Co-reporter:Simon Parsons
Tetrahedron: Asymmetry 2017 Volume 28, Issue 10(Issue 10) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.tetasy.2017.08.018
Methods for determination of absolute structure using X-ray crystallography are described, with an emphasis on applications for absolute configuration assignment of enantiopure light-atom organic compounds. The ability to distinguish between alternative absolute structures by X-ray crystallography is the result of a physical phenomenon called resonant scattering, which introduces small deviations from the inherent inversion symmetry of single-crystal X-ray diffraction patterns. The magnitude of the effect depends on the elements present in the crystal and the wavelength of the X-rays used to collect the diffraction data, but it is always very weak for crystals of compounds containing no element heavier than oxygen. The precision of absolute structure determination by conventional least squares refinement appears to be unduly pessimistic for light-atom materials. Recent developments based on Bijvoet differences, quotients and Bayesian statistics enable better and more realistic precision to be obtained. The new methods are sensitive to statistical outliers, and techniques for identifying these are summarised.Methods for determination of absolute structure using X-ray crystallography are described, with an emphasis on applications for absolute configuration assignment of enantiopure light-atom organic compounds.Download high-res image (61KB)Download full-size image
Co-reporter:Jack Binns, Konstantin V. Kamenev, Katie E. R. Marriott, Garry J. McIntyre, Stephen A. Moggach, Mark Murrie and Simon Parsons
Chemical Communications 2016 vol. 52(Issue 47) pp:7486-7489
Publication Date(Web):13 May 2016
DOI:10.1039/C6CC02489K
Negative linear compressibility (NLC), the increase in a unit cell length with pressure, is a rare phenomenon in which hydrostatic compression of a structure promotes expansion along one dimension. It is usually a consequence of crystal structure topology. We show that the source of NLC in the Co(II) citrate metal–organic framework UTSA-16 lies not in framework topology, but in the relative torsional flexibility of Co(II)-centred tetrahedra compared to more rigid octahedra.
Co-reporter:Andrew G. P. Maloney, Peter A. Wood and Simon Parsons
CrystEngComm 2016 vol. 18(Issue 18) pp:3273-3281
Publication Date(Web):12 Apr 2016
DOI:10.1039/C6CE00555A
PIXEL has been used to perform calculations of adsorbate-adsorbent interaction energies between a range of metal–organic frameworks (MOFs) and simple guest molecules. Interactions have been calculated for adsorption between MOF-5 and Ar, H2, and N2; Zn2(BDC)2(TED) (BDC = 1,4-benzenedicarboxylic acid, TED = triethylenediamine) and H2; and HKUST-1 and CO2. The locations of the adsorption sites and the calculated energies, which show differences in the Coulombic or dispersion characteristic of the interaction, compare favourably to experimental data and literature energy values calculated using density functional theory.
Co-reporter:Stephen A. Moggach, William G. Marshall, David M. Rogers and Simon Parsons
CrystEngComm 2015 vol. 17(Issue 28) pp:5315-5328
Publication Date(Web):26 May 2015
DOI:10.1039/C5CE00327J
The crystal structures of amino acids, which are composed of molecules in their zwitterionic tautomers, are usually interpreted in terms of strong NH⋯O hydrogen bond formation between the ammonium and carboxylate groups supported by weaker dispersion or CH⋯O interactions. This view of the factors which promote thermodynamic stability in the crystalline amino acids has been re-examined in two phases of glycine, the trigonal γ-form, which is the thermodynamically most stable form under ambient conditions, and the ε-form, which is generated from γ-glycine at high pressure. A combination of Hirshfeld surface analysis, periodic DFT, PIXEL and symmetry-adapted perturbation theory calculations indicates that the conventional interpretation of intermolecular interactions in crystalline amino acids phases fails to recognise the over-whelming significance of Coulombic attraction and repulsion. There are no intermolecular interactions in either phase that can plausibly be described as dispersion-based. The interaction energies of molecules connected by so-called CH⋯O H-bonds are far in excess of accepted values for such interactions. Of the 14 closest intermolecular contacts in both phases, six have destabilizing interaction energies: in γ-glycine a hydrogen bond with ‘text-book’ NH⋯O contact geometry is part of a destabilising molecule–molecule interaction. The relative stabilities of the phases are best understood not in terms of a series of stabilising atom–atom contacts, but rather as a balance between efficient filling of space in the high-pressure ε-phase, and more weakly repulsive electrostatic whole–molecule interactions in the γ-phase.
Co-reporter:Andrew G. P. Maloney, Peter A. Wood and Simon Parsons
CrystEngComm 2015 vol. 17(Issue 48) pp:9300-9310
Publication Date(Web):23 Sep 2015
DOI:10.1039/C5CE01522G
Parameters required to perform PIXEL energy calculations, a semi-empirical method for evaluating intermolecular interactions, have been defined for the transition metals. Using these parameters, lattice energies of thirty-two 1st row, five 2nd row and six 3rd row transition metal complexes have been calculated and compared to experimental values giving correlations of calculated sublimation enthalpies comparable to those obtained for organic crystal structures. Applications of the method are illustrated by analysis of the intermolecular interactions in chromium hexacarbonyl, stacking interactions in bis(acetylacetonato)-oxo-vanadium(IV) and dihydrogen bonding. The results extend the applicability of the PIXEL method from organic materials (ca. 40% of the Cambridge Structural Database (CSD)) to a much wider range of organic and organometallic systems (ca. 85% of the CSD).
Co-reporter:Andrew G. P. Maloney, Peter A. Wood and Simon Parsons
CrystEngComm 2014 vol. 16(Issue 19) pp:3867-3882
Publication Date(Web):03 Mar 2014
DOI:10.1039/C3CE42639D
The crystal structures of the primary amines from ethylamine to decylamine have been determined by X-ray diffraction following in situ crystallisation from the liquids. In the series from propylamine to decylamine structures remain in the same phase on cooling from the melting point to 150 K, and the structures of these compounds were determined by single-crystal methods. By contrast, ethylamine undergoes a slow reconstructive phase transition on cooling to 150 K. The structure of the high-temperature form was determined by single-crystal methods at 180 K, while that of the low-temperature form was determined by powder diffraction at 150 K. The stability of the low-temperature form can be ascribed in part to more energetic hydrogen bond formation. PIXEL calculations indicate that hydrogen bonding and methyl–methyl interactions at the chain ends are optimised in the early members of the series, with particularly inefficient inter-chain interactions observed for propylamine and pentylamine. In the later members of the series dispersion interactions become the principal structure-directing interaction and the energies of the hydrogen bonds and methyl–methyl interactions become weaker to accommodate more efficient inter-chain packing. The weakest methyl–methyl interactions occur in heptyl- and nonyl-amines. Overall, intermolecular interactions in the even membered amines are stronger and the packing more efficient than in the odd members, leading to an alternation in melting points along the series, an effect reminiscent of results obtained for the alkanes, carboxylic acids and several α–ω alkyl derivatives.
Co-reporter:Nicholas P. Funnell, Alice Dawson, William G. Marshall and Simon Parsons
CrystEngComm 2013 vol. 15(Issue 6) pp:1047-1060
Publication Date(Web):24 Sep 2012
DOI:10.1039/C2CE26403J
