Co-reporter:Grazia Malta;Simon J. Freakley;Simon A. Kondrat
Chemical Communications 2017 vol. 53(Issue 86) pp:11733-11746
Publication Date(Web):2017/10/26
DOI:10.1039/C7CC05986H
The replacement of mercuric chloride in the production of vinyl chloride monomer, a precursor to PVC, would greatly reduce the environmental impact of this large scale industrial process. The validation of single Au cations supported on carbon as the best catalyst for this reaction at an industrial scale has resulted from nearly 35 years of research. In this feature article we review the development of this catalyst system and address the limitations of a range of characterisation techniques used previously which may induce damage to the fresh catalyst. Following our latest findings using X-ray absorption spectroscopy, we show that under operating conditions the catalyst is atomically dispersed and can be classed as a single site catalyst, we give a perspective on future directions in single atom catalysis.
Co-reporter:Paul J. Smith;Simon A. Kondrat;Philip A. Chater;Benjamin R. Yeo;Greg M. Shaw;Li Lu;Jonathan K. Bartley;Stuart H. Taylor;Michael S. Spencer;Christopher J. Kiely;Gordon J. Kelly;Colin W. Park
Chemical Science (2010-Present) 2017 vol. 8(Issue 3) pp:2436-2447
Publication Date(Web):2017/02/28
DOI:10.1039/C6SC04130B
Zincian georgeite, an amorphous copper–zinc hydroxycarbonate, has been prepared by co-precipitation using acetate salts and ammonium carbonate. Incorporation of zinc into the georgeite phase and mild ageing conditions inhibits crystallisation into zincian malachite or aurichalcite. This zincian georgeite precursor was used to prepare a Cu/ZnO catalyst, which exhibits a superior performance to a zincian malachite derived catalyst for methanol synthesis and the low temperature water–gas shift (LTS) reaction. Furthermore, the enhanced LTS activity and stability in comparison to that of a commercial Cu/ZnO/Al2O3 catalyst, indicates that the addition of alumina as a stabiliser may not be required for the zincian georgeite derived Cu/ZnO catalyst. The enhanced performance is partly attributed to the exclusion of alkali metals from the synthesis procedure, which are known to act as catalyst poisons. The effect of residual sodium on the microstructural properties of the catalyst precursor was investigated further from preparations using sodium carbonate.
Co-reporter:Satoshi Ishikawa;Daniel R. Jones;Sarwat Iqbal;Christian Reece;David J. Morgan;David J. Willock;Peter J. Miedziak;Jonathan K. Bartley;Jennifer K. Edwards;Toru Murayama;Wataru Ueda
Green Chemistry (1999-Present) 2017 vol. 19(Issue 1) pp:225-236
Publication Date(Web):2017/01/03
DOI:10.1039/C6GC02598F
Cu–ZrO2 catalysts were synthesized by the methanothermal (Me) and oxalate gel precipitation (Og) methods. Detailed characterization of the catalysts synthesized by the Me method shows that these contain only Cu substituted into the tetragonal ZrO2 lattice. For catalysts prepared using the Og method Cu is found not only in the tetragonal ZrO2 lattice but also in the form of CuO particles on the zirconia surface. When these materials were tested for the hydrogenation of levulinic acid (LA) to γ-valerolactone (GVL) it was found that Me materials show no catalytic activity, whereas GVL was formed using Og catalysts. A reduction treatment of the Og catalysts prior to use resulted in a marked increase in the catalytic activity, however, no activity increase was observed when the Me material was exposed to a similar treatment before testing. Based on these results and characterization data, we conclude that the catalytically active component of Cu–ZrO2 catalysts for the hydrogenation of LA is reduced Cu particles dispersed on the catalyst surface with strong interaction with the Cu incorporated zirconia support, while the role of Cu in the zirconia lattice is to improve the adhesion of these particles and maintain their dispersion.
Co-reporter:Raiedhah Alsaiari;Luke T. Perrott;Ewa Nowicka;Rebecca V. Engel;Peter J. Miedziak;Simon A. Kondrat;Jennifer K. Edwards;David J. Willock
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 6) pp:1433-1439
Publication Date(Web):2017/03/20
DOI:10.1039/C6CY02448C
Carbon dioxide utilisation technology can contribute to the reduction of atmospheric CO2 levels both through its sequestration from flue gases and indirectly by relieving pressure on conventional feedstocks in chemical manufacturing. A promising approach is to employ CO2 to produce valuable cyclic carbonates (CCs) in reaction with suitable epoxides. This also has the advantage that carbon dioxide replaces toxic and hazardous reactants such as phosgene. In earlier work we have investigated the synthesis of epoxides from cycloalkenes using supported gold and gold–palladium nanoparticles as catalysts and oxygen from air as the oxidant under solvent free conditions. A strong dependence of epoxide selectivity on ring size was observed with C5 < C6 < C7 ≪ C8. In this study we extend this work to the investigation of cycloaddition of CO2 to different cycloalkene oxides with the ultimate aim of designing a process in which both epoxidation of an alkene and incorporation of CO2 could be achieved in a single process. However, we have found the opposite trend for the selectivity to carbonates: smaller ring cycloalkene oxides giving the highest carbonate selectivities while large rings do not yield CCs at all. The product distributions suggest that an alternative ring opening of the epoxides to yield alcohols and ketones is preferred under all the experimental conditions explored for larger ring systems. Additionally, the mechanism of the CC synthesis using a quaternary ammonium salt and ZnBr2 as the catalyst system was investigated using DFT methods. The results of the calculations support the experimental findings.
Co-reporter:Mark Douthwaite;Xiaoyang Huang;Sarwat Iqbal;Peter J. Miedziak;Gemma L. Brett;Simon A. Kondrat;Jennifer K. Edwards;Meenakshisundaram Sankar;David W. Knight;Donald Bethell
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 22) pp:5284-5293
Publication Date(Web):2017/11/14
DOI:10.1039/C7CY01025G
The emphasis of modern chemistry is to satisfy the needs of consumers by using methods that are sustainable and economical. Using a 1% AuPd/Mg(OH)2 catalyst in the presence of NaOH and under specific reaction conditions furfural; a platform chemical formed from lignocellulosic biomass, can be selectively oxidised to furoic acid, and the catalyst displays promising reusability for this reaction. The mechanism of this conversion is complex with multiple competing pathways possible. The experimental conditions and AuPd metal ratio can be fine-tuned to provide enhanced control of the reaction selectivity. Activation energies were derived for the homogeneous Cannizzaro pathway and the catalytic oxidation of furfural using the initial rates methodology. This work highlights the potential of using a heterogeneous catalyst for the oxidation of furfural to furoic acid that has potential for commercial application.
Co-reporter:Moataz Morad;Ewa Nowicka;Mark Douthwaite;Sarwat Iqbal;Peter Miedziak;Jennifer K. Edwards;Gemma L. Brett;Qian He;David Morgan;Hamed Alshammari;Donald Bethell;David W. Knight;Meenakshisundaram Sankar
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 9) pp:1928-1936
Publication Date(Web):2017/05/09
DOI:10.1039/C7CY00184C
We report the one-pot tandem synthesis of 4-(4-methoxyphenyl)butan-2-one directly from 4-methoxybenzyl alcohol and acetone using a multifunctional supported AuPd nanoalloy catalyst. This one-pot synthesis involves dehydrogenation, aldol condensation and hydrogenation of CC. In this supported AuPd catalyst, the bimetallic sites catalyse the dehydrogenation and hydrogenation steps and, in combination with the support, catalyse the C–C coupling (aldol) process. This supported bimetallic catalyst is also effective in utilizing hydrogen from the dehydrogenation reaction for the hydrogenation of 4-(4-methoxyphenyl)but-3-en-2-one to 4-(4-methoxyphenyl)butane-2-one via a hydrogen auto transfer route. These multifunctional catalysts were characterised using transmission electron microscopy, X-ray diffraction and X-ray photoelectron spectroscopy.
Co-reporter:Robert D. Armstrong;Benson M. Kariuki;David W. Knight
European Journal of Organic Chemistry 2017 Volume 2017(Issue 45) pp:6811-6814
Publication Date(Web):2017/12/08
DOI:10.1002/ejoc.201701343
Glucaric acid has potential applications in food, pharmaceutical and polymer industries yet no methodology exists within the public domain for isolation of this key bio-derived platform molecule as a pure, crystalline solid. Here we demonstrate the difficulties, which arise in doing so and report development of a process for derivation of free-glucaric acid from its Ca2+/K+ glucarate salts, which are both commercially available. Employing Amberlyst-15 (H+) exchange resin and azeotrope drying, powdered glucaric acid is prepared at > 99.96 % purity in 98.7 % dry yield.
Co-reporter:Dr. James H. Carter;Dr. Xi Liu;Dr. Qian He;Sultan Althahban;Dr. Ewa Nowicka;Dr. Simon J. Freakley;Dr. Liwei Niu;Dr. David J. Morgan;Dr. Yongwang Li; J. W. (Hans) Niemantsverdriet; Stanislaw Golunski; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie 2017 Volume 129(Issue 50) pp:16253-16257
Publication Date(Web):2017/12/11
DOI:10.1002/ange.201709708
AbstractGold (Au) on ceria–zirconia is one of the most active catalysts for the low-temperature water–gas shift reaction (LTS), a key stage of upgrading H2 reformate streams for fuel cells. However, this catalyst rapidly deactivates on-stream and the deactivation mechanism remains unclear. Using stop–start scanning transmission electron microscopy to follow the exact same area of the sample at different stages of the LTS reaction, as well as complementary X-ray photoelectron spectroscopy, we observed the activation and deactivation of the catalyst at various stages. During the heating of the catalyst to reaction temperature, we observed the formation of small Au nanoparticles (NPs; 1–2 nm) from subnanometer Au species. These NPs were then seen to agglomerate further over 48 h on-stream, and most rapidly in the first 5 h when the highest rate of deactivation was observed. These findings suggest that the primary deactivation process consists of the loss of active sites through the agglomeration and possible dewetting of Au NPs.
