Co-reporter:Rajwana Jahan, Michael Rajesh Stephen, Gloria S. Forkuo, Revathi Kodali, Margaret L. Guthrie, Amanda N. Nieman, Nina Y. Yuan, Nicolas M. Zahn, Michael M. Poe, Guanguan Li, Olivia B. Yu, Gene T. Yocum, Charles W. Emala, Douglas C. Stafford, James M. Cook, Leggy A. Arnold
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.11.045
•Deuterated and non-deuterated compounds were compared using preclinical assays.•Microsomal stability assays can be used to exclude drug candidates but fall short in selection of compounds stable in vivo.•A novel analog of XHE-III-74, compound 16, was identified as a potent inhibitor of airway smooth muscle contraction ex vivo.•Reduction of airway hyperresponsiveness by 16 in a murine model of asthma was observed despite suboptimal pharmacokinetics.We describe the synthesis of analogs of XHE-III-74, a selective α4β3γ2 GABAAR ligand, shown to relax airway smooth muscle ex vivo and reduce airway hyperresponsiveness in a murine asthma model. To improve properties of this compound as an asthma therapeutic, a series of analogs with a deuterated methoxy group in place of methoxy group at C-8 position was evaluated for isotope effects in preclinical assays; including microsomal stability, cytotoxicity, and sensorimotor impairment. The deuterated compounds were equally or more metabolically stable than the corresponding non-deuterated analogs and increased sensorimotor impairment was observed for some deuterated compounds. Thioesters were more cytotoxic in comparison to other carboxylic acid derivatives of this compound series. The most promising compound 16 identified from the in vitro screens also strongly inhibited smooth muscle constriction in ex vivo guinea pig tracheal rings. Smooth muscle relaxation, determined by reduction of airway hyperresponsiveness with a murine ovalbumin sensitized and challenged model, showed that 16 was efficacious at low methacholine concentrations. However, this effect was limited due to suboptimal pharmacokinetics of 16. Based on these findings, further analogs of XHE-III-74 will be investigated to improve in vivo metabolic stability while retaining the efficacy at lung tissues involved in asthma pathology.Download high-res image (253KB)Download full-size image
Co-reporter:M. Toufiqur Rahman, Jeffrey R. Deschamps, Gregory H. Imler, Alan W. Schwabacher, and James M. Cook
Organic Letters 2016 Volume 18(Issue 17) pp:4174-4177
Publication Date(Web):August 16, 2016
DOI:10.1021/acs.orglett.6b01526
An enolate driven copper-mediated cross-coupling process enabled cheaper and greener access to the key pentacyclic intermediates required for the enantiospecific total synthesis of a number of C-19 methyl substituted sarpagine/macroline indole alkaloids. Replacement of palladium (60–68%) with copper iodide (82–89%) resulted in much higher yields. The formation of an unusual 7-membered cross-coupling product was completely inhibited by using TEMPO as a radical scavenger. Further functionalization led to the first enantiospecific total synthesis of macrocarpines D and E.
Co-reporter:V. V. N. Phani Babu Tiruveedhula, Kashi Reddy Methuku, Jeffrey R. Deschamps and James M. Cook
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 43) pp:10705-10715
Publication Date(Web):03 Sep 2015
DOI:10.1039/C5OB01572C
A novel two step protocol was developed to gain regiospecific access to 3-substituted β- and aza-β-carbolines, 3-PBC (1), 3-ISOPBC (2), βCCt (3), 6-aza-3-PBC (4) and 6-aza-3-ISOPBC (5). These β-carbolines (1–3) are potential clinical agents to reduce alcohol self-administration, especially 3-ISOPBC·HCl (2·HCl) which appears to be a potent anti-alcohol agent active against binge drinking in a rat model of maternally deprived (MD) rats. The method consists of two consecutive palladium-catalyzed reactions: a Buchwald–Hartwig amination followed by an intramolecular Heck-type cyclization in high yield.
Co-reporter:German O. Fonseca, Zhi-Jian Wang, Ojas A. Namjoshi, Jeffrey R. Deschamps, James M. Cook
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3052-3056
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.11.036
The first synthesis of (−)-affinisine oxindole was completed in an enantiospecific fashion from commercially available d-(+)-tryptophan in 10% overall yield. The asymmetric Pictet–Spengler reaction, diastereospecific oxidative-rearrangement of a tetrahydro-β-carboline, and stereospecific enolate-mediated palladium-catalyzed cross coupling process were key steps in the sequence. This represents the first example of a total synthesis via stereospecific entry into either the alstonisine related or epimeric chitosenine related oxindole stereochemistry depending on the presence or absence of an Nb-benzyl protecting group.The first synthesis of (−)-affinisine oxindole was completed in an enantiospecific fashion from commercially available d-(+)-tryptophan in 10% overall yield. The asymmetric Pictet–Spengler reaction, diastereospecific oxidative-rearrangement of a tetrahydro-β-carboline, and stereospecific enolate-mediated palladium-catalyzed cross coupling process were key steps in the sequence. This represents the first example of a total synthesis via stereospecific entry into either the alstonisine related or epimeric chitosenine related oxindole stereochemistry depending on the presence or absence of an Nb-benzyl protecting group.
