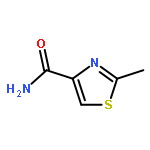
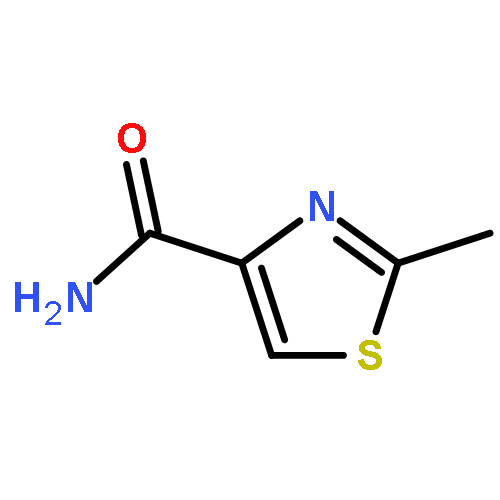
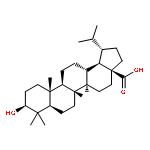
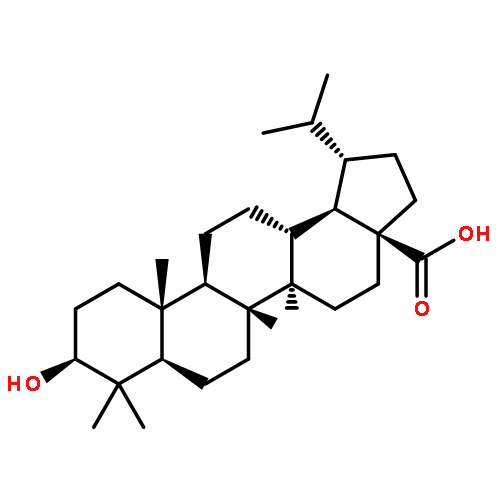
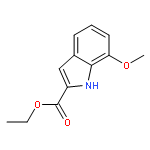
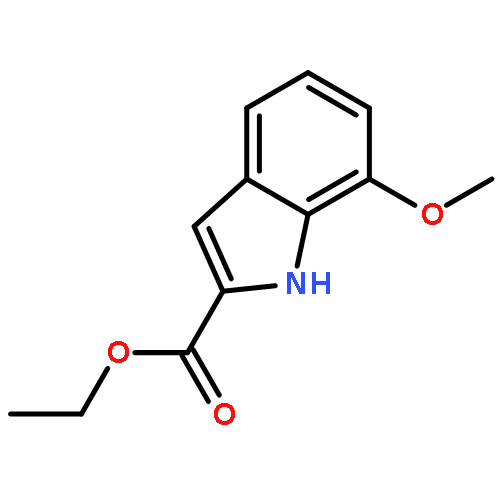
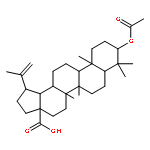
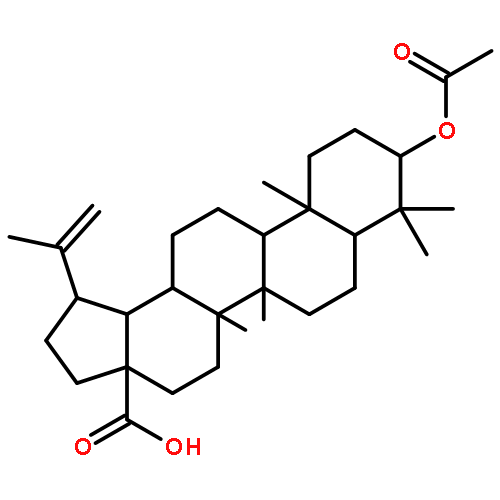
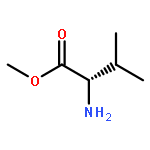
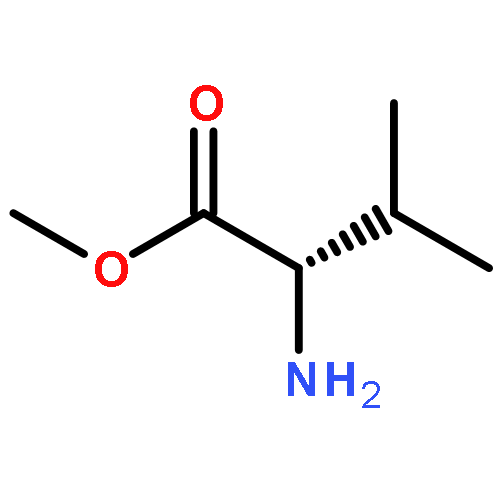
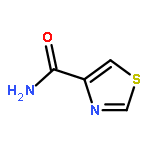
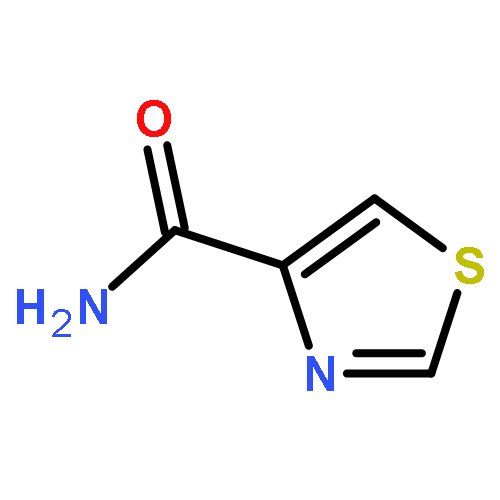
Co-reporter:Suwen Hu, Zhilong Wang, Tingjun Hou, Xiaodong Ma, Jing Li, Tao Liu, Xin Xie, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2015 23(5) pp: 1157-1168
Publication Date(Web):
DOI:10.1016/j.bmc.2014.12.052
Co-reporter:Suwen Hu, Quan Gu, Zhilong Wang, Zhiyong Weng, Yunrui Cai, Xiaowu Dong, Yongzhou Hu, Tao Liu, Xin Xie
European Journal of Medicinal Chemistry 2014 Volume 71() pp:259-266
Publication Date(Web):7 January 2014
DOI:10.1016/j.ejmech.2013.11.013
•We designed and synthesized a series of novel piperidine-4-carboxamide derivatives.•Two compounds showed similar activity to that of the positive control based on calcium mobilization assay.•Two compounds displayed nanomolar activity by HIV-1 single cycle antiviral assay.•The pharmacokinetic properties and cardiovascular safety (hERG) of 16g were evaluated.Based on a putative ‘Y shape’ pharmacophore model of CCR5 inhibitors, a series of novel piperidine-4-carboxamide derivatives were designed and synthesized using a group-reverse strategy. Among synthesized target compounds, 16g (IC50 = 25.73 nM) and 16i (IC50 = 25.53 nM) showed equivalent inhibitory activity against CCR5 to that of the positive control maraviroc (IC50 = 25.43 nM) in calcium mobilization assay. Selected compounds were further tested for their antiviral activity in HIV-1 single cycle assay. Two compounds, 16g and 16i, displayed antiviral activity with IC50 values of 73.01 nM and 94.10 nM, respectively. Additionally, the pharmacokinetic properties and inhibitory potency against hERG of 16g were evaluated, providing a foundation for ongoing optimization.16g and 16i showed equivalent inhibitory activity to that of the positive control maraviroc.