Co-reporter:James H. Carter, Sultan Althahban, Ewa Nowicka, Simon J. Freakley, David J. Morgan, Parag M. Shah, Stanislaw Golunski, Christopher J. Kiely, and Graham J. Hutchings
ACS Catalysis 2016 Volume 6(Issue 10) pp:6623
Publication Date(Web):August 29, 2016
DOI:10.1021/acscatal.6b01275
Highly active and stable bimetallic Au–Pd catalysts have been extensively studied for several liquid-phase oxidation reactions in recent years, but there are far fewer reports on the use of these catalysts for low-temperature gas-phase reactions. Here we initially established the presence of a synergistic effect in a range of bimetallic Au–Pd/CeZrO4 catalysts, by measuring their activity for selective oxidation of benzyl alcohol. The catalysts were then evaluated for low-temperature WGS, CO oxidation, and formic acid decomposition, all of which are believed to be mechanistically related. A strong anti-synergy between Au and Pd was observed for these reactions, whereby the introduction of Pd to a monometallic Au catalyst resulted in a significant decrease in catalytic activity. Furthermore, monometallic Pd was more active than Pd-rich bimetallic catalysts. The nature of the anti-synergy was probed by several ex situ techniques, which all indicated a growth in metal nanoparticle size with Pd addition. However, the most definitive information was provided by in situ CO-DRIFTS, in which CO adsorption associated with interfacial sites was found to vary with the molar ratio of the metals and could be correlated with the catalytic activity of each reaction. As a similar correlation was observed between activity and the presence of Au0* (as detected by XPS), it is proposed that peripheral Au0* species form part of the active centers in the most active catalysts for the three gas-phase reactions. In contrast, the active sites for the selective oxidation of benzyl alcohol are generally thought to be electronically modified gold atoms at the surface of the nanoparticles.Keywords: ceria−zirconia; CO oxidation; formic acid; gold; gold−palladium alloy; palladium; water-gas shift
Co-reporter:Adeeba Akram, Simon J. Freakley, Christian Reece, Marco Piccinini, Greg Shaw, Jennifer K. Edwards, Frédérique Desmedt, Pierre Miquel, Eero Seuna, David. J. Willock, Jacob A. Moulijn and Graham J. Hutchings
Chemical Science 2016 vol. 7(Issue 9) pp:5833-5837
Publication Date(Web):11 May 2016
DOI:10.1039/C6SC01332E
Hydrogen peroxide synthesis from hydrogen and oxygen in the gas phase is postulated to be a key reaction step in the gas phase epoxidation of propene using gold–titanium silicate catalysts. During this process H2O2 is consumed in a secondary step to oxidise an organic molecule so is typically not observed as a reaction product. We demonstrate that using AuPd nanoparticles, which are known to have high H2O2 synthesis rates in the liquid phase, it is possible to not only oxidise organic molecules in the gas phase but to detect H2O2 for the first time as a reaction product in both a fixed bed reactor and a pulsed Temporal Analysis of Products (TAP) reactor without stabilisers present in the gas feed. This observation opens up possibility of synthesising H2O2 directly using a gas phase reaction.
Co-reporter:Ren Su, Nikolaos Dimitratos, Jinjia Liu, Emma Carter, Sultan Althahban, Xueqin Wang, Yanbin Shen, Stefan Wendt, Xiaodong Wen, J. W. (Hans) Niemantsverdriet, Bo B. Iversen, Christopher J. Kiely, Graham J. Hutchings, and Flemming Besenbacher
ACS Catalysis 2016 Volume 6(Issue 7) pp:4239
Publication Date(Web):May 24, 2016
DOI:10.1021/acscatal.6b00982
Understanding the cocatalyst/semiconductor interaction is of key importance for the design and synthesis of next generation photocatalytic materials for efficient hydrogen production and environmental cleanup applications. Here we investigate preformed Pd nanoparticles (NPs) supported on a series of anatase TiO2 having well-controlled but varying degrees of crystallinity and crystallite size, and explore their photocatalytic performance for H2 production and phenol decomposition. While tuning the anatase crystallite size significantly influences the photocatalytic performance, varying the TiO2 crystallinity shows a negligible effect. Interestingly, the optimum quantum efficiency (∼78%) for H2 evolution is achieved with anatase having medium crystallite size (∼16 nm), whereas for phenol decomposition, a promotional effect is only observed for anatase with larger crystallite sizes (>20 nm). Surface radical species and radical densities study reveal that the photogenerated charge carriers have been trapped at different sites depending on the crystallite size of anatase. While the excited electrons are only trapped in bulk lattice sites in small anatase (<16 nm), larger anatase particles provide extra surface sites for charge trapping, which benefit charge storage and transportation to Pd surface sites, leading to a more efficient utilization of charge carriers for photocatalysis. Additionally, Pd supported on medium sized anatase (∼16 nm) hinders the formation of O2•– radicals on TiO2 surfaces, thus preventing unwanted reoxidation of photogenerated H2.Keywords: density functional theory; electron spin resonance; hydrogen evolution; metal−semiconductor interaction; phenol decomposition; photocatalysis; TiO2
Co-reporter:Mohd Hasbi Ab Rahim, Robert D. Armstrong, Ceri Hammond, Nikolaos Dimitratos, Simon J. Freakley, Michael M. Forde, David J. Morgan, Georgi Lalev, Robert L. Jenkins, Jose Antonio Lopez-Sanchez, Stuart H. Taylor and Graham J. Hutchings
Catalysis Science & Technology 2016 vol. 6(Issue 10) pp:3410-3418
Publication Date(Web):15 Dec 2015
DOI:10.1039/C5CY01586C
The selective oxidation of methane to methanol has been studied using trimetallic AuPdCu/TiO2 catalysts prepared by incipient wetness impregnation. They are able to catalyse the selective oxidation of methane to methanol under mild aqueous reaction conditions using H2O2 as the oxidant. When compared with bimetallic, Au–Pd/TiO2 analogues, the new trimetallic catalysts present productivities which are up to 5 times greater under the same test conditions, and this is coupled with methanol selectivity of up to 83%. Characterisation shows that whilst Au–Pd is present as Au-core–Pd-shell nanoparticles, copper is present as either Cu or Cu2O in <5 nm particles.
Co-reporter:Yueling Cao, Xi Liu, Sarwat Iqbal, Peter J. Miedziak, Jennifer K. Edwards, Robert D. Armstrong, David J. Morgan, Junwei Wang and Graham J. Hutchings
Catalysis Science & Technology 2016 vol. 6(Issue 1) pp:107-117
Publication Date(Web):25 Aug 2015
DOI:10.1039/C5CY00732A
1% Au/TiO2 catalysts prepared by a range of preparation methods were studied for the base-free oxidation of glucose. The highest catalytic activity was observed with the catalyst prepared by the sol-immobilization method. Furthermore we have studied the effect of the post-synthesis treatments of treatment with water, or heating in air on the activity. The catalyst calcined at 250 °C showed optimal activity and selectivity. Additionally, we studied the effect of the amount of stabilising ligand in the sol-immobilisation method and observed that this is a key parameter with respect to determining the catalyst's activity.
Co-reporter:Alberto Villa, Simon J. Freakley, Marco Schiavoni, Jennifer K. Edwards, Ceri Hammond, Gabriel M. Veith, Wu Wang, Di Wang, Laura Prati, Nikolaos Dimitratos and Graham J. Hutchings
Catalysis Science & Technology 2016 vol. 6(Issue 3) pp:694-697
Publication Date(Web):03 Dec 2015
DOI:10.1039/C5CY01880C
In this work, we show that the introduction of acidic oxygen functionalities to the surface of carbon nanofibers serves to depress the hydrogenation and the decomposition of hydrogen peroxide during the direct synthesis of H2O2. Moreover, the presence of acidic groups enhances the H2O2 productivity in the case of supported AuPd nanoparticles.
Co-reporter:Obaid F. Aldosari, Sarwat Iqbal, Peter J. Miedziak, Gemma L. Brett, Daniel R. Jones, Xi Liu, Jennifer K. Edwards, David J. Morgan, David K. Knight and Graham J. Hutchings
Catalysis Science & Technology 2016 vol. 6(Issue 1) pp:234-242
Publication Date(Web):03 Nov 2015
DOI:10.1039/C5CY01650A
The selective hydrogenation of furfural at ambient temperature has been investigated using a Pd/TiO2 catalyst. The effect of the solvent was studied and high activity and selectivity to 2-methylfuran and furfuryl alcohol was observed using octane as solvent but a number of byproducts were observed. The addition of Ru to the PdTiO2 catalyst decreased the catalytic activity but improved the selectivity towards 2-methylfuran and furfuryl alcohol with decreased byproduct formation. Variation of the Ru/Pd ratio has shown an interesting effect on the selectivity. The addition of a small amount of Ru (1 wt%) shifted the selectivity towards furfuryl alcohol and 2-methylrofuran. Further increasing the Ru ratio decreased the catalytic activity and also showed a very poor selectivity to 2-methylfuran.
Co-reporter:Daniel R. Jones, Sarwat Iqbal, Satoshi Ishikawa, Christian Reece, Liam M. Thomas, Peter J. Miedziak, David J. Morgan, Jennifer K. Edwards, Jonathon K. Bartley, David J. Willock and Graham J. Hutchings
Catalysis Science & Technology 2016 vol. 6(Issue 15) pp:6022-6030
Publication Date(Web):03 May 2016
DOI:10.1039/C6CY00382F
A series of Cu–ZrO2 catalysts prepared by a co-precipitation method were studied for the hydrogenation of levulinic acid to give γ-valerolactone (GVL). The effects of a range of catalyst preparation parameters, namely molar Cu/Zr ratio, calcination temperature and the ageing time of the precipitates, were systematically investigated. The molar Cu/Zr ratio was found to have a strong influence on the BET surface area of the material leading to a high activity for catalysts prepared with a Cu/Zr molar ratio of unity. Using this molar ratio the calcination temperature was varied from 300 °C to 800 °C, the material calcined at 400 °C showed the highest activity. Increasing the ageing time used in the catalyst preparation identified 6 h as the optimum to achieve the highest activity for LA conversion. Based on characterisation of all materials we conclude that the active Cu species is present in only low concentration suggesting that it should be possible to produce a catalyst of high activity with much lower Cu content.
Co-reporter:Daniel R. Jones, Sarwat Iqbal, Simon A. Kondrat, Giacomo M. Lari, Peter J. Miedziak, David J. Morgan, Stewart F. Parker and Graham J. Hutchings
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 26) pp:17259-17264
Publication Date(Web):01 Apr 2016
DOI:10.1039/C6CP01311B
A series of ruthenium catalysts supported on two different carbons were tested for the hydrogenation of lactic acid to 1,2-propanediol and butanone to 2-butanol. The properties of the carbon supports were investigated by inelastic neutron scattering and correlated with the properties of the ruthenium deposited onto the carbons by wet impregnation or sol-immobilisation. It was noted that the rate of butanone hydrogenation was highly dependent on the carbon support, while no noticeable difference in rates was observed between different catalysts for the hydrogenation of lactic acid.
Co-reporter:Benjamin R. Yeo, Geoffrey J.F. Pudge, Keith G. Bugler, Alice V. Rushby, Simon Kondrat, Jonathan Bartley, Stanislaw Golunski, Stuart H. Taylor, Emma Gibson, Peter.P. Wells, Catherine Brookes, Michael Bowker, Graham J. Hutchings
Surface Science 2016 Volume 648() pp:163-169
Publication Date(Web):June 2016
DOI:10.1016/j.susc.2015.11.010
•The surface of iron molybdate catalysts is all Mo.•Core–shell catalysts of Mo at the surface of iron oxide can be prepared for surface studies.•Ensemble effects dominate selectivity.•High selectivity catalysts can be prepared without aqueous waste.The oxidation of methanol to formaldehyde is a major chemical process carried out catalytically and iron molybdate is one of the major catalysts for this process. In this paper we explore the nature of the active and selective surfaces of iron molybdate catalysts and show that the effective catalysts comprise molybdenum rich surfaces. We conclude that it is therefore important to maximise the surface area of these active catalysts and to this end we have studied catalysts made using a new physical grinding method with oxalic acid. For super-stoichiometric materials (Fe:Mo = 1:2.2) the reaction data show that physical mixing produces effective catalysts, possibly offering an improvement over the conventional co-precipitation method.