Co-reporter:Sundari K. Rallapalli, Ojas A. Namjoshi, V. V. N. Phani Babu Tiruveedhula, Jeffrey R. Deschamps, and James M. Cook
The Journal of Organic Chemistry 2014 Volume 79(Issue 9) pp:3776-3780
Publication Date(Web):April 3, 2014
DOI:10.1021/jo402692u
The total synthesis of the indole alkaloid ervincidine (3) is reported. This research provides a general entry into C-6 hydroxy-substituted indole alkaloids with either an α or a β configuration. This study corrects the errors in Glasby’s book (Glasby, J. S. Encyclopedia of the Alkaloids; Plenum Press: New York, 1975) and Lounasmaa et al.’s review (Lounasmaa, M.; Hanhinen, P.; Westersund, M. In The Alkaloids; Cordell, G. A., Ed.; Academic Press: San Diego, CA, 1999; Vol. 52, pp 103–195) as well as clarifies the work of Yunusov et al. (Malikov, V. M.; Sharipov, M. R.; Yunusov, S. Yu. Khim. Prir. Soedin. 1972, 8, 760−761. Rakhimov, D. A.; Sharipov, M. R.; Aripov, Kh. N.; Malikov, V. M.; Shakirov, T. T.; Yunusov, S. Yu. Khim. Prir. Soedin. 1970, 6, 724–725). It establishes the correct absolute configuration of the C-6 hydroxyl function in ervincidine. This serves as a structure proof and corrects the misassigned structure reported in the literature.
Co-reporter:Rahul V. Edwankar, Chitra R. Edwankar, Jeffrey R. Deschamps, and James M. Cook
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:10030-10048
Publication Date(Web):September 23, 2014
DOI:10.1021/jo5016163
A detailed account of the development of a general strategy for synthesis of the C-19 methyl-substituted alkaloids including total synthesis of 19(S),20(R)-dihydroperaksine-17-al (1), 19(S),20(R)-dihydroperaksine (2), and peraksine (6) is presented. Efforts directed toward the total synthesis of macrosalhine chloride (5) are also reported. Important to success is the sequence of chemical reactions which include a critical haloboration reaction, regioselective hydroboration, and controlled oxidation (to provide sensitive enolizable aldehydes at C-20). In addition, the all-important Pd-catalyzed α-vinylation reaction has been extended to a chiral C-19 alkyl-substituted substrate for the first time. Synthesis of the advanced intermediate 64 completes an improved formal total synthesis of talcarpine (26) and provides a starting point for synthesis of macroline-related alkaloids 27–31. Similarly, extension of this synthetic strategy in the ring A oxygenated series should provide easy access to the northern hemisphere 32b of the bisindoles angustricraline, alstocraline, and foliacraline (Figure 4).
Co-reporter:V.V.N. Phani Babu Tiruveedhula, Christopher M. Witzigmann, Ranjit Verma, M. Shahjahan Kabir, Marc Rott, William R. Schwan, Sara Medina-Bielski, Michelle Lane, William Close, Rebecca L. Polanowski, David Sherman, Aaron Monte, Jeffrey R. Deschamps, James M. Cook
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7830-7840
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.011
The alarming increase in bacterial resistance over the last decade along with a dramatic decrease in new treatments for infections has led to problems in the healthcare industry. Tuberculosis (TB) is caused mainly by Mycobacterium tuberculosis which is responsible for 1.4 million deaths per year. A world-wide threat with HIV co-infected with multi and extensively drug-resistant strains of TB has emerged. In this regard, herein, novel acrylic acid ethyl ester derivatives were synthesized in simple, efficient routes and evaluated as potential agents against several Mycobacterium species. These were synthesized via a stereospecific process for structure activity relationship (SAR) studies. Minimum inhibitory concentration (MIC) assays indicated that esters 12, 13, and 20 exhibited greater in vitro activity against Mycobacterium smegmatis than rifampin, one of the current, first-line anti-mycobacterial chemotherapeutic agents. Based on these studies the acrylic ester 20 has been developed as a potential lead compound which was found to have an MIC value of 0.4 μg/mL against Mycobacterium tuberculosis. The SAR and biological activity of this series is presented; a Michael-acceptor mechanism appears to be important for potent activity of this series of analogs.
Co-reporter:Ojas A. Namjoshi, Zhi-jian Wang, Sundari K. Rallapalli, Edward Merle Johnson Jr., Yun-Teng Johnson, Hanna Ng, Joachim Ramerstorfer, Zdravko Varagic, Werner Sieghart, Samarpan Majumder, Bryan L. Roth, James K. Rowlett, James M. Cook
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 1) pp:93-101
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmc.2012.10.057
Selective modulation of specific benzodiazepine receptor (BzR) gamma amino butyric acid-A (GABAA) receptor ion channels has been identified as an important method for separating out the variety of pharmacological effects elicited by BzR-related drugs. Importantly, it has been demonstrated that both α2β(2/3)γ2 (α2BzR) and α3BzR (and/or α2/α3) BzR subtype selective ligands exhibit anxiolytic effects with little or no sedation. Previously we have identified several such ligands; however, three of our parent ligands exhibited significant metabolic liability in rodents in the form of a labile ester group. Here eight analogs are reported which were designed to circumvent this liability by utilizing a rational replacement of the ester moiety based on medicinal chemistry precedents. In a metabolic stability study using human liver microsomes, four compounds were found to undergo slower metabolic transformation, as compared to their corresponding ester analogs. These compounds were also evaluated in in vitro efficacy assays. Additionally, bioisostere 11 was evaluated in a rodent model of anxiety. It exhibited anxiolytic activity at doses of 10 and 100 mg/kg and was devoid of sedative properties.
Co-reporter:Chitra R. Edwankar, Rahul V. Edwankar, Ojas A. Namjoshi, Xuebin Liao, and James M. Cook
The Journal of Organic Chemistry 2013 Volume 78(Issue 13) pp:6471-6487
Publication Date(Web):May 30, 2013
DOI:10.1021/jo400469t
The first regio- and stereocontrolled total synthesis of the bisphenolic, bisquaternary alkaloid (+)-dispegatrine (1) has been accomplished in an overall yield of 8.3% (12 reaction vessels) from 5-methoxy-d-tryptophan ethyl ester (17). A crucial late-stage thallium(III) mediated intermolecular oxidative dehydrodimerization was employed in the formation of the C9–C9′ biaryl axis in 1. The complete stereocontrol observed in this key biaryl coupling step is due to the asymmetric induction by the natural sarpagine configuration of the monomer lochnerine (6) and was confirmed by both the Suzuki and the oxidative dehydrodimerization model studies on the tetrahydro β-carboline (35). The axial chirality of the lochnerine dimer (40) and in turn dispegatrine (1) was established by X-ray crystallography and was determined to be P(S). Additionally, the first total synthesis of the monomeric indole alkaloids (+)-spegatrine (2), (+)-10-methoxyvellosimine (5), (+)-lochnerine (6), lochvinerine (7), (+)-sarpagine (8), and (+)-lochneram (11) were also achieved via the common pentacyclic intermediate 16.
