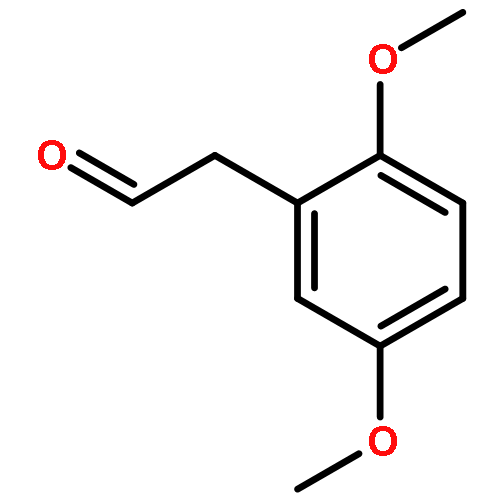
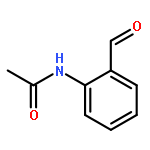
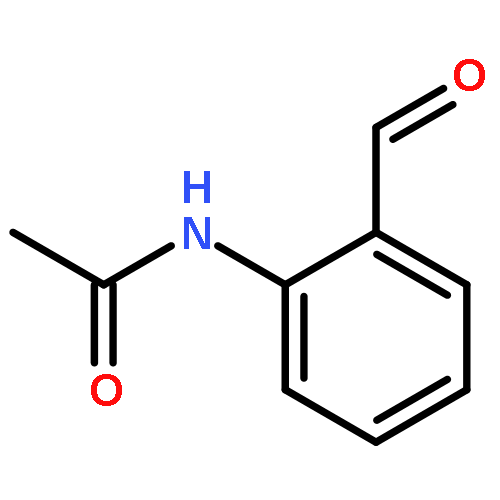
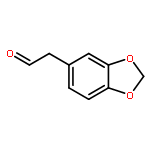
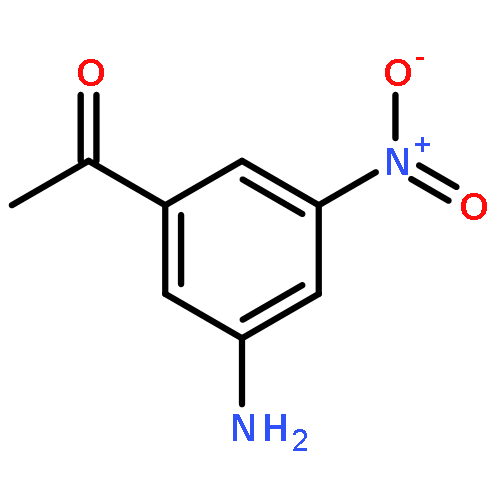
Co-reporter:Liu Chen, Danqi Chen, Le Tang, Jing Ren, Jiaojiao Chen, Xuechu Zhen, Yu-Chih Liu, Chenhua Zhang, Haibin Luo, Jingkang Shen, Bing Xiong
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmc.2017.04.019
Phosphodiesterases are important enzymes regulating signal transduction mediated by second messenger molecules cAMP or cGMP. PDE10A is a unique member in the PDE family because of its selective expression in medium spiny neurons. It is recognized as anti-psychotic drug target. Based on the structural similarity between our previous chemistry work on 8-aminoimidazo[1,2-a]pyrazines and the PDE10A inhibitors reported by Bartolome-Nebreda et al., we initialized a project for developing PDE10A inhibitors. After several rounds of optimization, we were able to obtain a few compounds with good PDE10A enzymatic activity. And after further PDE enzymatic selectivity study, metabolic stability assay and in vivo pharmacological tests we identified two inhibitors as interesting lead compounds with the potential for further PDE10A lead optimizatioin.Download high-res image (135KB)Download full-size image
Co-reporter:Jianping Hu, Yingqing Wang, Yanlian Li, Lin Xu, Danyan Cao, ShanShan Song, Mohammadali Soleimani Damaneh, Xin Wang, Tao Meng, Yue-Lei Chen, Jingkang Shen, Zehong Miao, Bing Xiong
European Journal of Medicinal Chemistry 2017 Volume 137(Volume 137) pp:
Publication Date(Web):8 September 2017
DOI:10.1016/j.ejmech.2017.05.049
•Using scaffold modification to design BRD4 inhibitors based on the PLK1-BRD4 dual inhibitor BI-2536.•Structure-based lead optimization and identify potent BRD4 inhibitors.•Selectivity profile on 32 bromodomains showed that this series is selective BET inhibitors.•In vitro study confirmed the compounds have profound effects on down-regulation of c-Myc.•In vivo study confirmed the efficacy on MM.1S CDX model.Recent years have seen much effort to discover new chemotypes of BRD4 inhibitors. Interestingly, some kinase inhibitors have been demonstrated to be potent bromodomain inhibitors, especially the PLK1 inhibitor BI-2536 and the JAK2 inhibitor TG101209, which can bind to BRD4 with IC50 values of 0.025 μM and 0.13 μM, respectively. Although the concept of dual inhibition is intriguing, selective BRD4 inhibitors are preferred as they may diminish off-target effects and provide more flexibility in anticancer drug combination therapy. Inspired by BI-2536, we designed and prepared a series of dihydroquinoxalin-2(1H)-one derivatives as selective bromodomain inhibitors. We found compound 54 had slightly higher activity than (+)-JQ1 in the fluorescence anisotropy assay and potent antiproliferative cellular activity in the MM.1S cell line. We have successfully solved the cocrystal structure of 52 in complex with BRD4-BD1, providing a solid structural basis for the binding mode of compounds of this series. Compound 54 exhibited high selectivity over most non-BET subfamily members and did not show bioactivity towards the PLK1 kinase at 10 or 1 μM. From in vivo studies, compound 54 demonstrated a good PK profile, and the results from in vivo pharmacological studies clearly showed the efficacy of 54 in the mouse MM.1S xenograft model.Download high-res image (156KB)Download full-size image
Co-reporter:Weiteng An, Wei Wang, Ting Yu, Yongliang Zhang, Zehong Miao, Tao Meng, Jingkang Shen
European Journal of Medicinal Chemistry 2016 Volume 112() pp:367-372
Publication Date(Web):13 April 2016
DOI:10.1016/j.ejmech.2016.02.004
