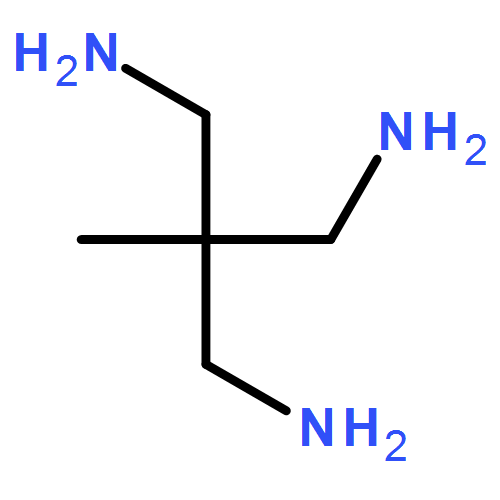
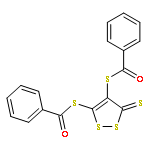
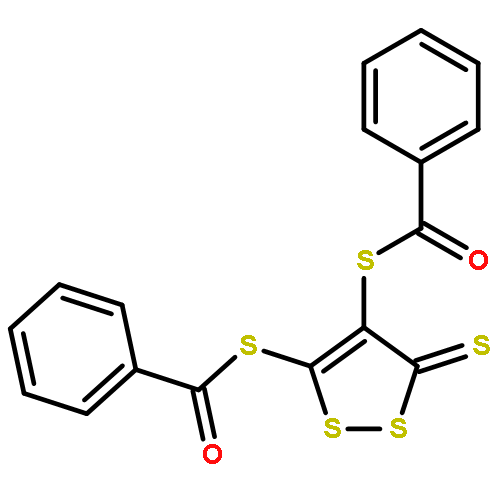
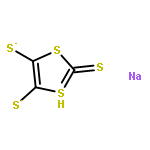
Co-reporter:Joseph M. Zadrozny, Audrey T. Gallagher, T. David Harris, and Danna E. Freedman
Journal of the American Chemical Society May 24, 2017 Volume 139(Issue 20) pp:7089-7089
Publication Date(Web):April 28, 2017
DOI:10.1021/jacs.7b03123
Realizing atomic-level spatial control over qubits, the fundamental units of both quantum information processing systems and quantum sensors, constitutes a crucial cross-field challenge. Toward this end, embedding electronic-spin-based qubits within the framework of a crystalline porous material is a promising approach to create precise arrays of qubits. Realizing porous hosts for qubits would also impact the emerging field of quantum sensing, whereby porosity would enable analytes to infuse into a sensor matrix. However, building viable qubits into a porous material is an appreciable challenge because of the extreme sensitivity of qubits to local magnetic noise. To insulate these frameworks from ambient magnetic signals, we borrowed from atomic physics the idea to exploit clock transitions at avoided level crossings. Here, sensitivity to magnetic noise is inherently limited by the flat slope of the so-called clock transition. More specifically, we created an array of clocklike qubits within a metal–organic framework by combining coordination chemistry considerations with the fundamental concept of atomic clock transitions. Electron paramagnetic resonance studies verify a clocklike transition for the hosted cobalt(II) spins in the framework [(TCPP)Co0.07Zn0.93]3[Zr6O4(OH)4(H2O)6]2, the first demonstration in any porous material. The clocklike qubits display lifetimes of up to 14 μs despite abundant local nuclear spins, illuminating a new path toward proof-of-concept quantum sensors and processors with high inherent structural precision.
