Co-reporter:Jian-Feng Zheng, Xiu-Ning Hu, Zhen Xu, Dong-Cheng Cai, Tai-Long Shen, and Pei-Qiang Huang
The Journal of Organic Chemistry September 15, 2017 Volume 82(Issue 18) pp:9693-9693
Publication Date(Web):August 29, 2017
DOI:10.1021/acs.joc.7b01768
Versatile and chemoselective C–C bond forming methods for the one-pot transformation of amides into other classes of compounds are highly demanding. In this report, we demonstrate the reductive addition of isocyanoacetates to common amides and lactams to produce 5-methoxyoxazoles or bicyclic imidazolines. This one-pot procedure involves partial reduction of amides with Schwartz reagent and chemoselective addition of the carbon of isocyanide group or α-carbon in isocyanoacetates. The quite different reactivity of the isocyanoacetate is due to the different steric hindrance of the amides and lactams.
Co-reporter:Zhongyi Mao, Elisabetta Martini, Guillaume Prestat, Julie Oble, Pei-Qiang Huang, Giovanni Poli
Tetrahedron Letters 2017 Volume 58, Issue 44(Issue 44) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.tetlet.2017.09.046
•New analogues of the 2-carboxyl-6-hydroxyoctahydroindole (CHOI) building block.•Key bicyclic lactams synthesized by diverging Pd-catalyzed allylations.•Selectivity of introducing 1C unity as a function of the double bond position.•Stereoselective epoxidation and dihydroxylation of 2-methoxycarbonyl hexahydroindoles.Pd-catalyzed allylations of cyclic bis-allylic substrates, carried out either as two separate steps or in a pseudo-domino fashion, can generate 2-carboxyl-hexahydroindoles bearing an unsaturation in different positions. Sequential homologation, and epoxidation or syn-dihydroxylation steps were investigated to access analogues of the bicyclic 2-carboxyl-6-hydroxyoctahydroindole motif of aeruginosins, a family of peptides displaying serine protease inhibitor activity.Download high-res image (130KB)Download full-size image
Co-reporter:Yan-Jiao Gao, Shi-Peng Luo, Jian-Liang Ye, Pei-Qiang Huang
Chinese Chemical Letters 2017 Volume 28, Issue 6(Volume 28, Issue 6) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.cclet.2017.04.016
We describe the design and execution of a novel synthetic route to the tricyclic core of haliclonin A, a tetracyclic marine natural product. The approach features Bachi’s thiol-medicated free radical cyclization of alkenyl isocyanide to build the bridged ring system, and ring-closing metathesis (RCM) reaction to form the macrocycle. Execution of the synthetic plan ultimately resulted in a diazatricyclic compound. By means of 2D NMR techniques, the structure of this compound was revealed to an unexpected product 8. Analysis of the synthetic pathways allowed concluding that the unexpected product is a result of an “unexpected” migration of olefinic bond during dioxolanation of the 2-cyclohexenone derivative 7. This investigation also resulted in a concise construction of the functionalized hexahydro-1H-isoindole-1,5(4H)-dione 12 and the macrocyclic tricyclic ring system 8.Download high-res image (92KB)Download full-size imageAs revealed by means of 2D NMR techniques, a synthetic approach designed for the concise construction of the tricyclic core (2) of haliclonin A, resulted in the unexpected di-aza-tricyclic compound 8.
Co-reporter:Pei-Qiang Huang;Hang Chen
Chemical Communications 2017 vol. 53(Issue 93) pp:12584-12587
Publication Date(Web):2017/11/21
DOI:10.1039/C7CC07457C
The catalytic conversion of amides to ketones is highly desirable yet challenging in organic synthesis. We herein report the first Ni/bis-NHC-catalyzed cross-coupling of N-acylpyrrole-type amides with arylboronic esters to obtain diarylketones. This method is facilitated by a new chelating bis-NHC ligand. The reaction tolerates diverse functional groups on both arylamide and arylboronic ester partners including sensitive ester and ketone groups.
Co-reporter:Pei-Qiang Huang;Ying-Hong Huang
Chinese Journal of Chemistry 2017 Volume 35(Issue 5) pp:613-620
Publication Date(Web):2017/05/01
DOI:10.1002/cjoc.201600700
AbstractDescribed in this paper are the results of an investigation on the extension of the C-H alkyliminylation and acylation of alkenes with secondary amides. The nucleophilic partner has been extended to cover a series of functionalized alkenes bearing functional groups including ester, α,β-unsaturated ester, uncongested ketone groups, as well as enol derivatives of acetaldehyde such as enol ether and enamides. The electrophilic partner has been extended from N-(2,6-dimethylphenyl) and N-methyl amides to N-n-butyl, and N-cyclohexyl amides. The results demonstrated that the method can be used to synthesize a number of functionalized α,β-unsaturated ketimines and α,β-enones in an efficient, high yielding, and one-pot manner. The method was applied to a concise synthesis of (E)-6-styryltetrahydro-2H-pyran-2-one (5), an immediate intermediate in the syntheses of a series of styryllactone natural products including (±)-goniothalamine (6), (±)-goniothalamine oxide (7), (±)-goniodiol (8), (±)-leiocarpin A (9), (±)-9-deoxygoniopypyrone (10), and (±)-7-epi-goniodiol (11).
Co-reporter:Pei-Qiang Huang;Ting Fan
European Journal of Organic Chemistry 2017 Volume 2017(Issue 43) pp:6369-6374
Publication Date(Web):2017/11/24
DOI:10.1002/ejoc.201701060
Although the aldol condensation is a well-known reaction, the aza-type aldol condensation, namely, the intramolecular condensation of common keto lactams leading to bicyclic vinylogous lactams, is a highly demanding transformation, with potential applications in both organic synthesis and medicinal chemistry. The known methods for this type of cyclization require several steps. In this paper, we disclose a straightforward approach that consists of the in-situ formation of silyl enol ethers with tert-butyldimethylsilyl trifluoromethanesulfonate (TBDMSOTf), lactam activation with triflic anhydride (Tf2O), and a tandem cyclocondensation reaction. The reaction can be run in a one-pot manner or in a two-step fashion, both under mild conditions. The yields for the one-pot version were 52–80 %. In some cases, the two-step version gave higher overall yields (44–85 %) than the one-pot version.
Co-reporter:Pei-Qiang Huang;Wei Ou
European Journal of Organic Chemistry 2017 Volume 2017(Issue 3) pp:582-592
Publication Date(Web):2017/01/18
DOI:10.1002/ejoc.201601326
Full details of our investigations into aza-Knoevenagel-type reactions with common tertiary amides as the electrophilic partner are reported. The method is based on the addition of stabilized carbanionic nucleophiles to the amides, which are activated in situ with triflic anhydride (Tf2O). The reaction proceeds under mild conditions and tolerates several sensitive functional groups including enamide and tert-butyldimethylsilyloxy (TBSO) groups. Significantly, with amides bearing more reactive ester, cyano, and aldehyde groups, the reaction occurs chemoselectively at the least reactive amide group. Such “umpolung” of the chemoselectivity in C=C bond formation is challenging, rare, and of synthetic value. Vinylogous bis-urethanes (aza-Knoevenagel-type condensation products), vinylogous urethanes, and vinylogous amides can be synthesized by employing enolates/carbanions generated from methyl ketones, malonic acid monoester, 2-phenylacetate, or (benzylsulfonyl)benzene. Moreover, when higher homologues of acetate were used, β-keto esters were obtained directly from amides. The method has been applied to the one-step syntheses of several known key intermediates in the total syntheses of alkaloids and 4-quinolone antibiotics. An efficient and mild intramolecular Friedel–Crafts-type cyclization of an anilinyl ester has also been achieved with Tf2O as an effective promoter. This amide-based method is a direct and umpoled alternative to the versatile but stepwise one based on thionation and the Eschenmoser sulfide contraction.
Co-reporter:Pei-Qiang Huang;Ying-Hong Huang;Shu-Ren Wang
Organic Chemistry Frontiers 2017 vol. 4(Issue 3) pp:431-444
Publication Date(Web):2017/02/28
DOI:10.1039/C6QO00720A
We report two efficient and versatile alkenylative cyclization methods for the one-pot synthesis of substituted pyrrolidine, piperidine, indolizidine, and quinolizidine ring systems, and enimino carbocycles, respectively. The first method consists of amide activation (Tf2O) induced dehydrative coupling of halogenated secondary amides with alkenes and the NaBH4 reduction triggered tandem cyclization reaction, while the second one features the Tf2O-promoted novel modes of extended Bischler–Napieralski cyclization reactions of olefinic secondary amides. Taking advantage of triflic anhydride (Tf2O) as the amide activating reagent, the hitherto failed two-step process for the construction of the quinolizidine ring system via intramolecular vinylogous Bischler–Napieralski cyclization has been realized in one pot. The first method was applied to the protecting-group-free one-pot synthesis of the cytotoxic natural product caulophyllumine B (5) and its bioactive derivatives, and to the synthesis of δ-coniceine and 6-styrylpiperidin-2-one. When ethyl vinyl ether and enamides were used as functionalized alkenes, saturated 1,3-amino-ether/amido products 4A1–4A3 were obtained in 73%–74% yields.
Co-reporter:Jian-Liang Ye, Yang Liu, Zhi-Ping Yang and Pei-Qiang Huang
Chemical Communications 2016 vol. 52(Issue 3) pp:561-563
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5CC07480K
The asymmetric total synthesis of (+)-N-acetyl norloline, the putative biogenic precursor of all known loline alkaloids, has been achieved in 12 steps from commercially available (R)-glyceraldehyde acetonide. The synthesis relies on the Rassu/Casiraghi's vinylogous aldol reaction, an intramolecular oxa-heteroconjugate addition and a reductive amination to establish the four contiguous stereogenic centers and construct the strained oxygen-bridge under mild conditions.
Co-reporter:Pei-Qiang Huang, Su-Yu Huang, Long-Hui Gao, Zhong-Yi Mao, Zong Chang and Ai-E Wang
Chemical Communications 2016 vol. 52(Issue 26) pp:4840-4840
Publication Date(Web):10 Mar 2016
DOI:10.1039/C6CC90127A
Correction for ‘Enantioselective total synthesis of (+)-methoxystemofoline and (+)-isomethoxystemofoline’ by Pei-Qiang Huang et al., Chem. Commun., 2015, 51, 4576–4578.
Co-reporter:Qi-Wei Lang;Xiu-Ning Hu
Science China Chemistry 2016 Volume 59( Issue 12) pp:1638-1644
Publication Date(Web):2016/12/01
DOI:10.1007/s11426-016-0224-5
The direct partial reduction of highly stable secondary amides to more reactive aldimines and aldehydes is a challenging yet highly demanding transformation. In this context, only three methods have been reported. We report herein an improved version of the Charette’s method. Our protocol consists of activation of secondary amides with triflic anhydride/2-fluoropyridine, and partial reduction of the resulting intermediates with 1,1,3,3-tetramethyldisiloxane (TMDS), which delivered aldimines or aldehydes upon acidic hydrolysis. Aromatic amides were reduced to the corresponding aldimines in 85%–100% NMR yields, and yields (NMR) from aliphatic amides were 72%–86%. Acidic hydrolysis of the aldimine intermediates afforded, in one-pot, the corresponding aldehydes in 80%–96% yields. A simple protocol was established to isolate labile aldimines in pure form in 92%–96% yields. The improved method gave generally higher yields as compared to the known ones, and features the use of cheaper and more atom-economical TMDS as a chemoselective reducing agent. In addition, a convenient extraction protocol has been established to allow the isolation of amines, which constitutes a mild method for the N-deacylation of amides, another highly desirable transformation. The extended method retains the advantages of the original method of Charette in terms of mild conditions, good functional group tolerance, and excellent chemoselectivity.
Co-reporter:Pei-Qiang Huang, Qi-Wei Lang, and Yan-Rong Wang
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4235-4243
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.joc.6b00572
The combination of amide activation by Tf2O with B(C6F5)3-catalyzed hydrosilylation with TMDS constitutes a method for the one-pot reduction of secondary amides to amines under mild conditions. The method displays a broad applicability for the reduction of many types of substrates, and shows good compatibility and excellent chemoselectivity for many sensitive functional groups. Reductions of a multifunctionalized α,β-unsaturated amide obtained from another synthetic methodology, and a C–H functionalization product produced the corresponding amines in good to excellent yield. Chemoselective reduction of enantiomeric pure (ee >99%) tetrahydro-5-oxo-2-furaneamides yielded 5-(aminomethyl)dihydrofuran-2(3H)-ones in a racemization-free manner. The latter were converted in one pot to N-protected 5-hydroxypiperidin-2-ones, which are building blocks for the synthesis of many natural products. Further elaboration of an intermediate led to a concise four-step synthesis of (−)-epi-pseudoconhydrine.
Co-reporter:Pei-Qiang Huang, Qi-Wei Lang, and Xiu-Ning Hu
The Journal of Organic Chemistry 2016 Volume 81(Issue 21) pp:10227-10235
Publication Date(Web):June 10, 2016
DOI:10.1021/acs.joc.6b01080
The one-pot reductive 1,3-dipolar cycloaddition of secondary aromatic N-(trimethylsilylmethyl)amides with reactive dipolarophiles is reported. The method relies on the in situ generation of nonstabilized NH azomethine ylide dipoles via amide activation with triflic anhydride, partial reduction with 1,1,3,3-tetramethyldisiloxane (TMDS), and desilylation with cesium fluoride (CsF). Running under mild conditions, the reaction tolerated several sensitive functional groups and provided cycloadducts in 71–93% yields. The use of less reactive dipolarophile methyl acrylate led to the cycloadduct in only 40% yield. A (Z) geometric intermediate of NH-azomethine 1,3-dipole was postulated to account for the observed higher yields and higher cis diastereoselectivity for the substrates bearing an electron-withdrawing group. This model features an unconventional cyclic transition state via carbanion–aryl ring interaction. Because the starting secondary amides can be prepared from common primary amides, the current method also constitutes a two-step transformation of primary amides.
Co-reporter:Pei-Qiang Huang, Ying-Hong Huang, and Kai-Jiong Xiao
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:9020-9027
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.joc.6b01647
The direct transformation of common secondary amides into aromatic ketimines and aromatic ketones with C–C bond formation is described. The reaction can also be used for N-deacylation of secondary amides to release amines. This method consists of in situ amide activation with triflic anhydride and intermolecular capture of the resulting highly electrophilic nitrilium intermediate with an arene. The reaction is applicable to various kinds of secondary amides (electrophiles), but only electron-rich and moderately electron-rich arenes can be used as nucleophiles. Thanks to the use of bench stable arenes instead of reactive and basic organometallics as nucleophiles, the reaction proceeded with high chemoselectivity at the secondary amido group in the presence of a series of sensitive functional groups such as aldehyde, ketone, ester, cyano, nitro, and tertiary amido groups. The reaction can be viewed as a Friedel–Crafts-type reaction using secondary amides as acylating agents or as an intermolecular version of the Bischler–Napieralski reaction.
Co-reporter:Lian-Dong Guo;Xiong-Zhi Huang;Shi-Peng Luo;Wen-Sen Cao; Yuan-Ping Ruan;Dr. Jian-Liang Ye;Dr. Pei-Qiang Huang
Angewandte Chemie 2016 Volume 128( Issue 12) pp:4132-4136
Publication Date(Web):
DOI:10.1002/ange.201512005
Abstract
The first total synthesis of the alkaloid (−)-haliclonin A is reported. The asymmetric synthesis relied on a novel organocatalytic asymmetric conjugate addition of nitromethane with 3-alkenyl cyclohex-2-enone to set the stereochemistry of the all-carbon quaternary stereogenic center. The synthesis also features a Pd-promoted cyclization to form the 3-azabicyclo[3,3,1]nonane core, a SmI2-mediated intermolecular reductive coupling of enone with aldehyde to form the requisite secondary chiral alcohol, ring-closing alkene and alkyne metathesis reactions to build the two aza-macrocyclic ring systems, and an unprecedented direct transformation of enol into enone.
Co-reporter:Lian-Dong Guo;Xiong-Zhi Huang;Shi-Peng Luo;Wen-Sen Cao; Yuan-Ping Ruan;Dr. Jian-Liang Ye;Dr. Pei-Qiang Huang
Angewandte Chemie International Edition 2016 Volume 55( Issue 12) pp:4064-4068
Publication Date(Web):
DOI:10.1002/anie.201512005
Abstract
