Co-reporter:Shahrina Alam, Kaelyn Skye Lingenfelter, Aaron M. Bender, and Craig W. Lindsley
ACS Chemical Neuroscience September 20, 2017 Volume 8(Issue 9) pp:1823-1823
Publication Date(Web):July 24, 2017
DOI:10.1021/acschemneuro.7b00270
Memantine was the first breakthrough medication for the treatment of moderate to severe Alzheimer’s disease (AD) patients and represents a fundamentally new mechanism of action (moderate-affinity, uncompetitive, voltage-dependent, N-methyl-d-aspartate (NMDA) receptor antagonist that exhibits fast on/off kinetics) to modulate glutamatergic dysfunction. Since its approval by the FDA in 2003, memantine, alone and in combination with donepezil, has improved patient outcomes in terms of cognition, behavioral disturbances, daily functioning, and delaying time to institutionalization. In this review, we will highlight the historical significance of memantine to AD (and other neuropsychiatric disorders) as well as provide an overview of the synthesis, pharmacology, and drug metabolism of this unique NMDA uncompetitive antagonist that clearly secures its place among the Classics in Chemical Neuroscience.Keywords: 3,5-dimethyladamantan-1-amine hydrochloride; Alzheimer’s disease; glutamatergic dysfunction; Memantine; N-methyl-d-aspartate (NMDA) receptor; uncompetitive antagonist;
Co-reporter:Julie L. Engers, Katrina A. Bollinger, Rebecca L. Weiner, Alice L. Rodriguez, Madeline F. Long, Megan M. Breiner, Sichen Chang, Sean R. Bollinger, Michael Bubser, Carrie K. Jones, Ryan D. Morrison, Thomas M. Bridges, Anna L. Blobaum, Colleen M. Niswender, P. Jeffrey Conn, Kyle A. Emmitte, and Craig W. Lindsley
ACS Medicinal Chemistry Letters September 14, 2017 Volume 8(Issue 9) pp:925-925
Publication Date(Web):August 15, 2017
DOI:10.1021/acsmedchemlett.7b00249
Herein, we detail the optimization of the mGlu3 NAM, VU0650786, via a reductionist approach to afford a novel, simplified mGlu3 NAM scaffold that engenders potent and selective mGlu3 inhibition (mGlu3 IC50 = 245 nM, mGlu2 IC50 > 30 μM) with excellent central nervous system penetration (rat brain/plasma Kp = 1.2, Kp,uu = 0.40). Moreover, this new chemotype, exemplified by VU6010572, requires only four synthetic steps and displays improved physiochemical properties and in vivo efficacy in a mouse tail suspension test (MED = 3 mg/kg i.p.).Keywords: depression; metabotropic glutamate receptor 3 (mGlu3); Negative allosteric modulator (NAM); physiochemical properties; VU6010572;
Co-reporter:Katrina A. Bollinger, Andrew S. Felts, Christopher J. Brassard, Julie L. Engers, Alice L. Rodriguez, Rebecca L. Weiner, Hyekyung P. Cho, Sichen Chang, Michael Bubser, Carrie K. Jones, Anna L. Blobaum, Colleen M. Niswender, P. Jeffrey Conn, Kyle A. Emmitte, and Craig W. Lindsley
ACS Medicinal Chemistry Letters September 14, 2017 Volume 8(Issue 9) pp:919-919
Publication Date(Web):August 3, 2017
DOI:10.1021/acsmedchemlett.7b00279
Herein, we detail the optimization of the mGlu2 negative allosteric modulator (NAM), VU6001192, by a reductionist approach to afford a novel, simplified mGlu2 NAM scaffold. This new chemotype not only affords potent and selective mGlu2 inhibition, as exemplified by VU6001966 (mGlu2 IC50 = 78 nM, mGlu3 IC50 > 30 μM), but also excellent central nervous system (CNS) penetration (Kp = 1.9, Kp,uu = 0.78), a feature devoid in all previously disclosed mGlu2 NAMs (Kps ≈ 0.3, Kp,uus ≈ 0.1). Moreover, this series, based on overall properties, represents an exciting lead series for potential mGlu2 PET tracer development.Keywords: CNS penetration; depression; metabotropic glutamate receptor 2 (mGlu2); Negative allosteric modulator (NAM); VU6001966;
Co-reporter:Craig W. Lindsley and Corey R. Hopkins
Journal of Medicinal Chemistry September 14, 2017 Volume 60(Issue 17) pp:7233-7233
Publication Date(Web):May 10, 2017
DOI:10.1021/acs.jmedchem.7b00151
The dopamine D4 receptor garnered a great deal of interest in the early 1990s when studies showed the atypical antipsychotic clozapine possessed higher affinity for D4, relative to other dopamine receptor subtypes, and that this activity might underlie the unique clinical efficacy of clozapine. Unfortunately, D4 antagonists that were developed for schizophrenia failed in the clinic. Thus, D4 fell out of favor as a therapeutic target, and work in this area was silent for decades. Recently, D4 ligands with improved selectivity for D4 against not only D1–3,5 but also other biogenic amine targets have emerged, and D4 is once again in the spotlight as a novel target for both addiction and Parkinson’s disease (PD), as well as other emerging diseases. This report will review the historical data for D4, review the known D4 ligands, and then highlight new data supporting a role for D4 inhibition in addiction, PD, and cancer.
Co-reporter:Aaron M. Bender, Trevor C. Chopko, Thomas M. Bridges, and Craig W. Lindsley
Organic Letters October 20, 2017 Volume 19(Issue 20) pp:5693-5693
Publication Date(Web):October 4, 2017
DOI:10.1021/acs.orglett.7b02868
N-Alkyl substituted chlorotetrazines were coupled with various boronic acids under Suzuki conditions in high yield at room temperature, giving a mild and straightforward synthetic route toward diverse unsymmetrical 1,2,4,5-tetrazines, a rare heteroarene. This chemistry not only expands the known substrate scope of tetrazine cross-coupling reactions but also allows for the synthesis of novel, tetrazine-containing biologically active molecules with improved DMPK properties.
Co-reporter:Masahito Abe, Mabel Seto, Rocco G. Gogliotti, Matthew T. Loch, Katrina A. Bollinger, Sichen Chang, Eileen M. Engelberg, Vincent B. Luscombe, Joel M. Harp, Michael Bubser, Darren W. Engers, Carrie K. Jones, Alice L. Rodriguez, Anna L. Blobaum, P. Jeffrey Conn, Colleen M. Niswender, and Craig W. Lindsley
ACS Medicinal Chemistry Letters October 12, 2017 Volume 8(Issue 10) pp:1110-1110
Publication Date(Web):September 1, 2017
DOI:10.1021/acsmedchemlett.7b00317
Herein, we report the structure–activity relationships within a series of mGlu7 PAMs based on a pyrazolo[1,5-a]pyrimidine core with excellent CNS penetration (Kps > 1 and Kp,uus > 1). Analogues in this series proved to display a range of Group III mGlu receptor selectivity, but VU6005649 emerged as the first dual mGlu7/8 PAM, filling a void in the Group III mGlu receptor PAM toolbox and demonstrating in vivo efficacy in a mouse contextual fear conditioning model.Keywords: cognition; metabotropic glutamate receptor 7 (mGlu7); Positive allosteric modulator (PAM); Rett syndrome; VU6005649;
Co-reporter:Kellie D. Nance;C. David Weaver;Emily L. Days;Anastasia Coldren;Tiffany D. Farmer;Hyekyung P. Cho;Colleen M. Niswender;Anna L. Blobaum;Kevin D. Niswender
Journal of Medicinal Chemistry February 23, 2017 Volume 60(Issue 4) pp:1611-1616
Publication Date(Web):January 19, 2017
DOI:10.1021/acs.jmedchem.6b01706
A duplexed, functional multiaddition high throughput screen and subsequent optimization effort identified the first orally bioavailable and CNS penetrant glucagon-like peptide-1 receptor (GLP-1R) noncompetitive antagonist. Antagonist 5d not only blocked exendin-4-stimulated insulin release in islets but also lowered insulin levels while increasing blood glucose in vivo.
Co-reporter:Daniel E. Jeffries, Craig W. Lindsley
Tetrahedron Letters 2017 Volume 58(Issue 1) pp:112-116
Publication Date(Web):4 January 2017
DOI:10.1016/j.tetlet.2016.11.120
•A one-pot, multi-component reaction cascade for the rapid synthesis of diversely functionalized heteroaryl methyl substrates.•A tandem SN2/Suzuki-Miyaura reaction with yields ranging between 50 and 66%•A tandem SN2 Sonogashira reaction across a breadth of chemical diversity with yields ranging between 31 and 87%•Rapid access to biologically relevant molecules.A novel one-pot, “green” protocol to rapidly access pharmaceutically relevant heteroaryl methyl substrates is described. This process allows for a tandem SN2/Suzuki-Miyaura reaction or Sonogashira reaction across a breadth of chemical diversity with yields ranging between 31 and 87% for the tandem Suzuki-Miyaura process and 50–66% for the tandem Sonogashira process. This procedure tolerates S, N, and O heteroatom linkers and is amenable for both rapid and robust lead development screening. In addition, T-type and N-type calcium channel blocker (15) was synthesized in 43% yield using this methodology which stands as an improvement in both yield and reaction time of the previously reported synthesis. The one-pot protocol also allows for the inclusion of greater chemical diversity within the scaffold of 15.
Co-reporter:Daniel E. Jeffries and Craig W. Lindsley
The Journal of Organic Chemistry 2017 Volume 82(Issue 1) pp:431-437
Publication Date(Web):December 5, 2016
DOI:10.1021/acs.joc.6b02534
Here, we report the first total synthesis of hybrubin A, a bipyrrole tetramic acid alkaloid representing a new carbon framework derived from convergent (truncated red cluster and exogenous hbn cluster) biosynthetic pathways. A highly convergent synthesis was developed, employing 4-methoxy-1,5-dihydro-2H-pyrrol-2-one (13) as a single starting material to provide hybrubin A in three steps from 13 and 20.8% overall yield. As no biological activity was prescribed to hybrubin A except for a lack of cytotoxicity, we further profiled this unique alkaloid across panels of discrete molecular targets. Interestingly, hybrubin A was found to be a ligand for a variety of GPCRs with a propensity for potent binding across therapeutically relevant adenosine receptors (A1, A2a, and A3) as well as a potent activity at a kinase, FLT3. This pattern of biological activity is distinct from other related prodigiosin natural and unnatural products and is even more intriguing in the absence of cytotoxicity.
Co-reporter:Aaron M. Bender, Rebecca L. Weiner, Vincent B. Luscombe, Sonia Ajmera, Hyekyung P. Cho, Sichen Chang, Xiaoyan Zhan, Alice L. Rodriguez, Colleen M. Niswender, Darren W. Engers, Thomas M. Bridges, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.05.042
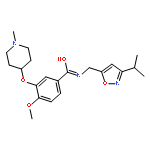
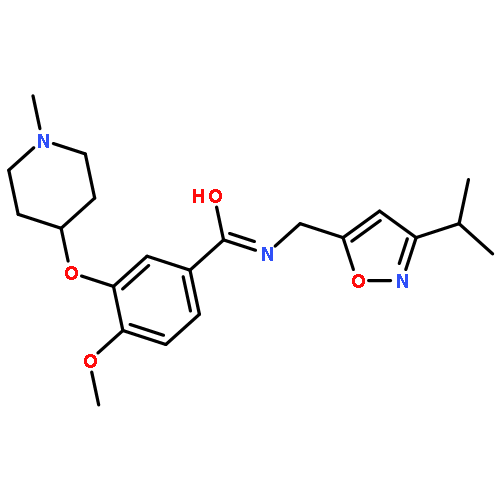
This letter describes the synthesis and structure activity relationship (SAR) studies of structurally novel M4 antagonists, based on a 3-(4-aryl/heteroarylsulfonyl)piperazin-1-yl)-6-(piperidin-1-yl)pyridazine core, identified from a high-throughput screening campaign. A multi-dimensional optimization effort enhanced potency at human M4 (hM4 IC50s < 200 nM), with only moderate species differences noted, and with enantioselective inhibition. Moreover, CNS penetration proved attractive for this series (rat brain:plasma Kp = 2.1, Kp,uu = 1.1). Despite the absence of the prototypical mAChR antagonist basic or quaternary amine moiety, this series displayed pan-muscarinic antagonist activity across M1-5 (with 9- to 16-fold functional selectivity at best). This series further expands the chemical diversity of mAChR antagonists.Download high-res image (121KB)Download full-size image
Co-reporter:James C. Tarr, Michael R. Wood, Meredith J. Noetzel, Jeanette L. Bertron, Rebecca L. Weiner, Alice L. Rodriguez, Atin Lamsal, Frank W. Byers, Sichen Chang, Hyekyung P. Cho, Carrie K. Jones, Colleen M. Niswender, Michael W. Wood, Nicholas J. Brandon, Mark E. Duggan, P. Jeffrey Conn, Thomas M. Bridges, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmcl.2017.05.014
This letter details the continued chemical optimization of a novel series of M4 positive allosteric modulators (PAMs) based on a 5-amino-thieno[2,3-c]pyridazine core by incorporating a 3-amino azetidine amide moiety. The analogs described within this work represent the most potent M4 PAMs reported for this series to date. The SAR to address potency, clearance, subtype selectivity, CNS exposure, and P-gp efflux are described. This work culminated in the discovery of VU6000918, which demonstrated robust efficacy in a rat amphetamine-induced hyperlocomotion reversal model at a minimum efficacious dose of 0.3 mg/kg.Download high-res image (98KB)Download full-size image
Co-reporter:Michael R. Wood, Meredith J. Noetzel, Bruce J. Melancon, Michael S. Poslusney, Kellie D. Nance, Miguel A. Hurtado, Vincent B. Luscombe, Rebecca L. Weiner, Alice L. Rodriguez, Atin Lamsal, Sichen Chang, Michael Bubser, Anna L. Blobaum, Darren W. Engers, Colleen M. Niswender, Carrie K. Jones, Nicholas J. Brandon, Michael W. Wood, Mark E. Duggan, P. Jeffrey Conn, Thomas M. Bridges, and Craig W. Lindsley
ACS Medicinal Chemistry Letters 2017 Volume 8(Issue 2) pp:
Publication Date(Web):December 16, 2016
DOI:10.1021/acsmedchemlett.6b00461
Herein, we report the structure–activity relationships within a series of potent, selective, and orally bioavailable muscarinic acetylcholine receptor 4 (M4) positive allosteric modulators (PAMs). Compound 6c (VU0467485) possesses robust in vitro M4 PAM potency across species and in vivo efficacy in preclinical models of schizophrenia. Coupled with an attractive DMPK profile and suitable predicted human PK, 6c (VU0467485) was evaluated as a preclinical development candidate.Keywords: muscarinic acetylcholine receptor 4 (M4); Positive allosteric modulator (PAM); schizophrenia; VU0467485;
Co-reporter:Douglas J. Sheffler, Michael T. Nedelcovych, Richard Williams, Stephen C. Turner, Brittany B. Duerk, Megan R. Robbins, Sataya B. Jadhav, Colleen M. Niswender, Carrie K. Jones, P. Jeffrey Conn, R. Nathan Daniels, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 9(Issue 9) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bmcl.2017.03.046
Co-reporter:Aaron M. Bender, Rebecca L. Weiner, Vincent B. Luscombe, Hyekyung P. Cho, Colleen M. Niswender, Darren W. Engers, Thomas M. Bridges, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 11(Issue 11) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bmcl.2017.04.009
This letter describes the synthesis and structure activity relationship (SAR) studies of structurally novel M4 antagonists, based on a 4,6-disubstituted core, identified from a high-throughput screening campaign. A multi-dimensional optimization effort enhanced potency at both human and rat M4 (IC50s < 300 nM), with no substantial species differences noted. Moreover, CNS penetration proved attractive for this series (brain:plasma Kp,uu = 0.87), while other DMPK attributes were addressed in the course of the optimization effort, providing low in vivo clearance in rat (CLp = 5.37 mL/min/kg). Surprisingly, this series displayed pan-muscarinic antagonist activity across M1–5, despite the absence of the prototypical basic or quaternary amine moiety, thus offering a new chemotype from which to develop a next generation of pan-muscarinic antagonist agents.Download high-res image (158KB)Download full-size image
Co-reporter:James C. Tarr, Michael R. Wood, Meredith J. Noetzel, Bruce J. Melancon, Atin Lamsal, Vincent B. Luscombe, Alice L. Rodriguez, Frank W. Byers, Sichen Chang, Hyekyung P. Cho, Darren W. Engers, Carrie K. Jones, Colleen M. Niswender, Michael W. Wood, Nicholas J. Brandon, Mark E. Duggan, P. Jeffrey Conn, Thomas M. Bridges, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 23(Issue 23) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.bmcl.2017.10.053
Herein we describe the continued optimization of M4 positive allosteric modulators (PAMs) within the 5-amino-thieno[2,3-c]pyridazine series of compounds. In this letter, we disclose our studies on tertiary amides derived from substituted azetidines. This series provided excellent CNS penetration, which had been challenging to consistently achieve in other amide series. Efforts to mitigate high clearance, aided by metabolic softspot analysis, were unsuccessful and precluded this series from further consideration as a preclinical candidate. In the course of this study, we found that potassium tetrafluoroborate salts could be engaged in a tosyl hydrazone reductive cross coupling reaction, a previously unreported transformation, which expands the synthetic utility of the methodology.Download high-res image (57KB)Download full-size image
Co-reporter:Blake R. Bewley, Paul K. Spearing, Rebecca L. Weiner, Vincent B. Luscombe, Xiaoyan Zhan, Sichen Chang, Hyekyung P. Cho, Alice L. Rodriguez, Colleen M. Niswender, P. Jeffrey Conn, Thomas M. Bridges, Darren W. Engers, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 18(Issue 18) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.bmcl.2017.08.043
This Letter details the discovery and subsequent optimization of a novel M4 PAM scaffold based on an 6-fluoro-4-(piperidin-1-yl)quinoline-3-carbonitrile core, which represents a distinct departure from the classical M4 PAM chemotypes. Optimized compounds in this series demonstrated improved M4 PAM potency on both human and rat M4 (4 to 5-fold relative to HTS hit), and displayed attractive physicochemical and DMPK profiles, including good CNS penetration (rat brain:plasma Kp = 5.3, Kp,uu = 2.4; MDCK-MDR1 (P-gp) ER = 1.1).Download high-res image (147KB)Download full-size image
Co-reporter:Madeline F. Long, Julie L. Engers, Sichen Chang, Xiaoyan Zhan, Rebecca L. Weiner, Vincent B. Luscombe, Alice L. Rodriguez, Hyekyung P. Cho, Colleen M. Niswender, Thomas M. Bridges, P. Jeffrey Conn, Darren W. Engers, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 22(Issue 22) pp:
Publication Date(Web):15 November 2017
DOI:10.1016/j.bmcl.2017.10.016
This Letter details our efforts to replace the 3-amino moiety, an essential pharmacophore for M4 PAM activity in most M4 PAMs to date, within the thieno[2,3-b]pyridine core, as the β-amino carboxamide motif has been shown to engender poor solubility, varying degrees of P-gp efflux and represents a structural alert. A scaffold hopping exercise identified a novel 2,4-dimethylquinoline carboxamide core that provided M4 PAM activity and good CNS penetration without an amino moiety. In addition, MacMillan photoredox catalysis chemistry was essential for construction of the 2,4-dimethylquinoline core.Download high-res image (145KB)Download full-size image
Co-reporter:Thomas K. David, Nayak K. Prashanth, M. Rajendraswami, Devendrareddy Pallalu, Arlindo L. Castelhano, Michael J. Kates, Anna L. Blobaum, Carrie K. Jones, Kyle A. Emmitte, P. Jeffrey Conn, Craig W. Lindsley
Tetrahedron Letters 2017 Volume 58, Issue 36(Issue 36) pp:
Publication Date(Web):6 September 2017
DOI:10.1016/j.tetlet.2017.07.100
•Scalable 5 step GMP synthesis of auglurant in >99.8% purity.•Elimination of chromatography steps and replacement with recrystallizations.•Addressed thermal instability of pyridine N-oxide.•Efficient cyanation and SNAr reactions.This communication details the kilogram-scale synthesis of N-(5-fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (VU0424238, auglurant), a novel mGlu5 negative allosteric modulator (NAM) developed as an alternative treatment for depression. The process highlights a challenging pyridine N-oxidation sequence, an SNAr reaction, and the elimination of all chromatography steps (required in the medicinal chemistry route) with replacement by highly efficient recrystallizations (save one silica plug). The improved process was utilized for the preparation of a 1.2 kg toxicology batch, as well as a 2.82 kg GMP batch to support the Phase I trial, in very high purity (99.8%).Download high-res image (51KB)Download full-size image
Co-reporter:Craig W. Lindsley, Kyle A. Emmitte, Corey R. Hopkins, Thomas M. Bridges, Karen J. Gregory, Colleen M. Niswender, and P. Jeffrey Conn
Chemical Reviews 2016 Volume 116(Issue 11) pp:6707
Publication Date(Web):February 16, 2016
DOI:10.1021/acs.chemrev.5b00656
Allosteric modulation of GPCRs has initiated a new era of basic and translational discovery, filled with therapeutic promise yet fraught with caveats. Allosteric ligands stabilize unique conformations of the GPCR that afford fundamentally new receptors, capable of novel pharmacology, unprecedented subtype selectivity, and unique signal bias. This review provides a comprehensive overview of the basics of GPCR allosteric pharmacology, medicinal chemistry, drug metabolism, and validated approaches to address each of the major challenges and caveats. Then, the review narrows focus to highlight recent advances in the discovery of allosteric ligands for metabotropic glutamate receptor subtypes 1–5 and 7 (mGlu1–5,7) highlighting key concepts (“molecular switches”, signal bias, heterodimers) and practical solutions to enable the development of tool compounds and clinical candidates. The review closes with a section on late-breaking new advances with allosteric ligands for other GPCRs and emerging data for endogenous allosteric modulators.
Co-reporter:Pedro M. Garcia-Barrantes and Craig W. Lindsley
Organic Letters 2016 Volume 18(Issue 15) pp:3810-3813
Publication Date(Web):July 21, 2016
DOI:10.1021/acs.orglett.6b01825
The first total synthesis of Gombamide A (1), a cytotoxic cyclic thiopeptide from the sponge Clathria gombawuiensis, has been achieved. Highlights of the convergent synthesis feature a disulfide bond forming cascade to close the 17-membered macrocycle and a selenoazidylation procedure to access the unusual para-hydroxystyrlyamide (pHSA) moiety. The synthesis required 18 steps, 11 steps in its longest linear sequence, and proceeded in 9.1% overall yield. This work will facilitate the study of the biological effects of Gombamide A and provide groundwork to explore the structure–activity relationship around this rare natural product.
Co-reporter:Kayla J. Temple, Matthew T. Duvernay, Summer E. Young, Wandong Wen, Wenjun Wu, Jae G. Maeng, Anna L. Blobaum, Shaun R. Stauffer, Heidi E. Hamm, and Craig W. Lindsley
Journal of Medicinal Chemistry 2016 Volume 59(Issue 16) pp:7690-7695
Publication Date(Web):August 2, 2016
DOI:10.1021/acs.jmedchem.6b00928
Here, we describe the development of a series of highly selective PAR4 antagonists with nanomolar potency and selectivity versus PAR1, derived from the indole-based 3. Of these, 9j (PAR4 IC50 = 445 nM, PAR1 response IC50 > 30 μM) and 10h (PAR4 IC50 = 179 nM, PAR1 response IC50 > 30 μM) maintained an overall favorable in vitro DMPK profile, encouraging rat/mouse in vivo pharmacokinetics (PK) and activity against γ-thrombin.
Co-reporter:Pedro M. Garcia-Barrantes, Hyekyung P. Cho, Tahj M. Starr, Anna L. Blobaum, Colleen M. Niswender, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2289-2292
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.044
This letter describes the re-exploration of the mGlu1 PAM Ro 07-11401 scaffold through a multi-dimensional, iterative parallel synthesis approach. Unlike recent series of mGlu1 PAMs with robust SAR, the SAR around the Ro 07-11401 structure was incredibly steep (only ∼6 of 200 analogs displayed mGlu1 PAM activity), and reminiscent of the CPPHA mGlu5 PAM scaffold. Despite the steep SAR, two new thiazole derivatives were discovered with improved in vitro DMPK profiles and ∼3- to 4-fold improvement in CNS exposure (Kps 1.01–1.19); albeit, with a ∼3-fold diminution in mGlu1 PAM potency, yet comparable efficacy (∼5-fold leftward shift of the glutamate concentration–response curve at 10 μM). Thus, this effort has provided additional CNS penetrant mGlu1 PAM tools in a different chemotype than the VU0486321 scaffold. These compounds will permit a better understanding of the pharmacology and therapeutic potential of selective mGlu1 activation, while highlighting the steep SAR challenges that can often be encountered in GPCR allosteric modulator discovery.
Co-reporter:Joseph D. Panarese, Hykeyung P. Cho, Jeffrey J. Adams, Kellie D. Nance, Pedro M. Garcia-Barrantes, Sichen Chang, Ryan D. Morrison, Anna L. Blobaum, Colleen M. Niswender, Shaun R. Stauffer, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3822-3825
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.04.083
This Letter describes the continued chemical optimization of the VU0453595 series of M1 positive allosteric modulators (PAMs). By surveying alternative 5,6- and 6,6-heterobicylic cores for the 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine-5-one core of VU453595, we found new cores that engendered not only comparable or improved M1 PAM potency, but significantly improved CNS distribution (Kps 0.3–3.1). Moreover, this campaign provided fundamentally distinct M1 PAM chemotypes, greatly expanding the available structural diversity for this valuable CNS target, devoid of hydrogen-bond donors.
Co-reporter:Pedro M. Garcia-Barrantes, Hyekyung P. Cho, Anna L. Blobaum, Colleen M. Niswender, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 8) pp:1869-1872
Publication Date(Web):15 April 2016
DOI:10.1016/j.bmcl.2016.03.031
This Letter describes the further lead optimization of the VU0486321 series of mGlu1 positive allosteric modulators (PAMs), focused on addressing the recurrent issue of plasma instability of the phthalimide moiety. Here, we evaluated a number of phthalimide bioisosteres, and ultimately identified isoindolinones as the ideal replacement that effectively address plasma instability, while maintaining acceptable mGlu1 PAM potency, DMPK profile, CNS penetration and mGluR selectivity.
Co-reporter:Michael R. Wood, Meredith J. Noetzel, James C. Tarr, Alice L. Rodriguez, Atin Lamsal, Sichen Chang, Jarrett J. Foster, Emery Smith, Peter Chase, Peter S. Hodder, Darren W. Engers, Colleen M. Niswender, Nicholas J. Brandon, Michael W. Wood, Mark E. Duggan, P. Jeffrey Conn, Thomas M. Bridges, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 17) pp:4282-4286
Publication Date(Web):1 September 2016
DOI:10.1016/j.bmcl.2016.07.042
This Letter describes the chemical optimization of a novel series of M4 PAMs based on a non-enolizable ketone core, identified from an MLPCN functional high-throughput screen. The HTS hit was potent, selective and CNS penetrant; however, the compound was highly cleared in vitro and in vivo. SAR provided analogs for which M4 PAM potency and CNS exposure were maintained; yet, clearance remained high. Metabolite identification studies demonstrated that this series was subject to rapid, and near quantitative, reductive metabolism to the corresponding secondary alcohol metabolite that was devoid of M4 PAM activity.
Co-reporter:Rocco D. Gogliotti, Darren W. Engers, Pedro M. Garcia-Barrantes, Joseph D. Panarese, Patrick R. Gentry, Anna L. Blobaum, Ryan D. Morrison, J. Scott Daniels, Analisa D. Thompson, Carrie K. Jones, P. Jeffrey Conn, Colleen M. Niswender, Craig W. Lindsley, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 12) pp:2915-2919
Publication Date(Web):15 June 2016
DOI:10.1016/j.bmcl.2016.04.041
This letter describes the further chemical optimization of the picolinamide-derived family of mGlu4 PAMs wherein we identified a 3-amino substituent to the picolinamide warhead that engendered potency, CNS penetration and in vivo efficacy. From this optimization campaign, VU0477886 emerged as a potent (EC50 = 95 nM, 89% Glu Max) mGlu4 PAM with an attractive DMPK profile (brain:plasma Kp = 1.3), rat CLp = 4.0 mL/min/kg, t1/2 = 3.7 h) and robust efficacy in our standard preclinical Parkinson’s disease model, haloperidol-induced catalepsy (HIC).
Co-reporter:Pedro M. Garcia-Barrantes, Joel R. Harp, Craig W. Lindsley
Tetrahedron Letters 2016 Volume 57(Issue 20) pp:2194-2196
Publication Date(Web):18 May 2016
DOI:10.1016/j.tetlet.2016.04.019
•First total synthesis of actinophenanthroline A.•A 6-step, protecting-group-free synthesis with a 24.2% overall yield.•Structure conforms to spectral data and confirmed by single X-ray crystallography.In this Letter, we describe the first total synthesis of actinophenanthroline A, an alkaloid from a marine actinobacterium with an unprecedented 1,7-phenanthroline core, that has been achieved. Highlights of the protecting-group-free synthesis feature two regioselective nitrosylation/reduction sequences and a Brønsted acid-catalyzed Doebner–Miller reaction. The synthesis required only six linear steps and proceeded in 24.2% overall yield, paving the way for future analog synthesis and biological studies.
Co-reporter:Pedro M. Garcia-Barrantes, Hyekyung P. Cho, Adam M. Metts, Anna L. Blobaum, Colleen M. Niswender, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 3) pp:751-756
Publication Date(Web):1 February 2016
DOI:10.1016/j.bmcl.2015.12.104
This Letter describes the further lead optimization of the VU0486321 series of mGlu1 positive allosteric modulators (PAMs), driven by recent genetic data linking loss of function GRM1 to schizophrenia. Steep and caveat-laden SAR plagues the series, but ultimately potent mGlu1 PAMs (EC50s ∼5 nM) have resulted with good DMPK properties (low intrinsic clearance, clean CYP profile, modest Fu) and CNS penetration (Kps 0.25–0.97), along with up to >450-fold selectivity versus mGlu4 and mGlu5.
Co-reporter:Jeanette L. Bertron, Elizabeth A. Ennis, Christopher J. Tarr, Jane Wright, Jonathan W. Dickerson, Charles W. Locuson, Anna L. Blobaum, Jerri M. Rook, Randy D. Blakely, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 19) pp:4637-4640
Publication Date(Web):1 October 2016
DOI:10.1016/j.bmcl.2016.08.062
This Letter describes the further lead optimization of the CHT inhibitor probe, ML352 (VU0476201), and the development of VU6001221, an improved in vivo tool. A multi-dimensional optimization effort encountered steep SAR, and ultimately, subtle tuning of the electronics of the central phenyl core provided VU6001221, a CHT inhibitor with comparable potency for choline uptake inhibition as ML352, yet improved PK and CNS penetration. Moreover, VU6001221 enabled evaluation, for the first time, of a CHT inhibitor in a standard preclinical rodent cognition model, novel object recognition (NOR). We observed VU6001221 to elicit a dose-responsive increase in NOR, raising the possibility of agonism of synaptic α7 nicotinic ACh receptors by elevated extracellular choline, that if confirmed would represent a novel molecular strategy to enhance cognition.
Co-reporter:Susana Conde-Ceide, Jesús Alcázar, Sergio A. Alonso de Diego, Silvia López, María Luz Martín-Martín, Carlos M. Martínez-Viturro, Miguel-Angel Pena, Han Min Tong, Hilde Lavreysen, Claire Mackie, Thomas M. Bridges, J. Scott Daniels, Colleen M. Niswender, Carrie K. Jones, Gregor J. Macdonald, Thomas Steckler, P. Jeffrey Conn, Shaun R. Stauffer, Craig W. Lindsley, José Manuel Bartolomé-Nebreda
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:429-434
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.11.098
As part of our efforts to identify a suitable back-up compound to our recently disclosed mGlu5 positive allosteric modulator (PAM) clinical candidate VU0490551/JNJ-46778212, this letter details the investigation and challenges of a novel series of 6,7-dihydropyrazolo[1,5-a]pyrazin-4-one derivatives. From these efforts, compound 4k emerged as a potent and selective mGlu5 PAM displaying overall attractive in vitro (pharmacological and ADMET) and PK profiles combined with in vivo efficacy in preclinical models of schizophrenia. However, further advancement of the compound was precluded due to severely limiting CNS-related side-effects confirming the previously reported association between excessive mGlu5 activation and target-related toxicities.
Co-reporter:Alexander R. Geanes, Hykeyung P. Cho, Kellie D. Nance, Kevin M. McGowan, P. Jeffrey Conn, Carrie K. Jones, Jens Meiler, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 18) pp:4487-4491
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmcl.2016.07.071
This Letter describes a ligand-based virtual screening campaign utilizing SAR data around the M5 NAMs, ML375 and VU6000181. Both QSAR and shape scores were employed to virtually screen a 98,000-member compound library. Neither approach alone proved productive, but a consensus score of the two models identified a novel scaffold which proved to be a modestly selective, but weak inhibitor (VU0549108) of the M5 mAChR (M5 IC50 = 6.2 μM, M1–4 IC50s >10 μM) based on an unusual 8-((1,3,5-trimethyl-1H-pyrazol-4-yl)sulfonyl)-1-oxa-4-thia-8-azaspiro[4,5]decane scaffold. [3H]-NMS binding studies showed that VU0549108 interacts with the orthosteric site (Ki of 2.7 μM), but it is not clear if this is negative cooperativity or orthosteric binding. Interestingly, analogs synthesized around VU0549108 proved weak, and SAR was very steep. However, this campaign validated the approach and warranted further expansion to identify additional novel chemotypes.
Co-reporter:Kayla J. Temple, Matthew T. Duvernay, Jae G. Maeng, Anna L. Blobaum, Shaun R. Stauffer, Heidi E. Hamm, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 22) pp:5481-5486
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmcl.2016.10.020
This letter describes the further deconstruction of the known PAR4 inhibitor chemotypes (MWs 490–525 and with high plasma protein binding) to identify a minimum PAR4 pharmacophore devoid of metabolic liabilities and improved properties. This exercise identified a greatly simplified 2-methoxy-6-arylimidazo[2,1-b][1,3,4]thiadiazole scaffold that afforded nanomolar inhibition of both activating peptide and γ-thrombin mediated PAR4 stimulation, while reducing both molecular weight and the number of hydrogen bond donors/acceptors by ∼50%. This minimum PAR4 pharmacophore, with competitive inhibition, versus non-competitive of the larger chemotypes, allows an ideal starting point to incorporate desired functional groups to engender optimal DMPK properties towards a preclinical candidate.
Co-reporter:Michael R. Wood, Meredith J. Noetzel, Julie L. Engers, Katrina A. Bollinger, Bruce J. Melancon, James C. Tarr, Changho Han, Mary West, Alison R. Gregro, Atin Lamsal, Sichen Chang, Sonia Ajmera, Emery Smith, Peter Chase, Peter S. Hodder, Michael Bubser, Carrie K. Jones, Corey R. Hopkins, Kyle A. Emmitte, Colleen M. Niswender, Michael W. Wood, et al.
Bioorganic & Medicinal Chemistry Letters 2016 26(13) pp: 3029-3033
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmcl.2016.05.010
This Letter describes the chemical optimization of a novel series of M4 positive allosteric modulators (PAMs) based on a 5,6-dimethyl-4-(piperidin-1-yl)thieno[2,3-d]pyrimidine core, identified from an MLPCN functional high-throughput screen. The HTS hit was potent and selective, but not CNS penetrant. Potency was maintained, while CNS penetration was improved (rat brain:plasma Kp = 0.74), within the original core after several rounds of optimization; however, the thieno[2,3-d]pyrimidine core was subject to extensive oxidative metabolism. Ultimately, we identified a 6-fluoroquinazoline core replacement that afforded good M4 PAM potency, muscarinic receptor subtype selectivity and CNS penetration (rat brain:plasma Kp > 10). Moreover, this campaign provided fundamentally distinct M4 PAM chemotypes, greatly expanding the available structural diversity for this exciting CNS target.
Co-reporter:Pedro M. Garcia-Barrantes; Hyekyung P. Cho; Colleen M. Niswender; Frank W. Byers; Charles W. Locuson; Anna L. Blobaum; Zixiu Xiang; Jerri M. Rook; P. Jeffrey Conn
Journal of Medicinal Chemistry 2015 Volume 58(Issue 20) pp:7959-7971
Publication Date(Web):October 1, 2015
DOI:10.1021/acs.jmedchem.5b00727
The therapeutic potential of selective mGlu1 activation is vastly unexplored relative to the other group I mGlu receptor, mGlu5; therefore, our lab has focused considerable effort toward developing mGlu1 positive allosteric modulators (PAMs) suitable as in vivo proof of concept tool compounds. Optimization of a series of mGlu1 PAMs based on an N-(3-chloro-4-(1,3-dioxoisoindolin-2-yl)phenyl)-3-methylfuran-2-carboxamide scaffold provided 17e, a potent (mGlu1 EC50 = 31.8 nM) and highly CNS penetrant (brain to plasma ratio (Kp) of 1.02) mGlu1 PAM tool compound, that potentiated not only wild-type human mGlu1 but also mutant mGlu1 receptors derived from deleterious GRM1 mutations found in schizophrenic patients. Moreover, both electrophysiological and in vivo studies indicate the mGlu1 ago-PAMs/PAMs do not possess the same epileptiform adverse effect liability as mGlu5 ago-PAMs/PAMs and maintain temporal activity suggesting a broader therapeutic window.
Co-reporter:Sarah A. Scott, Cierra T. Spencer, Matthew C. O’Reilly, Kyle A. Brown, Robert R. Lavieri, Chul-Hee Cho, Dai-Il Jung, Richard C. Larock, H. Alex Brown, and Craig W. Lindsley
ACS Chemical Biology 2015 Volume 10(Issue 2) pp:421
Publication Date(Web):November 10, 2014
DOI:10.1021/cb500828m
Phospholipase D (PLD) hydrolyses cellular lipids to produce the important lipid second messenger phosphatidic acid. A PLD enzyme expressed by Pseudomonas aeruginosa (PldA) has been shown to be important in bacterial infection, and NAPE-PLD has emerged as being key in the synthesis of endocannabinoids. In order to better understand the biology and therapeutic potential of these less explored PLD enzymes, small molecule tools are required. Selective estrogen receptor modulators (SERMs) have been previously shown to inhibit mammalian PLD (PLD1 and PLD2). By targeted screening of a library of SERM analogues, additional parallel synthesis, and evaluation in multiple PLD assays, we discovered a novel desketoraloxifene-based scaffold that inhibited not only the two mammalian PLDs but also structurally divergent PldA and NAPE-PLD. This finding represents an important first step toward the development of small molecules possessing universal inhibition of divergent PLD enzymes to advance the field.
Co-reporter:Susana Conde-Ceide, Carlos M. Martínez-Viturro, Jesús Alcázar, Pedro M. Garcia-Barrantes, Hilde Lavreysen, Claire Mackie, Paige N. Vinson, Jerri M. Rook, Thomas M. Bridges, J. Scott Daniels, Anton Megens, Xavier Langlois, Wilhelmus H. Drinkenburg, Abdellah Ahnaou, Colleen M. Niswender, Carrie K. Jones, Gregor J. Macdonald, Thomas Steckler, P. Jeffrey Conn, Shaun R. Stauffer, José Manuel Bartolomé-Nebreda, and Craig W. Lindsley
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 6) pp:716
Publication Date(Web):May 20, 2015
DOI:10.1021/acsmedchemlett.5b00181
Herein, we report the structure–activity relationship of a novel series of (2(phenoxymethyl)-6,7-dihydrooxazolo[5,4-c]pyridine-5(4H)-yl(aryl)methanones as potent, selective, and orally bioavailable metabotropic glutamate receptor subtype 5 (mGlu5) positive allosteric modulators (PAMs). On the basis of its robust in vitro potency and in vivo efficacy in multiple preclinical models of multiple domains of schizophrenia, coupled with a good DMPK profile and an acceptable therapeutic window, 17a (VU0409551/JNJ-46778212) was selected as a candidate for further development.Keywords: 4,5,6,7-tetrahydrooxazolo[5,4-c]pyridine; JNJ-46778212; metabotropic glutamate receptor 5 (mGlu5); Positive allosteric modulator (PAM); schizophrenia; VU0409551
Co-reporter:Haruto Kurata, Patrick R. Gentry, Masaya Kokubo, Hyekyung P. Cho, Thomas M. Bridges, Colleen M. Niswender, Frank W. Byers, Michael R. Wood, J. Scott Daniels, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 3) pp:690-694
Publication Date(Web):1 February 2015
DOI:10.1016/j.bmcl.2014.11.082
This Letter describes the continued optimization of the MLPCN probe ML375, a highly selective M5 negative allosteric modulator (NAM), through a combination of matrix libraries and iterative parallel synthesis. True to certain allosteric ligands, SAR was shallow, and the matrix library approach highlighted the challenges with M5 NAM SAR within in this chemotype. Once again, enantiospecific activity was noted, and potency at rat and human M5 were improved over ML375, along with slight enhancement in physiochemical properties, certain in vitro DMPK parameters and CNS distribution. Attempts to further enhance pharmacokinetics with deuterium incorporation afforded mixed results, but pretreatment with a pan-P450 inhibitor (1-aminobenzotriazole; ABT) provided increased plasma exposure.
Co-reporter:Ya Zhou, Chrysa Malosh, Susana Conde-Ceide, Carlos Manuel Martínez-Viturro, Jesus Alcázar, Hilde Lavreysen, Claire Mackie, Thomas M. Bridges, J. Scott Daniels, Colleen M. Niswender, Carrie K. Jones, Gregor J. Macdonald, Thomas Steckler, P. Jeffrey Conn, Shaun R. Stauffer, José Manuel Bartolomé-Nebreda, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3515-3519
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.06.096
This Letter describes the progress and challenges in the continued optimization of the mGlu5 positive allosteric modulator (PAM) clinical candidate VU0490551/JNJ-46778212. While many analogs addressed key areas for improvement, no one compound possessed the amalgamation of improvements needed within the (2(phenoxymethyl)-6,7-dihydrooxazolo[5,4-c]pyridine-5(4H)-yl(aryl)methanone scaffold to advance as a back-up clinical candidate. However, many analogs displayed excellent solubility and physiochemical properties, and were active in the amphetamine-induced hyperlocomotion (AHL) model. Moreover, the SAR was robust for this series of PAMs, and both polar and hydrogen-bond donors were found to be tolerated, leading to analogs with overall attractive profiles and good ligand efficiencies.
Co-reporter:Timothy J. Senter, Matthew C. O’Reilly, Katherine M. Chong, Gary A. Sulikowski, Craig W. Lindsley
Tetrahedron Letters 2015 Volume 56(Issue 10) pp:1276-1279
Publication Date(Web):4 March 2015
DOI:10.1016/j.tetlet.2015.01.140
A short, high-yielding protocol involving the enantioselective α-chlorination of aldehydes has been developed for the enantioselective synthesis of C2-functionalized aziridines and N-alkyl terminal azetidines from a common intermediate. This methodology allows for the rapid preparation of functionalized aziridines in 50–73% overall yields and 88–94% ee, and azetidines in 22–32% overall yields and 84–92% ee. Moreover, we developed a scalable and cost-effective route to the key organocatalyst (54% overall yield, >95% dr).
Co-reporter:Pedro M. Garcia-Barrantes, Hyekyung P. Cho, Anna L. Blobaum, Colleen M. Niswender, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5107-5110
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.10.013
Co-reporter:Lindsey C. Morris ; Kellie D. Nance ; Patrick R. Gentry ; Emily L. Days ; C. David Weaver ; Colleen M. Niswender ; Analisa D. Thompson ; Carrie K. Jones ; Chuck W. Locuson ; Ryan D. Morrison ; J. Scott Daniels ; Kevin D. Niswender
Journal of Medicinal Chemistry 2014 Volume 57(Issue 23) pp:10192-10197
Publication Date(Web):November 25, 2014
DOI:10.1021/jm501375c
A duplexed, functional multiaddition high throughput screen and subsequent iterative parallel synthesis effort identified the first highly selective and CNS penetrant glucagon-like peptide-1R (GLP-1R) positive allosteric modulator (PAM). PAM (S)-9b potentiated low-dose exenatide to augment insulin secretion in primary mouse pancreatic islets, and (S)-9b alone was effective in potentiating endogenous GLP-1R to reverse haloperidol-induced catalepsy.
Co-reporter:Craig W. Lindsley
Journal of Medicinal Chemistry 2014 Volume 57(Issue 18) pp:7485-7498
Publication Date(Web):September 2, 2014
DOI:10.1021/jm5011786
The identification of sites on receptors topographically distinct from the orthosteric sites, so-called allosteric sites, has heralded novel approaches and modes of pharmacology for target modulation. Over the past 20 years, our understanding of allosteric modulation has grown significantly, and numerous advantages, as well as caveats (e.g., flat structure–activity relationships, species differences, “molecular switches”), have been identified. For multiple receptors and proteins, numerous examples have been described where unprecedented levels of selectivity are achieved along with improved physiochemical properties. While not a panacea, these novel approaches represent exciting opportunities for tool compound development to probe the pharmacology and therapeutic potential of discrete molecular targets, as well as new medicines. In this Perspective, in commemoration of the 2013 Philip S. Portoghese Medicinal Chemistry Lectureship (Lindsley, C. W.Adventures in allosteric drug discovery. Presented at the 246th National Meeting of the American Chemical Society, Indianapolis, IN, September 10, 2013; The 2013 Portoghese Lectureship), several vignettes of drug discovery campaigns targeting novel allosteric mechanisms will be recounted, along with lessons learned and guidelines that have emerged for successful lead optimization.
Co-reporter:Hyekyung P. Cho, Pedro M. Garcia-Barrantes, John T. Brogan, Corey R. Hopkins, Colleen M. Niswender, Alice L. Rodriguez, Daryl F. Venable, Ryan D. Morrison, Michael Bubser, J. Scott Daniels, Carrie K. Jones, P. Jeffrey Conn, and Craig W. Lindsley
ACS Chemical Biology 2014 Volume 9(Issue 10) pp:2334
Publication Date(Web):August 19, 2014
DOI:10.1021/cb500560h
Schizophrenia is a complex and highly heterogeneous psychiatric disorder whose precise etiology remains elusive. While genome-wide association studies (GWAS) have identified risk genes, they have failed to determine if rare coding single nucleotide polymorphisms (nsSNPs) contribute in schizophrenia. Recently, two independent studies identified 12 rare, deleterious nsSNPS in the GRM1 gene, which encodes the metabotropic glutamate receptor subtype 1 (mGlu1), in schizophrenic patients. Here, we generated stable cell lines expressing the mGlu1 mutant receptors and assessed their pharmacology. Using both the endogenous agonist glutamate and the synthetic agonist DHPG, we found that several of the mutant mGlu1 receptors displayed a loss of function that was not due to a loss in plasma membrane expression. Due to a lack of mGlu1 positive allosteric modulators (PAM) tool compounds active at human mGlu1, we optimized a known mGlu4 PAM/mGlu1 NAM chemotype into a series of potent and selective mGlu1 PAMs by virtue of a double “molecular switch”. Employing mGlu1 PAMs from multiple chemotypes, we demonstrate that the mutant receptors can be potentiated by small molecules and in some cases efficacy restored to that comparable to wild type mGlu1 receptors, suggesting deficits in patients with schizophrenia due to these mutations may be amenable to intervention with an mGlu1 PAM. However, in wild type animals, mGlu1 negative allosteric modulators (NAMs) are efficacious in classic models predictive of antipsychotic activity, whereas we show that mGlu1 PAMs have no effect to slight potentiation in these models. These data further highlight the heterogeneity of schizophrenia and the critical role of patient selection strategies in psychiatric clinical trials to match genotype with therapeutic mechanism.
Co-reporter:Cody J. Wenthur, Megan R. Bennett, and Craig W. Lindsley
ACS Chemical Neuroscience 2014 Volume 5(Issue 1) pp:14
Publication Date(Web):November 9, 2013
DOI:10.1021/cn400186j
Fluoxetine (Prozac) was the first major breakthrough for the treatment of depression since the introduction of tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) nearly 30 years earlier. It was the first selective serotonin reuptake inhibitor (SSRI) approved by the United States Food and Drug Administration, offering superior efficacy and reduced side effects relative to TCAs and MAOIs. Though a debate remains regarding the exact mechanism by which the clinical efficacy of fluoxetine is manifested, the importance of fluoxetine and related SSRIs to the field is unquestionable. The trade name Prozac has permeated popular culture, helping to raise awareness of depression and to diminish the prevalence of long-standing stigmas associated with this illness. In this Review, we will showcase the history and importance of fluoxetine to neuroscience in general, as well as for the treatment of depression, and review the synthesis, pharmacology, drug metabolism, and adverse effects of fluoxetine.Keywords: antidepressant; depression; Fluoxetine; Prozac; serotonin; SSRI
Co-reporter:Craig W. Lindsley
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 10) pp:1066
Publication Date(Web):September 5, 2014
DOI:10.1021/ml500351u
Pharmacoeconomics is a rational, scientific approach to compare the value (in terms of both cost and patient outcome) of one medication or drug therapy regimen to another. The impact of this new approach on both the practicing medicinal chemist and broader drug discovery efforts is considered.Keywords: cost benefit; outcomes; Pharmacoeconomics
Co-reporter:Matthew C. O’Reilly, Sarah A. Scott, H. Alex Brown, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 24) pp:5553-5557
Publication Date(Web):15 December 2014
DOI:10.1016/j.bmcl.2014.11.017
Co-reporter:Wandong Wen, Wenjun Wu, C. David Weaver, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 21) pp:5102-5106
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmcl.2014.08.061
This Letter describes the on-going SAR efforts based on ML297, a potent, efficacious and selective GIRK1/2 activator (∼10-fold vs GIRK1/4 and inactive on GIRK2/3) via an iterative parallel synthesis approach. The chemical optimization at the 3-position of pyrazole within ML297 indicated that various functionalized 3-cyclopropyl moieties modulated GIRK pharmacology between inhibitor/activator within a series of 1-(3-cyclopropyl-1-phenyl-1H-pyrazol-5-yl)ureas. Importantly, novel ‘molecular switches’ that modulated the mode of pharmacology from inhibitor to activator was discovered on both the 3-cyclopropyl and N-phenyl moiety of the pyrazole core, providing the first highly selective GIRK1/2 activator.
Co-reporter:Cody J. Wenthur, Ryan D. Morrison, J. Scott Daniels, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 12) pp:2693-2698
Publication Date(Web):15 June 2014
DOI:10.1016/j.bmcl.2014.04.051
Herein we report the design and synthesis of a series of substituted pyrazolo[1,5-a]quinazolin-5(4H)-ones as negative allosteric modulators of metabotropic glutamate receptors 2 and 3 (mGlu2 and mGlu3, respectively). Development of this series was initiated by reports that pyrazolo[1,5-a]quinazoline-derived scaffolds can yield compounds with activity at group II mGlu receptors which are prone to molecular switching following small structural changes. Several potent analogues, including 4-methyl-2-phenyl-8-(pyrimidin-5-yl)pyrazolo[1,5-a]quinazolin-5(4H)-one (10b), were discovered with potent in vitro activity as dual mGlu2/mGlu3 NAMs, with excellent selectivity versus the other mGluRs.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Carrie K. Jones, Douglas J. Sheffler, Richard Williams, Sataya B. Jadhav, Andrew S. Felts, Ryan D. Morrison, Colleen M. Niswender, J. Scott Daniels, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1067-1070
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2014.01.013
This Letter describes the development and SAR of a novel series of GlyT1 inhibitors derived from a scaffold hopping approach that provided a robust intellectual property position, in lieu of a traditional, expensive HTS campaign. Members within this new [3.1.0]-based series displayed excellent GlyT1 potency, selectivity, free fraction, CNS penetration and efficacy in a preclinical model of schizophrenia (prepulse inhibition).
Co-reporter:Douglas J. Sheffler, Michael T. Nedelovych, Richard Williams, Stephen C. Turner, Brittany B. Duerk, Megan R. Robbins, Sataya B. Jadhav, Colleen M. Niswender, Carrie K. Jones, P. Jeffrey Conn, R. Nathan Daniels, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 4) pp:1062-1066
Publication Date(Web):15 February 2014
DOI:10.1016/j.bmcl.2014.01.011
This Letter describes the development and SAR of a novel series of GlyT1 inhibitors derived from a scaffold hopping approach, in lieu of an HTS campaign, which provided intellectual property position. Members within this new [3.3.0]-based series displayed excellent GlyT1 potency, selectivity, free fraction, and modest CNS penetration. Moreover, enantioselective GlyT1 inhibition was observed, within this novel series and a number of other piperidine bioisosteric cores.
Co-reporter:Patrick R. Gentry;Dr. Masaya Kokubo;Dr. Thomas M. Bridges;Dr. Hyekyung P. Cho;Emery Smith;Peter Chase;Dr. Peter S. Hodder;Thomas J. Utley;Anuruddha Rajapakse;Frank Byers; Colleen M. Niswender;Ryan D. Morrison; J. Scott Daniels; Michael R. Wood; P. Jeffrey Conn; Craig W. Lindsley
ChemMedChem 2014 Volume 9( Issue 8) pp:1677-1682
Publication Date(Web):
DOI:10.1002/cmdc.201402051
Abstract
Of the five G-protein-coupled muscarinic acetylcholine receptors (mAChRs; M1–M5), M5 is the least explored and understood due to a lack of mAChR subtype-selective ligands. We recently performed a high-throughput functional screen and identified a number of weak antagonist hits that are selective for the M5 receptor. Here, we report an iterative parallel synthesis and detailed molecular pharmacologic profiling effort that led to the discovery of the first highly selective, central nervous system (CNS)-penetrant M5-orthosteric antagonist, with sub-micromolar potency (hM5 IC50=450 nM, hM5 Ki=340 nM, M1–M4 IC50 >30 μM), enantiospecific inhibition, and an acceptable drug metabolism and pharmacokinetics (DMPK) profile for in vitro and electrophysiology studies. This compound will be a powerful tool and molecular probe for the further investigation into the role of M5 in addiction and other diseases.
Co-reporter:Matthew C. O'Reilly;Thomas H. Oguin III;Dr. Sarah A. Scott; Paul G. Thomas;Dr. Charles W. Locuson;Ryan D. Morrison; J. Scott Daniels; H. Alex Brown; Craig W. Lindsley
ChemMedChem 2014 Volume 9( Issue 12) pp:2633-2637
Publication Date(Web):
DOI:10.1002/cmdc.201402333
Abstract
Further chemical optimization of the halopemide-derived family of dual phospholipase D1/2 (PLD1/2) inhibitors afforded ML395 (VU0468809), a potent, >80-fold PLD2 selective allosteric inhibitor (cellular PLD1, IC50>30 000 nM; cellular PLD2, IC50=360 nM). Moreover, ML395 possesses an attractive in vitro DMPK profile, improved physiochemical properties, ancillary pharmacology (Eurofins Panel) cleaner than any other reported PLD inhibitor, and has been found to possess interesting activity as an antiviral agent in cellular assays against a range of influenza strains (H1, H3, H5 and H7).
Co-reporter:Matthew C. O'Reilly;Thomas H. Oguin III;Dr. Sarah A. Scott; Paul G. Thomas;Dr. Charles W. Locuson;Ryan D. Morrison; J. Scott Daniels; H. Alex Brown; Craig W. Lindsley
ChemMedChem 2014 Volume 9( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201490047
Co-reporter:Matthew C. O’Reilly ; Sarah A. Scott ; Kyle A. Brown ; Thomas H. Oguin ; III; Paul G. Thomas ; J. Scott Daniels ; Ryan Morrison ; H. Alex Brown
Journal of Medicinal Chemistry 2013 Volume 56(Issue 6) pp:2695-2699
Publication Date(Web):February 27, 2013
DOI:10.1021/jm301782e
An iterative parallel synthesis effort identified a PLD2 selective inhibitor, ML298 (PLD1 IC50 > 20 000 nM, PLD2 IC50 = 355 nM) and a dual PLD1/2 inhibitor, ML299 (PLD1 IC50 = 6 nM, PLD2 IC50 = 20 nM). SAR studies revealed that a small structural change (incorporation of a methyl group) increased PLD1 activity within this classically PLD2-preferring core and that the effect was enantiospecific. Both probes decreased invasive migration in U87-MG glioblastoma cells.
Co-reporter:Cody J. Wenthur ; Ryan Morrison ; Andrew S. Felts ; Katrina A. Smith ; Julie L. Engers ; Frank W. Byers ; J. Scott Daniels ; Kyle A. Emmitte ; P. Jeffrey Conn
Journal of Medicinal Chemistry 2013 Volume 56(Issue 12) pp:5208-5212
Publication Date(Web):May 29, 2013
DOI:10.1021/jm400439t
A multidimensional, iterative parallel synthesis effort identified a series of highly selective mGlu3 NAMs with submicromolar potency and good CNS penetration. Of these, ML337 resulted (mGlu3 IC50 = 593 nM, mGlu2 IC50 >30 μM) with B:P ratios of 0.92 (mouse) to 0.3 (rat). DMPK profiling and shallow SAR led to the incorporation of deuterium atoms to address a metabolic soft spot, which subsequently lowered both in vitro and in vivo clearance by >50%.
Co-reporter:Patrick R. Gentry ; Masaya Kokubo ; Thomas M. Bridges ; Nathan R. Kett ; Joel M. Harp ; Hyekyung P. Cho ; Emery Smith ψ; Peter Chase ψ; Peter S. Hodder ψ; Colleen M. Niswender ; J. Scott Daniels ; P. Jeffrey Conn ; Michael R. Wood
Journal of Medicinal Chemistry 2013 Volume 56(Issue 22) pp:9351-9355
Publication Date(Web):October 28, 2013
DOI:10.1021/jm4013246
A functional high throughput screen and subsequent multidimensional, iterative parallel synthesis effort identified the first muscarinic acetylcholine receptor (mAChR) negative allosteric modulator (NAM) selective for the M5 subtype. ML375 is a highly selective M5 NAM with submicromolar potency (human M5 IC50 = 300 nM, rat M5 IC50 = 790 nM, M1–M4 IC50 > 30 μM), excellent multispecies PK, high CNS penetration, and enantiospecific inhibition.
