Co-reporter:Isabella Panovic, James R. D. Montgomery, Christopher S. Lancefield, Dhivya Puri, Tomas Lebl, and Nicholas J. Westwood
ACS Sustainable Chemistry & Engineering November 6, 2017 Volume 5(Issue 11) pp:10640-10640
Publication Date(Web):September 13, 2017
DOI:10.1021/acssuschemeng.7b02575
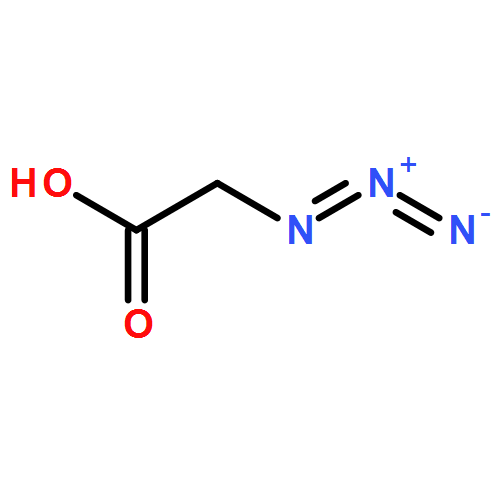
A new method has been developed to enable the modification of the organosolv technical lignins. Using a walnut shell butanol alkoxasolv lignin as a source of high β-O-4 content material, the β-O-4 γ-position has been selectively modified via tosylation, azidation, and copper-catalyzed azide–alkyne triazole formation. In addition, extensive model studies were used to aid the detailed characterization of the modified lignin structure. The copper-catalyzed click reaction was used to attach modified PEG chains, and the resulting lignin-based copolymer displayed improved thermal stability. This protocol was also used to incorporate a novel BODIPY-type fluorophore, generating a fluorescent lignin. Copper catalytic loadings were effective as low as 0.3 wt % and were found to catalyze the cycloaddition efficiently. This efficient and generic approach to preparing lignin-derived polymers is relevant to the core societal challenge of improving biorefinery efficiency.Keywords: Click; Copolymer; Fluorescent; Lignin modification;
Co-reporter:Daniel M. Miles-Barrett, James R. D. Montgomery, Christopher S. Lancefield, David B. CordesAlexandra M. Z. Slawin, Tomas Lebl, Reuben Carr, Nicholas J. Westwood
ACS Sustainable Chemistry & Engineering 2017 Volume 5(Issue 2) pp:
Publication Date(Web):December 22, 2016
DOI:10.1021/acssuschemeng.6b02566
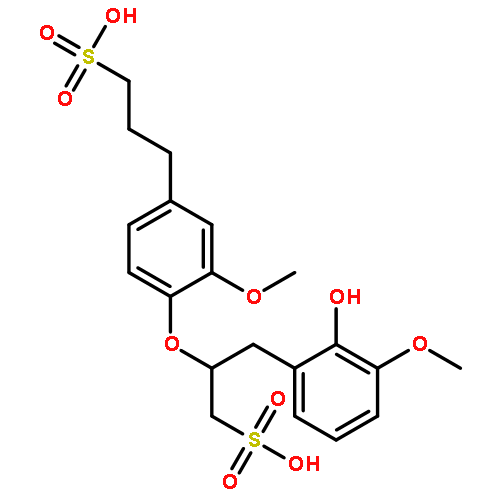
With lignin-first biorefineries likely to become a reality, controlled depolymerization of high-quality lignin streams to high-value products has become a priority. Using bisulfite chemistry, access to a high-β-O-4 content water-soluble lignosulfonate has been achieved, allowing follow-up procedures in water to be conducted. We show that phenolic β-O-4 units preferentially react under acidic bisulfite conditions, while nonphenolic β-O-4 units react much more slowly. Exploiting this improved chemical understanding and inherent selectivity, we have prepared a softwood lignosulfonate in which phenolic β-O-4 α-sulfonation has occurred, leaving significant native β-O-4 content. Use of an O-benzoylation protocol with lignin coupled with advanced two-dimensional nuclear magnetic resonance methods has allowed detailed analysis of this and other commercial and industrial lignosulfonates. Conversion of the native β-O-4 to benzylic, oxidized β-O-4 units was followed by a selective reductive cleavage to give a premium aromatic monomer in pure form.Keywords: Biorefineries; Bisulfite chemistry; Chemical feedstocks; Controlled depolymerization; O-Benzoylation;
Co-reporter:Ciaran W. Lahive; Peter J. Deuss; Christopher S. Lancefield; Zhuohua Sun; David B. Cordes; Claire M. Young; Fanny Tran; Alexandra M. Z. Slawin; Johannes G. de Vries; Paul C. J. Kamer; Nicholas J. Westwood;Katalin Barta
Journal of the American Chemical Society 2016 Volume 138(Issue 28) pp:8900-8911
Publication Date(Web):June 16, 2016
DOI:10.1021/jacs.6b04144
The development of fundamentally new approaches for lignin depolymerization is challenged by the complexity of this aromatic biopolymer. While overly simplified model compounds often lack relevance to the chemistry of lignin, the direct use of lignin streams poses significant analytical challenges to methodology development. Ideally, new methods should be tested on model compounds that are complex enough to mirror the structural diversity in lignin but still of sufficiently low molecular weight to enable facile analysis. In this contribution, we present a new class of advanced (β-O-4)-(β-5) dilinkage models that are highly realistic representations of a lignin fragment. Together with selected β-O-4, β-5, and β–β structures, these compounds provide a detailed understanding of the reactivity of various types of lignin linkages in acid catalysis in conjunction with stabilization of reactive intermediates using ethylene glycol. The use of these new models has allowed for identification of novel reaction pathways and intermediates and led to the characterization of new dimeric products in subsequent lignin depolymerization studies. The excellent correlation between model and lignin experiments highlights the relevance of this new class of model compounds for broader use in catalysis studies. Only by understanding the reactivity of the linkages in lignin at this level of detail can fully optimized lignin depolymerization strategies be developed.
Co-reporter:Alan R. Healy, Douglas R. Houston, Lucy Remnant, Anne-Sophie Huart, Veronika Brychtova, Magda M. Maslon, Olivia Meers, Petr Muller, Adam Krejci, Elizabeth A. Blackburn, Borek Vojtesek, Lenka Hernychova, Malcolm D. Walkinshaw, Nicholas J. Westwood and Ted R. Hupp
Chemical Science 2015 vol. 6(Issue 5) pp:3109-3116
Publication Date(Web):20 Mar 2015
DOI:10.1039/C4SC03885A
Developing approaches to discover protein–protein interactions (PPIs) remains a fundamental challenge. A chemical biology platform is applied here to identify novel PPIs for the AAA+ superfamily oncoprotein reptin. An in silico screen coupled with chemical optimization provided Liddean, a nucleotide-mimetic which modulates reptin's oligomerization status, protein-binding activity and global conformation. Combinatorial peptide phage library screening of Liddean-bound reptin with next generation sequencing identified interaction motifs including a novel reptin docking site on the p53 tumor suppressor protein. Proximity ligation assays demonstrated that endogenous reptin forms a predominantly cytoplasmic complex with its paralog pontin in cancer cells and Liddean promotes a shift of this complex to the nucleus. An emerging view of PPIs in higher eukaryotes is that they occur through a striking diversity of linear peptide motifs. The discovery of a compound that alters reptin's protein interaction landscape potentially leads to novel avenues for therapeutic development.
Co-reporter:F. Tran, C. S. Lancefield, P. C. J. Kamer, T. Lebl and N. J. Westwood
Green Chemistry 2015 vol. 17(Issue 1) pp:244-249
Publication Date(Web):11 Aug 2014
DOI:10.1039/C4GC01012D
The depolymerisation of the biopolymer lignin has the potential to provide access to a range of high value and commodity chemicals. However, research in this increasingly important area of green chemistry is hindered by the lack of analytical methods. The key challenge in using NMR is the throughput that can be achieved without the need for high field spectrometers fitted with cryoprobes. Here, we report the use of a relatively fast 2D HSQC NMR experiment performed on a 500 MHz spectrometer fitted with a BBFO+ probe to obtain high quality spectra. The use of the developed protocol to study the selective modification of the β–β linkage in Kraft lignin is also reported.
Co-reporter:C. S. Lancefield and N. J. Westwood
Green Chemistry 2015 vol. 17(Issue 11) pp:4980-4990
Publication Date(Web):03 Aug 2015
DOI:10.1039/C5GC01334H
If the lignin-first biorefinery concept becomes a reality, high quality lignins close in structure to native lignins will become available in large quantities. One potential way to utilise this renewable material is through depolymerisation to aromatic chemicals. This will require the development of new chemical methods. Here, we report the synthesis and characterisation of advanced lignin model polymers to be used as tools to develop these methods. The controlled incorporation of the major linkages in lignin is demonstrated to give complex hardwood and softwood lignin model polymers. These polymers have been characterised by 2D HSQC NMR and GPC analysis and have been compared to isolated lignins.
Co-reporter:Alan R. Healy, Francesco Vinale, Matteo Lorito, and Nicholas J. Westwood
Organic Letters 2015 Volume 17(Issue 3) pp:692-695
Publication Date(Web):January 28, 2015
DOI:10.1021/ol503717r
The bioactive natural product harzianic acid was prepared for the first time in just six steps (longest linear sequence) with an overall yield of 22%. The identification of conditions to telescope amide bond formation and a Lacey–Dieckmann reaction into one pot proved important. The three stereoisomers of harzianic acid were also prepared, providing material for comparison of their biological activity. While all of the isomers promoted root growth, improved antifungal activity was unexpectedly associated with isomers in the enantiomeric series opposite that of harzianic acid.
Co-reporter:A. R. Healy and N. J. Westwood
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 42) pp:10527-10531
Publication Date(Web):02 Sep 2015
DOI:10.1039/C5OB01771H
A late stage Diels–Alder reaction is used to prepare a mixture of JBIR-22, a natural product from the Equisetin family of tetramic acids, and one of its diastereomers. This is achieved in just 8 steps from pyruvate. The success of the late stage DA approach is discussed in the context of the biosynthesis of JBIR-22 (and perhaps related natural products).
Co-reporter:Christopher S. Lancefield;Alexra M. Z. Slawin;Tomas Lebl
Magnetic Resonance in Chemistry 2015 Volume 53( Issue 6) pp:467-475
Publication Date(Web):
DOI:10.1002/mrc.4213
Determining the conformational preferences of molecules in solution remains a considerable challenge. Recently, the use of residual dipolar coupling (RDC) analysis has emerged as a key method to address this. Whilst to date the majority of the applications have focused on biomolecules including proteins and DNA, the use of RDCs for studying small molecules is gaining popularity. Having said that, the method continues to develop, and here, we describe an early case study of the quantification of conformer populations in small molecules using RDC analysis. Having been inspired to study conformational preferences by unexpected differences in the NMR spectra and the reactivity of related natural products, we showed that the use of more established techniques was unsatisfactory in explaining the experimental observations. The use of RDCs provided an improved understanding that, following use of methods to quantify conformer populations using RDCs, culminated in a rationalisation of the contrasting diastereoselectivities observed in a ketone reduction reaction. Copyright © 2015 John Wiley & Sons, Ltd.
Co-reporter:Alan R. Healy;Dr. Miho Izumikawa; Alexra M. Z. Slawin;Dr. Kazuo Shin-ya; Nicholas J. Westwood
Angewandte Chemie 2015 Volume 127( Issue 13) pp:4118-4122
Publication Date(Web):
DOI:10.1002/ange.201411141
Abstract
Recent reports have highlighted the biological activity associated with a subfamily of the tetramic acid class of natural products. Despite the fact that members of this subfamily act as protein–protein interaction inhibitors that are of relevance to proteasome assembly, no synthetic work has been reported. This may be due to the fact that this subfamily contains an unnatural 4,4-disubstitued glutamic acid, the synthesis of which provides a key challenge. A highly stereoselective route to a masked form of this unnatural amino acid now enabled the synthesis of two of the possible diastereomers of JBIR-22 and allowed the assignment of its relative and absolute stereochemistry.
Co-reporter:Alan R. Healy;Dr. Miho Izumikawa; Alexra M. Z. Slawin;Dr. Kazuo Shin-ya; Nicholas J. Westwood
Angewandte Chemie International Edition 2015 Volume 54( Issue 13) pp:4046-4050
Publication Date(Web):
DOI:10.1002/anie.201411141
Abstract
Recent reports have highlighted the biological activity associated with a subfamily of the tetramic acid class of natural products. Despite the fact that members of this subfamily act as protein–protein interaction inhibitors that are of relevance to proteasome assembly, no synthetic work has been reported. This may be due to the fact that this subfamily contains an unnatural 4,4-disubstitued glutamic acid, the synthesis of which provides a key challenge. A highly stereoselective route to a masked form of this unnatural amino acid now enabled the synthesis of two of the possible diastereomers of JBIR-22 and allowed the assignment of its relative and absolute stereochemistry.
Co-reporter:Christopher S. Lancefield;Dr. O. Stephen Ojo;Dr. Fanny Tran ; Nicholas J. Westwood
Angewandte Chemie 2015 Volume 127( Issue 1) pp:260-264
Publication Date(Web):
DOI:10.1002/ange.201409408
Abstract
Functionalized phenolic monomers have been generated and isolated from an organosolv lignin through a two-step depolymerization process. Chemoselective catalytic oxidation of β-O-4 linkages promoted by the DDQ/tBuONO/O2 system was achieved in model compounds, including polymeric models and in real lignin. The oxidized β-O-4 linkages were then cleaved on reaction with zinc. Compared to many existing methods, this protocol, which can be achieved in one pot, is highly selective, giving rise to a simple mixture of products that can be readily purified to give pure compounds. The functionality present in these products makes them potentially valuable building blocks.
Co-reporter:Christopher S. Lancefield;Dr. O. Stephen Ojo;Dr. Fanny Tran ; Nicholas J. Westwood
Angewandte Chemie International Edition 2015 Volume 54( Issue 1) pp:258-262
Publication Date(Web):
DOI:10.1002/anie.201409408
Abstract
Functionalized phenolic monomers have been generated and isolated from an organosolv lignin through a two-step depolymerization process. Chemoselective catalytic oxidation of β-O-4 linkages promoted by the DDQ/tBuONO/O2 system was achieved in model compounds, including polymeric models and in real lignin. The oxidized β-O-4 linkages were then cleaved on reaction with zinc. Compared to many existing methods, this protocol, which can be achieved in one pot, is highly selective, giving rise to a simple mixture of products that can be readily purified to give pure compounds. The functionality present in these products makes them potentially valuable building blocks.
Co-reporter:Linna Zhou ; Gavin Stewart ; Emeline Rideau ; Nicholas J. Westwood ;Terry K. Smith
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:796-806
Publication Date(Web):January 3, 2013
DOI:10.1021/jm301215e