•Based on the iMHB approach, a novel series of 2-phenyl-imidazo[1,2-a]pyridine analogues were designed and synthesized.•Compounds 1a, 3b, 3d were found to have excellent antiproliferative activities against HeLa cell.•These compounds showed good tubulin polymerization inhibition activities.•These studies provided a new class of antitubulin agents with the potential to be clinically developed for cancer treatment.A series of 2-aryl-imidazo-pyridines/pyrazines derivatives has been designed, synthesized and evaluated for their biological activities. Among them, several investigated compounds (1a, 3b and 3d) displayed potent antiproliferative activity against HeLa cell, and also displayed comparable tubulin polymerization inhibitory activity to colchicine. These studies provided a new molecular scaffold for the further development of antitumor agents that target tubulin.The discovery of a series of 2-phenyl-imidazo[1,2-a]pyridines as a new class of antitubulin agents. SARs observed for members of this series revealed that the presence of intramolecular hydrogen bonding formed by the phenolic group and nitrogen atom, is crucial for antiproliferative activity.
Co-reporter:Jianping Hu, Xin Wang, Lin Chen, Min Huang, Wei Tang, Jianping Zuo, Yu-Chih Liu, Zhe Shi, Rongfeng Liu, Jingkang Shen, Bing Xiong
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 3) pp:721-725
Publication Date(Web):1 February 2016
DOI:10.1016/j.bmcl.2016.01.006
The histone methylation on lysine residues is one of the most studied post-translational modifications, and its aberrant states have been associated with many human diseases. In 2012, Kruidenier et al. reported GSK-J1 as a selective Jumonji H3K27 demethylase (JMJD3 and UTX) inhibitor. However, there is limited information on the structure–activity relationship of this series of compounds. Moreover, there are few scaffolds reported as chelating groups for Fe(II) ion in Jumonji demethylase inhibitors development. To further elaborate the structure–activity relationship of selective JMJD3 inhibitors and to explore the novel chelating groups for Fe(II) ion, we initialized a medicinal chemistry modification based on the GSK-J1 structure. Finally, we found that several compounds bearing different chelating groups showed similar activities with respect to GSK-J1 and excellent metabolic stability in liver microsomes. The ethyl ester prodrugs of these inhibitors also showed a better activity than GSK-J4 for inhibition of TNF-α production in LPS-stimulated murine macrophage cell line Raw 264.7 cells. Taking together, the current study not only discovered alternative potent JMJD3 inhibitors, but also can benefit other researchers to design new series of Jumonji demethylase inhibitors based on the identified chelating groups.
Co-reporter:Lele Zhao; Yingqing Wang; Danyan Cao; Tiantian Chen; Qi Wang; Yanlian Li; Yechun Xu; Naixia Zhang; Xin Wang; Danqi Chen; Lin Chen; Yue-Lei Chen; Guangxin Xia; Zhe Shi; Yu-Chih Liu; Yijyun Lin; Zehong Miao; Jingkang Shen;Bing Xiong
Journal of Medicinal Chemistry 2015 Volume 58(Issue 3) pp:1281-1297
Publication Date(Web):January 5, 2015
DOI:10.1021/jm501504k
The signal transduction of acetylated histone can be processed through a recognition module, bromodomain. Several inhibitors targeting BRD4, one of the bromodomain members, are in clinical trials as anticancer drugs. Hereby, we report our efforts on discovery and optimization of a new series of 2-thiazolidinones as BRD4 inhibitors along our previous study. In this work, guided by crystal structure analysis, we reversed the sulfonamide group and identified a new binding mode. A structure–activity relationship study on this new series led to several potent BRD4 inhibitors with IC50 of about 0.05–0.1 μM in FP binding assay and GI50 of 0.1–0.3 μM in cell based assays. To complete the lead-like assessment of this series, we further checked its effects on BRD4 downstream protein c-Myc, investigated its selectivity among five different bromodomain proteins, as well as the metabolic stability test, and reinforced the utility of 2-thiazolidinone scaffold as BET bromodomain inhibitors in novel anticancer drug development.