The first total synthesis of the alkaloid (−)-haliclonin A is reported. The asymmetric synthesis relied on a novel organocatalytic asymmetric conjugate addition of nitromethane with 3-alkenyl cyclohex-2-enone to set the stereochemistry of the all-carbon quaternary stereogenic center. The synthesis also features a Pd-promoted cyclization to form the 3-azabicyclo[3,3,1]nonane core, a SmI2-mediated intermolecular reductive coupling of enone with aldehyde to form the requisite secondary chiral alcohol, ring-closing alkene and alkyne metathesis reactions to build the two aza-macrocyclic ring systems, and an unprecedented direct transformation of enol into enone.
Co-reporter:Pei-Qiang Huang, Su-Yu Huang, Long-Hui Gao, Zhong-Yi Mao, Zong Chang and Ai-E Wang
Chemical Communications 2015 vol. 51(Issue 22) pp:4576-4578
Publication Date(Web):08 Jan 2015
DOI:10.1039/C4CC09598G
The first enantioselective total synthesis of (+)-methoxystemofoline (2) and (+)-isomethoxystemofoline (3) has been reported. The synthesis employed the halide-assisted bromotropanonation method that we developed recently to construct the core structure, and Overman's strategy for the implementation of the butenolide moiety. Through this work, the structure of methoxystemofoline was revised as 2 with an E-alkene, and its absolute configuration was established.
Co-reporter:Pei-Qiang Huang, Qi-Wei Lang, Ai-E Wang and Jian-Feng Zheng
Chemical Communications 2015 vol. 51(Issue 6) pp:1096-1099
Publication Date(Web):25 Nov 2014
DOI:10.1039/C4CC08330J
We report the first one-pot reductive homocoupling reaction of secondary amides and cross-coupling reaction of secondary amides with ketones to give secondary vicinal diamines and amino alcohols. This method relies on the direct generation of α-amino carbon radicals from secondary amides by activation with trifluoromethanesulfonic anhydride, partial reduction with triethylsilane and samarium diiodide-mediated single-electron transfer. The reactions were run under mild conditions and tolerated several functional groups.
Co-reporter:Ai-E Wang, Zong Chang, Wei-Ting Sun, and Pei-Qiang Huang
Organic Letters 2015 Volume 17(Issue 3) pp:732-735
Publication Date(Web):January 27, 2015
DOI:10.1021/acs.orglett.5b00004
With Tf2O as the activation reagent, a mild and general method has been developed for the bisphosphonylation of both secondary and tertiary amides. The protocol is highly efficient and chemoselective, and it tolerates a number of sensitive functional groups such as cyano, ester, and aldehyde groups.
Co-reporter:Pei-Qiang Huang;Wei Ou ;Jian-Liang Ye
Chinese Journal of Chemistry 2015 Volume 33( Issue 6) pp:655-662
Publication Date(Web):
DOI:10.1002/cjoc.201400762
Abstract
A versatile and divergent two-step transformation of malimides to racemic tetramates and tetramic acids is described. The method consists of Grignard reagent addition with malimides to give hemiaminals and concentrated HCl-promoted chemoselective transformations of the latter. When running the reaction in CH2Cl2 and in the presence of 2.5 molar equiv. of conc. HCl, 5-alkyltetramates were formed, while in neat conc. HCl, 5-alkyltetramic acids were obtained. Using this method, a variety of title compounds were prepared in good to excellent yields (82%–99%, and 76%/85%). The work also constitutes a formal racemic total synthesis of reutericyclin. On the basis of the experimental evidences and of a deuterium labeling experiment, a plausible reaction mechanism was proposed. This work thus demonstrated two new types of step economical and chemodivergent transformations starting from malic acid.
Co-reporter:Shi-Peng Luo;Hui Geng;Yu Wang
Chinese Journal of Chemistry 2015 Volume 33( Issue 6) pp:646-654
Publication Date(Web):
DOI:10.1002/cjoc.201400849
Abstract
We report the concise and protecting-group-free enantioselective total syntheses of circumdatins F and H. In view of the extreme importance of analogs of quinazolinone alkaloids in drug research and discovery, four analogs of bioactive quinazolinobenzodiazepine alkaloids, including demethoxycircumdatin H (12) and N-demethylbenzomalvin A (13), have been synthesized. The method is based on the low-valent titanium-promoted intramolecular reductive coupling of imides with o-nitrobenzimides, which yielded quinazolino[3,2-a][1,4]benzodiazepines under mild conditions. In addition, heptacyclic dehydraasperlicin E (16) has been synthesized from asperlicin C by a NCS-mediated dehydra-cyclization reaction.
Co-reporter:Pei-Qiang Huang, Yu Wang, Shi-Peng Luo, Hui Geng, Yuan-Ping Ruan, Ai-E Wang
Tetrahedron Letters 2015 Volume 56(Issue 10) pp:1255-1258
Publication Date(Web):4 March 2015
DOI:10.1016/j.tetlet.2015.01.084
We report a procedure—economical method for the highly enantioselective and protecting-group free total syntheses of nonpeptidal CCK antagonists asperlicins C and E. Starting from l-tryptophan, the synthesis of asperlicin C has been achieved in three steps, which features the low-valent titanium (LVT: TiCl4–Zn combination)-mediated reductive cyclization of o-nitrobenzamide to construct the (3H)-quinazolin-4-one moiety. This is the first employment of LVT for the synthesis of asperlicin C, which allowed accessing asperlicin C in >99% enantioselectivity. Asperlicin C was converted, in one-pot, into asperlicin E and 2,3-di-epi-asperlicin E by dimethyl dioxirane (DMDO)-mediated tandem reactions. The use of DMDO as a green, cheap, and easily available oxidant to replace the photochemical method renders the synthesis of asperlicin E experimentally convenient.
Co-reporter:Pei-Qiang Huang;Hui Geng;Yong-Song Tian;Qiu-Ran Peng
Science China Chemistry 2015 Volume 58( Issue 3) pp:478-482
Publication Date(Web):2015 March
DOI:10.1007/s11426-014-5270-0
The first enantioselective total synthesis of (+)-preussin B and an improved synthesis of the antifungal alkaloid (+)-preussin are described. Our approach relied on the four step-economical synthetic methods developed in our laboratory: (1) the cis-diastereoselective reductive dehydroxylation of hemiaminals; (2) the direct amide/lactam reductive alkylation; (3) the one-pot N,O-bisdebenzylation-N-methylation; and (4) the one-step synthesis of malimide from malic acid. Both total syntheses are quite concise, which have been achieved in six steps, and gave overall yields of 25.7% and 27.6%, respectively.
Co-reporter:Pei-Qiang Huang, Yu Wang, Kai-Jiong Xiao, Ying-Hong Huang
Tetrahedron 2015 Volume 71(Issue 24) pp:4248-4254
Publication Date(Web):17 June 2015
DOI:10.1016/j.tet.2015.04.074
The direct transformation of amides into ketones by addition of organometallic reagents has attracted the attention of organic chemists for a long time. However limited methods are reliable for common amides and have found synthetic applications. Here we report a method featuring in situ activation of tertiary amides with triflic anhydride (Tf2O) followed by addition of Grignard reagents. The method displays a good generality in scope for both amides and Grignard reagents, and it can be viewed as the acylation of Grignard reagents using amides as stable and selective acylating agents. Moreover, this deaminative alkylation reaction provides a mild method for the N-Deacylation of amides to give free amines.
Co-reporter:Hui Geng, Pei-Qiang Huang
Tetrahedron 2015 Volume 71(Issue 23) pp:3795-3801
Publication Date(Web):10 June 2015
DOI:10.1016/j.tet.2015.03.094
Triflic anhydride in combination with 2-fluoropyridine effectively dehydrates secondary amides to afford nitriles under mild reaction conditions. The reaction is general in scope and compatible with the use of aliphatic, α,β-unsaturated, aromatic, and heteroaromatic amides bearing either secondary, tertiary, or benzylic N-alkyl groups. The reaction also shows good to excellent chemoselectivity for the secondary amide and tolerates several labile functional groups.
Co-reporter:Pei-Qiang Huang, Ying-Hong Huang, Kai-Jiong Xiao, Yu Wang, and Xiao-Er Xia
The Journal of Organic Chemistry 2015 Volume 80(Issue 5) pp:2861-2868
Publication Date(Web):February 5, 2015
DOI:10.1021/jo502929x
A one-pot reaction for the transformation of common secondary amides into amines with C–C bond formation is described. This method consists of in situ amide activation with Tf2O–partial reduction–addition of C-nucleophiles. The method is general in scope, which allows employing both hard nucleophiles (RMgX, RLi) and soft nucleophiles, as well as enolates. With the use of soft nucleophiles, the reaction proceeded with high chemoselectivity at a secondary amide in the presence of ester, cyano, nitro, and tertiary amide groups.
Co-reporter:Qi-Long Peng, Shi-Peng Luo, Xiao-Er Xia, Liang-Xian Liu and Pei-Qiang Huang
Chemical Communications 2014 vol. 50(Issue 16) pp:1986-1988
Publication Date(Web):12 Dec 2013
DOI:10.1039/C3CC48833K
The total synthesis of the alkaloid (−)-chaetominine (1) has been achieved in four steps with an overall yield of 33.4%. Key features of our strategy include a one-pot cascade indole epoxidation – epoxide ring-opening cyclization – lactamization reaction sequence, and the use of a nitro group as a latent amino group for the one-pot construction of the quinazolinone ring. This constitutes a step economical, redox economical and protecting group-free total synthesis.
Co-reporter:Pei-Qiang Huang, Wei Ou, Kai-Jiong Xiao and Ai-E Wang
Chemical Communications 2014 vol. 50(Issue 63) pp:8761-8763
Publication Date(Web):19 Jun 2014
DOI:10.1039/C4CC03826F
We report one-pot and chemoselective Knoevenagel-type reactions using highly stable amides and lactams as the electrophilic substrates. The method is based on the in situ activation of amide carbonyl with triflic anhydride and a subsequent reaction with carbanions generated in situ from carbonyl compounds. The amide-based method is an alternative to the versatile thioamide-based Eschenmoser sulfide contraction.
Co-reporter:Chu-Pei Xu, Shi-Peng Luo, Ai-E Wang and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 18) pp:2859-2863
Publication Date(Web):13 Feb 2014
DOI:10.1039/C4OB00314D
We demonstrated, for the first time, that on the basis of chemistry principles, the hexacyclic peptidyl alkaloid (−)-chaetominine (1) can be synthesized in a straightforward manner from L-Trp. The approach features the efficient generation of molecular complexity via a tandem C3/C14 syn-selective epoxidation (dr = 3:2)–annulative ring-opening reaction and a regioselective epimerization at C14. The successful production of (−)-chaetominine (1) from L-Trp could be helpful for revealing how the configuration of L-tryptophan becomes inverted in the biosynthetic pathway of (−)-chaetominine (1).
Co-reporter:Xiao-Gang Wang, Ai-E Wang, Pei-Qiang Huang
Chinese Chemical Letters 2014 Volume 25(Issue 2) pp:193-196
Publication Date(Web):February 2014
DOI:10.1016/j.cclet.2013.12.003
A short formal stereoselective synthesis of (−)-swainsonine (1) is described. Our synthesis started with the versatile building block (R)-3-benzyloxyglutarimide 5. Through controlled regioselective reduction, Ley's-sulfone chemistry (N-α-sulfonylation and ZnCl2-catalyzed N-α-amidovinylation), an RCM reaction, and an amide reduction, the synthesis of unsaturated indolizidine (8R,8aS)-3 has been achieved in five steps. The indolizidine (8R,8aS)-3 is an advanced intermediate toward the synthesis of (−)-swainsonine (1).The synthesis of unsaturated indolizidine (8R,8aS)-3, an advanced intermediate toward the synthesis of (−)-swainsonine (1), has been achieved in five steps from the versatile building block (R)-3-benzyloxyglutarimide 5, via Ley's-sulfone-based α-amidovinylation and the RCM reaction as the key steps.
Co-reporter:Shi-Peng Luo;Qi-Long Peng;Chu-Pei Xu;Ai-E Wang
Chinese Journal of Chemistry 2014 Volume 32( Issue 8) pp:757-770
Publication Date(Web):
DOI:10.1002/cjoc.201400413
Abstract
Full details of the enantioselective four-step and five-step total syntheses of (−)-chaetominine from D-Trp and L-Trp are described. Featuring an oxidative double cyclization reaction, and tandem C14 epimerization-lactamization reactions as key steps, the method provides a rapid access to (−)-chaetominine (6a) and analogues. The total syntheses of (−)-chaetominine (6a) are so far the most concise and efficient. Through comprehensive investigation, the stereochemical requirements for the double cyclization reaction were revealed, and the confusion regarding physicochemical properties of this natural product was clarified. Moreover, short pathways to complexity generation, a scenarios revealed for the biosynthesis of fungal peptidyl alkaloid multi-cyclic scaffolds, have been validated through the chemical synthesis. On the basis of these findings, a plausible biosynthetic pathway for (−)-chaetominine (6a) was suggested.
Co-reporter:ZhongYi Mao;SuYu Huang;LongHui Gao;AiE Wang;PeiQiang Huang
Science China Chemistry 2014 Volume 57( Issue 2) pp:252-264
Publication Date(Web):2014 February
DOI:10.1007/s11426-013-4998-2
A full account of the novel and flexible approach to hydroxylated 8-azabicyclo[3,2,1]octan-3-ones and 9-azabicyclo[3,3,1] nonan-3-ones is presented. Using keto-lactams as the starting materials, this two-step method consists of silyl enol ether formation with TBDMSOTf, lactam activation with Tf2O/DTBMP, and halide-promoted cyclization. Radical dechlorination of the resulting 1-halotropan-3-ones led to the corresponding hydroxylated tropan-3-ones, which can be hydrogenated to yield 3α,6β-dihydroxytropanes. Starting from optically active keto-lactams, the method has been applied to the enantioselective syntheses of (+)-(1S,3S,5R,6S)-pervilleine C (6), (+)-(1S,3R,5S,6R)-valeroidine (3), (+)-(1S,3S,5R,6S)-dibenzoyloxytropane (8), and (+)-(1S,3S,5R,6S)-merredissine (9).
Co-reporter:Su-Yu Huang, Zong Chang, Shi-Chuan Tuo, Long-Hui Gao, Ai-E Wang and Pei-Qiang Huang
Chemical Communications 2013 vol. 49(Issue 63) pp:7088-7090
Publication Date(Web):13 Jun 2013
DOI:10.1039/C3CC43665A
The halo-assisted intramolecular addition of silyl enol ethers with in situ activated lactams yielded (hydroxylated) 1-halo-8-azabicyclo[3,2,1]octane and 1-halo-9-azabicyclo[3,3,1]nonane ring systems, which provided an easy enantioselective access to 6β-silyloxytropane-3-one, 3α,6β-dihydroxytropane, and pervilleine B. The absolute configuration of the natural (−)-pervilleine B was determined to be 1R,3R,5S,6R.
Co-reporter:Zhi-Gang Wang, Liqun Chen, Jiebo Chen, Jian-Feng Zheng, Weiwei Gao, Zhiping Zeng, Hu Zhou, Xiao-kun Zhang, Pei-Qiang Huang, Ying Su
European Journal of Medicinal Chemistry 2013 Volume 62() pp:632-648
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2013.01.012
RXRα represents an intriguing and unique target for pharmacologic interventions. We recently showed that Sulindac and a designed analog could bind to RXRα and modulate its biological activity, including inhibition of the interaction of an N-terminally truncated RXRα (tRXRα) with the p85α regulatory subunit of phosphatidylinositol-3-OH kinase (PI3K). Here we report the synthesis, testing and SAR of a series of novel analogs of Sulindac as potential modulators for inhibiting tRXRα-dependent AKT activation. A new compound 30 was identified to have improved biological activity.Graphical abstractHighlights► 28 New Sulindac analogs as modulators of tRXRα-dependent AKT activation were synthesized. ► Binding assay was used to test the 28 compounds for SAR study. ► A new scaffold was found with improved biological properties.
Co-reporter:Lian-Dong Guo;Pan Liang;Jian-Feng Zheng
European Journal of Organic Chemistry 2013 Volume 2013( Issue 11) pp:2230-2236
Publication Date(Web):
DOI:10.1002/ejoc.201201618
Abstract
The vinylogous Mannich reaction (VMR) between 2-(tert-butyldimethylsilyloxy)furan (TBSOF) and (RS)-t-BS-imine 12a and the application of the VMR adduct butenolide 13a as a versatile chiral building block for the synthesis of hydroxylated piperidine alkaloids and azasugars were investigated. Firstly, both the anti diastereoselectivity and the chemical yield of the asymmetric VMR between TBSOF and (RS)-t-BS-imine 12a were improved by the use of Sm(OTf)3/H2O (1.5 equiv.) as the promoter. Similar diastereoselectivities were also obtained with Yb(OTf)3/H2O, Cu(OTf)2/H2O, Zn(OTf)2/H2O, or the Brønsted acids TfOH or MsOH as the promotors. Secondly, an efficient four-step procedure for the elaboration of butenolide 13a into piperidine alkaloid (–)-deoxoprosophylline (2) was established. Thirdly, by taking advantage of the olefin functionality in the butenolide 13a, polyhydroxylated δ-lactams 23 and 21, which are ready precursors of azasugars L-deoxyallonojirimycin (ent-7) and L-3-epi-fagomine (ent-6), were obtained in two and three steps, respectively, via dihydroxylated lactone 17. The easily available synthetic intermediate 17 can also serve as a key intermediate for the synthesis of the glycosyl nucleoside amino acid cores of polyoxins and nikkomycins.
Co-reporter:Shichuan Tuo;Xuekui Liu ;Peiqiang Huang
Chinese Journal of Chemistry 2013 Volume 31( Issue 1) pp:55-62
Publication Date(Web):
DOI:10.1002/cjoc.201200904
Abstract