Co-reporter:Scott C. Coste, Bess Vlaisavljevich, and Danna E. Freedman
Inorganic Chemistry July 17, 2017 Volume 56(Issue 14) pp:8195-8195
Publication Date(Web):June 29, 2017
DOI:10.1021/acs.inorgchem.7b00923
Precise modulation of the magnetic anisotropy of a transition-metal center would affect physical properties ranging from photoluminescence to magnetism. Over the past decade, exerting nuanced control over ligand fields enabled the incorporation of significant magnetic anisotropy in a number of mononuclear transition-metal complexes. An alternate approach to increasing spin–orbit coupling relies upon using heavy diamagnetic main-group elements as sources of magnetic anisotropy. Interacting first-row transition metals with main-group elements enables the transfer of magnetic anisotropy to the paramagnetic metal center without restricting coordination geometry. We sought to study the effect of covalency on this anisotropy transfer by probing the effect of halides in comparison to early main-group elements. Toward that end, we synthesized a series of four isostructural heterobimetallic complexes, with germanium or tin covalently bound to a triplet spin Fe(II) center. These complexes are ligated by a halide (Br– or I–) in the apical position to yield a series of complexes with variation in the mass of the main-group elements. This series enabled us to interrogate which electronic structure factors influence the heavy-atom effect. Using a suite of approaches including magnetometry, computation, and Mössbauer spectroscopy, we probed the electronic structure and the spin–orbit coupling, as parametrized by axial zero-field splitting across the series of complexes, and found an increase in zero-field splitting from −11.8 to −17.9 cm–1 by increasing the axial ligand mass. Through direct comparison between halides and group 14 elements, we observe a greater impact on magnetic anisotropy from the halide interaction. We attribute this counterintuitive effect to a larger spin population on the halide elements, despite greater covalency in the group 14 interactions. These results recommend modification of the intuitive design principle of increasing covalency toward a deeper focus on the interactions of the spin-bearing orbitals.
Co-reporter:Michael J. Graham, Matthew D. Krzyaniak, Michael R. Wasielewski, and Danna E. Freedman
Inorganic Chemistry July 17, 2017 Volume 56(Issue 14) pp:8106-8106
Publication Date(Web):June 28, 2017
DOI:10.1021/acs.inorgchem.7b00794
Quantum information processing (QIP) has the potential to transform numerous fields from cryptography, to finance, to the simulation of quantum systems. A promising implementation of QIP employs unpaired electronic spins as qubits, the fundamental units of information. Though molecular electronic spins offer many advantages, including chemical tunability and facile addressability, the development of design principles for the synthesis of complexes that exhibit long qubit superposition lifetimes (also known as coherence times, or T2) remains a challenge. As nuclear spins in the local qubit environment are a primary cause of shortened superposition lifetimes, we recently conducted a study which employed a modular spin-free ligand scaffold to place a spin-laden propyl moiety at a series of fixed distances from an S = 1/2 vanadium(IV) ion in a series of vanadyl complexes. We found that, within a radius of 4.0(4)–6.6(6) Å from the metal center, nuclei did not contribute to decoherence. To assess the generality of this important design principle and test its efficacy in a different coordination geometry, we synthesized and investigated three vanadium tris(dithiolene) complexes with the same ligand set employed in our previous study: K2[V(C5H6S4)3] (1), K2[V(C7H6S6)3] (2), and K2[V(C9H6S8)3] (3). We specifically interrogated solutions of these complexes in DMF-d7/toluene-d8 with pulsed electron paramagnetic resonance spectroscopy and electron nuclear double resonance spectroscopy and found that the distance dependence present in the previously synthesized vanadyl complexes holds true in this series. We further examined the coherence properties of the series in a different solvent, MeCN-d3/toluene-d8, and found that an additional property, the charge density of the complex, also affects decoherence across the series. These results highlight a previously unknown design principle for augmenting T2 and open new pathways for the rational synthesis of complexes with long coherence times.
Co-reporter:K. M. Powderly;S. M. Clarke;M. Amsler;C. Wolverton;C. D. Malliakas;Y. Meng;S. D. Jacobsen;D. E. Freedman
Chemical Communications 2017 vol. 53(Issue 81) pp:11241-11244
Publication Date(Web):2017/10/10
DOI:10.1039/C7CC06471C
Herein, we present the discovery of a new high-pressure phase in the Ni–Bi system, β-NiBi, which crystallizes in the TlI structure type. The powerful technique of in situ high-pressure and high-temperature powder X-ray diffraction enabled observation of the formation of β-NiBi and its reversible reconversion to the ambient pressure phase, α-NiBi.