Two crystalline phases of aniline have been investigated by a combination of single crystal X-ray diffraction data on aniline-h7 and neutron powder diffraction data on aniline-d7. Phase-I, which is formed on cooling the liquid at ambient pressure, is monoclinic (P21/c). Orthorhombic (Pna21) phase-II was crystallised at 0.84 GPa at room temperature and structurally characterised at pressures up to 7.3 GPa. The strongest intermolecular interactions in both structures are NH⋯π contacts and NH⋯N H-bonds. These interactions occur within layers in both phases, and the phases differ in the way the layers are stacked. The structures of both phases have been obtained under two sets of identical conditions, at 0.84 GPa and 0.35 GPa and studied at room temperature by neutron powder and X-ray single-crystal diffraction. At 0.84 GPa phase-II is the thermodynamically stable form because it has a lower molar volume than phase-I, but as the pressure is reduced the volume of phase-I becomes less than that of phase-II, and at 0.35 GPa phase-II partially transformed into phase-I. PIXEL calculations indicate that the intermolecular interaction energy for pairs of molecules connected by H-bonds is −9 to −16 kJ mol−1 in phase-I and II at 0.84 GPa, but one of these becomes destabilising in phase-II at 7.3 GPa, with an energy of +1 kJ mol−1, making it similar to several compressed CH⋯π contacts. The results demonstrate how the hierarchy of intermolecular interaction energies can be manipulated with pressure, driving a H-bond beyond its ambient-pressure distance limit into repulsive region of its potential, and trapping it within a compressed crystal structure.
Co-reporter:Dr. Alessro Prescimone;Chelsey Morien;Dr. David Allan;Dr. John A. Schlueter;Dr. Stan W. Tozer;Dr. Jamie L. Manson; Simon Parsons; Euan K. Brechin; Stephen Hill
Angewandte Chemie 2012 Volume 124( Issue 30) pp:7608-7612
Publication Date(Web):
DOI:10.1002/ange.201202367
Co-reporter:Dr. Alessro Prescimone;Chelsey Morien;Dr. David Allan;Dr. John A. Schlueter;Dr. Stan W. Tozer;Dr. Jamie L. Manson; Simon Parsons; Euan K. Brechin; Stephen Hill
Angewandte Chemie International Edition 2012 Volume 51( Issue 30) pp:7490-7494
Publication Date(Web):
DOI:10.1002/anie.201202367
Co-reporter:Dr. Peter J. Byrne;Dr. Patricia J. Richardson;Dr. John Chang;Dr. Anna F. Kusmartseva;Dr. David R. Allan;Dr. Anita C. Jones;Dr. Konstantin V. Kamenev; Peter A. Tasker; Simon Parsons
Chemistry - A European Journal 2012 Volume 18( Issue 25) pp:7738-7748
Publication Date(Web):
DOI:10.1002/chem.201200213
Abstract
The crystal structures of bis(3-fluoro-salicylaldoximato)nickel(II) and bis(3-methoxy-salicylaldoximato)nickel(II) have been determined at room temperature between ambient pressure and approximately 6 GPa. The principal effect of pressure is to reduce intermolecular contact distances. In the fluoro system molecules are stacked, and the Ni⋅⋅⋅Ni distance decreases from 3.19 Å at ambient pressure to 2.82 Å at 5.4 GPa. These data are similar to those observed in bis(dimethylglyoximato)nickel(II) over a similar pressure range, though contrary to that system, and in spite of their structural similarity, the salicyloximato does not become conducting at high pressure. Ni–ligand distances also shorten, on average by 0.017 and 0.011 Å for the fluoro and methoxy complexes, respectively. Bond compression is small if the bond in question is directed towards an interstitial void. A band at 620 nm, which occurs in the visible spectrum of each derivative, can be assigned to a transition to an antibonding molecular orbital based on the metal 3d(x2−y2) orbital. Time-dependent density functional theory calculations show that the energy of this orbital is sensitive to pressure, increasing in energy as the Ni–ligand distances are compressed, and consequently increasing the energy of the transition. The resulting blueshift of the UV-visible band leads to piezochromism, and crystals of both complexes, which are green at ambient pressure, become red at 5 GPa.
Co-reporter:Nicholas P. Funnell, William G. Marshall and Simon Parsons
CrystEngComm 2011 vol. 13(Issue 19) pp:5841-5848
Publication Date(Web):01 Aug 2011
DOI:10.1039/C1CE05487B
L-Alanine is the smallest chiral amino acid, crystallising in the space groupP212121. By comparison with other amino acids its crystal structure is extremely resilient to pressure, the ambient-pressure crystalline phase persisting until 13.6 GPa. At 15.46 GPa, L-alanine undergoes a reversible transformation to an amorphous phase. Hirshfeld fingerprint analysis and PIXEL calculations show that there is no development of destabilising contacts as pressure increases, so minimisation of volume is the likely driving force for this transition. Void-space analysis suggests that interstitial voids are all but absent in alanine at 13.6 GPa, and further compression is only possible with a change of phase. With the exception of benzene, 15.46 GPa is the highest pressure for which crystal structure data have been obtained for an organic system.
Co-reporter:Pascal Parois, Stephen A. Moggach, Javier Sanchez-Benitez, Konstantin V. Kamenev, Alistair R. Lennie, John E. Warren, Euan K. Brechin, Simon Parsons and Mark Murrie
Chemical Communications 2010 vol. 46(Issue 11) pp:1881-1883
Publication Date(Web):18 Jan 2010
DOI:10.1039/B923962F
Pressure-induced switching of a fast-relaxing single-molecule magnet to a slow-relaxing isomer is observed for the first time by using a combination of high pressure single-crystal X-ray diffraction and high pressure magnetic measurements.
Co-reporter:Lynne A. Crawford, Maria Ieva, Hamish McNab and Simon Parsons
Dalton Transactions 2010 vol. 39(Issue 30) pp:7147-7152
Publication Date(Web):02 Jul 2010
DOI:10.1039/C0DT00029A
X-Ray crystal structures, and calculated structures (at B3LYP/6-31G level) are reported for seven N-arylbenzazoles (two carbazoles, indoles and benzimidazoles, and one indazole) bearing electron withdrawing groups in the 2-position of the N-aryl ring. The structures are markedly non-planar by rotation around the N-aryl bond, with the substituent in most cases lying s-E in relation to the N-aryl bond; intermolecular electrostatic interactions in the crystal rationalise the two examples in which an s-Z conformation is observed. A large interplanar angle between the benzazole and the N-aryl planes is associated with a small interplanar angle between the planes of the N-aryl group and the substituent and vice versa.
Co-reporter:Pascal Parois, Stephen A. Moggach, Alistair R. Lennie, John E. Warren, Euan K. Brechin, Mark Murrie and Simon Parsons
Dalton Transactions 2010 vol. 39(Issue 30) pp:7004-7011
Publication Date(Web):23 Jun 2010
DOI:10.1039/C0DT00046A
The effect of pressure on the crystal structure of the coordination polymer [Gd(PhCOO)3(DMF)]n has been studied to 5.0 GPa. At ambient pressure the structure is tetragonal (space group P42/n) with the polymers extending along the c-direction of the unit cell; successive Gd atoms are alternately bridged by four benzoates and by two benzoates; the coordination spheres of the metal atoms are completed by DMF ligands. This results in two different Gd⋯Gd repeats, measuring 3.8953(3) and 5.3062(3) Å, respectively. The polymer chains interact with each other via dispersion interactions, including a number of CH⋯π contacts to phenyl rings in which the H⋯ring-centroid distances are 3.19 to 3.28 Å. Up to 3.7 GPa the crystal remains in a compressed form of its ambient-pressure phase. The a-axis shortens by 7.7%, and the c-axis by 2.9%, the difference reflecting the greater ease of compression along the crystallographic directions mediated by weak intermolecular interactions. At ambient pressure the Gd–O distances span 2.290(2)–2.559(2) Å, with an average of 2.39(3) Å. At 3.7 GPa the corresponding parameters are 2.259(3) to 2.509(4) and 2.36(3) Å. The Gd⋯Gd distances shortened by 0.0467(4) and 0.1851(4) Å, and the CH⋯π distances span the range 2.76–2.90 Å. During compression a number of H⋯H contacts develop, the shortest measuring 1.84 Å at 3.7 GPa. On increasing the pressure to 5.0 GPa a phase transition occurred in which the shortest H⋯H contact is relieved by conversion of an edge-to-edge phenyl-phenyl contact into a π⋯π stacking interaction. The new phase is also tetragonal, space group P, the inversion symmetry present in phase-I being lost in phase-II. The phase transition allows more efficient packing of ligands, and while the a-axis decreases in length the c-axis increases. This leads to Gd⋯Gd distances of 3.8373(4) and 5.3694(4) Å, the latter being longer than at ambient pressure. Gd–O distances at 5.0 GPa span the range 2.265(5) to 2.516(5) Å, with a mean of 2.36(2) Å.