Co-reporter:Rahul V. Edwankar, Chitra R. Edwankar, Ojas A. Namjoshi, Jeffrey R. Deschamps, and James M. Cook
Journal of Natural Products 2012 Volume 75(Issue 2) pp:181-188
Publication Date(Web):January 18, 2012
DOI:10.1021/np200759h
The development of an efficient diastereoselective method that permits rapid construction of the tetracyclic core 17 of the Strychnos-Aspidosperma alkaloids is described. Enaminone 16, synthesized in high yield, has been cyclized under the influence of a Brønsted acid to provide the core tetracyclic framework 17 of the Strychnos alkaloids in optically active form or alternatively to the β-ketoester tetrahydro-β-carboline (THBC) unit 18, by varying the equivalents of acid and the molar concentration. Attempts to utilize 18 to form the C(7)–C(16) bond of the akuammiline related alkaloids represented by strictamine (22), using metal-carbenoid chemistry, are also described.
Co-reporter:M. Shahjahan Kabir, Ojas A. Namjoshi, Ranjit Verma, Michael Lorenz, V. V. N. Phani Babu Tiruveedhula, Aaron Monte, Steven H. Bertz, Alan W. Schwabacher, and James M. Cook
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:300-310
Publication Date(Web):November 10, 2011
DOI:10.1021/jo201948e
The stereospecific synthesis of aryloxy and amino substituted E- and Z-ethyl-3-acrylates is of interest because of their potential in the polymer industry and in medicinal chemistry. During work on a copper-catalyzed cross-coupling reaction of ethyl (E)- and (Z)-3-iodoacrylates with phenols and N-heterocycles, we discovered a very simple (nonmetallic) method for the stereospecific synthesis of aryloxy and amino substituted acrylates. To study this long-standing problem on the stereoselectivity of aryloxy and amino substituted acrylates, a series of O- and N-substituted nucleophiles was allowed to react with ethyl (E)- and (Z)-3-iodoacrylates. Screening of different bases indicated that DABCO (1,4-diazabicyclo[2.2.2]octane) afforded successful conversion of ethyl (E)- and (Z)-3-iodoacrylates into aryloxy and amino substituted ethyl acrylates in a stereospecific manner. Herein are the details of this DABCO-mediated stereospecific synthesis of aryloxy and amino substituted E- or Z-acrylates.
Co-reporter:Dr. Chitra R. Edwankar;Dr. Rahul V. Edwankar;Dr. Jeffrey R. Deschamps;Dr. James M. Cook
Angewandte Chemie 2012 Volume 124( Issue 47) pp:11932-11935
Publication Date(Web):
DOI:10.1002/ange.201206015
Co-reporter:Dr. Chitra R. Edwankar;Dr. Rahul V. Edwankar;Dr. Jeffrey R. Deschamps;Dr. James M. Cook
Angewandte Chemie International Edition 2012 Volume 51( Issue 47) pp:11762-11765
Publication Date(Web):
DOI:10.1002/anie.201206015
Co-reporter:Ojas A. Namjoshi, Angelica Gryboski, German O. Fonseca, Michael L. Van Linn, Zhi-jian Wang, Jeffrey R. Deschamps, and James M. Cook
The Journal of Organic Chemistry 2011 Volume 76(Issue 11) pp:4721-4727
Publication Date(Web):April 15, 2011
DOI:10.1021/jo200425m
To gain access to 3-propoxy-β-carboline hydrochloride (3-PBC·HCl) (1·HCl) and β-carboline-3-carboxylate-tert-butyl ester (βCCt) (2), potential clinical agents active against alcohol self-administration, a two-step route was developed. This process involves a palladium-catalyzed Buchwald–Hartwig coupling and an intramolecular Heck reaction. This two-step route provides rapid access to multigram quantities of 3-PBC (1) and βCCt (2), as well as analogues for studies of alcohol self-administration. The overall yield of 3-PBC (1) was improved from 8% to 50% by this route.
Co-reporter:M. Shahjahan Kabir, Michael Lorenz, Ojas A. Namjoshi and James M. Cook
Organic Letters 2010 Volume 12(Issue 3) pp:464-467
Publication Date(Web):December 29, 2009
DOI:10.1021/ol9026446
2-Pyridin-2-yl-1H-benzoimidazole L3 is presented as a new, efficient, and versatile bidentate N-donor ligand suitable for the copper-catalyzed formation of vinyl C−N and C−O bonds. This inexpensive and easily prepared ligand facilitates copper-catalyzed cross-coupling reactions of alkenyl bromides and iodides with N-heterocycles and phenols to afford the desired cross-coupled products in good to excellent yields with full retention of stereochemistry. This method is particularly noteworthy given its efficiency, that is, mild reaction conditions, low catalyst loading, simplicity, versatility, and exceptional level of functional group tolerance.