Co-reporter:Chung-Jui Yu, Michael J. Graham, Joseph M. Zadrozny, Jens Niklas, Matthew D. Krzyaniak, Michael R. Wasielewski, Oleg G. Poluektov, and Danna E. Freedman
Journal of the American Chemical Society 2016 Volume 138(Issue 44) pp:14678-14685
Publication Date(Web):October 31, 2016
DOI:10.1021/jacs.6b08467
Quantum information processing (QIP) offers the potential to create new frontiers in fields ranging from quantum biology to cryptography. Two key figures of merit for electronic spin qubits, the smallest units of QIP, are the coherence time (T2), the lifetime of the qubit, and the spin–lattice relaxation time (T1), the thermally defined upper limit of T2. To achieve QIP, processable qubits with long coherence times are required. Recent studies on (Ph4P-d20)2[V(C8S8)3], a vanadium-based qubit, demonstrate that millisecond T2 times are achievable in transition metal complexes with nuclear spin-free environments. Applying these principles to vanadyl complexes offers a route to combine the previously established surface compatibility of the flatter vanadyl structures with a long T2. Toward those ends, we investigated a series of four qubits, (Ph4P)2[VO(C8S8)2] (1), (Ph4P)2[VO(β-C3S5)2] (2), (Ph4P)2[VO(α-C3S5)2] (3), and (Ph4P)2[VO(C3S4O)2] (4), by pulsed electron paramagnetic resonance (EPR) spectroscopy and compared the performance of these species with our recently reported set of vanadium tris(dithiolene) complexes. Crucially we demonstrate that solutions of 1–4 in SO2, a uniquely polar nuclear spin-free solvent, reveal T2 values of up to 152(6) μs, comparable to the best molecular qubit candidates. Upon transitioning to vanadyl species from the tris(dithiolene) analogues, we observe a remarkable order of magnitude increase in T1, attributed to stronger solute–solvent interactions with the polar vanadium-oxo moiety. Simultaneously, we detect a small decrease in T2 for the vanadyl analogues relative to the tris(dithiolene) complexes. We attribute this decrease to the absence of one nuclear spin-free ligand, which served to shield the vanadium centers against solvent nuclear spins. Our results highlight new design principles for long T1 and T2 times by demonstrating the efficacy of ligand-based tuning of solute–solvent interactions.
Co-reporter:Majed S. Fataftah; Joseph M. Zadrozny; Scott C. Coste; Michael J. Graham; Dylan M. Rogers
Journal of the American Chemical Society 2016 Volume 138(Issue 4) pp:1344-1348
Publication Date(Web):January 7, 2016
DOI:10.1021/jacs.5b11802
The implementation of quantum computation (QC) would revolutionize scientific fields ranging from encryption to quantum simulation. One intuitive candidate for the smallest unit of a quantum computer, a qubit, is electronic spin. A prominent proposal for QC relies on high-spin magnetic molecules, where multiple transitions between the many MS levels are employed as qubits. Yet, over a decade after the original notion, the exploitation of multiple transitions within a single manifold for QC remains unrealized in these high-spin species due to the challenge of accessing forbidden transitions. To create a proof-of-concept system, we synthesized the novel nuclear spin-free complex [Cr(C3S5)3]3– with precisely tuned zero-field splitting parameters that create two spectroscopically addressable transitions, with one being a forbidden transition. Pulsed electron paramagnetic resonance (EPR) measurements enabled the investigation of the coherent lifetimes (T2) and quantum control (Rabi oscillations) for two transitions, one allowed and one forbidden, within the S = 3/2 spin manifold. This investigation represents a step forward in the development of high-spin species as a pathway to scalable QC systems within magnetic molecules.
Co-reporter:Majed S. Fataftah, Scott C. Coste, Bess Vlaisavljevich, Joseph M. Zadrozny and Danna E. Freedman
Chemical Science 2016 vol. 7(Issue 9) pp:6160-6166
Publication Date(Web):21 Jun 2016
DOI:10.1039/C6SC02170K
Mononuclear transition metal complexes demonstrate significant potential in the divergent applications of spintronics and quantum information processing. The facile tunability of these complexes enables structure function correlations for a plethora of relevant magnetic quantities. We present a series of pseudotetrahedral [Co(C3S5)2]2− complexes with varying deviations from D2d symmetry to investigate the influence of structural distortions on spin relaxation dynamics and qubit viability, as tuned by the variable transverse magnetic anisotropy, E. To overcome the traditional challenge of measuring E in species where D ≫ E, we employed a different approach of harnessing ac magnetic susceptibility to probe the emergence of quantum tunneling of magnetization as a proxy for E. Across the range of values for E in the series, we observe magnetic hysteresis for the smallest value of E. The hysteresis disappears with increasing E, concomitant with the appearance of an observable, low frequency (L-band) electron paramagnetic resonance (EPR) signal, indicating the potential to controllably shift the molecule's utilization from classical to quantum information processing applications. The development of design principles for molecular magnet information processing requires separate design principles for classical versus quantum regimes. Here we show for the first time how subtle structural changes can switch the utility of a complex between these two types of applications.