Recently, the World Health Organization approved the nifurtimox–eflornithine combination therapy for the treatment of human African trypanosomiasis, renewing interest in nitroheterocycle therapies for this and associated diseases. In this study, we have synthesized a series of novel 5-nitro-2-furancarboxylamides that show potent trypanocidal activity, ∼1000-fold more potent than nifurtimox against in vitro Trypanosoma brucei with very low cytotoxicity against human HeLa cells. More importantly, the most potent analogue showed very limited cross-resistance to nifurtimox-resistant cells and vice versa. This implies that our novel, relatively easy to synthesize and therefore cheap, 5-nitro-2-furancarboxylamides are targeting a different, but still essential, biochemical process to those targeted by nifurtimox or its metabolites in the parasites. The significant increase in potency (smaller dose probably required) has the potential for greatly reducing unwanted side effects and also reducing the likelihood of drug resistance. Collectively, these findings have important implications for the future therapeutic treatment of African sleeping sickness.
Co-reporter:Christopher S. Lancefield, Linna Zhou, Tomas Lébl, Alexandra M. Z. Slawin, and Nicholas J. Westwood
Organic Letters 2012 Volume 14(Issue 24) pp:6166-6169
Publication Date(Web):December 5, 2012
DOI:10.1021/ol302859j
A concise synthesis of melohenine B and O-ethyl-14-epimelohenine B, from eburnamonine, was achieved via a biomimetic diastereoselective singlet oxygen-mediated oxidative cleavage of the indole C2–C7 bond. These studies enabled the assignment of the absolute configuration of the natural products. In line with a proposed biosynthetic pathway, the resulting nine-membered ring containing products could be converted to the corresponding quinolones.
Co-reporter:Linna Zhou, Hironori Ishizaki, Michaela Spitzer, Kerrie L. Taylor, Nicholas D. Temperley, Stephen L. Johnson, Paul Brear, Philippe Gautier, Zhiqiang Zeng, Amy Mitchell, Vikram Narayan, Ewan M. McNeil, David W. Melton, Terry K. Smith, Mike Tyers, Nicholas J. Westwood, E. Elizabeth Patton
Chemistry & Biology 2012 Volume 19(Issue 7) pp:883-892
Publication Date(Web):27 July 2012
DOI:10.1016/j.chembiol.2012.05.017
Understanding how drugs work in vivo is critical for drug design and for maximizing the potential of currently available drugs. 5-nitrofurans are a class of prodrugs widely used to treat bacterial and trypanosome infections, but despite relative specificity, 5-nitrofurans often cause serious toxic side effects in people. Here, we use yeast and zebrafish, as well as human in vitro systems, to assess the biological activity of 5-nitrofurans, and we identify a conserved interaction between aldehyde dehydrogenase (ALDH) 2 and 5-nitrofurans across these species. In addition, we show that the activity of nifurtimox, a 5-nitrofuran anti-trypanosome prodrug, is dependent on zebrafish Aldh2 and is a substrate for human ALDH2. This study reveals a conserved and biologically relevant ALDH2-5-nitrofuran interaction that may have important implications for managing the toxicity of 5-nitrofuran treatment.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (133 K)Download as PowerPoint slideHighlights► Zebrafish provide a viable assay for the biological toxicity of 5-nitrofurans ► ALDH2 inhibitors prevent 5-nitrofuran toxicity in zebrafish and yeast ► Genetic dependence on ALDH2 for 5-nitrofuran toxicity in zebrafish and yeast systems ► 5-Nitrofurans bind to and are substrates of human ALDH2
Co-reporter:Anna R. McCarthy, Lisa Pirrie, Jonathan J. Hollick, Sebastien Ronseaux, Johanna Campbell, Maureen Higgins, Oliver D. Staples, Fanny Tran, Alexandra M.Z. Slawin, Sonia Lain, Nicholas J. Westwood
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 5) pp:1779-1793
Publication Date(Web):1 March 2012
DOI:10.1016/j.bmc.2012.01.001
The tenovins are small molecule inhibitors of the NAD+-dependent family of protein deacetylases known as the sirtuins. There remains considerable interest in inhibitors of this enzyme family due to possible applications in both cancer and neurodegenerative disease therapy. Through the synthesis of novel tenovin analogues, further insights into the structural requirements for activity against the sirtuins in vitro are provided. In addition, the activity of one of the analogues in cells led to an improved understanding of the function of SirT1 in cells.
Co-reporter:Paul Brear;Judith Telford; Garry L. Taylor;Dr. Nicholas J. Westwood
ChemBioChem 2012 Volume 13( Issue 16) pp:2374-2383
Publication Date(Web):
DOI:10.1002/cbic.201200433
Abstract
The major human pathogen Streptococcus pneumoniae plays a key role in several disease states including septicaemia, meningitis and community-acquired pneumonia. Although vaccines against S. pneumoniae are available as prophylactics, there remains a need to identify and characterise novel chemical entities that can treat the diseases caused by this pathogen. S. pneumoniae expresses three sialidases, enzymes that cleave sialic acid from carbohydrate-based surface molecules. Two of these enzymes, NanA and NanB, have been implicated in the pathogenesis of S. pneumoniae and are considered to be validated drug targets. Here we report our studies on the synthesis and structural characterisation of novel NanB-selective inhibitors that are inspired by the β-amino-sulfonic acid family of buffers.
Co-reporter:Christopher P. A. T. Lawson, Alexandra M. Z. Slawin and Nicholas J. Westwood
Chemical Communications 2011 vol. 47(Issue 3) pp:1057-1059
Publication Date(Web):15 Nov 2010
DOI:10.1039/C0CC03624B
A robust protocol for the CuI-catalysed arylation of amidines is presented. Whilst the initially identified conditions were useful for benzamidine-derived substrates, difficulties were encountered with more complex substrates. This problem was overcome following a change in ligand type, enabling the synthesis of analogues of the chemical tool, blebbistatin.
Co-reporter:Federico Medda, Thomas L. Joseph, Lisa Pirrie, Maureen Higgins, Alexandra M. Z. Slawin, Sonia Lain, Chandra Verma and Nicholas J. Westwood
MedChemComm 2011 vol. 2(Issue 7) pp:611-615
Publication Date(Web):05 May 2011
DOI:10.1039/C1MD00023C
The human deacetylase SIRT2 is believed to promote neurodegeneration with recent studies demonstrating that a reduction in the activity of SIRT2 can rescue alpha synuclein toxicity in Parkinson's disease models. In contrast, a second member of the sirtuin family, SIRT1, is believed to play a neuroprotective role. This dichotomy places an additional challenge in the path of sirtuin inhibitor development as a need for isozyme selectivity arises. By combining computational methods with assessment of the biological activity of novel N1-substituted cambinol analogues, further insights that are relevant to this challenge are obtained.
Co-reporter:Dr. Jeffrey G. A. Walton;Dr. Deuan C. Jones;Dr. Paula Kiuru;Dr. Alastair J. Durie;Dr. Nicholas J. Westwood; Alan H. Fairlamb;Daniel Spinks;Dr. Han B. Ong;Dr. Chidochangu P. Mpamhanga;Dr. Emma J. Shanks;Dr. David A. Robinson;Iain T. Collie;Dr. Kevin D. Read; Julie A. Frearson; Paul G. Wyatt;Dr. Ruth Brenk; Alan H. Fairlamb; Ian H. Gilbert;Dr. Gian Filippo Ruda;Dr. Corinne Nguyen;Dr. Przemys&x142;aw Ziemkowski;Dr. Krzysztof Felczak;Dr. Ganasan Kasinathan;Alexer Musso-Buendia;Christian Sund;Xiao Xiong Zhou;Marcel Kaiser; Luis M. Ruiz-Pérez;Dr. Reto Brun; Tadeusz Kulikowski;Dr. Nils Gunnar Johansson; Dolores González-Pacanowska; Ian H. Gilbert
ChemMedChem 2011 Volume 6( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201190000
Co-reporter:Dr. Jeffrey G. A. Walton;Dr. Deuan C. Jones;Dr. Paula Kiuru;Dr. Alastair J. Durie;Dr. Nicholas J. Westwood; Alan H. Fairlamb
ChemMedChem 2011 Volume 6( Issue 2) pp:321-328
Publication Date(Web):
DOI:10.1002/cmdc.201000442
Abstract
The search for novel compounds of relevance to the treatment of diseases caused by trypanosomatid protozoan parasites continues. Screening of a large library of known bioactive compounds has led to several drug-like starting points for further optimisation. In this study, novel analogues of the monoamine uptake inhibitor indatraline were prepared and assessed both as inhibitors of trypanothione reductase (TryR) and against the parasite Trypanosoma brucei. Although it proved difficult to significantly increase the potency of the original compound as an inhibitor of TryR, some insight into the preferred substituent on the amine group and in the two aromatic rings of the parent indatraline was deduced. In addition, detailed mode of action studies indicated that two of the inhibitors exhibit a mixed mode of inhibition.
Co-reporter:Dr. Alan M. Jones;Dr. Gu Liu;Dr. Magali M. Lorion;Dr. Stephen Patterson;Dr. Tomas Lebl; Alexra M. Z. Slawin ;Dr. Nicholas J. Westwood
Chemistry - A European Journal 2011 Volume 17( Issue 20) pp:5714-5718
Publication Date(Web):
DOI:10.1002/chem.201003188
Abstract
Oxidative cleavage of internal double bonds in polycyclic systems can give access to compounds containing medium- to large-sized rings. In this example, the nine- and ten-membered ring containing compounds that resulted from the mCPBA-mediated (mCPBA=meta-chloroperoxybenzoic acid) oxidative cleavage reaction were shown to exhibit atropisomerism. The reaction of the polycyclic system with catalytic amounts of ruthenium tetraoxide followed by diol cleavage achieved the same synthetic goal. Use of the Nishiyama–Beller ruthenium-based catalysts enabled the synthesis of optically-enriched samples, providing the first example of an atropselective oxidative cleavage reaction.
Co-reporter:Neil A. McIntyre ; Campbell McInnes ; Gary Griffiths ; Anna L. Barnett ; George Kontopidis ; Alexandra M. Z. Slawin ; Wayne Jackson ; Mark Thomas ; Daniella I. Zheleva ; Shudong Wang ∞; David G. Blake ; Nicholas J. Westwood ;Peter M. Fischer ∞
Journal of Medicinal Chemistry 2010 Volume 53(Issue 5) pp:2136-2145
Publication Date(Web):February 10, 2010
DOI:10.1021/jm901660c
Following the recent discovery and development of 2-anilino-4-(thiazol-5-yl)pyrimidine cyclin dependent kinase (CDK) inhibitors, a program was initiated to evaluate related ring-constrained analogues, specifically, 2-methyl- and 2-amino-N-aryl-4,5-dihydrothiazolo[4,5-h]quinazolin-8-amines for inhibition of CDKs. Here we report the rational design, synthesis, structure−activity relationships (SARs), and cellular mode-of-action profile of these second generation CDK inhibitors. Many of the analogues from this chemical series inhibit CDKs with very low nanomolar Ki values. The most potent compound reported in this study inhibits CDK2 with an IC50 of 0.7 nM ([ATP] = 100 μM). Furthermore, an X-ray crystal structure of 2-methyl-N-(3-(nitro)phenyl)-4,5-dihydrothiazolo[4,5-h]quinazolin-8-amine (11g), a representative from the chemical series in complex with cyclin A−CDK2, is reported, confirming the design rationale and expected binding mode within the CDK2 ATP binding pocket.
Co-reporter:Nicholas Voûte, Douglas Philp, Alexandra M. Z. Slawin and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 2) pp:442-450
Publication Date(Web):26 Nov 2009
DOI:10.1039/B915677A
A study of the effect of substrate structure on a Claisen-aza-Cope reaction is presented including a rationalisation of the reaction outcome using DFT calculations. An asymmetric version of the reaction is also described that is of relevance to a proposed approach to the communesin family of natural products.
Co-reporter:Tomas Lebl, Magali M. Lorion, Alan M. Jones, Douglas Philp, Nicholas J. Westwood
Tetrahedron 2010 66(51) pp: 9694-9702
Publication Date(Web):
DOI:10.1016/j.tet.2010.10.043
Co-reporter:Alan M. Jones, Magali M. Lorion, Tomas Lebl, Alexandra M.Z. Slawin, Douglas Philp, Nicholas J. Westwood
Tetrahedron 2010 66(51) pp: 9667-9674
Publication Date(Web):
DOI:10.1016/j.tet.2010.10.044
Co-reporter:Gu Liu;CatherineH. Botting;KathrynM. Evans;JeffreyA.G. Walton;Guogang Xu;AlexraM.Z. Slawin ;NicholasJ. Westwood
ChemMedChem 2010 Volume 5( Issue 1) pp:41-45
Publication Date(Web):
DOI:10.1002/cmdc.200900391
Co-reporter:Federico Medda ; Rupert J. M. Russell ; Maureen Higgins ; Anna R. McCarthy ; Johanna Campbell ; Alexandra M. Z. Slawin ; David P. Lane ; Sonia Lain
Journal of Medicinal Chemistry 2009 Volume 52(Issue 9) pp:2673-2682
Publication Date(Web):April 1, 2009
DOI:10.1021/jm8014298
The tenovins and cambinol are two classes of sirtuin inhibitor that exhibit antitumor activity in preclinical models. This report describes modifications to the core structure of cambinol, in particular by incorporation of substitutents at the N1-position, which lead to increased potency and modified selectivity. These improvements have been rationalized using molecular modeling techniques. The expected functional selectivity in cells was also observed for both a SIRT1 and a SIRT2 selective analog.
Co-reporter:Jeralyn D. Haraldsen, Gu Liu, Catherine H. Botting, Jeffrey G. A. Walton, Janet Storm, Timothy J. Phalen, Lai Yu Kwok, Dominique Soldati-Favre, Nicholas H. Heintz, Sylke Müller, Nicholas J. Westwood and Gary E. Ward
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 15) pp:3040-3048
Publication Date(Web):21 May 2009
DOI:10.1039/B901735F
Conoidin A (1) is an inhibitor of host cell invasion by the protozoan parasite Toxoplasma gondii. In the course of studies aimed at identifying potential targets of this compound, we determined that it binds to the T. gondii enzyme peroxiredoxin II (TgPrxII). Peroxiredoxins are a widely conserved family of enzymes that function in antioxidant defense and signal transduction, and changes in PrxII expression are associated with a variety of human diseases, including cancer. Disruption of the TgPrxII gene by homologous recombination had no effect on the sensitivity of the parasites to 1, suggesting that TgPrxII is not the invasion-relevant target of 1. However, we showed that 1 binds covalently to the peroxidatic cysteine of TgPrxII, inhibiting its enzymatic activity in vitro. Studies with human epithelial cells showed that 1 also inhibits hyperoxidation of human PrxII. These data identify Conoidin A as a novel inhibitor of this important class of antioxidant and redox signaling enzymes.
Co-reporter:Jeffrey G. A. Walton, Stephen Patterson, Gu Liu, Jeralyn D. Haraldsen, Jonathan J. Hollick, Alexandra M. Z. Slawin, Gary E. Ward and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 15) pp:3049-3060
Publication Date(Web):17 Jun 2009
DOI:10.1039/B902319D
Techniques for the identification of the protein target(s) of small molecules are proving very important following an increase in the use of phenotype-based screening in chemical biology and drug discovery. One approach, known as the yeast-3-hybrid approach, has shown considerable potential. A key factor in the success of this approach is the preparation of a complex molecule referred to as a chemical inducer of dimerisation (CID). The synthesis of two CIDs based on a bioactive tetrahydro-β-carboline core structure is reported and evidence presented that shows the CIDs are of utility in this approach. A series of chemo- and bioinformatic studies coupled with SAR development inspired the choice of CIDs.