Co-reporter:Cody J. Wenthur and Craig W. Lindsley
ACS Chemical Neuroscience 2013 Volume 4(Issue 7) pp:1018
Publication Date(Web):July 17, 2013
DOI:10.1021/cn400121z
Clozapine was the first true breakthrough in schizophrenia treatment since the discovery of chlorpromazine in 1950, effectively treating positive, negative, and some cognitive symptoms, as well as possessing unprecedented efficacy in treatment-resistant patients. Despite over 30 years of intense study, the precise molecular underpinnings that account for clozapine’s unique efficacy remain elusive. In this Viewpoint, we will showcase the history and importance of clozapine to neuroscience in general, as well as for the treatment of schizophrenia, and review the synthesis, pharmacology, drug metabolism, and adverse events of clozapine.Keywords: clozapine; N-desmethylclozapine; pharmacology; schizophrenia
Co-reporter:Leslie N. Aldrich;Cynthia B. Berry;Brittney S. Bates;Leah C. Konkol;Mira So
European Journal of Organic Chemistry 2013 Volume 2013( Issue 20) pp:4215-4218
Publication Date(Web):
DOI:10.1002/ejoc.201300643
Abstract
Herein, we describe the enantioselective construction of the 12-membered macrocyclic pyrrole core of marineosin A in 5.1 % overall yield from (S)-propylene oxide. The route features a key Stetter reaction to install a 1,4-diketone, which is subjected to Paal–Knorr pyrrole synthesis and ring-closing metathesis to afford the macrocycle. A divergence point in the synthetic scheme also enabled access to a highly functionalized spiroaminal model system through an acid-mediated hydroxy oxo amide cyclization strategy.
Co-reporter:Matthew C. O’Reilly, Craig W. Lindsley
Tetrahedron Letters 2013 Volume 54(Issue 28) pp:3627-3629
Publication Date(Web):10 July 2013
DOI:10.1016/j.tetlet.2013.04.116
In this Letter, we describe a short, high yielding protocol for the enantioselective (87–96% ee) and general synthesis of β-fluoroamines and previously difficult to access γ-fluoroamines from commercial aldehydes via organocatalysis.
Co-reporter:Timothy J. Senter, Michael L. Schulte, Leah C. Konkol, Tyler E. Wadzinski, Craig W. Lindsley
Tetrahedron Letters 2013 Volume 54(Issue 13) pp:1645-1648
Publication Date(Web):27 March 2013
DOI:10.1016/j.tetlet.2013.01.041
In this Letter, we describe a novel approach for the general and enantioselective synthesis of a diverse array of small to large 1-azabicyclo[m.n.0]alkyl ring systems with an embedded olefin handle for further functionalization. The stereochemistry is established via a highly diastereoselective indium-mediated allylation of an Ellman sulfinimine in greater than 9:1 dr, which is readily separable by column chromatography to afford a single diastereomer. This methodology allows for the rapid preparation of 1-azabicyclo[m.n.0]alkane ring systems that are not readily accessible through any other chemistry in excellent overall yields and, for many systems, the only enantioselective preparation reported to date.
Co-reporter:Joseph D. Panarese, Leah C. Konkol, Cynthia B. Berry, Brittney S. Bates, Leslie N. Aldrich, Craig W. Lindsley
Tetrahedron Letters 2013 Volume 54(Issue 18) pp:2231-2234
Publication Date(Web):1 May 2013
DOI:10.1016/j.tetlet.2013.02.059
In this Letter, we describe a short, six step enantioselective route to spiroaminal lactam model systems reminiscent of marineosins A and B that has been developed starting from either (R)- or (S)-hydroxysuccinic acid, respectively, in ∼9% overall yield. This route enables late stage incorporation of the pyrrole ring at C5 via nucleophilic displacement of an iminium triflate salt.
Co-reporter:Wandong Wen, Wenjun Wu, Ian M. Romaine, Kristian Kaufmann, Yu Du, Gary A. Sulikowski, C. David Weaver, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 16) pp:4562-4566
Publication Date(Web):15 August 2013
DOI:10.1016/j.bmcl.2013.06.023
This letter describes a multi-dimensional SAR campaign based on a potent, efficacious and selective GIRK1/2 activator (∼10-fold versus GIRK1/4 and inactive on nonGIRK 1-containing GIRKs, GIRK 2 or GIRK2/3). Further chemical optimization through an iterative parallel synthesis effort identified multiple ‘molecular switches’ that modulated the mode of pharmacology from activator to inhibitor, as well as engendering varying selectivity profiles for GIRK1/2 and GIRK1/4. Importantly, these compounds were all inactive on nonGIRK1 containing GIRK channels. However, SAR was challenging as subtle structural modifications had large effects on both mode of pharmacology and GIRK1/2 and GIRK1/4 channel selectivity.
Co-reporter:Patrick R. Gentry, Thomas M. Bridges, Atin Lamsal, Paige N. Vinson, Emery Smith, Peter Chase, Peter S. Hodder, Julie L. Engers, Colleen M. Niswender, J. Scott Daniels, P. Jeffrey Conn, Michael R. Wood, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 10) pp:2996-3000
Publication Date(Web):15 May 2013
DOI:10.1016/j.bmcl.2013.03.032
This Letter describes the further chemical optimization of the M5 PAM MLPCN probes ML129 and ML172. A multi-dimensional iterative parallel synthesis effort quickly explored isatin replacements and a number of southern heterobiaryl variations with no improvement over ML129 and ML172. An HTS campaign identified several weak M5 PAMs (M5 EC50 >10 μM) with a structurally related isatin core that possessed a southern phenethyl ether linkage. While SAR within the HTS series was very shallow and unable to be optimized, grafting the phenethyl ether linkage onto the ML129/ML172 cores led to the first sub-micromolar M5 PAM, ML326 (VU0467903), (human and rat M5 EC50s of 409 nM and 500 nM, respectively) with excellent mAChR selectivity (M1–M4 EC50s >30 μM) and a robust 20-fold leftward shift of the ACh CRC.
Co-reporter:Douglas J. Sheffler, Christian Sevel, Uyen Le, Kimberly M. Lovell, James C. Tarr, Sheridan J.S. Carrington, Hyekyung P. Cho, Gregory J. Digby, Colleen M. Niswender, P. Jeffrey Conn, Corey R. Hopkins, Michael R. Wood, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 1) pp:223-227
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmcl.2012.10.132
This letter describes the further exploration of two series of M1 allosteric agonists, TBPB and VU0357017, previously reported from our lab. Within the TPBP scaffold, either electronic or steric perturbations to the central piperidine ring led to a loss of selective M1 allosteric agonism and afforded pan-mAChR antagonism, which was demonstrated to be mediated via the orthosteric site. Additional SAR around a related M1 allosteric agonist family (VU0357017) identified similar, subtle ‘molecular switches’ that modulated modes of pharmacology from allosteric agonism to pan-mAChR orthosteric antagonism. Therefore, all of these ligands are best classified as bi-topic ligands that possess high affinity binding at an allosteric site to engender selective M1 activation, but all bind, at higher concentrations, to the orthosteric ACh site, leading to non-selective orthosteric site binding and mAChR antagonism.
Co-reporter:Dr. Michael L. Schulte;Dr. Mark L. Turlington;Dr. Sharangdhar S. Phatak;Dr. Joel M. Harp;Dr. Shaun R. Stauffer; Craig W. Lindsley
Chemistry - A European Journal 2013 Volume 19( Issue 36) pp:11847-11852
Publication Date(Web):
DOI:10.1002/chem.201302669
Co-reporter:Timothy J. Senter, Olugbeminiyi O. Fadeyi, and Craig W. Lindsley
Organic Letters 2012 Volume 14(Issue 7) pp:1869-1871
Publication Date(Web):March 20, 2012
DOI:10.1021/ol300466a
The first total synthesis of (+)-amabiline, an unsaturated pyrrolizidine alkaloid from Cynoglossum amabile, is reported. This convergent, enantioselective synthesis proceeds in 15 steps (10-step longest linear sequence) in 6.2% overall yield and features novel methodology to construct the unsaturated pyrrolizidine or (−)-supinidine core.
Co-reporter:Matthew C. O’Reilly and Craig W. Lindsley
Organic Letters 2012 Volume 14(Issue 11) pp:2910-2913
Publication Date(Web):May 22, 2012
DOI:10.1021/ol301203z
A short, high yielding protocol has been developed for the enantioselective and general synthesis of C2-functionalized, benzyl protected morpholines and orthogonally N,N′-protected piperazines from a common intermediate.
Co-reporter:Joseph D. Panarese and Craig W. Lindsley
Organic Letters 2012 Volume 14(Issue 22) pp:5808-5810
Publication Date(Web):October 29, 2012
DOI:10.1021/ol3024665
The first total synthesis of Aplidiopsamine A, a rare 3H-pyrrolo[2,3-c]quinoline alkaloid from the Aplidiopsis confluata, has been achieved following the proposed biosynthesis. This biomimetic synthesis requires only five steps and proceeds in 20.8% overall yield. Biological evaluation across large panels of discrete molecular targets identified that Aplidiopsamine A is a highly selective PDE4 inhibitor, a target for numerous CNS disorders.
Co-reporter:Bruce J. Melancon ; Corey R. Hopkins ; Michael R. Wood ; Kyle A. Emmitte ; Colleen M. Niswender ; Arthur Christopoulos ; P. Jeffrey Conn
Journal of Medicinal Chemistry 2012 Volume 55(Issue 4) pp:1445-1464
Publication Date(Web):December 9, 2011
DOI:10.1021/jm201139r
Co-reporter:John T. Brogan, Sydney L. Stoops, and Craig W. Lindsley
ACS Chemical Neuroscience 2012 Volume 3(Issue 9) pp:658
Publication Date(Web):June 27, 2012
DOI:10.1021/cn300064r
The synthesis of phidianidines A and B, the first 1,2,4-oxadiazole-containing alkaloid, from the marine opisthobranch mollusk Phidiana militaris is reported. The synthesis proceeds in six steps from known indole acetic acids in 39.9% (phidianidine A) and 21% (phidianidine B) overall yields from commercially available materials. Biological characterization found that phidianidines A and B are selective inhibitors of DAT (versus SERT and NET) and a selective, potent ligand and partial agonist of the μ opioid receptor (versus δ- and κ-opioid receptors). Moreover, neither phidianidines A and B are cytotoxic, and thus represent an attractive starting point for chemical optimization; therefore, we piloted a number of chemistries and prepared a diverse series of unnatural analogs.
Co-reporter:Kristopher N. Hahn, Olugbeminiyi O. Fadeyi, Hyekyung P. Cho, Craig W. Lindsley
Tetrahedron Letters 2012 Volume 53(Issue 28) pp:3577-3580
Publication Date(Web):11 July 2012
DOI:10.1016/j.tetlet.2012.05.009
In this Letter, we describe the first total synthesis of cremastrine, a pyrrolizidine alkaloid from Cremastra appendiculata, with anticholinergic activity as well as an unnatural analogue. The streamlined synthesis proceeds in nine steps, seven steps longest linear sequence, in 25.2% overall yield, and features novel methodology to construct the pyrrolizidine core. Biological evaluation of cremastrine and the unnatural analogue indicated that both are pan-mAChR functional antagonists.
Co-reporter:Matthew C. O’Reilly, Craig W. Lindsley
Tetrahedron Letters 2012 Volume 53(Issue 13) pp:1539-1542
Publication Date(Web):28 March 2012
DOI:10.1016/j.tetlet.2011.12.063
In this Letter, we describe a novel three-step, one-pot procedure for the enantioselective synthesis of N-benzyl protected morpholines and orthogonally N,N′-protected piperazines with chiral alkyl groups installed at the C2 position of each heterocyclic core via organocatalysis. This methodology allows for the rapid preparation of functionalized morpholines and piperazines that are not readily accessible through any other chemistry in good to excellent % ee (55–98% ee).
Co-reporter:Eric Delpire, Aleksandra Baranczak, Alex G. Waterson, Kwangho Kim, Nathan Kett, Ryan D. Morrison, J. Scott Daniels, C. David Weaver, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4532-4535
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.126
Further chemical optimization of the MLSCN/MLPCN probe ML077 (KCC2 IC50 = 537 nM) proved to be challenging as the effort was characterized by steep SAR. However, a multi-dimensional iterative parallel synthesis approach proved productive. Herein we report the discovery and SAR of an improved novel antagonist (VU0463271) of the neuronal-specific potassium-chloride cotransporter 2 (KCC2), with an IC50 of 61 nM and >100-fold selectivity versus the closely related Na-K-2Cl cotransporter 1 (NKCC1) and no activity in a larger panel of GPCRs, ion channels and transporters.
Co-reporter:Douglas J. Sheffler, Cody J. Wenthur, Joshua A. Bruner, Sheridan J.S. Carrington, Paige N. Vinson, Kiran K. Gogi, Anna L. Blobaum, Ryan D. Morrison, Mitchell Vamos, Nicholas D.P. Cosford, Shaun R. Stauffer, J. Scott Daniels, Colleen M. Niswender, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:3921-3925
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.112
Herein we report the discovery and SAR of a novel metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) with 15-fold selectivity versus mGlu2. The mGlu3 NAM was discovered via a ‘molecular switch’ from a closely related, potent mGlu5 positive allosteric modulator (PAM), VU0092273. This NAM (VU0463597, ML289) displays an IC50 value of 0.66 μM and is inactive against mGlu5.
Co-reporter:Dr. Olugbeminiyi O. Fadeyi;Timothy J. Senter;Kristopher N. Hahn; Craig W. Lindsley
Chemistry - A European Journal 2012 Volume 18( Issue 19) pp:5826-5831
Publication Date(Web):
DOI:10.1002/chem.201200629
Co-reporter:Dr. Ralf Mueller;Dr. Eric S. Dawson; Jens Meiler;Dr. Alice L. Rodriguez;Dr. Brian A. Chauder;Brittney S. Bates;Dr. Andrew S. Felts;Jeffrey P. Lamb;Usha N. Menon;Dr. Sataywan B. Jadhav;Dr. Alexer S. Kane;Dr. Carrie K. Jones;Dr. Karen J. Gregory; Colleen M. Niswender; P. Jeffrey Conn;Dr. Christopher M. Olsen; Danny G. Winder; Kyle A. Emmitte; Craig W. Lindsley
ChemMedChem 2012 Volume 7( Issue 3) pp:406-414
Publication Date(Web):
DOI:10.1002/cmdc.201100510
Co-reporter:Sydney L. Stoops, A. Scott Pearson, Connie Weaver, Alex G. Waterson, Emily Days, Chris Farmer, Suzanne Brady, C. David Weaver, R. Daniel Beauchamp, and Craig W. Lindsley
ACS Chemical Biology 2011 Volume 6(Issue 5) pp:452
Publication Date(Web):January 18, 2011
DOI:10.1021/cb100305h
E-cadherin is a transmembrane protein that maintains intercellular contacts and cell polarity in epithelial tissue. The down-regulation of E-cadherin contributes to the induction of the epithelial-to-mesenchymal transition (EMT), resulting in an increased potential for cellular invasion of surrounding tissues and entry into the bloodstream. Loss of E-cadherin has been observed in a variety of human tumors as a result of somatic mutations, chromosomal deletions, silencing of the CDH1 gene promoter, and proteolytic cleavage. To date, no compounds directly targeting E-cadherin restoration have been developed. Here, we report the development and use of a novel high-throughput immunofluorescent screen to discover lead compounds that restore E-cadherin expression in the SW620 colon adenocarcinoma cell line. We confirmed restoration of E-cadherin using immunofluorescent microscopy and were able to determine the EC50 for selected compounds using an optimized In-Cell Western assay. The profiled compounds were also shown to have a minimal effect on cell proliferation but did decrease cellular invasion. We have also conducted preliminary investigations to elucidate a discrete molecular target to account for the phenotypic behavior of these small molecules and have noted a modest increase in E-cadherin mRNA transcripts, and RNA-Seq analysis demonstrated that potent analogues elicited a 10-fold increase in CDH1 (E-cadherin) gene expression.
Co-reporter:Hao-Ran Wang, Meng Wu, Haibo Yu, Shunyou Long, Amy Stevens, Darren W. Engers, Henry Sackin, J. Scott Daniels, Eric S. Dawson, Corey R. Hopkins, Craig W. Lindsley, Min Li, and Owen B. McManus
ACS Chemical Biology 2011 Volume 6(Issue 8) pp:845
Publication Date(Web):May 26, 2011
DOI:10.1021/cb200146a
The Kir inward rectifying potassium channels have a broad tissue distribution and are implicated in a variety of functional roles. At least seven classes (Kir1–Kir7) of structurally related inward rectifier potassium channels are known, and there are no selective small molecule tools to study their function. In an effort to develop selective Kir2.1 inhibitors, we performed a high-throughput screen (HTS) of more than 300,000 small molecules within the MLPCN for modulators of Kir2.1 function. Here we report one potent Kir2.1 inhibitor, ML133, which inhibits Kir2.1 with an IC50 of 1.8 μM at pH 7.4 and 290 nM at pH 8.5 but exhibits little selectivity against other members of Kir2.x family channels. However, ML133 has no effect on Kir1.1 (IC50 > 300 μM) and displays weak activity for Kir4.1 (76 μM) and Kir7.1 (33 μM), making ML133 the most selective small molecule inhibitor of the Kir family reported to date. Because of the high homology within the Kir2 family—the channels share a common design of a pore region flanked by two transmembrane domains—identification of site(s) critical for isoform specificity would be an important basis for future development of more specific and potent Kir inhibitors. Using chimeric channels between Kir2.1 and Kir1.1 and site-directed mutagenesis, we have identified D172 and I176 within M2 segment of Kir2.1 as molecular determinants critical for the potency of ML133 mediated inhibition. Double mutation of the corresponding residues of Kir1.1 to those of Kir2.1 (N171D and C175I) transplants ML133 inhibition to Kir1.1. Together, the combination of a potent, Kir2 family selective inhibitor and identification of molecular determinants for the specificity provides both a tool and a model system to enable further mechanistic studies of modulation of Kir2 inward rectifier potassium channels.
Co-reporter:John T. Brogan, Sydney L. Stoops, Brenda C. Crews, Lawrence J. Marnett, and Craig W. Lindsley
ACS Chemical Neuroscience 2011 Volume 2(Issue 11) pp:633
Publication Date(Web):August 13, 2011
DOI:10.1021/cn200075n
The first total synthesis of (+)-7-bromotrypargine, a β-carboline alkaloid from Ancornia sp. is reported. The synthesis proceeds in nine steps, eight steps longest linear sequence, in 36.9% overall yield. Biological characterization found that (+)-7-bromotrypargine is a H3 antagonist, and a selective inhibitor of the dopamine transporter (DAT) and norepinephrine transporter (NET), without inhibiting the serotonin transporter (SERT). Moreover, unlike electron rich congeners, (+)-7-bromotrypargine is not cytotoxic and thus represents an attractive starting point for chemical optimization; therefore, we piloted a number of chemistries for the synthesis of unnatural analogues.
Co-reporter:Zixiu Xiang, Analisa D. Thompson, John T. Brogan, Michael L. Schulte, Bruce J. Melancon, Debbie Mi, L. Michelle Lewis, Bende Zou, Liya Yang, Ryan Morrison, Tammy Santomango, Frank Byers, Katrina Brewer, Jonathan S. Aldrich, Haibo Yu, Eric S. Dawson, Min Li, Owen McManus, Carrie K. Jones, J. Scott Daniels, Corey R. Hopkins, Ximin Simon Xie, P. Jeffrey Conn, C. David Weaver, and Craig W. Lindsley
ACS Chemical Neuroscience 2011 Volume 2(Issue 12) pp:730
Publication Date(Web):October 17, 2011
DOI:10.1021/cn200090z
T-Type Ca2+ channel inhibitors hold tremendous therapeutic potential for the treatment of pain, epilepsy, sleep disorders, essential tremor, and other neurological disorders; however, a lack of truly selective tools has hindered basic research, and selective tools from the pharmaceutical industry are potentially burdened with intellectual property (IP) constraints. Thus, an MLPCN high-throughput screen (HTS) was conducted to identify novel T-type Ca2+ channel inhibitors free from IP constraints, and freely available through the MLPCN, for use by the biomedical community to study T-type Ca2+ channels. While the HTS provided numerous hits, these compounds could not be optimized to the required level of potency to be appropriate tool compounds. Therefore, a scaffold hopping approach, guided by SurflexSim, ultimately afforded ML218 (CID 45115620), a selective T-type Ca2+ (Cav3.1, Cav3.2, Cav3.3) inhibitor (Cav3.2, IC50 = 150 nM in Ca2+ flux; Cav3.2 IC50 = 310 nM; and Cav3.3 IC50 = 270 nM, respectively in patch clamp electrophysiology) with good DMPK properties, acceptable in vivo rat PK, and excellent brain levels. Electrophysiology studies in subthalamic nucleus (STN) neurons demonstrated robust effects of ML218 on the inhibition of T-type calcium current, inhibition of low threshold spike, and rebound burst activity. Based on the basal ganglia circuitry in Parkinson’s disease (PD), the effects of ML218 in STN neurons suggest a therapeutic role for T-type Ca2+ channel inhibitors, and ML218 was found to be orally efficacious in haloperidol-induced catalepsy, a preclinical PD model, with comparable efficacy to an A2A antagonist, a clinically validated PD target. ML218 proves to be a powerful new probe to study T-type Ca2+ function in vitro and in vivo, and freely available.Keywords: electrophysiology; inhibitor; Parkinson’s disease; T-Type calcium channel
Co-reporter:Michael R. Wood, Corey R. Hopkins, John T. Brogan, P. Jeffrey Conn, and Craig W. Lindsley
Biochemistry 2011 Volume 50(Issue 13) pp:
Publication Date(Web):February 22, 2011
DOI:10.1021/bi200129s
G-Protein-coupled receptors (GPCRs) represent the largest class of drug targets, accounting for more than 40% of marketed drugs; however, discovery efforts for many GPCRs have failed to provide viable drug candidates. Historically, drug discovery efforts have focused on developing ligands that act at the orthosteric site of the endogenous agonist. Recently, efforts have focused on functional assay paradigms and the discovery of ligands that act at allosteric sites to modulate receptor function in either a positive, negative, or neutral manner. Allosteric modulators have numerous advantages over orthosteric ligands, including high subtype selectivity; the ability to mimic physiological conditions; the lack of densensitization, downregulation, and internalization; and reduced side effects. Despite these virtues, challenging issues have now arisen for allosteric modulators of metabotropic glutamate receptors (mGluRs): shallow SAR, ligand-directed trafficking, and the identification of subtle “molecular switches” that modulate the modes of pharmacology. Here, we will discuss the impact of modest structural changes to multiple mGluR allosteric ligands scaffolds that unexpectedly modulate pharmacology and raise concerns over metabolism and the pharmacology of metabolites.
Co-reporter:Evan P. Lebois, Gregory J. Digby, Douglas J. Sheffler, Bruce J. Melancon, James C. Tarr, Hyekyung P. Cho, Nicole R. Miller, Ryan Morrison, Thomas M. Bridges, Zixiu Xiang, J. Scott Daniels, Michael R. Wood, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6451-6455
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.08.084
Herein we report the discovery and SAR of a novel series of M1 agonists based on the MLPCN probe, ML071. From this, VU0364572 emerged as a potent, orally bioavailable and CNS penetrant M1 agonist with high selectivity, clean ancillary pharmacology and enantiospecific activity.
Co-reporter:Jeffrey P. Lamb, Darren W. Engers, Colleen M. Niswender, Alice L. Rodriguez, Daryl F. Venable, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 9) pp:2711-2714
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmcl.2010.11.119
This Letter describes a chemical lead optimization campaign directed at a weak mGlu5 NAM discovered while developing SAR for the mGlu5 PAM, ADX-47273. An iterative parallel synthesis effort discovered multiple, subtle molecular switches that afford potent mGlu5 NAMs, mGlu5 PAMs as well as mGlu5 partial antagonists.This Letter describes a chemical lead optimization campaign directed at a weak mGlu5 NAM discovered while developing SAR for the mGlu5 PAM, ADX-47273. An iterative parallel synthesis effort discovered multiple, subtle molecular switches that afford potent mGlu5 NAMs, mGlu5 PAMs as well as mGlu5 partial antagonists.
Co-reporter:Leslie N. Aldrich, Eric S. Dawson and Craig W. Lindsley
Organic Letters 2010 Volume 12(Issue 5) pp:1048-1051
Publication Date(Web):February 8, 2010
DOI:10.1021/ol100034p
The first synthetic efforts toward marineosins A and B, novel spiroaminals from a Streptomyces actinomycete, are described by evaluation of the proposed biosynthesis. The hypothesized biosynthetic C1−C25 Diels−Alder substrate was prepared in 8 steps in 5.1% overall yield; however, the proposed biomimetic inverse-electron-demand hetero-Diels−Alder reaction failed to deliver the marineosin core. Molecular mechanics supports this observation.
Co-reporter:Olugbeminiyi O. Fadeyi, Michael L. Schulte and Craig W. Lindsley
Organic Letters 2010 Volume 12(Issue 14) pp:3276-3278
Publication Date(Web):June 16, 2010
DOI:10.1021/ol101276x
A three step, one-pot protocol involving enantioselective α-chlorination of aldehydes, subsequent reductive amination with a primary amine, and SN2 displacement to afford chiral N-alkyl terminal aziridines in 40−65% yield (74−87%/step) and, in most cases, >90% ee is reported.
Co-reporter:Robert R. Lavieri ; Sarah A. Scott ; Paige E. Selvy ; Kwangho Kim ; Satyawan Jadhav ; Ryan D. Morrison ; J. Scott Daniels ; H. Alex Brown
Journal of Medicinal Chemistry 2010 Volume 53(Issue 18) pp:6706-6719
Publication Date(Web):August 24, 2010
DOI:10.1021/jm100814g
Phospholipase D (PLD) catalyzes the conversion of phosphatidylcholine to the lipid second messenger phosphatidic acid. Two mammalian isoforms of PLD have been identified, PLD1 and PLD2, which share 53% sequence identity and are subject to different regulatory mechanisms. Inhibition of PLD enzymatic activity leads to increased cancer cell apoptosis, decreased cancer cell invasion, and decreased metastasis of cancer cells; therefore, the development of isoform-specific, PLD inhibitors is a novel approach for the treatment of cancer. Previously, we developed potent dual PLD1/PLD2, PLD1-specific (>1700-fold selective), and moderately PLD2-preferring (>10-fold preferring) inhibitors. Here, we describe a matrix library strategy that afforded the most potent (PLD2 IC50 = 20 nM) and selective (75-fold selective versus PLD1) PLD2 inhibitor to date, N-(2-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)-2-naphthamide (22a), with an acceptable DMPK profile. Thus, these new isoform-selective PLD inhibitors will enable researchers to dissect the signaling roles and therapeutic potential of individual PLD isoforms to an unprecedented degree.
Co-reporter:Evan P Lebois, Thomas M Bridges, L. Michelle Lewis, Eric S Dawson, Alexander S. Kane, Zixiu Xiang, Satyawan B Jadhav, Huiyong Yin, J. Phillip Kennedy, Jens Meiler, Colleen M. Niswender, Carrie K Jones, P Jeffrey Conn, C David Weaver and Craig W Lindsley
ACS Chemical Neuroscience 2010 Volume 1(Issue 2) pp:104
Publication Date(Web):September 25, 2009
DOI:10.1021/cn900003h
Cholinergic transmission in the forebrain is mediated primarily by five subtypes of muscarinic acetylcholine receptors (mAChRs), termed M1−M5. Of the mAChR subtypes, M1 is among the most heavily expressed in regions that are critical for learning and memory and has been viewed as the most critical mAChR subtype for memory and attention mechanisms. Unfortunately, it has been difficult to develop selective activators of M1 and other individual mAChR subtypes, which has prevented detailed studies of the functional roles of selective activation of M1. Using a functional high-throughput screening and subsequent diversity-oriented synthesis approach, we have discovered a novel series of highly selective M1 allosteric agonists. These compounds activate M1 with EC50 values in the 150−500 nM range and have unprecedented, clean ancillary pharmacology (no substantial activity at 10 μM across a large panel of targets). Targeted mutagenesis revealed a potentially novel allosteric binding site in the third extracellular loop of the M1 receptor for these allosteric agonists. Optimized compounds, such as VU0357017, provide excellent brain exposure after systemic dosing and have robust in vivo efficacy in reversing scopolamine-induced deficits in a rodent model of contextual fear conditioning. This series of selective M1 allosteric agonists provides critical research tools to allow dissection of M1-mediated effects in the CNS and potential leads for novel treatments for Alzheimer’s disease and schizophrenia. Keywords (keywords): agonist; allosteric; cognition; mAChR; muscarinic
Co-reporter:J. Phillip Kennedy, Craig W. Lindsley
Tetrahedron Letters 2010 Volume 51(Issue 18) pp:2493-2496
Publication Date(Web):5 May 2010
DOI:10.1016/j.tetlet.2010.02.168
This Letter describes the synthesis of the five non-proteogenic amino acids required for the total synthesis of piperazimycin A, and synthetic elaboration into multiple dipeptides. Importantly, this Letter details the first example of an elusive piperazic acid–piperazic acid coupling to form this key C5–C14 dipeptide.
Co-reporter:Leslie N. Aldrich, Sydney L. Stoops, Brenda C. Crews, Lawrence J. Marnett, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:5207-5211
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.06.154
Herein we disclose the first total synthesis of tambjamine K and a library of unnatural analogs. Unnatural analogs were shown to be more potent in viability, proliferation, and invasion assays than the natural product in multiple cancer cell lines, with minimal to no cytotoxicity on non-transformed cell lines.The first total synthesis of tambjamine K and a library of unnatural analogs is reported. Unnatural analogs were shown to be more potent in viability, proliferation, and invasion assays than the natural product in multiple cancer cell lines, with minimal to no cytotoxicity on non-transformed cell lines.
Co-reporter:Thomas M. Bridges, J. Phillip Kennedy, Corey R. Hopkins, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 19) pp:5617-5622
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmcl.2010.08.042
This Letter describes a chemical lead optimization campaign directed at VU0238429, the first M5-preferring positive allosteric modulator (PAM), discovered through analog work around VU0119498, a pan Gq mAChR M1, M3, M5 PAM. An iterative parallel synthesis approach was employed to incorporate basic heterocycles to improve physiochemical properties.This Letter describes a chemical lead optimization campaign directed at VU0238429, the first M5-preferring positive allosteric modulator (PAM), discovered through analog work around VU0119498, a pan Gq mAChR M1, M3, M5 PAM. An iterative parallel synthesis approach was employed to incorporate basic heterocycles to improve physiochemical properties.
Co-reporter:Thomas M. Bridges, J. Phillip Kennedy, Meredith J. Noetzel, Micah L. Breininger, Patrick R. Gentry, P.J. Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 6) pp:1972-1975
Publication Date(Web):15 March 2010
DOI:10.1016/j.bmcl.2010.01.109
This Letter describes a chemical lead optimization campaign directed at VU0119498, a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) with the goal of developing a selective M1 PAM. An iterative library synthesis approach delivered a potent (M1 EC50 = 830 nM) and highly selective M1 PAM (>30 μM vs M2–M5).This Letter describes a chemical lead optimization campaign directed around VU0119498, a pan Gq mAChR M1, M3, M5 PAM, which previously afforded the first selective M5 PAM. An iterative library synthesis approach delivered a novel chemotype with selective M1 PAM activity (no activity at M2–M5 @ 30 μM), and comparable M1 potency (EC50 = 830 nM) to the prototypical M1 PAM, BQCA.
Co-reporter:Thomas M. Bridges, J. Phillip Kennedy, Hyekyung P. Cho, Micah L. Breininger, Patrick R. Gentry, Corey R. Hopkins, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 2) pp:558-562
Publication Date(Web):15 January 2010
DOI:10.1016/j.bmcl.2009.11.089
This Letter describes a chemical lead optimization campaign directed at VU0238429, the first M5-preferring positive allosteric modulator (PAM), discovered through analog work around VU0119498, a pan Gq mAChR M1, M3, M5 PAM. An iterative library synthesis approach delivered the first selective M5 PAM (no activity at M1–M4 @ 30 μM), and an important tool compound to study the role of M5 in the CNS.This Letter describes a chemical lead optimization campaign directed at VU0238429, the first M5-preferring positive allosteric modulator (PAM), discovered through analog work around VU0119498, a pan Gq mAChR M1, M3, M5 PAM. An iterative library synthesis approach delivered the first selective M5 PAM (no activity at M1–M4 @ 30 μM), and an important tool compound to study the role of M5 in the CNS.
Co-reporter:Nicole R. Miller, R. Nathan Daniels, David Lee, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 7) pp:2174-2177
Publication Date(Web):1 April 2010
DOI:10.1016/j.bmcl.2010.02.041
This Letter describes the synthesis and SAR, developed through an iterative analog library approach, of a novel series of selective M1 mAChR antagonists, based on an N-(4-(4-alkylpiperazin-1-yl)phenyl)benzamide scaffold for the potential treatment of Parkinson’s disease, dystonia and other movement disorders. Compounds in this series possess M1 antagonist IC50s in the 350 nM to >10 μM range with varying degrees of functional selectivity versus M2–M5.This Letter describes the synthesis and SAR, developed through an iterative analog library approach, of a novel series of selective M1 mAChR antagonists, based on an N-(4-(4-alkylpiperazin-1-yl)phenyl)benzamide scaffold for the potential treatment of Parkinson’s disease, dystonia and other movement disorders. Compounds in this series possess M1 antagonist IC50s in the 350 nM to >10 μM range with varying degrees of functional selectivity versus M2–M5.
Co-reporter:Olugbeminiyi O. Fadeyi and Craig W. Lindsley
Organic Letters 2009 Volume 11(Issue 17) pp:3950-3952
Publication Date(Web):July 29, 2009
DOI:10.1021/ol9015755
The second total synthesis of Brevisamide, a marine cyclic ether alkaloid from Karenia brevis, is reported. This streamlined synthesis proceeds in 21 steps, 14 steps longest linear sequence, in 5.2% overall yield and features a key SmI2 reductive cyclization step to access the tetrasubstituted pyran core.
Co-reporter:Thomas M. Bridges ; Joy E. Marlo ; Colleen M. Niswender ; Carrie K. Jones ; Satyawan B. Jadhav ; Patrick R. Gentry ; Hyekyung C. Plumley ; C. David Weaver ; P. Jeffrey Conn
Journal of Medicinal Chemistry 2009 Volume 52(Issue 11) pp:3445-3448
Publication Date(Web):May 13, 2009
DOI:10.1021/jm900286j
This report describes the discovery and initial characterization of the first positive allosteric modulator of muscarinic acetylcholine receptor subtype 5 (mAChR5 or M5). Functional HTS, identified VU0119498, which displayed micromolar potencies for potentiation of acetylcholine at M1, M3, and M5 receptors in cell-based Ca2+ mobilization assays. Subsequent optimization led to the discovery of VU0238429, which possessed an EC50 of approximately 1.16 μM at M5 with >30-fold selectivity versus M1 and M3, with no M2 or M4 potentiator activity.
Co-reporter:Sameer Sharma ; Jeffrey Kedrowski ; Jerri M. Rook ; Randy L. Smith ; Carrie K. Jones ; Alice L. Rodriguez ; P. Jeffrey Conn
Journal of Medicinal Chemistry 2009 Volume 52(Issue 14) pp:4103-4106
Publication Date(Web):June 19, 2009
DOI:10.1021/jm900654c
We describe the synthesis and SAR of a series of analogues of the mGlu5 partial antagonist 5-(phenylethynyl)pyrimidine. New molecular switches are identified that modulate the pharmacological activity of the lead compound. Slight structural modifications around the proximal pyrimidine ring change activity of the partial antagonist lead to that of potent and selective full negative allosteric modulators and positive allosteric modulators, which demonstrate in vivo efficacy in rodent models for anxiolytic and antipsychotic activity, respectively.
Co-reporter:Jana A. Lewis, Sarah A. Scott, Robert Lavieri, Jason R. Buck, Paige E. Selvy, Sydney L. Stoops, Michelle D. Armstrong, H. Alex Brown, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 7) pp:1916-1920
Publication Date(Web):1 April 2009
DOI:10.1016/j.bmcl.2009.02.057
This Letter describes the synthesis and structure–activity-relationships (SAR) of isoform-selective PLD inhibitors. By virtue of the installation of alternative halogenated piperidinyl benzimidazolone privileged structures, in combination with a key (S)-methyl group, novel PLD inhibitors with low nM potency and unprecedented levels of PLD1 isoform selectivity (∼1700-fold) over PLD2 were developed.The synthesis and SAR of isoform-selective PLD inhibitors is described. By virtue of the installation of alternative halogenated piperidinyl benzimidazolone privileged structures, in combination with a key (S)-methyl group, novel PLD inhibitors with low nM potency and unprecedented levels of isoform selectivity for PLD1 (∼1700-fold) over PLD2 were developed.
Co-reporter:Alice L. Rodriguez, Richard Williams, Ya Zhou, Stacey R. Lindsley, Uyen Le, Mark D. Grier, C. David Weaver, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 12) pp:3209-3213
Publication Date(Web):15 June 2009
DOI:10.1016/j.bmcl.2009.04.110
This Letter describes the discovery and SAR of three novel series of mGluR5 non-competitive antagonists/negative allosteric modulators (NAMs) not based on manipulation of an MPEP/MTEP chemotype. This work demonstrates fundamentally new mGluR5 NAM chemotypes with submicromolar potencies, and the first example of a mode of pharmacology ‘switch’ to provide PAMs with a non-MPEP scaffold.This Letter describes the discovery and SAR of three novel series of mGluR5 non-competitive antagonists/negative allosteric modulators (NAMs) not based on manipulation of an MPEP/MTEP chemotype. This work demonstrates fundamentally new mGluR5 NAM chemotypes with submicromolar potencies, and the first example of a mode of pharmacology ‘switch’ to provide PAMs with a non-MPEP scaffold.
Co-reporter:J. Phillip Kennedy, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 12) pp:3204-3208
Publication Date(Web):15 June 2009
DOI:10.1016/j.bmcl.2009.04.106
This Letter describes the natural product guided synthesis of unnatural analogs of the marine bromopyrrole alkaloid dispyrin, and the resulting SAR of H3 antagonism. Multiple rounds of iterative parallel synthesis improved human H3 IC50 ∼33-fold, and afforded a new class of H3 antagonists based on the novel bromotyramine core of dispyrin.The natural product guided synthesis of analogs of the marine alkaloid dispyrin, and the resulting SAR of H3 antagonism, is described. Three rounds of iterative parallel synthesis generated 24d, a potent H3 antagonist (IC50 = 30 nM, Ki = 70 nM) with a novel chemotype based on the bromotyramine motif of dispyrin.
Co-reporter:Robert Lavieri, Sarah A. Scott, Jana A. Lewis, Paige E. Selvy, Michelle D. Armstrong, H. Alex Brown, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 8) pp:2240-2243
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmcl.2009.02.125
This Letter describes the synthesis and structure–activity relationships (SAR) of isoform-selective PLD inhibitors. By virtue of the installation of a 1,3,8-triazaspiro[4,5]decan-4-one privileged structure, PLD inhibitors with nanomolar potency and an unprecedented 40-fold selectivity for PLD2 over PLD1 were developed. Interestingly, SAR for this diverged from our earlier efforts, and dual PLD1/2 inhibitors were also discovered within this series.The synthesis and SAR of isoform-selective PLD inhibitors is described. By virtue of the installation of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure, inhibitors with up to an unprecedented 40-fold selectivity for PLD2 over PLD1 were developed. Interestingly, SAR for this series diverged considerably from earlier efforts, and also provided potent dual PLD1/2 inhibitors.
Co-reporter:Richard Williams, Colleen M. Niswender, Qingwei Luo, Uyen Le, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 3) pp:962-966
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmcl.2008.11.104
This Letter describes the synthesis and SAR of two mGluR4 positive allosteric modulator leads, 6 and 7. VU001171 (6) represents the most potent (EC50 = 650 nM), efficacious (141% Glu Max) and largest fold shift (36-fold) of any mGluR4 PAM reported to date. However, this work highlights the challenges in hit-to-lead for mGluR4 PAMs, with multiple confirmed HTS hits displaying little or no tractable SAR.The synthesis and SAR of two mGluR4 positive allosteric modulator leads 6 and 7 is described. VU001171 (6) represents the most potent (EC50 = 650 nM), efficacious (141% Glu Max) and largest fold shift (36-fold) of any mGluR4 PAM ever described. However, this work highlights the challenges in hit-to-lead for mGluR4 PAMs, with multiple confirmed HTS hits displaying little or no tractable SAR.
Co-reporter:Leslie N. Aldrich, Evan P. Lebois, L. Michelle Lewis, Natalia T. Nalywajko, Colleen M. Niswender, C. David Weaver, P. Jeffrey Conn, Craig W. Lindsley
Tetrahedron Letters 2009 50(2) pp: 212-215
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.10.127
Co-reporter:Olugbeminiyi O. Fadeyi, R. Nathan Daniels, Sean M. DeGuire, Craig W. Lindsley
Tetrahedron Letters 2009 50(25) pp: 3084-3087
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.043
Co-reporter:J. Phillip Kennedy, Micah L. Breininger, Craig W. Lindsley
Tetrahedron Letters 2009 50(50) pp: 7067-7069
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.09.180
Co-reporter:DarrenW. Engers Dr.;AliceL. Rodriguez Dr.;Richard Williams Dr.;AlexisS. Hammond;Daryl Venable;Oluwatomi Oluwatola;GaryA. Sulikowski Dr.;P.Jeffrey Conn Dr.;CraigW. Lindsley Dr.
ChemMedChem 2009 Volume 4( Issue 4) pp:505-511
Publication Date(Web):
DOI:10.1002/cmdc.200800357
Co-reporter:J. Phillip Kennedy;ThomasM. Bridges;PatrickR. Gentry;JohnT. Brogan;AlexerS. Kane;CarrieK. Jones Dr.;AshleyE. Brady Dr.;JanaK. Shirey;P. Jeffrey Conn ;CraigW. Lindsley
ChemMedChem 2009 Volume 4( Issue 10) pp:1600-1607
Publication Date(Web):
DOI:10.1002/cmdc.200900231
Co-reporter:Jana A Lewis, Evan P Lebois, Craig W Lindsley
Current Opinion in Chemical Biology 2008 Volume 12(Issue 3) pp:269-280
Publication Date(Web):June 2008
DOI:10.1016/j.cbpa.2008.02.014
Allosteric binding sites, as opposed to traditional orthosteric binding sites, offer unparalleled opportunities for drug discovery by providing high levels of selectivity, mimicking physiological conditions, affording fewer side effects because of desensitization/downregulation, and engendering ligands with chemotypes divergent from orthosteric ligands. For kinases, allosteric mechanisms described to date include alteration of protein kinase conformation blocking productive ATP binding which appear ‘ATP competitive’ or blocking kinase activation by conformational changes that are ‘ATP non-competitive’. For GPCRs, allosteric mechanisms impart multiple modes of target modulation (positive allosteric modulation (PAM), negative allosteric modulation (NAM), neutral cooperativity, partial antagonism (PA), allosteric agonism and allosteric antagonism). Here, we review recent developments in the design principles and structural diversity of allosteric ligands for kinases and GPCRs.
Co-reporter:Thomas M. Bridges and Craig W. Lindsley
ACS Chemical Biology 2008 Volume 3(Issue 9) pp:530
Publication Date(Web):July 25, 2008
DOI:10.1021/cb800116f
Heterotrimeric G-protein-coupled receptors (GPCRs) represent a large protein family responsible for mediating extracellular to intracellular signaling within a broad range of physiological contexts. Various conventional models have been used to describe their interactions with ligands and G-proteins. In recent years, however, numerous novel ligand−receptor interactions not adequately addressed by classical receptor theory have been recognized. In addition to traditional orthosteric ligands, many GPCRs can bind allosteric ligands that modulate receptor activity by interacting with distinct or overlapping receptor sites. Such ligands include positive allosteric modulators, which have become the focus of pharmaceutical drug discovery programs and have gained the attention of a growing body of basic and translational researchers within the academic community. Here, we review the fundamental aspects of allosteric GPCR modulation by small-molecule ligands, with particular focus on the emerging position of positive allosteric modulators in modern drug discovery.Keywords: Ago-allosteric modulator: An allosteric ligand that functions as both an allosteric modulator and as an agonist on its own (though the latter is usually only at higher concentrations.; Allosteric agonist: A ligand that is capable of receptor activation on its own by binding to a recognition site that is distinct from the orthosteric site.; Allosteric modulator: A ligand that increases or decreases the action of an orthosteric ligand (agonist or antagonist) by binding at an allosteric site. The modulator may enhance the affinity and/or efficacy of the orthosteric ligand while exerting no effects on its own.; Allosteric site: A ligand binding site that is distinct from the orthosteric binding site. In truest form, there should be no overlap with the orthosteric binding site.; Orthosteric site: The binding site of the endogenous agonist.
Co-reporter:J. Phillip Kennedy, John T. Brogan and Craig W. Lindsley
Journal of Natural Products 2008 Volume 71(Issue 10) pp:1783-1786
Publication Date(Web):September 19, 2008
DOI:10.1021/np800339e
The first total synthesis of dispyrin, a recently reported bromopyrrole alkaloid from Agelas dispar with an unprecedented bromopyrrole tyramine motif, was achieved in three steps on a gram scale (68.4% overall). No biological activity was reported for dispyrin, so we evaluated synthetic dispyrin against >200 discrete molecular targets in radioligand binding and functional assays. Unlike most marine natural products, dispyrin (1) possesses no antibacterial or anticancer activity, but was found to be a potent ligand and antagonist of several therapeutically relevant GPCRs, the α1D and α2A adrenergic receptors and the H2 and H3 histamine receptors.
Co-reporter:J. Phillip Kennedy, Lyndsey Williams, Thomas M. Bridges, R. Nathan Daniels, David Weaver and Craig W. Lindsley
ACS Combinatorial Science 2008 Volume 10(Issue 3) pp:345
Publication Date(Web):January 26, 2008
DOI:10.1021/cc700187t
Co-reporter:Nicole R. Miller, R. Nathan Daniels, Thomas M. Bridges, Ashley E. Brady, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 20) pp:5443-5447
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmcl.2008.09.032
This letter describes the further synthesis and SAR, developed through an iterative analog library approach, of analogs of the highly selective M1 allosteric agonist TBPB by deletion of the distal basic piperidine nitrogen by the formation of amides, sulfonamides and ureas. Despite the large change in basicity and topology, M1 selectivity was maintained.This letter describes the further synthesis and SAR, developed through an iterative analog library approach, of analogs of the highly selective M1 allosteric agonist TBPB by deletion of the distal basic piperidine nitrogen by the formation of amides, sulfonamides and ureas. Despite the large change in basicity and topology, M1 selectivity was maintained.
Co-reporter:Thomas M. Bridges, Ashley E. Brady, J. Phillip Kennedy, R. Nathan Daniels, Nicole R. Miller, Kwango Kim, Micah L. Breininger, Patrick R. Gentry, John T. Brogan, Carrie K. Jones, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 20) pp:5439-5442
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmcl.2008.09.023
This Letter describes the first account of the synthesis and SAR, developed through an iterative analogue library approach, of analogues of the highly selective M1 allosteric agonist TBPB. With slight structural changes, mAChR selectivity was maintained, but the degree of partial M1 agonism varied considerably.The synthesis and SAR of analogues of the M1 allosteric agonist TBPB is described. With slight structural changes, mAChR selectivity was maintained, but the degree of partial agonism varied considerably.
Co-reporter:Colleen M. Niswender, Evan P. Lebois, Qingwei Luo, Kwangho Kim, Hubert Muchalski, Huiyong Yin, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 20) pp:5626-5630
Publication Date(Web):15 October 2008
DOI:10.1016/j.bmcl.2008.08.087
This letter describes the synthesis and SAR, developed through an iterative analogue library approach, of an mGluR4 positive allosteric modulator lead based on a pyrazolo[3,4-d]pyrimidine scaffold. Despite tremendous therapeutic potential, Compound 7, VU0080421, and related congeners represent only a handful of mGluR4 positive allosteric modulators ever described.The synthesis and SAR of a novel mGluR4 positive allosteric modulator 7, VU0080241, is described. VU0080241 displayed an EC50 of 4.6 μM and shifted the glutamate response curve 11.8- to 27-fold to the left.
Co-reporter:Sameer Sharma, Alice L. Rodriguez, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 14) pp:4098-4101
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmcl.2008.05.091
This Letter describes the synthesis and SAR, developed through an iterative analogue library approach, of a mGluR5 allosteric partial antagonist lead based on a 5-(phenylethynyl)pyrimidine scaffold. With slight structural modifications to the distal phenyl ring, analogues demonstrated a range of pharmacological activities from mGluR5 partial antagonism to full antagonism/negative allosteric modulation to positive allosteric modulation.The synthesis and SAR of a mGluR5 allosteric partial antagonist lead 8 is described. We have identified ‘molecular switches’ on the distal phenyl ring that modulate modes of efficacy from mGluR5 partial antagonism in 8, to full non-competitive antagonism 12a to positive allosteric modulation 12h by the addition of a 3- or 4-methyl group, respectively.
Co-reporter:L. Michelle Lewis, Douglas Sheffler, Richard Williams, Thomas M. Bridges, J. Phillip Kennedy, J.T. Brogan, Matthew J. Mulder, Lyndsey Williams, Natalia T. Nalywajko, Colleen M. Niswender, Charles D. Weaver, P. Jeffrey Conn, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 3) pp:885-890
Publication Date(Web):1 February 2008
DOI:10.1016/j.bmcl.2007.12.051
This Letter describes the synthesis and SAR, developed through an iterative analogue library approach, of a novel series of selective M1 mAChR antagonists for the potential treatment of Parkinson’s disease, dystonia and other movement disorders. Compounds in this series possess M1 antagonist IC50s in the 441 nM–19 μM range with 8- to >340-fold functional selectivity versus rM2–rM5.The synthesis and SAR of selective rM1 antagonists, such as 9g (>45-fold selective versus rM2–rM5) and 9i (>340-fold selective versus rM4), are described as part of a chemical probe development program for the Molecular Library Screening Network.
Co-reporter:Craig W. Lindsley, David Weaver, Carrie Jones, Larry Marnett and P. Jeffrey Conn
ACS Chemical Biology 2007 Volume 2(Issue 1) pp:17
Publication Date(Web):January 19, 2007
DOI:10.1021/cb6004867
Co-reporter:Darren W. Engers, Craig W. Lindsley
Drug Discovery Today: Technologies (Summer 2013) Volume 10(Issue 2) pp:e269-e276
Publication Date(Web):1 June 2013
DOI:10.1016/j.ddtec.2012.10.007
Allosteric modulation has emerged as an innovative pharmacological approach to selectively activate or inhibit several Class C GPCRs. Of the Class C GPCRs, metabotropic glutamate (mGlu) receptors represent the most promising candidates for clinical success, and both positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) of mGluRs have demonstrated therapeutic potential for a range of psychiatric and neurological disorders such as pain, depression, anxiety, cognition, Fragile X syndrome, Parkinson's disease and schizophrenia.
Co-reporter:Bruce J. Melancon, James C. Tarr, Joseph D. Panarese, Michael R. Wood, Craig W. Lindsley
Drug Discovery Today (December 2013) Volume 18(Issues 23–24) pp:1185-1199
Publication Date(Web):1 December 2013
DOI:10.1016/j.drudis.2013.09.005
•Dysfunction in glutamatergic and cholinergic signaling in schizophrenia is discussed.•The advantages of allosteric modulation are discussed.•Activation of the M1 mAChR targets the cognitive and negative symptoms of schizophrenia.•We provide an overview of M1 allosteric agonists and issues hindering development.•M1 positive allosteric modulators (PAMs) represent a productive path forward.Allosteric modulation of AMPA, NR2B, mGlu2, mGlu5 and M1, targeting glutamatergic dysfunction, represents a significant area of research for the treatment of schizophrenia. Of these targets, clinical promise has been demonstrated using muscarinic activators for the treatment of Alzheimer's disease (AD) and schizophrenia. These diseases have inspired researchers to determine the effects of modulating cholinergic transmission in the forebrain, which is primarily regulated by one of five subtypes of muscarinic acetylcholine receptor (mAChR), a subfamily of G-protein-coupled receptors (GPCRs). Of these five subtypes, M1 is highly expressed in brain regions responsible for learning, cognition and memory. Xanomeline, an orthosteric muscarinic agonist with modest selectivity, was one of the first compounds that displayed improvements in behavioral disturbances in AD patients and efficacy in schizophrenics. Since these initial clinical results, many scientists, including those in our laboratories, have strived to elucidate the role of M1 with compounds that display improved selectivity for this receptor by targeting allosteric modes of receptor activation. A survey of selected compounds in this area will be presented.
Co-reporter:Kevin M. McGowan, Kellie D. Nance, Hykeyung P. Cho, Thomas M. Bridges, P. Jeffrey Conn, Carrie K. Jones, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters (15 March 2017) Volume 27(Issue 6) pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.bmcl.2017.02.020
This letter describes the continued optimization of M5 NAM ML375 (VU0483253). While a valuable in vivo tool compound, ML375 has an excessively long elimination half-life in rat (t1/2 = 80 h), which can be problematic in certain rodent addiction paradigms (e.g., reinstatement). Thus, we required an M5 NAM of comparable potency to ML375, but with a rat t1/2 of less than 4 h. Steep SAR plagued this chemotype, and here we detail aniline replacements that offered some improvements over ML375, but failed to advance. Ultimately, incorporation of a single methyl group to the 9b-phenyl ring acted as a metabolic shunt, providing (S)-11 (VU6008667), an equipotent M5 NAM, with high CNS penetration, excellent selectivity versus M1–4 and the desired short half-life (t1/2 = 2.3 h) in rat.
Co-reporter:Michael R. Wood, Meredith J. Noetzel, Michael S. Poslusney, Bruce J. Melancon, James C. Tarr, Atin Lamsal, Sichen Chang, Vincent B. Luscombe, Rebecca L. Weiner, Hyekyung P. Cho, Michael Bubser, Carrie K. Jones, Colleen M. Niswender, Michael W. Wood, Darren W. Engers, Nicholas J. Brandon, Mark E. Duggan, P. Jeffrey Conn, Thomas M. Bridges, Craig W. Lindsley
Bioorganic & Medicinal Chemistry Letters (15 January 2017) Volume 27(Issue 2) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.bmcl.2016.11.086
This letter describes the chemical optimization of a novel series of M4 positive allosteric modulators (PAMs) based on a 5-amino-thieno[2,3-c]pyridazine core, developed via iterative parallel synthesis, and culminating in the highly utilized rodent in vivo tool compound, VU0467154 (5). This is the first report of the optimization campaign (SAR and DMPK profiling) that led to the discovery of VU0467154, and details all of the challenges faced in allosteric modulator programs (steep SAR, species differences in PAM pharmacology and subtle structural changes affecting CNS penetration).
Co-reporter:Pedro M. Garcia-Barrantes, Kevin McGowan, Steven W. Ingram, Craig W. Lindsley
Tetrahedron Letters (1 March 2017) Volume 58(Issue 9) pp:898-901
Publication Date(Web):1 March 2017
DOI:10.1016/j.tetlet.2017.01.064
.jpg)
![3-Amino-5-chloro-N-cyclopropyl-6-methoxy-4-methylthieno[2,3-b]pyridine-2-carboxamide](http://img.cochemist.com/ccimg/886100/886047-13-8.png)
![3-Amino-5-chloro-N-cyclopropyl-6-methoxy-4-methylthieno[2,3-b]pyridine-2-carboxamide](http://img.cochemist.com/ccimg/886100/886047-13-8_b.png)