Some Stemona alkaloids belonging to the tuberostemospironine group possess a spirolactone moiety with anti-configuration (C-9/C-9a). In this paper, we describe two approaches to this structural unity. By using bromine atom as a traceless directing group, the SmI2-mediated reductive coupling of ketone 6 and β-bromomethacrylate proceeded with complete anti-diastereoselectivity. In the absence of an α-directing (chelation) group, the one-pot reaction of the ketone derived from alcohol 15 with the organozinc reagent generated from bromomethacrylate afforded spiro-α-methylene-γ-lactone derivative 16 as a single diastereomer. These two highly diastereoselective methods would find application in the synthesis of stemona alkaloids containing anti-configured spiro-lactone/pyrrolidine moieties. In addition, on the basis of our previous work, the total synthesis of (−)-9-epi-11-demethylsessilifoliamide J (11), and an improved synthesis of (−)-9,11-di-epi-sessilifoliamide J (9) were accomplished.
Co-reporter:Xue-Kui Liu, Jian-Liang Ye, Yuan-Ping Ruan, Yu-Xiu Li, and Pei-Qiang Huang
The Journal of Organic Chemistry 2013 Volume 78(Issue 1) pp:35-41
Publication Date(Web):September 4, 2012
DOI:10.1021/jo3014484
An efficient synthesis of the Stemona alkaloid (−)-sessilifoliamide J (1) in 12 steps and 7.7% overall yield from the known building block 8 is presented. The synthesis features the Corey lactonization reaction and a highly diastereoselective α-methylation reaction to build the spiro-lactone moiety.
Co-reporter:Hao-Hua Huo, Xiao-Er Xia, Hong-Kui Zhang, and Pei-Qiang Huang
The Journal of Organic Chemistry 2013 Volume 78(Issue 2) pp:455-465
Publication Date(Web):December 5, 2012
DOI:10.1021/jo302362b
The enantioselective total syntheses of the potent immunosuppressant FR901483 (1) and its 8-epimer (47) have been accomplished. Our approach features the use of building block 6 as the chiron, the application of the one-pot amide reductive bis-alkylation method to construct the chiral aza-quaternary center (dr = 9:1), regio- and diastereoselective intramolecular aldol reaction to build the bridged ring, and RCM to form the 3-pyrrolin-2-one ring.
Co-reporter:Kong-Zhen Hu, Jie Ma, Shi Qiu, Xiao Zheng, and Pei-Qiang Huang
The Journal of Organic Chemistry 2013 Volume 78(Issue 5) pp:1790-1801
Publication Date(Web):August 1, 2012
DOI:10.1021/jo301277n
We report, for the first time, the synthesis of 8-aza-analogues of PGE2. The SmI2-mediated cross coupling reactions of γ-lactam-hemiaminal 9, lactam 2-pyridyl sulfide 17, and lactam 2-pyridyl sulfone 18 with activated alkenes/alkyne were first developed, giving the corresponding γ-lactams in 49–78%, 45–75%, and 75–90%, respectively. The reactions of lactam 2-pyridyl sulfide and 2-pyridyl sulfone proceeded with ≥12:1 trans-diastereoselectivities. This represents the first intermolecular coupling reaction of the γ-lactam N-α-alkyl radicals of types B, B1, and B2 with activated alkenes. Two radical-based mechanisms were suggested. The asymmetric synthesis of the 11-hydroxylated analogue of the highly selective EP4 receptor agonist PF-04475270 (30), the 11-hydroxylated analogue of ocular hypotensive CP-734432 (31), compounds 35 and 36 have been achieved on the basis of this method.
Co-reporter:Kai-Jiong Xiao, Yu Wang, Ying-Hong Huang, Xiao-Gang Wang, and Pei-Qiang Huang
The Journal of Organic Chemistry 2013 Volume 78(Issue 17) pp:8305-8311
Publication Date(Web):August 2, 2013
DOI:10.1021/jo4007656
Full details of the direct and general method for the reductive alkylation of tertiary lactams and amides to give tertiary sec-alkylamines are presented. This one-pot method consists of in situ activation of a lactam or an amide with Tf2O/DTBMP, addition of a Grignard reagent, and reduction of the resulting iminium intermediates. Alkyl, benzyl, and aryl Grignard reagents and several reductants or reducing conditions (LiAlH4, NaBH4, Hantzsch ester, Bu3SnH, Pd(OH)2/C, H2) could be used effectively. Reductive alkylations of substituted lactams demonstrated good to excellent 1,3-asymmetric induction to provide the corresponding di- or trisubstituted pyrrolidine/piperidine in 6:1 (LiAlH4), 11:1 (Et3SiH), and 20:1 (catalytic hydrogenation) cis/trans diastereoselectivity, respectively. The versatility of this methodology was demonstrated by its application in the concise stereoselective synthesis of piperidine alkaloid (−)-morusimic acid.
Co-reporter:Xiao-Gang Wang, Ai-E Wang, Yi Hao, Yuan-Ping Ruan, and Pei-Qiang Huang
The Journal of Organic Chemistry 2013 Volume 78(Issue 18) pp:9488-9493
Publication Date(Web):August 19, 2013
DOI:10.1021/jo401412g
We report herein for the first time the enantioselective synthesis of 8-aza-PGE1. The synthesis used the cross olefin metathesis reaction to connect the 5-vinyl-γ-lactam subunit, prepared from (R)-malic acid via the Ley’s sulfone-based α-amidalkylation protocol (dr = 6.8:1), with the chiral pre-ω-chain. The latter was synthesized in high enantioselectivity from (E)-2-octenol by the Sharpless asymmetric epoxidation and the titanocene-mediated epoxide opening. This modular approach is quite concise and flexible, and requires only eight steps from commercially available reagents.
Co-reporter:Shi-Peng Luo;Lian-Dong Guo;Long-Hui Gao;Shuang Li;Dr. Pei-Qiang Huang
Chemistry - A European Journal 2013 Volume 19( Issue 1) pp:87-91
Publication Date(Web):
DOI:10.1002/chem.201203203
Co-reporter:Dr. Kai-Jiong Xiao;Dr. Jie-Min Luo;Xiao-Er Xia;Yu Wang ;Dr. Pei-Qiang Huang
Chemistry - A European Journal 2013 Volume 19( Issue 39) pp:13075-13086
Publication Date(Web):
DOI:10.1002/chem.201302096
Abstract
Amides are a class of highly stable and readily available compounds. The amide functional group constitutes a class of powerful directing/activating and protecting group for CC bond formation. Tertiary tert-alkylamine, including 1-azaspirocycle is a key structural feature found in many bioactive natural products and pharmaceuticals. The transformation of amides into tert-alkylamines generally requires several steps. In this paper, we report the full details of the first general method for the direct transformation of tertiary lactams/amides into tert-alkylamines. The method is based on in situ activation of amide with triflic anhydride/2,6-di-tert-butyl-4-methylpyridine (DTBMP), followed by successive addition of two organometallic reagents of the same or different kinds to form two CC bonds. Both alkyl and functionalized organometallic reagents and enolates can be used as the nucleophiles. The method displayed excellent 1,2- and good 1,3-asymmetric induction. Construction of 1-azaspirocycles from lactams required only two steps or even one-step by direct spiroannelation of lactams. The power of the method was demonstrated by a concise formal total synthesis of racemic cephalotaxine.
Co-reporter:Hao-Hua Huo, Hong-Kui Zhang, Xiao-Er Xia, and Pei-Qiang Huang
Organic Letters 2012 Volume 14(Issue 18) pp:4834-4837
Publication Date(Web):August 31, 2012
DOI:10.1021/ol302165d
A formal enantioselective total synthesis of the potent immunosuppressant FR901483 (1) has been accomplished. Our approach features the use of chiron 6 as the starting material, the application of the one-pot amide reductive bisalkylation method to construct the chiral aza-quaternary center (dr = 9:1), regio- and diastereoselective intramolecular aldol reaction to build the bridged ring, and ring closing metathesis to form the 3-pyrrolin-2-one ring.
Co-reporter:Chu-Pei Xu, Pei-Qiang Huang, and Sandrine Py
Organic Letters 2012 Volume 14(Issue 8) pp:2034-2037
Publication Date(Web):April 10, 2012
DOI:10.1021/ol300550x
Nitrones and tert-butanesulfinyl imines undergo conjugate addition to alkyl allenoates under SmI2-mediated reductive coupling conditions to produce novel β-methylenyl-substituted γ-amino esters. The latter were readily transformed into the corresponding β-methylenyl-γ-lactams by simple zinc reduction (N-hydroxy amines) or by acid hydrolysis (sulfinamides). The diastereoselective preparation of various β-methylenyl-γ-lactams offers a route to tetramic acids, the key structural features of an important class of bioactive natural products.
Co-reporter:Yu-Huang Wang, Jian-Liang Ye, Ai-E Wang and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 32) pp:6504-6511
Publication Date(Web):14 Jun 2012
DOI:10.1039/C2OB25901J
We have developed a one-pot method for the direct intermolecular reductive hydroxyalkylation or alkylation of amines using lactones or esters as the hydroxyalkylating/alkylating reagents. The method is based on the in situ amidation of lactones/esters with DIBAL-H–amine complex (for primary amines) or DIBAL-H–amine hydrochloride salt complex (for secondary amines), followed by reduction of the amides with an excess of DIBAL-H. Different from the reduction of Weinreb amides with DIBAL-H where aldehydes are formed, the reduction of the in situ formed Weinreb amides yielded amines. Moreover, this method is not limited to Weinreb amides, instead, it also works for other amides in general. A plausible mechanism is suggested to account for the outcome of the reactions.
Co-reporter:Xue-Kui Liu, Xiao Zheng, Yuan-Ping Ruan, Jie Ma and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 6) pp:1275-1284
Publication Date(Web):17 Nov 2011
DOI:10.1039/C1OB06697H
The one-pot reductive coupling of N-acylcarbamates with activated alkenes is described. The method is based on partial reduction of N-acylcarbamates with DIBAL-H, followed by N-acyliminium ion formation and SmI2-mediated radical coupling with activated alkenes. Both acyclic and cyclic N-acylcarbamates can be used as stable substrates, and a range of activated alkenes serve as effective radical receptors. The reductive coupling of L-N-acylcarbamates 12/13 gave 2,5-disubstituted pyrrolidine derivatives in high trans-diastereoselectivities. The reductive coupling with penta-2,4-dienoate proceeded exclusively in a 1,6-addition fashion, producing a single non-conjugated E-isomer. On the basis of this method, a three-step construction of pyrrolo[1,2-a]azepin-5-one 16, the skeleton of many stemona alkaloids and lehmizidine alkaloids, and a seven-step synthesis of (−)-xenovenine (pyrrolizidinecis-223H, ent-6), the unnatural enantiomer of the frog/ant venom alkaloid possessing potent inhibitory activity towards nAChR channel, were achieved starting from L-12.
Co-reporter:Kai-Jiong Xiao;Dr. Ai-E Wang;Ying-Hong Huang ;Dr. Pei-Qiang Huang
Asian Journal of Organic Chemistry 2012 Volume 1( Issue 2) pp:130-132
Publication Date(Web):
DOI:10.1002/ajoc.201200066
Co-reporter:Kai-Jiong Xiao;Dr. Ai-E Wang;Ying-Hong Huang ;Dr. Pei-Qiang Huang
Asian Journal of Organic Chemistry 2012 Volume 1( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/ajoc.201290005
Co-reporter:Yu-Huang Wang;Wei Ou;Dr. Linfeng Xie;Dr. Jian-Liang Ye ;Dr. Pei-Qiang Huang
Asian Journal of Organic Chemistry 2012 Volume 1( Issue 4) pp:359-365
Publication Date(Web):
DOI:10.1002/ajoc.201200113
Abstract
A chemo-, regio-, and stereoselectively controlled reaction is highly desirable, yet challenging in organic synthesis. Diversely substituted cis and trans isomers of 2-alkyl-3-pyrrolidinols, 5-alkyl-4-hydroxy-2-pyrrolidinones, β-hydroxy-γ-amino acids, and their higher homologues are key structural units found in numerous drugs, drug candidates, and bioactive natural products. Previously, we established a flexible approach to trans-5-alkyl-4-benzyloxy-2-pyrrolidinones 14 and trans-6-alkyl-5-benzyloxy-2-piperidinones 15. Herein, we report a direct, flexible, moisture insensitive, and highly diastereoselective approach to the corresponding cis diastereomers 16. This stereocontrolled method is based on the MsOH-mediated (Ms=methane sulfonyl) reductive dehydroxylation of hemiaminal 12 with NaBH(OAc)3. cis-5-Alkyl-4-benzyloxy-2-pyrrolidinones 16 are useful building blocks for the syntheses of natural products such as (+)-preussin (4) and streptopyrrolidine (5) as well as (3S,4S)-γ-alkyl-β-hydroxy-γ-amino acids (6).
Co-reporter:Xijie Dai ;Peiqiang Huang
Chinese Journal of Chemistry 2012 Volume 30( Issue 9) pp:1953-1956
Publication Date(Web):
DOI:10.1002/cjoc.201200660
Abstract
A novel strategy was developed for a rapid access to the naturally occurring racemic neoclausenamide and its analogs, which featured a highly erythro-selective vinylogous Mukaiyama type reaction (dr=12:1) and a highly diastereoselective tandem conjugate addition-Davis oxidation of N-Boc-pyrrol-2(5H)-one 5 (dr=10:1). Remarkably, the skeleton of neoclausenamide, namely 8a, an analog of neoclausenamide, was built in just two steps with all the four stereogenic centers (relative stereochemistry) established correctly and in excellent diastereoselectivities.
Co-reporter:Jian-Feng Zheng;Hong-Qiao Lan;Rui-Feng Yang;Qi-Long Peng;Zhen-Hua Xiao;Shi-Chuan Tuo;Kong-Zhen Hu;Yong-Gang Xiang;Zhen Wei;Zhen Zhang
Helvetica Chimica Acta 2012 Volume 95( Issue 10) pp:1799-1808
Publication Date(Web):
DOI:10.1002/hlca.201200341
Abstract
We describe efficient and flexible enantioselective syntheses of the active enantiomers of the pheromones of pine sawflies, including the species Diprion jingyuanensis, their homologs and, stereoisomers, as well as those identified from the Chinese species Diprion jingyuanensis, i.e., 126. A total of 48 compounds, including acetates 78–101 and propanoates 102–125, have been synthesized. Our general approach towards these compounds originated from the commercially available chirons diethyl (S)- and (R)-malates, as well as ethyl (R)-3-hydroxybutanoate. The Seebach asymmetric methylation was employed in a key step to control additional configuration.
Co-reporter:Jie Chen;AiE Wang;HaoHua Huo;PeiQiang Huang
Science China Chemistry 2012 Volume 55( Issue 7) pp:1175-1212
Publication Date(Web):2012 July
DOI:10.1007/s11426-012-4534-9
This paper summarizes the progress on the total syntheses of natural products accomplished in mainland China during the period from 2006 to 2010. The overview focuses on the first total synthesis of natural products of contemporary interest including alkaloids, cyclopeptides and cyclic depsipeptides, macrolides, terpenoids and steroids, saponins and glycosides. The development of novel synthetic strategies and methodologies, and application of new selective synthetic methods in the total syntheses of natural products are included as well.
Co-reporter:Kai-Jiong Xiao;Dr. Ai-E Wang ;Dr. Pei-Qiang Huang
Angewandte Chemie International Edition 2012 Volume 51( Issue 33) pp:8314-8317
Publication Date(Web):
DOI:10.1002/anie.201204098
Co-reporter:Kai-Jiong Xiao;Dr. Ai-E Wang ;Dr. Pei-Qiang Huang
Angewandte Chemie 2012 Volume 124( Issue 33) pp:8439-8442
Publication Date(Web):
DOI:10.1002/ange.201204098
Co-reporter:Jin-Cheng Liao, Kai-Jiong Xiao, Xiao Zheng, Pei-Qiang Huang
Tetrahedron 2012 68(26) pp: 5297-5302
Publication Date(Web):
DOI:10.1016/j.tet.2012.01.085
Co-reporter:Gui-Yang Chen;Huang Huang;Dr. Jian-Liang Ye;Dr. Ai-E Wang;Hui-Ying Huang;Dr. Hong-Kui Zhang ;Dr. Pei-Qiang Huang
Chemistry – An Asian Journal 2012 Volume 7( Issue 3) pp:504-518
Publication Date(Web):
DOI:10.1002/asia.201100809
Abstract
The first enantioselective synthesis of cytotoxic natural products rigidiusculamides A (ent-21) and B (8) has been achieved by two synthetic routes. The first one is convergent based on the common intermediate 11, obtained through a high yielding SmI2-mediated Reformatsky-type reaction. A highly diastereoselective one-pot Dess–Martin periodinane-mediated bis-oxidation allowed the direct conversion of the diastereomeric mixture of 11 into rigidiusculamide B (8). Isolation of minor diastereomer 21, in combination with computational work, allowed us to suggest the structure of the natural rigidiusculamide A to be 21, as synthesized by the second route. Four diastereomers (7, 7, 22a, and 22b) and an enantiomer (21) of rigidiusculamide A (21) have been synthesized. On the basis of literature precedents and computational work, a biosynthetic pathway for rigidiusculamides A and B was proposed to account for the opposite configuration at C-5 of those two congeners.
Co-reporter:Shu-Tang Ruan, Jie-Min Luo, Yu Du, and Pei-Qiang Huang
Organic Letters 2011 Volume 13(Issue 18) pp:4938-4941
Publication Date(Web):August 19, 2011
DOI:10.1021/ol2020384
Asymmetric vinylogous Mannich reaction (VMR) of 2-(tert-butyldimethylsilyloxy)furan (TBSOF, 1) with (RS)- or (SS)-t-BS-imines (3) furnished 5-aminoalkylbutenolides 7a–k in 75–87% yields with anti/syn ratios ranging from 75:25 to 97:3. Butenolides 7a–f,k were readily converted into substituted lactones 8 and 5 and 6-substituted 5-hydroxypiperidin-2-ones 11a–g, which are, in turn, key intermediates for the synthesis of many bioactive compounds.
Co-reporter:Geng-Jie Lin, Xiao Zheng and Pei-Qiang Huang
Chemical Communications 2011 vol. 47(Issue 5) pp:1545-1547
Publication Date(Web):25 Nov 2010
DOI:10.1039/C0CC04371K
An efficient and highly diastereoselective method for the construction of the hydroxylated tropane skeleton is described. The method features a new intramolecular reductive coupling reaction of N-acyl N,O-acetal with aldehyde, cooperatively mediated by BF3·OEt2 and SmI2. On the basis of this method, a new enantioselective total synthesis of (−)-Bao Gong Teng A has been accomplished.