Co-reporter:Edward F. Makiyi;Raquel F. M. Frade;Tomas Lebl;Ellis G. Jaffray;Susan E. Cobb;Alan L. Harvey;Alexra M. Z. Slawin;Ronald T. Hay
European Journal of Organic Chemistry 2009 Volume 2009( Issue 33) pp:5711-5715
Publication Date(Web):
DOI:10.1002/ejoc.200901016
Abstract
The isolation, identification and total synthesis of two plant-derived inhibitors of the NF-κB signaling pathway from the iso-seco-tanapartholide family of natural products is described. A key step in the efficient reaction sequence is a late-stage oxidative cleavage reaction that was carried out in the absence of protecting groups to give the natural products directly. A detailed comparison of the synthetic material with samples of the natural products proved informative. Biological studies on synthetic material confirmed that these compounds act late in the NF-κB signaling pathway. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Alan M. Jones, Tomas Lebl, Stephen Patterson, Tanja van Mourik, Herbert A. Früchtl, Douglas Philp, Alexandra M.Z. Slawin, Nicholas J. Westwood
Tetrahedron 2009 65(2) pp: 563-578
Publication Date(Web):
DOI:10.1016/j.tet.2008.10.049
Co-reporter:Cristina Lucas-Lopez, John S. Allingham, Tomas Lebl, Christopher P. A. T. Lawson, Ruth Brenk, James R. Sellers, Ivan Rayment and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2008 vol. 6(Issue 12) pp:2076-2084
Publication Date(Web):21 Apr 2008
DOI:10.1039/B801223G
The small molecule blebbistatin is now a front line tool in the study of myosin function. Chemical modification of the tricyclic core of blebbistatin could deliver the next generation of myosin inhibitors and to help address this we report here on the impact of structural changes in the methyl-substituted aromatic ring of blebbistatin on its biological activity. Chemical methods for the preparation of isomeric methyl-containing analogues are reported and a series of co-crystal structures are used to rationalise the observed variations in their biological activity. These studies further support the view that the previously identified binding mode of blebbistatin to Dictyostelium discoideum myosin II is of relevance to its mode of action. A discussion of the role that these observations have on planning the synthesis of focused libraries of blebbistatin analogues is also provided including an assessment of possibilities by computational methods. These studies are ultimately directed at the development of novel myosin inhibitors with improved affinity and different selectivity profiles from blebbistatin itself.
Co-reporter:Kimberly L. Carey;Timothy J. Mitchison;Gary E. Ward
PNAS 2004 Volume 101 (Issue 19 ) pp:7433-7438
Publication Date(Web):2004-05-11
DOI:10.1073/pnas.0307769101
Toxoplasma gondii is the most common protozoan parasite of humans. Infection with T. gondii can lead to life-threatening disease as a result of repeated cycles of host cell invasion, parasite replication, and host
cell lysis. Relatively little is known about the invasive mechanisms of T. gondii and related parasites within the Phylum Apicomplexa (including Plasmodium spp., the causative agents of malaria), due to difficulties associated with studying genes essential to invasion in haploid
obligate intracellular organisms. To circumvent this problem, we have developed a high-throughput microscope-based assay,
which we have used to screen a collection of 12,160 structurally diverse small molecules for inhibitors of T. gondii invasion. A total of 24 noncytotoxic invasion inhibitors were identified. Secondary assays demonstrated that different inhibitors
perturb different aspects of invasion, including gliding motility, secretion of host cell adhesins from apical organelles
(the micronemes), and extension of a unique tubulin-based structure at the anterior of the parasite (the conoid). Unexpectedly,
the screen also identified six small molecules that dramatically enhance invasion, gliding motility, and microneme secretion.
The small molecules identified here reveal a previously unrecognized complexity in the control of parasite motility and microneme
secretion, and they constitute a set of useful probes for dissecting the invasive mechanisms of T. gondii and related parasites. Small-molecule-based approaches provide a powerful means to address experimentally challenging problems
in host-pathogen interaction, while simultaneously identifying new potential targets for drug development.
Co-reporter:Anna R. McCarthy, Jonathan J. Hollick, Nicholas J. Westwood
Seminars in Cancer Biology (February 2010) Volume 20(Issue 1) pp:40-45
Publication Date(Web):1 February 2010
DOI:10.1016/j.semcancer.2010.02.007
The reactivation of mutant forms of the transcriptional regulator p53 or artificially raising activated p53 levels in a controlled nongenotoxic manner are seen as two of the grand challenges in anti-cancer drug discovery. Recent reports suggest that these demanding goals are achievable. This review article focuses on the use of cell-based high-throughput screening to discover novel nongenotoxic activators of endogenous p53. This challenging approach to the early phases of drug discovery prioritises the discovery of compounds with activity in cells in the hope that the compounds discovered will ultimately be of more direct relevance to therapeutic development. However, this approach also requires that protein target identification studies are carried out. We, and others, have shown that whilst a sometimes daunting proposition, it is possible to identify the targets of compounds that activate p53.
Co-reporter:Cristina Lucas-Lopez, John S. Allingham, Tomas Lebl, Christopher P. A. T. Lawson, Ruth Brenk, James R. Sellers, Ivan Rayment and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2008 - vol. 6(Issue 12) pp:NaN2084-2084
Publication Date(Web):2008/04/21
DOI:10.1039/B801223G
The small molecule blebbistatin is now a front line tool in the study of myosin function. Chemical modification of the tricyclic core of blebbistatin could deliver the next generation of myosin inhibitors and to help address this we report here on the impact of structural changes in the methyl-substituted aromatic ring of blebbistatin on its biological activity. Chemical methods for the preparation of isomeric methyl-containing analogues are reported and a series of co-crystal structures are used to rationalise the observed variations in their biological activity. These studies further support the view that the previously identified binding mode of blebbistatin to Dictyostelium discoideum myosin II is of relevance to its mode of action. A discussion of the role that these observations have on planning the synthesis of focused libraries of blebbistatin analogues is also provided including an assessment of possibilities by computational methods. These studies are ultimately directed at the development of novel myosin inhibitors with improved affinity and different selectivity profiles from blebbistatin itself.
Co-reporter:Ross. P. Wilkie, Andrew R. Neal, Craig A. Johnston, Nicholas Voute, Christopher S. Lancefield, Matthew D. Stell, Federico Medda, Edward F. Makiyi, Emma M. Turner, O. Stephen Ojo, Alexandra M. Z. Slawin, Tomas Lebl, Peter Mullen, David J. Harrison, Chris M. Ireland and Nicholas J. Westwood
Chemical Communications 2016 - vol. 52(Issue 71) pp:NaN10750-10750
Publication Date(Web):2016/08/05
DOI:10.1039/C6CC05747K
The synthesis of dehaloperophoramidine, a non-halogenated derivative of the marine natural product perophoramidine, and its biological activity towards HCT116, HT29 and LoVo colorectal carcinoma cells is reported. A [3,3]-Claisen rearrangement and an epoxide opening/allylsilylation reaction installed the contiguous all-carbon quaternary stereocentres with the required relative stereochemistry.
Co-reporter:A. R. Healy and N. J. Westwood
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 42) pp:NaN10531-10531
Publication Date(Web):2015/09/02
DOI:10.1039/C5OB01771H
A late stage Diels–Alder reaction is used to prepare a mixture of JBIR-22, a natural product from the Equisetin family of tetramic acids, and one of its diastereomers. This is achieved in just 8 steps from pyruvate. The success of the late stage DA approach is discussed in the context of the biosynthesis of JBIR-22 (and perhaps related natural products).
Co-reporter:Christopher P. A. T. Lawson, Alexandra M. Z. Slawin and Nicholas J. Westwood
Chemical Communications 2011 - vol. 47(Issue 3) pp:NaN1059-1059
Publication Date(Web):2010/11/15
DOI:10.1039/C0CC03624B
A robust protocol for the CuI-catalysed arylation of amidines is presented. Whilst the initially identified conditions were useful for benzamidine-derived substrates, difficulties were encountered with more complex substrates. This problem was overcome following a change in ligand type, enabling the synthesis of analogues of the chemical tool, blebbistatin.
Co-reporter:Nicholas Voûte, Douglas Philp, Alexandra M. Z. Slawin and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 2) pp:NaN450-450
Publication Date(Web):2009/11/26
DOI:10.1039/B915677A
A study of the effect of substrate structure on a Claisen-aza-Cope reaction is presented including a rationalisation of the reaction outcome using DFT calculations. An asymmetric version of the reaction is also described that is of relevance to a proposed approach to the communesin family of natural products.
Co-reporter:Jeffrey G. A. Walton, Stephen Patterson, Gu Liu, Jeralyn D. Haraldsen, Jonathan J. Hollick, Alexandra M. Z. Slawin, Gary E. Ward and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 15) pp:NaN3060-3060
Publication Date(Web):2009/06/17
DOI:10.1039/B902319D
Techniques for the identification of the protein target(s) of small molecules are proving very important following an increase in the use of phenotype-based screening in chemical biology and drug discovery. One approach, known as the yeast-3-hybrid approach, has shown considerable potential. A key factor in the success of this approach is the preparation of a complex molecule referred to as a chemical inducer of dimerisation (CID). The synthesis of two CIDs based on a bioactive tetrahydro-β-carboline core structure is reported and evidence presented that shows the CIDs are of utility in this approach. A series of chemo- and bioinformatic studies coupled with SAR development inspired the choice of CIDs.
Co-reporter:Alan R. Healy, Douglas R. Houston, Lucy Remnant, Anne-Sophie Huart, Veronika Brychtova, Magda M. Maslon, Olivia Meers, Petr Muller, Adam Krejci, Elizabeth A. Blackburn, Borek Vojtesek, Lenka Hernychova, Malcolm D. Walkinshaw, Nicholas J. Westwood and Ted R. Hupp
Chemical Science (2010-Present) 2015 - vol. 6(Issue 5) pp:NaN3116-3116
Publication Date(Web):2015/03/20
DOI:10.1039/C4SC03885A
Developing approaches to discover protein–protein interactions (PPIs) remains a fundamental challenge. A chemical biology platform is applied here to identify novel PPIs for the AAA+ superfamily oncoprotein reptin. An in silico screen coupled with chemical optimization provided Liddean, a nucleotide-mimetic which modulates reptin's oligomerization status, protein-binding activity and global conformation. Combinatorial peptide phage library screening of Liddean-bound reptin with next generation sequencing identified interaction motifs including a novel reptin docking site on the p53 tumor suppressor protein. Proximity ligation assays demonstrated that endogenous reptin forms a predominantly cytoplasmic complex with its paralog pontin in cancer cells and Liddean promotes a shift of this complex to the nucleus. An emerging view of PPIs in higher eukaryotes is that they occur through a striking diversity of linear peptide motifs. The discovery of a compound that alters reptin's protein interaction landscape potentially leads to novel avenues for therapeutic development.
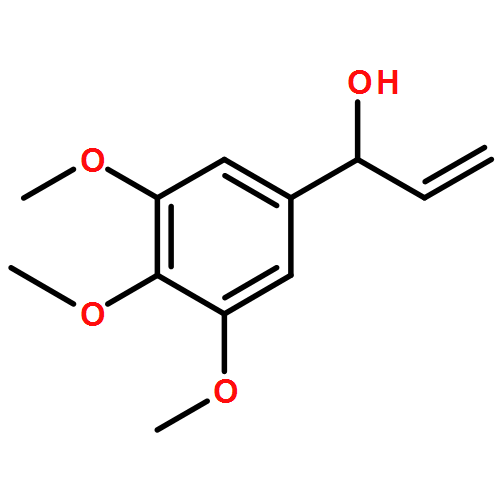
Co-reporter:Daniel M. Miles-Barrett, Andrew R. Neal, Calum Hand, James R. D. Montgomery, Isabella Panovic, O. Stephen Ojo, Christopher S. Lancefield, David B. Cordes, Alexandra M. Z. Slawin, Tomas Lebl and Nicholas J. Westwood
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 42) pp:NaN10030-10030
Publication Date(Web):2016/09/22
DOI:10.1039/C6OB01915C
Understanding the structure of technical lignins resulting from acid-catalysed treatment of lignocellulosic biomass is important for their future applications. Here we report an investigation into the fate of lignin under acidic aqueous organosolv conditions. In particular we examine in detail the formation and reactivity of non-native Hibbert ketone structures found in isolated organosolv lignins from both Douglas fir and beech woods. Through the use of model compounds combined with HSQC, HMBC and HSQC-TOCSY NMR experiments we demonstrate that, depending on the lignin source, both S and G lignin-bound Hibbert ketone units can be present. We also show that these units can serve as a source of novel mono-aromatic compounds following an additional lignin depolymerisation reaction.
Co-reporter:Jeralyn D. Haraldsen, Gu Liu, Catherine H. Botting, Jeffrey G. A. Walton, Janet Storm, Timothy J. Phalen, Lai Yu Kwok, Dominique Soldati-Favre, Nicholas H. Heintz, Sylke Müller, Nicholas J. Westwood and Gary E. Ward
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 15) pp:NaN3048-3048
Publication Date(Web):2009/05/21
DOI:10.1039/B901735F
Conoidin A (1) is an inhibitor of host cell invasion by the protozoan parasite Toxoplasma gondii. In the course of studies aimed at identifying potential targets of this compound, we determined that it binds to the T. gondii enzyme peroxiredoxin II (TgPrxII). Peroxiredoxins are a widely conserved family of enzymes that function in antioxidant defense and signal transduction, and changes in PrxII expression are associated with a variety of human diseases, including cancer. Disruption of the TgPrxII gene by homologous recombination had no effect on the sensitivity of the parasites to 1, suggesting that TgPrxII is not the invasion-relevant target of 1. However, we showed that 1 binds covalently to the peroxidatic cysteine of TgPrxII, inhibiting its enzymatic activity in vitro. Studies with human epithelial cells showed that 1 also inhibits hyperoxidation of human PrxII. These data identify Conoidin A as a novel inhibitor of this important class of antioxidant and redox signaling enzymes.