Co-reporter:Fei Chen, Hui Chai, Ming-Bo Su, Yang-Ming Zhang, Jia Li, Xin Xie, and Fa-Jun Nan
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 6) pp:628
Publication Date(Web):April 4, 2014
DOI:10.1021/ml400470s
Inspired by the thiazole–thiazoline cap group in natural product largazole, a series of structurally simplified bisthiazole-based histone deacetylase inhibitors were prepared and evaluated. Compound 8f was evaluated in vivo in an experimental autoimmune encephalomyelitis (EAE) model and found to be orally efficacious in ameliorating clinical symptoms of EAE mice.Keywords: bisthiazole; Histone deacetylase inhibitors; largazole
Co-reporter:Xinxiu Xu, Quan Wang, Yuan Long, Ru Zhang, Xiaoyuan Wei, Mingzhe Xing, Haifeng Gu and Xin Xie
Cell Research 2013 23(1) pp:131-141
Publication Date(Web):October 9, 2012
DOI:10.1038/cr.2012.143
Environmental stress-mediated adaptation plays essential roles in the evolution of life. Cellular adaptation mechanisms usually involve the regulation of chromatin structure, transcription, mRNA stability and translation, which eventually lead to efficient changes in gene expression. Global epigenetic change is also involved in the reprogramming of somatic cells into induced pluripotent stem (iPS) cells by defined factors. Here we report that environmental stress such as hyperosmosis not only facilitates four factor-mediated reprogramming, but also enhances two or one factor-induced iPS cell generation. Hyperosmosis-induced p38 activation plays a critical role in this process. Constitutive active p38 mimics the positive effect of hyperosmosis, while dominant negative p38 and p38 inhibitor block the effect of hyperosmosis. Further study indicates stress-mediated p38 activation may promote reprogramming by reducing the global DNA methylation level and enhancing the expression of pluripotency genes. Our results demonstrate how simple environmental stress like hyperosmosis helps to alter the fate of cells via intracellular signaling and epigenetic modulation.