Co-reporter:Bi-Shuang Chen, Long-He Yang, Jian-Liang Ye, Tao Huang, Yuan-Ping Ruan, Jin Fu, Pei-Qiang Huang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 11) pp:5480-5486
Publication Date(Web):November 2011
DOI:10.1016/j.ejmech.2011.09.010
An improved four-step approach for the stereoselective synthesis of long-chain anti-2-amino-3-alkanols is described. Using this method, the syntheses of antiproliferative (antitumoral) compounds, spisulosine (ES-285, 2), clavaminols A and B (3 and 4), the deacetylated products of clavaminols H and N (7 and 8), as well as (2S,3R)-2-aminododecan-3-ol (9) and xestoaminol C (10), have been achieved in excellent diastereoselectivities. In vitro study showed that these compounds induced cell death and dose-dependently inhibited cell proliferation in human glioblastoma cell line SHG-44, indicating the anti-tumor property of this series of compounds.Highlights► An improved diastereoselective synthesis of anti-2-amino-3-alkanols is reported. ► Seven long-chain anti-2-amino-3-alkanol natural products have been synthesized. ► In vitro study in human glioblastoma cell line SHG-44 undertook. ► These compounds induced cell death and inhibited cell proliferation.
Co-reporter:Bo Teng;Jianfeng Zheng;Huiying Huang ;Peiqiang Huang
Chinese Journal of Chemistry 2011 Volume 29( Issue 7) pp:1312-1318
Publication Date(Web):
DOI:10.1002/cjoc.201180248
Abstract
Five glutarimide alkaloids cordiarimide A (5), cordiarimide B (6), crotonimide A (3), crotonimide B (4), and julocrotine (2) have been synthesized starting from Boc-L-glutamine (7). The benzylic alcohol chiral centre of cordiarimides B (6) has been established in 6:1 diastereoselectivity by catalytic asymmetric hydrogenation using Zhou's catalytic system Pd(CF3CO2)2/(R,R)-Me-DuPhos.
Co-reporter:HongKui Zhang;Xin Li;Huang Huang;PeiQiang Huang
Science China Chemistry 2011 Volume 54( Issue 5) pp:
Publication Date(Web):2011 May
DOI:10.1007/s11426-011-4256-4
Starting from the oxygenated piperidine building block 20, two synthetic approaches to new building blocks (8R,8aS)- and (8R,8aR)-8-hydroxy-5-indolizidinones 19a/19b and 15a/15b have been developed, respectively. The first one is based on the trans-diastereoselective reductive alkylation (dr = 93:7), followed by a four-step procedure; and the second one called for the RCM reaction on the N,O-acetal derived from a vinylation, which was followed by a pyrrole formation, and a stereocontrolled cis-selective (dr = 91:9) catalytic hydrogenation. Reduction of the diastereomer 15a produced (8R,8aR)-8-indolizidinol (18).
Co-reporter:Jin-Li Zheng, Hui Liu, Yu-Feng Zhang, Wei Zhao, Jin-Shuan Tong, Yuan-Ping Ruan, Pei-Qiang Huang
Tetrahedron: Asymmetry 2011 Volume 22(Issue 3) pp:257-263
Publication Date(Web):10 February 2011
DOI:10.1016/j.tetasy.2011.01.012
Analytical HPLC methods for the determination of the enantiomeric excess of N-protected malimides 1 as well as the corresponding pyrrolidinol 5 and tartarimides 2 and 3 have been established. On this basis, a study to reveal the racemization step in the synthesis of pyrrolidinols from α-hydroxyacids, via chiral cyclic α-hydroxyimides, has been undertaken. It was confirmed that the known, one-step method for the synthesis of the N-protected chiral cyclic imides from α-hydroxydiacids proceeded with little racemization, and partial racemization has been proven to occur during the reduction of the resultant imide 1a with LAH to yield the corresponding pyrrolidinol 5. Conditions have been defined in order to avoid racemization in the LAH reduction step.(S)-N-BenzylmalimideC11H11NO3[α]D25=-43.0 (c 1.0, MeOH)Source of chirality: (S)-malic acidAbsolute configuration: (S)(S)-N-Benzylpyrrolidin-3-olC11H15NO[α]D25=-5.3 (c 0.5, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (S)(R)-4-Hydroxy-1-(4-methoxybenzyl)pyrrolidine-2,5-dioneC12H13NO4[α]D25=+69.2 (c 0.8, MeOH)Source of chirality: (R)-malic acidAbsolute configuration: (R)(3S,4S)-3,4-Dihydroxy-1-(4-methoxybenzyl)pyrrolidine-2,5-dioneC12H13NO5[α]D25=-97.4 (c 1.0, MeOH)Source of chirality: d-tartaric acidAbsolute configuration: (2S,3S)(3R,4R)-3,4-Dihydroxy-1-benzylpyrrolidine-2,5-dioneC11H11NO4[α]D25=+121.3 (c 1.0, MeOH)Source of chirality: l-tartaric acidAbsolute configuration: (2R,3R)
Co-reporter:Dr. Hong-Qiao Lan;Dr. Jian-Liang Ye;Dr. Ai-E Wang; Yuan-Ping Ruan;Dr. Pei-Qiang Huang
Chemistry - A European Journal 2011 Volume 17( Issue 3) pp:958-968
Publication Date(Web):
DOI:10.1002/chem.201002063
Abstract
By using a methyl tetramate derivative (R)- or (S)-9 as a novel chiral building block, a direct, flexible, and highly enantioselective approach to methyl (R)- or (S)-5-alkyltetramates (2) is disclosed. Among the synthesized methyl 5-alkyltetramates 2, methyl 5-methyltetramate (2 a) is found in cytotoxic mirabimide E (4) and dysideapyrrolidone (5), and methyl 5-benzyltetramate (2 g) is a substructure in the potent antineoplastic dolastatin 15 (3). On the basis of this method, the first asymmetric synthesis of the antimitotic tetrapeptide belamide A (7) has been achieved in seven steps from (S)-9, with an overall yield of 23.8 %. Not only have the structure and absolute configuration of (+)-belamide A (7) been confirmed, but also the solvent used for recording the 13C NMR spectrum, the 13C NMR spectrum data correlation, and optical rotation data of natural belamide A (7) have been revised.
Co-reporter:Chao Yang, Yu-Hui Bao, Pan Liang, Jian-Liang Ye, Ai-E. Wang, Pei-Qiang Huang
Tetrahedron 2011 67(34) pp: 6281-6288
Publication Date(Web):
DOI:10.1016/j.tet.2011.06.023
Co-reporter:Xue-Kui Liu, Shi Qiu, Yong-Gang Xiang, Yuan-Ping Ruan, Xiao Zheng, and Pei-Qiang Huang
The Journal of Organic Chemistry 2011 Volume 76(Issue 12) pp:4952-4963
Publication Date(Web):May 16, 2011
DOI:10.1021/jo200600n
The SmI2-mediated radical coupling reactions of β-hydroxylated pyrrolidine/piperidine aza-hemiacetals 8 and 9 and N,S-acetals 6 and 33 with α,β-unsaturated compounds are described. This method allows a rapid access to β-hydroxylated pyrrolidines, piperidines, pyrrolizidinones, and indolizidinones. Starting from N,S-acetal 33 and via a common intermediate 27, the alkaloids hyacinthacine A2 (2), uniflorine A (3, 6-epi-casuarine), and the unnatural epimer 7-epi-casuarine (37) have been synthesized in four and five steps with overall yields of 34%, 16%, and 13%, respectively. The radical mechanism of the coupling reactions has been confirmed by controlled experiments, which also allowed deducing the anionic mechanism in the coupling between N,S-acetal 6 and carbonyl compounds. This demonstrates that the mechanisms of these SmI2-mediated reactions are switchable from Barbier-type anionic to radical by cooperative action of BF3·OEt2 and t-BuOH.
Co-reporter:Chu-Pei Xu, Zhen-Hua Xiao, Bi-Qin Zhuo, Yu-Huang Wang and Pei-Qiang Huang
Chemical Communications 2010 vol. 46(Issue 41) pp:7834-7836
Publication Date(Web):09 Sep 2010
DOI:10.1039/C0CC01487G
We report a mild and environmentally benign method for the synthesis of tertiary amines using alcohols as the alkylating reagents. Not only secondary amines such as piperazines but also amino acids and amino alcohols can be N-alkylated selectively. For N,O-benzyl protected amino alcohols, both N,O-de-benzylation and N-methylation were achieved in one-pot.
Co-reporter:Hong-Qiao Lan, Yuan-Ping Ruan and Pei-Qiang Huang
Chemical Communications 2010 vol. 46(Issue 29) pp:5319-5321
Publication Date(Web):16 Jun 2010
DOI:10.1039/C0CC00452A
Methyl tetramate derivative 6 has been developed as a new building block for the flexible and racemization-free synthesis of methyl 5-benzyl-3-methyltetramate via alkylation, and used in the first asymmetric synthesis of palau’imide (1). This allowed the establishment of the hitherto unknown stereochemistry at the C-20 of palau’imide as S.
Co-reporter:Wen-Jun Liu, Jian-Liang Ye and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 9) pp:2085-2091
Publication Date(Web):03 Mar 2010
DOI:10.1039/B926741G
A concise and flexible approach for the asymmetric syntheses of polyhydroxylated pyrrolizidine alkaloids hyacinthacines A2 and A3 has been developed using iterative reductive alkylation of O,O′-dibenzyltartarimide (5) as key steps. The ambiguity about the structure of synthetic hyacinthacine A3 due to the differences in the NMR data of the synthetic material (2) and the natural product (hyacinthacine A3) was eliminated jointly by comprehensive 1D and 2D-NMR studies, and by analysis of the 1H and 13C NMR spectra of a mixed synthetic product and natural hyacinthacine A3. The latter method also allowed a confirmation of the structure of the natural hyacinthacine A3, and may be useful for structural confirmation of other hydroxylated alkaloids.
Co-reporter:Hongkui Zhang;Zhijie Lin;Huang Huang;Haohua Huo;Yanju Huang;Jianliang Ye ;Peiqiang Huang
Chinese Journal of Chemistry 2010 Volume 28( Issue 9) pp:1717-1724
Publication Date(Web):
DOI:10.1002/cjoc.201090290
Abstract
A concise enantioselective synthesis of the diazatricyclic core of alkaloid TAN1251C is reported. The method featured a stereoselective formation of the iodide intermediate 16b (in 2:1 ratio) from homoallylic amine 11b by iodine-promoted iodoaminocyclization, a previously claimed to be unsuccessful reaction. The intermediate 16b was converted to the tricyclic core of TAN1251C 28 in five steps. During this study, two unexpected products 22 and 25 were obtained, and compound 23, derived from 11a was shown to be unsuitable for the oxidative formation of the enamide of TAN1251C.
Co-reporter:Rui-Feng Yang;Dr. Pei-Qiang Huang
Chemistry - A European Journal 2010 Volume 16( Issue 34) pp:10319-10322
Publication Date(Web):
DOI:10.1002/chem.201001582
Co-reporter:Kai-Jiong Xiao;Yu Wang;Ke-Yin Ye ; Pei-Qiang Huang
Chemistry - A European Journal 2010 Volume 16( Issue 43) pp:12792-12796
Publication Date(Web):
DOI:10.1002/chem.201002054
Co-reporter:Li-Jiao Jiang, Bo Teng, Jian-Feng Zheng, Jian-Liang Ye, Pei-Qiang Huang
Tetrahedron 2010 66(1) pp: 172-175
Publication Date(Web):
DOI:10.1016/j.tet.2009.11.003
Co-reporter:Shao-Feng Wu, Yuan-Ping Ruan, Xiao Zheng, Pei-Qiang Huang
Tetrahedron 2010 66(9) pp: 1653-1660
Publication Date(Web):
DOI:10.1016/j.tet.2010.01.011
Co-reporter:Kai-Jiong Xiao;Jie-Min Luo;Ke-Yin Ye;Yu Wang
Angewandte Chemie International Edition 2010 Volume 49( Issue 17) pp:3037-3040
Publication Date(Web):
DOI:10.1002/anie.201000652
Co-reporter:Zhao-Bao Ye, Jie Chen, Wei-Hua Meng, Pei-Qiang Huang
Tetrahedron: Asymmetry 2010 Volume 21(Issue 8) pp:895-902
Publication Date(Web):30 April 2010
DOI:10.1016/j.tetasy.2010.04.063
O-Benzyl-N-benzyloxymalimide 12 has been synthesized as a useful variant of the chiral building blocks 9. The advantage of the malimide 12 over 9 was demonstrated by mild and high-yielding reductive N-deprotection of N-benzyloxylactams 18a–i to give a series of (4R,5S)-5-alkyl-4-hydroxy-pyrrolidin-2-ones, which are key intermediates for the asymmetric synthesis of pyrrolidines, pyrrolizidines, and indolizidines, as well as β-hydroxy γ-amino acids. Lactam ent-11a has been applied to the synthesis of the mixed imide 25, a key intermediate for the total synthesis of microcolin B, which also demonstrated that lactam ent-11a or 11a can serve as a latent form of the 5-methyl-3-pyrrolin-2-one substructure of majusculamide D 4, deoxymajusculamide D, and the jamaicamides A–C.Diethyl (S)-2-(benzyloxy)succinateC15H20O5[α]D20=-59.9 (c 1.2, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (S)Diethyl (R)-2-(benzyloxy)succinateC15H20O5[α]D20=+59.9 (c 1.0, CHCl3)Source of chirality: (R)-malic acidAbsolute configuration: (R)(S)-2-(Benzyloxy)succinic acidC11H12O5[α]D20=-56.9 (c 1.9, CH3OH)Source of chirality: (S)-malic acidAbsolute configuration: (S)(R)-2-(Benzyloxy)succinic acidC11H12O5[α]D20=+49.6 (c 1.2, CH3OH)Source of chirality: (R)-malic acidAbsolute configuration: (R)(S)-1,3-Dibenzyloxypyrrolidin-2,5-dioneC18H17NO4[α]D20=-90.2 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (S)(R)-1,3-Dibenzyloxypyrrolidin-2,5-dioneC18H17NO4[α]D20=+90.0 (c 1.2, CHCl3)Source of chirality: (R)-malic acidAbsolute configuration: (R)(4S,5R)-1,4-Dibenzyloxy-5-methylpyrrolidin-2-oneC19H21NO3[α]D20=+89.7 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4R,5S)-1,4-Dibenzyloxy-5-methylpyrrolidin-2-oneC19H21NO3[α]D20=-95.5 (c 1.2, CHCl3)Source of chirality: (R)-malic acidAbsolute configuration: (4R,5S)(4S,5R)-1,4-Dibenzyloxy-5-ethylpyrrolidin-2-oneC20H23NO3[α]D20=+86.8 (c 1.2, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-propylpyrrolidin-2-oneC21H25NO3[α]D20=+88.7 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-isopropylpyrrolidin-2-oneC21H25NO3[α]D20=+69.5 (c 1.3, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-butylpyrrolidin-2-oneC22H27NO3[α]D20=+84.9 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-isobutylpyrrolidin-2-oneC22H27NO3[α]D20=+90.7 (c 1.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-heptylpyrrolidin-2-oneC25H33NO3[α]D20=+62.7 (c 0.6, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-octylpyrrolidin-2-oneC26H35NO3[α]D20=+64.3 (c 1.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-1,4-Dibenzyloxy-5-phenylpyrrolidin-2-oneC24H23NO3[α]D20=+53.3 (c 1.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-methylpyrrolidin-2-oneC12H15NO2[α]D20=+58.2 (c 1.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4R,5S)-4-Benzyloxy-5-methylpyrrolidin-2-oneC12H15NO2[α]D20=-59.4 (c 1.2, CHCl3)Source of chirality: (R)-malic acidAbsolute configuration: (4R,5S)(4S,5R)-4-Benzyloxy-5-ethylpyrrolidin-2-oneC13H17NO2[α]D20=+44.9 (c 1.5, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-ethylpyrrolidin-2-oneC14H19NO2[α]D20=+45.5 (c 2.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-isopropylpyrrolidin-2-oneC14H19NO2[α]D20=+42.6 (c 2.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-butylpyrrolidin-2-oneC15H21NO2[α]D20=+44.8 (c 1.2, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-isobutylpyrrolidin-2-oneC15H21NO2[α]D20=+47.8 (c 0.9, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-heptylpyrrolidin-2-oneC18H27NO2[α]D20=+39.3 (c 1.3, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-octylpyrrolidin-2-oneC19H29NO2[α]D20=+38.3 (c 1.3, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-phenylpyrrolidin-2-oneC17H17NO2[α]D20=+46.4 (c 2.2, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)1-tert-Butyl 2-perfluorophenyl (S)-pyrrolidine-1,2-dicarboxylateC16H16F5NO4[α]D20=-58 (c 1.2, CHCl3)Source of chirality: L-prolineAbsolute configuration: (S)(4R,5S,1′S)-4-Benzyloxy-1-(N-butyloxycarbonylprolyl)-5-methylpyrrolidin-2-oneC22H30N2O5[α]D20=-49 (c 0.9, CHCl3)Source of chirality: (S)-malic acid and l-prolineAbsolute configuration: (4R,5S,1′S)(4R,5S,1′S)-1-(N-butyloxycarbonylprolyl)-4-hydroxyl-5-methylpyrrolidin-2-oneC15H24N2O5[α]D20=-40.0 (c 1.3, CHCl3)Source of chirality: (S)-malic acid and l-prolineAbsolute configuration: (4R,5S,1′S)(5S,1′S)-1-(N-Butyloxycarbonylprolyl)-5-methylpyrrol-2(5H)-oneC15H22N2O4[α]D20=-80 (c 1.0, CHCl3)Source of chirality: (S)-malic acid and l-prolineAbsolute configuration: (4R,5S,1′S)
Co-reporter:Gang Liu;Tian-Jun Wu Dr.;Yuan-Ping Ruan Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 19) pp:5755-5768
Publication Date(Web):
DOI:10.1002/chem.200903490
Abstract
The asymmetric total synthesis of natural azasugars (+)-castanospermine, (+)-7-deoxy-6-epi-castanospermine, and synthetic (+)-1-epi-castanospermine has been accomplished in nine to ten steps from a common chiral building block (S)-8. The method features a powerful chiral relay strategy consisting of a highly diastereoselective vinylogous Mukaiyama-type reaction with either chiral or achiral aldehydes (≥95 % de; de=diastereomeric excess) and a diastereodivergent reduction of tetramic acids, which allows formation of three continuous stereogenic centers with high diastereoselectivities. The method also provides a flexible access to structural arrays of 5-(α-hydroxyalkyl)tetramic acids, such as 17/34, and 5-(α-hydroxyalkyl)-4-hydroxyl-2-pyrrolidinones, such as 18 and 25/35 a. The method constitutes the first realization of the challenging chiral synthons A and D and thus of the conceptually attractive retrosynthetic analysis shown in Scheme 1 in a highly enantioselective manner.
Co-reporter:Kai-Jiong Xiao;Jie-Min Luo;Ke-Yin Ye;Yu Wang
Angewandte Chemie 2010 Volume 122( Issue 17) pp:3101-3104
Publication Date(Web):
DOI:10.1002/ange.201000652
Co-reporter:Yong-Gang Xiang, Xiang-Wu Wang, Xiao Zheng, Yuan-Ping Ruan and Pei-Qiang Huang
Chemical Communications 2009 (Issue 45) pp:7045-7047
Publication Date(Web):13 Oct 2009
DOI:10.1039/B915488D
The synergistic action of BF3·OEt2 and SmI2 allowed a series of intermolecular cross-couplings of readily available N-acyl N,O-acetals with α,β-unsaturated compounds to be performed in high yields, which was applied to the stereoselective synthesis of pyrrolizidine alkaloid (+)-xenovenine.