Co-reporter:Alessandro Prescimone, Javier Sanchez-Benitez, Konstantin K. Kamenev, Stephen A. Moggach, John E. Warren, Alistair R. Lennie, Mark Murrie, Simon Parsons and Euan K. Brechin
Dalton Transactions 2010 vol. 39(Issue 1) pp:113-123
Publication Date(Web):05 Nov 2009
DOI:10.1039/B918287J
A combination of high pressure single crystal X-ray diffraction and high pressure SQUID magnetometry has been used to study three hydroxo-bridged copper(II) dimers. [Cu2(OH)2(H2O)2(tmen)2](ClO4)2 (1; tmen = tetramethylethylenediamine), [Cu2(OH)2(tben)2](ClO4)2 (2; tben = di-tbutylethylenediamine) and [Cu2(OH)2(bpy)2](BF4)2 (3; bpy = 2,2′-bipyridine) have been structurally determined to 2.5, 0.9 and 4.7 GPa, respectively. The application of hydrostatic pressure imposes significant distortions and modifications in the structures of all three complexes. This is particularly true of the bond distances and angles between the metal centres and the bridging hydroxo groups. Compound 1 undergoes a phase transition between 1.2 and 2.5 GPa caused by the loss of a coordinated water molecule. This leads to a loss of symmetry and dramatic changes in the molecular structure of the complex. The structural changes are manifested in changes in the magnetic behaviour of the complexes as seen in dc susceptibility measurements up to ∼0.9 GPa for 1, 2 and 3: the exchange becomes less antiferromagnetic in 1 and 2 and more ferromagnetic in 3.
Co-reporter:Nicholas P. Funnell, Alice Dawson, Duncan Francis, Alistair R. Lennie, William G. Marshall, Stephen A. Moggach, John E. Warren and Simon Parsons
CrystEngComm 2010 vol. 12(Issue 9) pp:2573-2583
Publication Date(Web):28 Apr 2010
DOI:10.1039/C001296C
L-Alanine crystallises as a zwitterion in space group P212121 at ambient pressure. The strongest intermolecular interactions are three N–H⋯O hydrogen bonds. The H-bonds link the molecules into puckered layers in the ac plane, which are then stacked along the b axis. PIXEL calculations indicate that the H-bond mediated intermolecular energies within the layers are 118 and 145 kJ mol−1, while the stacking interactions are considerably weaker (31 kJ mol−1). Neutron powder diffraction data on L-alanine-d7 to 9.87 GPa show that the effects of pressure are most prominent along a, the direction which is least well aligned with the H-bonds. The a axis decreases in length more rapidly than the c axis, and the two axis lengths are equal at ca. 2 GPa. Although the structure is metrically tetragonal at this point, the true symmetry is still orthorhombic. In fact, the structure remains in a compressed form of its starting orthorhombic phase throughout the pressure range studied, in contradiction of previous Raman and energy dispersive power diffraction studies, in which data were interpreted in terms of phase transitions at ca. 2 GPa and 9 GPa. It is likely that the spectroscopic signature of the apparent transition at 2 GPa is the result of a conformational change at the ammonium group. The molecular coordination number in L-alanine is 14, and the packing topology is a distorted form of body-centred cubic at ambient pressure. As pressure increases this topology becomes more regular, until at 9.87 GPa it is very similar to the perfect BCC topology of a structure such as tungsten. It is suggested that this behaviour can be traced to the sphericity of the alanine molecule.
Co-reporter:Russell D. L. Johnstone, Maria Ieva, Alistair R. Lennie, Hamish McNab, Elna Pidcock, John E. Warren and Simon Parsons
CrystEngComm 2010 vol. 12(Issue 9) pp:2520-2523
Publication Date(Web):19 Jan 2010
DOI:10.1039/B917290D
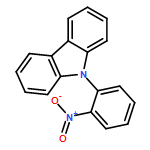
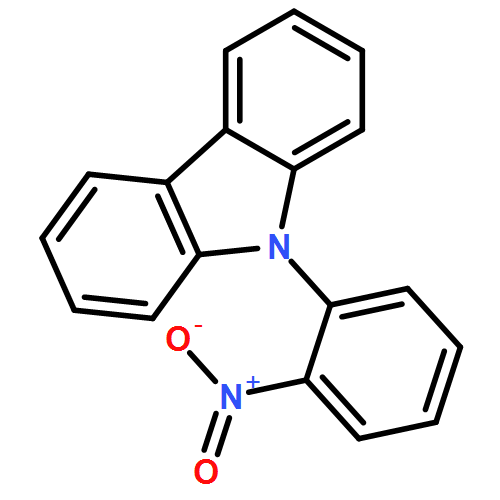
Methyl 2-(carbazol-9-yl)benzoate is unusual in crystallising with eight molecules in the crystallographic asymmetric unit (Z′ = 8). Under high pressure it transforms to a Z′ = 2 structure. The molecules in the Z′ = 2 phase have unfavourable conformations, but these are stabilised in the crystal by their efficient packing at high pressure.
Co-reporter:Russell D. L. Johnstone, Alistair R. Lennie, Stewart F. Parker, Simon Parsons, Elna Pidcock, Patricia R. Richardson, John E. Warren and Peter A. Wood
CrystEngComm 2010 vol. 12(Issue 4) pp:1065-1078
Publication Date(Web):20 Jan 2010
DOI:10.1039/B921288D
We report the compression of a single crystal of salicylamide to 5.1 GPa. Between ambient pressure and 5.1 GPa the structure remains in a compressed form of the ambient-pressure phase, referred to as salicylamide-I. This phase has been investigated twice previously, but the coordinates appear to have been reported with respect to a non-standard space group origin, though no comment to this effect is made in either of the original reports. Short H⋯H contacts implied by the previously published coordinates are strongly destabilising according to PIXEL packing energy calculations, but are absent in the structure reported here. A new high-pressure polymorph, salicylamide-II, is formed if salicylamide is crystallised in situ from a saturated solution in a 4 : 1 mixture of methanol and ethanol at 0.2 GPa. Crystal growth yielded three crystallites within the pressure cell, and combination of single-crystal X-ray diffraction intensity data from all three yielded a dataset which was >90% complete. PIXEL calculations indicate that salicylamide-II exhibits weaker H-bonding but stronger dispersion interactions than phase-I. Harmonic frequencies calculated using periodic DFT (and validated by inelastic neutron scattering data) indicate that phase-II is favoured at high pressure by its lower volume, its lower zero-point energy and higher entropy, and we estimate that at 0.2 GPa the free energy of phase-II is lower than that of phase-I by about 3 kJ mol−1.
Co-reporter:Alessandro Prescimone, Javier Sanchez-Benitez, Konstantin V. Kamenev, Stephen A. Moggach, Alistair R. Lennie, John E. Warren, Mark Murrie, Simon Parsons and Euan K. Brechin
Dalton Transactions 2009 (Issue 36) pp:7390-7395
Publication Date(Web):20 Jul 2009
DOI:10.1039/B908718D
A combined study of the high pressure crystallography and high pressure magnetism of the complex [Mn3(Hcht)2(bpy)4](ClO4)3·Et2O·2MeCN (1·Et2O·2MeCN) (H3cht is cis,cis-1,3,5-cyclohexanetriol) is presented in an attempt to observe and correlate pressure induced changes in its structural and physical properties. At 0.16 GPa the complex 1·Et2O·2MeCN loses all associated solvent in the crystal lattice, becoming 1. At higher pressures structural distortions occur changing the distances between the metal centres and the bridging oxygen atoms making the magnetic exchange between the manganese ions weaker. No significant variations are observed in the Jahn–Teller axis of the only MnIII present in the structure. High pressure dc χMT plots display a gradual decrease in both the low temperature value and slope. Simulations show a decrease in J with increasing pressure although the ground state is preserved. Magnetisation data do not show any change in |D|.
Co-reporter:Alessandro Prescimone, Constantinos J. Milios, Javier Sanchez-Benitez, Konstantin V. Kamenev, Claudia Loose, Jens Kortus, Stephen Moggach, Mark Murrie, John E. Warren, Alistair R. Lennie, Simon Parsons and Euan K. Brechin
Dalton Transactions 2009 (Issue 25) pp:4858-4867
Publication Date(Web):07 May 2009
DOI:10.1039/B902485A
