Co-reporter:Gopi Kumar Mittapalli, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:430-441
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.11.061
Neuropeptide Y (NPY) is one of the most abundant neuropeptides in the mammalian brain and exerts a variety of physiological processes in humans via four different receptor subtypes Y1, Y2, Y4 and Y5. Y2 receptor is the most abundant Y subtype receptor in the central nervous system and implicated with food intake, bone formation, affective disorders, alcohol and drugs of abuse, epilepsy, pain, and cancer. The lack of small molecule non-peptidic Y2 receptor modulators suitable as in vivo pharmacological tools hampered the progress to uncover the precise pharmacological role of Y2. Only in recent years, several potent, selective and non-peptidic Y2 antagonists have been discovered providing the tools to validate Y2 receptor as a therapeutic target. This Letter reviews Y2 receptor modulators mainly non-peptidic antagonists and their structure–activity relationships.Neuropeptide Y (NPY) is one of the most abundant neuropeptides in the mammalian brain and exerts a variety of physiological processes in humans via four different receptor subtypes Y1, Y2, Y4 and Y5. Y2 receptor is the most abundant Y subtype receptor in the central nervous system and implicated with food intake, bone formation, affective disorders, alcohol and drugs of abuse, epilepsy, pain, and cancer. The lack of small molecule non-peptidic Y2 receptor modulators suitable as in vivo pharmacological tools hampered the progress to uncover the precise pharmacological role of Y2. Only in recent years, several potent, selective and non-peptidic Y2 antagonists have been discovered providing the tools to validate Y2 receptor as a therapeutic target. This Letter reviews Y2 receptor modulators mainly non-peptidic antagonists and their structure–activity relationships.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Mariangela Urbano, Miguel Guerrero, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 9) pp:2021-2032
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmcl.2014.03.040
The research community has increasingly focused on the development of OPRK antagonists as pharmacotherapies for the treatment of depression, anxiety, addictive disorders and other psychiatric conditions produced or exacerbated by stress. Short-acting OPRK antagonists have been recently developed as a potential improvement over long-acting prototypic ligands including nor-BNI and JDTic. Remarkably the short-acting LY2456302 is undergoing phase II clinical trials for the augmentation of the antidepressant therapy in treatment-resistant depression. This Letter reviews relevant chemical and pharmacological advances in the identification and development of OPRK antagonists.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Miguel Guerrero, Mariangela Urbano, Marie-Therese Schaeffer, Steven Brown, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:614-619
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.12.030
In this Letter we report on the advances in our NPBWR1 antagonist program aimed at optimizing the 5-chloro-2-(3,5-dimethylphenyl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one lead molecule previously obtained from a high-throughput screening (HTS)-derived hit. Synthesis and structure–activity relationships (SAR) studies around the 3,5-dimethylphenyl and 4-methoxyphenyl regions resulted in the identification of a novel series of non-peptidic submicromolar NPBWR1 antagonists based on a 5-chloro-4-(4-alkoxyphenoxy)-2-(benzyl)pyridazin-3(2H)-one chemotype. Amongst them, 5-chloro-2-(9H-fluoren-9-yl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one 9h (CYM50769) inhibited NPW activation of NPBWR1 with a submicromolar IC50, and displayed high selectivity against a broad array of off-targets with pharmaceutical relevance. Our medicinal chemistry study provides innovative non-peptidic selective NPBWR1 antagonists that may enable to clarify the biological role and therapeutic utility of the target receptor in the regulation of feeding behavior, pain, stress, and neuroendocrine function.Synthesis and structure–activity relationships (SAR) studies around the 3,5-dimethylphenyl and 4-methoxyphenyl regions of our previously identified 5-chloro-2-(3,5-dimethylphenyl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one lead CYM50557 resulted in the identification of a novel series of non-peptidic submicromolar NPBWR1 (GPR7) antagonists based on a 5-chloro-4-(4-alkoxyphenoxy)-2-(benzyl)pyridazin-3(2H)-one chemotype. 5-chloro-2-(9H-flouren-9-yl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one 9h (CYM50769) inhibited NPW activation of NPBWR1 with a submicromolar IC50, and displayed high selectivity against a broad array of off-targets with pharmaceutical relevance. The molecules herein reported provide innovative non-peptidic selective NPBWR1 antagonists that may enable to clarify the biological role and therapeutic utility of the target receptor in the regulation of feeding behavior, pain, stress, and neuroendocrine function.
Co-reporter:Mariangela Urbano, Miguel Guerrero, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 23) pp:6377-6389
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmcl.2013.09.058
The Sphingosine 1-phosphate receptor (S1P-R) signaling system has proven to be of biological and medical importance in autoimmune settings. S1P1-R is a validated drug target for multiple sclerosis (MS) for which FTY720 (Fingolimod), a S1P1,3–5-R pan-agonist, was recently approved as the first orally active drug for the treatment of relapsing-remitting MS. Transient bradycardia and long half-life are the FTY720 critical pitfalls. This review provides the latest advances on next-generation S1P1-R modulators from 2012 up to date, with an overview of the chemical structures, structure–activity relationships, and relevant biological and clinical properties.