Co-reporter:Rui Fu, Jie Chen, Lu-Chuan Guo, Jian-Liang Ye, Yuan-Ping Ruan and Pei-Qiang Huang
Organic Letters 2009 Volume 11(Issue 22) pp:5242-5245
Publication Date(Web):October 20, 2009
DOI:10.1021/ol902180t
The first asymmetric total synthesis of the unnatural enantiomer of cytotoxic awajanomycin (1) is reported. The synthetic approach features first a convergent strategy using the cross-olefin metathesis reaction to link the lipid side chain 2 and the piperidinone core structure 3. The second feature of the synthesis resides on the construction of segment 3 from the building block 5 via a three-component tandem reaction on the mixed imide 12. Through this work, the stereochemistry at C-11 and the absolute configuration of awajanomycin were established as 3R,5R,6S,8S,11S.
Co-reporter:Geng-Jie Lin and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 21) pp:4491-4495
Publication Date(Web):24 Aug 2009
DOI:10.1039/B912190K
A seven-step synthesis of (3S,5R,8S,9S)-3-butyl-5-propyl-8-hydroxyindolizidine (2), an ant venom alkaloid isolated from Myrmicaria melanogaster, is disclosed with an overall yield of 28.9%. The key feature of the synthesis is the use of the iodocyclization for the introduction of the hydroxyl group of the 3-piperidinol. Remarkably, all the reaction steps proceeded with excellent chemo-, regio- and/or diastereoselectivities.
Co-reporter:Shao-Feng Wu, Xiao Zheng, Yuan-Ping Ruan and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 14) pp:2967-2975
Publication Date(Web):02 Jun 2009
DOI:10.1039/B906224F
A flexible diastereoselective approach to trans-(3S)-hydroxyprolinol derivatives is described, which is based on the samarium diiodide-mediated reductive coupling of the chiral 1-pyrroline N-oxide (nitrone)(S)-10 with carbonyl compounds. The reductive hydroxyalkylation of nitrone 10 with ketones and aromatic aldehydes is highly diastereoselective in establishing the C-2 chiral center of the pyrrolidine ring.
Co-reporter:Liang-Xian Liu, Kai-Jiong Xiao, Pei-Qiang Huang
Tetrahedron 2009 65(19) pp: 3834-3841
Publication Date(Web):
DOI:10.1016/j.tet.2009.03.021
Co-reporter:Rui Fu, Yu Du, Zhao-Ying Li, Wei-Xuan Xu, Pei-Qiang Huang
Tetrahedron 2009 65(47) pp: 9765-9771
Publication Date(Web):
DOI:10.1016/j.tet.2009.09.083
Co-reporter:Jie-Min Luo;Chao-Feng Dai Dr.;Shu-Yong Lin Dr.
Chemistry – An Asian Journal 2009 Volume 4( Issue 2) pp:328-335
Publication Date(Web):
DOI:10.1002/asia.200800355
Co-reporter:Shao-Hua Xiang, Hong-Qiu Yuan, Pei-Qiang Huang
Tetrahedron: Asymmetry 2009 Volume 20(Issue 17) pp:2021-2026
Publication Date(Web):8 September 2009
DOI:10.1016/j.tetasy.2009.08.018
A concise, flexible, and highly diastereoselective approach to cis-5-alkyl-4-hydroxy-2-pyrrolidinones 1 is described. The key step is an ammonium acetate-assisted catalytic hydrogenation of the enamides 9, derived in two steps from malimides 6a,b as we have described previously. The method was applied to the asymmetric synthesis of streptopyrrolidine 5, a natural product, which exhibited significant anti-angiogenesis activity.(4S,5S)-1-Benzyl-4-(benzyloxy)-5-methylpyrrolidin-2-oneC19H21NO2[α]D20=-19.8 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-1-Benzyl-4-(benzyloxy)-5-ethylpyrrolidin-2-oneC20H23NO2[α]D20=-2.7 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-1-Benzyl-4-(benzyloxy)-5-butylpyrrolidin-2-oneC22H27NO2[α]D20=+7.8 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-1-Benzyl-4-(benzyloxy)-5-pentylpyrrolidin-2-oneC23H29NO2[α]D20=+7.9 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-1-Benzyl-4-(benzyloxy)-5-isobutylpyrrolidin-2-oneC22H27NO2[α]D20=+7.3 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-1,5-Dibenzyl-4-(benzyloxy)pyrrolidin-2-oneC25H25NO2[α]D20=-4.2 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(S,E)-1-Benzyl-4-(benzyloxy)-5-octylidenepyrrolidin-2-oneC26H33NO2[α]D20=+80.0 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S)(4S,5S)-1-Benzyl-4-(benzyloxy)-5-octylpyrrolidin-2-oneC26H35NO2[α]D20=+10.4 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(S,E)-1-Benzyl-4-(benzyloxy)-5-dodecylidenepyrrolidin-2-oneC30H43NO2[α]D20=+80.6 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S)(4S,5S)-1-Benzyl-4-(benzyloxy)-5-dodecylpyrrolidin-2-oneC30H45NO2[α]D20=+10.5 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-1-(4-methoxybenzyl)-4-(benzyloxy)-5-Methylpyrrolidin-2-oneC20H23NO3[α]D20=-19.7 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(S,E)-5-Benzylidene-4-(benzyloxy)-1-(4-methoxybenzyl)pyrrolidin-2-oneC26H25NO3[α]D20=+291.0 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S)(4S,5S)-5-Benzyl-4-(benzyloxy)-1-(4-methoxybenzyl)pyrrolidin-2-oneC26H27NO3[α]D20=-5.7 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-5-Benzyl-4-(benzyloxy)pyrrolidin-2-oneC18H19NO2[α]D20=-35.6 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)(4S,5S)-5-Benzyl-4-hydroxypyrrolidin-2-oneC11H13NO2[α]D20=-43.5 (c 1.0, MeOH)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5S)
Co-reporter:Kai-Jiong Xiao, Liang-Xian Liu, Pei-Qiang Huang
Tetrahedron: Asymmetry 2009 Volume 20(Issue 10) pp:1181-1184
Publication Date(Web):5 June 2009
DOI:10.1016/j.tetasy.2009.04.008
Herein we report a concise enantioselective synthesis of (+)-azimic acid starting from (5S,6S)-6-methyl-5-benzyloxy-2-piperidinone 8a, which was prepared from protected (S)-3-hydroxyglutarimide 6 according to a method recently disclosed in our laboratory. The key step is a stepwise regioselective reductive alkylation of the imide 10, which established the 2,6-cis-stereochemistry in excellent diastereoselectivity.(5S,6S)-5-Benzyloxy-6-methyl-2-piperidinoneC13H17NO2[α]D20=+9.4 (c 0.5, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (5S,6S)(5S,6S)-5-Benzyloxy-1-(tert-butyloxycarbonyl)-6-methyl-2-piperidinoneC18H25NO4[α]D20=+8.9 (c 1.1, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (5S,6S)tert-Butyl [(2S,3S)-3-benzyloxy-12-(tert-butyldimethylsilyloxy)-6-oxododecan]-2-yl carbamateC30H53NO5Si[α]D20=-13.5 (c 0.9, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (2S,3S)tert-Butyl [(2S,3S)-3-benzyloxy-12-hydroxy-6-oxododecan]-2-yl carbamateC24H39NO5[α]D20=-24.6 (c 0.8, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (2S,3S)(10S,11S)-10-Benzyloxy-11-(tert-butoxycarbonylamino)-7-oxododecanoic acidC24H37NO6[α]D20=-15.9 (c 1.0, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (10S,11S)(+)-(2S,3S,6R)-Azimic acidC12H23NO3[α]D20=+7.7 (c 0.5, MeOH)Source of chirality: (S)-glutamic acidAbsolute configuration: (2S,3S,6R)
Co-reporter:Gang Liu, Jie Meng, Chen-Guo Feng, Pei-Qiang Huang
Tetrahedron: Asymmetry 2008 Volume 19(Issue 11) pp:1297-1303
Publication Date(Web):16 June 2008
DOI:10.1016/j.tetasy.2008.05.002
The asymmetric syntheses of (−)-epi-pseudoconhydrine and (−)-5-hydroxysedamine are reported. The key to these syntheses is an unusual highly cis-diastereoselective 1,4-asymmetric induction in the α-amidoallylation of the new chiral building block 15.(S)-N-(4-Methoxybenzyl)-2,5-dihydroxypentanamideC13H19NO4[α]D20=-23.8 (c 1.0, CH3OH)Source of chirality: (S)-glutamic acidAbsolute configuration: (S)(3S,6S)-1-(4-Methoxybenzyl)-6-allyl-2-oxopiperidin-3-yl acetateC18H23NO4[α]D20=+44.0 (c 1.0, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)(3S,6S)-1-(4-Methoxybenzyl)-6-allylpiperidin-3-olC16H23NO2[α]D20=-73.9 (c 1.1, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)(−)-(2R,5S)-epi-Pseudoconhydrine hydrochloride saltC8H17NO·HCl[α]D20=-10.2 (c 1.0, EtOH)Source of chirality: (S)-glutamic acidAbsolute configuration: (2R,5S)(3S,6S)-1-(4-Methoxybenzyl)-6-(2-oxo-2-phenylethyl)-3-(tetrahydro-2H-pyran-2-yloxy)piperidin-2-oneC26H31NO5[α]D20=-65.4 (c 1.0, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)(3S,6S)-1-(4-Methoxybenzyl)-3-hydroxy-6-(2-oxo-2-phenylethyl)piperidin-2-oneC21H23NO4[α]D20=-10.6 (c 1.1, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)(3S,6S)-1-(4-Methoxybenzyl)-6-((S)-2- hydroxy-2-phenylethyl)piperidin-3-olC21H27NO3[α]D20=-59.3 (c 1.0, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)(2S,5S)-tert-Butyl 5-hydroxy-2-((S)-2-hydroxy-2-phenylethyl)piperidine-1-carboxylateC18H27NO4[α]D20=-62.4 (c 0.8, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (2S,5S,2′S)(3S,6S)-6-((S)-2-Hydroxy-2-phenylethyl)-1-methylpiperidin-3-olC14H21NO2[α]D20=-53.4 (c 0.5, MeOH)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S,2′S)(3S,6S)-1-(4-Methoxybenzyl)-2-oxo-6-(2-oxoethyl)piperidin-3-yl acetateC17H21NO5[α]D20=+51.0 (c 1.0, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)(3S,6S)-1-(4-Methoxybenzyl)-6-(2-oxo-2-phenylethyl)-2-oxopiperidin-3-yl acetateC23H25NO5[α]D20=+6.9 (c 1.4, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (3S,6S)
Co-reporter:Liang-Xian Liu, Pei-Qiang Huang
Tetrahedron: Asymmetry 2007 Volume 18(Issue 1) pp:155
Publication Date(Web):31 January 2007
DOI:10.1016/j.tetasy.2007.01.016
Co-reporter:Liang-Xian Liu, Pei-Qiang Huang
Tetrahedron: Asymmetry 2006 Volume 17(Issue 23) pp:3265-3272
Publication Date(Web):11 December 2006
DOI:10.1016/j.tetasy.2006.12.009
Starting from protected (S)-3-hydroxyglutarimide 2a, the asymmetric synthesis of (2R,3S)-CP-99,994 8 was achieved. The crucial steps were a neighboring group participation leading to pyrrolidino-aziridinium intermediate 25 and the subsequent regioselective ring-opening reaction. In the case where neighboring group participation was not involved, only the eliminated product 15 was obtained.(5S,6R)-5-Hydroxyl-1-(4-methoxybenzyl)-6-phenyl-2-piperidinoneC19H21NO3[α]D20=+53.0 (c 1.0, CHCl3)Source of chirality:(S)-glutamic acidAbsolute configuration:(5S,6R)(2R,3S)-3-Hydroxyl-1-(4-methoxybenzyl)-2-phenylpiperidineC19H23NO2[α]D20=-18.5 (c 1.0, CHCl3)Source of chirality:(S)-glutamic acidAbsolute configuration:(2R,3S)(2R,3S)-3-[N-(2-Methoxybenzyl)-N-(2-nitrobenzenesulfonyl)]amino-1-(4-methoxybenzyl)-2-phenylpiperidineC33H35N3O6S[α]D20=-7.1 (c 1.0, CHCl3)Source of chirality:(S)-glutamic acidAbsolute configuration:(2R,3S)(2R,3S)-3-[N-(2-Aminobenzenesulfonyl)-N-(2-methoxybenzyl)]amino-2-phenyl piperidineC25H29N3O3S[α]D20=+91.7 (c 0.2, CHCl3)Source of chirality:(S)-glutamic acidAbsolute configuration:(2R,3S)(2R,3S)-3-(2-Methoxybenzyl)amino-2-phenylpiperidineC19H24N2O[α]D20=+73.4 (c 0.4, MeOH, dihydrochloride)Source of chirality:(S)-glutamic acidAbsolute configuration:(2R,3S)(5S,6R)-5-Hydroxyl-1-(4-methoxybenzyl)-6-methyl-2-piperidinoneC14H19NO3[α]D20=+64.1 (c 1.6, CHCl3)Source of chirality:(S)-glutamic acidAbsolute configuration:(5S,6R)(2R,3S)-3-Hydroxyl-1-(4-methoxybenzyl)-2-methylpiperidineC14H21NO2[α]D20=-32.7 (c 2.1, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (2R,3S)(2R,3S)-3-[N-(2-Methoxybenzyl)-N-(2-nitrobenzenesulfonyl)]amino-1-(4-methoxybenzyl)-2-methylpiperidineC28H33N3O6S[α]D20=-50.4 (c 1.4, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (2R,3S)(2R,3R)-3-[N-(2-Methoxybenzyl)-N-(2-nitrobenzenesulfonyl)]amino-1-(4-methoxybenzyl)-2-methylpiperidineC28H33N3O6S[α]D20=+10.2 (c 2.3, CHCl3)Source of chirality: (S)-glutamic acidAbsolute configuration: (2R,3R)
Co-reporter:Pei-Qiang Huang, Zhao-Ying Li
Tetrahedron: Asymmetry 2005 Volume 16(Issue 20) pp:3367-3370
Publication Date(Web):17 October 2005
DOI:10.1016/j.tetasy.2005.09.009
Co-reporter:Yuan-Ping Ruan;Xiu-Qing Xu;Bang-Guo Wei;Gang Liu;De-Sheng Yu;Liang-Xian Liu
Chirality 2005 Volume 17(Issue 9) pp:595-599
Publication Date(Web):30 SEP 2005
DOI:10.1002/chir.20205
An analytical HPLC method using CHIREX (S)-LEU/(S)-α-NEA column was developed for the determination of the enantiomeric excesses of N-protected (S)-3-hydroxyglutarimides. Using this method, detailed studies on the base-promoted ring-expansion reaction of the amidolactones, derived from l-glutamic acid, were undertaken. © 2005 Wiley-Liss, Inc. Chirality 17:595–599, 2005.
Co-reporter:Jing-Xing Du, Hui-Ying Huang, Pei-Qiang Huang
Tetrahedron: Asymmetry 2004 Volume 15(Issue 21) pp:3461-3466
Publication Date(Web):1 November 2004
DOI:10.1016/j.tetasy.2004.09.020
Based on the highly regio- and diastereoselective reductive-ethoxycarbonylmethylation of protected (S)-malimide 4, a new approach to the Geissman–Waiss lactone 1 and a key intermediate 3 for the synthesis of pyrrolidine trans-lactones (e.g., 2) is described. The synthesis features a one-step and a two-step chemoselective reduction of an amide carbonyl in the presence of an ester group as the key steps.Ethyl 2-[(4S,5R)-4-benzyloxy-1-(4-methoxybenzyl)-2-oxopyrrolidin-5-yl]acetateC23H27NO5[α]D20=+24.5 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)Ethyl 2-[(4S,5R)-4-hydroxy-1-(4-methoxybenzyl)-2-oxopyrrolidin-5-yl]acetateC16H21NO5[α]D20=+1.6 (c 1.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(1R,5R)-6-Aza-6-(4-methoxybenzyl)-2-oxabicyclo[3.3.0]octan-3,7-dioneC14H15NO4[α]D20=+48.2 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (1R,5R)(1R,5R)-6-(4-Methoxybenzyl)-6-aza-2-oxabicyclo[3.3.0]octan-3-oneC14H17NO3[α]D20=-3.65 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (1R,5R)(1R,5R)-6-Aza-2-oxabicyclo[3.3.0]octan-3-one hydrochlorideC6H10ClNO2[α]D20=+45.0 (c 0.4, MeOH)Source of chirality: (S)-malic acidAbsolute configuration: (1R,5R)Ethyl 2-[(4S,5R)-4-(benzyloxy)-1-(4-methoxybenzyl)-2-thioxopyrrolidin-5-yl]acetateC23H27NO4S[α]D20=+99.5 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)Ethyl 2-[(2R,3S)-3-(benzyloxy)-1-(4-methoxybenzyl)-pyrrolidin-2-yl]acetateC23H29NO4[α]D20=-19.4 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (2R,3S)Benzyl [(2R,3S)-2-ethoxycarbonylmethyl-3-hydroxypyrrolidin-1-yl]carboxylateC16H21NO5[α]D20=-33.9 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (2R,3S)
Co-reporter:Pei-Qiang Huang;Tian-Jun Wu;Jian-Liang Ye
Chinese Journal of Chemistry 2003 Volume 21(Issue 7) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20030210704
A flexible approach to ethyl (3R.4S)-N-Boc-4-amino-3-hydroxy-S-phenylpentanoate (N-Boc-AHPPA-OEt), the γ-aminoβ-hydroxy add moiety of hapalosin is described. The synthetic method features a ring-opening ethanolysis of an activated N-Boc-lactam, which is obtained via a diastereoselective reductive-alkylation of (R)-malimide derivative. The flexibility of the method resides in the introduction of the alkyl side chain by Grignard reagent addition.
Project supported by the National Science Foundation for Distinguished Young Investigators, the National Natural Science Foundation of China and the Fund for doctoral sites of the Ministry of Education.
Co-reporter:Bi-Yan He, Tian-Jun Wu, Xian-Yong Yu, Pei-Qiang Huang
Tetrahedron: Asymmetry 2003 Volume 14(Issue 14) pp:2101-2108
Publication Date(Web):18 July 2003
DOI:10.1016/S0957-4166(03)00442-7
A flexible non-amino acid-based formal asymmetric synthesis of naturally occurring antimicrobial (2R,3S)-2-aminotetradeca-5,7-dien-3-ol is reported. The method features a flexible and highly regioselective Grignard addition to (S)-malimide followed by a trans-diastereoselective reductive deoxygenation. The scope and limitations of the highly regio and diastereoselective reductive alkylation of malimides were defined. A remarkable protecting group effect on the regio and diastereoselective reductive alkylation of malimides was observed.Graphic(4S,5R)-4-Benzyloxy-1-(4-methoxybenzyl)-5-methyl-2-pyrrolidinoneC20H23NO3[α]D20=+91.2 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-methyl-2-pyrrolidinoneC12H15NO2[α]D20=+60.5 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-1-(tert-butyloxycarbonyl)-5-methyl-2-pyrrolidinoneC17H23NO4[α]D20=−21.1 (c 1.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Hydroxy-1-(tert-butyloxycarbonyl)-5-methyl-2-pyrrolidinoneC10H17NO4[α]D20=−48.4 (c 0.7, MeOH)[α]D20=−36.4 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(3S,4R)-4-(tert-Butyloxycarbonylamino)-3-hydroxypentanoic acid ethyl esterC12H23NO5[α]D20=+10.0 (c 0.58, MeOH)Source of chirality: (S)-malic acidAbsolute configuration: (3S,4R)(4S,5R)-4-Benzyloxy-5-(cyclohexylmethyl)-1-(4-methoxybenzyl)-2-pyrrolidinoneC26H33NO3[α]D20=+33.5 (c 2.1, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-5-(cyclohexylmethyl)-2-pyrrolidinoneC16H25NO2[α]D20=+50.0 (c 1.0, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Benzyloxy-1-(tert-butyloxycarbonyl)-5-(cyclohexylmethyl)-2-pyrrolidinoneC23H33NO4[α]D20=−34.8 (c 0.9, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(4S,5R)-4-Hydroxy-1-(tert-butyloxycarbonyl)-5-(cyclohexylmethyl)-2-pyrrolidinoneC16H27NO4[α]D20=−48.4 (c 1.1, MeOH)Source of chirality: (S)-malic acidAbsolute configuration: (4S,5R)(3S,4R)-4-[(tert-Butyloxycarbonyl)amino]-5-cyclohexyl-3-hydroxypentanoic acid ethyl esterC12H23NO5[α]D20=+21.5 (c 1.0, MeOH)Source of chirality: (S)-malic acidAbsolute configuration: (3S,4R)(S)-1-Benzyl-3-(tert-butyldimethylsilyloxy)pyrrolidine-2,5-dioneC17H25NO3Si[α]D20=−42.1 (c 1.2, CHCl3)Source of chirality: (S)-malic acidAbsolute configuration: (S)
Co-reporter:Ming-De Chen;Ming-Zhu He;Li-Qiang Huang;Yuan-Ping Ruan