Co-reporter:Yuchi Ma; Guangqiang Sun; Danqi Chen; Xia Peng; Yue-Lei Chen; Yi Su; Yinchun Ji; Jin Liang; Xin Wang; Lin Chen; Jian Ding; Bing Xiong; Jing Ai; Meiyu Geng
Journal of Medicinal Chemistry 2015 Volume 58(Issue 5) pp:2513-2529
Publication Date(Web):February 10, 2015
DOI:10.1021/jm502018y
c-Met has emerged as an attractive target for targeted cancer therapy because of its abnormal activation in many cancer cells. To identify high potent and selective c-Met inhibitors, we started with profiling the potency and in vitro metabolic stability of a reported hit 7. By rational design, a novel sulfonylpyrazolo[4,3-b]pyridine 9 with improved DMPK properties was discovered. Further elaboration of π–π stacking interactions and solvent accessible polar moieties led to a series of highly potent and selective type I c-Met inhibitors. On the basis of in vitro and in vivo pharmacological and pharmacokinetics studies, compound 46 was selected as a preclinical candidate for further anticancer drug development.
Co-reporter:Fengxia Yang, Weiteng An, Zhiwei Qian, Ting Yu, Yongli Du, Lanping Ma, Xin Wang, Tao Meng and Jingkang Shen
RSC Advances 2015 vol. 5(Issue 40) pp:32015-32019
Publication Date(Web):23 Mar 2015
DOI:10.1039/C5RA02809D
An efficient route to the synthesis of 3-amino-2-ethoxycarbonyl imidazo[1,2-a]pyridine derivatives starting from ethyl α-benzotriazolyl-α-morpholinoacetate has been developed. By the reactions between primary heterocyclic amidines and isocyanides catalyzed by Ga(OTf)3, target compounds were obtained in moderate to good yields. This in turn will set the stage for the wide application of this useful reaction for the synthesis of 3-amino-2-carbonylimidazo[1,2-a]pyridines based on imidazo[1,2-a]pyridine privileged structures.
Co-reporter:Tao Meng ; Dadong Zhang ; Zuoquan Xie ; Ting Yu ; Shuchao Wu ; Lorenza Wyder ; Urs Regenass ; Kurt Hilpert ; Min Huang ; Meiyu Geng
Journal of Medicinal Chemistry 2014 Volume 57(Issue 23) pp:9832-9843
Publication Date(Web):November 10, 2014
DOI:10.1021/jm5010144
Upregulation of pyruvate dehydrogenase kinase (PDHK) has been observed in a variety of cancers. Inhibition of PDHK offers an attractive opportunity for the development of novel cancer therapies. To obtain novel PDHK inhibitors, we took advantage of the homology of the ATP-binding pocket between Heat Shock Protein 90 (HSP90) and PDHK, and utilized 4,5-diarylisoxazole based HSP90 inhibitor for structural design. Our efforts led to the identification of 5k that inhibited PDHK1 with an IC50 value of 17 nM, which, however, showed marginal cellular activity. Further structural optimization resulted in compound 11a with improved cellular activity which could effectively modulate the metabolic profile of cancer cells and lead to the inhibition of cancer cell proliferation, evidenced by the increased oxidative phosphorylation and decreased glycolysis and associated oxidative stress. Our results suggested 11a as an excellent lead compound and a favorable biological tool to further evaluate the therapeutic potential of PDHK and HSP90 dual inhibitors in the treatment of cancer.
Co-reporter:Danqi Chen, Aijun Shen, Jian Li, Feng Shi, Wuyan Chen, Jing Ren, Hongchun Liu, Yechun Xu, Xin Wang, Xinying Yang, Yiming Sun, Min Yang, Jianhua He, Yueqin Wang, Liping Zhang, Min Huang, Meiyu Geng, Bing Xiong, Jingkang Shen
European Journal of Medicinal Chemistry 2014 Volume 87() pp:765-781
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.065
•A new scaffold of HSP90 inhibitors was developed with aids of fragment screening.•Comprehensive assessment was taken.•Compound 108 exhibited excellent in vivo activities and pharmacokinetic properties.HSP90 is ubiquitously overexpressed in a broad spectrum of human cancers and has been recognized as an attractive target for cancer treatment. Here, we described the fragment screening, synthesis and structure-activity relationship studies of small molecule inhibitors with 4,5-diarylisoxazole scaffold targeting HSP90. Among them, the compound N-(3-(2,4-dihydroxy-5-isopropylphenyl)-4-(4-((4-morpholinopiperidin-1-yl)methyl)phenyl)isoxazol-5-yl)cyclopropanecarboxamide (108) showed high affinity for binding to HSP90 (FP binding assay, IC50 = 0.030 μM) and inhibited the proliferation of various human cancer cell lines with averaging GI50 about 88 nM. Compound 108 exhibited its functional inhibition of HSP90 by depleting key signaling pathways and concomitantly elevating of HSP70 and HSP27 in U-87MG cells. Further in vivo studies showed that compound 108 strongly suppressed the tumor growth of human glioblastoma xenograft model U-87MG with T/C = 18.35% at 50 mg/kg q3w/2.5w. Moreover, compound 108 also exhibited good pharmacokinetic properties. Together, our study implicates that compound 108 is a promising candidate of HSP90 inhibitor and is currently advanced to preclinical study.By fragment screening and x-ray crystallography techniques a scaffold of 4,5-diarylisoxazole was selected and developed to a novel series of HSP90 inhibitors with excellent in vivo activities.