Co-reporter:Zeng Li, Jing Li, Nan Yang, Ying Chen, Yu Zhou, Xun Ji, Lei Zhang, Jinfang Wang, Xin Xie, and Hong Liu
The Journal of Organic Chemistry 2013 Volume 78(Issue 21) pp:10802-10811
Publication Date(Web):October 7, 2013
DOI:10.1021/jo4017887
An efficient and facile gold(I)-catalyzed one-pot cascade protocol has been developed for the synthesis of tryptamine-fused polycyclic privileged structures through the treatment of substituted tryptamines and 2-ethynylbenzoic acids or 2-ethynylphenylacetic acids. This strategy features the formation of one C–C bond and two C–N bonds with high yields and broad substrate tolerance. The selected reduced target molecules are validated to perform as α1-adrenergic receptors antagonists. The most potent one, 4bh, exhibits an IC50 value of 277 nM on α1A subtype with a selectivity ratio of 15.8 over α1B subtype.
Co-reporter:Ru Zhang;Li-hong Zhang;Xin Xie
Acta Pharmacologica Sinica 2013 34(6) pp:765-776
Publication Date(Web):2013-04-22
DOI:10.1038/aps.2013.21
The revolutionary induced pluripotent stem cell (iPSC) technology provides a new path for cell replacement therapies and drug screening. Patient-specific iPSCs and subsequent differentiated cells manifesting disease phenotypes will finally position human disease pathology at the core of drug discovery. Cells used to test the toxic effects of drugs can also be generated from normal iPSCs and provide a much more accurate and cost-effective system than many animal models. Here, we highlight the recent progress in iPSC-based cell therapy, disease modeling and drug evaluations. In addition, we discuss the use of small molecule drugs to improve the generation of iPSCs and understand the reprogramming mechanism. It is foreseeable that the interplay between iPSC technology and small molecule compounds will push forward the applications of iPSC-based therapy and screening systems in the real world and eventually revolutionize the methods used to treat diseases.
Co-reporter:Changsheng Du and Xin Xie
Cell Research 2012 22(7) pp:1108-1128
Publication Date(Web):June 5, 2012
DOI:10.1038/cr.2012.87
G protein-coupled receptors (GPCRs) mediate most of our physiological responses to hormones, neurotransmitters and environmental stimulants. They are considered as the most successful therapeutic targets for a broad spectrum of diseases. Multiple sclerosis (MS) is an inflammatory disease that is characterized by immune-mediated demyelination and degeneration of the central nervous system (CNS). It is the leading cause of non-traumatic disability in young adults. Great progress has been made over the past few decades in understanding the pathogenesis of MS. Numerous data from animal and clinical studies indicate that many GPCRs are critically involved in various aspects of MS pathogenesis, including antigen presentation, cytokine production, T-cell differentiation, T-cell proliferation, T-cell invasion, etc. In this review, we summarize the recent findings regarding the expression or functional changes of GPCRs in MS patients or animal models, and the influences of GPCRs on disease severity upon genetic or pharmacological manipulations. Hopefully some of these findings will lead to the development of novel therapies for MS in the near future.
Co-reporter:Lixin Cui ; Jing Li
Journal of Natural Products 2012 Volume 75(Issue 6) pp:1058-1062
Publication Date(Web):May 31, 2012
DOI:10.1021/np3000359
In order to identify small-molecule antagonists of Methuselah (Mth), a Drosophila G-protein-coupled receptor (GPCR) involved in life-span control, a library of natural compounds was screened, and it was found that rediocide A (1), a daphnane ester from the roots of Trigonostemon reidioides and used currently for flea control, potently inhibited calcium mobilization mediated by this receptor. Compound 1 inhibited calcium mobilization in GPCRs other than Mth, indicating that the inhibitory effect was not due to receptor antagonism but rather to a more general mechanism. It was found that 1 can induce GPCR desensitization and internalization, and such effects were mediated by the activation of conventional protein kinase C.