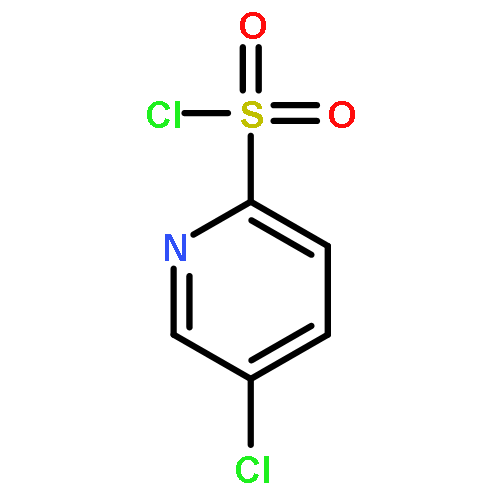
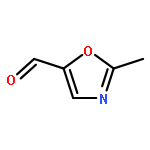
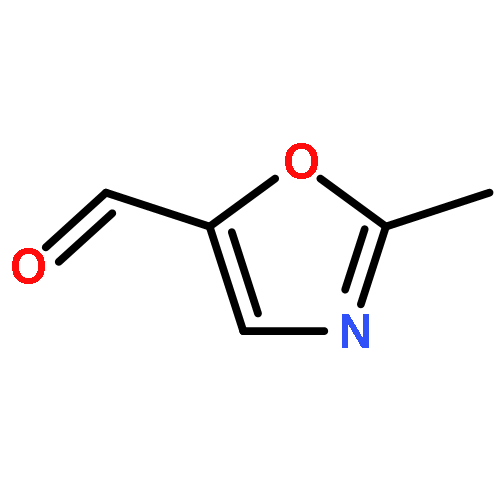
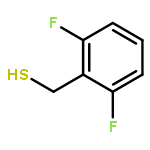
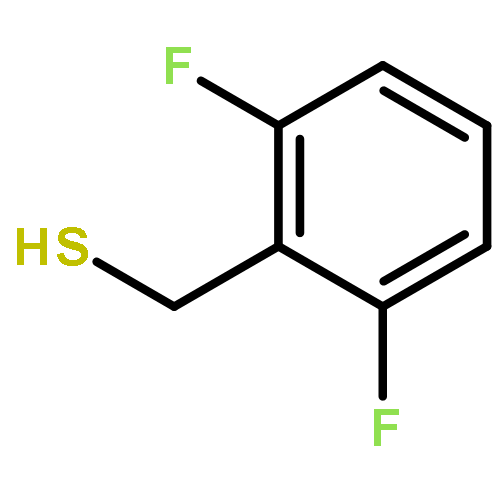
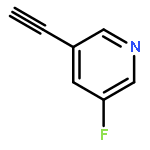
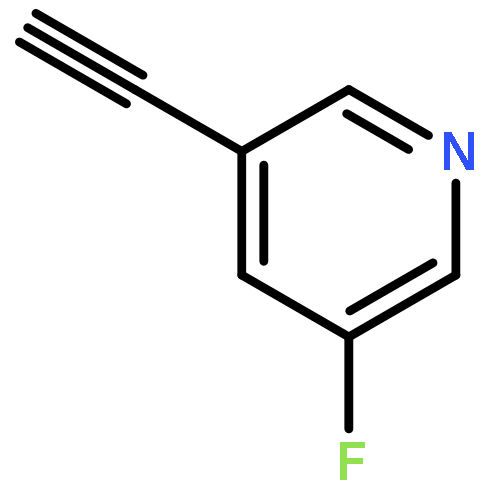
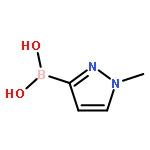
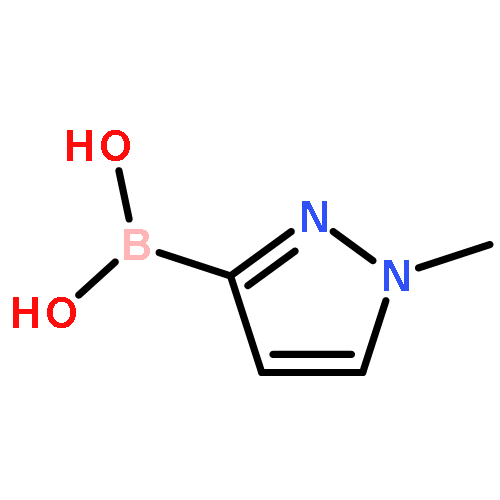
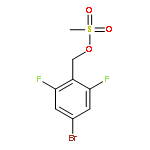
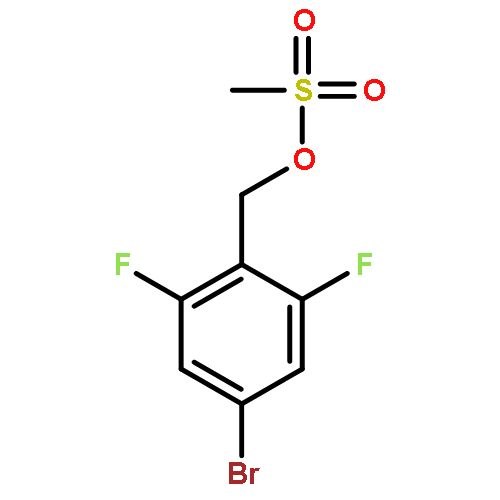
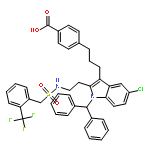
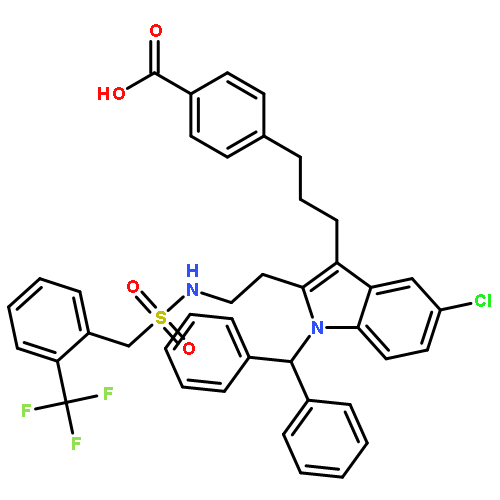
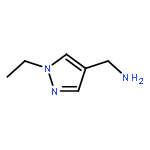