Chinese Journal of Chemistry 2002 Volume 20(Issue 11) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20020201105
A flexible approach to (R)-3-alkyl-isoindolin-1-ones and (R)-3-aryl-isoindolin-l-ones via a diastereoselective reductive-alkylation is described. Present method is versatile in scope, allowing the easy introduction of various C-3 substituents by Grignard addition to phthalimide derived from (R)-phenylglycinol. 3-Alkyl-3-hydroxy-isoindolin-1-ones can also be obtained in the first step of the present method.
Co-reporter:Pei Qiang Huang, Quan Feng Chen, Chang Lin Chen, Hong Kui Zhang
Tetrahedron: Asymmetry 2000 Volume 11(Issue 8) pp:1843
Publication Date(Web):5 May 2000
DOI:10.1016/S0957-4166(00)00152-X
Co-reporter:Pei-Qiang Huang, Wei Ou and Jian-Liang Ye
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 9) pp:NaN1106-1106
Publication Date(Web):2015/07/20
DOI:10.1039/C5QO00191A
An efficient approach to N-monosubstituted β,β-difunctionalized enamines, a class of versatile building blocks for the synthesis of bioactive compounds, is reported. The method is based on the triflic anhydride-mediated direct aza-Knoevenagel-type condensation of secondary amides with active methylene compounds. The reaction showed good chemoselectivity and functional group tolerance. A number of title compounds have been synthesized in good to excellent yields in one pot from readily available starting materials.
Co-reporter:Pei-Qiang Huang, Su-Yu Huang, Long-Hui Gao, Zhong-Yi Mao, Zong Chang and Ai-E Wang
Chemical Communications 2016 - vol. 52(Issue 26) pp:NaN4840-4840
Publication Date(Web):2016/03/10
DOI:10.1039/C6CC90127A
Correction for ‘Enantioselective total synthesis of (+)-methoxystemofoline and (+)-isomethoxystemofoline’ by Pei-Qiang Huang et al., Chem. Commun., 2015, 51, 4576–4578.
Co-reporter:Pei-Qiang Huang, Su-Yu Huang, Long-Hui Gao, Zhong-Yi Mao, Zong Chang and Ai-E Wang
Chemical Communications 2015 - vol. 51(Issue 22) pp:NaN4578-4578
Publication Date(Web):2015/01/08
DOI:10.1039/C4CC09598G
The first enantioselective total synthesis of (+)-methoxystemofoline (2) and (+)-isomethoxystemofoline (3) has been reported. The synthesis employed the halide-assisted bromotropanonation method that we developed recently to construct the core structure, and Overman's strategy for the implementation of the butenolide moiety. Through this work, the structure of methoxystemofoline was revised as 2 with an E-alkene, and its absolute configuration was established.
Co-reporter:Qi-Long Peng, Shi-Peng Luo, Xiao-Er Xia, Liang-Xian Liu and Pei-Qiang Huang
Chemical Communications 2014 - vol. 50(Issue 16) pp:NaN1988-1988
Publication Date(Web):2013/12/12
DOI:10.1039/C3CC48833K
The total synthesis of the alkaloid (−)-chaetominine (1) has been achieved in four steps with an overall yield of 33.4%. Key features of our strategy include a one-pot cascade indole epoxidation – epoxide ring-opening cyclization – lactamization reaction sequence, and the use of a nitro group as a latent amino group for the one-pot construction of the quinazolinone ring. This constitutes a step economical, redox economical and protecting group-free total synthesis.
Co-reporter:Pei-Qiang Huang, Wei Ou, Kai-Jiong Xiao and Ai-E Wang
Chemical Communications 2014 - vol. 50(Issue 63) pp:NaN8763-8763
Publication Date(Web):2014/06/19
DOI:10.1039/C4CC03826F
We report one-pot and chemoselective Knoevenagel-type reactions using highly stable amides and lactams as the electrophilic substrates. The method is based on the in situ activation of amide carbonyl with triflic anhydride and a subsequent reaction with carbanions generated in situ from carbonyl compounds. The amide-based method is an alternative to the versatile thioamide-based Eschenmoser sulfide contraction.
Co-reporter:Xue-Kui Liu, Xiao Zheng, Yuan-Ping Ruan, Jie Ma and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 6) pp:NaN1284-1284
Publication Date(Web):2011/11/17
DOI:10.1039/C1OB06697H
The one-pot reductive coupling of N-acylcarbamates with activated alkenes is described. The method is based on partial reduction of N-acylcarbamates with DIBAL-H, followed by N-acyliminium ion formation and SmI2-mediated radical coupling with activated alkenes. Both acyclic and cyclic N-acylcarbamates can be used as stable substrates, and a range of activated alkenes serve as effective radical receptors. The reductive coupling of L-N-acylcarbamates 12/13 gave 2,5-disubstituted pyrrolidine derivatives in high trans-diastereoselectivities. The reductive coupling with penta-2,4-dienoate proceeded exclusively in a 1,6-addition fashion, producing a single non-conjugated E-isomer. On the basis of this method, a three-step construction of pyrrolo[1,2-a]azepin-5-one 16, the skeleton of many stemona alkaloids and lehmizidine alkaloids, and a seven-step synthesis of (−)-xenovenine (pyrrolizidinecis-223H, ent-6), the unnatural enantiomer of the frog/ant venom alkaloid possessing potent inhibitory activity towards nAChR channel, were achieved starting from L-12.
Co-reporter:Geng-Jie Lin and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 21) pp:NaN4495-4495
Publication Date(Web):2009/08/24
DOI:10.1039/B912190K
A seven-step synthesis of (3S,5R,8S,9S)-3-butyl-5-propyl-8-hydroxyindolizidine (2), an ant venom alkaloid isolated from Myrmicaria melanogaster, is disclosed with an overall yield of 28.9%. The key feature of the synthesis is the use of the iodocyclization for the introduction of the hydroxyl group of the 3-piperidinol. Remarkably, all the reaction steps proceeded with excellent chemo-, regio- and/or diastereoselectivities.
Co-reporter:Yu-Huang Wang, Jian-Liang Ye, Ai-E Wang and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 32) pp:NaN6511-6511
Publication Date(Web):2012/06/14
DOI:10.1039/C2OB25901J
We have developed a one-pot method for the direct intermolecular reductive hydroxyalkylation or alkylation of amines using lactones or esters as the hydroxyalkylating/alkylating reagents. The method is based on the in situ amidation of lactones/esters with DIBAL-H–amine complex (for primary amines) or DIBAL-H–amine hydrochloride salt complex (for secondary amines), followed by reduction of the amides with an excess of DIBAL-H. Different from the reduction of Weinreb amides with DIBAL-H where aldehydes are formed, the reduction of the in situ formed Weinreb amides yielded amines. Moreover, this method is not limited to Weinreb amides, instead, it also works for other amides in general. A plausible mechanism is suggested to account for the outcome of the reactions.
Co-reporter:Wen-Jun Liu, Jian-Liang Ye and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 9) pp:NaN2091-2091
Publication Date(Web):2010/03/03
DOI:10.1039/B926741G
A concise and flexible approach for the asymmetric syntheses of polyhydroxylated pyrrolizidine alkaloids hyacinthacines A2 and A3 has been developed using iterative reductive alkylation of O,O′-dibenzyltartarimide (5) as key steps. The ambiguity about the structure of synthetic hyacinthacine A3 due to the differences in the NMR data of the synthetic material (2) and the natural product (hyacinthacine A3) was eliminated jointly by comprehensive 1D and 2D-NMR studies, and by analysis of the 1H and 13C NMR spectra of a mixed synthetic product and natural hyacinthacine A3. The latter method also allowed a confirmation of the structure of the natural hyacinthacine A3, and may be useful for structural confirmation of other hydroxylated alkaloids.
Co-reporter:Chu-Pei Xu, Shi-Peng Luo, Ai-E Wang and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 18) pp:NaN2863-2863
Publication Date(Web):2014/02/13
DOI:10.1039/C4OB00314D
We demonstrated, for the first time, that on the basis of chemistry principles, the hexacyclic peptidyl alkaloid (−)-chaetominine (1) can be synthesized in a straightforward manner from L-Trp. The approach features the efficient generation of molecular complexity via a tandem C3/C14 syn-selective epoxidation (dr = 3:2)–annulative ring-opening reaction and a regioselective epimerization at C14. The successful production of (−)-chaetominine (1) from L-Trp could be helpful for revealing how the configuration of L-tryptophan becomes inverted in the biosynthetic pathway of (−)-chaetominine (1).
Co-reporter:Chu-Pei Xu, Zhen-Hua Xiao, Bi-Qin Zhuo, Yu-Huang Wang and Pei-Qiang Huang
Chemical Communications 2010 - vol. 46(Issue 41) pp:NaN7836-7836
Publication Date(Web):2010/09/09
DOI:10.1039/C0CC01487G
We report a mild and environmentally benign method for the synthesis of tertiary amines using alcohols as the alkylating reagents. Not only secondary amines such as piperazines but also amino acids and amino alcohols can be N-alkylated selectively. For N,O-benzyl protected amino alcohols, both N,O-de-benzylation and N-methylation were achieved in one-pot.
Co-reporter:Hong-Qiao Lan, Yuan-Ping Ruan and Pei-Qiang Huang
Chemical Communications 2010 - vol. 46(Issue 29) pp:NaN5321-5321
Publication Date(Web):2010/06/16
DOI:10.1039/C0CC00452A
Methyl tetramate derivative 6 has been developed as a new building block for the flexible and racemization-free synthesis of methyl 5-benzyl-3-methyltetramate via alkylation, and used in the first asymmetric synthesis of palau’imide (1). This allowed the establishment of the hitherto unknown stereochemistry at the C-20 of palau’imide as S.
Co-reporter:Yong-Gang Xiang, Xiang-Wu Wang, Xiao Zheng, Yuan-Ping Ruan and Pei-Qiang Huang
Chemical Communications 2009(Issue 45) pp:NaN7047-7047
Publication Date(Web):2009/10/13
DOI:10.1039/B915488D
The synergistic action of BF3·OEt2 and SmI2 allowed a series of intermolecular cross-couplings of readily available N-acyl N,O-acetals with α,β-unsaturated compounds to be performed in high yields, which was applied to the stereoselective synthesis of pyrrolizidine alkaloid (+)-xenovenine.
Co-reporter:Geng-Jie Lin, Xiao Zheng and Pei-Qiang Huang
Chemical Communications 2011 - vol. 47(Issue 5) pp:NaN1547-1547
Publication Date(Web):2010/11/25
DOI:10.1039/C0CC04371K
An efficient and highly diastereoselective method for the construction of the hydroxylated tropane skeleton is described. The method features a new intramolecular reductive coupling reaction of N-acyl N,O-acetal with aldehyde, cooperatively mediated by BF3·OEt2 and SmI2. On the basis of this method, a new enantioselective total synthesis of (−)-Bao Gong Teng A has been accomplished.
Co-reporter:Su-Yu Huang, Zong Chang, Shi-Chuan Tuo, Long-Hui Gao, Ai-E Wang and Pei-Qiang Huang
Chemical Communications 2013 - vol. 49(Issue 63) pp:NaN7090-7090
Publication Date(Web):2013/06/13
DOI:10.1039/C3CC43665A
The halo-assisted intramolecular addition of silyl enol ethers with in situ activated lactams yielded (hydroxylated) 1-halo-8-azabicyclo[3,2,1]octane and 1-halo-9-azabicyclo[3,3,1]nonane ring systems, which provided an easy enantioselective access to 6β-silyloxytropane-3-one, 3α,6β-dihydroxytropane, and pervilleine B. The absolute configuration of the natural (−)-pervilleine B was determined to be 1R,3R,5S,6R.
Co-reporter:Pei-Qiang Huang, Qi-Wei Lang, Ai-E Wang and Jian-Feng Zheng
Chemical Communications 2015 - vol. 51(Issue 6) pp:NaN1099-1099
Publication Date(Web):2014/11/25
DOI:10.1039/C4CC08330J
We report the first one-pot reductive homocoupling reaction of secondary amides and cross-coupling reaction of secondary amides with ketones to give secondary vicinal diamines and amino alcohols. This method relies on the direct generation of α-amino carbon radicals from secondary amides by activation with trifluoromethanesulfonic anhydride, partial reduction with triethylsilane and samarium diiodide-mediated single-electron transfer. The reactions were run under mild conditions and tolerated several functional groups.
Co-reporter:Jian-Liang Ye, Yang Liu, Zhi-Ping Yang and Pei-Qiang Huang
Chemical Communications 2016 - vol. 52(Issue 3) pp:NaN563-563
Publication Date(Web):2015/10/26
DOI:10.1039/C5CC07480K
The asymmetric total synthesis of (+)-N-acetyl norloline, the putative biogenic precursor of all known loline alkaloids, has been achieved in 12 steps from commercially available (R)-glyceraldehyde acetonide. The synthesis relies on the Rassu/Casiraghi's vinylogous aldol reaction, an intramolecular oxa-heteroconjugate addition and a reductive amination to establish the four contiguous stereogenic centers and construct the strained oxygen-bridge under mild conditions.
Co-reporter:Hui-Qiong Deng, Xiang-Yang Qian, Yu-Xiu Li, Jian-Feng Zheng, Linfeng Xie and Pei-Qiang Huang
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 3) pp:NaN266-266
Publication Date(Web):2014/02/11
DOI:10.1039/C3QO00065F
The reductive alkylation of secondary lactams is an important transformation in the total synthesis of alkaloids and pharmaceuticals. Methods for this transformation are scarce. We report in this paper a versatile two-step approach consisting of N-benzylation and one-pot reductive alkylation–debenzylation. With suitably functionalized Grignard reagents, a formal [4 + 2] annulation reaction has been achieved. Using this method, we have successfully synthesized several N-α-alkyl and N-α,α′-dialkyl heterocycles, including (±)-coniine (7) and the ant venom alkaloids (2R,5S)-cis-2-butyl-5-propylpyrrolidine (8) and (+)-monomorine I (10) each in two steps starting from readily available secondary lactams.
Co-reporter:Pei-Qiang Huang and Hui Geng
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 2) pp:NaN158-158
Publication Date(Web):2015/01/06
DOI:10.1039/C4QO00317A
The reduction of secondary amides to amines is an important transformation for the total synthesis of alkaloids and pharmaceuticals. General and chemoselective direct methods for this transformation are scarce. We report in this paper a simple method for the direct reduction of secondary amides, which uses only two reagents, triflic anhydride for amide activation, and NaBH4 for reduction. Running under mild conditions (0 °C to r.t.), the reaction works well with several types of secondary amides including aromatic amides, aliphatic amides, and α,β-unsaturated amides. The reaction displays a good functional group tolerance for a series of reducible functional groups. The method is applicable to the direct reduction of C–H lithiation-functionalization products, and C–H activation-functionalization products. Moreover, chemoselective reduction of secondary lactams has been achieved using Cp2ZrHCl–NaBH4 combination.
Co-reporter:Zhong-Yi Mao, Hui Geng, Tian-Tian Zhang, Yuan-Ping Ruan, Jian-Liang Ye and Pei-Qiang Huang
Inorganic Chemistry Frontiers 2016 - vol. 3(Issue 1) pp:NaN37-37
Publication Date(Web):2015/10/30
DOI:10.1039/C5QO00298B
The first enantioselective and stereodivergent total syntheses of (−)-isochaetominines A–C and all eight 2,3-cis-stereoisomers of (−)-isochaetominine C, including the natural (+)-14-epi-isochaetominine C, and the proposed structures of (−)-pseudofischerine (2) and (−)-aniquinazoline D (3), have been achieved. The stereodivergent approach relies on the DMDO-initiated divergent tandem reaction to give a separable mixture of two products, a monocyclization product and a diastereomer of isochaetominine C (or a homologue) as a result of double cyclization. An epimerization-free two-step protocol has been developed for the highly diastereoselective transformation of the former product into an isochaetominine-type compound with characteristic 3,14-cis-stereochemistry. As a result of our synthetic efforts, the structures of the natural (−)-pseudofischerine and (−)-aniquinazoline D have been revised both as (−)-isochaetominine C (6).
Co-reporter:Pei-Qiang Huang, Ying-Hong Huang and Shu-Ren Wang
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 3) pp:
Publication Date(Web):
DOI:10.1039/C6QO00720A
Co-reporter:Pei-Qiang Huang, Wei Ou and Feng Han
Chemical Communications 2016 - vol. 52(Issue 80) pp:NaN11970-11970
Publication Date(Web):2016/09/05
DOI:10.1039/C6CC05318A
We report herein a convenient and versatile method for the direct reductive alkynylation of tertiary amides to give propargylic amines through sequential Ir-catalysed hydrosilylation–Cu(I)-catalysed alkynylation. The reactions proceed chemoselectively at the amide group in the presence of several sensitive functional groups including the very reactive aldehyde group on either the amide or the alkyne coupling partner. The method is general for tert-amides with or without α-hydrogen.
Co-reporter:Shao-Feng Wu, Xiao Zheng, Yuan-Ping Ruan and Pei-Qiang Huang
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 14) pp:NaN2975-2975
Publication Date(Web):2009/06/02
DOI:10.1039/B906224F
A flexible diastereoselective approach to trans-(3S)-hydroxyprolinol derivatives is described, which is based on the samarium diiodide-mediated reductive coupling of the chiral 1-pyrroline N-oxide (nitrone)(S)-10 with carbonyl compounds. The reductive hydroxyalkylation of nitrone 10 with ketones and aromatic aldehydes is highly diastereoselective in establishing the C-2 chiral center of the pyrrolidine ring.




