Co-reporter:Zhiyong Weng;Wei Wei;Xiaowu Dong
Monatshefte für Chemie - Chemical Monthly 2012 Volume 143( Issue 2) pp:303-308
Publication Date(Web):2012 February
DOI:10.1007/s00706-011-0645-9
A series of novel piperidin-4-ol derivatives were designed, synthesized, and evaluated for potential treatment of HIV. The compounds were obtained via an efficient synthetic route in excellent yields and have been characterized by 1H NMR, 13C NMR, MS, and elemental analysis. The CCR5 antagonistic activities of the compounds have also been evaluated.
Co-reporter:Ru Zhang;Xin Xie
Acta Pharmacologica Sinica 2012 33(3) pp:372-384
Publication Date(Web):2012-01-23
DOI:10.1038/aps.2011.173
G-protein-coupled receptors (GPCRs) mediate many important physiological functions and are considered as one of the most successful therapeutic targets for a broad spectrum of diseases. The design and implementation of high-throughput GPCR assays that allow the cost-effective screening of large compound libraries to identify novel drug candidates are critical in early drug discovery. Early functional GPCR assays depend primarily on the measurement of G-protein-mediated 2nd messenger generation. Taking advantage of the continuously deepening understanding of GPCR signal transduction, many G-protein-independent pathways are utilized to detect the activity of GPCRs, and may provide additional information on functional selectivity of candidate compounds. With the combination of automated imaging systems and label-free detection systems, such assays are now suitable for high-throughput screening (HTS). In this review, we summarize the most widely used GPCR assays and recent advances in HTS technologies for GPCR drug discovery.
Co-reporter:Quan Wang, Xinxiu Xu, Jun Li, Jing Liu, Haifeng Gu, Ru Zhang, Jiekai Chen, Yin Kuang, Jian Fei, Cong Jiang, Ping Wang, Duanqing Pei, Sheng Ding and Xin Xie
Cell Research 2011 21(10) pp:1424-1435
Publication Date(Web):July 5, 2011
DOI:10.1038/cr.2011.108
Somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) by defined factors. The low efficiency of reprogramming and genomic integration of oncogenes and viral vectors limited the potential application of iPSCs. Here we report that Lithium (Li), a drug used to treat mood disorders, greatly enhances iPSC generation from both mouse embryonic fibroblast and human umbilical vein endothelial cells. Li facilitates iPSC generation with one (Oct4) or two factors (OS or OK). The effect of Li on promoting reprogramming only partially depends on its major target GSK3β. Unlike other GSK3β inhibitors, Li not only increases the expression of Nanog, but also enhances the transcriptional activity of Nanog. We also found that Li exerts its effect by promoting epigenetic modifications via downregulation of LSD1, a H3K4-specific histone demethylase. Knocking down LSD1 partially mimics Li's effect in enhancing reprogramming. Our results not only provide a straightforward method to improve the iPSC generation efficiency, but also identified a histone demethylase as a critical modulator for somatic cell reprogramming.
Co-reporter:Tao Meng, Jue Wang, Hongli Peng, Guanghua Fang, Min Li, Bing Xiong, Xin Xie, Yongliang Zhang, Xin Wang, Jingkang Shen
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 3) pp:1133-1139
Publication Date(Web):March 2010
DOI:10.1016/j.ejmech.2009.12.018
The present study describes the identification via privileged structure-based approach of the benzhydrylpiperazine moiety as a potential scaffold to develop novel CB1 receptor modulators. Efficient structural optimization of the initial four hit compounds led to a high quality lead series, represented by compound 6c. Compound 6c is a highly potent and selective CB1 receptor inverse agonist that is able to reduce body weight in diet-induced obese Sprague–Dawley rats. The preparation of privileged structure-based library, the progression from hit to lead, the structure–activity relationships in the lead series and in vitro and in vivo activity of compound 6c are discussed.A series of benzhydrylpiperazine derivatives was identified as a novel class of CB1 receptor inverse agonists utilizing privileged structure approach.