The first combined high pressure single-crystal X-ray diffraction and high pressure magnetism study of two polymetallic clusters is presented in an attempt to correlate the observed changes in structure with changes in magnetic response without the need for changes in external ligation. At 1.5 GPa the structure of [Mn6O2(Et-sao)6(O2CPh(Me)2)2(EtOH)6] (1; Et-saoH2 = 2-hydroxyphenylpropanone)—a single molecule magnet (SMM) with an effective anisotropy barrier of ∼86 K—and of [Mn6O2(Et-sao)6(O2C-naphth)2(EtOH)4(H2O)2] 2 both undergo significant structural distortions of their metallic skeletons, which has a direct effect upon the observed magnetic response. The application of hydrostatic pressure on the two compounds (up to 1.5 GPa) flattens the Mn–N–O–Mn torsion angles weakening the magnetic exchange between the metal centres. In both compounds one interaction switches from ferro- to antiferromagnetic, with the Jahn–Teller (JT) axes compressing (on average) and re-aligning differently with respect to the plane of the three metal centres. High pressure dc χMT plots display a gradual decrease in the low temperature peak height and slope, simulations showing a decrease in |J| with increasing pressure with a second antiferromagnetic J value required to simulate the data. The “ground states” change from S = 12 to S = 11 for 1 and to S = 10 for 2. Magnetisation data for both 1 and 2 suggest a small decrease in |D|, while out-of-phase (χM//) ac data show a large decrease in the effective energy barrier for magnetisation reversal.
Co-reporter:Stephen Crawford;MichaelT. Kirchner Dr.;Dieter Bläser;Rol Boese Dr.;WilliamI.F. David ;Alice Dawson Dr.;Annette Gehrke Dr.;RichardM. Ibberson Dr.;WilliamG. Marshall Dr. ;Osamu Yamamuro Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/anie.200990000
Co-reporter:Stephen Crawford;MichaelT. Kirchner Dr.;Dieter Bläser;Rol Boese Dr.;WilliamI.F. David ;Alice Dawson Dr.;Annette Gehrke Dr.;RichardM. Ibberson Dr.;WilliamG. Marshall Dr. ;Osamu Yamamuro Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 4) pp:755-757
Publication Date(Web):
DOI:10.1002/anie.200803589
Co-reporter:Peter A. Wood, Delia A. Haynes, Alistair R. Lennie, W. D. Samuel Motherwell, Simon Parsons, Elna Pidcock and John E. Warren
Crystal Growth & Design 2008 Volume 8(Issue 2) pp:549
Publication Date(Web):January 5, 2008
DOI:10.1021/cg0705815
The crystal structure of 3-aza-bicyclo(3.3.1)nonane-2,4-dione has been determined at room temperature between ambient pressure and 7.1 GPa. The structure consists of chains formed by NH···O hydrogen bonds, which are then connected through CH···O contacts to form sheets. CH···O interactions also connect pairs of sheets into slabs. The dicarboximide moiety of the molecule becomes nonplanar at elevated pressure. This crystal structure has been regarded as anomalous because it fails to conform to expectations regarding a preference for packing based on dimers. However, no phase transition to a dimer-based structure was identified in this study. Analysis of the Hirshfeld surfaces identified the largest intermolecular voids at ambient conditions, and the distribution of these voids is consistent with the direction of largest linear strain developed on compression. The surfaces also facilitated identification of the short contacts which appeared at pressure, including two close CH···O interactions and three contacts between alkyl groups. PIXEL analysis shows that the hydrogen bond is relatively weak (14.6 kJ mol−1) and that a pair of CH···O contacts and a van der Waals interaction are of comparable energy (between 8.7 and 6.0 kJ mol−1). The energies of the CH···O interactions and various H···H contacts in the structure are more affected by pressure than the hydrogen bond, but it appears that the response to pressure is dominated by dispersion interactions rather than hydrogen bonds. This study illustrates the combined use of Hirshfeld surfaces and the PIXEL method is a particularly effective combination for analyzing changes in crystal structures.
Co-reporter:Tobias Borrmann, Enno Lork, Rüdiger Mews, Simon Parsons, Jan Petersen, Wolf-Dieter Stohrer, Paul G. Watson
Inorganica Chimica Acta 2008 Volume 361(Issue 2) pp:479-486
Publication Date(Web):15 January 2008
DOI:10.1016/j.ica.2007.05.016
The synthesis of the first unequivocally characterised bis(difluorothiazyne), [NSF2N(CH3)CH2–]2 is reported. The crystal structures of this and NSF3 are also reported. NSF3 has the same geometrical parameters, within error, as it does in the gas phase. PIXEL calculations show that the principal interactions in its crystal structure are SN⋯SN dipolar contacts, which form chains with S⋯N = 3.533(2) Å. These contacts are reminiscent of those observed in the crystal structures of ketones. The exchange of a fluorine by a dialkylamino group has almost no influence on the NS bond distance while the SF bonds are significantly elongated. This behaviour is explained by negative hyperconjugation and confirmed by experimental data (as far as available) and quantum chemical calculations for NSFn(NMe2)3−n and NSFnPh3−n (n = 1–3).NSF3 is considered the archetype of a compound with a sulfur(VI)–nitrogen triple bond. We report the first determination of its crystal structure. The SN bond distance in [NSF2N(Me)–CH2–]2 is essentially the same as that in NSF3, but the SF bonds are longer. Ab initio calculations reveal that this can be explained by negative hyperconjugation.
Co-reporter:Alessro Prescimone;ConstantinosJ. Milios Dr.;Stephen Moggach;JohnE. Warren Dr.;AlistairR. Lennie Dr.;Javier Sanchez-Benitez Dr.;Konstantin Kamenev Dr.;Rol Bircher Dr.;Mark Murrie Dr. ;EuanK. Brechin Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 15) pp:2828-2831
Publication Date(Web):
DOI:10.1002/anie.200705819
Co-reporter:Jennifer A. J. Pardoe;Nicholas C. Norman Dr.;Peter L. Timms Dr. Dr.;Iain Mackie;Colin R. Pulham Dr.;David W. H. Rankin Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 5) pp:
Publication Date(Web):30 JAN 2003
DOI:10.1002/anie.200390164
Unstable, reactive, and yellow: Compound B8F12, assumed for 30 years to have a diborane-like structure B2(BF2)6, has now been shown also to contain a short, central BB bond. Across the BB bond are two bridging BF2 groups, which are bent out of the plane by long-range, intramolecular F⋅⋅⋅B interactions. The B-(μ-BF2) bonds are long and, surprisingly, asymmetric, but calculations confirm that this structure is intrinsic to the molecule.
Co-reporter:Jennifer A. J. Pardoe;Nicholas C. Norman Dr.;Peter L. Timms Dr. Dr.;Iain Mackie;Colin R. Pulham Dr.;David W. H. Rankin Dr.
Angewandte Chemie 2003 Volume 115(Issue 5) pp:
Publication Date(Web):30 JAN 2003
DOI:10.1002/ange.200390132
Instabil, reaktiv und gelb: Mehr als 30 Jahre nahm man an, dass B8F12 als B2(BF2)6 mit einer Struktur ähnlich der des Diborans vorliege, doch wurde jetzt eine kurze, zentrale B-B-Bindung nachgewiesen. Darüber befinden sich zwei verbrückende BF2-Gruppen, die durch intramolekulare F⋅⋅⋅B-Wechselwirkungen aus der Ebene herausgedrückt werden. Die B-(μ-BF2)-Bindungen sind lang und – überraschenderweise – asymmetrisch, doch Rechnungen bestätigen, dass diese Struktur dem Molekül eigen ist.
Co-reporter:Pascal Parois, Stephen A. Moggach, Javier Sanchez-Benitez, Konstantin V. Kamenev, Alistair R. Lennie, John E. Warren, Euan K. Brechin, Simon Parsons and Mark Murrie
Chemical Communications 2010 - vol. 46(Issue 11) pp:NaN1883-1883
Publication Date(Web):2010/01/18
DOI:10.1039/B923962F
Pressure-induced switching of a fast-relaxing single-molecule magnet to a slow-relaxing isomer is observed for the first time by using a combination of high pressure single-crystal X-ray diffraction and high pressure magnetic measurements.
Co-reporter:Alessandro Prescimone, Constantinos J. Milios, Javier Sanchez-Benitez, Konstantin V. Kamenev, Claudia Loose, Jens Kortus, Stephen Moggach, Mark Murrie, John E. Warren, Alistair R. Lennie, Simon Parsons and Euan K. Brechin
Dalton Transactions 2009(Issue 25) pp:NaN4867-4867
Publication Date(Web):2009/05/07
DOI:10.1039/B902485A
