acidity. ► Guest molecule is the main reason to cause violation of traditional acidity–activity. ► The enhanced “activity” is not always correlated with enhanced “acidity”.
Co-reporter:Li-juan Fu, Lin Jin, Chang-bin Shao and Yi-hong Ding
Inorganic Chemistry 2010 Volume 49(Issue 11) pp:5276-5284
Publication Date(Web):May 3, 2010
DOI:10.1021/ic100429b
Contrasting the boranes BnHn+4 with rich chemistry, the alanes AlnHn+4 remain largely unknown in laboratory, except for the simplest Al2H6. Though recent experimental and theoretical studies have proved AlnHn+2 to be the borane analogues, whether or not the borane analogy can exist for the more complicated AlnHn+4 is still unclear. In this paper, we find that at the B3PW91/TZVP level, AlnHn+4 each has a nido-single cluster ground structure as BnHn+4 for n < 12. For n ≥ 12, the fusion cluster becomes energetically more competitive than the single cluster also as BnHn+4. Thus, concerning the ground structures, the alanes AlnHn+4 (n = 5−19) could be considered as the borane analogues. Remarkably, Al8H12 has a novel closo(4)−closo(4) cluster fused by two Td-like subunits Al4H6, lying only 0.49 kcal/mol above the single cluster. The Born−Oppenheimer molecular dynamic simulation shows that the closo(4)−closo(4) fusion cluster intrinsically has high kinetic stability, which can be ascribed to the rigidity of the Td-Al4H6 subunit. Since Td-Al4H6 has been experimentally characterized in a gas phase very recently, we strongly recommend that the unprecedented non-Wade−Mingos alane Al8H12 can be effectively formed via the direct dimerization between two Td-Al4H6, with the reaction energy (−39.65 kcal/mol) very similar to that of the known dialane (2AlH3 → Al2H6, −35.27 kcal/mol).
Co-reporter:Zhong-hua Cui, Chang-bin Shao, Si-meng Gao and Yi-hong Ding
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 41) pp:13637-13645
Publication Date(Web):20 Sep 2010
DOI:10.1039/C0CP00296H
Among the fascinating planar tetracoordinate carbon (ptC) species, pentaatomic molecules belong to the smallest class, well-known as “pptC”. It has been generally accepted that the planarity of pptC structure is realized via the “delocalization” of the pz lone pair at the central carbon and the ligand–ligand bonding interaction. Although “localization” is as key driving force in organic chemistry as “delocalization”, the “localization” concept has not been applied to the design of pptC molecules, to the best of our knowledge. In this paper, we apply the “localization” strategy to design computationally a series of new pptC. It is shown that the central carbon atom and one “electronegative” ligand atom X (compared to the Al ligand) effectively form a highly localized C–X multiple bond, converting the lone pair at the central carbon to a two-center two-electron π-bond. At the aug-cc-pVTZ-B3LYP, MP2 and CCSD(T) levels, the designed 18-valence-electron pptC species [XCAl3]q; [(X,q) = (B,−2), (C,−1), (N,0)] are found to each possess a stable ptC structure bearing a C–X double bond, indicated by the structural, molecular orbital, Wiberg bonding, potential energy surface and Born–Oppenheimer molecular dynamics (BOMD) analysis. Moreover, our OVGF calculations showed that the presently disclosed (yet previously unconsidered) pptC structure of [C2Al3]− could well account for the observed photoelectron spectrum (previously only ascribed to a close-energy fan-like structure). Therefore, [C2Al3]− could be the first pptC that bears the highly localized C–X double bond that has been experimentally generated. Notably, the pptC structure is the respective global minimum point for [BCAl3]2− and [NCAl3], and the counterion(s) would further stabilize [BCAl3]2− and [C2Al3]−. Thus, these newly designed pptC species with interesting bonding structure should be viable for future experimental characterization. The presently applied “localization” approach complements well the previous “delocalization” one, indicating that the general “localization vs. delocalization” concept in organic chemistry can be effectively transplanted to exotic pptC chemistry.