Co-reporter:Ya Qiong Chen
Molecules and Cells 2010 Volume 29( Issue 1) pp:41-50
Publication Date(Web):2010 January
DOI:10.1007/s10059-010-0015-1
CRE-driven luciferase reporter is commonly used in drug screening systems involving G protein-coupled receptors (GPCRs). In a screen campaign designed to search for melanocortin-4 receptor (MC4R) agonists, podophyllotoxin, a microtubules disruptor, was found to induce cAMPresponsive element (CRE)-driven reporter expression. MC4R was not involved because podophyllotoxin induced CREB activation and CRE-driven transcription in cells not expressing MC4R. Previous studies indicated that intracellular calcium, PKA, and MAPKs are involved in CREB phosphorylation and activation. Our studies revealed that podophyllotoxin did not affect intracellular calcium level and the phosphorylation state of p38. Podophyllotoxin induced JNK and ERK activation, but blockade of JNK and ERK activation with specific inhibitors had no effect on podophyllotoxininduced CREB activation and CRE-regulated gene expression. Further experiments revealed that H89, a specific inhibitor of PKA, significantly inhibited podophyllotoxin-induced CREB activation. Podophyllotoxin itself did not alter intracellular cAMP level. Taken together, podophyllotoxin induces CREB activation and CRE-driven gene expression via PKA activation by a cAMP-independent mechanism.
Co-reporter:Fuying Li, Linghuan Gaob, Chenlei Yin, Jie Chen, Jinggen Liu, Xin Xie, Ao Zhang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 16) pp:4603-4606
Publication Date(Web):15 August 2009
DOI:10.1016/j.bmcl.2009.06.093
A series of skeletal rearranged indolomorphinans 7a–d were obtained by N-demethylation of 3-methoxy-N-methyl-14-hydroxymorphinan-6-one 12 followed by N-realkylation, reduction and Fischer indole cyclization. The structure of the novel skeleton was confirmed by X-ray analysis. These new indoles displayed moderate binding affinity and selectivity at the μ receptor, with compound 7b showing the highest affinity at this receptor with a Ki value of 40 nM, and 6- and 25-fold selectivity against δ and κ receptors, respectively. Function assays showed that indolopropellanes 7b and 7c possessed full agonistic activity at all the opioid receptors indicating a different interaction model existed.A series of skeletal rearranged indolomorphinans 7a–d was synthesized, and their binding affinity and functional activity for opioid receptors were evaluated.
Co-reporter:Fuying Li;Chenlei Yin;Jie Chen;Jinggen Liu ;Ao Zhang
ChemMedChem 2009 Volume 4( Issue 12) pp:2103-2110
Publication Date(Web):
DOI:10.1002/cmdc.200900308
Abstract
A series of new 14-hydroxymorphinan analogues with a thiazole or imidazo[2,1-b]thiazole fragment as the heterocyclic function fused to ring C were designed and synthesized. These compounds can be viewed as the result of a direct modification at ring C of the 14-hydroxymorphinan scaffold. Among these compounds, three were identified as having potent binding affinity (∼1 nM) at both κ and μ receptors, and acting as agonists at κ and partial agonists or antagonists at μ receptors. In view of the promising results from studies on compounds with mixed κ and μ receptor activities, these new compounds warrant further investigation.