The first combined high pressure single-crystal X-ray diffraction and high pressure magnetism study of two polymetallic clusters is presented in an attempt to correlate the observed changes in structure with changes in magnetic response without the need for changes in external ligation. At 1.5 GPa the structure of [Mn6O2(Et-sao)6(O2CPh(Me)2)2(EtOH)6] (1; Et-saoH2 = 2-hydroxyphenylpropanone)—a single molecule magnet (SMM) with an effective anisotropy barrier of ∼86 K—and of [Mn6O2(Et-sao)6(O2C-naphth)2(EtOH)4(H2O)2] 2 both undergo significant structural distortions of their metallic skeletons, which has a direct effect upon the observed magnetic response. The application of hydrostatic pressure on the two compounds (up to 1.5 GPa) flattens the Mn–N–O–Mn torsion angles weakening the magnetic exchange between the metal centres. In both compounds one interaction switches from ferro- to antiferromagnetic, with the Jahn–Teller (JT) axes compressing (on average) and re-aligning differently with respect to the plane of the three metal centres. High pressure dc χMT plots display a gradual decrease in the low temperature peak height and slope, simulations showing a decrease in |J| with increasing pressure with a second antiferromagnetic J value required to simulate the data. The “ground states” change from S = 12 to S = 11 for 1 and to S = 10 for 2. Magnetisation data for both 1 and 2 suggest a small decrease in |D|, while out-of-phase (χM//) ac data show a large decrease in the effective energy barrier for magnetisation reversal.
Co-reporter:Alessandro Prescimone, Javier Sanchez-Benitez, Konstantin V. Kamenev, Stephen A. Moggach, Alistair R. Lennie, John E. Warren, Mark Murrie, Simon Parsons and Euan K. Brechin
Dalton Transactions 2009(Issue 36) pp:NaN7395-7395
Publication Date(Web):2009/07/20
DOI:10.1039/B908718D
A combined study of the high pressure crystallography and high pressure magnetism of the complex [Mn3(Hcht)2(bpy)4](ClO4)3·Et2O·2MeCN (1·Et2O·2MeCN) (H3cht is cis,cis-1,3,5-cyclohexanetriol) is presented in an attempt to observe and correlate pressure induced changes in its structural and physical properties. At 0.16 GPa the complex 1·Et2O·2MeCN loses all associated solvent in the crystal lattice, becoming 1. At higher pressures structural distortions occur changing the distances between the metal centres and the bridging oxygen atoms making the magnetic exchange between the manganese ions weaker. No significant variations are observed in the Jahn–Teller axis of the only MnIII present in the structure. High pressure dc χMT plots display a gradual decrease in both the low temperature value and slope. Simulations show a decrease in J with increasing pressure although the ground state is preserved. Magnetisation data do not show any change in |D|.
Co-reporter:Pascal Parois, Stephen A. Moggach, Alistair R. Lennie, John E. Warren, Euan K. Brechin, Mark Murrie and Simon Parsons
Dalton Transactions 2010 - vol. 39(Issue 30) pp:NaN7011-7011
Publication Date(Web):2010/06/23
DOI:10.1039/C0DT00046A
The effect of pressure on the crystal structure of the coordination polymer [Gd(PhCOO)3(DMF)]n has been studied to 5.0 GPa. At ambient pressure the structure is tetragonal (space group P42/n) with the polymers extending along the c-direction of the unit cell; successive Gd atoms are alternately bridged by four benzoates and by two benzoates; the coordination spheres of the metal atoms are completed by DMF ligands. This results in two different Gd⋯Gd repeats, measuring 3.8953(3) and 5.3062(3) Å, respectively. The polymer chains interact with each other via dispersion interactions, including a number of CH⋯π contacts to phenyl rings in which the H⋯ring-centroid distances are 3.19 to 3.28 Å. Up to 3.7 GPa the crystal remains in a compressed form of its ambient-pressure phase. The a-axis shortens by 7.7%, and the c-axis by 2.9%, the difference reflecting the greater ease of compression along the crystallographic directions mediated by weak intermolecular interactions. At ambient pressure the Gd–O distances span 2.290(2)–2.559(2) Å, with an average of 2.39(3) Å. At 3.7 GPa the corresponding parameters are 2.259(3) to 2.509(4) and 2.36(3) Å. The Gd⋯Gd distances shortened by 0.0467(4) and 0.1851(4) Å, and the CH⋯π distances span the range 2.76–2.90 Å. During compression a number of H⋯H contacts develop, the shortest measuring 1.84 Å at 3.7 GPa. On increasing the pressure to 5.0 GPa a phase transition occurred in which the shortest H⋯H contact is relieved by conversion of an edge-to-edge phenyl-phenyl contact into a π⋯π stacking interaction. The new phase is also tetragonal, space group P, the inversion symmetry present in phase-I being lost in phase-II. The phase transition allows more efficient packing of ligands, and while the a-axis decreases in length the c-axis increases. This leads to Gd⋯Gd distances of 3.8373(4) and 5.3694(4) Å, the latter being longer than at ambient pressure. Gd–O distances at 5.0 GPa span the range 2.265(5) to 2.516(5) Å, with a mean of 2.36(2) Å.
Co-reporter:Alessandro Prescimone, Javier Sanchez-Benitez, Konstantin K. Kamenev, Stephen A. Moggach, John E. Warren, Alistair R. Lennie, Mark Murrie, Simon Parsons and Euan K. Brechin
Dalton Transactions 2010 - vol. 39(Issue 1) pp:NaN123-123
Publication Date(Web):2009/11/05
DOI:10.1039/B918287J
A combination of high pressure single crystal X-ray diffraction and high pressure SQUID magnetometry has been used to study three hydroxo-bridged copper(II) dimers. [Cu2(OH)2(H2O)2(tmen)2](ClO4)2 (1; tmen = tetramethylethylenediamine), [Cu2(OH)2(tben)2](ClO4)2 (2; tben = di-tbutylethylenediamine) and [Cu2(OH)2(bpy)2](BF4)2 (3; bpy = 2,2′-bipyridine) have been structurally determined to 2.5, 0.9 and 4.7 GPa, respectively. The application of hydrostatic pressure imposes significant distortions and modifications in the structures of all three complexes. This is particularly true of the bond distances and angles between the metal centres and the bridging hydroxo groups. Compound 1 undergoes a phase transition between 1.2 and 2.5 GPa caused by the loss of a coordinated water molecule. This leads to a loss of symmetry and dramatic changes in the molecular structure of the complex. The structural changes are manifested in changes in the magnetic behaviour of the complexes as seen in dc susceptibility measurements up to ∼0.9 GPa for 1, 2 and 3: the exchange becomes less antiferromagnetic in 1 and 2 and more ferromagnetic in 3.
Co-reporter:Jack Binns, Konstantin V. Kamenev, Katie E. R. Marriott, Garry J. McIntyre, Stephen A. Moggach, Mark Murrie and Simon Parsons
Chemical Communications 2016 - vol. 52(Issue 47) pp:NaN7489-7489
Publication Date(Web):2016/05/13
DOI:10.1039/C6CC02489K
Negative linear compressibility (NLC), the increase in a unit cell length with pressure, is a rare phenomenon in which hydrostatic compression of a structure promotes expansion along one dimension. It is usually a consequence of crystal structure topology. We show that the source of NLC in the Co(II) citrate metal–organic framework UTSA-16 lies not in framework topology, but in the relative torsional flexibility of Co(II)-centred tetrahedra compared to more rigid octahedra.
Co-reporter:Lynne A. Crawford, Maria Ieva, Hamish McNab and Simon Parsons
Dalton Transactions 2010 - vol. 39(Issue 30) pp:NaN7152-7152
Publication Date(Web):2010/07/02
DOI:10.1039/C0DT00029A
X-Ray crystal structures, and calculated structures (at B3LYP/6-31G level) are reported for seven N-arylbenzazoles (two carbazoles, indoles and benzimidazoles, and one indazole) bearing electron withdrawing groups in the 2-position of the N-aryl ring. The structures are markedly non-planar by rotation around the N-aryl bond, with the substituent in most cases lying s-E in relation to the N-aryl bond; intermolecular electrostatic interactions in the crystal rationalise the two examples in which an s-Z conformation is observed. A large interplanar angle between the benzazole and the N-aryl planes is associated with a small interplanar angle between the planes of the N-aryl group and the substituent and vice versa.