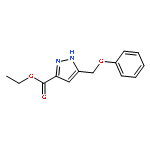
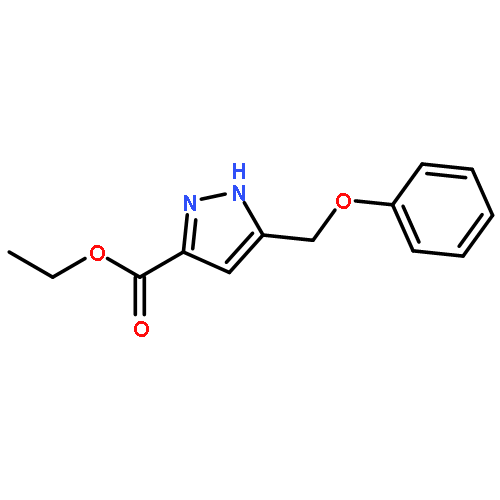
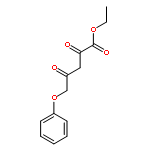
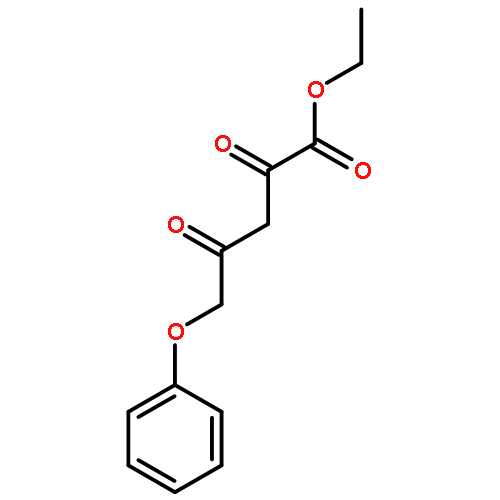
![1-(5,6-Dimethyl-thieno[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxylic acid](http://img.cochemist.com/ccimg/843000/842971-60-2.png)
![1-(5,6-Dimethyl-thieno[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxylic acid](http://img.cochemist.com/ccimg/843000/842971-60-2_b.png)
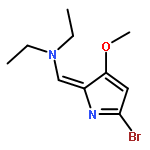
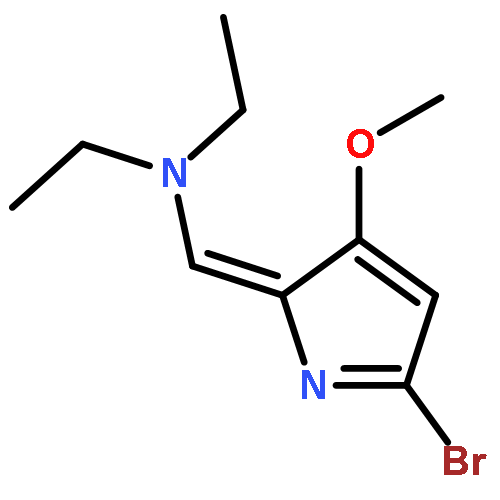
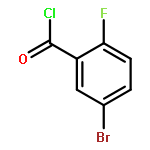