Co-reporter:Dr. Xi Liu;Dr. Viktoria Fabos;Stuart Taylor; David W. Knight;Dr. Keith Whiston; Graham J. Hutchings
Chemistry - A European Journal 2016 Volume 22( Issue 35) pp:12290-12294
Publication Date(Web):
DOI:10.1002/chem.201602390
Abstract
We report the direct production of 1,3-butadiene from the dehydration of 2,3-butandiol by using alumina as catalyst. Under optimized kinetic reaction conditions, the production of methyl ethyl ketone and isobutyraldehyde, formed via the pinacol–pinacolone rearrangement, was markedly reduced and almost 80 % selectivity to 1,3-butadiene and 1,3-butadiene could be achieved. The presence of water plays a critical role in the inhibition of oligomerization. The amphoteric nature of γ-Al2O3 was identified as important and this contributed to the improved catalytic selectivity when compared with other acidic catalysts.
Co-reporter:Alberto Villa, Nikolaos Dimitratos, Carine E. Chan-Thaw, Ceri Hammond, Laura Prati, and Graham J. Hutchings
Accounts of Chemical Research 2015 Volume 48(Issue 5) pp:1403
Publication Date(Web):April 17, 2015
DOI:10.1021/ar500426g
Glycerol is an important byproduct of biodiesel production, and it is produced in significant amounts by transesterification of triglycerides with methanol. Due to the highly functionalized nature of glycerol, it is an important biochemical that can be utilized as a platform chemical for the production of high-added-value products. At present, research groups in academia and industry are exploring potential direct processes for the synthesis of useful potential chemicals using catalytic processes. Over the last 10 years, there has been huge development of potential catalytic processes using glycerol as the platform chemical.One of the most common processes investigated so far is the catalytic oxidation of glycerol at mild conditions for the formation of valuable oxygenated compounds used in the chemical and pharmaceutical industry. The major challenges associated with the selective oxidation of glycerol are (i) the control of selectivity to the desired products, (ii) high activity and resistance to poisoning, and (iii) minimizing the usage of alkaline conditions. To address these challenges, the most common catalysts used for the oxidation of glycerol are based on supported metal nanoparticles. The first significant breakthrough was the successful utilization of supported gold nanoparticles for improving the selectivity to specific products, and the second was the utilization of supported bimetallic nanoparticles based on gold, palladium, and platinum for improving activity and controlling the selectivity to the desired products. Moreover, the utilization of base-free reaction conditions for the catalytic oxidation of glycerol has unlocked new pathways for the production of free-base products, which facilitates potential industrial application.The advantages of using gold-based catalysts are the improvement of the catalyst lifetime, stability, and reusability, which are key factors for potential commercialization. In this Account, we discuss the advantages of the using supported gold-based nanoparticles, preparation methods for achieving highly active gold-based catalysts, and parameters such as particle size, morphology of the bimetallic particle, and metal–support interactions, which can influence activity and selectivity to the desired products.
Co-reporter:Sarwat Iqbal, Simon A. Kondrat, Daniel R. Jones, Daniël C. Schoenmakers, Jennifer K. Edwards, Li Lu, Benjamin R. Yeo, Peter P. Wells, Emma K. Gibson, David J. Morgan, Christopher J. Kiely, and Graham J. Hutchings
ACS Catalysis 2015 Volume 5(Issue 9) pp:5047
Publication Date(Web):July 15, 2015
DOI:10.1021/acscatal.5b00625
The hydrogenation of lactic acid to form 1,2-propanediol has been investigated using Ru nanoparticles supported on carbon as a catalyst. Two series of catalysts which were prepared by wet impregnation and sol-immobilization were investigated. Their activity was contrasted with that of a standard commercial Ru/C catalyst (all catalysts comprise 5 wt % Ru). The catalyst prepared using sol-immobilization was found to be more active than the wet impregnation materials. In addition, the catalyst made by sol-immobilization was initially more active than the standard commercial catalyst. However, when reacted for an extended time or with successive reuse cycles, the sol-immobilized catalyst became less active, whereas the standard commercial catalyst became steadily more active. Furthermore, both catalysts exhibited an induction period during the first 1000 s of reaction. Detailed scanning transmission electron microscopy, X-ray photoelectron spectroscopy and X-ray absorption fine structure analysis data, when correlated with the catalytic performance results, showed that the high activity can be ascribed to highly dispersed Ru nanoparticles. Although the sol-immobilization method achieved these optimal discrete Ru nanoparticles immediately, as can be expected from this preparation methodology, the materials were unstable upon reuse. In addition, surface lactide species were detected on these particles using X-ray photoelectron spectroscopy, which could contribute to their deactivation. The commercial Ru/C catalysts, on the other hand, required treatment under reaction conditions to change from raft-like morphologies to the desired small nanoparticle morphology, during which time the catalytic performance progressively improved.Keywords: catalyst reuse; Catalytic hydrogenation; lactic acid; propane diol; propylene glycol; Ru/C
Co-reporter:Jiacheng Wang, Simon A. Kondrat, Yingyu Wang, Gemma L. Brett, Cicely Giles, Jonathan K. Bartley, Li Lu, Qian Liu, Christopher J. Kiely, and Graham J. Hutchings
ACS Catalysis 2015 Volume 5(Issue 6) pp:3575
Publication Date(Web):April 29, 2015
DOI:10.1021/acscatal.5b00480
The control over both the dispersion and the particle size distribution of supported precious metal nanoparticles used in heterogeneous catalysts is of paramount importance. Here, we demonstrate the successful formation of highly accessible and well dispersed gold–palladium nanoparticles, stabilized with two-dimensional graphene oxide, that itself is dispersed by intercalated titania particles to form ternary hybrid catalysts. In this application, graphene oxide acts as an effective substitute for the more conventional polymer ligands that are used to stabilize nanoparticles in a sol-immobilization procedure. The particle size distribution can be adjusted by varying the graphene oxide-to-metal mass ratio. The addition of titania efficiently hinders the stacking and agglomeration of the supported metal on graphene oxide sheets, facilitating diffusion of oxygen and reactants to the catalyst surface. This gold–palladium/graphene oxide/titania “ternary” catalyst has been tested for the selective oxidation of a range of alcohols. The resulting optimized catalyst exhibits a comparable activity to a sol-immobilized derived catalyst where the metal nanoparticles are stabilized by poly(vinyl alcohol) ligands, with the graphene oxide-stabilized hybrid catalyst having enhanced stability. We consider that the novel strategy of supporting metal nanoparticles described here can also be adopted to synthesize a wide range of high activity, stable heterogeneous catalysts for other reactions.Keywords: bimetallic nanoparticles; gold−palladium nanoalloy; graphene oxide nanoparticle stabilizer; graphene oxide/titania catalyst composites; selective alcohol oxidation
Co-reporter:Upendra Nath Gupta, Hamed Alshammari, Nicholas F. Dummer, Robert L. Jenkins, Donald Bethell and Graham J. Hutchings
Catalysis Science & Technology 2015 vol. 5(Issue 2) pp:1307-1313
Publication Date(Web):12 Nov 2014
DOI:10.1039/C4CY01355G
The oxidation of dec-1-ene is investigated under solvent-free conditions using gold nanoparticles supported on graphite and in a batch reactor in the presence of a radical initiator using oxygen from air as the terminal oxidant. The evolution of the products with reaction time shows that there is an initial induction period and during this time very little epoxide is fomed and the products of allylic oxidation are dominant. Subsequently the epoxide becomes the major product prior to the diol being formed from hydrolysis due to the presence of by-product water formed from the selective oxidation reaction. It is considered that the allylic oxidation products are in part converted in situ into aldehydes which form peracids during the induction period; the peracid leads to epoxide formation as the major product as the conversion is increased. The effect of addition of a number of aldehydes is investigated, all leading to enhanced epoxide formation when added in small amounts. Molar enhancements of epoxide yield can approach twice the amount of aldehyde initially added. This behaviour is in contrast to earlier studies which utilise aldehydes in greater than stoichiometric amounts as sacrificial reactants. The importance of in situ aldehyde formation is also demonstrated by the addition of benzyl alcohol which under the reaction conditions rapidly gives benzaldehyde and enhanced epoxide formation. Possible mechanistic interpretations of the observations are discussed.
Co-reporter:Xi Liu, Marco Conte, Weihao Weng, Qian He, Robert L. Jenkins, Masashi Watanabe, David J. Morgan, David W. Knight, Damien M. Murphy, Keith Whiston, Christopher J. Kiely and Graham J. Hutchings
Catalysis Science & Technology 2015 vol. 5(Issue 1) pp:217-227
Publication Date(Web):23 Oct 2014
DOI:10.1039/C4CY01213E
Molybdenum blue (MB), a multivalent molybdenum oxide with a nano-ring morphology is well-known in analytical chemistry but, to date it has been largely ignored in other applications. In the present work, MB has been characterized by STEM-HAADF imaging for the first time, showing the nano-ring morphology of this complex molybdenum oxide and the ordered super-molecular framework crystals that can result from the self-assembly of these MB nano-ring units. The potential of MB as an oxidation catalyst has also been investigated, where it is shown to have excellent catalytic activity and stability in the selective oxidation of cyclohexane to cyclohexanol and cyclohexanone which are important intermediates in the production of nylon.
Co-reporter:Giacomo M. Lari, Ewa Nowicka, David J. Morgan, Simon A. Kondrat and Graham J. Hutchings
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 35) pp:23236-23244
Publication Date(Web):05 Aug 2015
DOI:10.1039/C5CP02512E
The sol immobilisation technique, in which a stabilising ligand (such as polyvinyl alcohol or polyvinyl pyridine) can be used to tune metal particle size and composition, has become a valuable method of making supported nanoparticle catalysts. An unfortunate consequence of the stabilising ligand is that often access of reactant molecules to the metal nanoparticle surface is impeded. Several methods have been proposed for the removal of these ligands, though determination of the degree of their success is difficult. Here, we demonstrate the use of in situ infrared and UV-Vis spectroscopy to elucidate the access of carbon monoxide to the surface of Au/TiO2 catalysts before and after various ligand removal treatments. These were contrasted with a catalyst prepared by deposition precipitation prepared in the absence of stabilising ligand as a control. Changes were observed in the infrared spectrum, with the wavenumber of carbon monoxide linearly bonded to Au for catalysts shifting before and after ligand removal, which correlated well with the activity of the catalyst for carbon monoxide oxidation. Also the extent of shifting of the Au surface resonance plasmon band on the addition of carbon monoxide, observed by UV-Vis, also correlated well with catalyst activity. These simple methods can be used to determine the quantity of exposed metal sites after a ligand removal treatment and so determine the treatments effectiveness.
Co-reporter:Gavin M. King;Dr. Sarwat Iqbal;Dr. Peter J. Miedziak;Dr. Gemma L. Brett;Dr. Simon A. Kondrat;Benjamin R. Yeo;Dr. Xi Liu;Dr. Jennifer K. Edwards;Dr. David J. Morgan; David K. Knight ; Graham J. Hutchings
ChemCatChem 2015 Volume 7( Issue 14) pp:2122-2129
Publication Date(Web):
DOI:10.1002/cctc.201500242
Abstract
The selective hydrogenation of furfuryl alcohol was investigated at room temperature by using supported palladium catalysts. The catalysts are very selective to the formation of 2-methylfuran. Furthermore, the addition of tin to palladium showed similar catalytic activity, but was more selective to tetrahydrofurfuryl alcohol. Variation of the Sn/Pd ratio has shown a considerable and interesting effect on the selectivity pattern. Addition of a small amount of Sn (1 wt %) shifted the selectivity towards tetrahydrofurfuryl alcohol and methyltetrahydrofuran, which are ring-saturated molecules. Increasing the tin ratio further decreased the catalytic activity and also showed very poor selectivity to either of these products.