Co-reporter:Joseph M. Zadrozny, Samuel M. Greer, Stephen Hill and Danna E. Freedman
Chemical Science 2016 vol. 7(Issue 1) pp:416-423
Publication Date(Web):19 Oct 2015
DOI:10.1039/C5SC02477C
The relationship between electronic structure and zero-field splitting dictates key design parameters for magnetic molecules. In particular, to enable the directed synthesis of new electronic spin based qubits, developing complexes where zero-field splitting energies are invariant to structural changes is a critical challenge. Toward those ends, we report three salts of a new compound, a four-coordinate iron(II) complex [Fe(C3S5)2]2− ([(18-crown-6)K]+ (1), Ph4P+ (2), Bu4N+ (3)) with a continuous structural variation in a single parameter, the dihedral angle (θd) between the two C3S52− ligands, as a function of counterion (θd = 89.98(4)° for 1 to 72.41(2)° for 3). Electron paramagnetic resonance data for 1–3 reveal zero-field splitting parameters that are unusually robust to the structural variation. Mössbauer spectroscopic measurements indicate that the structural variation in θd primarily affects the highest-energy 3d-orbitals (dxz and dyz) of the iron(II) ion. These orbitals have the smallest impact on the zero-field splitting parameters, thus the distortion has a minor effect on D and E. These results represent the first part of a directed effort to understand how spin state energies may be fortified against structural distortions for future applications of qubits in non-crystalline environments.
Co-reporter:Joseph M. Zadrozny, Michael J. Graham, Matthew D. Krzyaniak, Michael R. Wasielewski and Danna E. Freedman
Chemical Communications 2016 vol. 52(Issue 66) pp:10175-10178
Publication Date(Web):27 Jul 2016
DOI:10.1039/C6CC05094H
A counterintuitive three-order of magnitude slowing of the spin–lattice relaxation rate is observed in a high spin qubit at high magnetic field via multifrequency pulsed electron paramagnetic resonance measurements.
Co-reporter:James P. S. Walsh, Samantha M. Clarke, Yue Meng, Steven D. Jacobsen, and Danna E. Freedman
ACS Central Science 2016 Volume 2(Issue 11) pp:867
Publication Date(Web):October 26, 2016
DOI:10.1021/acscentsci.6b00287
Recent advances in high-pressure techniques offer chemists access to vast regions of uncharted synthetic phase space, expanding our experimental reach to pressures comparable to the core of the Earth. These newfound capabilities enable us to revisit simple binary systems in search of compounds that for decades have remained elusive. The most tantalizing of these targets are systems in which the two elements in question do not interact even as molten liquids—so-called immiscible systems. As a prominent example, immiscibility between iron and bismuth is so severe that no material containing Fe–Bi bonds is known to exist. The elusiveness of Fe–Bi bonds has a myriad of consequences; crucially, it precludes completing the iron pnictide superconductor series. Herein we report the first iron–bismuth binary compound, FeBi2, featuring the first Fe–Bi bond in the solid state. We employed geologically relevant pressures, similar to the core of Mars, to access FeBi2, which we synthesized at 30 GPa and 1500 K. The compound crystallizes in the Al2Cu structure type (space group I4/mcm) with a = 6.3121(3) Å and c = 5.4211(4) Å. The new binary intermetallic phase persists from its formation pressure of 30 GPa down to 3 GPa. The existence of this phase at low pressures suggests that it might be quenchable to ambient pressure at low temperatures. These results offer a pathway toward the realization of new exotic materials.