Co-reporter:Wenyuan Yin, Samarpan Majumder, Terry Clayton, Steven Petrou, Michael L. VanLinn, Ojas A. Namjoshi, Chunrong Ma, Brett A. Cromer, Bryan L. Roth, Donna M. Platt, James M. Cook
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 21) pp:7548-7564
Publication Date(Web):1 November 2010
DOI:10.1016/j.bmc.2010.08.049
A series of 3,6-disubstituted β-carbolines was synthesized and evaluated for their in vitro affinities at αxβ3γ2 GABAA/benzodiazepine receptor subtypes by radioligand binding assays in search of α1 subtype selective ligands to treat alcohol abuse. Analogues of β-carboline-3-carboxylate-t-butyl ester (βCCt, 1) were synthesized via a CDI-mediated process and the related 6-substituted β-carboline-3-carboxylates 6 including WYS8 (7) were synthesized via a Sonogashira or Stille coupling processes from 6-iodo-βCCt (5). The bivalent ligands of βCCt (32 and 33) were also designed and prepared via a palladium-catalyzed homocoupling process to expand the structure–activity relationships (SAR) to larger ligands. Based on the pharmacophore/receptor model, a preliminary SAR study on 34 analogues illustrated that large substituents at position-6 of the β-carbolines were well tolerated. As expected, these groups are proposed to project into the extracellular domain (LDi region) of GABAA/Bz receptors (see 32 and 33). Moreover, substituents located at position-3 of the β-carboline nucleus exhibited a conserved stereo interaction in lipophilic pocket L1, while N(2) presumably underwent a hydrogen bonding interaction with H1. Three novel β-carboline ligands (βCCt, 3PBC and WYS8), which preferentially bound to α1 BzR subtypes permitted a comparison of the pharmacological efficacies with a range of classical BzR antagonists (flumazenil, ZK93426) from several different structural groups and indicated these β-carbolines were ‘near GABA neutral antagonists’. Based on the SAR, the most potent (in vitro) α1 selective ligand was the 6-substituted acetylenyl βCCt (WYS8, 7). Earlier both βCCt and 3PBC had been shown to reduce alcohol self-administration in alcohol preferring (P) and high alcohol drinking (HAD) rats but had little or no effect on sucrose self-administration. , and Moreover, these two β-carbolines were orally active, and in addition, were anxiolytic in P rats but were only weakly anxiolytic in rodents. These data prompted the synthesis of the β-carbolines presented here.A series of 3,6-disubstituted β-carbolines was synthesized and evaluated for their in vitro affinities at αxβ3γ2 GABAA/benzodiazepine receptor subtypes by radioligand binding assays in search of α1 subtype selective ligands to treat alcohol abuse. Analogues of β-carboline-3-carboxylate-t-butyl ester (βCCt, 1) were synthesized via a CDI-mediated process and the related 6-substituted β-carboline-3-carboxylates 6 including WYS8 (7) were synthesized via a Sonogashira or Stille coupling processes from 6-iodo-βCCt (5). The bivalent ligands of βCCt (32 and 33) were also designed and prepared via a palladium-catalyzed homocoupling process to expand the structure–activity relationships (SAR) to larger ligands. Based on the pharmacophore/receptor model, a preliminary SAR study on 34 analogues illustrated that large substituents at position-6 of the β-carbolines were well tolerated. As expected, these groups are proposed to project into the extracellular domain (LDi region) of GABAA/Bz receptors (see 32 and 33). Moreover, substituents located at position-3 of the β-carboline nucleus exhibited a conserved stereo interaction in lipophilic pocket L1, while N(2) presumably underwent a hydrogen bonding interaction with H1. Three novel β-carboline ligands (βCCt, 3PBC and WYS8), which preferentially bound to α1 BzR subtypes permitted a comparison of the pharmacological efficacies with a range of classical BzR antagonists (flumazenil, ZK93426) from several different structural groups and indicated these β-carbolines were ‘near GABA neutral antagonists’. Based on the SAR, the most potent (in vitro) α1 selective ligand was the 6-substituted acetylenyl βCCt (WYS8, 7). Earlier both βCCt and 3PBC had been shown to reduce alcohol self-administration in alcohol preferring (P) and high alcohol drinking (HAD) rats but had little or no effect on sucrose self-administration. , and Moreover, these two β-carbolines were orally active, and in addition, were anxiolytic in P rats but were only weakly anxiolytic in rodents. These data prompted the synthesis of the β-carbolines presented here.
Co-reporter:M. Shahjahan Kabir, Ojas A. Namjoshi, Ranjit Verma, Rebecca Polanowski, Sarah M. Krueger, David Sherman, Marc A. Rott, William R. Schwan, Aaron Monte, James M. Cook
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 12) pp:4178-4186
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmc.2010.05.016
Novel acrylic acid ethyl ester derivatives were synthesized and evaluated as potential agents against Mycobacterium species. A versatile and efficient copper-catalyzed coupling process was developed and used to prepare a library of substituted acrylic acid ethyl ester analogs. Minimum inhibitory concentration assays indicated that two of these compounds 3 and 4 have greater in vitro activity against Mycobacterium smegmatis than rifampin, one of the current, first-line anti-mycobacterial chemotherapeutic agents. Moreover, members of this new class of compounds appear to exhibit a specific anti-mycobacterial effect and do not inhibit the growth of the other Gram-positive or Gram-negative species tested.
Co-reporter:Michael Lorenz, M. Shahjahan Kabir, James M. Cook
Tetrahedron Letters 2010 Volume 51(Issue 7) pp:1095-1098
Publication Date(Web):17 February 2010
DOI:10.1016/j.tetlet.2009.12.107
A general route for the synthesis of biologically important flavones is reported via a two step sequence employing a catalytic Wacker–Cook oxidation4b as the key step.
Co-reporter:Jie Yang, Sundari K. Rallapalli, James M. Cook
Tetrahedron Letters 2010 Volume 51(Issue 5) pp:815-817
Publication Date(Web):3 February 2010
DOI:10.1016/j.tetlet.2009.12.002
The first enantiospecific total synthesis of the 3-oxygenated sarpagine indole alkaloids affinine (1) and 16-epiaffinine (2) as well as the synthesis of vobasinediol (3) and 16-epivobasinediol (4) was accomplished from d-(+)-tryptophan methyl ester.