Co-reporter:Jennifer K. Edwards, Simon J. Freakley, Albert F. Carley, Christopher J. Kiely, and Graham J. Hutchings
Accounts of Chemical Research 2014 Volume 47(Issue 3) pp:845
Publication Date(Web):October 31, 2013
DOI:10.1021/ar400177c
Hydrogen peroxide is a widely used chemical but is not very efficient to make in smaller than industrial scale. It is an important commodity chemical used for bleaching, disinfection, and chemical manufacture. At present, manufacturers use an indirect process in which anthraquinones are sequentially hydrogenated and oxidized in a manner that hydrogen and oxygen are never mixed. However, this process is only economic at a very large scale producing a concentrated product. For many years, the identification of a direct process has been a research goal because it could operate at the point of need, producing hydrogen peroxide at the required concentration for its applications. Research on this topic has been ongoing for about 100 years.Until the last 10 years, catalyst design was solely directed at using supported palladium nanoparticles. These catalysts require the use of bromide and acid to arrest peroxide decomposition, since palladium is a very active catalyst for hydrogen peroxide hydrogenation. Recently, chemists have shown that supported gold nanoparticles are active when gold is alloyed with palladium because this leads to a significant synergistic enhancement in activity and importantly selectivity. Crucially, bimetallic gold-based catalysts do not require the addition of bromide and acids, but with carbon dioxide as a diluent its solubility in the reaction media acts as an in situ acid promoter, which represents a greener approach for peroxide synthesis. The gold catalysts can operate under intrinsically safe conditions using dilute hydrogen and oxygen, yet these catalysts are so active that they can generate peroxide at commercially significant rates.The major problem associated with the direct synthesis of hydrogen peroxide concerns the selectivity of hydrogen usage, since in the indirect process this factor has been finely tuned over decades of operation. In this Account, we discuss how the gold–palladium bimetallic catalysts have active sites for the synthesis and hydrogenation of hydrogen peroxide that are different, in contrast to monometallic palladium in which synthesis and hydrogenation operate at the same sites. Through treatment of the support with acids prior to the deposition of the gold–palladium bimetallic particles, we can obtain a catalyst that can make hydrogen peroxide at a very high rate without decomposing or hydrogenating the product. This innovation opens up the way to design improved catalysts for the direct synthesis process, and these possibilities are described in this Account.
Co-reporter:Peter J. Miedziak, Hamed Alshammari, Simon A. Kondrat, Tomos J. Clarke, Thomas E. Davies, Moataz Morad, David J. Morgan, David J. Willock, David W. Knight, Stuart H. Taylor and Graham J. Hutchings
Green Chemistry 2014 vol. 16(Issue 6) pp:3132-3141
Publication Date(Web):10 Apr 2014
DOI:10.1039/C4GC00087K
We report the selective oxidation of glucose to gluconic acid under mild conditions and show that if a basic support is used then the reaction can be carried out without the addition of sacrificial base or pH control. The use of sol-immobilisation prepared catalysts supported on magnesium oxide facilitates the use of ambient air as an oxidant source. These mild conditions resulted in an excellent selectivity towards gluconic acid. Different heat treatments result in an improvement in the activity of the catalyst, these improvements are discussed in terms of XRD, DRIFTD and TEM analysis of the catalysts, despite significant particle growth and phase segregation occurring during the thermal treatments.
Co-reporter:Ren Su, Lokesh Kesavan, Mads M. Jensen, Ramchandra Tiruvalam, Qian He, Nikolaos Dimitratos, Stefan Wendt, Marianne Glasius, Christopher J. Kiely, Graham J. Hutchings and Flemming Besenbacher
Chemical Communications 2014 vol. 50(Issue 84) pp:12612-12614
Publication Date(Web):11 Jul 2014
DOI:10.1039/C4CC04024D
The selectivity of photocatalytic phenol production from the direct oxidation of benzene can be enhanced by fine adjustment of the morphology and composition of Au–Pd metal nanoparticles supported on titanium dioxide thereby suppressing the decomposition of benzene and evolution of phenolic compounds.
Co-reporter:Raimon P. Marin, Simon A. Kondrat, Thomas E. Davies, David J. Morgan, Dan I. Enache, Gary B. Combes, Stuart H. Taylor, Jonathan K. Bartley and Graham J. Hutchings
Catalysis Science & Technology 2014 vol. 4(Issue 7) pp:1970-1978
Publication Date(Web):08 Apr 2014
DOI:10.1039/C4CY00044G
Cobalt zinc oxide catalysts have been prepared by anti-solvent precipitation in supercritical CO2 and investigated for CO hydrogenation. Here we show how the textural and catalytic properties of the catalyst can be tailored by the addition of water to the initial solution of cobalt and zinc acetates in methanol. Characterization of the catalysts by powder X-ray diffraction, infra-red and Raman spectroscopy showed that in the absence of water a high surface area mixed acetate was produced which upon calcination formed wurtzite type Zn1−xCoxO and spinel type ZnxCo3−xO4. The addition of 5 vol.% water resulted in a phase separated Co3O4/ZnO catalyst and enhanced active cobalt surface area as a result of disruption of the solvent/CO2 phase equilibrium during precipitation.
Co-reporter:Moataz Morad, Meenakshisundaram Sankar, Enhong Cao, Ewa Nowicka, Thomas E. Davies, Peter J. Miedziak, David J. Morgan, David W. Knight, Donald Bethell, Asterios Gavriilidis and Graham J. Hutchings
Catalysis Science & Technology 2014 vol. 4(Issue 9) pp:3120-3128
Publication Date(Web):22 May 2014
DOI:10.1039/C4CY00387J
The synthesis of stable, supported, bimetallic nanoalloys with controlled size, morphology and composition is of great practical importance. Compared to their monometallic analogues, such materials exhibit remarkable enhancement in functional properties, which can be exploited in various fields including catalysis. Recently, we have reported a simple excess anion modification of the impregnation method to prepare supported gold–palladium catalysts which gives very good control over the particle sizes and the composition without using any stabilizer ligands in the preparation. Here, we report the results from a comparative study of using this modified impregnation catalyst for the solvent-free aerobic oxidation of alcohols in two different reactors: a glass stirred reactor and a micro packed bed reactor under batch and continuous mode respectively. These modified impregnation catalysts are exceptionally active and more importantly, when tested in a micro packed bed reactor under flow conditions, are observed to be stable for several days without any sign of deactivation in contrast to the same catalyst prepared by the sol immobilization method in the presence of stabilizer ligands which showed a 3–5% decrease in conversion over 10–12 h.
Co-reporter:Sarwat Iqbal, Xi Liu, Obaid F. Aldosari, Peter J. Miedziak, Jennifer K. Edwards, Gemma L. Brett, Adeeba Akram, Gavin M. King, Thomas E. Davies, David J. Morgan, David K. Knight and Graham J. Hutchings
Catalysis Science & Technology 2014 vol. 4(Issue 8) pp:2280-2286
Publication Date(Web):14 Apr 2014
DOI:10.1039/C4CY00184B
The selective hydrogenation of furfuryl alcohol into 2-methylfuran was investigated at room temperature using palladium supported catalysts. We have shown that Pd–TiO2 catalysts can be very effective for the synthesis of 2-methylfuran at room temperature and low pressure of hydrogen (1–3 bar). The effect of various reaction conditions (pressure, catalyst amount, and solvent) was studied.
Co-reporter:Wilm Jones, Ren Su, Peter P. Wells, Yanbin Shen, Nikolaos Dimitratos, Michael Bowker, David Morgan, Bo B. Iversen, Arunabhiram Chutia, Flemming Besenbacher and Graham Hutchings
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 48) pp:26638-26644
Publication Date(Web):27 Oct 2014
DOI:10.1039/C4CP04693E
The development of efficient photocatalytic routines for producing hydrogen is of great importance as society moves away from energy sources derived from fossil fuels. Recent studies have identified that the addition of metal nanoparticles to TiO2 greatly enhances the photocatalytic performance of these materials towards the reforming of alcohols for hydrogen production. The core–shell structured Au–Pd bimetallic nanoparticle supported on TiO2 has being of interest as it exhibited extremely high quantum efficiencies for hydrogen production. However, the effect of shell composition and thickness on photocatalytic performance remains unclear. Here we report the synthesis of core–shell structured AuPd NPs with the controlled deposition of one and two monolayers (ML) equivalent of Pd onto Au NPs by colloidal and photodeposition methods. We have determined the shell composition and thickness of the nanoparticles by a combination of X-ray absorption fine structure and X-ray photoelectron spectroscopy. Photocatalytic ethanol reforming showed that the core–shell structured Au–Pd promoters supported on TiO2 exhibit enhanced activity compared to that of monometallic Au and Pd as promoters, whilst the core–shell Au–Pd promoters containing one ML equivalent Pd provide the optimum reactivity.
Co-reporter:Ren Su, Michael M. Forde, Qian He, Yanbin Shen, Xueqin Wang, Nikolaos Dimitratos, Stefan Wendt, Yudong Huang, Bo B. Iversen, Christopher J. Kiely, Flemming Besenbacher and Graham J. Hutchings
Dalton Transactions 2014 vol. 43(Issue 40) pp:14976-14982
Publication Date(Web):19 Jun 2014
DOI:10.1039/C4DT01309C
As co-catalyst materials, metal nanoparticles (NPs) play crucial roles in heterogeneous photocatalysis. The photocatalytic performance strongly relies on the physical properties (i.e., composition, microstructure, and surface impurities) of the metal NPs. Here we report a convenient chemical vapour impregnation (CVI) approach for the deposition of monometallic-, alloyed, and core–shell structured metal co-catalysts onto the TiO2 photocatalyst. The as-synthesised metal NPs are highly dispersed on the support and show narrow size distributions, which suit photocatalysis applications. More importantly, the surfaces of the as-synthesised metal NPs are free of protecting ligands, enabling the photocatalysts to be ready to use without further treatment. The effect of the metal identity, the alloy chemical composition, and the microstructure on the photocatalytic performance has been investigated for hydrogen production and phenol decomposition. Whilst the photocatalytic H2 production performance can be greatly enhanced by using the core–shell structured co-catalyst (Pdshell–Aucore and Ptshell–Aucore), the Ptshell–Aucore modified TiO2 yields enhanced quantum efficiency but a reduced effective decomposition of phenol to CO2 compared to that of the monometallic counterparts. We consider the CVI approach provides a feasible and elegant process for the decoration of photocatalyst materials.
Co-reporter:Marco Santonastaso, Simon J. Freakley, Peter J. Miedziak, Gemma L. Brett, Jennifer K. Edwards, and Graham J. Hutchings
Organic Process Research & Development 2014 Volume 18(Issue 11) pp:1455-1460
Publication Date(Web):August 29, 2014
DOI:10.1021/op500195e
Catalysts containing bimetallic gold–palladium nanoparticles are extremely active and selective for the oxidation of alcohols to aldehydes and the direct synthesis of hydrogen peroxide from molecular hydrogen and oxygen. We show that the oxidation of benzyl alcohol can be carried out at 50 °C and below by generating hydrogen peroxide in situ. The oxidation of benzyl alcohol to benzaldehyde has been achieved with high selectivity (>85%) at temperatures where no reaction is observed with only molecular oxygen in an autoclave. The effect of temperature, catalyst support, and solvent are studied in an autoclave system and reactions were carried out in a fixed bed reactor at a range of gas flow rates where the catalysts demonstrated stable conversion and selectivity.