Co-reporter:Tao Meng, Wei Wang, Zhixiang Zhang, Lanping Ma, Yongliang Zhang, Zehong Miao, Jingkang Shen
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 2) pp:848-855
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmc.2013.12.004
A series of 6H-pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5(6H)-ones have been synthesized and evaluated for their antiproliferative activities. Among them, compounds 2j and 4d displayed potent cytotoxic activities in vitro against HeLa cell line with IC50 values of 0.07 and 0.06 μM, respectively. In general, the antiproliferative activities are correlated with the inhibitory effect on tubulin polymerization and binding property of the colchicine binding site. In addition, flow cytometry and immunofluorescence analysis revealed selected compounds caused G2/M phase arrest of the cell cycle and disruption of the mitotic spindle assembly, which had correlation with proliferation inhibitory activity.
Co-reporter:Xiao Liu, Bingbing Zhang, Xiangying Gu, Guohua Chen, Lin Chen, Xin Wang, Bing Xiong, Qi-Dong You, Yue-Lei Chen, Jingkang Shen
Carbohydrate Research 2014 Volume 398() pp:45-49
Publication Date(Web):29 October 2014
DOI:10.1016/j.carres.2014.05.010
•1,2-trans-1-Dihydroxyboryl benzyl S-glycosides were readily prepared.•From these compounds, glycosylation with simple alcohols gave pure 1,2-cis-O-glycoside outcome.•Boronic acid moiety was found essential for the product formation and good anomeric ratio.Activated by NBS, readily available 1,2-trans-1-dihydroxyboryl benzyl S-glycosides served as glycosyl donors and reacted with certain simple alcohol acceptors to produce pure 1,2-cis-O-glycosides in moderate yields. The boronic acid moiety was revealed essential in the glycosylation for product formation and good anomeric ratio. The preliminary model reactions suggested that glycosyl aryl boronic acids could be used for stereoselective glycosylation.
Co-reporter:Xiangying Gu, Lin Chen, Xin Wang, Xiao Liu, Qidong You, Wenwei Xi, Li Gao, Guohua Chen, Yue-Lei Chen, Bing Xiong, and Jingkang Shen
The Journal of Organic Chemistry 2014 Volume 79(Issue 3) pp:1100-1110
Publication Date(Web):January 10, 2014
DOI:10.1021/jo402551x
A new strategy for diversity-oriented direct glycosylation of bioactive small molecules was developed. This reaction features (−)-β-pinene as acid scavenger and work with glycosyl iodides under mild conditions. With the aid of RP-HPLC and chiral SFC separation techniques, the new direct glycosylation proved effective at gram scale on bioactive small molecules including AZD6244, podophyllotoxin, paclitaxel, and docetaxel. Interesting glycoside derivatives were efficiently created with good yields and 1,2-cis selectivity.
Co-reporter:Linrong Zhu;Dr. Yuanyuan Li;Dr. Ling Qiu;Dr. Mingbo Su; Xin Wang;Chunmei Xia;Dr. Yi Qu; Jingya Li; Jia Li; Bing Xiong; Jingkang Shen
ChemMedChem 2013 Volume 8( Issue 7) pp:1104-1116
Publication Date(Web):
DOI:10.1002/cmdc.201300104
Abstract
The worldwide prevalence of diabetes has spurred numerous studies on the development of new antidiabetic medicines. As a result, dipeptidyl peptidase IV (DPP4) has been recognized as a validated target. In our efforts to discover new DPP4 inhibitors, we analyzed the complexed structures of DPP4 available in Protein Data Bank and designed a series of triazole compounds. After enzyme activity assays and crystallographic verification of the binding interaction patterns, we found that the triazole compounds can inhibit DPP4 with micromolar IC50 values. Liver microsome stability and cytochrome P450 metabolic tests were performed on this series, revealing undesirable pharmacokinetic profiles for the triazole compounds. To overcome this liability, we substituted the triazole ring with an amide or urea group to produce a new series of DPP4 inhibitors. Based on its enzyme activity, metabolic stability, and selectivity over DPP8 and DPP9, we selected compound 21 r for further study of its in vivo effects in mice using an oral glucose tolerance test (OGTT). The results show that 21 r has efficacy similar to that of sitagliptin at a dose of 3 mg kg−1. The crystal structure of 21 r bound to DPP4 also reveals that the trifluoromethyl group is directed toward a subpocket different from the subsite bound by sitagliptin, providing clues for the design of new DPP4 inhibitors.