Co-reporter:Miguel Guerrero, Ramulu Poddutoori, Mariangela Urbano, Xuemei Peng, Timothy P. Spicer, Peter S. Chase, Peter S. Hodder, Marie-Therese Schaeffer, Steven Brown, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 23) pp:6346-6349
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmcl.2013.09.075
Potent and selective S1P3 receptor (S1P3-R) agonists may represent important proof-of-principle tools used to clarify the receptor biological function and assess the therapeutic potential of the S1P3-R in cardiovascular, inflammatory and pulmonary diseases. N,N-Dicyclohexyl-5-propylisoxazole-3-carboxamide was identified by a high-throughput screening of MLSMR library as a promising S1P3-R agonist. Rational chemical modifications of the hit allowed the identification of N,N-dicyclohexyl-5-cyclopropylisoxazole-3-carboxamide, a S1P3-R agonist endowed with submicromolar activity and exquisite selectivity over the remaining S1P1,2,4,5-R family members. A combination of ligand competition, site-directed mutagenesis and molecular modeling studies showed that the N,N-dicyclohexyl-5-cyclopropylisoxazole-3-carboxamide is an allosteric agonist and binds to the S1P3-R in a manner that does not disrupt the S1P3-R–S1P binding. The lead molecule herein disclosed constitutes a valuable pharmacological tool to explore the molecular basis of the receptor function, and provides the bases for further rational design of more potent and drug-like S1P3-R allosteric agonists.
Co-reporter:Gopi Kumar Mittapalli, Danielle Vellucci, Jun Yang, Marion Toussaint, Shaun P. Brothers, Claes Wahlestedt, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:3916-3920
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.107
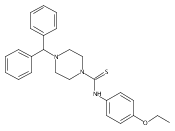
Highly potent and selective small molecule neuropeptide Y Y2 receptor antagonists are reported. The systematic SAR exploration of a hit molecule N-(4-ethoxyphenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide, identified from HTS, led to the discovery of highly potent NPY Y2 antagonists 16 (CYM 9484) and 54 (CYM 9552) with IC50 values of 19 nM and 12 nM respectively.Highly potent and selective small molecule neuropeptide Y Y2 receptor antagonists are reported. The systematic SAR exploration of a hit molecule N-(4-ethoxyphenyl)-4-[hydroxy(diphenyl)methyl]piperidine-1-carbothioamide, identified from HTS, led to the discovery of highly potent NPY Y2 antagonists 16 (CYM 9484) and 54 (CYM 9552) with IC50 values of 19 nM and 12 nM respectively.
Co-reporter:Mariangela Urbano, Miguel Guerrero, Jian Zhao, Subash Velaparthi, S. Adrian Saldanha, Peter Chase, Zhiwei Wang, Olivier Civelli, Peter Hodder, Marie-Therese Schaeffer, Steven Brown, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7135-7141
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.074
Novel small molecule antagonists of NPBWR1 (GPR7) are herein reported. A high-throughput screening (HTS) of the Molecular Libraries-Small Molecule Repository library identified 5-chloro-4-(4-methoxyphenoxy)-2-(p-tolyl)pyridazin-3(2H)-one as a NPBWR1 hit antagonist with micromolar activity. Design, synthesis and structure–activity relationships study of the HTS-derived hit led to the identification of 5-chloro-2-(3,5-dimethylphenyl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one lead molecule with submicromolar antagonist activity at the target receptor and high selectivity against a panel of therapeutically relevant off-target proteins. This lead molecule may provide a pharmacological tool to clarify the molecular basis of the in vivo physiological function and therapeutic utility of NPBWR1 in diverse disease areas including inflammatory pain and eating disorders.Design, synthesis and SAR analysis of the 5-chloro-4-(4-methoxyphenoxy)-2-(p-toly)pyridazin-3(2H)-one hit, identified through high-through-put screening (HTS) of the Molecular Libraries-Small Molecule Repository, led to the discovery of novel small molecule antagonists of NPBWR1 (GPR7). The lead molecule 5-chloro-2-(3,5-dimethylphenyl)-4-(4-methoxyphenoxy)pyridazin-3(2H)-one 1z (CYM50557) display submicromolar antagonist activity at the target receptor and high selectivity against a panel of therapeutically relevant off-targets. 1z may provide a pharmacological tool to probe the molecular basis of the in vivo physiological function and therapeutic utility of the target receptor in diverse disease areas including inflammatory pain and eating disorders.
Co-reporter:Miguel Guerrero, Mariangela Urbano, Subash Velaparthi, Jian Zhao, Marie-Therese Schaeffer, Steven Brown, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3632-3636
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.04.097
Selective S1P4 receptor antagonists could be novel therapeutic agents for the treatment of influenza infection in addition to serving as a useful tool for understanding S1P4 receptor biological functions. 5-(2,5-Dichlorophenyl)-N-(2,6-dimethylphenyl)furan-2-carboxamide was identified from screening the Molecular Libraries-Small Molecule Repository (MLSMR) collection and selected as a promising S1P4 antagonist hit with moderate in vitro potency and high selectivity against the other family receptor subtypes (S1P1–3,5). Rational chemical modifications of the hit allowed the disclosure of the first reported highly selective S1P4 antagonists with low nanomolar activity and adequate physicochemical properties suitable for further lead-optimization studies.SAR analysis of a HTS-derived hit allowed the identification of the first reported potent 5-aryl furan-2-arylcarboxamide S1P receptor family subtypes (S1P1–3,5). The lead molecules (4v, 16) represent a significant pharmacological tool to explore the biological functions of the target receptor in the fundamental immunological processes.