![Benzene, 1,1'-[(1E,3E)-3-(1-bromoethylidene)-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/94300/94223-24-2.png)
![Benzene, 1,1'-[(1E,3E)-3-(1-bromoethylidene)-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/94300/94223-24-2_b.png)




![Dibenz[c,h]anthyridine](http://img.cochemist.com/ccimg/40500/40455-54-7.png)
![Dibenz[c,h]anthyridine](http://img.cochemist.com/ccimg/40500/40455-54-7_b.png)
![2-Pyridinemethanamine, 6-amino-N,N-bis[(6-amino-2-pyridinyl)methyl]-](http://img.cochemist.com/ccimg/401600/401560-62-1.png)
![2-Pyridinemethanamine, 6-amino-N,N-bis[(6-amino-2-pyridinyl)methyl]-](http://img.cochemist.com/ccimg/401600/401560-62-1_b.png)
![Dibenzo[c,h][1,5]naphthyridine](http://img.cochemist.com/ccimg/38900/38895-31-7.png)
![Dibenzo[c,h][1,5]naphthyridine](http://img.cochemist.com/ccimg/38900/38895-31-7_b.png)


![6bH-2a,4a,6a-Triazacyclopenta[cd]pentalene, hexahydro-](http://img.cochemist.com/ccimg/67800/67705-38-8.png)
![6bH-2a,4a,6a-Triazacyclopenta[cd]pentalene, hexahydro-](http://img.cochemist.com/ccimg/67800/67705-38-8_b.png)
![DIBENZO[C,F][1,8]NAPHTHYRIDINE](http://img.cochemist.com/ccimg/86900/86864-74-6.png)
![DIBENZO[C,F][1,8]NAPHTHYRIDINE](http://img.cochemist.com/ccimg/86900/86864-74-6_b.png)