Co-reporter:Danqi Chen, Aijun Shen, Guanghua Fang, Hongchun Liu, Minmin Zhang, Shuai Tang, Bing Xiong, Lanping Ma, Meiyu Geng, Jingkang Shen
Acta Pharmaceutica Sinica B (January 2016) Volume 6(Issue 1) pp:93-99
Publication Date(Web):January 2016
DOI:10.1016/j.apsb.2015.11.002

![N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3h-3-benzazepin-3-yl)-4- Pyrimidinyl]-β-alanine](http://img.cochemist.com/ccimg/1373500/1373422-53-7.png)
![N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3h-3-benzazepin-3-yl)-4- Pyrimidinyl]-β-alanine](http://img.cochemist.com/ccimg/1373500/1373422-53-7_b.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2_b.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8_b.png)
![Carbamic acid, [3-(3-formylphenoxy)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/897500/897404-12-5.png)
![Carbamic acid, [3-(3-formylphenoxy)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/897500/897404-12-5_b.png)
![Benzonitrile, 4-[3-(1-piperazinyl)propoxy]-](http://img.cochemist.com/ccimg/360600/360553-67-9.png)
![Benzonitrile, 4-[3-(1-piperazinyl)propoxy]-](http://img.cochemist.com/ccimg/360600/360553-67-9_b.png)
![4-(1-methylethyl)-7H-Pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/1309500/1309430-81-6.png)