Co-reporter:Lin Jin, Li-juan Fu and Yi-hong Ding
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 36) pp:10956-10962
Publication Date(Web):27 Jul 2010
DOI:10.1039/C003503C
Recently, a combined laser ablation and density functional theory study (Jiang and Xu, J. Am. Chem. Soc. 2005, 127, 8906) claimed the existence of the long-sought 18-electron member of the first-row transition metal carbonyl complex, Zn(CO)3. In this paper, we systematically investigate the thermodynamic and kinetic stability of Zn(CO)3 towards CO-extrusion at the BP86, B3PW91, BPW91, PBEPBE, BH&HLYP, B3LYP, MP2, MP4SDQ, QCISD, CCSD and CASPT2 levels as well as the Born–Oppenheimer molecular dynamic (BOMD) simulation. All these calculations consistently reveal that the 18e Zn(0) complex Zn(CO)3 is neither a genuine minimum point nor kinetically stable with negligibly low barriers. In particular, Zn(CO)3 is quite thermodynamically unstable with respect to the fragments 1Zn + 3CO by around 40 kcal mol−1 at all the three sophisticated correlation levels, i.e., MP4SDQ, QCISD and CCSD. We thus conclude that the tricarbonyl Zn(0) complex, Zn(CO)3, should not exist even for spectroscopic characterization. Interestingly, our extensive structural search predicts that two triplet di-zinc carbonyls, i.e., 3(CO)ZnZn and 3(CO)2ZnZn, have noticeable kinetic stability (10.41 and 8.11 kcal mol−1 at the CCSD level) against the respective CO- and Zn-extrusion, which can be compared with the value 8.70 kcal mol−1 for the already detected 3Zn(CO)2 (Jiang and Xu, J. Phys. Chem. A2006,110, 7092). Our designed 3(CO)ZnZn and 3(CO)2ZnZn together with the experimentally known 3ZnCO and 3Zn(CO)2 are formally associated with the zinc (0) “spin-based zinc carbonyls” and should be considered as remarkable, since most of the known zinc complexes usually contain +2 or +1 oxidation state Zn.
Co-reporter:Bo Xiao, Jing-xiang Zhao, Yi-hong Ding, Chia-chung Sun
Surface Science 2010 Volume 604(21–22) pp:1882-1888
Publication Date(Web):October 2010
DOI:10.1016/j.susc.2010.07.020
Silicon carbide (SiC) nanotubes have attracted extensive attention due to the unique properties. Modifying the electronic properties of SiC nanotubes is helpful for further widening their potential applications. In this paper, we have studied the chemisorption of NO2 molecules at different coverage on a series of SiC nanotubes through density functional theory (DFT) calculations. The results indicate that changes in energetic, structural and electronic properties of the SiC nanotubes are significantly dependent on the coverage of adsorbed NO2 molecules: (1) a nitrite-like structure is obtained for an odd number of NO2 molecules adsorption on the SiC nanotube, while an even number of NO2 molecules adsorption leads to a nitro-like configuration; (2) the adsorption energy per NO2 molecule for even number adsorption is larger than that of odd number, suggesting that the NO2 groups prefer the pair arrangement due to the coupling of two radicals; (3) with the increase of the coverage of the adsorbed NO2, the band-gaps of SiC nanotubes are decreased, thus leading to the enhancement of the electro-conductivity of SiC nanotubes. Our results might provide an alternative strategy to modify the properties of SiC nanotubes, which might be useful for the design of SiC nanotubes-based nanodevices.