![5-[(2R)-2-[2-(2-ETHOXYPHENOXY)ETHYLAMINO]PROPYL]-2-METHOXYBENZENESULFONAMIDE](/data/chemimg/307000/106133-20-4.png)
![5-[(2R)-2-[2-(2-ETHOXYPHENOXY)ETHYLAMINO]PROPYL]-2-METHOXYBENZENESULFONAMIDE](/data/chemimg/307000/106133-20-4_b.png)










![1H-Indole-3-acetamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/13500/13433-48-2.png)
![1H-Indole-3-acetamide, N-[2-(3,4-dimethoxyphenyl)ethyl]-](http://img.cochemist.com/ccimg/13500/13433-48-2_b.png)







![5,8,13,13a-tetrahydro-9,10-dimethoxy-6H-benzo[g]benzo-1,3-dioxolo[5,6-a]quinolizine](http://img.cochemist.com/ccimg/600/522-97-4.png)
![5,8,13,13a-tetrahydro-9,10-dimethoxy-6H-benzo[g]benzo-1,3-dioxolo[5,6-a]quinolizine](http://img.cochemist.com/ccimg/600/522-97-4_b.png)












![N-[2-Chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl]-N'-(5-methyl-3-isoxazolyl)urea](http://img.cochemist.com/ccimg/475200/475108-18-0.png)
![N-[2-Chloro-4-[(6,7-dimethoxy-4-quinolyl)oxy]phenyl]-N'-(5-methyl-3-isoxazolyl)urea](http://img.cochemist.com/ccimg/475200/475108-18-0_b.png)






![4-[3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6-quinolinecarboxamide](http://img.cochemist.com/ccimg/417800/417716-92-8.png)
![4-[3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6-quinolinecarboxamide](http://img.cochemist.com/ccimg/417800/417716-92-8_b.png)
![4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1h-benzimidazol-2-yl]quinolin-2(1h)-one](http://img.cochemist.com/ccimg/405200/405169-16-6.png)
![4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1h-benzimidazol-2-yl]quinolin-2(1h)-one](http://img.cochemist.com/ccimg/405200/405169-16-6_b.png)






![8-Azabicyclo[3.2.1]octane-3-carbonitrile, 8-(phenylmethyl)-](http://img.cochemist.com/ccimg/186000/185985-38-0.png)
![8-Azabicyclo[3.2.1]octane-3-carbonitrile, 8-(phenylmethyl)-](http://img.cochemist.com/ccimg/186000/185985-38-0_b.png)
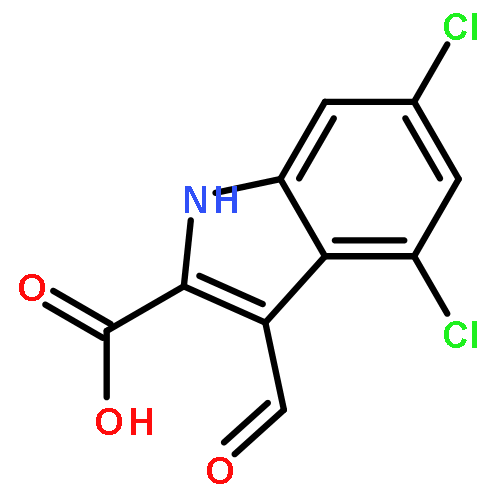



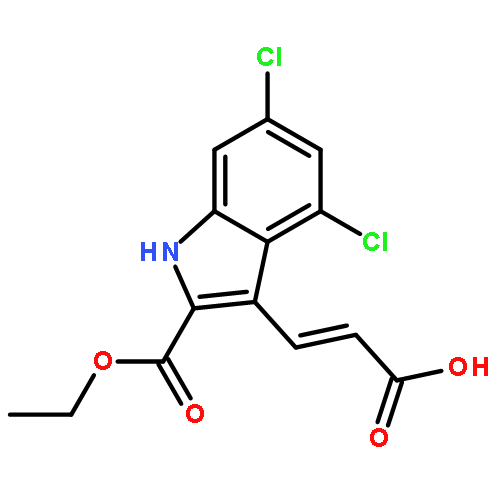








![Benzenemethanol, 3-[2-(7-chloro-2-quinolinyl)ethenyl]-, (E)-](/data/chemimg/1714400/139425-04-0.png)