![4-(1-methylethyl)-7H-Pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/1309500/1309430-81-6_b.png)
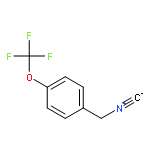
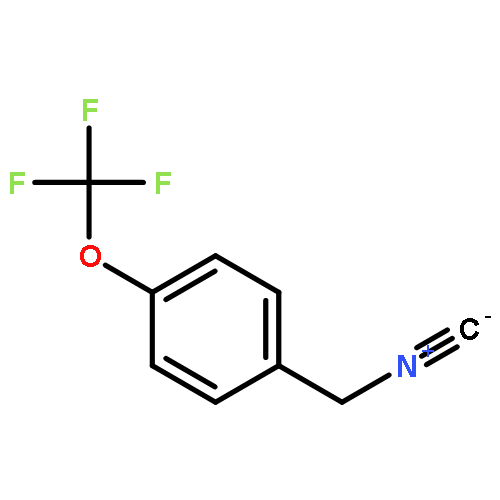
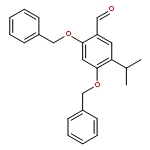
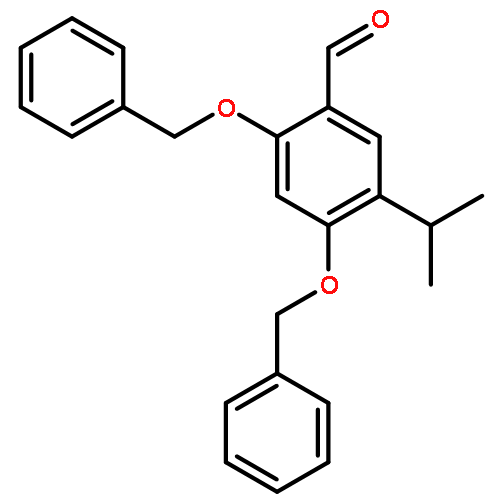
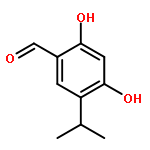
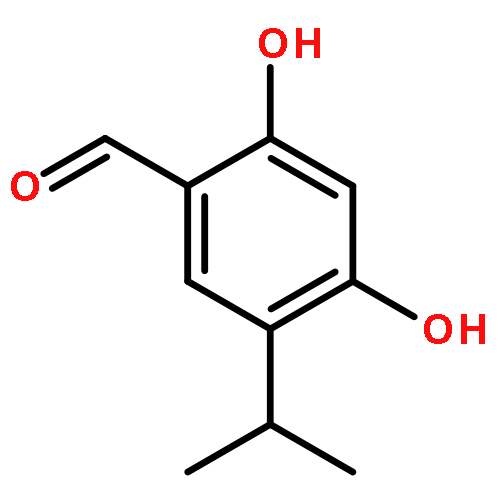
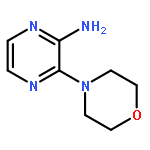
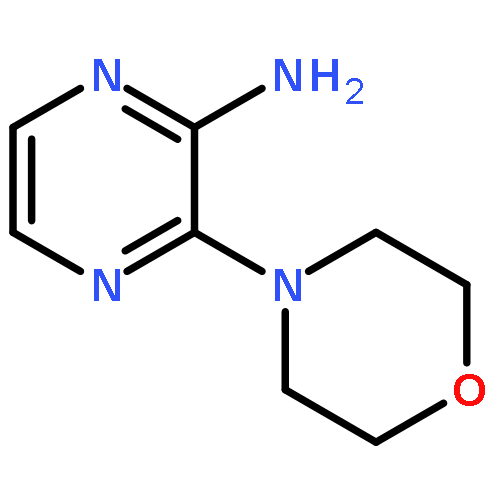
![6-Methoxyimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/955400/955376-51-9.png)
![6-Methoxyimidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/955400/955376-51-9_b.png)
![9-[[[(2R)-1,4-dioxan-2-yl]methyl-methylsulfamoyl]amino]-2-(1-methylpyrazol-4-yl)-11-oxobenzo[1,2]cyclohepta[2,4-b]pyridine](http://img.cochemist.com/ccimg/917900/917879-39-1.png)
![9-[[[(2R)-1,4-dioxan-2-yl]methyl-methylsulfamoyl]amino]-2-(1-methylpyrazol-4-yl)-11-oxobenzo[1,2]cyclohepta[2,4-b]pyridine](http://img.cochemist.com/ccimg/917900/917879-39-1_b.png)
![(3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/905900/905854-02-6.png)
![(3R,4R)-3-(5,6-Dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1H-indol-3-yl)pyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/905900/905854-02-6_b.png)