![Carbamic acid, [4-[[(1,1-dimethylethoxy)carbonyl]amino]phenyl]methyl-, phenylmethyl ester](http://img.cochemist.com/ccimg/113300/113283-96-8.png)
![Carbamic acid, [4-[[(1,1-dimethylethoxy)carbonyl]amino]phenyl]methyl-, phenylmethyl ester](http://img.cochemist.com/ccimg/113300/113283-96-8_b.png)












![2H-Pyrrolo[3,4-b]quinoxaline, 2-butyl-, 4-oxide](http://img.cochemist.com/ccimg/857700/857686-08-9.png)
![2H-Pyrrolo[3,4-b]quinoxaline, 2-butyl-, 4-oxide](http://img.cochemist.com/ccimg/857700/857686-08-9_b.png)
![Benzo[g]quinoline-5,10-dione, 3-methyl-](http://img.cochemist.com/ccimg/83700/83669-73-2.png)
![Benzo[g]quinoline-5,10-dione, 3-methyl-](http://img.cochemist.com/ccimg/83700/83669-73-2_b.png)
![Pentanamide, 5-bromo-2-ethyl-N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/54000/53908-55-7.png)
![Pentanamide, 5-bromo-2-ethyl-N-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/54000/53908-55-7_b.png)
![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 3,6-bis(4-chlorophenyl)-](http://img.cochemist.com/ccimg/502500/502423-29-2.png)
![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 3,6-bis(4-chlorophenyl)-](http://img.cochemist.com/ccimg/502500/502423-29-2_b.png)


