![Benzenemethanol, 3-[2-(7-chloro-2-quinolinyl)ethenyl]-, (E)-](/data/chemimg/1714400/139425-04-0_b.png)
![b-Alanine, N,N-dimethyl-,(3R,4aR,5S,6S,6aS,10S,10aR,10bS)-5-(acetyloxy)-3-ethenyldodecahydro-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-1H-naphtho[2,1-b]pyran-6-ylester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/138700/138605-00-2.png)
![b-Alanine, N,N-dimethyl-,(3R,4aR,5S,6S,6aS,10S,10aR,10bS)-5-(acetyloxy)-3-ethenyldodecahydro-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-1H-naphtho[2,1-b]pyran-6-ylester, hydrochloride (1:1)](http://img.cochemist.com/ccimg/138700/138605-00-2_b.png)




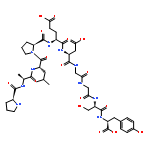
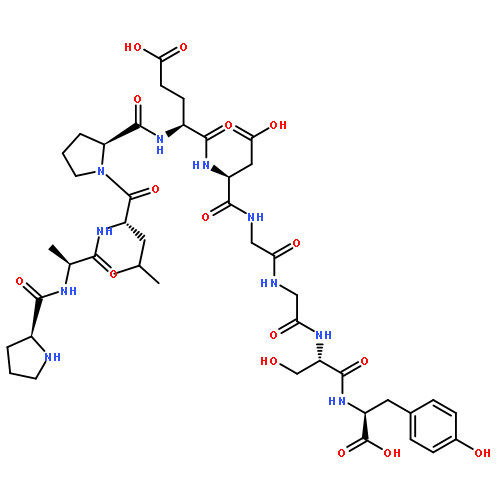




![ACETAMIDE, N-[3-METHYL-1-(PHENYLMETHYL)-3-PYRROLIDINYL]-](http://img.cochemist.com/ccimg/96600/96567-94-1.png)
![ACETAMIDE, N-[3-METHYL-1-(PHENYLMETHYL)-3-PYRROLIDINYL]-](http://img.cochemist.com/ccimg/96600/96567-94-1_b.png)
![[2,4'-Bithiazole]-4-carboxylic acid, 2'-methyl-](http://img.cochemist.com/ccimg/91600/91520-83-1.png)
![[2,4'-Bithiazole]-4-carboxylic acid, 2'-methyl-](http://img.cochemist.com/ccimg/91600/91520-83-1_b.png)


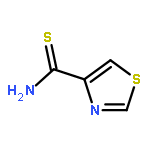
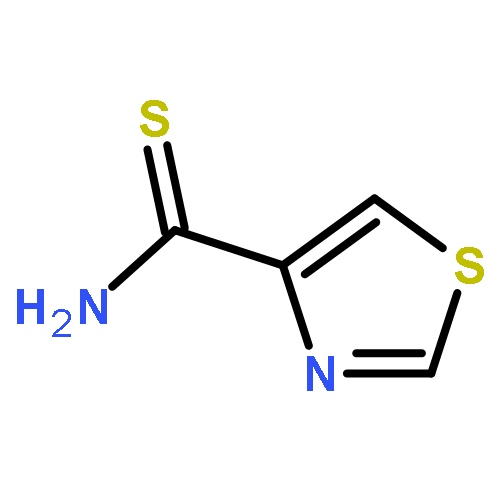
![Benzoic acid,4-[(1E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/71500/71441-28-6.png)
![Benzoic acid,4-[(1E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/71500/71441-28-6_b.png)
![2-(3,4-dichlorophenyl)-N-methyl-N-[2-(pyrrolidin-1-yl)cyclohexyl]acetamide](http://img.cochemist.com/ccimg/67200/67198-13-4.png)
![2-(3,4-dichlorophenyl)-N-methyl-N-[2-(pyrrolidin-1-yl)cyclohexyl]acetamide](http://img.cochemist.com/ccimg/67200/67198-13-4_b.png)
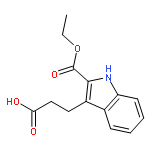
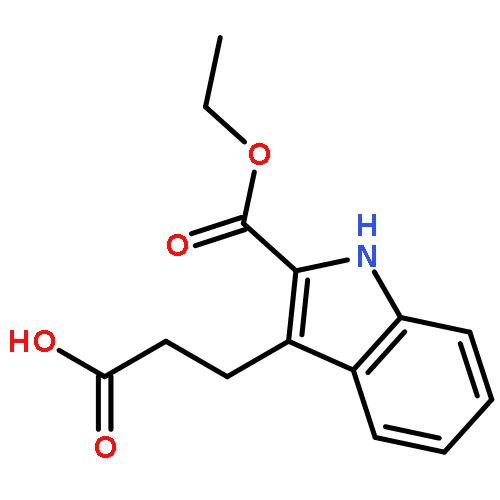
![Propanoic acid, 2-[(2-methoxyphenyl)hydrazono]-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/33000/32979-69-4.png)
![Propanoic acid, 2-[(2-methoxyphenyl)hydrazono]-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/33000/32979-69-4_b.png)