Co-reporter:Dr. Bo Xiao;Dr. Jing-xiang Zhao; Yi-hong Ding; Chia-chung Sun
ChemPhysChem 2010 Volume 11( Issue 16) pp:3505-3510
Publication Date(Web):
DOI:10.1002/cphc.201000325
Abstract
Density functional theory calculations are used to study the healing process of a defective CNT (i.e. (8,0) CNT) by CO molecules. The healing undergoes three evolutionary steps: 1) the chemisorption of the first CO molecule, 2) the incorporation of the C atom of CO into the CNT, accompanied by the adsorption of the leaving O atom on the CNT surface, 3) the removal of the adsorbed O atom from the CNT surface by a second CO molecule to form CO2 and the perfect CNT. Overall, adsorption of the first CO reveals a barrier of 2.99 kcal mol−1 and is strongly exothermal by 109.11 kcal mol−1, while adsorption of a second CO has an intrinsic barrier of 32.37 kcal mol−1and is exothermal by 62.34 kcal mol−1. In light of the unique conditions of CNT synthesis, that is, high temperatures in a closed container, the healing of the defective CNT could be effective in the presence of CO molecules. Therefore, we propose that among the available CNT synthesis procedures, the good performance of chemical vapor decomposition of CO on metal nanoparticles might be ascribed to the dual role of CO, that is, CO acts both as a carbon source and a defect healer. The present results are expected to help a deeper understanding of CNT growth.
Co-reporter:Li-juan Fu, Hong-bin Xie and Yi-hong Ding
Inorganic Chemistry 2009 Volume 48(Issue 12) pp:5370-5375
Publication Date(Web):May 11, 2009
DOI:10.1021/ic900401f
In this work, we report the first comparative study directly between AlnHn2− and BnHn2− (5 ≤ n ≤ 12), AlnHn+2 and BnHn+2 (4 ≤ n ≤ 12) covering diverse structural forms. It was shown that AlnHn2− each have a closo ground structure as BnHn2−, nicely consistent with the Wade−Mingos rule. However, AlnHn+2 adopt the closo-nido ground structures following the even−odd alternation in the number of Al atoms, showing distinct violation of the Wade−Mingos rule for the odd-numbered Al-atoms. Interestingly, the corresponding BnHn+2 also have similar closo (even)-nido (odd) alternation. Therefore, our direct comparison showed that AlnHn2− (5 ≤ n ≤ 12) and AlnHn+2 (4 ≤ n ≤ 12) can be viewed as the borane analogues, though not all AlnHn+2 have the ability of being explained by Wade−Mingos rule. So, the analogy between alanes and boranes in form of XnHn+2 should not be simply judged by the Wade−Mingos rule because of the significant influence of the additional two hydrogen atoms on the closo-structure.
Co-reporter:Jing-xiang Zhao and Yi-hong Ding
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 4) pp:1099-1105
Publication Date(Web):March 5, 2009
DOI:10.1021/ct9000069
Detection of carbon dioxide (CO2) is very important in environmental, biological, and industrial processes. Recent experiment showed that carbon nanotubes can act as chemical sensors for detecting certain gaseous molecules such as NH3, NO2, and O2. Unfortunately, the intrinsic stability of CO2 makes its sensing by CNTs unsuccessful due to the rather weak adsorption energy on the tube surface. In the present Article, we study the CO2 adsorption on various zigzag (n,0) (n = 6, 8, 10, 12, and 18) single-walled SiC nanotubes to explore the possibility of the SiC tube as potential gas sensors for CO2-detection by density functional theory (DFT) calculations. It is found that tube diameter and CO2 coverage play important roles in the tube−CO2 interaction. A single CO2 can be chemisorbed to the Si−C bonds of SiCNT with appreciable adsorption energy and can draw significant charge transfer from the SiCNT. The adsorption energy decreases gradually with increased tube diameter. The addition of more CO2 molecules in different patterns has been considered for the exemplified (8,0) tube, and CO2 molecules prefer to be as far from each other as possible. With the increase of CO2 coverage, the interaction between CO2 molecules and tube becomes weaker, and up to eight CO2 molecules can be adsorbed on the tube. In addition, we find that the band gap is lowered to a different degree due to the different adsorption. Because of the sufficient charge transfer and high concentration of CO2, SiCNT could be a perfect material for efficiently detecting the CO2 molecule.