![1H-Pyrrolo[3,4-b]quinoxalinium, 2,2-diethyl-2,3-dihydro-, bromide](http://img.cochemist.com/ccimg/40200/40197-25-9.png)
![1H-Pyrrolo[3,4-b]quinoxalinium, 2,2-diethyl-2,3-dihydro-, bromide](http://img.cochemist.com/ccimg/40200/40197-25-9_b.png)
![ETHANESULFONIC ACID, 2-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/39800/39775-17-2.png)
![ETHANESULFONIC ACID, 2-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/39800/39775-17-2_b.png)


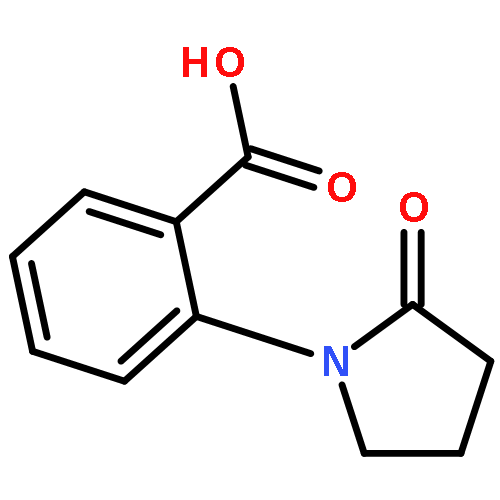
![BENZOIC ACID, 2-[[2-(2-OXO-1-PYRROLIDINYL)BENZOYL]AMINO]-, METHYL ESTER](http://img.cochemist.com/ccimg/865200/865190-71-2.png)
![BENZOIC ACID, 2-[[2-(2-OXO-1-PYRROLIDINYL)BENZOYL]AMINO]-, METHYL ESTER](http://img.cochemist.com/ccimg/865200/865190-71-2_b.png)


![Cyclohexanone, 2,6-bis[(4-methylphenyl)methylene]-, (E,E)-](http://img.cochemist.com/ccimg/42800/42792-79-0.png)
![Cyclohexanone, 2,6-bis[(4-methylphenyl)methylene]-, (E,E)-](http://img.cochemist.com/ccimg/42800/42792-79-0_b.png)




![3H-Naphtho[2,1-b]pyran-3-one, 2-benzoyl-1,2-dihydro-](http://img.cochemist.com/ccimg/14200/14103-22-1.png)
![3H-Naphtho[2,1-b]pyran-3-one, 2-benzoyl-1,2-dihydro-](http://img.cochemist.com/ccimg/14200/14103-22-1_b.png)









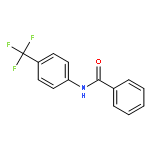
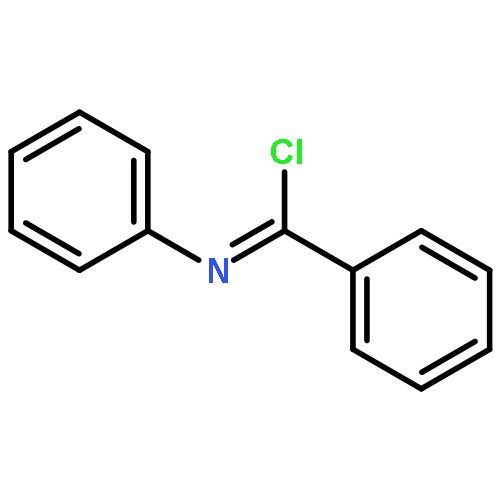
![BENZENECARBOXIMIDOYL CHLORIDE, N-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/80900/80819-62-1.png)
![BENZENECARBOXIMIDOYL CHLORIDE, N-[4-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/80900/80819-62-1_b.png)
![1H-FURO[3,4-C]PYRROLE-1,4(5H)-DIONE, 3,5,6-TRIPHENYL-](http://img.cochemist.com/ccimg/128400/128318-56-9.png)
![1H-FURO[3,4-C]PYRROLE-1,4(5H)-DIONE, 3,5,6-TRIPHENYL-](http://img.cochemist.com/ccimg/128400/128318-56-9_b.png)


![6H,9H,15H-BENZO[6,7][1,8]NAPHTHYRIDINO[1,8-AB]QUINAZOLINE-9,15-DIONE, 7,8-DIHYDRO-](http://img.cochemist.com/ccimg/49900/49854-18-4.png)
![6H,9H,15H-BENZO[6,7][1,8]NAPHTHYRIDINO[1,8-AB]QUINAZOLINE-9,15-DIONE, 7,8-DIHYDRO-](http://img.cochemist.com/ccimg/49900/49854-18-4_b.png)


![3H-NAPHTHO[2,1-B]PYRAN-3-ONE, 2-(4-METHYLBENZOYL)-](http://img.cochemist.com/ccimg/82600/82531-51-9.png)
![3H-NAPHTHO[2,1-B]PYRAN-3-ONE, 2-(4-METHYLBENZOYL)-](http://img.cochemist.com/ccimg/82600/82531-51-9_b.png)


![Ethanesulfonic acid, 2-[[(2-hydroxyphenyl)methyl]amino]-](http://img.cochemist.com/ccimg/162200/162127-01-7.png)
![Ethanesulfonic acid, 2-[[(2-hydroxyphenyl)methyl]amino]-](http://img.cochemist.com/ccimg/162200/162127-01-7_b.png)



![1,2-Cyclohexanedione, mono[(4-bromophenyl)hydrazone]](http://img.cochemist.com/ccimg/108700/108606-59-3.png)
![1,2-Cyclohexanedione, mono[(4-bromophenyl)hydrazone]](http://img.cochemist.com/ccimg/108700/108606-59-3_b.png)






![methyl 2-[(2-iodobenzoyl)amino]benzoate](http://img.cochemist.com/ccimg/75600/75541-70-7.png)
![methyl 2-[(2-iodobenzoyl)amino]benzoate](http://img.cochemist.com/ccimg/75600/75541-70-7_b.png)




![5,7,8,9,10,11-hexahydro-6H-Cyclooct[b]indol-6-one](http://img.cochemist.com/ccimg/16300/16244-20-5.png)
![5,7,8,9,10,11-hexahydro-6H-Cyclooct[b]indol-6-one](http://img.cochemist.com/ccimg/16300/16244-20-5_b.png)