Co-reporter:Michael L. Van Linn and James M. Cook
The Journal of Organic Chemistry 2010 Volume 75(Issue 11) pp:3587-3599
Publication Date(Web):April 29, 2010
DOI:10.1021/jo1003778
It is well-known that Nb-benzyltryptophan alkyl esters undergo the Pictet−Spengler reaction with aldehydes to furnish both cis- and trans-1,2,3,4-tetrahydro-β-carbolines, with the trans isomer predominating. Epimerization at C-1 took place under acidic conditions to produce, exclusively, the thermodynamically more stable trans diastereomer via internal asymmetric induction. Recent kinetic experiments provided insight into the cis to trans epimerization mechanism involved in the Pictet−Spengler reaction of 1,2,3-trisubsituted tetrahydro-β-carbolines. Since the epimerization reaction had been shown to be sensitive to electronic effects at C-1, the rate data for a series of 1-phenyl-substituted 1,2,3,4-tetrahydro-β-carbolines was investigated via a Hammett study. Analysis of the data supported the presence of a positively charged intermediate with a ρ value of −1.4, although the existence of an iminium ion intermediate or a carbocationic intermediate could not be determined from this data alone. Analysis of the rate of epimerization demonstrated first-order kinetics with respect to TFA following the initial protonation of the substrate. This observation was consistent with the formation of a doubly protonated intermediate as the rate-determining step in the carbocation-mediated cis to trans epimerization process. In addition, the observed first-order rate dependence was inconsistent with the retro-Pictet−Spengler mechanism since protonation at the indole-2 position was not rate determining as demonstrated by kinetic isotope effects. Based on this kinetic data, the retro-Pictet−Spengler pathway was ruled out for the cis to trans epimerization of 1,2,3-trisubstituted 1,2,3,4-tetrahydro-β-carbolines, while the olefinic mechanism had been ruled out by experiments carried out in TFA-d.
Co-reporter:M. Shahjahan Kabir, Michael Lorenz, Michael L. Van Linn, Ojas A. Namjoshi, Shamim Ara and James M. Cook
The Journal of Organic Chemistry 2010 Volume 75(Issue 11) pp:3626-3643
Publication Date(Web):April 29, 2010
DOI:10.1021/jo1004179
cis-1,2-Cyclohexanediol (L3) has been shown to be an efficient and versatile bidentate O-donor ligand that provides a highly active Cu-catalytic system. It was more effective than diols such as trans-1,2-cyclohexanediol or ethylene glycol. This commercially available cis-1,2-cyclohexanediol ligand facilitated the Cu-catalyzed cross-coupling reactions of alkyl, aryl, or heterocyclic thiols with either alkyl, aryl, heterocyclic, or substituted vinyl halides. This new catalytic system promoted the mild and efficient stereo- and regiospecific synthesis of biologically important vinyl sulfides. The yields obtained using electron-rich substituted vinyl sulfides with this catalyst system are generally 75−98%. Most importantly, this singular catalyst system is extremely versatile and provides entry into a wide range of sulfides. This method is particularly noteworthy given its mild reaction conditions, simplicity, generality, and exceptional level of functional group tolerance.
Co-reporter:Felix M. Rivas, James P. Stables, Lauren Murphree, Rahul V. Edwankar, Chitra R. Edwankar, Shengming Huang, Hiteshkumar D. Jain, Hao Zhou, Samarpan Majumder, Subramanian Sankar, Bryan L. Roth, Joachim Ramerstorfer, Roman Furtmüller, Werner Sieghart and James M. Cook
Journal of Medicinal Chemistry 2009 Volume 52(Issue 7) pp:1795-1798
Publication Date(Web):March 10, 2009
DOI:10.1021/jm801652d
The antiseizure activity of benzodiazepines (BDZs) 1−5 in mice and rats as animal models is described. These BDZs have selective efficacy for α2β3γ2 and α3β3γ2 GABAA-receptors. Significant anticonvulsant activity with little or no motor impairment and therapeutic indexes (TI) of 2.8−44 (mice, ip) were observed for compounds 2−4 in the subcutaneous metrazole seizure (scMET) test. In rats, orally (po) the TI was >5 to 105. These compounds represent novel leads in the search for anticonvulsants devoid of sedative, ataxic, and amnestic side effects.
Co-reporter:Jie Yang, Xiangyu Z. Wearing, Philip W. Le Quesne, Jeffrey R. Deschamps and James M. Cook
Journal of Natural Products 2008 Volume 71(Issue 8) pp:1431-1440
Publication Date(Web):July 9, 2008
DOI:10.1021/np800269k
The first enantiospecific total synthesis of (+)-alstonisine has been accomplished from d-tryptophan methyl ester 13 in 12% overall yield (in 17 reaction vessels). A diastereospecific osmylation process has been employed as a key step to convert indole 18 into spirocyclic oxindole 19. Mechanistic studies of the stereoselective osmylation of the 2,3-indole double bond of indole alkaloids has been carried out. Compelling evidence for the intramolecular delivery of OsO4 via Nb-complexation was obtained for the osmylation process. The correct structure of (+)-alstonisine (1) was determined by NOE spectroscopic experiments and further confirmed by single-crystal X-ray analysis.