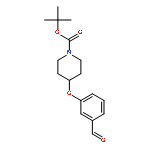
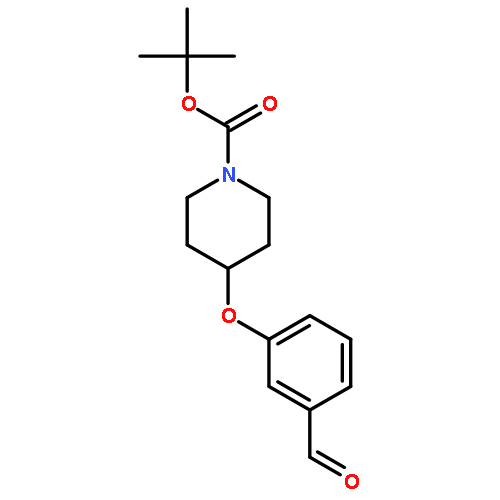
![(1S,11S,13S,14R,15R)-14,15-DIMETHOXY-20-METHYL-5,7,21-TRIOXA-20-AZAHEXACYCLO[11.4.3.1<SUP>11,14</SUP>.0<SUP>1,13</SUP>.0<SUP>2,10</SUP>.0<SUP>4,8</SUP>]HENICOSA-2(10),3,8-TRIEN-16-ONE](http://img.cochemist.com/ccimg/747500/747414-16-0.png)
![(1S,11S,13S,14R,15R)-14,15-DIMETHOXY-20-METHYL-5,7,21-TRIOXA-20-AZAHEXACYCLO[11.4.3.1<SUP>11,14</SUP>.0<SUP>1,13</SUP>.0<SUP>2,10</SUP>.0<SUP>4,8</SUP>]HENICOSA-2(10),3,8-TRIEN-16-ONE](http://img.cochemist.com/ccimg/747500/747414-16-0_b.png)
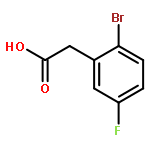
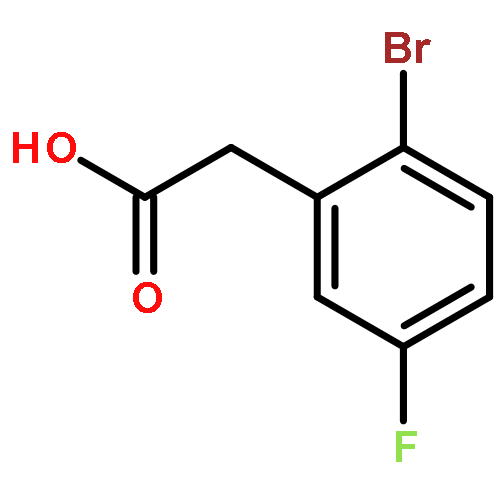
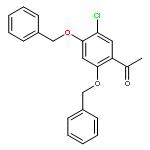
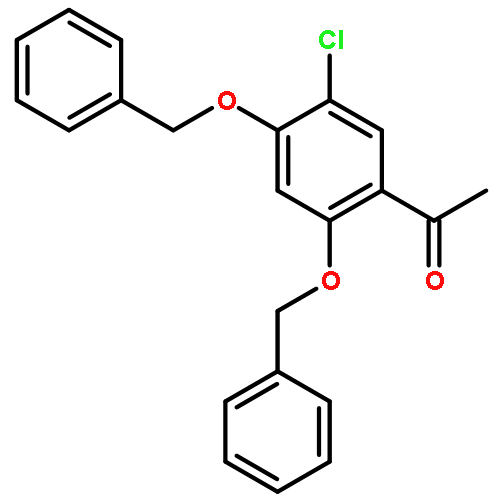
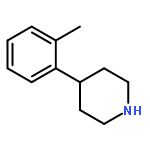
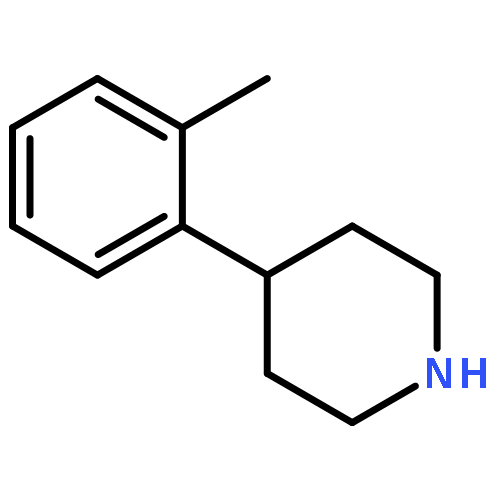
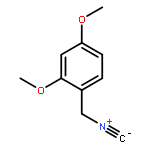
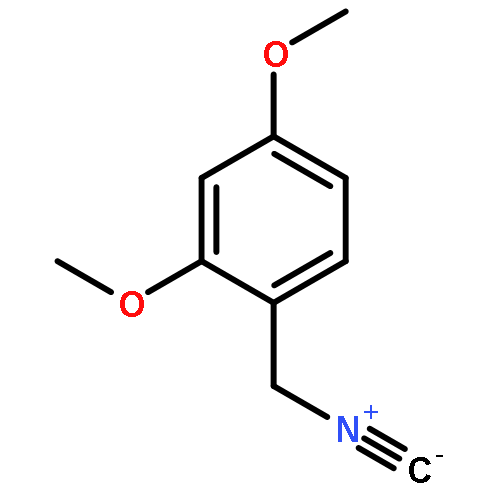
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[4-(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/502700/502649-34-5.png)
![1,3,2-Dioxaborolane, 4,4,5,5-tetramethyl-2-[4-(1-methylethoxy)phenyl]-](http://img.cochemist.com/ccimg/502700/502649-34-5_b.png)