![3H-NAPHTHO[2,1-B]PYRAN-3-ONE, 2-(2-FURANYLCARBONYL)-](http://img.cochemist.com/ccimg/578000/577988-70-6.png)
![3H-NAPHTHO[2,1-B]PYRAN-3-ONE, 2-(2-FURANYLCARBONYL)-](http://img.cochemist.com/ccimg/578000/577988-70-6_b.png)








![Cyclohexanone, 2,6-bis[(4-methoxyphenyl)methylene]-, (E,E)-](http://img.cochemist.com/ccimg/62100/62085-71-6.png)
![Cyclohexanone, 2,6-bis[(4-methoxyphenyl)methylene]-, (E,E)-](http://img.cochemist.com/ccimg/62100/62085-71-6_b.png)
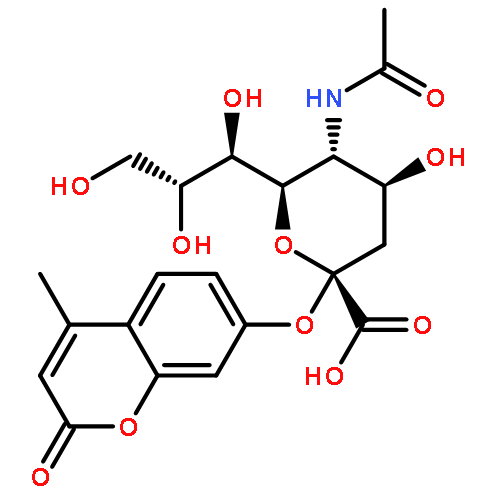
![(2S,4S,5R,6R)-5-acetamido-4-hydroxy-2-(4-methyl-2-oxo-chromen-7-yl)oxy-6-[(2R)-1,2,3-trihydroxypropyl]tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/59400/59322-44-0.png)
![(2S,4S,5R,6R)-5-acetamido-4-hydroxy-2-(4-methyl-2-oxo-chromen-7-yl)oxy-6-[(2R)-1,2,3-trihydroxypropyl]tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/59400/59322-44-0_b.png)
![5-nitro-N-[3-(trifluoromethyl)phenyl]-2-furamide](http://img.cochemist.com/ccimg/15300/15212-60-9.png)
![5-nitro-N-[3-(trifluoromethyl)phenyl]-2-furamide](http://img.cochemist.com/ccimg/15300/15212-60-9_b.png)
![N-[2-chloro-5-(trifluoromethyl)phenyl]-5-nitro-2-furamide](http://img.cochemist.com/ccimg/215800/215779-26-3.png)
![N-[2-chloro-5-(trifluoromethyl)phenyl]-5-nitro-2-furamide](http://img.cochemist.com/ccimg/215800/215779-26-3_b.png)
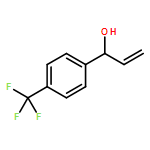
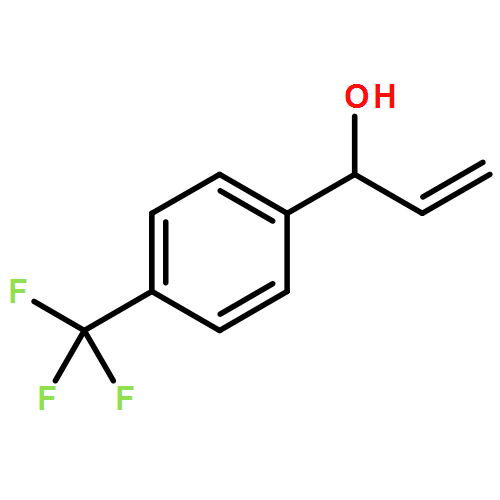
![1-[3-(trifluoromethyl)phenyl]prop-2-en-1-ol](http://img.cochemist.com/ccimg/154400/154309-48-5.png)
![1-[3-(trifluoromethyl)phenyl]prop-2-en-1-ol](http://img.cochemist.com/ccimg/154400/154309-48-5_b.png)


![Benzenemethanol, 3,4-dimethoxy-a-[(2-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/17100/17078-88-5.png)
![Benzenemethanol, 3,4-dimethoxy-a-[(2-methoxyphenoxy)methyl]-](http://img.cochemist.com/ccimg/17100/17078-88-5_b.png)







![Pyrrolo[1,2-a]quinazolin-5(1H)-one, 2,3-dihydro-](http://img.cochemist.com/ccimg/16200/16198-97-3.png)
![Pyrrolo[1,2-a]quinazolin-5(1H)-one, 2,3-dihydro-](http://img.cochemist.com/ccimg/16200/16198-97-3_b.png)









![7,8,9,10-tetrahydro-Cyclohept[b]indol-6(5H)-one](http://img.cochemist.com/ccimg/7300/7257-25-2.png)
![7,8,9,10-tetrahydro-Cyclohept[b]indol-6(5H)-one](http://img.cochemist.com/ccimg/7300/7257-25-2_b.png)




![2-[4-(dimethylamino)phenyl]guanidine](http://img.cochemist.com/ccimg/67500/67453-82-1.png)
![2-[4-(dimethylamino)phenyl]guanidine](http://img.cochemist.com/ccimg/67500/67453-82-1_b.png)




![4-Thiazolecarboxamide,N-[5-[[(3-amino-3-iminopropyl)amino]carbonyl]-1-methyl-1H-pyrrol-3-yl]-2-(formylamino)-](http://img.cochemist.com/ccimg/1300/1264-57-9.png)
![4-Thiazolecarboxamide,N-[5-[[(3-amino-3-iminopropyl)amino]carbonyl]-1-methyl-1H-pyrrol-3-yl]-2-(formylamino)-](http://img.cochemist.com/ccimg/1300/1264-57-9_b.png)
![Ethanesulfonic acid,2-[(2-hydroxyethyl)amino]-](http://img.cochemist.com/ccimg/29800/29706-49-8.png)
![Ethanesulfonic acid,2-[(2-hydroxyethyl)amino]-](http://img.cochemist.com/ccimg/29800/29706-49-8_b.png)
![2(3H)-Furanone,dihydro-5-[(3S)-3-hydroxy-2-methyl-5-oxo-1-cyclopenten-1-yl]-3-methylene-4-(3-oxobutyl)-,(4S,5S)-](http://img.cochemist.com/ccimg/103000/102926-01-2.png)
![2(3H)-Furanone,dihydro-5-[(3S)-3-hydroxy-2-methyl-5-oxo-1-cyclopenten-1-yl]-3-methylene-4-(3-oxobutyl)-,(4S,5S)-](http://img.cochemist.com/ccimg/103000/102926-01-2_b.png)
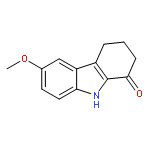
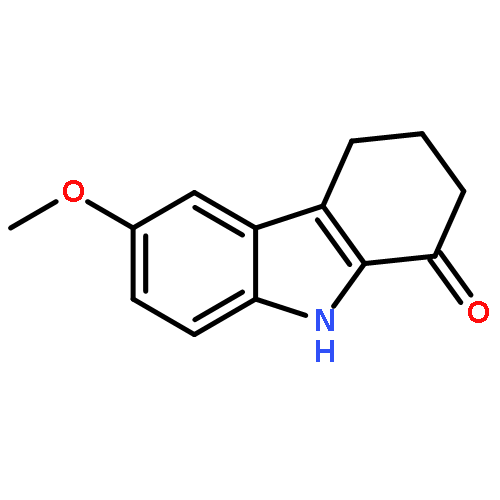
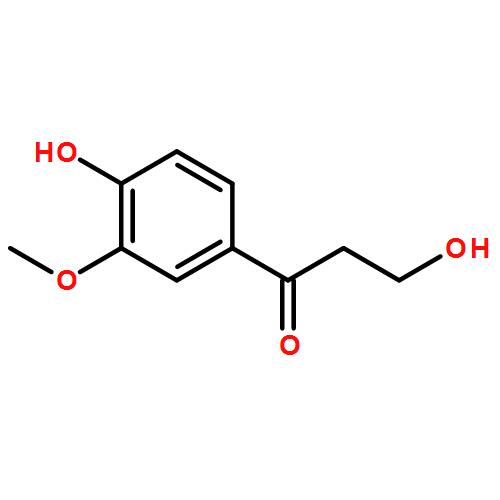
![4-Hydroxy[1,1'-biphenyl]-3-carbaldehyde](http://img.cochemist.com/ccimg/1800/1761-63-3.png)
![4-Hydroxy[1,1'-biphenyl]-3-carbaldehyde](http://img.cochemist.com/ccimg/1800/1761-63-3_b.png)


![Carbamic acid, [(1S,2S)-1-ethynyl-2-methylbutyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/181800/181760-14-5.png)
![Carbamic acid, [(1S,2S)-1-ethynyl-2-methylbutyl]-, 1,1-dimethylethyl ester (9CI)](http://img.cochemist.com/ccimg/181800/181760-14-5_b.png)












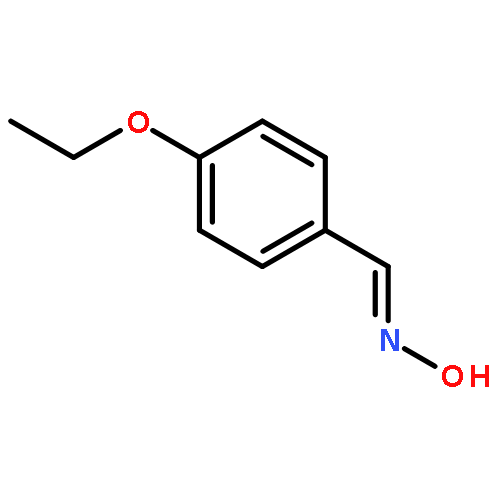
![N-[(E)-(5-nitrofuran-2-yl)methylidene]aniline](http://img.cochemist.com/ccimg/200/156-44-5.png)
![N-[(E)-(5-nitrofuran-2-yl)methylidene]aniline](http://img.cochemist.com/ccimg/200/156-44-5_b.png)












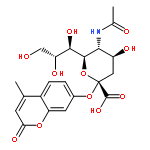
![Benzaldehyde, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methoxy-](http://img.cochemist.com/ccimg/69500/69404-94-0.png)
![Benzaldehyde, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3-methoxy-](http://img.cochemist.com/ccimg/69500/69404-94-0_b.png)