Co-reporter:Dr. Muhammad H. Haider;Dr. Carmine D'Agostino;Dr. Nicholas F. Dummer;Dr. Mick D. Mantle; Lynn F. Gladden; David W. Knight;Dr. David J. Willock;Dr. David J. Morgan; Stuart H. Taylor; Graham J. Hutchings
Chemistry - A European Journal 2014 Volume 20( Issue 6) pp:1743-1752
Publication Date(Web):
DOI:10.1002/chem.201302348
Abstract
The effect of ceria and zirconia grafting onto alumina (α and θ–δ phases) as supports for silicotungstic acid for the dehydration of glycerol to acrolein was studied. 30 % Silicotungstic acid (STA) supported on 5 % zirconia/δ,θ-alumina was the best catalyst, producing 85 % selectivity to acrolein at 100 % glycerol conversion, and it showed stable activity without using oxygen as a co-feed. The catalyst produced a STA of 90 g(acrolein) kg(cat)−1 h−1, which was greater than the STA simply supported on δ,θ-alumina, which only demonstrated 75 % selectivity towards acrolein. The effect of grafting on the support material was investigated by means of nitrogen adsorption, ammonia temperature-programmed desorption, thermogravimetric analysis, Raman spectroscopy, and powder X-ray diffraction. A pulsed-field gradient (PFG) NMR technique was also used to study diffusion processes associated with the catalysts. Diffusion studies of the grafted catalysts showed that zirconia contributes to the formation of more tortuous pathways within the pore structure, leading to the diminution of acid strength and making the catalyst less susceptible to coke formation.
Co-reporter:Dr. Mosaed Alhumaimess;Dr. Zhongjie Lin;Dr. Qian He;Dr. Li Lu;Dr. Nickolaos Dimitratos;Dr. Nicholas F. Dummer;Dr. Marco Conte;Dr. Stuart H. Taylor;Dr. Jonathan K. Bartley; Christopher J. Kiely; Graham J. Hutchings
Chemistry - A European Journal 2014 Volume 20( Issue 6) pp:1701-1710
Publication Date(Web):
DOI:10.1002/chem.201303355
Abstract
MnO2 was synthesised as a catalyst support material using a hydrothermal method. This involved reacting MnSO4⋅H2O and (NH4)2S2O8 at 120 °C for a range of crystallisation times, which affords control over the morphology and phase composition of the MnO2 formed. Gold was deposited on these supports using sol-immobilisation, impregnation and deposition precipitation methods, and the resultant materials were used for the oxidation of benzyl alcohol and carbon monoxide. The effect of the support morphology on the dispersion of the gold nanoparticles and the consequent effect on the catalytic performance is described and discussed.
Co-reporter:Dr. Simon A. Kondrat;Dr. Peter J. Miedziak;Mark Douthwaite;Dr. Gemma L. Brett;Dr. Thomas E. Davies;Dr. David J. Morgan;Dr. Jennifer K. Edwards;David W. Knight; Christopher J. Kiely; Stuart H. Taylor; Graham J. Hutchings
ChemSusChem 2014 Volume 7( Issue 5) pp:1326-1334
Publication Date(Web):
DOI:10.1002/cssc.201300834
Abstract
Base-free selective oxidation of glycerol has been investigated using trimetallic Au–Pd–Pt nanoparticles supported on titania and their corresponding bimetallic catalysts. Catalysts were prepared by the sol-immobilization method and characterized by means of TEM, UV/Vis spectroscopy, diffuse reflectance infrared fourier transform spectroscopy, X-ray photoelectron spectroscopy, and microwave plasma–atomic emission spectroscopy. It was found that of the bimetallic catalysts, Pd–Pt/TiO2 was the most active with high selectivity to C3 products. The addition of Au to this catalyst to form the trimetallic Au–Pd–Pt/TiO2, resulted in an increase in activity relative to Pd–Pt/TiO2. The turnover frequency increased from 210 h−1 with the Pd–Pt/TiO2 catalyst to378 h−1 for the trimetallic Au–Pd–Pt/TiO2 catalyst with retention of selectivity towards C3 products.
Co-reporter:Michael M. Forde, Lokesh Kesavan, Mohd Izham bin Saiman, Qian He, Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, Robert L. Jenkins, Stuart H. Taylor, Christopher J. Kiely, and Graham J. Hutchings
ACS Nano 2014 Volume 8(Issue 1) pp:957
Publication Date(Web):December 16, 2013
DOI:10.1021/nn405757q
The use of precious metals in heterogeneous catalysis relies on the preparation of small nanoparticles that are stable under reaction conditions. To date, most conventional routes used to prepare noble metal nanoparticles have drawbacks related to surface contamination, particle agglomeration, and reproducibility restraints. We have prepared titania-supported palladium (Pd) and platinum (Pt) catalysts using a simplified vapor deposition technique termed chemical vapor impregnation (CVI) that can be performed in any standard chemical laboratory. These materials, composed of nanoparticles typically below 3 nm in size, show remarkable activity under mild conditions for oxidation and hydrogenation reactions of industrial importance. We demonstrate the preparation of bimetallic Pd–Pt homogeneous alloy nanoparticles by this new CVI method, which show synergistic effects in toluene oxidation. The versatility of our CVI methodology to be able to tailor the composition and morphology of supported nanoparticles in an easily accessible and scalable manner is further demonstrated by the synthesis of Pdshell–Aucore nanoparticles using CVI deposition of Pd onto preformed Au nanoparticles supported on titania (prepared by sol immobilization) in addition to the presence of monometallic Au and Pd nanoparticles.Keywords: bimetallic nanoparticle; catalysis; core−shell structures; gold; hydrogenation; nanoalloy; oxidation; palladium; platinum
Co-reporter:Ren Su, Ramchandra Tiruvalam, Andrew J. Logsdail, Qian He, Christopher A. Downing, Mikkel T. Jensen, Nikolaos Dimitratos, Lokesh Kesavan, Peter P. Wells, Ralf Bechstein, Henrik H. Jensen, Stefan Wendt, C. Richard A. Catlow, Christopher J. Kiely, Graham J. Hutchings, and Flemming Besenbacher
ACS Nano 2014 Volume 8(Issue 4) pp:3490
Publication Date(Web):March 7, 2014
DOI:10.1021/nn500963m
Photocatalytic hydrogen evolution may provide one of the solutions to the shift to a sustainable energy society, but the quantum efficiency of the process still needs to be improved. Precise control of the composition and structure of the metal nanoparticle cocatalysts is essential, and we show that fine-tuning the Au–Pd nanoparticle structure modifies the electronic properties of the cocatalyst significantly. Specifically, Pdshell–Aucore nanoparticles immobilized on TiO2 exhibit extremely high quantum efficiencies for H2 production using a wide range of alcohols, implying that chemical byproducts from the biorefinery industry can be used as feedstocks. In addition, the excellent recyclability of our photocatalyst material indicates a high potential in industrial applications. We demonstrate that this particular elemental segregation provides optimal positioning of the unoccupied d-orbital states, which results in an enhanced utilization of the photoexcited electrons in redox reactions. We consider that the enhanced activity observed on TiO2 is generic in nature and can be transferred to other narrow band gap semiconductor supports for visible light photocatalysis.Keywords: cocatalysts; density functional theory; hydrogen evolution; metal nanoparticles; photocatalysis; TiO2
Co-reporter:Marco Conte;Catherine J. Davies;David J. Morgan;Albert F. Carley
Catalysis Letters 2014 Volume 144( Issue 1) pp:1-8
Publication Date(Web):2014 January
DOI:10.1007/s10562-013-1138-8
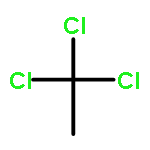
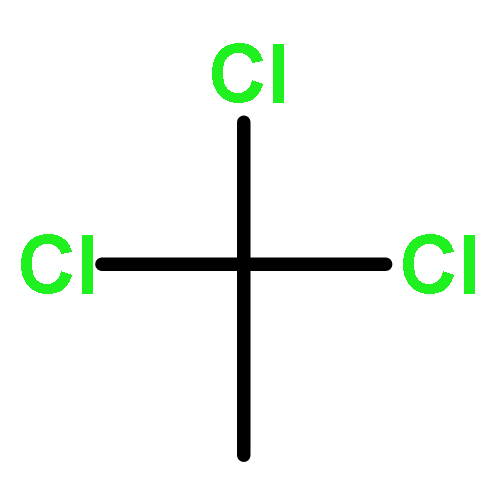
A set of Au/C catalysts for the gas phase hydrochlorination of acetylene to vinyl chloride monomer were prepared using a range of strong acids as impregnating solvents and varying the preparation drying temperature. The most active catalyst is the material prepared using aqua regia as solvent with an intermediate drying temperature of 140 °C. The effects of the catalyst preparation parameters on the catalytic activity are examined using XPS and TPR as analytical tools. In particular, the use of thermal reduction methods allows the determination of kinetic parameters for the reduction of Au3+ to Au0 by means of H2. These data support the existence of a redox cycle between Au3+/Au0 when carbon is used as support.
Co-reporter:Graham J. Hutchings and Christopher J. Kiely
Accounts of Chemical Research 2013 Volume 46(Issue 8) pp:1759
Publication Date(Web):April 15, 2013
DOI:10.1021/ar300356m
The discovery that supported gold nanoparticles are exceptionally effectivecatalysts for redox reactions has led to an explosion of interest in gold nanoparticles. In addition, incorporating a second metal as an alloy with gold can enhance the catalyst performance even more. The addition of small amounts of gold to palladium, in particular, and vice versa significantly enhances the activity of supported gold–palladium nanoparticles as redox catalysts through what researchers believe is an electronic effect.In this Account, we describe and discuss methodologies for the synthesisof supported gold–palladium nanoparticles and their use as heterogeneous catalysts. In general, three key challenges need to be addressed in the synthesis of bimetallic nanoparticles: (i) control of the particle morphology, (ii) control of the particle size distribution, and (iii) control of the nanoparticle composition. We describe three methodologies to address these challenges. First, we discuss the relatively simple method of coimpregnation. Impregnation allows control of particle morphology during alloy formation but does not control the particle compositions or the particle size distribution. Even so, we contend that this method is the best preparation method in the catalyst discovery phase of any project, since it permits the investigation of many different catalyst structures in one experiment, which may aid the identification of new catalysts. A second approach, sol-immobilization, allows enhanced control of the particle size distribution and the particle morphology, but control of the composition of individual nanoparticles is not possible. Finally, a modified impregnation method can allow the control of all three of these crucial parameters. We discuss the effect of the different methodologies on three redox reactions: benzyl alcohol oxidation, toluene oxidation, and the direct synthesis of hydrogen peroxide. We show that the coimpregnation method provides the best reaction selectivity for benzyl alcohol oxidation and the direct synthesis of hydrogen peroxide. However, because of the reaction mechanism, the sol-immobilzation method gives very active and selective catalysts for toluene oxidation. We discuss the possible nature of the preferred active structures of the supported nanoparticles for these reactions. This paper is based on the IACS Heinz Heinemann Award Lecture entitled “Catalysis using gold nanoparticles” which was given in Munich in July 2012.