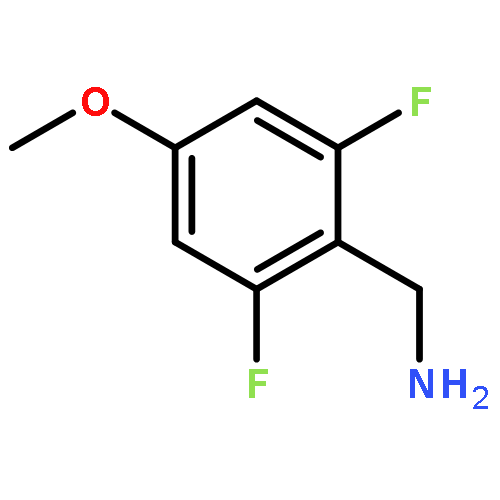
![Benzenemethanamine, 4-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/771500/771473-17-7.png)
![Benzenemethanamine, 4-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/771500/771473-17-7_b.png)
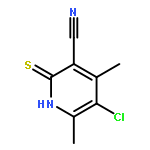
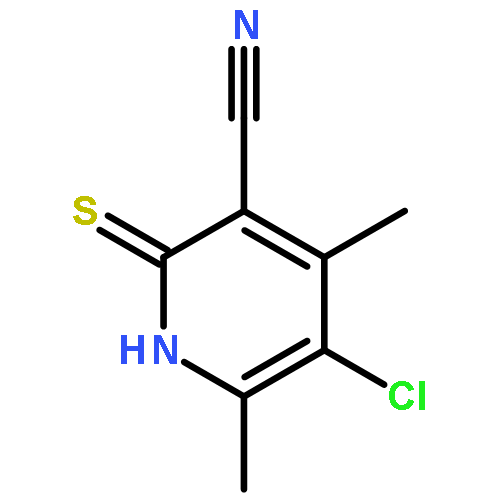