![TERT-BUTYL [(1S,3S)-1-FORMYL-3-METHYLPENTYL]CARBAMATE](http://img.cochemist.com/ccimg/87700/87694-55-1.png)
![TERT-BUTYL [(1S,3S)-1-FORMYL-3-METHYLPENTYL]CARBAMATE](http://img.cochemist.com/ccimg/87700/87694-55-1_b.png)





![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dihydro-2-methyl-3,5,6-triphenyl-](/data/chemimg/1342000/836624-08-9.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dihydro-2-methyl-3,5,6-triphenyl-](/data/chemimg/1342000/836624-08-9_b.png)






![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 3,6-diphenyl-5-(phenylmethyl)-](http://img.cochemist.com/ccimg/502500/502423-27-0.png)
![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 3,6-diphenyl-5-(phenylmethyl)-](http://img.cochemist.com/ccimg/502500/502423-27-0_b.png)








![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 3,6-diphenyl-](http://img.cochemist.com/ccimg/502200/502183-99-5.png)
![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 3,6-diphenyl-](http://img.cochemist.com/ccimg/502200/502183-99-5_b.png)
![Benzoic acid, 4-[[(phenylmethoxy)carbonyl]amino]-](/data/chemimg/1139400/5330-71-2.png)
![Benzoic acid, 4-[[(phenylmethoxy)carbonyl]amino]-](/data/chemimg/1139400/5330-71-2_b.png)
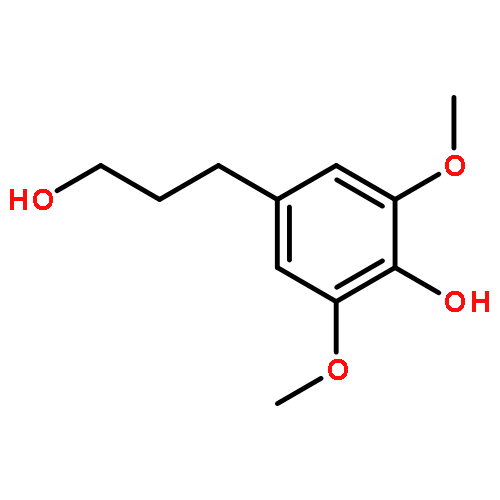
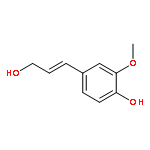
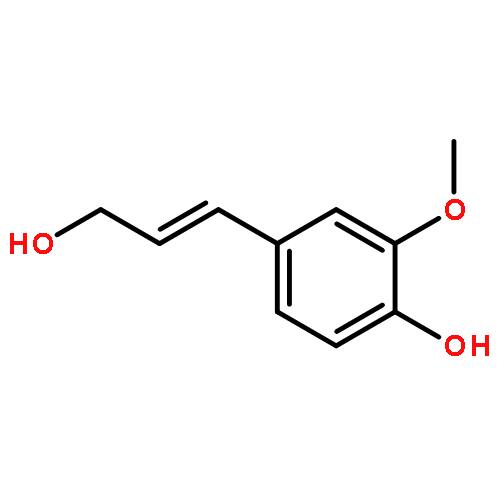
![Phenol,4-[(1E)-3-hydroxy-1-propenyl]-2,6-dimethoxy-](http://img.cochemist.com/ccimg/20700/20675-96-1.png)
![Phenol,4-[(1E)-3-hydroxy-1-propenyl]-2,6-dimethoxy-](http://img.cochemist.com/ccimg/20700/20675-96-1_b.png)


![4H-Pyrrolo[2,3-b]quinolin-4-one,1,2,3,3a-tetrahydro-3a-hydroxy-6-methyl-1-phenyl-, (3aS)-](http://img.cochemist.com/ccimg/857000/856925-71-8.png)
![4H-Pyrrolo[2,3-b]quinolin-4-one,1,2,3,3a-tetrahydro-3a-hydroxy-6-methyl-1-phenyl-, (3aS)-](http://img.cochemist.com/ccimg/857000/856925-71-8_b.png)



![1H-Indolo[3,2,1-de]pyrido[3,2,1-ij][1,5]naphthyridin-12(5H)-one,13a-ethyl-2,3,6,13,13a,13b-hexahydro-, (13aR,13bR)-rel-](http://img.cochemist.com/ccimg/2600/2580-88-3.png)
![1H-Indolo[3,2,1-de]pyrido[3,2,1-ij][1,5]naphthyridin-12(5H)-one,13a-ethyl-2,3,6,13,13a,13b-hexahydro-, (13aR,13bR)-rel-](http://img.cochemist.com/ccimg/2600/2580-88-3_b.png)








![Phenol, 4,4'-(tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl)bis[2,6-dimethoxy-, (1R,3aS,4R,6aS)-rel-](/data/chemimg/379600/1177-14-6.png)
![Phenol, 4,4'-(tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl)bis[2,6-dimethoxy-, (1R,3aS,4R,6aS)-rel-](/data/chemimg/379600/1177-14-6_b.png)


![10-(3-chlorophenyl)-6,8,9,10-tetrahydrobenzo[b][1,8]naphthyridin-5(7H)-one](http://img.cochemist.com/ccimg/110600/110545-79-4.png)
![10-(3-chlorophenyl)-6,8,9,10-tetrahydrobenzo[b][1,8]naphthyridin-5(7H)-one](http://img.cochemist.com/ccimg/110600/110545-79-4_b.png)

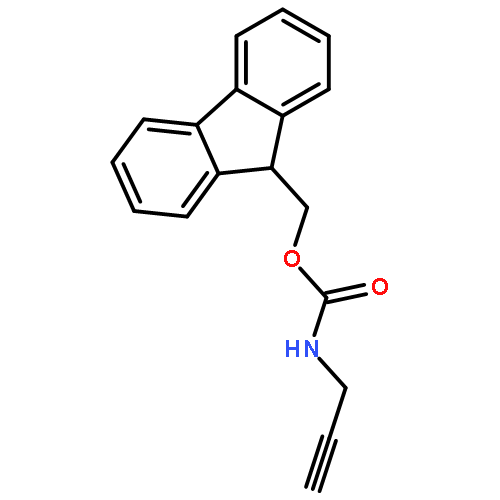
![Carbamic acid,N-[(1S)-1-methyl-2-propyn-1-yl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/118100/118080-79-8.png)
![Carbamic acid,N-[(1S)-1-methyl-2-propyn-1-yl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/118100/118080-79-8_b.png)


![4H-Pyrrolo[2,3-b]quinolin-4-one,1,2,3,9-tetrahydro-6-methyl-1-phenyl-](http://img.cochemist.com/ccimg/857000/856925-72-9.png)
![4H-Pyrrolo[2,3-b]quinolin-4-one,1,2,3,9-tetrahydro-6-methyl-1-phenyl-](http://img.cochemist.com/ccimg/857000/856925-72-9_b.png)



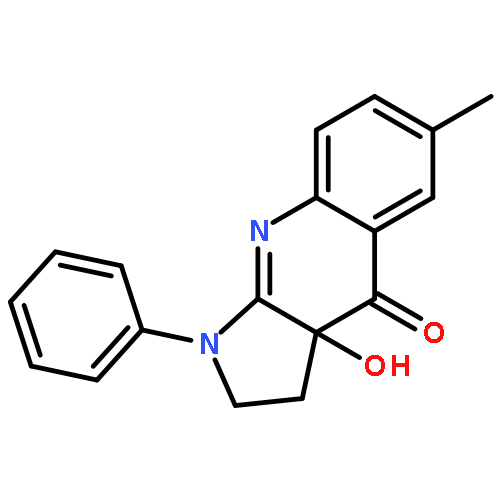
![Ethanesulfonic acid, 2-[(1,1-dimethylethyl)amino]-](http://img.cochemist.com/ccimg/24400/24368-03-4.png)
![Ethanesulfonic acid, 2-[(1,1-dimethylethyl)amino]-](http://img.cochemist.com/ccimg/24400/24368-03-4_b.png)
![3H-Naphtho[2,1-b]pyran-3-one, 2-(4-methoxybenzoyl)-](http://img.cochemist.com/ccimg/86600/86548-41-6.png)
![3H-Naphtho[2,1-b]pyran-3-one, 2-(4-methoxybenzoyl)-](http://img.cochemist.com/ccimg/86600/86548-41-6_b.png)






![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 5-methyl-3,6-diphenyl-](http://img.cochemist.com/ccimg/502500/502423-30-5.png)
![1H-Furo[3,4-c]pyrrole-1,4(5H)-dione, 5-methyl-3,6-diphenyl-](http://img.cochemist.com/ccimg/502500/502423-30-5_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione,2,5-dihydro-2,3,6-triphenyl-5-(phenylmethyl)-](/data/chemimg/1250800/502423-25-8.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione,2,5-dihydro-2,3,6-triphenyl-5-(phenylmethyl)-](/data/chemimg/1250800/502423-25-8_b.png)
![Benzoicacid, 5-methyl-2-[(1-phenyl-2-pyrrolidinylidene)amino]-, methyl ester](http://img.cochemist.com/ccimg/857000/856925-73-0.png)
![Benzoicacid, 5-methyl-2-[(1-phenyl-2-pyrrolidinylidene)amino]-, methyl ester](http://img.cochemist.com/ccimg/857000/856925-73-0_b.png)
![1H,3H-Furo[3,4-c]furan,1,4-bis(3,4-dimethoxyphenyl)tetrahydro-, (1R,3aS,4R,6aS)-](http://img.cochemist.com/ccimg/600/526-06-7.png)
![1H,3H-Furo[3,4-c]furan,1,4-bis(3,4-dimethoxyphenyl)tetrahydro-, (1R,3aS,4R,6aS)-](http://img.cochemist.com/ccimg/600/526-06-7_b.png)


































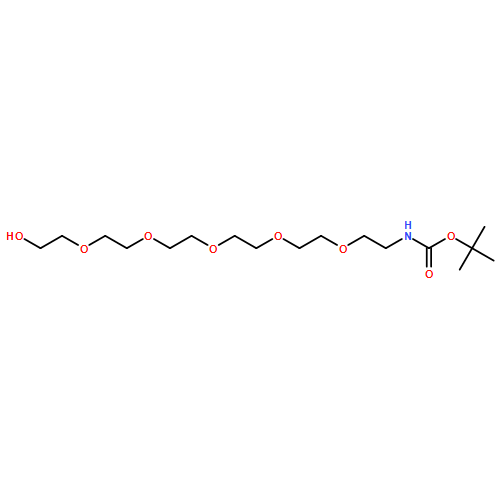
![O-(2-Aminoethyl)-O’-[2-(Boc-amino)ethyl]tetraethylene Glycol](http://img.cochemist.com/ccimg/189300/189209-27-6.png)
![O-(2-Aminoethyl)-O’-[2-(Boc-amino)ethyl]tetraethylene Glycol](http://img.cochemist.com/ccimg/189300/189209-27-6_b.png)
