Co-reporter:Hiteshkumar D. Jain, Chunchun Zhang, Shuo Zhou, Hao Zhou, Jun Ma, Xiaoxiang Liu, Xuebin Liao, Amy M. Deveau, Christine M. Dieckhaus, Michael A. Johnson, Kirsten S. Smith, Timothy L. Macdonald, Hideaki Kakeya, Hiroyuki Osada, James M. Cook
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 8) pp:4626-4651
Publication Date(Web):15 April 2008
DOI:10.1016/j.bmc.2008.02.050
Tryprostatin A is an inhibitor of breast cancer resistance protein, consequently a series of structure–activity studies on the cell cycle inhibitory effects of tryprostatin A analogues as potential antitumor antimitotic agents have been carried out. These analogues were assayed for their growth inhibition properties and their ability to perturb the cell cycle in tsFT210 cells. SAR studies resulted in the identification of the essential structural features required for cytotoxic activity. The absolute configuration l-Tyr-l-pro in the diketopiperazine ring along with the presence of the 6-methoxy substituent on the indole moiety of 1 was shown to be essential for dual inhibition of topoisomerase II and tubulin polymerization. Biological evaluation also indicated the presence of the 2-isoprenyl moiety on the indole scaffold of 1 was essential for potent inhibition of cell proliferation. Substitution of the indole Na–H in 1 with various alkyl or aryl groups, incorporation of various l-amino acids into the diketopiperazine ring in place of l-proline, and substitution of the 6-methoxy group in 1 with other functionality provided active analogues. The nature of the substituents present on the indole Na–H or the indole C-2 position influenced the mechanism of action of these analogues. Analogues 68 (IC50 = 10 μM) and 67 (IC50 = 19 μM) were 7-fold and 3.5-fold more potent, respectively, than 1 (IC50 = 68 μM) in the inhibition of the growth of tsFT210 cells. Diastereomer-2 of tryprostatin B 8 was a potent inhibitor of the growth of three human carcinoma cell lines: H520 (IC50 = 11.9 μM), MCF-7 (IC50 = 17.0 μM) and PC-3 (IC50 = 11.1 μM) and was equipotent with etoposide, a clinically used anticancer agent. Isothiocyanate analogue 71 and 6-azido analogue 72 were as potent as 1 in the tsFT210 cell proliferation and may be useful tools in labeling BCRP.Analogues of tryprostatin A modified in the regions A–D were synthesized and evaluated for their antitumor activity.
Co-reporter:Dongmei Han, F. Holger Försterling, Xiaoyan Li, Jeffrey R. Deschamps, Damon Parrish, Hui Cao, Sundari Rallapalli, Terry Clayton, Yun Teng, Samarpan Majumder, Subramaniam Sankar, Bryan L. Roth, Werner Sieghart, Roman Furtmuller, James K. Rowlett, Michael R. Weed, James M. Cook
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 19) pp:8853-8862
Publication Date(Web):1 October 2008
DOI:10.1016/j.bmc.2008.08.072
The stable conformations of GABAA–benzodiazepine receptor bivalent ligands were determined by low temperature NMR spectroscopy and confirmed by single crystal X-ray analysis. The stable conformations in solution correlated well with those in the solid state. The linear conformation was important for these dimers to access the binding site and exhibit potent in vitro affinity and was illustrated for α5 subtype selective ligands. Bivalent ligands with an oxygen-containing linker folded back upon themselves both in solution and the solid state. Dimers which are folded do not bind to Bz receptors.Linear conformation is important for subtype selective benzodiazepine dimers to access the Bz binding site and exhibit potent in vitro affinity.
Co-reporter:M. Shahjahan Kabir, Kathleen Engelbrecht, Rebecca Polanowski, Sarah M. Krueger, Rachel Ignasiak, Marc Rott, William R. Schwan, Mary E. Stemper, Kurt D. Reed, David Sherman, James M. Cook, Aaron Monte
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 21) pp:5745-5749
Publication Date(Web):1 November 2008
DOI:10.1016/j.bmcl.2008.09.085
An antimicrobial phenolic stilbene, (E)-3-hydroxy-5-methoxystilbene, 1 was recently isolated from the leaves of Comptonia peregrina (L.) Coulter and shown to possess inhibitory activity against several Gram-positive bacteria, including isolates of methicillin-resistant Staphylococcus aureus (MRSA), Mycobacterium bovis BCG, and avirulent Bacillusanthracis (Sterne strain), among others. These results prompted the design and synthesis of two new classes of compounds, phenoxystyrenes and phenothiostyrenes, as analogs of the natural antimicrobial stilbene. These and additional stilbenoid analogs were synthesized using new, efficient, copper-mediated coupling strategies. Minimum inhibitory concentration (MIC) antimicrobial assays were performed on all compounds prepared. These preliminary structure–activity relationship studies indicated that both new classes of synthetic analogs, as well as the stilbenes, show promising activity against Gram-positive bacteria when at least one phenolic moiety is present, but not when absent. The potencies of the phenolic phenoxystyrenes and phenothiostyrenes were found to be comparable to those of the phenolic stilbenes tested.Substituted stilbene, phenoxystyrene, and phenothiostyrene analogs of an antimicrobial natural product E-stilbene were synthesized and assayed for the ability to inhibit the growth of clinically significant bacteria.
Co-reporter:Dongmei Han, F.Holger Försterling, Xiaoyan Li, Jeffrey R. Deschamps, Hui Cao, James M. Cook
Bioorganic & Medicinal Chemistry Letters 2004 Volume 14(Issue 6) pp:1465-1469
Publication Date(Web):22 March 2004
DOI:10.1016/j.bmcl.2004.01.018
The stable conformations of GABAA-benzodiazepine receptor bivalent ligands 2 and 3 were determined by low temperature NMR spectroscopy and confirmed by single crystal X-ray analysis. The linear conformation was important for these dimers to access the binding site and exhibit potent in vitro affinity as illustrated for α5 subtype selective ligand 2 (15 nM). Bivalent ligand 3 with the 5 atom linker folded back upon itself both in solution and in the solid state, moreover, it did not bind to Bz receptors.The stable conformations of GABAA-benzodiazepine receptor bivalent ligands were determined by low temperature NMR spectroscopy and confirmed by single crystal X-ray analysis, moreover the effect on in vitro affinities was discussed.