![TRICYCLO[3.3.1.13,7]DECANE-1-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/42900/42862-36-2.png)
![TRICYCLO[3.3.1.13,7]DECANE-1-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/42900/42862-36-2_b.png)
![BICYCLO[2.2.1]HEPTANE-2,6-DIONE, 1,7,7-TRIMETHYL-](http://img.cochemist.com/ccimg/2000/1935-17-7.png)
![BICYCLO[2.2.1]HEPTANE-2,6-DIONE, 1,7,7-TRIMETHYL-](http://img.cochemist.com/ccimg/2000/1935-17-7_b.png)



![Benzenamine, 4-[tris[4-(1,1-dimethylethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/149800/149704-55-2.png)
![Benzenamine, 4-[tris[4-(1,1-dimethylethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/149800/149704-55-2_b.png)



![[2,2'-Bipyridine]-3,3'-dicarboxylic acid, diethyl ester](http://img.cochemist.com/ccimg/1800/1762-36-3.png)
![[2,2'-Bipyridine]-3,3'-dicarboxylic acid, diethyl ester](http://img.cochemist.com/ccimg/1800/1762-36-3_b.png)











![Propanamide, 2,2-dimethyl-N-[6-(4-thiomorpholinylmethyl)-2-pyridinyl]-](http://img.cochemist.com/ccimg/674400/674312-42-6.png)
![Propanamide, 2,2-dimethyl-N-[6-(4-thiomorpholinylmethyl)-2-pyridinyl]-](http://img.cochemist.com/ccimg/674400/674312-42-6_b.png)



![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)
![Dipyrido[3,2-a:2',3'-c]phenazine](/data/chemimg/1778900/19535-47-8.png)
![Dipyrido[3,2-a:2',3'-c]phenazine](/data/chemimg/1778900/19535-47-8_b.png)
![Bicyclo[2.2.1]heptan-2-one, 6-hydroxy-1,7,7-trimethyl-, (1S,4R,6R)-](http://img.cochemist.com/ccimg/2000/1935-16-6.png)
![Bicyclo[2.2.1]heptan-2-one, 6-hydroxy-1,7,7-trimethyl-, (1S,4R,6R)-](http://img.cochemist.com/ccimg/2000/1935-16-6_b.png)






![6-[(4-methyl-1-piperazinyl)methyl]-2-Pyridinamine](http://img.cochemist.com/ccimg/639100/639009-18-0.png)
![6-[(4-methyl-1-piperazinyl)methyl]-2-Pyridinamine](http://img.cochemist.com/ccimg/639100/639009-18-0_b.png)
![3-Azabicyclo[3.3.1]nonane-2,4-dione](http://img.cochemist.com/ccimg/4400/4355-17-3.png)
![3-Azabicyclo[3.3.1]nonane-2,4-dione](http://img.cochemist.com/ccimg/4400/4355-17-3_b.png)
![3-[(acetyloxy)methyl]-1H-Pyrrole-2-carboxaldehyde](http://img.cochemist.com/ccimg/154400/154367-57-4.png)
![3-[(acetyloxy)methyl]-1H-Pyrrole-2-carboxaldehyde](http://img.cochemist.com/ccimg/154400/154367-57-4_b.png)






![(6Z,6'Z)-6,6'-{(1S,2S)-cyclohexane-1,2-diylbis[imino(Z)methylylidene]}bis[4-tert-butyl-2-(morpholin-4-ylmethyl)cyclohexa-2,4-dien-1-one]](http://img.cochemist.com/ccimg/252800/252735-72-1.png)
![(6Z,6'Z)-6,6'-{(1S,2S)-cyclohexane-1,2-diylbis[imino(Z)methylylidene]}bis[4-tert-butyl-2-(morpholin-4-ylmethyl)cyclohexa-2,4-dien-1-one]](http://img.cochemist.com/ccimg/252800/252735-72-1_b.png)

![5H-Pyrrolo[2,1-a]isoindol-5-one](http://img.cochemist.com/ccimg/74200/74117-38-7.png)
![5H-Pyrrolo[2,1-a]isoindol-5-one](http://img.cochemist.com/ccimg/74200/74117-38-7_b.png)




![(2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)borane](http://img.cochemist.com/ccimg/64300/64234-27-1.png)
![(2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)borane](http://img.cochemist.com/ccimg/64300/64234-27-1_b.png)


![Propanamide, N-[6-(aminomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/161100/161041-56-1.png)
![Propanamide, N-[6-(aminomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/161100/161041-56-1_b.png)


![Benzenemethanamine, N-[2-(diphenylphosphino)ethyl]-](http://img.cochemist.com/ccimg/134300/134296-11-0.png)
![Benzenemethanamine, N-[2-(diphenylphosphino)ethyl]-](http://img.cochemist.com/ccimg/134300/134296-11-0_b.png)