Co-reporter:Jing-xiang Zhao, Yi-hong Ding
Materials Chemistry and Physics 2009 Volume 116(Issue 1) pp:21-27
Publication Date(Web):15 July 2009
DOI:10.1016/j.matchemphys.2009.02.014
Recently, many investigations have shown that functionalization of carbon nanotube (CNT) with porphyrin or its metal complex is an efficient way to widen its application areas. As its structural counterparts, boron nitride nanotube (BNNT) exhibits strikingly different properties. In this work, through density functional theory calculations, we report the first study on the functionalization of an (8,0) BNNT with several metalloporphyrin complexes MP (M = Fe, Co, Ni, Cu, and Zn). The results show that adsorption of the MP complex on the N site is the most energetically preferable and the binding energy ranges from 0.17 eV (NiP) to 0.91 eV (FeP). Upon the FeP adsorption, the sp2 hybridization of the adsorbed N atom is converted to sp3 hybridization, while the hybridization of the N atom is not changed for the adsorption of other MP complexes. Moreover, the ground state of the BNNT–FeP is predicted to be low-spin (S = 0), although the intermediate-spin (S = 1) state is the most stable state of the free FeP complex. Finally, the effects of the adsorption of the BNNT by these MP complexes on electronic and magnetic properties of the pure BNNT are further elucidated.
Co-reporter:Jing-xiang Zhao, Yi-hong Ding
Journal of Physics and Chemistry of Solids 2009 Volume 70(Issue 6) pp:1030-1033
Publication Date(Web):June 2009
DOI:10.1016/j.jpcs.2009.05.016
The nanotube with open edges is an excellent candidate for designing efficient tip for atomistic scanning probes or field emission display (FED) devices. In the present work, we have studied the functionalization of an open-ended boron nitride nanotube (BNNT) with a series of transition metal rings and the effects on the properties of open-ended BNNT through density functional theory (DFT) calculations. The results show that the TM-BNNT complexes are energetically favorable. Moreover, it is found that the functionalization (a) significantly decreases the band gap of BNNT to different degrees, which might effectively modify the electronic properties of the open-ended BNNT; and (b) efficiently lowers the work function, which might improve the field emission properties. Our results might be helpful not only to design specific BNNT-based tips but also to further discuss the chemical vapor deposition (CVD) growth of BNNT on nanoparticles.
Co-reporter:Bo Xiao;JingXiang Zhao;YiHong Ding;ChiaChung Sun
Science China Chemistry 2009 Volume 52( Issue 11) pp:
Publication Date(Web):2009 November
DOI:10.1007/s11426-009-0282-x
Recent studies have shown that the inner phase of carbon nanotubes (CNTs) can not only change the properties of molecules inside the tube, but also enhance or restrain the SN2 reactions. Thus, the CNTs can be considered a form of solid solvent. In this paper, we study the [2+2] cycloaddition reaction between CH2O and PH3CH2 in the gas phase, benzene solution and inner phase of CNT using the density functional theory (DFT). The results indicate that the inner phase of CNT has little effect on the [2+2] cycloaddition reaction. This can be explained as that while taking the linear arrangement for SN2 reaction, the reactants do not possess the axial symmetry for the studied [2+2] cycloaddition reaction. Therefore, although the CNT has large axial polarizability, it can exert little influence on the [2+2] cycloaddition reaction. Our studies will be helpful for further understanding of the inner phase chemistry of CNTs.