Co-reporter:Mariangela Urbano, Miguel Guerrero, Jian Zhao, Subash Velaparthi, Marie-Therese Schaeffer, Steven Brown, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 18) pp:5470-5474
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmcl.2011.06.132
Recent evidence suggests an innovative application of chemical modulators targeting the S1P4 receptor as novel mechanism-based drugs for the treatment of influenza virus infection. Modulation of the S1P4 receptor may also represent an alternative therapeutic approach for clinical conditions where reactive thrombocytosis is an undesired effect or increased megakaryopoiesis is required. With the exception of our recent research program disclosure, we are not aware of any selective S1P4 antagonists reported in the literature to date. Herein, we describe complementary structure–activity relationships (SAR) of the high-throughput screening (HTS)-derived hit 5-(2,5-dichlorophenyl)-N-(2,6-dimethylphenyl)furan-2-carboxamide and its 2,5-dimethylphenyl analog. Systematic structural modifications of the furan ring showed that both steric and electronic factors in this region have a significant impact on the potency. The furan moiety was successfully replaced with a thiophene or phenyl ring maintaining potency in the low nanomolar range and high selectivity against the other S1P receptor subtypes. By expanding the molecular diversity within the hit-derived class, our SAR study provides innovative small molecule potent and selective S1P4 antagonists suitable for in vivo pharmacological validation of the target receptor.SAR analysis of innovative HTS-derived 5-aryl furan-2-arylcarboxamide antagonists of the S1P4 receptor allowed the elucidation of the putative binding requirements of the central furan moiety. Molecular diversity within the hit class was increased, and novel nanomolar affinity S1P4 antagonists with high selectivity against the SIP1–3,5 family members were developed. Remarkably, thiophene and phenyl derivative 19, 47 (CYM50333, CYM50367), represent valuable small molecule tools for in vivo pharmacological validation of the target receptor in megakaryocyte differentiation and immunopathological response to airway viral infections.
Co-reporter:Vasudeva Naidu Sagi, Tianyu Liu, Xiaoying Lu, Tamas Bartfai, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:7210-7215
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.033
GalR1 and GalR2 represent unique pharmacological targets for treatment of seizures and epilepsy. A novel series of 2,4,6-triaminopyrimidine derivatives were synthesized and found to have sub-micromolar affinity for GalR2. Optimization of a series of 2,4,6-triaminopyrimidines led to the discovery of several analogs with IC50 values ranging from 0.3 to 1 μM.
Co-reporter:Mariangela Urbano, Miguel Guerrero, Subash Velaparthi, Melissa Crisp, Peter Chase, Peter Hodder, Marie-Therese Schaeffer, Steven Brown, Hugh Rosen, Edward Roberts
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 22) pp:6739-6745
Publication Date(Web):15 November 2011
DOI:10.1016/j.bmcl.2011.09.049
High affinity and selective S1P4 receptor (S1P4-R) small molecule agonists may be important proof-of-principle tools used to clarify the receptor biological function and effects to assess the therapeutic potential of the S1P4-R in diverse disease areas including treatment of viral infections and thrombocytopenia. A high-throughput screening campaign of the Molecular Libraries-Small Molecule Repository was carried out by our laboratories and identified (2Z,5Z)-5-((1-(2-fluorophenyl)-2,5-dimethyl-1H-pyrrol-3-yl)methylene)-3-methyl-2-(methylimino) thiazolidin-4-one as a promising S1P4-R agonist hit distinct from literature S1P4-R modulators. Rational chemical modifications of the hit allowed the identification of a promising lead molecule with low nanomolar S1P4-R agonist activity and exquisite selectivity over the other S1P1–3,5-Rs family members. The lead molecule herein disclosed constitutes a valuable pharmacological tool to explore the effects of the S1P4-R signaling cascade and elucidate the molecular basis of the receptor function.Systematic SAR studies of the S1P4-R agonist hit (4) identified through high-throughtput screeening (HTS) of the Molecular Libraries-Small Molecule Respository led to the discovery of novel selective small molecule S1P4-R agonists based on a (2Z,5Z)-5-((pyrrol-3-yl)methylene)-3-alkyl-2-(alkylimino)thiazolidin-4-one chemotype. Noteworthy, 24f (CYM50308) displayed low nanomolar S1P4-R agonist activity and exquisite selectivity over the other S1P1-3,5-Rs family members. The lead molecule herein disclosed constitutes a valuable pharmacological tool to deeper investigate the biological function and the therapeutic potential of the S1P4-R.
.jpg)
![5-[3-[(1S)-2,3-Dihydro-1-[(2-hydroxyethyl)amino]-1H-inden-4-yl]-1,2,4-oxadiazol-5-yl]-2-(1-methylethoxy)benzonitrile](http://img.cochemist.com/ccimg/1306800/1306760-87-1.png)
![5-[3-[(1S)-2,3-Dihydro-1-[(2-hydroxyethyl)amino]-1H-inden-4-yl]-1,2,4-oxadiazol-5-yl]-2-(1-methylethoxy)benzonitrile](http://img.cochemist.com/ccimg/1306800/1306760-87-1_b.png)
![4-Thiazolidinone, 5-[[3-chloro-4-[(2R)-2,3-dihydroxypropoxy]phenyl]methylene]-3-(2-methylphenyl)-2-(propylimino)-, (2Z,5Z)-](http://img.cochemist.com/ccimg/854200/854107-55-4.png)
![4-Thiazolidinone, 5-[[3-chloro-4-[(2R)-2,3-dihydroxypropoxy]phenyl]methylene]-3-(2-methylphenyl)-2-(propylimino)-, (2Z,5Z)-](http://img.cochemist.com/ccimg/854200/854107-55-4_b.png)
![1-Butanone,1-[5-[(3R)-3-amino-4-hydroxy-3-methylbutyl]-1-methyl-1H-pyrrol-2-yl]-4-(4-methylphenyl)-](http://img.cochemist.com/ccimg/827400/827344-05-8.png)
![1-Butanone,1-[5-[(3R)-3-amino-4-hydroxy-3-methylbutyl]-1-methyl-1H-pyrrol-2-yl]-4-(4-methylphenyl)-](http://img.cochemist.com/ccimg/827400/827344-05-8_b.png)