![5H-Imidazo[2,1-a]isoindol-5-one](http://img.cochemist.com/ccimg/67800/67792-81-8.png)
![5H-Imidazo[2,1-a]isoindol-5-one](http://img.cochemist.com/ccimg/67800/67792-81-8_b.png)
![Benzonitrile, 4,4'-[1,10-decanediylbis(oxy)]bis-](http://img.cochemist.com/ccimg/50400/50381-94-7.png)
![Benzonitrile, 4,4'-[1,10-decanediylbis(oxy)]bis-](http://img.cochemist.com/ccimg/50400/50381-94-7_b.png)




![Ethane,1,1'-thiobis[2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/51500/51472-73-2.png)
![Ethane,1,1'-thiobis[2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/51500/51472-73-2_b.png)
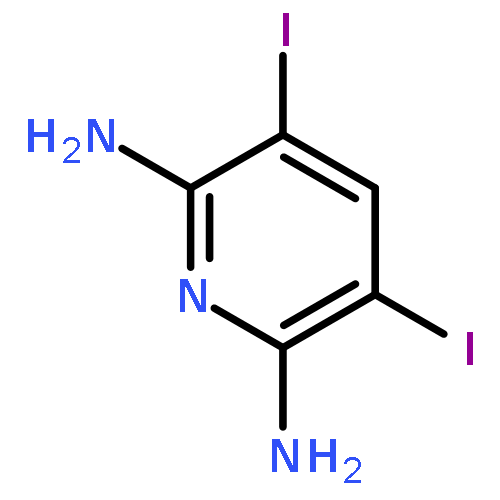



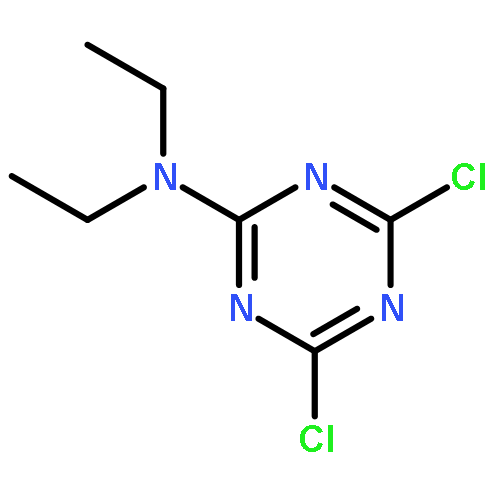
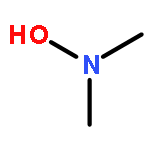






![Benzoic acid, 2-[(2-aminophenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/5900/5814-40-4.png)
![Benzoic acid, 2-[(2-aminophenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/5900/5814-40-4_b.png)










![Benzenamine,N-[[5-[(phenylamino)methylene]-1,3-cyclopentadien-1-yl]methylene]-](http://img.cochemist.com/ccimg/29900/29836-96-2.png)
![Benzenamine,N-[[5-[(phenylamino)methylene]-1,3-cyclopentadien-1-yl]methylene]-](http://img.cochemist.com/ccimg/29900/29836-96-2_b.png)









![6'-FLUORO-2',3'-DIHYDRO-2H,5H-SPIRO[IMIDAZOLIDINE-4,4'-THIOCHROMENE]-2,5-DIONE 1',1'-DIOXIDE](http://img.cochemist.com/ccimg/74300/74280-71-0.png)
![6'-FLUORO-2',3'-DIHYDRO-2H,5H-SPIRO[IMIDAZOLIDINE-4,4'-THIOCHROMENE]-2,5-DIONE 1',1'-DIOXIDE](http://img.cochemist.com/ccimg/74300/74280-71-0_b.png)







![Borane, dichloro[(1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-3-yl]-](http://img.cochemist.com/ccimg/121400/121326-42-9.png)
![Borane, dichloro[(1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]hept-3-yl]-](http://img.cochemist.com/ccimg/121400/121326-42-9_b.png)













































![[2,2'-Bipyridine]-3,3'-dicarboxylic acid](http://img.cochemist.com/ccimg/4500/4433-01-6.png)
![[2,2'-Bipyridine]-3,3'-dicarboxylic acid](http://img.cochemist.com/ccimg/4500/4433-01-6_b.png)







![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2.png)
![(2-ISOPROPYL-3-INDOLIZINYL)(4-{3-[(2-METHYL-2-PROPANYL)AMINO]PROP<WBR />OXY}PHENYL)METHANONE](http://img.cochemist.com/ccimg/3600/3546-21-2_b.png)



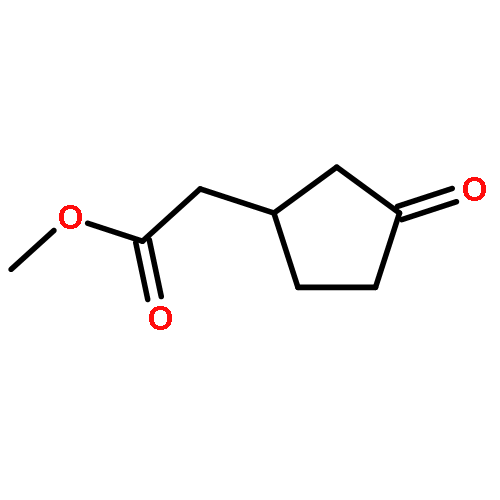


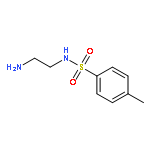
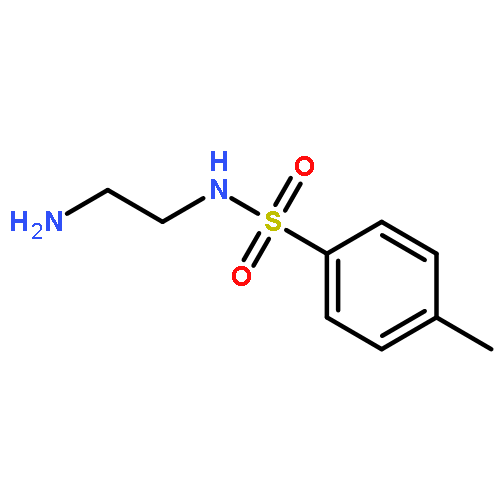


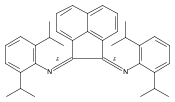



















![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5.png)
![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5_b.png)


![Propanamide,N-[6-[[bis(2-pyridinylmethyl)amino]methyl]-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/351500/351415-88-8.png)
![Propanamide,N-[6-[[bis(2-pyridinylmethyl)amino]methyl]-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/351500/351415-88-8_b.png)
![Propanamide, N-[6-(bromomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/111500/111477-43-1.png)
![Propanamide, N-[6-(bromomethyl)-2-pyridinyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/111500/111477-43-1_b.png)










![Bicyclo[2.2.2]octane-2,5-dione](http://img.cochemist.com/ccimg/57400/57346-05-1.png)
![Bicyclo[2.2.2]octane-2,5-dione](http://img.cochemist.com/ccimg/57400/57346-05-1_b.png)