![Imidazo[1,2-a]pyridine-3-sulfonyl chloride](http://img.cochemist.com/ccimg/499800/499770-78-4.png)
![Imidazo[1,2-a]pyridine-3-sulfonyl chloride](http://img.cochemist.com/ccimg/499800/499770-78-4_b.png)
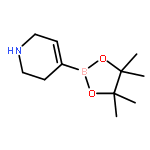
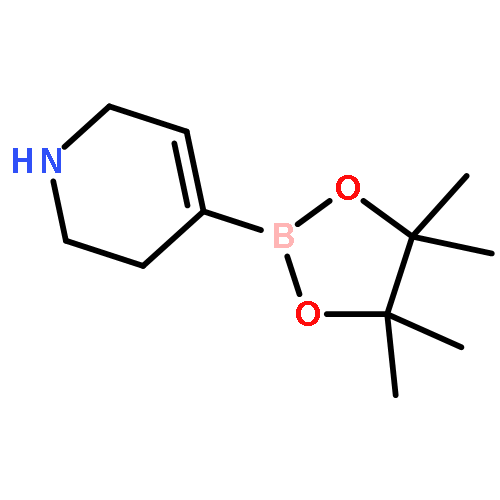








![Benzaldehyde, 3-[(1-methyl-4-piperidinyl)oxy]-](http://img.cochemist.com/ccimg/186200/186191-30-0.png)
![Benzaldehyde, 3-[(1-methyl-4-piperidinyl)oxy]-](http://img.cochemist.com/ccimg/186200/186191-30-0_b.png)




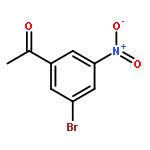
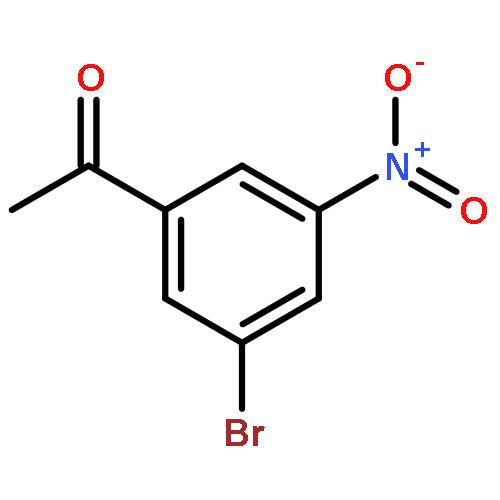
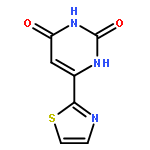
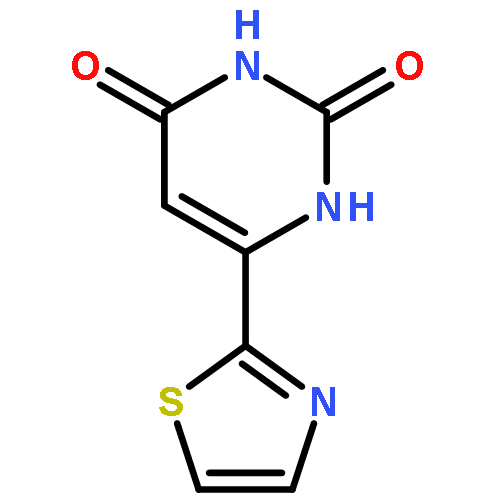




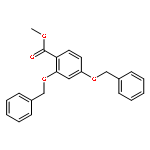
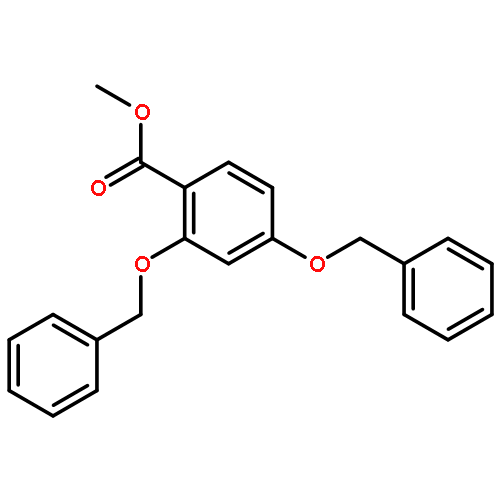
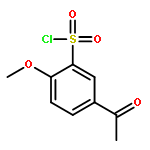
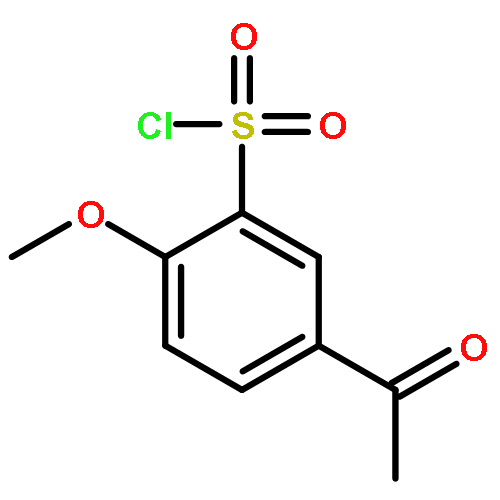
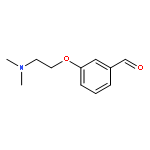
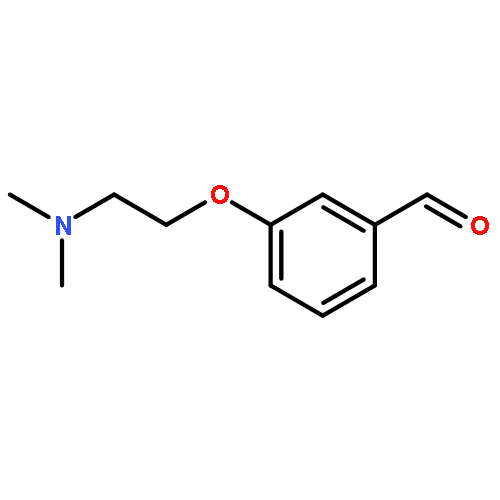
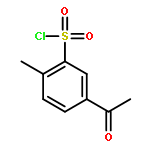
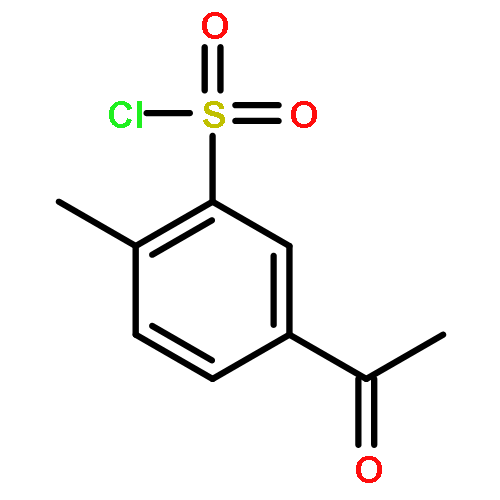
![5,7-DI(PROPAN-2-YL)PYRAZOLO[1,5-A]PYRIMIDINE](http://img.cochemist.com/ccimg/55600/55536-79-3.png)
![5,7-DI(PROPAN-2-YL)PYRAZOLO[1,5-A]PYRIMIDINE](http://img.cochemist.com/ccimg/55600/55536-79-3_b.png)