Co-reporter:Nina Y. Yuan, Michael M. Poe, Christopher Witzigmann, James M. Cook, ... Leggy A. Arnold
Journal of Pharmacological and Toxicological Methods (November–December 2016) Volume 82() pp:109-114
Publication Date(Web):1 November 2016
DOI:10.1016/j.vascn.2016.08.006
IntroductionAutomated patch clamp is a recent but widely used technology to assess pre-clinical drug safety. With the availability of human neurons derived from pluripotent stem cells, this technology can be extended to determine CNS effects of drug candidates, especially those acting on the GABAA receptor.MethodsiCell Neurons (Cellular Dynamics International, A Fujifilm Company) were cultured for ten days and analyzed by patch clamp in the presence of agonist GABA or in combination with positive allosteric GABAA receptor modulators. Both efficacy and affinity were determined. In addition, mRNA of GABAA receptor subunits were quantified by qRT-PCR.ResultsWe have shown that iCell Neurons are compatible with the IonFlux microfluidic system of the automated patch clamp instrument. Resistance ranging from 15 to 25 MΩ was achieved for each trap channel of patch clamped cells in a 96-well plate format. GABA induced a robust change of current with an EC50 of 0.43 μM. Positive GABAA receptor modulators diazepam, HZ-166, and CW-04-020 exhibited EC50 values of 0.42 μM, 1.56 μM, and 0.23 μM, respectively. The α2/α3/α5 selective compound HZ-166-induced the highest potentiation (efficacy) of 810% of the current induced by 100 nM GABA. Quantification of GABAA receptor mRNA in iCell Neurons revealed high levels of α5 and β3 subunits and low levels of α1, which is similar to the configuration in human neonatal brain.DiscussioniCell Neurons represent a new cellular model to characterize GABAergic compounds using automated patch clamp. These cells have excellent representation of cellular GABAA receptor distribution that enable determination of total small molecule efficacy and affinity as measured by cell membrane current change.
Co-reporter:William R. Schwan, Maren Kallaus, Sarah Krueger, Aaron Monte, M. Shahjahan Kabir, James M. Cook
Journal of Infection and Chemotherapy (2012) Volume 18(Issue 1) pp:124-126
Publication Date(Web):1 January 2012
DOI:10.1007/s10156-011-0273-7
Staphylococcus aureus causes hundreds of thousands of infections and thousands of deaths per year in the United States. The emergence of methicillin-resistant S. aureus (MRSA), including community-associated methicillin-resistant S. aureus (CA-MRSA), has added to the problem. As MRSA continue to evolve, they are becoming resistant to more classes of antibiotics. In the past 20 years, only three new antibiotics have been approved for human use (linezolid, daptomycin, and tigecycline), and resistance to these three drugs has already emerged. New antibiotics are needed, and we have developed a promising drug candidate that may be applicable to treating MRSA, among other gram-positive bacterial infections. We have identified a novel synthetic drug, coded SK-03-92, that shows broad in vitro efficacy against a variety of gram-positive bacterial strains that include a number of strains of S. aureus. Besides the activity against gram-positive bacteria, this new drug also exhibits activity against Mycobacterium strains.
Co-reporter:V. V. N. Phani Babu Tiruveedhula, Kashi Reddy Methuku, Jeffrey R. Deschamps and James M. Cook