![2-METHYL 6-(2-METHYL-2-PROPANYL) 1-OXA-6-AZASPIRO[3.4]OCTANE-2,6-<WBR />DICARBOXYLATE](http://img.cochemist.com/ccimg/3900/3851-81-8.png)
![2-METHYL 6-(2-METHYL-2-PROPANYL) 1-OXA-6-AZASPIRO[3.4]OCTANE-2,6-<WBR />DICARBOXYLATE](http://img.cochemist.com/ccimg/3900/3851-81-8_b.png)








![Pentanoic acid, 5-[(2,6-dimethylphenyl)amino]-5-oxo-, methyl ester](http://img.cochemist.com/ccimg/887400/887307-34-8.png)
![Pentanoic acid, 5-[(2,6-dimethylphenyl)amino]-5-oxo-, methyl ester](http://img.cochemist.com/ccimg/887400/887307-34-8_b.png)






![MAGNESIUM, [4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]BUTYL]IODO-](http://img.cochemist.com/ccimg/862100/862013-95-4.png)
![MAGNESIUM, [4-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]BUTYL]IODO-](http://img.cochemist.com/ccimg/862100/862013-95-4_b.png)
![Thiourea,N-[(1R,2R)-2-aminocyclohexyl]-N'-[3,5-bis(trifluoromethyl)phenyl]-](/data/chemimg/103900/860994-58-7.png)
![Thiourea,N-[(1R,2R)-2-aminocyclohexyl]-N'-[3,5-bis(trifluoromethyl)phenyl]-](/data/chemimg/103900/860994-58-7_b.png)


