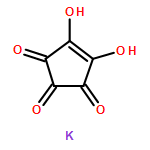
Co-reporter:Joseph M. Zadrozny
Inorganic Chemistry 2015 Volume 54(Issue 24) pp:12027-12031
Publication Date(Web):December 9, 2015
DOI:10.1021/acs.inorgchem.5b02429
High-spin transition metal complexes are of interest as candidates for quantum information processing owing to the tunability of the pairs of MS levels for use as quantum bits (qubits). Thus, the design of high-spin systems that afford qubits with stable superposition states is of primary importance. Nuclear spins are a potent instigator of superposition instability; thus, we probed the Ph4P+ salt of the nuclear spin-free complex [Fe(C5O5)3]3– (1) to see if long-lived superpositions were possible in such a system. Continuous-wave and pulsed electron paramagnetic resonance (EPR) spectroscopic measurements reveal a strong EPR transition at X-band that can be utilized as a qubit. However, at 5 K the coherent lifetime, T2, for this resonance is 721(3) ns and decreases rapidly with increasing temperature. Simultaneously, the spin–lattice relaxation time is extremely short, 11.33(1) μs, at 5 K, and also rapidly decreases with increasing temperature. The coincidence of these two temperature-dependent data sets suggests that T2 in 1 is strongly limited by the short T1. Importantly, these results highlight the need for new design parameters in pursuit of high-spin species with appreciable coherence times.
Co-reporter:Samantha M. Clarke
Inorganic Chemistry 2015 Volume 54(Issue 6) pp:2765-2771
Publication Date(Web):February 23, 2015
DOI:10.1021/ic5029178
Compounds containing both heavy main group elements and paramagnetic transition metals form a fertile area for the study of magnetic anisotropy. We pursued the synthesis, characterization, and magnetic measurements of Bi–Se–Cr compounds: a ternary system with no structurally characterized materials. Those efforts led to the isolation of two novel misfit layer compounds, namely, (BiSe)1.23CrSe2 (1) and (BiSe)1.22(Cr1.2Se2)2 (2). The crystal structure of 1 consists of alternating BiSe and CrSe2 layers along the c-axis, and 2 is composed of alternating BiSe and (Cr1.2Se2)2 layers along the c-axis. Lattice mismatch occurs in both compounds along the b-axis and leads to positional modulation of the atoms. Field- and temperature-dependent measurements were performed to assess the degree of magnetic anisotropy. Temperature-dependent susceptibility measurements on aligned crystals of 1 display increased bifurcation of zero-field cooled and field cooled data when crystals are oriented with H perpendicular to c than when the crystals are oriented with H parallel to c. Magnetic anisotropy is less pronounced in 2 where both crystallographic orientations exhibit bifurcation at 26 K. The complexity of the magnetic behavior in both compounds likely signifies a competition between CrSe2 intralayer ferromagnetic coupling and interlayer antiferromagnetic coupling. These results highlight the exciting magnetic properties that can arise from the exploration of new ternary phases.
Co-reporter:Joseph M. Zadrozny, Jens Niklas, Oleg G. Poluektov, and Danna E. Freedman
ACS Central Science 2015 Volume 1(Issue 9) pp:488
Publication Date(Web):December 2, 2015
DOI:10.1021/acscentsci.5b00338
Quantum information processing (QIP) could revolutionize areas ranging from chemical modeling to cryptography. One key figure of merit for the smallest unit for QIP, the qubit, is the coherence time (T2), which establishes the lifetime for the qubit. Transition metal complexes offer tremendous potential as tunable qubits, yet their development is hampered by the absence of synthetic design principles to achieve a long T2. We harnessed molecular design to create a series of qubits, (Ph4P)2[V(C8S8)3] (1), (Ph4P)2[V(β-C3S5)3] (2), (Ph4P)2[V(α-C3S5)3] (3), and (Ph4P)2[V(C3S4O)3] (4), with T2s of 1–4 μs at 80 K in protiated and deuterated environments. Crucially, through chemical tuning of nuclear spin content in the vanadium(IV) environment we realized a T2 of ∼1 ms for the species (d20-Ph4P)2[V(C8S8)3] (1′) in CS2, a value that surpasses the coordination complex record by an order of magnitude. This value even eclipses some prominent solid-state qubits. Electrochemical and continuous wave electron paramagnetic resonance (EPR) data reveal variation in the electronic influence of the ligands on the metal ion across 1–4. However, pulsed measurements indicate that the most important influence on decoherence is nuclear spins in the protiated and deuterated solvents utilized herein. Our results illuminate a path forward in synthetic design principles, which should unite CS2 solubility with nuclear spin free ligand fields to develop a new generation of molecular qubits.