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 43) pp:NaN10715-10715
Publication Date(Web):2015/09/03
DOI:10.1039/C5OB01572C
A novel two step protocol was developed to gain regiospecific access to 3-substituted β- and aza-β-carbolines, 3-PBC (1), 3-ISOPBC (2), βCCt (3), 6-aza-3-PBC (4) and 6-aza-3-ISOPBC (5). These β-carbolines (1–3) are potential clinical agents to reduce alcohol self-administration, especially 3-ISOPBC·HCl (2·HCl) which appears to be a potent anti-alcohol agent active against binge drinking in a rat model of maternally deprived (MD) rats. The method consists of two consecutive palladium-catalyzed reactions: a Buchwald–Hartwig amination followed by an intramolecular Heck-type cyclization in high yield.



![tert-butyl 5-[(2-chlorophenyl)amino]picolinate](http://img.cochemist.com/ccimg/1300800/1300728-31-7.png)
![tert-butyl 5-[(2-chlorophenyl)amino]picolinate](http://img.cochemist.com/ccimg/1300800/1300728-31-7_b.png)
![3-BUTYN-2-OL, 4-[TRIS(1-METHYLETHYL)SILYL]-](http://img.cochemist.com/ccimg/726300/726202-65-9.png)
![3-BUTYN-2-OL, 4-[TRIS(1-METHYLETHYL)SILYL]-](http://img.cochemist.com/ccimg/726300/726202-65-9_b.png)
![1H-Indole, 3,3'-[(2-chlorophenyl)methylene]bis[2-methyl-](http://img.cochemist.com/ccimg/618500/618406-36-3.png)
![1H-Indole, 3,3'-[(2-chlorophenyl)methylene]bis[2-methyl-](http://img.cochemist.com/ccimg/618500/618406-36-3_b.png)
















![3-Butyn-2-ol, 4-[tris(1-methylethyl)silyl]-, (2R)-](http://img.cochemist.com/ccimg/183900/183852-53-1.png)
![3-Butyn-2-ol, 4-[tris(1-methylethyl)silyl]-, (2R)-](http://img.cochemist.com/ccimg/183900/183852-53-1_b.png)












![9H-Imidazo[1,5-a]pyrrolo[2,1-c][1,4]benzodiazepine-1-carboxylicacid, 11,12,13,13a-tetrahydro-7-methoxy-9-oxo-, ethyl ester, (13aS)-](http://img.cochemist.com/ccimg/130500/130477-52-0.png)
![9H-Imidazo[1,5-a]pyrrolo[2,1-c][1,4]benzodiazepine-1-carboxylicacid, 11,12,13,13a-tetrahydro-7-methoxy-9-oxo-, ethyl ester, (13aS)-](http://img.cochemist.com/ccimg/130500/130477-52-0_b.png)
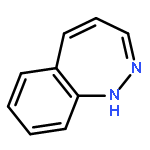
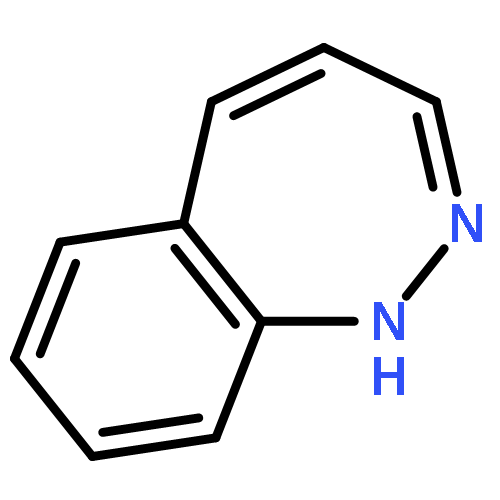
![9H-Pyrido[3,4-b]indole-3-carboxylic acid 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/93900/93835-05-3.png)
![9H-Pyrido[3,4-b]indole-3-carboxylic acid 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/93900/93835-05-3_b.png)
![9H-Pyrido[3,4-b]indole, 3-(1-methylethoxy)-](http://img.cochemist.com/ccimg/92000/91985-84-1.png)
![9H-Pyrido[3,4-b]indole, 3-(1-methylethoxy)-](http://img.cochemist.com/ccimg/92000/91985-84-1_b.png)
![9H-Pyrido[3,4-b]indole, 3-propoxy-](http://img.cochemist.com/ccimg/92000/91985-83-0.png)
![9H-Pyrido[3,4-b]indole, 3-propoxy-](http://img.cochemist.com/ccimg/92000/91985-83-0_b.png)



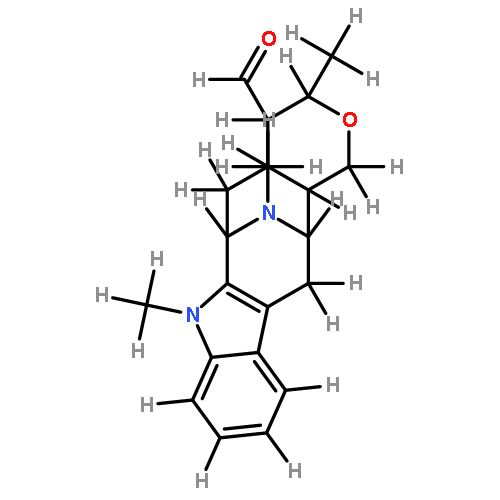
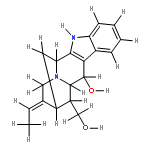
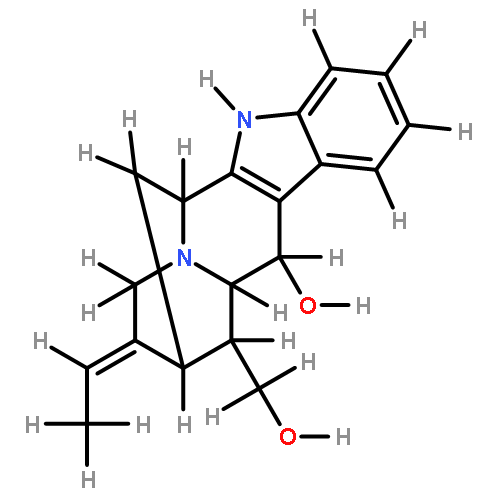


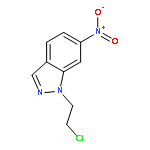
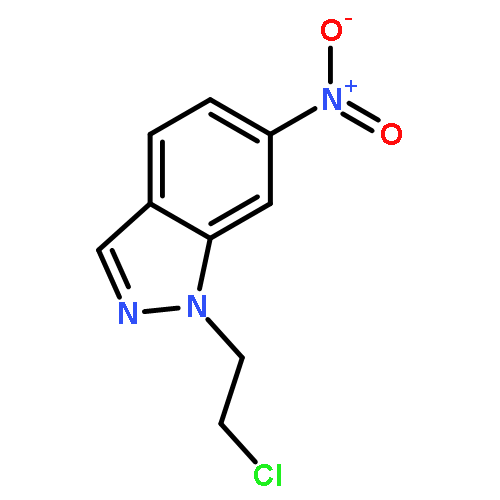
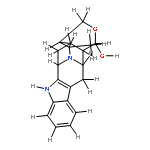
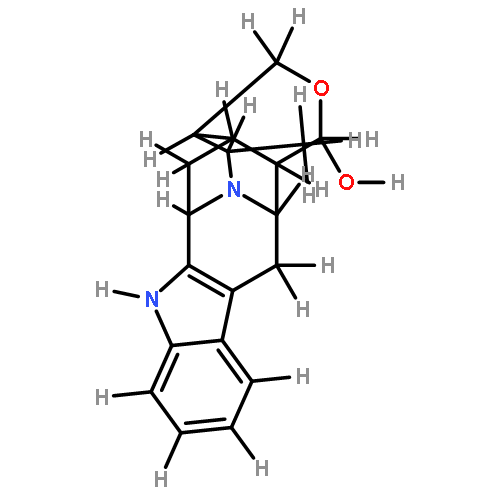
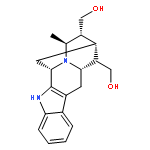












![(R)-Methyl 4-((3R,5R,8R,9S,10S,13R,14S,17R)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate](http://img.cochemist.com/ccimg/1300/1249-75-8.png)
![(R)-Methyl 4-((3R,5R,8R,9S,10S,13R,14S,17R)-3-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate](http://img.cochemist.com/ccimg/1300/1249-75-8_b.png)