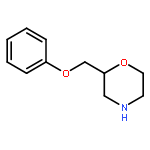

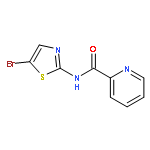
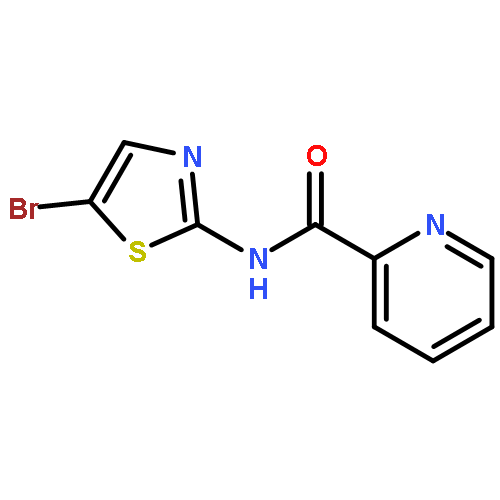
![Benzenemethanamine, 4-[(1-methylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/583900/583837-96-1.png)
![Benzenemethanamine, 4-[(1-methylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/583900/583837-96-1_b.png)
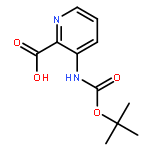
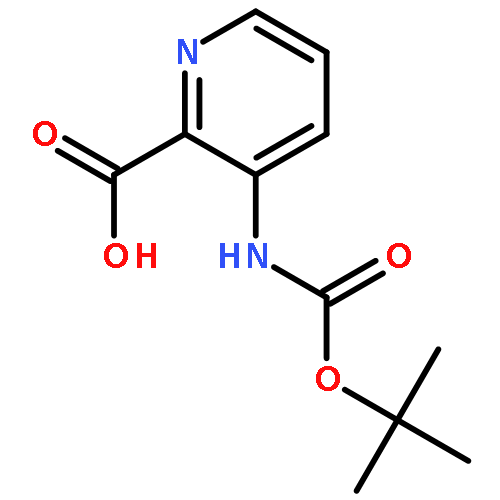
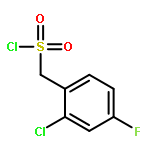
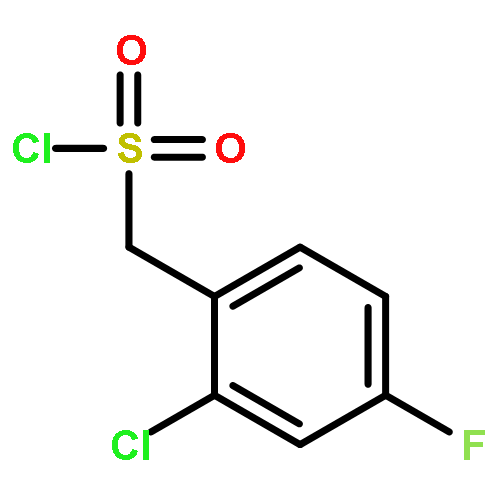
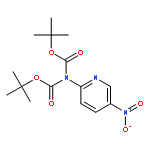
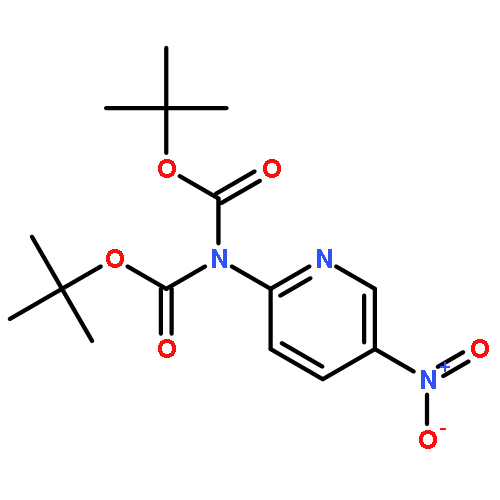
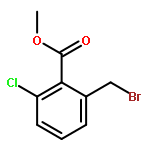
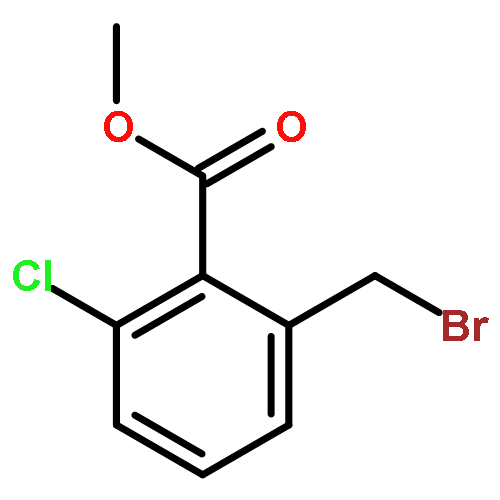
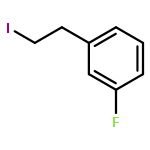

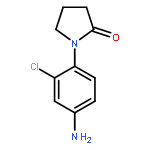
![TERT-BUTYL N-[(2-METHYLPROPAN-2-YL)OXYCARBONYL]-N-(4-NITROPHENYL)CARBAMATE](http://img.cochemist.com/ccimg/395700/395639-03-9.png)
![TERT-BUTYL N-[(2-METHYLPROPAN-2-YL)OXYCARBONYL]-N-(4-NITROPHENYL)CARBAMATE](http://img.cochemist.com/ccimg/395700/395639-03-9_b.png)
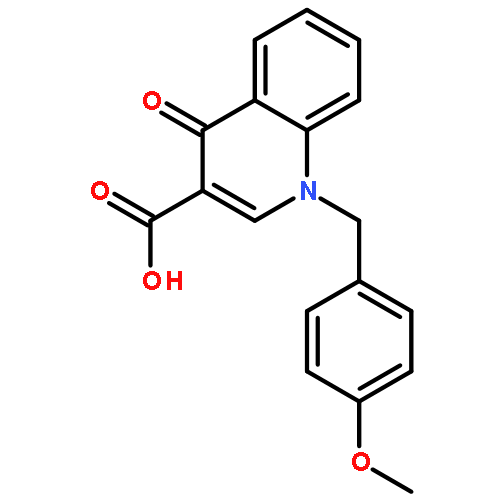
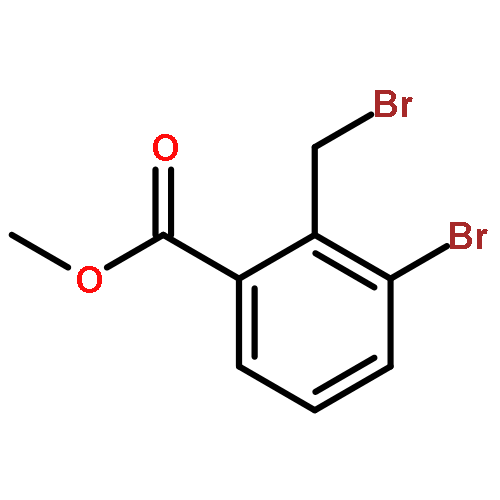




![Pyridine, 2-[5-(3-chlorophenyl)-1,2,4-oxadiazol-3-yl]-](http://img.cochemist.com/ccimg/327100/327056-08-6.png)
![Pyridine, 2-[5-(3-chlorophenyl)-1,2,4-oxadiazol-3-yl]-](http://img.cochemist.com/ccimg/327100/327056-08-6_b.png)
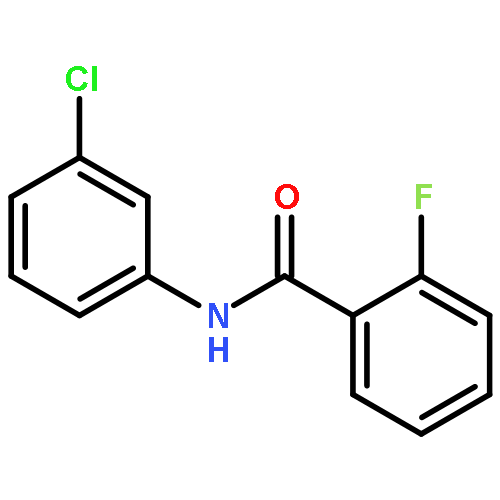
![Methanone,(3-amino-4,6-dimethylthieno[2,3-b]pyridin-2-yl)(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/300000/299932-44-8.png)
![Methanone,(3-amino-4,6-dimethylthieno[2,3-b]pyridin-2-yl)(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/300000/299932-44-8_b.png)