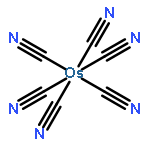
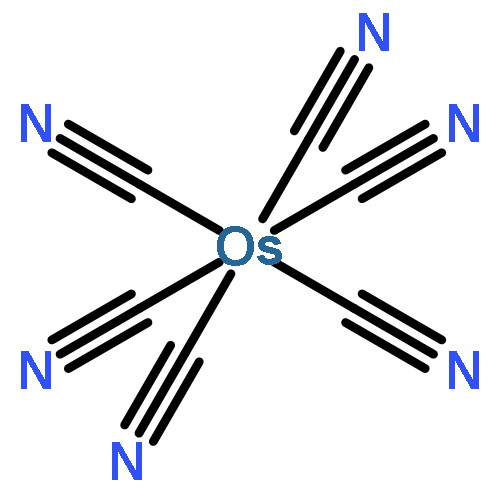
Co-reporter:Michael J. Graham ; Joseph M. Zadrozny ; Muhandis Shiddiq ; John S. Anderson ; Majed S. Fataftah ; Stephen Hill
Journal of the American Chemical Society 2014 Volume 136(Issue 21) pp:7623-7626
Publication Date(Web):May 16, 2014
DOI:10.1021/ja5037397
Enabling the rational synthesis of molecular candidates for quantum information processing requires design principles that minimize electron spin decoherence. Here we report a systematic investigation of decoherence via the synthesis of two series of paramagnetic coordination complexes. These complexes, [M(C2O4)3]3– (M = Ru, Cr, Fe) and [M(CN)6]3– (M = Fe, Ru, Os), were prepared and interrogated by pulsed electron paramagnetic resonance (EPR) spectroscopy to assess quantitatively the influence of the magnitude of spin (S = 1/2, 3/2, 5/2) and spin–orbit coupling (ζ = 464, 880, 3100 cm–1) on quantum decoherence. Coherence times (T2) were collected via Hahn echo experiments and revealed a small dependence on the two variables studied, demonstrating that the magnitudes of spin and spin–orbit coupling are not the primary drivers of electron spin decoherence. On the basis of these conclusions, a proof-of-concept molecule, [Ru(C2O4)3]3−, was selected for further study. The two parameters establishing the viability of a qubit are a long coherence time, T2, and the presence of Rabi oscillations. The complex [Ru(C2O4)3]3– exhibits both a coherence time of T2 = 3.4 μs and the rarely observed Rabi oscillations. These two features establish [Ru(C2O4)3]3– as a molecular qubit candidate and mark the viability of coordination complexes as qubit platforms. Our results illustrate that the design of qubit candidates can be achieved with a wide range of paramagnetic ions and spin states while preserving a long-lived coherence.
Co-reporter:Joseph M. Zadrozny ; Jens Niklas ; Oleg G. Poluektov
Journal of the American Chemical Society 2014 Volume 136(Issue 45) pp:15841-15844
Publication Date(Web):October 23, 2014
DOI:10.1021/ja507846k
We report a vanadium complex in a nuclear-spin free ligand field that displays two key properties for an ideal candidate qubit system: long coherence times that persist at high temperature, T2 = 1.2 μs at 80 K, and the observation of quantum coherences from multiple transitions. The electron paramagnetic resonance (EPR) spectrum of the complex [V(C8S8)3]2– displays multiple transitions arising from a manifold of states produced by the hyperfine coupling of the S = 1/2 electron spin and I = 7/2 nuclear spin. Transient nutation experiments reveal Rabi oscillations for multiple transitions. These observations suggest that each pair of hyperfine levels hosted within [V(C8S8)3]2– are candidate qubits. The realization of multiple quantum coherences within a transition metal complex illustrates an emerging method of developing scalability and addressability in electron spin qubits. This study presents a rare molecular demonstration of multiple Rabi oscillations originating from separate transitions. These results extend observations of multiple quantum coherences from prior reports in solid-state compounds to the new realm of highly modifiable coordination compounds.