Co-reporter:Michael M. Forde ; Robert D. Armstrong ; Ceri Hammond ; Qian He ; Robert L. Jenkins ; Simon A. Kondrat ; Nikolaos Dimitratos ; Jose Antonio Lopez-Sanchez ; Stuart H. Taylor ; David Willock ; Christopher J. Kiely ;Graham John Hutchings
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11087-11099
Publication Date(Web):June 26, 2013
DOI:10.1021/ja403060n
Iron and copper containing ZSM-5 catalysts are effective for the partial oxidation of ethane with hydrogen peroxide giving combined oxygenate selectivities and productivities of up to 95.2% and 65 mol kgcat–1 h–1, respectively. High conversion of ethane (ca. 56%) to acetic acid (ca. 70% selectivity) can be observed. Detailed studies of this catalytic system reveal a complex reaction network in which the oxidation of ethane gives a range of C2 oxygenates, with sequential C–C bond cleavage generating C1 products. We demonstrate that ethene is also formed and can be subsequently oxidized. Ethanol can be directly produced from ethane, and does not originate from the decomposition of its corresponding alkylperoxy species, ethyl hydroperoxide. In contrast to our previously proposed mechanism for methane oxidation over similar zeolite catalysts, the mechanism of ethane oxidation involves carbon-based radicals, which lead to the high conversions we observe.
Co-reporter:Hamed Alshammari, Peter J. Miedziak, David J. Morgan, David W. Knight and Graham J. Hutchings
Green Chemistry 2013 vol. 15(Issue 5) pp:1244-1254
Publication Date(Web):27 Mar 2013
DOI:10.1039/C3GC36828A
The oxidation of 2-hexen-1-ol and 1-hexen-3-ol with air has been studied using supported gold, palladium and gold–palladium catalysts. The main aim was to determine if either the alcohol or alkene functional group can be oxidised selectively. However, based on the reaction products observed (2-hexen-1-ol forms 2-hexene, hexanal, (E)-2-hexenal, (E)-3-hexen-1-ol, 4-hexen-1-ol and (E)-2-hexanoic acid. 1-Hexen-3-ol forms 1-hexene, 3-hexanone, 1-hexen-3-one and 3-hexenol), the main pathway in these reactions is isomerisation and, in addition, significant yields of the products are due to a disproportionation reaction. Controlling the selectivity in molecules with multiple function groups by manipulating the catalyst composition and reaction conditions can promote or hinder the various reaction pathways, thereby increasing the selectivity to the desired oxidation products.
Co-reporter:Ceri Hammond, Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, Robert L. Jenkins, Gareth Whiting, Simon A. Kondrat, Mohd Hasbi ab Rahim, Michael M. Forde, Adam Thetford, Henk Hagen, Eric E. Stangland, Jacob M. Moulijn, Stuart H. Taylor, David J. Willock, and Graham J. Hutchings
ACS Catalysis 2013 Volume 3(Issue 8) pp:1835
Publication Date(Web):July 2, 2013
DOI:10.1021/cs400288b
Fe- and Cu-containing zeolites have recently been shown to be efficient catalysts for the one-step selective transformation of methane into methanol in an aqueous medium at only 50 °C, using H2O2 as green oxidant. Previously, we have observed that Fe species alone are capable of catalyzing this highly selective transformation. However, further catalytic testing and spectroscopic investigations demonstrate that although these extra-framework Fe species are the active component of the catalyst, significant promotion is observed upon the incorporation of other trivalent cations, e.g., Al3+ or Ga3+, into the MFI-framework. While these additional framework species do not constitute active catalytic centers, promotion is observed upon their incorporation as they (1) facilitate the extraction of Fe from the zeolite framework and hence increase the formation of the active Fe species and (2) provide an associated negatively charged framework, which is capable of stabilizing and maintaining the dispersion of the cationic extra-framework Fe species responsible for catalytic activity. By understanding these phenomena and subsequently controlling the overall composition of the catalyst (Fe and Al), we have subsequently been able to prepare a catalyst of equal intrinsic activity (i.e., TOF) but five-times higher productivity (i.e., space-time-yield) compared with the best catalysts reported for this reaction to date.Keywords: methane oxidation; natural gas upgrading; selective oxidation; sustainable chemistry; zeolite catalysis
Co-reporter:Raimon P. Marin, Simon A. Kondrat, James R. Gallagher, Dan I. Enache, Paul Smith, Paul Boldrin, Thomas E. Davies, Jonathan K. Bartley, Garry B. Combes, Peter B. Williams, Stuart H. Taylor, John B. Claridge, Matthew J. Rosseinsky, and Graham J. Hutchings
ACS Catalysis 2013 Volume 3(Issue 4) pp:764
Publication Date(Web):March 6, 2013
DOI:10.1021/cs4000359
Cobalt and ruthenium-promoted cobalt Fischer–Tropsch catalysts supported on titania have been prepared for the first time by gas anti-solvent precipitation. The use of dense CO2 as an anti-solvent enables the precipitation of cobalt acetate and ruthenium acetylacetonate onto preformed titania. The gas anti-solvent process produces catalysts with the desired 20 wt % cobalt content as precursors, which on calcination give highly dispersed Co3O4. The addition of ruthenium to the gas anti-solvent prepared cobalt catalysts has been investigated by two methods (a) coprecipitation with cobalt acetate and (b) wet impregnation onto a precalcined cobalt titania catalyst, and these resulted in catalysts with distinctly different properties. These catalysts were compared with a standard ruthenium-promoted cobalt catalyst prepared by wet impregnation and were found to be substantially more active for the Fischer–Tropsch reaction.Keywords: cobalt; cobalt acetate; Fischer−Tropsch; ruthenium; supercritical anti-solvent precipitation
Co-reporter:Simon J. Freakley, Marco Piccinini, Jennifer K. Edwards, Edwin N. Ntainjua, Jacob A. Moulijn, and Graham J. Hutchings
ACS Catalysis 2013 Volume 3(Issue 4) pp:487
Publication Date(Web):February 6, 2013
DOI:10.1021/cs400004y
The direct synthesis of hydrogen peroxide (H2O2) represents a potential alternative to the currently industrially used anthraquinone process, and Au–Pd catalysts have been identified as effective catalysts. To obtain a direct process, a detailed understanding of the reaction conditions in a continuous flow system is needed. In this study, we use a flow reactor to study reaction conditions independently, including total gas flow rate, catalyst mass, reaction pressure, solvent flow rate, and H2/O2 molar ratio. The study was carried out without the addition of any halide or acid additives often used to suppress the sequential hydrogenation and decomposition reactions that allowed the kinetics of these reactions to be studied along with the synthesis reaction. A global kinetic model describing the net and gross synthesis rate is proposed, and on the basis of this model, we propose that the decomposition reaction suppresses the production of H2O2 to a greater extent than hydrogenation and that catalyst design studies should aim at blocking or generating catalysts without O–O dissociation sites.Keywords: flow reactor; gold; hydrogen peroxide; kinetics; palladium catalyst
Co-reporter:Ceri Hammond, Nikolaos Dimitratos, Robert L. Jenkins, Jose Antonio Lopez-Sanchez, Simon A. Kondrat, Mohd Hasbi ab Rahim, Michael M. Forde, Adam Thetford, Stuart H. Taylor, Henk Hagen, Eric E. Stangland, Joo H. Kang, Jacob M. Moulijn, David J. Willock, and Graham J. Hutchings
ACS Catalysis 2013 Volume 3(Issue 4) pp:689
Publication Date(Web):February 7, 2013
DOI:10.1021/cs3007999
The development of a catalytic, one-step route for the oxidation of methane to methanol remains one of the greatest challenges within catalysis. Of particular importance is the need to develop an efficient route that proceeds under mild reaction conditions so as to avoid deeper oxidation and the economic limitations of the currently practiced syngas route. Recently, it was demonstrated that a copper- and iron-containing zeolite is an efficient catalyst for such a one-step process. The catalyst in question (Cu–Fe–ZSM-5) is capable of selectively transforming methane to methanol in an aqueous medium with hydrogen peroxide as the terminal oxidant. Nevertheless, despite its high activity and unparalleled methanol selectivity, the origin of its activity and the precise nature of its active species are not yet fully understood. Through a combination of catalytic and spectroscopic studies, we hereby demonstrate that extraframework Fe species are the active component of the catalyst for methane oxidation, although the speciation of these sites from synthesis to catalysis significantly alters the observed activity and selectivity. The analogies and differences between this system and other iron-containing zeolite-catalyzed processes, such as N2O-mediated benzene hydroxylation, are also considered.Keywords: green chemistry; methane oxidation; selective oxidation; zeolite catalysis
Co-reporter:Marco Conte, Catherine J. Davies, David J. Morgan, Thomas E. Davies, Albert F. Carley, Peter Johnston and Graham J. Hutchings
Catalysis Science & Technology 2013 vol. 3(Issue 1) pp:128-134
Publication Date(Web):07 Aug 2012
DOI:10.1039/C2CY20478A
The effect of the gold oxidation state and carbon structure on the activity of Au/C catalysts for the hydrochlorination of acetylene was investigated by a combined approach using TPR, XPS and porosimetry determinations. The activity of the catalyst in the synthesis of vinyl chloride monomer was found to be dependent on the presence of Au3+ species in the catalyst. However, by preparing catalysts with different Au3+ content it was possible to determine the existence of a threshold Au3+ amount, beyond which the excess of Au3+ was not active for the reaction. This was explained by the existence of active sites at the Au/C interface, and not just by the presence of Au3+ species on top of Au nanoparticles, as explained by current models for these catalysts. It was also possible to determine the existence of a subset of Au nanoclusters which do not take part in the reaction, as well as changes in the textural properties of the carbon that can affect its long term reusability.
Co-reporter:Gerolamo Budroni, Simon A. Kondrat, Stuart H. Taylor, David J. Morgan, Albert F. Carley, Peter B. Williams and Graham J. Hutchings
Catalysis Science & Technology 2013 vol. 3(Issue 10) pp:2746-2754
Publication Date(Web):01 Aug 2013
DOI:10.1039/C3CY00449J
Alumina supported nickel palladium bimetallic catalysts were prepared by a selective redox deposition method and used for the hydrogenation of crotonaldehyde. Supported nickel catalysts, prepared by incipient wetness, were reduced to form a nickel hydride surface prior to contact with palladium(II) salt solutions. Palladium was successfully deposited selectively onto the nickel hydride surfaces, by a redox reaction. Catalysts were prepared using two different palladium(II) salt solutions; (a) pH 1 palladium chloride in hydrochloric acid solution or (b) pH 3 palladium nitrate in nitric acid. The deposition of palladium onto the supported nickel nanoparticles was strikingly different when using the two palladium solutions, with strong alloy formation with the pH 1 solution and a weaker segregated nickel palladium catalyst with the pH 3 solution. Both catalysts were compared with monometallic palladium and nickel supported catalysts for the hydrogenation of crotonaldehyde with the sample prepared at pH 1 being more active.