![N-[4-[(Dimethylamino)sulfonyl]phenyl]-4-(hydroxydiphenylmethyl)-1-piperidinecarbothioamide](http://img.cochemist.com/ccimg/1383500/1383478-94-1.png)
![N-[4-[(Dimethylamino)sulfonyl]phenyl]-4-(hydroxydiphenylmethyl)-1-piperidinecarbothioamide](http://img.cochemist.com/ccimg/1383500/1383478-94-1_b.png)
![(2z,5z)-5-{[1-(2,4-difluorophenyl)-2,5-dimethyl-1h-pyrrol-3-yl]me Thylene}-2-[(2-methoxyethyl)imino]-3-methyl-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/1345900/1345858-76-5.png)
![(2z,5z)-5-{[1-(2,4-difluorophenyl)-2,5-dimethyl-1h-pyrrol-3-yl]me Thylene}-2-[(2-methoxyethyl)imino]-3-methyl-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/1345900/1345858-76-5_b.png)












![2',5'-Dimethyl-[1,1'-biphenyl]-3-carboxylic acid](http://img.cochemist.com/ccimg/1181700/1181626-10-7.png)
![2',5'-Dimethyl-[1,1'-biphenyl]-3-carboxylic acid](http://img.cochemist.com/ccimg/1181700/1181626-10-7_b.png)