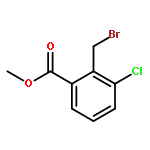
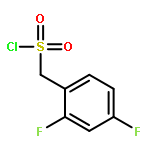
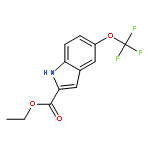
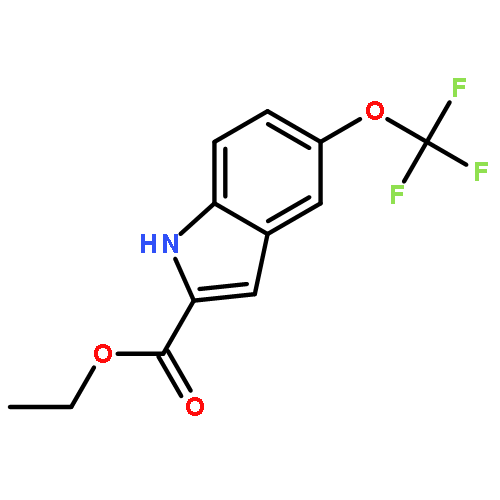
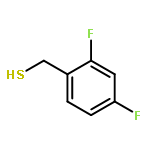
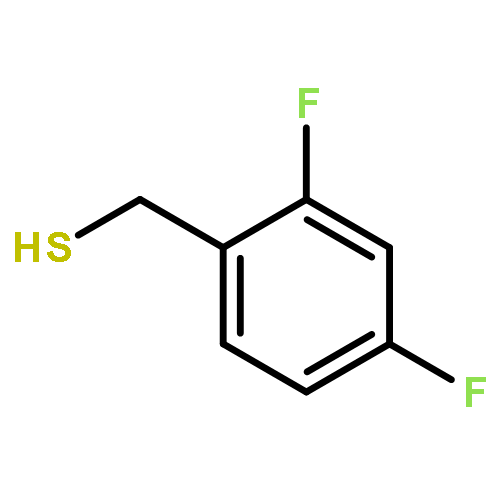
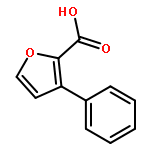
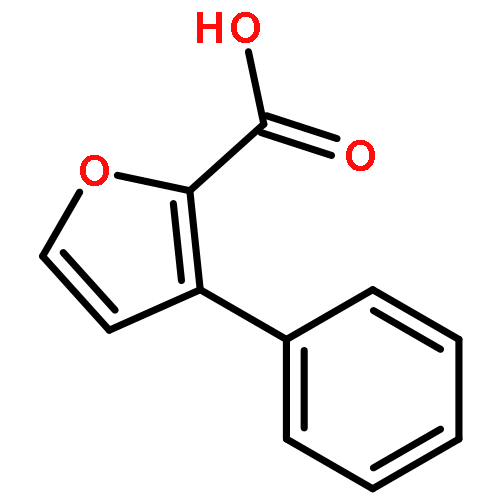


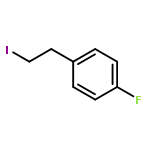
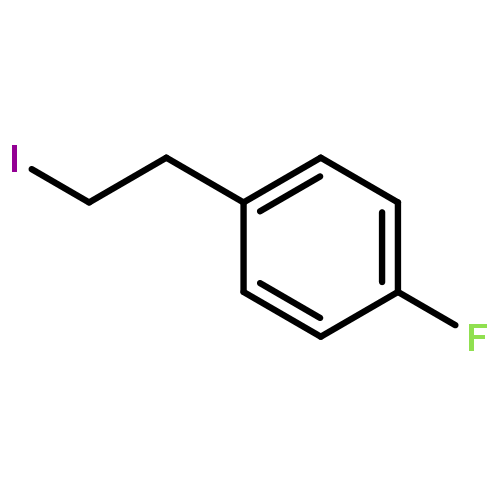


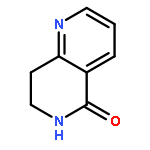
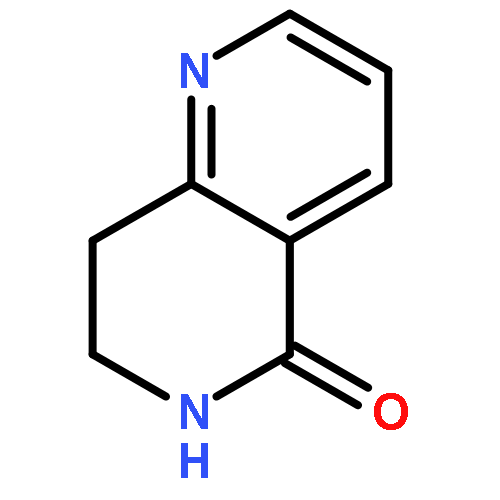
![6-Chloro-2-(chloromethyl)imidazo[1,2-b]pyridazine](http://img.cochemist.com/ccimg/154600/154578-23-1.png)
![6-Chloro-2-(chloromethyl)imidazo[1,2-b]pyridazine](http://img.cochemist.com/ccimg/154600/154578-23-1_b.png)
![Methyl pyrazolo[1,5-a]pyridine-2-carboxylate](http://img.cochemist.com/ccimg/151900/151831-21-9.png)
![Methyl pyrazolo[1,5-a]pyridine-2-carboxylate](http://img.cochemist.com/ccimg/151900/151831-21-9_b.png)


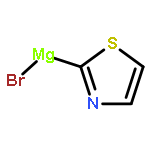
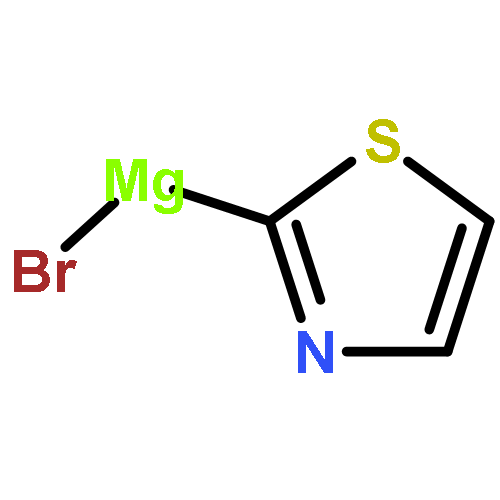





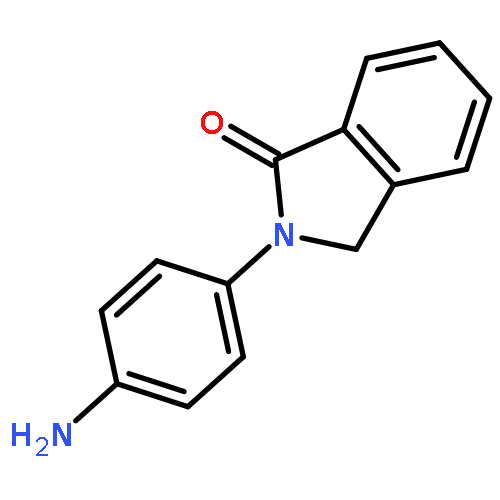
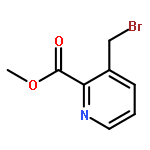
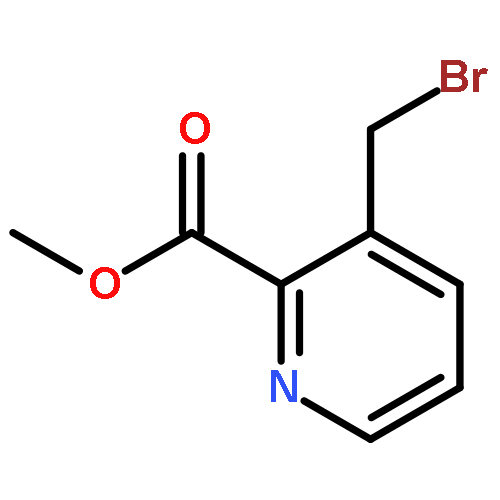
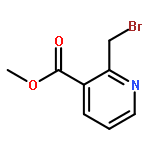
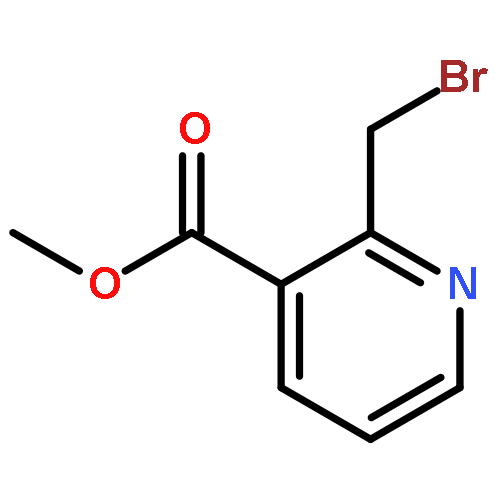







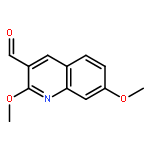
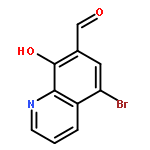
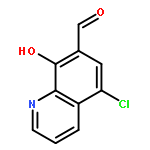
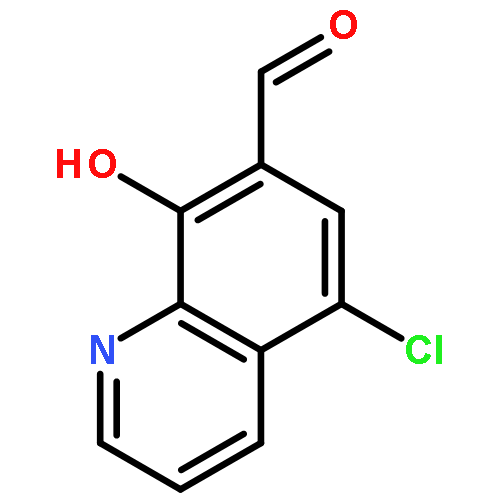
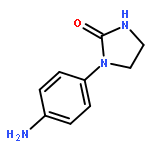
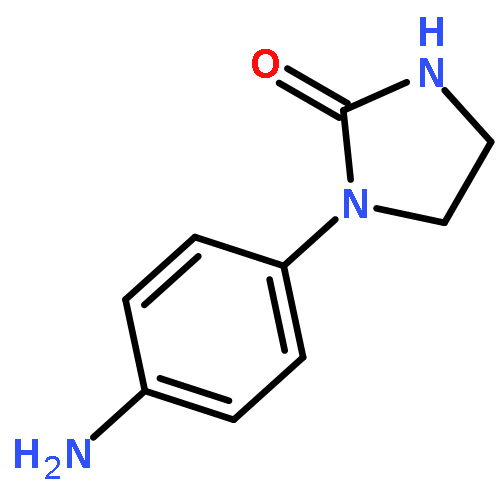
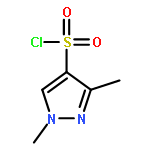
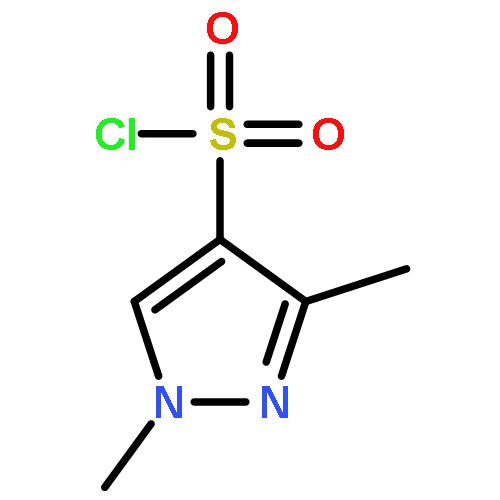
![Benzoic acid,2-[[2-[(methylsulfonyl)amino]benzoyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/89400/89369-45-9.png)
![Benzoic acid,2-[[2-[(methylsulfonyl)amino]benzoyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/89400/89369-45-9_b.png)
![IMIDAZO[2,1-B]-1,3,4-THIADIAZOLE, 2-BROMO-6-PHENYL-](http://img.cochemist.com/ccimg/88100/88013-15-4.png)
![IMIDAZO[2,1-B]-1,3,4-THIADIAZOLE, 2-BROMO-6-PHENYL-](http://img.cochemist.com/ccimg/88100/88013-15-4_b.png)
![2-(Chloromethyl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one](http://img.cochemist.com/ccimg/87600/87591-79-5.png)
![2-(Chloromethyl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one](http://img.cochemist.com/ccimg/87600/87591-79-5_b.png)
![(1S,2S,4R)-Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/84300/84235-33-6.png)
![(1S,2S,4R)-Bicyclo[2.2.1]heptan-2-amine](http://img.cochemist.com/ccimg/84300/84235-33-6_b.png)


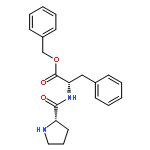
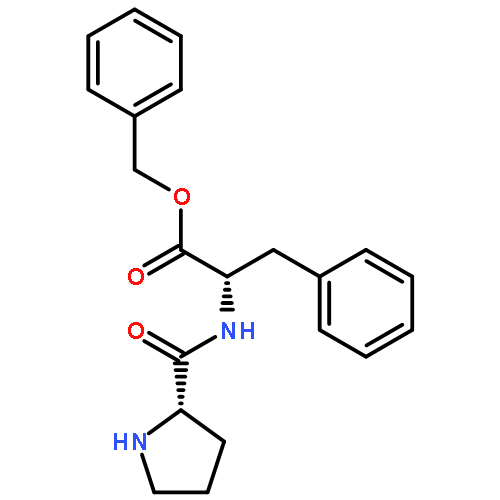
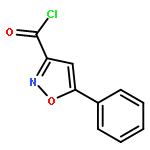
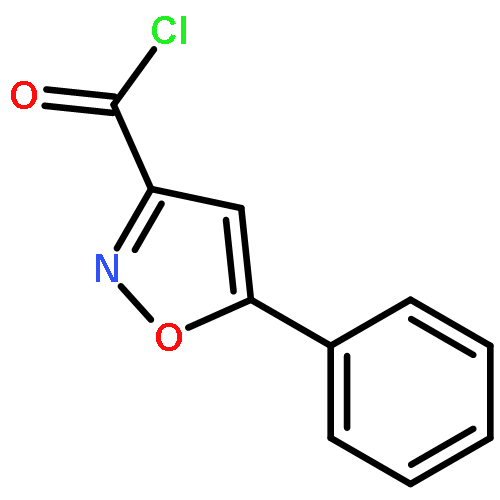



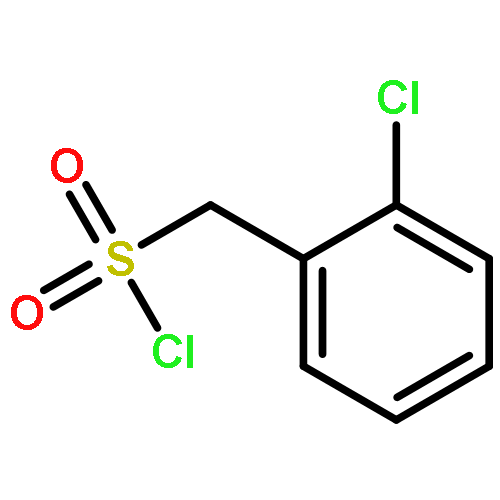


![2-[3-(3-CHLOROPHENYL)-1H-1,2,4-TRIAZOL-5-YL]PYRIDINE](http://img.cochemist.com/ccimg/76600/76591-82-7.png)
![2-[3-(3-CHLOROPHENYL)-1H-1,2,4-TRIAZOL-5-YL]PYRIDINE](http://img.cochemist.com/ccimg/76600/76591-82-7_b.png)
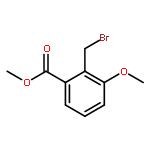
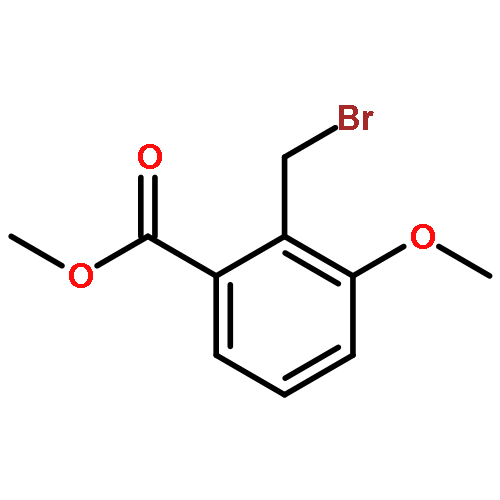
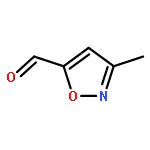
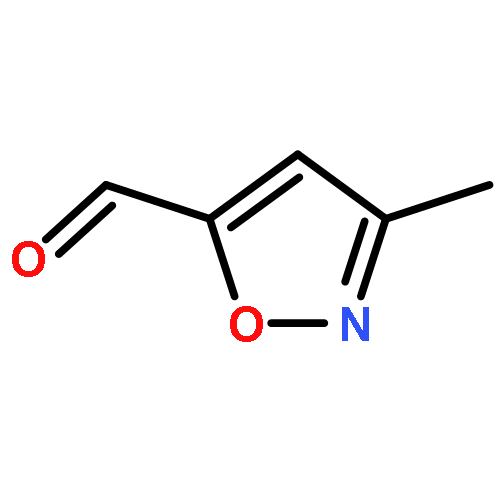


![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8.png)
![(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate](http://img.cochemist.com/ccimg/67600/67526-95-8_b.png)




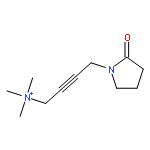
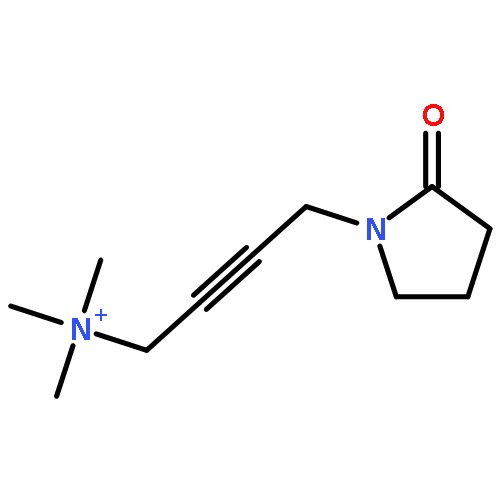
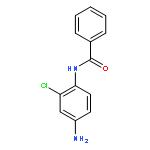
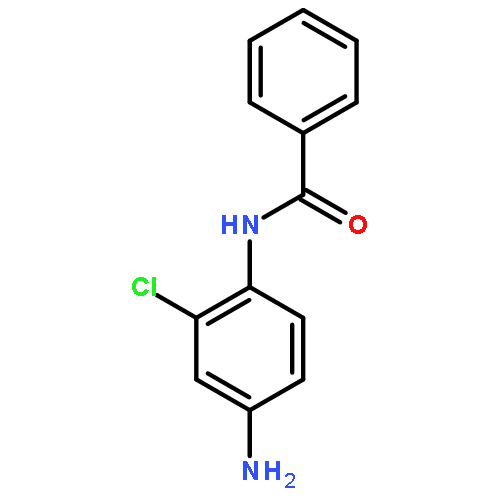
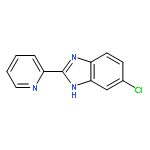
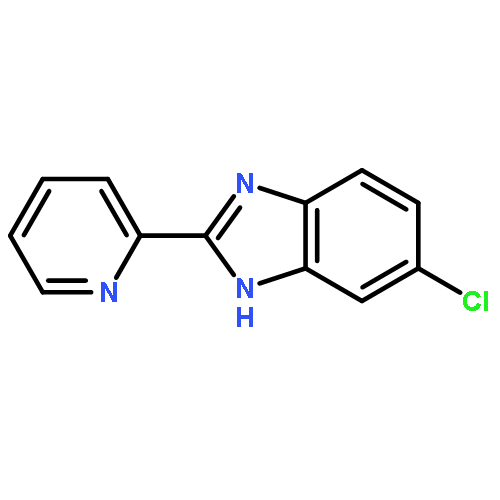



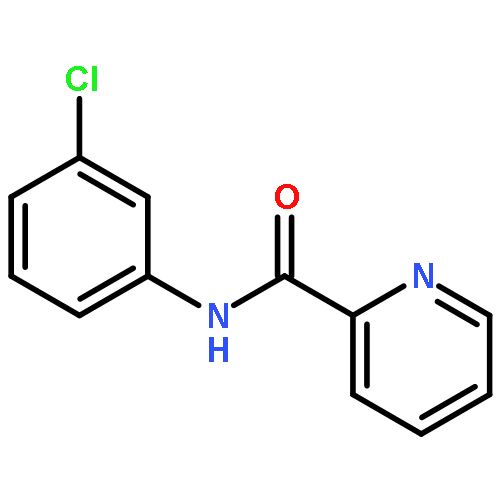
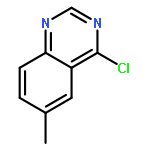
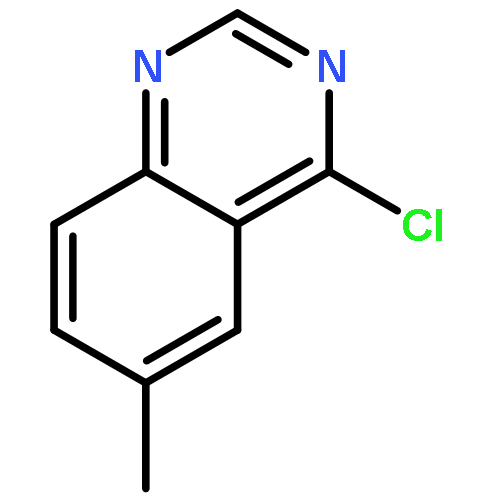
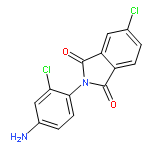
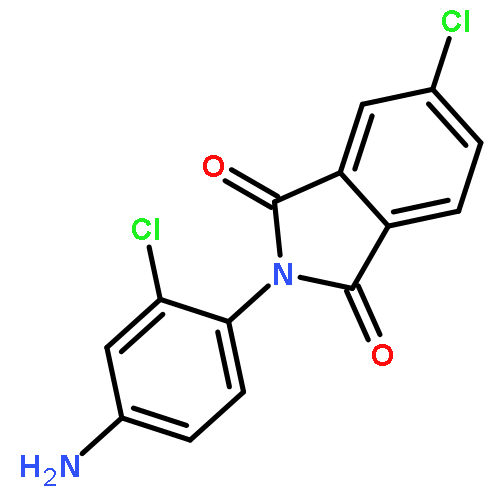
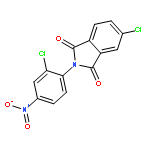
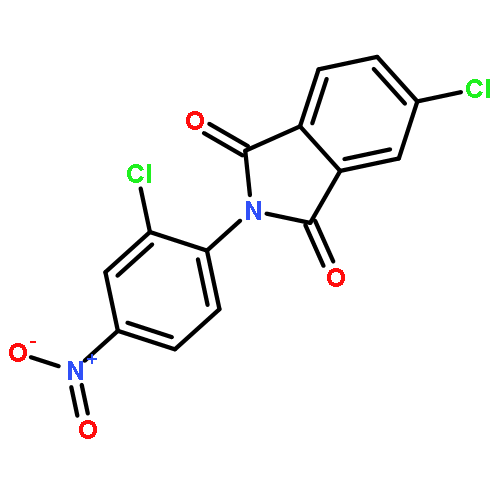
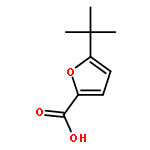
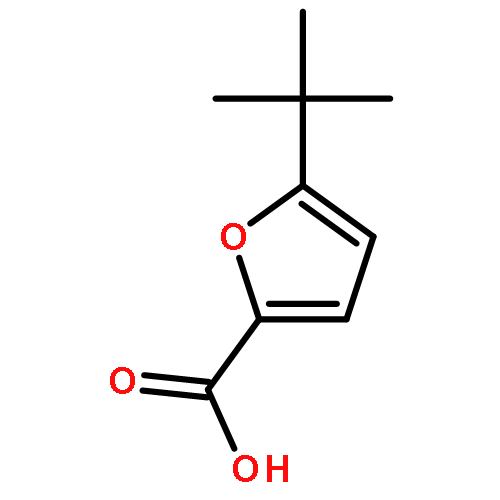
![Morpholine, 2-[(4-methoxyphenoxy)methyl]-, (S)- (9CI)](/data/chemimg/99700/56287-60-6.png)
![Morpholine, 2-[(4-methoxyphenoxy)methyl]-, (S)- (9CI)](/data/chemimg/99700/56287-60-6_b.png)