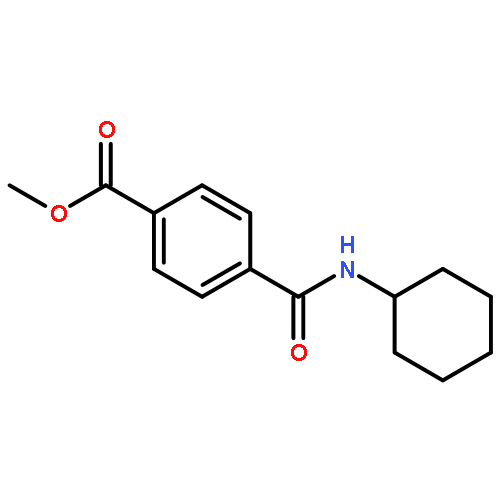

















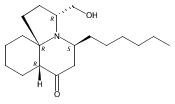

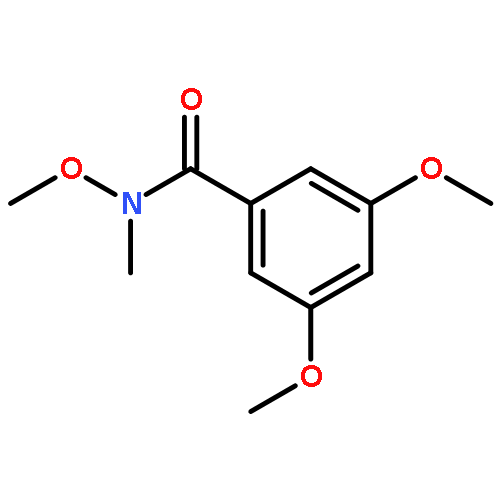




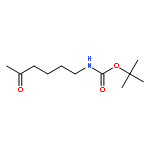










![Lithium, [(diethylamino)diphenylsilyl]-](http://img.cochemist.com/ccimg/140500/140438-35-3.png)
![Lithium, [(diethylamino)diphenylsilyl]-](http://img.cochemist.com/ccimg/140500/140438-35-3_b.png)




![Silane, (1,1-dimethylethyl)dimethyl[(3-methyl-2-furanyl)oxy]-](http://img.cochemist.com/ccimg/131500/131497-06-8.png)
![Silane, (1,1-dimethylethyl)dimethyl[(3-methyl-2-furanyl)oxy]-](http://img.cochemist.com/ccimg/131500/131497-06-8_b.png)

















![2-Propenamide,N-[(1R,7aS)-hexahydro-1H-pyrrolizin-1-yl]-3-(4-methoxyphenyl)-, (2E)-](http://img.cochemist.com/ccimg/112600/112513-33-4.png)
![2-Propenamide,N-[(1R,7aS)-hexahydro-1H-pyrrolizin-1-yl]-3-(4-methoxyphenyl)-, (2E)-](http://img.cochemist.com/ccimg/112600/112513-33-4_b.png)




























![Benzamide, N-[(1S)-1-(methoxymethyl)-2-methylpropyl]-](http://img.cochemist.com/ccimg/87800/87768-46-5.png)
![Benzamide, N-[(1S)-1-(methoxymethyl)-2-methylpropyl]-](http://img.cochemist.com/ccimg/87800/87768-46-5_b.png)










![Tricyclo[3.3.1.13,7]decane-1-carboxamide, N-cyclohexyl-](http://img.cochemist.com/ccimg/81400/81311-58-2.png)
![Tricyclo[3.3.1.13,7]decane-1-carboxamide, N-cyclohexyl-](http://img.cochemist.com/ccimg/81400/81311-58-2_b.png)


![METHYL 1-AZABICYCLO[2.2.1]HEPTANE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/73400/73325-61-8.png)
![METHYL 1-AZABICYCLO[2.2.1]HEPTANE-3-CARBOXYLATE](http://img.cochemist.com/ccimg/73400/73325-61-8_b.png)




















































![N-[(1S,6R,7S,7aS)-hexahydro-1H-1,6-epoxypyrrolizin-7-yl]acetamide](http://img.cochemist.com/ccimg/39000/38964-35-1.png)
![N-[(1S,6R,7S,7aS)-hexahydro-1H-1,6-epoxypyrrolizin-7-yl]acetamide](http://img.cochemist.com/ccimg/39000/38964-35-1_b.png)


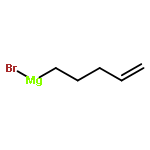
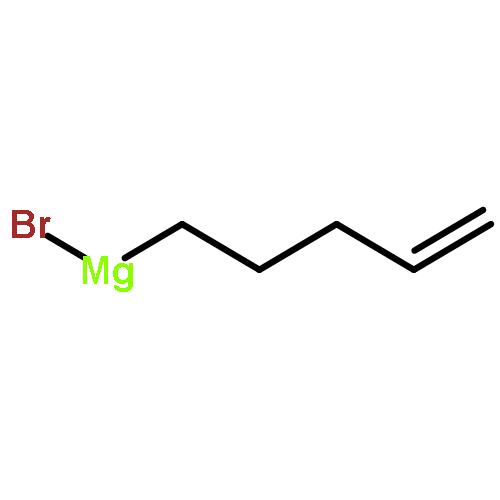

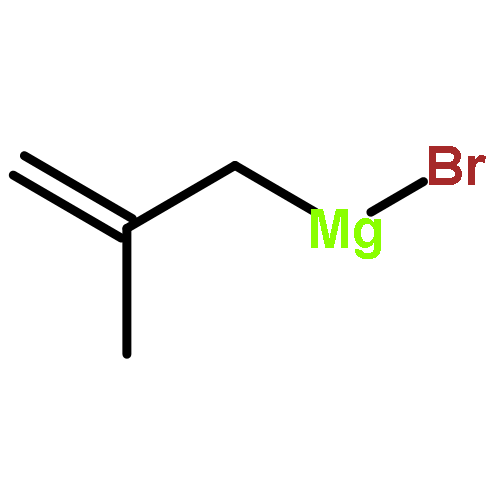


![2-Propen-1-one, 3-phenyl-1-[4-(trifluoromethyl)phenyl]-, (E)-](http://img.cochemist.com/ccimg/32200/32120-33-5.png)
![2-Propen-1-one, 3-phenyl-1-[4-(trifluoromethyl)phenyl]-, (E)-](http://img.cochemist.com/ccimg/32200/32120-33-5_b.png)
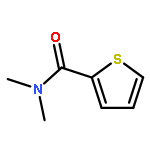

















![1H-Pyrrolo[2,1-c][1,4]benzodiazepine-5,11(10H,11aH)-dione,2,3-dihydro-, (11aS)-](http://img.cochemist.com/ccimg/18900/18877-34-4.png)
![1H-Pyrrolo[2,1-c][1,4]benzodiazepine-5,11(10H,11aH)-dione,2,3-dihydro-, (11aS)-](http://img.cochemist.com/ccimg/18900/18877-34-4_b.png)
























![8H-6,11b-Methanofuro[2,3-c]pyrido[1,2-a]azepin-2(6H)-one,9,10,11,11a-tetrahydro-, (6R,11aS,11bR)-](http://img.cochemist.com/ccimg/6800/6704-68-3.png)
![8H-6,11b-Methanofuro[2,3-c]pyrido[1,2-a]azepin-2(6H)-one,9,10,11,11a-tetrahydro-, (6R,11aS,11bR)-](http://img.cochemist.com/ccimg/6800/6704-68-3_b.png)






















![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4.png)
![Benzo[d][1,3]dioxole-5-carbonitrile](http://img.cochemist.com/ccimg/4500/4421-09-4_b.png)



























![Butanoic acid,3-methyl-, (1S,3R,5S,6R)-6-hydroxy-8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester](http://img.cochemist.com/ccimg/500/490-96-0.png)
![Butanoic acid,3-methyl-, (1S,3R,5S,6R)-6-hydroxy-8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester](http://img.cochemist.com/ccimg/500/490-96-0_b.png)








![N-{2-[(3-CYANO-5,7-DIMETHYL-2-QUINOLINYL)AMINO]ETHYL}-3-METHOXYBE<WBR />NZAMIDE](http://img.cochemist.com/ccimg/321700/321661-62-5.png)
![N-{2-[(3-CYANO-5,7-DIMETHYL-2-QUINOLINYL)AMINO]ETHYL}-3-METHOXYBE<WBR />NZAMIDE](http://img.cochemist.com/ccimg/321700/321661-62-5_b.png)
![2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1H-Pyrrole-1-carboxylic acid 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/141300/141293-28-9.png)
![2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1H-Pyrrole-1-carboxylic acid 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/141300/141293-28-9_b.png)


![Quinazolino[3,2-a][1,4]benzodiazepine-5,13-dione,6,7-dihydro-7-(1H-indol-3-ylmethyl)-, (7S)-](http://img.cochemist.com/ccimg/93500/93413-06-0.png)
![Quinazolino[3,2-a][1,4]benzodiazepine-5,13-dione,6,7-dihydro-7-(1H-indol-3-ylmethyl)-, (7S)-](http://img.cochemist.com/ccimg/93500/93413-06-0_b.png)





















![(5E)-1-(2-ethylphenyl)-5-{[5-(piperidin-1-yl)furan-2-yl]methylidene}-2-thioxodihydropyrimidine-4,6(1H,5H)-dione](http://img.cochemist.com/ccimg/6400/6335-52-0.png)
![(5E)-1-(2-ethylphenyl)-5-{[5-(piperidin-1-yl)furan-2-yl]methylidene}-2-thioxodihydropyrimidine-4,6(1H,5H)-dione](http://img.cochemist.com/ccimg/6400/6335-52-0_b.png)

















![N-[(1S)-1-phenylethyl]benzamide](http://img.cochemist.com/ccimg/4200/4108-58-1.png)
![N-[(1S)-1-phenylethyl]benzamide](http://img.cochemist.com/ccimg/4200/4108-58-1_b.png)