Co-reporter:Inés Moreno, Nicholas F. Dummer, Jennifer K. Edwards, Mosaed Alhumaimess, Meenakshisundaram Sankar, Raul Sanz, Patricia Pizarro, David P. Serrano and Graham J. Hutchings
Catalysis Science & Technology 2013 vol. 3(Issue 9) pp:2425-2434
Publication Date(Web):16 Jul 2013
DOI:10.1039/C3CY00493G
Benzyl alcohol was oxidized by an “in situ generated” hydrogen peroxy species, formed from a dilute mixture of hydrogen and oxygen, under mild conditions at a high rate over gold, palladium and gold–palladium nanoparticles supported on hierarchical titanium silicate materials. Hierarchical TS-1 supports were obtained from the crystallization of silanized protozeolitic units, being characterized by having a secondary porous system within supermicro/mesopore range and an enhanced surface area over a standard reference TS-1 material. The presence of the secondary porosity not only improves the accessibility to the active sites of the relatively large reactant molecules but also enhances the metal dispersion, leading to an improved catalytic performance for alcohol oxidation. The catalytic activity of metal loaded hierarchical TS-1 materials was found to be higher in reactions conducted in the presence of diluted hydrogen and oxygen, resulting in a 5-fold increase in the yield of benzaldehyde at 30 °C with an AuPd catalyst with secondary porosity. The improvement in rate observed is due to the oxidizing efficacy of in situ generated hydroperoxy species as compared to molecular oxygen alone as the terminal oxidant.
Co-reporter:Hamed Alshammari, Peter J. Miedziak, David W. Knight, David J. Willock and Graham J. Hutchings
Catalysis Science & Technology 2013 vol. 3(Issue 6) pp:1531-1539
Publication Date(Web):04 Mar 2013
DOI:10.1039/C3CY20864H
In this work we expand on our previous studies on the oxidation of cyclic alkenes using supported gold nanoparticles together with catalytic amounts of peroxides. We demonstrate that various sized cyclic alkenes can be oxidised by this catalyst, under green conditions, without solvent and using air as the oxidant gas. The effect of support, preparation method and choice of metal have been investigated, we demonstrate that supported gold is superior to palladium or a gold palladium alloy, we show that oxides provide the best support for these gold catalysts and the preparation methods that afford the smallest particles are the most active. We show that the reactivity of the cyclic alkenes is related to the ring size with the smaller rings more reactive than the larger rings at the same temperature. The selectivity to the epoxide is dependent on the size of the cyclic alkene ring. In particular, the epoxide selectivity is very low for rings containing fewer than seven carbon atoms. We discuss the origins of this selectivity effect, using DFT calculations to investigate the effect of ring strain on the intermediates and reaction products.
Co-reporter:James Pritchard, Marco Piccinini, Ramchandra Tiruvalam, Quian He, Nikolaos Dimitratos, Jose A. Lopez-Sanchez, David J. Morgan, Albert F. Carley, Jennifer K. Edwards, Christopher J. Kiely and Graham J. Hutchings
Catalysis Science & Technology 2013 vol. 3(Issue 2) pp:308-317
Publication Date(Web):16 May 2012
DOI:10.1039/C2CY20234D
We have investigated the effect of heat treatment in air on Au–Pd nanoparticles supported on titania and activated carbon prepared via the immobilisation of PVA-stabilised alloy nanoparticles. The catalytic activity of the gold–palladium nanoparticles was affected by both metal and PVA loading, as well as the degree of interaction of the nanoparticles with the support. The turnover frequency numbers for benzyl alcohol and hydrogen peroxide synthesis were also sensitive to the calcination procedure employed and find a doubling of catalytic activity when using activated carbon as opposed to TiO2 as the support material. These results illustrate the importance of understanding the precise metal–support interaction of catalyst systems designed for benzyl alcohol oxidation and hydrogen peroxide synthesis.
Co-reporter:Peter J. Miedziak, Simon A. Kondrat, Noreen Sajjad, Gavin M. King, Mark Douthwaite, Greg Shaw, Gemma L. Brett, Jennifer K. Edwards, David J. Morgan, Ghulam Hussain and Graham J. Hutchings
Catalysis Science & Technology 2013 vol. 3(Issue 11) pp:2910-2917
Publication Date(Web):09 May 2013
DOI:10.1039/C3CY00263B
We have investigated the optimisation of the catalytic parameters for the preparation of catalysts by the simple mixing and thermal treatment of a support and metal acetate precursors. We have studied the effect of metal ratio and metal loading to produce a catalyst which has the optimum activity for a variety of reactions including benzyl alcohol oxidation, glycerol oxidation and the direct synthesis of hydrogen peroxide. We have demonstrated the high activity of these catalyst's for a variety of substrates and performed XPS and XRD studies on the catalysts to help elucidate the origin of the catalysts improved activity.
Co-reporter:Junting Feng, Chao Ma, Peter J. Miedziak, Jennifer K. Edwards, Gemma L. Brett, Dianqing Li, Yiyun Du, David J. Morgan and Graham J. Hutchings
Dalton Transactions 2013 vol. 42(Issue 40) pp:14498-14508
Publication Date(Web):19 Aug 2013
DOI:10.1039/C3DT51855H
Au–Pd nanoalloys supported on Mg–Al mixed metal oxides prepared using sol-immobilisation are found to be highly efficient and reusable catalysts for the solvent-free oxidation of benzyl alcohol using molecular oxygen under low pressure. When using this support alloying Pd with Au resulted in an increase in both activity and selectivity to benzaldehyde and moreover an improved resistance to catalyst deactivation compared with the monometallic Pd and Au catalysts. The turnover number for the Au/Pd 1:1 molar ratio catalyst achieved 13000 after 240 min and the selectivity to benzaldehyde was maintained at 93%; this high catalytic activity can be retained in full after three successive uses. The ensemble and electronic effect of Au–Pd nanoalloys were studied by IR spectroscopy using CO chemisorption, XPS and HRTEM. Moreover, the bifunctional nature of the acid–base MgAl-MMO support was found to be important as the acid sites are considered to be responsible for the improvement of catalytic activity; while, the basic sites gave rise to high selectivity. A possible mechanism with Au–Pd nanoparticles as the active sites has been proposed, illustrating that the oxidation of benzyl alcohol can proceed through the cooperation between the Au–Pd nanoalloys and the base/acid sites on the surface of the support.
Co-reporter:Virginie Peneau, Qian He, Gregory Shaw, Simon A. Kondrat, Thomas E. Davies, Peter Miedziak, Michael Forde, Nikolaos Dimitratos, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 26) pp:10636-10644
Publication Date(Web):02 Apr 2013
DOI:10.1039/C3CP50361E
Supported nano-alloys have been prepared using the sol-immobilisation method for two bimetallic combinations, namely gold–platinum and palladium–platinum, using activated carbon and titania as supports. Some of the materials were prepared using a method where both metals are simultaneously reduced, thereby leading to homogeneous alloys being formed. In addition, sequential reduction of the metal combinations has also been investigated to facilitate the formation of core–shell structures. The materials have been characterized using X-ray photoelectron spectroscopy and aberration-corrected scanning transmission electron microscopy. The supported nanoparticles have been tested for a two selective oxidation reactions, namely the oxidation of toluene and benzyl alcohol using tertiary butyl hydroperoxide at 80 °C, in order to elucidate any potential structure–activity relationships.
Co-reporter:Ewa Nowicka, Jan P. Hofmann, Stewart F. Parker, Meenakshisundaram Sankar, Giacomo M. Lari, Simon A. Kondrat, David W. Knight, Donald Bethell, Bert M. Weckhuysen and Graham J. Hutchings
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 29) pp:12147-12155
Publication Date(Web):02 May 2013
DOI:10.1039/C3CP50710F
In the solvent free oxidation of benzyl alcohol, using supported gold–palladium nanoalloys, toluene is often one of major by-products and it is formed by the disproportionation of benzyl alcohol. Gold–palladium catalysts on acidic supports promote both the disproportionation of benzyl alcohol and oxidative dehydrogenation to form benzaldehyde. Basic supports completely switch off disproportionation and the gold–palladium nanoparticles catalyse the oxidative dehydrogenation reaction exclusively. In an attempt to provide further details on the course of these reactions, we have utilized in situ ATR-IR, in situ DRIFT and inelastic neutron scattering spectroscopic methods, and in this article we present the results of these in situ spectroscopic studies.
Co-reporter:Dr. Gemma L. Brett;Dr. Peter J. Miedziak;Dr. Qian He; David W. Knight;Dr. Jennifer K. Edwards;Dr. Stuart H. Taylor; Christopher J. Kiely; Graham J. Hutchings
ChemSusChem 2013 Volume 6( Issue 10) pp:1952-1958
Publication Date(Web):
DOI:10.1002/cssc.201300420
Abstract
The oxidation of 1,4-butanediol and butyrolactone have been investigated by using supported gold, palladium and gold–palladium nanoparticles. The products of such reactions are valuable chemical intermediates and, for example, can present a viable pathway for the sustainable production of polymers. If both gold and palladium were present, a significant synergistic effect on the selective formation of dimethyl succinate was observed. The support played a significant role in the reaction, with magnesium hydroxide leading to the highest yield of dimethyl succinate. Based on structural characterisation of the fresh and used catalysts, it was determined that small gold–palladium nanoalloys supported on a basic Mg(OH)2 support provided the best catalysts for this reaction.
Co-reporter:Dr. Mohd Hasbi AbRahim;Dr. Michael M. Forde;Dr. Robert L. Jenkins;Dr. Ceri Hammond;Dr. Qian He;Dr. Nikolaos Dimitratos;Dr. Jose Antonio Lopez-Sanchez;Dr. Albert F. Carley;Dr. Stuart H. Taylor;Dr. David J. Willock;Dr. Damien M. Murphy; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie 2013 Volume 125( Issue 4) pp:1318-1322
Publication Date(Web):
DOI:10.1002/ange.201207717
Co-reporter:Dr. Mohd Hasbi AbRahim;Dr. Michael M. Forde;Dr. Robert L. Jenkins;Dr. Ceri Hammond;Dr. Qian He;Dr. Nikolaos Dimitratos;Dr. Jose Antonio Lopez-Sanchez;Dr. Albert F. Carley;Dr. Stuart H. Taylor;Dr. David J. Willock;Dr. Damien M. Murphy; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie International Edition 2013 Volume 52( Issue 4) pp:1280-1284
Publication Date(Web):
DOI:10.1002/anie.201207717
Co-reporter:Meenakshisundaram Sankar, Nikolaos Dimitratos, Peter J. Miedziak, Peter P. Wells, Christopher J. Kiely and Graham J. Hutchings
Chemical Society Reviews 2012 vol. 41(Issue 24) pp:8099-8139
Publication Date(Web):23 Oct 2012
DOI:10.1039/C2CS35296F
This Critical Review provides an overview of the recent developments in the synthesis and characterization of bimetallic nanoparticles. Initially the review follows a materials science perspective on preparing bimetallic nanoparticles with designer morphologies, after which the emphasis shifts towards recent developments in using these bimetallic particles for catalysing either oxidation or reduction. In the final part of this review we present an overview of the utilization of bimetallic catalyst systems for the transformation of bio-renewable substrates and reactions related to the realization of a bio-refinery. Because of the sheer number of examples of transformations in this area, a few key examples, namely selective oxidation, hydrogenation/hydrogenolysis and reforming of biomass derived molecules, have been chosen for this review. Reports of bimetallic catalysts being used for the aforementioned transformations are critically analysed and the potential for exploiting such bimetallic catalysts have also been highlighted. A specific objective of this review article is to motivate researchers to synthesize some of the “designer” bimetallic catalysts with specific nanostructures, inspired from recent advances in the area of materials chemistry, and to utilize them for the transformation of biomass derived materials that are very complex and pose different challenges compared to those of simple organic molecules. We consider that supported bimetallic nanoparticles have an important role to play as catalysts in our quest for a more green and sustainable society.