Co-reporter:Majed S. Fataftah, Joseph M. Zadrozny, Dylan M. Rogers, and Danna E. Freedman
Inorganic Chemistry 2014 Volume 53(Issue 19) pp:10716-10721
Publication Date(Web):September 8, 2014
DOI:10.1021/ic501906z
The high-spin pseudotetrahedral complex [Co(C3S5)2]2– exhibits slow magnetic relaxation in the absence of an applied dc magnetic field, one of a small number of mononuclear complexes to display this property. Fits to low-temperature magnetization data indicate that this single-molecule magnet possesses a very large and negative axial zero-field splitting and small rhombicity. The presence of single-molecule magnet behavior in a zero-nuclear spin ligand field offers the opportunity to investigate the potential for this molecule to be a qubit, the smallest unit of a quantum information processing (QIP) system. However, simulations of electron paramagnetic resonance (EPR) spectra and the absence of EPR spectra demonstrate that this molecule is unsuitable as a qubit due to the same factors that promote single molecule magnet behavior. We discuss the influence of rhombic and axial zero-field splitting on QIP applications and the implications for future molecular qubit syntheses.
Co-reporter:Joseph M. Zadrozny, Samuel M. Greer, Stephen Hill and Danna E. Freedman
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN423-423
Publication Date(Web):2015/10/19
DOI:10.1039/C5SC02477C
The relationship between electronic structure and zero-field splitting dictates key design parameters for magnetic molecules. In particular, to enable the directed synthesis of new electronic spin based qubits, developing complexes where zero-field splitting energies are invariant to structural changes is a critical challenge. Toward those ends, we report three salts of a new compound, a four-coordinate iron(II) complex [Fe(C3S5)2]2− ([(18-crown-6)K]+ (1), Ph4P+ (2), Bu4N+ (3)) with a continuous structural variation in a single parameter, the dihedral angle (θd) between the two C3S52− ligands, as a function of counterion (θd = 89.98(4)° for 1 to 72.41(2)° for 3). Electron paramagnetic resonance data for 1–3 reveal zero-field splitting parameters that are unusually robust to the structural variation. Mössbauer spectroscopic measurements indicate that the structural variation in θd primarily affects the highest-energy 3d-orbitals (dxz and dyz) of the iron(II) ion. These orbitals have the smallest impact on the zero-field splitting parameters, thus the distortion has a minor effect on D and E. These results represent the first part of a directed effort to understand how spin state energies may be fortified against structural distortions for future applications of qubits in non-crystalline environments.
Co-reporter:Tyler J. Pearson, Majed S. Fataftah and Danna E. Freedman
Chemical Communications 2016 - vol. 52(Issue 76) pp:NaN11397-11397
Publication Date(Web):2016/09/01
DOI:10.1039/C6CC06369A
A novel Mn2+⋯Bi3+ heterobimetallic complex, featuring the closest Mn⋯Bi interaction for a paramagnetic molecular species, exhibits unusually large axial zero-field splitting. We attribute this enhancement to the proximity of Mn2+ to a heavy main group element, namely, bismuth.
Co-reporter:Majed S. Fataftah, Scott C. Coste, Bess Vlaisavljevich, Joseph M. Zadrozny and Danna E. Freedman
Chemical Science (2010-Present) 2016 - vol. 7(Issue 9) pp:
Publication Date(Web):
DOI:10.1039/C6SC02170K
Co-reporter:Joseph M. Zadrozny, Michael J. Graham, Matthew D. Krzyaniak, Michael R. Wasielewski and Danna E. Freedman
Chemical Communications 2016 - vol. 52(Issue 66) pp:NaN10178-10178
Publication Date(Web):2016/07/27
DOI:10.1039/C6CC05094H
A counterintuitive three-order of magnitude slowing of the spin–lattice relaxation rate is observed in a high spin qubit at high magnetic field via multifrequency pulsed electron paramagnetic resonance measurements.
.jpg)