Co-reporter:Lin Jin, Li-juan Fu and Yi-hong Ding
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 36) pp:NaN10962-10962
Publication Date(Web):2010/07/27
DOI:10.1039/C003503C
Recently, a combined laser ablation and density functional theory study (Jiang and Xu, J. Am. Chem. Soc. 2005, 127, 8906) claimed the existence of the long-sought 18-electron member of the first-row transition metal carbonyl complex, Zn(CO)3. In this paper, we systematically investigate the thermodynamic and kinetic stability of Zn(CO)3 towards CO-extrusion at the BP86, B3PW91, BPW91, PBEPBE, BH&HLYP, B3LYP, MP2, MP4SDQ, QCISD, CCSD and CASPT2 levels as well as the Born–Oppenheimer molecular dynamic (BOMD) simulation. All these calculations consistently reveal that the 18e Zn(0) complex Zn(CO)3 is neither a genuine minimum point nor kinetically stable with negligibly low barriers. In particular, Zn(CO)3 is quite thermodynamically unstable with respect to the fragments 1Zn + 3CO by around 40 kcal mol−1 at all the three sophisticated correlation levels, i.e., MP4SDQ, QCISD and CCSD. We thus conclude that the tricarbonyl Zn(0) complex, Zn(CO)3, should not exist even for spectroscopic characterization. Interestingly, our extensive structural search predicts that two triplet di-zinc carbonyls, i.e., 3(CO)ZnZn and 3(CO)2ZnZn, have noticeable kinetic stability (10.41 and 8.11 kcal mol−1 at the CCSD level) against the respective CO- and Zn-extrusion, which can be compared with the value 8.70 kcal mol−1 for the already detected 3Zn(CO)2 (Jiang and Xu, J. Phys. Chem. A2006,110, 7092). Our designed 3(CO)ZnZn and 3(CO)2ZnZn together with the experimentally known 3ZnCO and 3Zn(CO)2 are formally associated with the zinc (0) “spin-based zinc carbonyls” and should be considered as remarkable, since most of the known zinc complexes usually contain +2 or +1 oxidation state Zn.
Co-reporter:Jing-jing Sui, Jing Xu and Yi-hong Ding
Dalton Transactions 2016 - vol. 45(Issue 1) pp:NaN60-60
Publication Date(Web):2015/11/16
DOI:10.1039/C5DT03989D
Through a global isomeric study, we computationally identified the first structural template C2Si2X that could encompass a planar tetracoordinate X for all the heavier group 14 elements X in the 0, +1 or −1 charge state. We thus significantly expanded the traditional 16/17/18ve rules to 19/20/21ve for ptX.
Co-reporter:Zhong-hua Cui, Valentin Vassilev-Galindo, José Luis Cabellos, Edison Osorio, Mesías Orozco, Sudip Pan, Yi-hong Ding and Gabriel Merino
Chemical Communications 2017 - vol. 53(Issue 1) pp:NaN141-141
Publication Date(Web):2016/11/29
DOI:10.1039/C6CC08273D
Viable planar pentacoordinate carbon (ppC) systems with a ppC bonded to a transition metal and embedded in a metallocene framework are reported. Our detailed global minima search shows that CAl4MX2 (M = Zr and Hf; X = F–I and C5H5) clusters with ppCs are appropriate candidates for experimental realization in the gas phase. The fulfillment of the 18 electron rule and electron delocalization is found to be crucial for the stabilization of these ppC arrangements.
Co-reporter:Nan-nan Liu, Si-meng Gao and Yi-hong Ding
Dalton Transactions 2015 - vol. 44(Issue 1) pp:NaN350-350
Publication Date(Web):2014/09/23
DOI:10.1039/C4DT02282C
The inverse sandwich Ca–C8H8–Ca is predicted to be an open-shell singlet state. Since the C8H8 ligand prevents the spin-up and spin-down electrons of different calcium atoms from forming Ca–Ca bonds, the spin-coupling electrons lead to a singlet diradical character. The metal–ligand interaction contributes to the stability of Ca–C8H8–Ca against dissociation and isomerization. For the coordination complex (DME)3Ca–C8H8–Ca(DME)3, the open-shell singlet state is unavailable, while the closed-shell singlet state with direct Ca–Ca bonds is more favorable, because dimethyl ether molecules could push the spin-paired electrons of different calcium atoms to migrate towards the direction of Ca–Ca bonding. For Ca–C4H4–Ca, the ground state is an open-shell singlet state, of which the diradical character is very similar to that of Ca–C8H8–Ca. For (DME)3Ca–C4H4–Ca(DME)3, the lowest energy is the triplet state.