![Ethanol, 2-[[4-[5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-inden-1-yl]amino]-](http://img.cochemist.com/ccimg/1094100/1094042-01-9.png)
![Ethanol, 2-[[4-[5-(3,4-diethoxyphenyl)-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-inden-1-yl]amino]-](http://img.cochemist.com/ccimg/1094100/1094042-01-9_b.png)














































































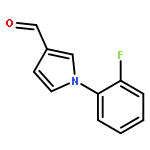









































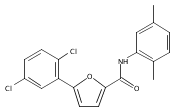




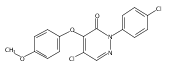
![1-[3-(Trifluoromethyl)pyrid-2-yl]-1,4-diazepane](/data/chemimg/19600/243666-15-1.png)
![1-[3-(Trifluoromethyl)pyrid-2-yl]-1,4-diazepane](/data/chemimg/19600/243666-15-1_b.png)









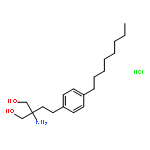
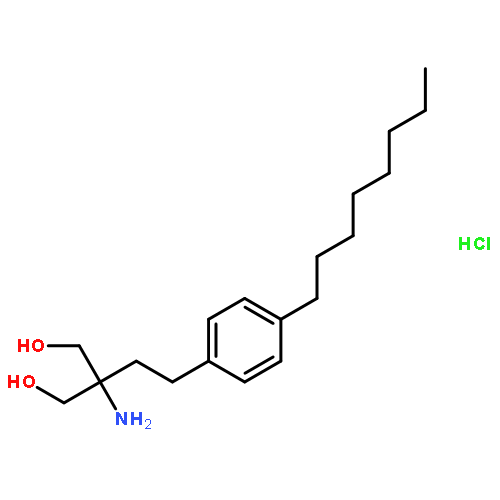













![Phenol, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126100/126070-20-0.png)
![Phenol, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126100/126070-20-0_b.png)


















![5H-Thiazolo[3,2-a]pyrimidin-3(2H)-one,6,7-dihydro-](http://img.cochemist.com/ccimg/92900/92897-32-0.png)
![5H-Thiazolo[3,2-a]pyrimidin-3(2H)-one,6,7-dihydro-](http://img.cochemist.com/ccimg/92900/92897-32-0_b.png)














![DIBENZ[B,F][1,4]OXAZEPINE, 8,11-DICHLORO-](http://img.cochemist.com/ccimg/80100/80012-58-4.png)
![DIBENZ[B,F][1,4]OXAZEPINE, 8,11-DICHLORO-](http://img.cochemist.com/ccimg/80100/80012-58-4_b.png)








![11-Chlorodibenzo[b,f][1,4]oxazepine](http://img.cochemist.com/ccimg/62500/62469-61-8.png)
![11-Chlorodibenzo[b,f][1,4]oxazepine](http://img.cochemist.com/ccimg/62500/62469-61-8_b.png)