![5-[(4-CHLOROPHENOXY)METHYL]-4-PHENYL-4H-1,2,4-TRIAZOLE-3-THIOL](http://img.cochemist.com/ccimg/7300/7218-43-1.png)
![5-[(4-CHLOROPHENOXY)METHYL]-4-PHENYL-4H-1,2,4-TRIAZOLE-3-THIOL](http://img.cochemist.com/ccimg/7300/7218-43-1_b.png)














![Carbamic acid, [(1S)-1-(1-methylethyl)-2-propynyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/143400/143327-78-0.png)
![Carbamic acid, [(1S)-1-(1-methylethyl)-2-propynyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/143400/143327-78-0_b.png)
![Cyclohexanone, 2,6-bis[(4-bromophenyl)methylene]-, (E,E)-](http://img.cochemist.com/ccimg/140500/140422-06-6.png)
![Cyclohexanone, 2,6-bis[(4-bromophenyl)methylene]-, (E,E)-](http://img.cochemist.com/ccimg/140500/140422-06-6_b.png)











![NSC-1125476, Tetrahydro-5-[(2-hydroxy-1-naphthalenyl)methyl]-6-phenyl-2-thioxo-4(1H)-Pyrimidinone, 5-(2-Hydroxynaphthalen-1-ylmethyl)-6-phenyl-2-thioxo-2,3-dihydro-1H-pyrimidin-4-one, 5-[(2-hydroxy-1-naphthyl)methyl]-2-mercapto-6-phenyl-4(3H)-Pyrimidinone](http://img.cochemist.com/ccimg/14600/14513-15-6.png)
![NSC-1125476, Tetrahydro-5-[(2-hydroxy-1-naphthalenyl)methyl]-6-phenyl-2-thioxo-4(1H)-Pyrimidinone, 5-(2-Hydroxynaphthalen-1-ylmethyl)-6-phenyl-2-thioxo-2,3-dihydro-1H-pyrimidin-4-one, 5-[(2-hydroxy-1-naphthyl)methyl]-2-mercapto-6-phenyl-4(3H)-Pyrimidinone](http://img.cochemist.com/ccimg/14600/14513-15-6_b.png)














![2-Pyrrolidinepropanoicacid, a-hydroxy-4-[(2Z,3E,5E)-1-hydroxy-2,4-octadien-1-ylidene]-1-methyl-a-(1-methylethyl)-3,5-dioxo-, (aS,2S)-](http://img.cochemist.com/ccimg/157200/157148-06-6.png)
![2-Pyrrolidinepropanoicacid, a-hydroxy-4-[(2Z,3E,5E)-1-hydroxy-2,4-octadien-1-ylidene]-1-methyl-a-(1-methylethyl)-3,5-dioxo-, (aS,2S)-](http://img.cochemist.com/ccimg/157200/157148-06-6_b.png)




![2-Propenoic acid, 3-phenyl-3-[(phenylmethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/14200/14191-09-4.png)
![2-Propenoic acid, 3-phenyl-3-[(phenylmethyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/14200/14191-09-4_b.png)


![2-Piperidinone, 3-ethyl-1-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/54000/53956-14-2.png)
![2-Piperidinone, 3-ethyl-1-[2-(1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/54000/53956-14-2_b.png)









![Phenol,4,4'-[(1S,3aR,4S,6aR)-tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl]bis[2-methoxy-](http://img.cochemist.com/ccimg/500/487-36-5.png)
![Phenol,4,4'-[(1S,3aR,4S,6aR)-tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl]bis[2-methoxy-](http://img.cochemist.com/ccimg/500/487-36-5_b.png)
![(4aR,7R,8aS)-8,8-Dichloro-9,9-dimethyltetrahydro-4H-4a,7-methanobenzo[c][1,2]oxazireno[2,3-b]isothiazole 3,3-dioxide](http://img.cochemist.com/ccimg/139700/139628-16-3.png)
![(4aR,7R,8aS)-8,8-Dichloro-9,9-dimethyltetrahydro-4H-4a,7-methanobenzo[c][1,2]oxazireno[2,3-b]isothiazole 3,3-dioxide](http://img.cochemist.com/ccimg/139700/139628-16-3_b.png)






![Benzaldehyde, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,5-dimethoxy-](http://img.cochemist.com/ccimg/106900/106852-80-6.png)
![Benzaldehyde, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,5-dimethoxy-](http://img.cochemist.com/ccimg/106900/106852-80-6_b.png)

















![3H-Naphtho[2,1-b]pyran-3-one,2-benzoyl-](http://img.cochemist.com/ccimg/4900/4852-81-7.png)
![3H-Naphtho[2,1-b]pyran-3-one,2-benzoyl-](http://img.cochemist.com/ccimg/4900/4852-81-7_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dihydro-2,3,5,6-tetraphenyl-](/data/chemimg/452000/128318-54-7.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-dihydro-2,3,5,6-tetraphenyl-](/data/chemimg/452000/128318-54-7_b.png)



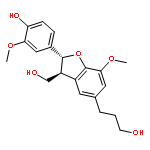
![3-[(2S,3R)-2-(3,4-DIMETHOXYPHENYL)-3-(HYDROXYMETHYL)-7-METHOXY-2,3-DIHYDRO-1-BENZOFURAN-5-YL]PROPAN-1-OL](http://img.cochemist.com/ccimg/127200/127179-41-3.png)
![3-[(2S,3R)-2-(3,4-DIMETHOXYPHENYL)-3-(HYDROXYMETHYL)-7-METHOXY-2,3-DIHYDRO-1-BENZOFURAN-5-YL]PROPAN-1-OL](http://img.cochemist.com/ccimg/127200/127179-41-3_b.png)





![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)




![N-[[5-[bis(diethylamino)phosphanyl]-9,9-dimethylxanthen-4-yl]-(diethylamino)phosphanyl]-n-ethylethanamine](http://img.cochemist.com/ccimg/349200/349100-75-0.png)
![N-[[5-[bis(diethylamino)phosphanyl]-9,9-dimethylxanthen-4-yl]-(diethylamino)phosphanyl]-n-ethylethanamine](http://img.cochemist.com/ccimg/349200/349100-75-0_b.png)




![Ethanamine, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/124000/166388-57-4.png)
![Ethanamine, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/124000/166388-57-4_b.png)


![1H,9H-Pyrrolo[2',3':2,3]pyrido[4,3-o]quinindoline,6-bromo-10,12-dichloro-2,3,14,15-tetrahydro-1-methyl-, (3aR,13bS)-rel-(+)-](http://img.cochemist.com/ccimg/474800/474779-75-4.png)
![1H,9H-Pyrrolo[2',3':2,3]pyrido[4,3-o]quinindoline,6-bromo-10,12-dichloro-2,3,14,15-tetrahydro-1-methyl-, (3aR,13bS)-rel-(+)-](http://img.cochemist.com/ccimg/474800/474779-75-4_b.png)

















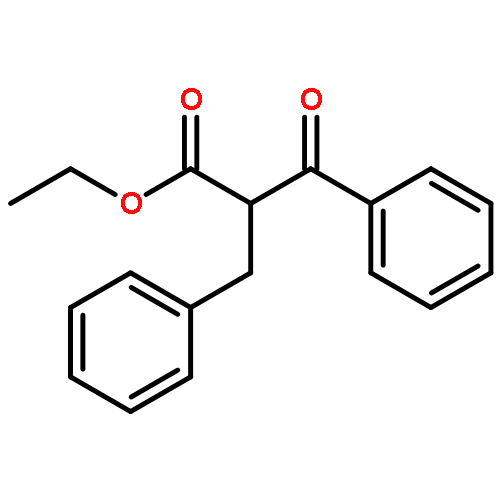
![4-Thiomorpholinamine,3-methyl-N-[(5-nitro-2-furanyl)methylene]-, 1,1-dioxide](http://img.cochemist.com/ccimg/23300/23256-30-6.png)
![4-Thiomorpholinamine,3-methyl-N-[(5-nitro-2-furanyl)methylene]-, 1,1-dioxide](http://img.cochemist.com/ccimg/23300/23256-30-6_b.png)








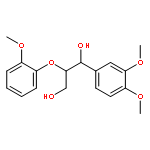
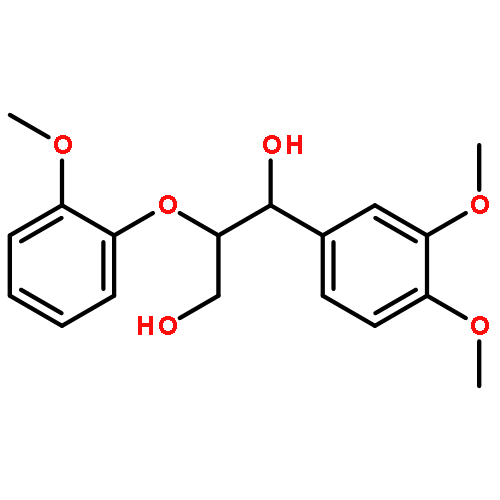












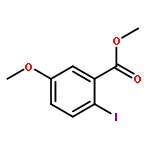
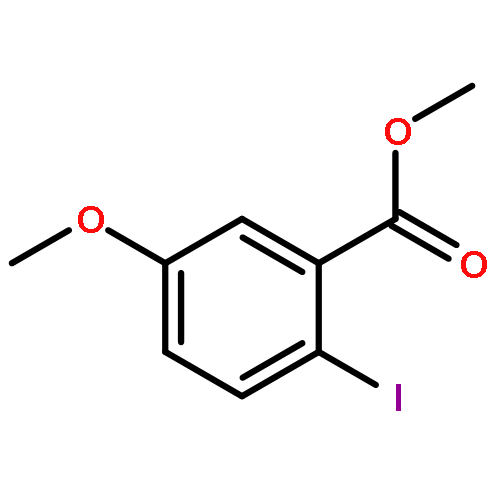
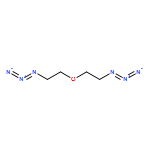

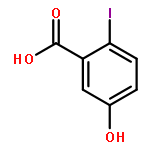

![1H-Benzo[f]chromen-3(2H)-one](http://img.cochemist.com/ccimg/5700/5690-03-9.png)
![1H-Benzo[f]chromen-3(2H)-one](http://img.cochemist.com/ccimg/5700/5690-03-9_b.png)