Co-reporter:Zhong-hua Cui, Chang-bin Shao, Si-meng Gao and Yi-hong Ding
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 41) pp:NaN13645-13645
Publication Date(Web):2010/09/20
DOI:10.1039/C0CP00296H
Among the fascinating planar tetracoordinate carbon (ptC) species, pentaatomic molecules belong to the smallest class, well-known as “pptC”. It has been generally accepted that the planarity of pptC structure is realized via the “delocalization” of the pz lone pair at the central carbon and the ligand–ligand bonding interaction. Although “localization” is as key driving force in organic chemistry as “delocalization”, the “localization” concept has not been applied to the design of pptC molecules, to the best of our knowledge. In this paper, we apply the “localization” strategy to design computationally a series of new pptC. It is shown that the central carbon atom and one “electronegative” ligand atom X (compared to the Al ligand) effectively form a highly localized C–X multiple bond, converting the lone pair at the central carbon to a two-center two-electron π-bond. At the aug-cc-pVTZ-B3LYP, MP2 and CCSD(T) levels, the designed 18-valence-electron pptC species [XCAl3]q; [(X,q) = (B,−2), (C,−1), (N,0)] are found to each possess a stable ptC structure bearing a C–X double bond, indicated by the structural, molecular orbital, Wiberg bonding, potential energy surface and Born–Oppenheimer molecular dynamics (BOMD) analysis. Moreover, our OVGF calculations showed that the presently disclosed (yet previously unconsidered) pptC structure of [C2Al3]− could well account for the observed photoelectron spectrum (previously only ascribed to a close-energy fan-like structure). Therefore, [C2Al3]− could be the first pptC that bears the highly localized C–X double bond that has been experimentally generated. Notably, the pptC structure is the respective global minimum point for [BCAl3]2− and [NCAl3], and the counterion(s) would further stabilize [BCAl3]2− and [C2Al3]−. Thus, these newly designed pptC species with interesting bonding structure should be viable for future experimental characterization. The presently applied “localization” approach complements well the previous “delocalization” one, indicating that the general “localization vs. delocalization” concept in organic chemistry can be effectively transplanted to exotic pptC chemistry.
Co-reporter:Zhong-hua Cui, Jing-jing Sui and Yi-hong Ding
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 47) pp:NaN32022-32022
Publication Date(Web):2015/10/30
DOI:10.1039/C5CP04776E
With the high preference in forming multi-center bonding, boron has been a miracle ligand in constructing diverse planar multi-coordinate (pM) (tetra/hyper) species. Unfortunately, the boron ligand usually dislikes encompassing a pM carbon (pMC) due to the high competition with pM boron (pMB), which makes the realization of boron-based pMC very difficult and quite challenging. Herein, we propose a strategy that by means of cooperative doping and charge-compensation, we can successfully improve and tune the stability of pMC relative to pMB for CB42−. In the free CBxEy2− (E = Al/Ga) species, ptC is thermodynamically less stable than the global ptB in mono- and di-substituted systems, in agreement with the results of Boldyrev and Wang. However, the thermodynamic preference of pMC increases along with the Al/Ga-doping. The pMC species can be further stabilized by the introduction of the alkaline-earth counterion (Mg2+). CB2E2Mg (E = Al, Ga) designed in the present study represents the first successful design of a boron-based planar penta-coordinate carbon (ppC) structures as the global minima. The strategy proposed in this study should be useful in the manipulation of competition between exotic pMC and pMB in B-based systems.