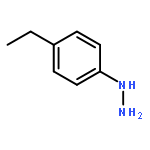






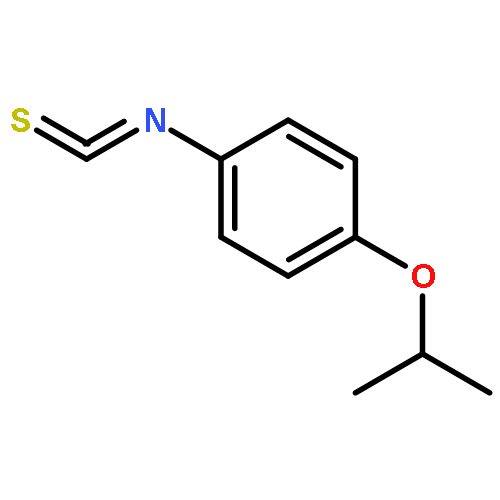










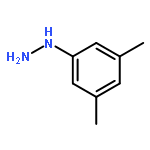




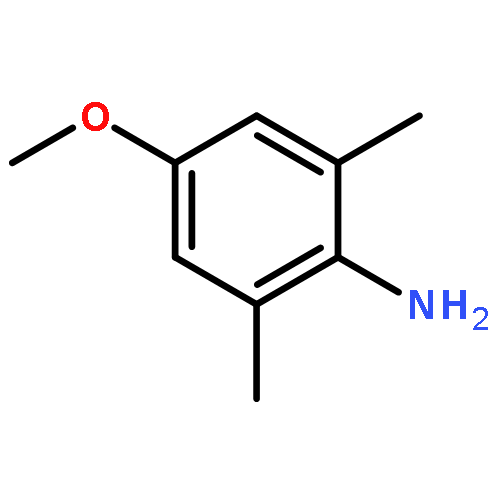










![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(3-METHOXYPHENYL)-1,2,3-THI<WBR />ADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/30200/30186-48-2.png)
![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(3-METHOXYPHENYL)-1,2,3-THI<WBR />ADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/30200/30186-48-2_b.png)












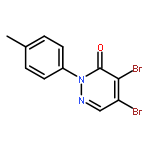

![11-Chlorodibenzo[b,f][1,4]thiazepine](http://img.cochemist.com/ccimg/13800/13745-86-3.png)
![11-Chlorodibenzo[b,f][1,4]thiazepine](http://img.cochemist.com/ccimg/13800/13745-86-3_b.png)







![5,6-Dihydro-imidazo[2,1-b]thiazol-3-one](http://img.cochemist.com/ccimg/6800/6703-51-1.png)
![5,6-Dihydro-imidazo[2,1-b]thiazol-3-one](http://img.cochemist.com/ccimg/6800/6703-51-1_b.png)


![(5Z)-5-(2-chlorobenzylidene)-3-[2-(morpholin-4-yl)-2-oxoethyl]-2-thioxo-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/6400/6335-34-8.png)
![(5Z)-5-(2-chlorobenzylidene)-3-[2-(morpholin-4-yl)-2-oxoethyl]-2-thioxo-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/6400/6335-34-8_b.png)










![Hydrazine,[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/2300/2217-77-8.png)
![Hydrazine,[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/2300/2217-77-8_b.png)





![5H-Dibenzo[b,e]azepine-6,11-dione](http://img.cochemist.com/ccimg/1200/1143-50-6.png)
![5H-Dibenzo[b,e]azepine-6,11-dione](http://img.cochemist.com/ccimg/1200/1143-50-6_b.png)






![11-hydroxy-5,11-dihydro-6H-dibenzo[b,e]azepin-6-one](http://img.cochemist.com/ccimg/800/723-87-5.png)
![11-hydroxy-5,11-dihydro-6H-dibenzo[b,e]azepin-6-one](http://img.cochemist.com/ccimg/800/723-87-5_b.png)




![[1,1'-Biphenyl]-4-ol,3-chloro-](http://img.cochemist.com/ccimg/100/92-04-6.png)
![[1,1'-Biphenyl]-4-ol,3-chloro-](http://img.cochemist.com/ccimg/100/92-04-6_b.png)


![1,3-Propanediol,2-amino-2-[2-(4-octylphenyl)ethyl]-](http://img.cochemist.com/ccimg/162400/162359-55-9.png)
![1,3-Propanediol,2-amino-2-[2-(4-octylphenyl)ethyl]-](http://img.cochemist.com/ccimg/162400/162359-55-9_b.png)