![(2s)-2-[(3-methoxyphenoxy)methyl]morpholine;hydrochloride](http://img.cochemist.com/ccimg/55300/55253-30-0.png)
![(2s)-2-[(3-methoxyphenoxy)methyl]morpholine;hydrochloride](http://img.cochemist.com/ccimg/55300/55253-30-0_b.png)
![5H-Imidazo[2,1-a]isoindol-5-one, 1,2,3,9b-tetrahydro-9b-(2-pyridinyl)-](http://img.cochemist.com/ccimg/55000/54941-70-7.png)
![5H-Imidazo[2,1-a]isoindol-5-one, 1,2,3,9b-tetrahydro-9b-(2-pyridinyl)-](http://img.cochemist.com/ccimg/55000/54941-70-7_b.png)
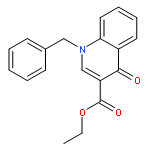
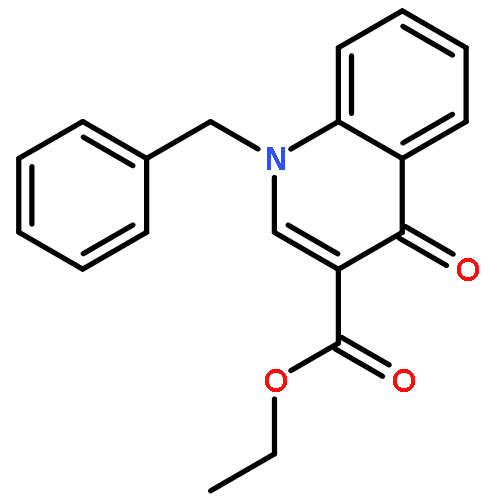
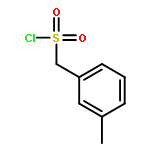
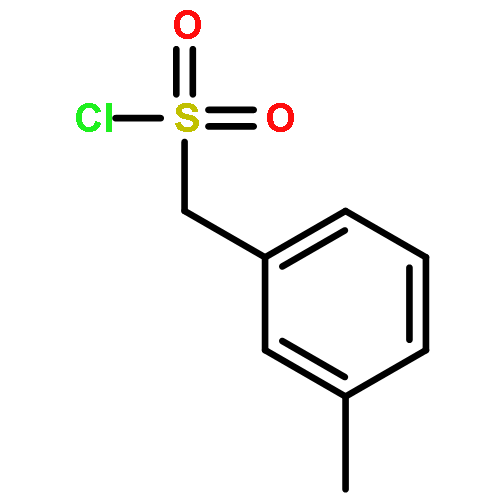
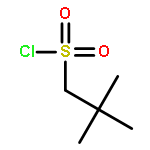
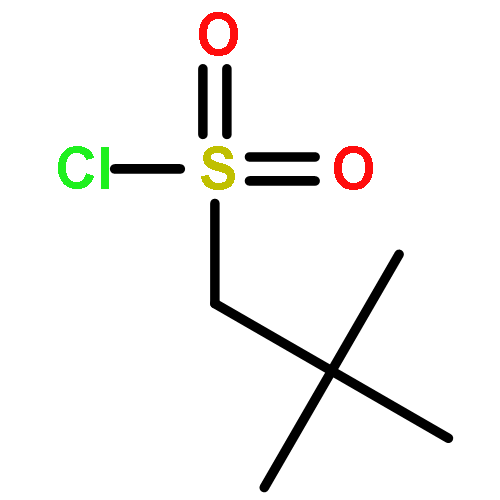
![Benzamide, 4-chloro-N-[2,2,2-trifluoro-1-(trifluoromethyl)ethylidene]-](http://img.cochemist.com/ccimg/52800/52786-52-4.png)
![Benzamide, 4-chloro-N-[2,2,2-trifluoro-1-(trifluoromethyl)ethylidene]-](http://img.cochemist.com/ccimg/52800/52786-52-4_b.png)
![Methanone, (3-amino-4,6-dimethylthieno[2,3-b]pyridin-2-yl)phenyl-](http://img.cochemist.com/ccimg/52600/52505-58-5.png)
![Methanone, (3-amino-4,6-dimethylthieno[2,3-b]pyridin-2-yl)phenyl-](http://img.cochemist.com/ccimg/52600/52505-58-5_b.png)
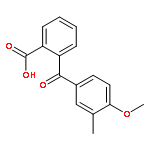
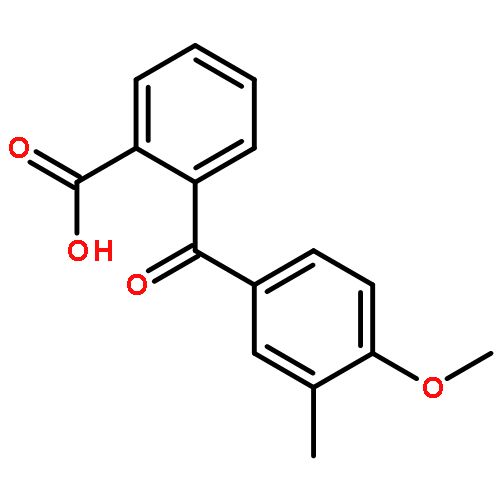
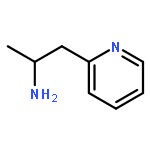






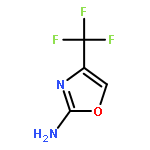
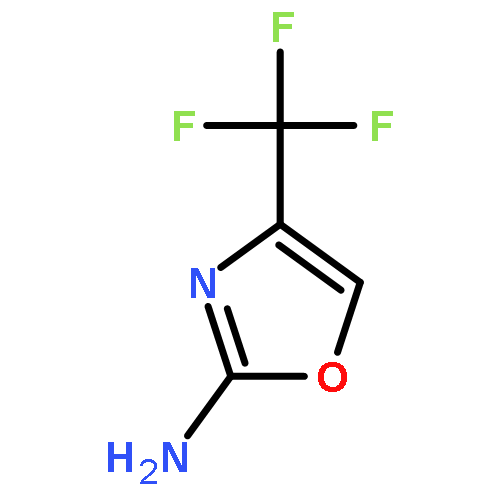
![PYRIDO[2,3-B]PYRAZIN-2(1H)-ONE](http://img.cochemist.com/ccimg/35300/35252-03-0.png)
![PYRIDO[2,3-B]PYRAZIN-2(1H)-ONE](http://img.cochemist.com/ccimg/35300/35252-03-0_b.png)





![1H,12H-Pyrano[4',3':3,4]pyrido[2,1-i]indol-12-one,2,3,5,6,8,9,10,13-octahydro-2-methoxy-, hydrobromide (1:1), (2S,13bS)-](http://img.cochemist.com/ccimg/29800/29734-68-7.png)
![1H,12H-Pyrano[4',3':3,4]pyrido[2,1-i]indol-12-one,2,3,5,6,8,9,10,13-octahydro-2-methoxy-, hydrobromide (1:1), (2S,13bS)-](http://img.cochemist.com/ccimg/29800/29734-68-7_b.png)







![Benzoic acid,2-[[(2-chloro-4-nitrophenyl)amino]carbonyl]-](http://img.cochemist.com/ccimg/19400/19368-46-8.png)
![Benzoic acid,2-[[(2-chloro-4-nitrophenyl)amino]carbonyl]-](http://img.cochemist.com/ccimg/19400/19368-46-8_b.png)

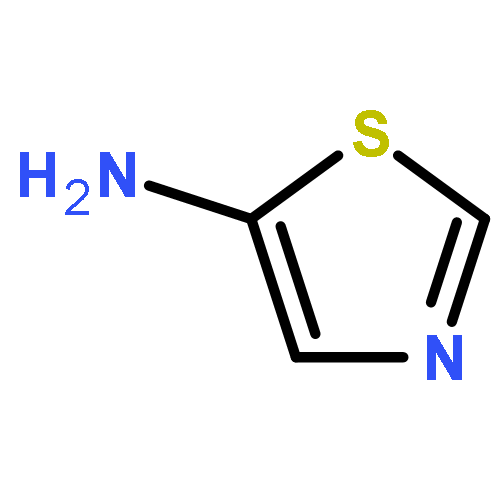
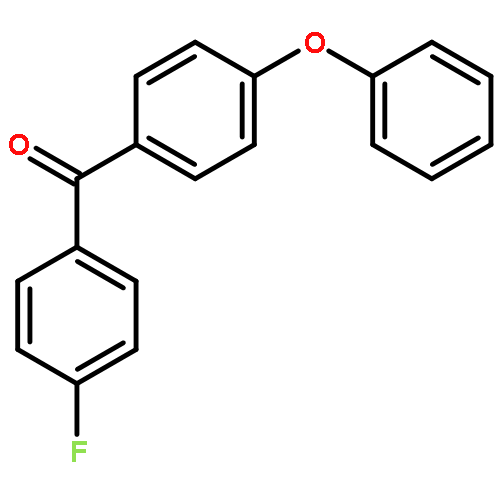


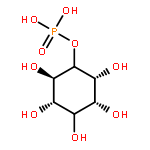

![Bicyclo[3.1.1]heptan-3-amine,2,6,6-trimethyl-, (1S,2S,3S,5R)-](http://img.cochemist.com/ccimg/13300/13293-47-5.png)
![Bicyclo[3.1.1]heptan-3-amine,2,6,6-trimethyl-, (1S,2S,3S,5R)-](http://img.cochemist.com/ccimg/13300/13293-47-5_b.png)









![5H-Imidazo[2,1-a]isoindol-5-one,9b-(4-chlorophenyl)-1,2,3,9b-tetrahydro-](http://img.cochemist.com/ccimg/6100/6038-49-9.png)
![5H-Imidazo[2,1-a]isoindol-5-one,9b-(4-chlorophenyl)-1,2,3,9b-tetrahydro-](http://img.cochemist.com/ccimg/6100/6038-49-9_b.png)
![N~2~-(3,3-dimethylbutanoyl)-N-[5-(3-methylphenyl)-1,3,4-thiadiazol-2-yl]isoleucinamide](http://img.cochemist.com/ccimg/6000/5983-82-4.png)
![N~2~-(3,3-dimethylbutanoyl)-N-[5-(3-methylphenyl)-1,3,4-thiadiazol-2-yl]isoleucinamide](http://img.cochemist.com/ccimg/6000/5983-82-4_b.png)






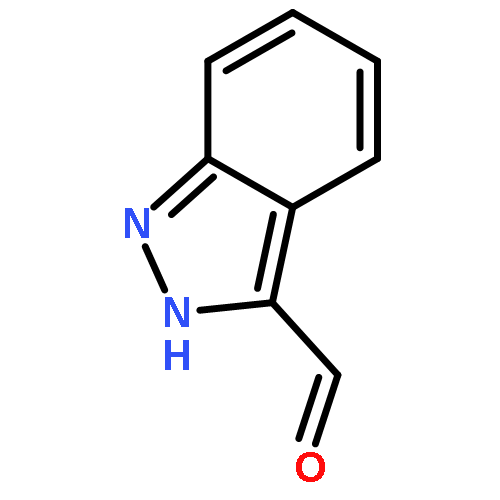
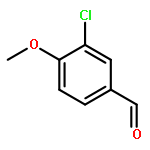

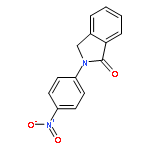
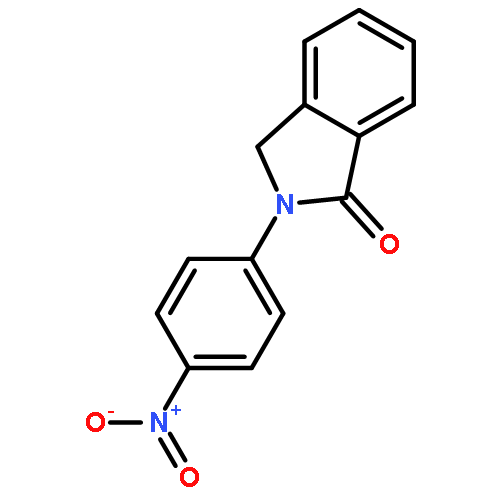





![2-Pyrazinecarboxamide,3,5-diamino-6-chloro-N-[imino[(phenylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/2900/2898-76-2.png)
![2-Pyrazinecarboxamide,3,5-diamino-6-chloro-N-[imino[(phenylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/2900/2898-76-2_b.png)


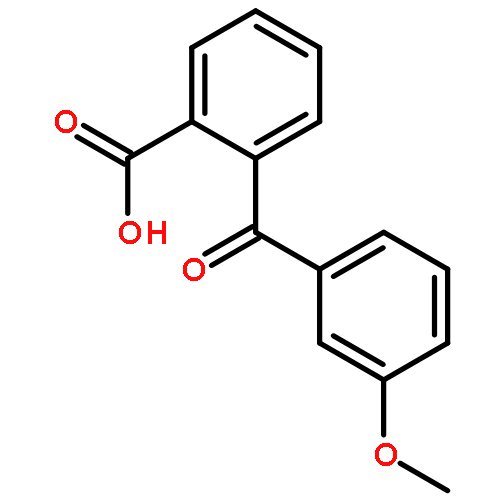








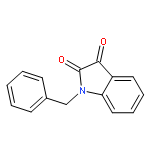
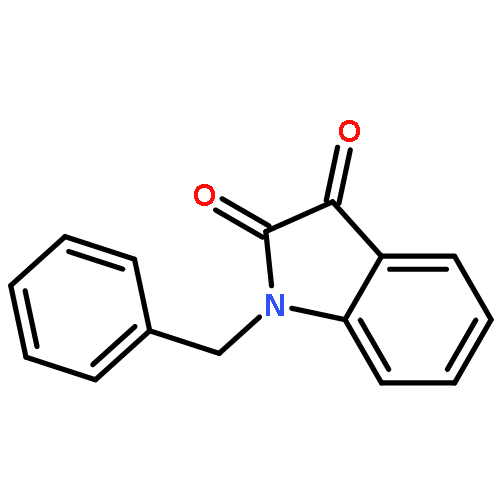




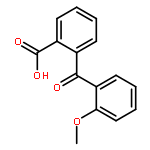





![3-Azaspiro[5.5]undecane](http://img.cochemist.com/ccimg/200/180-44-9.png)
![3-Azaspiro[5.5]undecane](http://img.cochemist.com/ccimg/200/180-44-9_b.png)












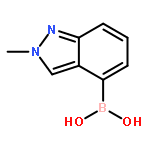
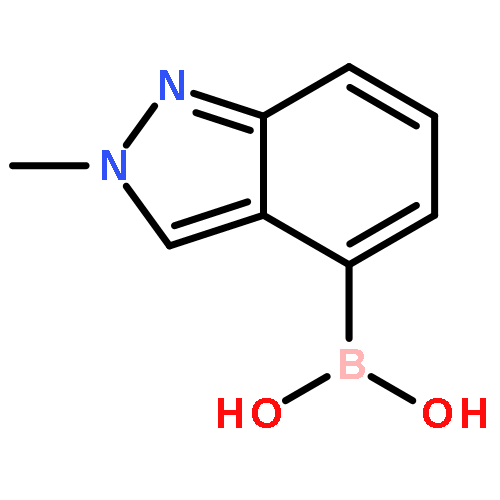
![3-amino-n-[(4-methoxyphenyl)methyl]-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide](http://img.cochemist.com/ccimg/409400/409351-28-6.png)
![3-amino-n-[(4-methoxyphenyl)methyl]-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide](http://img.cochemist.com/ccimg/409400/409351-28-6_b.png)


![9H-Xanthene-9-propanoicacid, a-amino-a-[(1S,2S)-2-carboxycyclopropyl]-,(aS)-](http://img.cochemist.com/ccimg/202000/201943-63-7.png)
![9H-Xanthene-9-propanoicacid, a-amino-a-[(1S,2S)-2-carboxycyclopropyl]-,(aS)-](http://img.cochemist.com/ccimg/202000/201943-63-7_b.png)






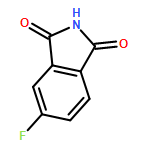
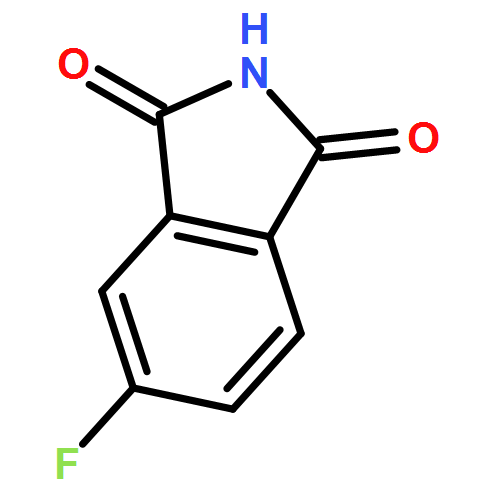
![Imidazo[1,5-a]pyridine-1-carbaldehyde](http://img.cochemist.com/ccimg/56700/56671-67-1.png)
![Imidazo[1,5-a]pyridine-1-carbaldehyde](http://img.cochemist.com/ccimg/56700/56671-67-1_b.png)


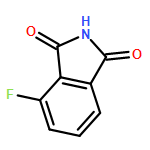
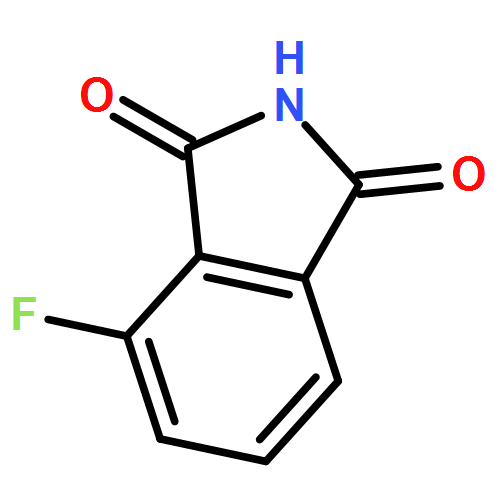
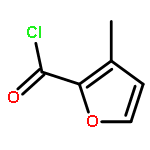
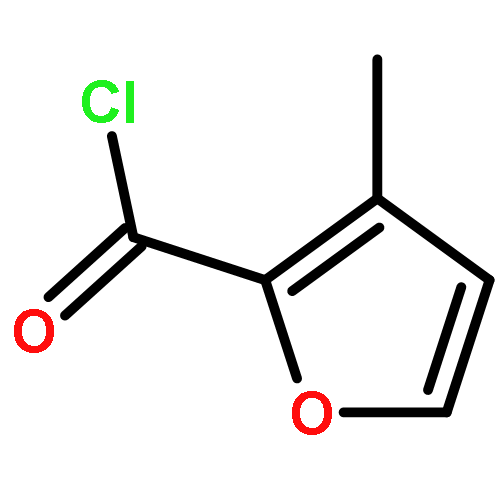
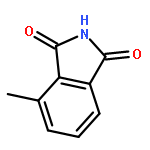
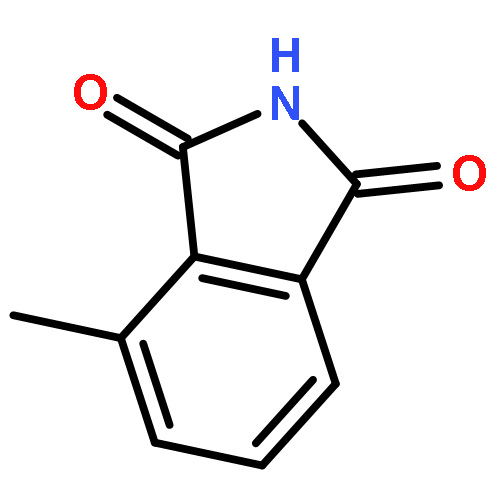
![Morpholinium,2,2'-[1,1'-biphenyl]-4,4'-diylbis[2-hydroxy-4,4-dimethyl-, bromide (1:2)](http://img.cochemist.com/ccimg/400/312-45-8.png)
![Morpholinium,2,2'-[1,1'-biphenyl]-4,4'-diylbis[2-hydroxy-4,4-dimethyl-, bromide (1:2)](http://img.cochemist.com/ccimg/400/312-45-8_b.png)


![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)