Co-reporter:Chong Wang and Yi-hong Ding
Journal of Materials Chemistry A 2013 - vol. 1(Issue 5) pp:NaN1891-1891
Publication Date(Web):2012/12/03
DOI:10.1039/C2TA00736C
Graphene with a perfect hexagonal network structure is desirable for various reasons, e.g., mechanical, thermal conductivity and transport properties. Yet, the embedded defects generated either in synthesis or usage stages have posed obstacles for graphene applications. Therefore, removal of the structural defects in graphene has remained an important task. Stone–Wales (SW) defects are one typical topological structure in the carbon nanomaterials. Unfortunately, the SW defects in graphene have to overcome a very high restoration barrier (ca. 6 eV). Very recent theoretical work has shown the promise to reduce the restoration barrier by the adsorbed transition metal atoms down to 2.86 eV (for W) (yet this is still too high). In the present density functional theory (DFT) study, we find that through a mechanically different process, the adsorption of carbon atoms can dramatically reduce the restoration barrier to hitherto the lowest value, i.e., 20.0 kcal mol−1 (0.87 eV), which could make the SW-healing experimentally accessible. Subsequently, the C-adatom can migrate very easily on the graphene surface. As a result, one carbon adatom could principally catalyze the healing of all the SW defects in a cascade mode if no termination steps exist. During the graphene growth, the presently proposed carbon-adatom catalytic mechanism could have played a role in healing the SW defect. Moreover, we propose that in the post-treatment of graphene, adsorption of the carbon adatom could be used as an effective catalyst for the SW-healing. The catalytic role of carbon atoms on the SW defect should be included in the modeling of graphene growth.

![Silylene, [1-(1,1-dimethylethyl)-2,2-dimethylpropylidene]-](http://img.cochemist.com/ccimg/186000/185953-02-0.png)
![Silylene, [1-(1,1-dimethylethyl)-2,2-dimethylpropylidene]-](http://img.cochemist.com/ccimg/186000/185953-02-0_b.png)
![1,2,5-Trisilatricyclo[2.1.0.01,3]penta-2,4-diene-2,5-diyl](http://img.cochemist.com/ccimg/162600/162521-10-0.png)
![1,2,5-Trisilatricyclo[2.1.0.01,3]penta-2,4-diene-2,5-diyl](http://img.cochemist.com/ccimg/162600/162521-10-0_b.png)







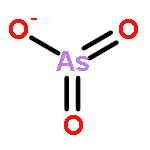
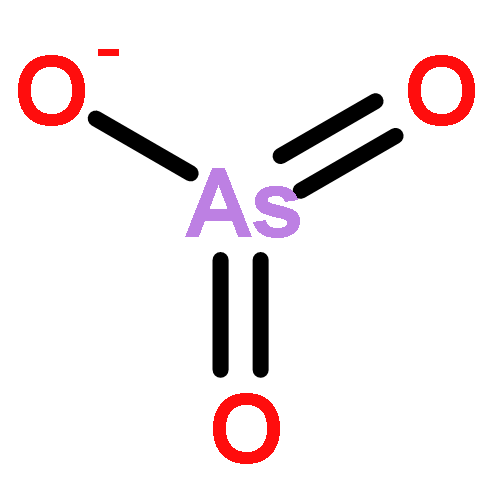
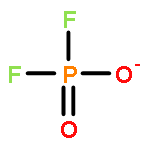
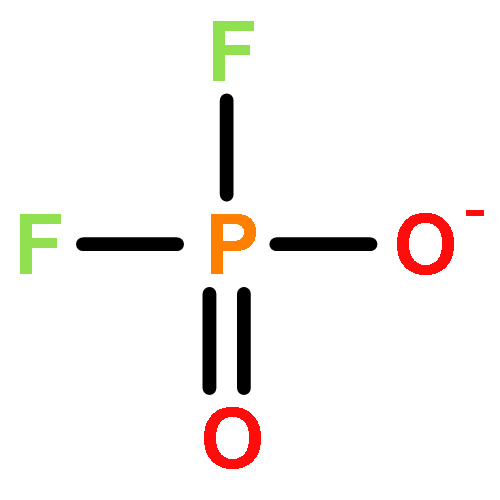
![2,4,6,8,9,10-Hexaoxa-1,3,5,7-tetraphosphatricyclo[3.3.1.13,7]decane,1,3,5,7-tetraoxide](http://img.cochemist.com/ccimg/16800/16752-60-6.png)
![2,4,6,8,9,10-Hexaoxa-1,3,5,7-tetraphosphatricyclo[3.3.1.13,7]decane,1,3,5,7-tetraoxide](http://img.cochemist.com/ccimg/16800/16752-60-6_b.png)