Co-reporter:Simon A. Kondrat, Greg Shaw, Simon J. Freakley, Qian He, Joanna Hampton, Jennifer K. Edwards, Peter J. Miedziak, Thomas E. Davies, Albert F. Carley, Stuart H. Taylor, Christopher. J. Kiely and Graham J. Hutchings
Chemical Science 2012 vol. 3(Issue 10) pp:2965-2971
Publication Date(Web):16 Jul 2012
DOI:10.1039/C2SC20450A
We have prepared supported gold, palladium and gold–palladium bimetallic catalysts by the physical mixing of the acetate salts of the metals followed by a simple heat treatment. The use of the acetates as the metal precursor eliminates chloride from the catalyst preparation step. Extensive characterisation shows the formation of bimetallic alloy particles. These catalysts are extremely active for alcohol oxidations and the direct formation of hydrogen peroxide.
Co-reporter:Sivaram Pradhan, Rhys Lloyd, Jonathan K. Bartley, Donald Bethell, Stan Golunski, Robert L. Jenkins and Graham J. Hutchings
Chemical Science 2012 vol. 3(Issue 10) pp:2958-2964
Publication Date(Web):02 Jul 2012
DOI:10.1039/C2SC20683H
Zeolite catalysts have been evaluated for the gas-phase conversion of decane, to study new routes for upgrading intermediate-length straight chain hydrocarbons. For a gas-feed of dilute n-decane in an inert carrier, at a contact time of 4 s, the initial activity of ZSM-5 and Ga/ZSM-5 was consistently high (>95% conversion) over the temperature range 300–460 °C. The parent zeolite produced almost equal yields of cracked hydrocarbons and aromatics, while the Ga-modified zeolite produced predominantly BTX and other heavier aromatics. This difference in product distribution is consistent with the short-chain alkanes formed within the internal pore structure of the zeolite being intermediates in a Cyclar-type aromatisation mechanism, while the direct conversion of decane to heavy (C10–C15) aromatics occurs at the unconstrained external acid sites. Under aerobic conditions, the rate of CO formation was negligible and CO2 was barely detectable over either the parent or the Ga-modified zeolite, even though all the O2 was consumed. The ability of Ga/ZSM-5 to catalyse selective oxidation, of the H2 released during the dehydrogenation steps, thus provides the prospect of the aromatisation reaction being operated autothermally. Although the external sites are preferentially blocked by carbon retention, rapid deactivation did not occur until after 65 h on line (under either anaerobic or aerobic conditions) when blocking of the internal pore structure became limiting.
Co-reporter:Gemma L. Brett, Peter J. Miedziak, Nikolaos Dimitratos, Jose A. Lopez-Sanchez, Nicholas F. Dummer, Ramchandra Tiruvalam, Christopher J. Kiely, David W. Knight, Stuart H. Taylor, David J. Morgan, Albert F. Carley and Graham J. Hutchings
Catalysis Science & Technology 2012 vol. 2(Issue 1) pp:97-104
Publication Date(Web):31 Aug 2011
DOI:10.1039/C1CY00254F
The oxidative esterification of 1,2-propanediol to methyl lactate and methyl pyruvate was investigated using gold and gold palladium nanoparticles supported on a variety of supports. Methyl lactate can be used in cosmetics and personal care products, whereas methyl pyruvate is useful in the treatment of diseases of the nervous system. We show that gold-palladium alloy catalysts can be very effective for the oxidative esterification of 1,2-propanediol to methyl lactate and methyl pyruvate. Five supports, titania, carbon, silica, iron oxide and ceria are contrasted. The addition of palladium to gold significantly enhances the activity and retains the high selectivity to methyl lactate using O2 as oxidant. Using ceria as support a significant improvement in the selectivity to methyl lactate was observed, whereas using silica as support high selectivity to methyl pyruvate was achieved. The use of colloidal methods and the effect of support and Au/Pd molar ratio are discussed.
Co-reporter:Marco Conte, Jose A. Lopez-Sanchez, Qian He, David J. Morgan, Yulia Ryabenkova, Jonathan K. Bartley, Albert F. Carley, Stuart H. Taylor, Christopher J. Kiely, Karim Khalid and Graham J. Hutchings
Catalysis Science & Technology 2012 vol. 2(Issue 1) pp:105-112
Publication Date(Web):27 Oct 2011
DOI:10.1039/C1CY00299F
Catalysts comprising zeolite ZSM-5 impregnated with precious metals including Ag, Cu, Ni, Pd, Ir and Ru, have been tested for the methanol to hydrocarbons reaction in a continuous flow fixed bed reactor. Comparison with the activity of unmodified ZSM-5 showed that Ag, Cu and Ni enhanced the selectivity to C6–C11 aromatic products by a factor of two or higher. Moreover, Ag/ZSM-5 showed improved selectivity for the C6–C7 fraction of aromatic products. Ni/ZSM-5 was found to be selective to naphthalene, while Cu/ZSM-5 was selective for C9–C11 aromatic products. It was ascertained that all the impregnated metals were present as metal oxides in the starting materials. It is therefore proposed that the enhanced selectivity to aromatic products is due to the interaction of the acid sites of the zeolite with the basic sites of the metal oxide at the edge of the zeolite crystals, as well as the possible coordination of propene molecules formed during the reaction, that are likely to be the building blocks for the formation of aromatics.
Co-reporter:Marco Piccinini, Jennifer K. Edwards, Jacob A. Moulijn and Graham J. Hutchings
Catalysis Science & Technology 2012 vol. 2(Issue 9) pp:1908-1913
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2CY20282D
The direct synthesis of hydrogen peroxide has been studied using a highly active AuPd/C catalyst, where the activated carbon support has been pretreated with dilute HNO3 prior to metal deposition and consequently using standard reaction conditions this catalyst does not hydrogenate H2O2. The effect of reaction variables has been investigated on the synthesis and hydrogenation activity over this catalyst. The effect of H2/O2 molar ratio, temperature, total pressure and solvent composition has been studied and optimised conditions identified. The effect of these conditions on the hydrogenation activity was also evaluated; thereby permitting an optimal set of reaction conditions to be identified for both the synthesis of H2O2 and its hydrogenation/decomposition.
Co-reporter:Mohd Hasbi Ab Rahim, Qian He, Jose A. Lopez-Sanchez, Ceri Hammond, Nikolaos Dimitratos, Meenakshisundaram Sankar, Albert F. Carley, Christopher J. Kiely, David W. Knight and Graham J. Hutchings
Catalysis Science & Technology 2012 vol. 2(Issue 9) pp:1914-1924
Publication Date(Web):23 May 2012
DOI:10.1039/C2CY20288C
Supported Au–Pd nanoparticles are shown to be effective catalysts for the transformation of glycerol into glycerol carbonate. The reaction of glycerol with urea to form glycerol carbonate is a very attractive reaction that utilises two inexpensive and readily available raw materials in a chemical cycle that, overall, results in the chemical fixation of carbon dioxide. Previous reports are largely based on the utilisation of high concentrations of metal sulphates or oxides, which suffer from low intrinsic activity and selectivity and limited recoverability due to the dissolution of the catalyst in the reaction media. We now report that magnesium oxide is an excellent support for gold and bimetallic gold–palladium nanoparticles for this reaction. The preparation method and pre-treatment affect the catalytic performance and a colloidal preparation route produces the most active catalysts.
Co-reporter:Marco Conte, Xi Liu, Damien M. Murphy, Keith Whiston and Graham J. Hutchings
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 47) pp:16279-16285
Publication Date(Web):29 Oct 2012
DOI:10.1039/C2CP43363J
The liquid phase oxidation of cyclohexane was undertaken using Au/MgO and the reaction mechanism was investigated by means of continuous wave (CW) EPR spectroscopy employing the spin trapping technique. Activity tests aimed to determine the conversion and selectivity of Au/MgO catalyst showed that Au was capable of selectivity control to cyclohexanol formation up to 70%, but this was accompanied by a limited enhancement in conversion when compared with the reaction in the absence of catalyst. In contrast, when radical initiators were used, in combination with Au/MgO, an activity comparable to that observed in industrial processes at ca. 5% conversion was found, with retained high selectivity. By studying the free radical autoxidation of cyclohexane and the cyclohexyl hydroperoxide decomposition in the presence of spin traps, we show that Au nanoparticles are capable of an enhanced generation of cyclohexyl alkoxy radicals, and the role of Au is identified as a promoter of the catalytic autoxidation processes, therefore demonstrating that the reaction proceeds via a radical chain mechanism.
Co-reporter:Marco Conte, Bin Xu, Thomas E. Davies, Jonathan K. Bartley, Albert F. Carley, Stuart H. Taylor, Kharim Khalid, Graham J. Hutchings
Microporous and Mesoporous Materials 2012 Volume 164() pp:207-213
Publication Date(Web):1 December 2012
DOI:10.1016/j.micromeso.2012.05.001
Composite co-crystalline zeolites ZSM-5/11 were synthesised and tested in the methanol to hydrocarbons reaction. The composite materials consist of crystals with intergrowth between the MFI framework typical of zeolite ZSM-5 and the MEL framework of zeolite ZSM-11 and displayed selectivities to propene of up to 50%, and a relative increase over ZSM-5 of ca. 80% under the same reaction conditions. Comparison with the activity of commercial ZSM-5 permits the construction of a model that correlates the structure of these materials to the final selectivity. It is proposed that the enhanced selectivity is due to changes in the MFI/MEL contribution to the final structure.Graphical abstractComposite co-crystalline zeolites ZSM-5/11 were effective catalysts in the synthesis of propene using methanol as a substrate. The enhanced selectivity to propene is due to changes in the zeolite framework as a consequence of crystal intergrowth.Highlights► Synthesis of co-crystalline zeolite ZSM-5/11. ► ZSM-5/11 consists of crystals with intergrowth between the MFI of zeolite ZSM-5 and the MEL framework of ZSM-11. ► Enhanced selectivity to propene is obtained in the methanol to hydrocarbons reaction. ► Crystals with changes in the MFI/MEL contribution to the final structure are obtained.
Co-reporter:Hamed Alshammari;Dr. Peter J. Miedziak;Dr. Salem Bawaked; David W. Knight ; Graham J. Hutchings
ChemCatChem 2012 Volume 4( Issue 10) pp:1565-1571
Publication Date(Web):
DOI:10.1002/cctc.201200273
Abstract
We report the solvent-free oxidation of 1-hexene with air by using supported gold catalysts with a catalytic amount of tert-butyl hydroperoxide (TBHP) as initiator. We confirm that gold supported on graphite is an effective catalyst for such oxidations and that graphite was the preferred support. Preparation of catalysts using modified sol-immobilisation was found to be effective, particularly when the PVA stabiliser was removed by a solvent treatment prior to the reaction.
Co-reporter:Dr. Ceri Hammond;Dr. Robert L. Jenkins;Dr. Nikolaos Dimitratos;Dr. Jose Antonio Lopez-Sanchez;Dr. Mohd Hasbi abRahim;Dr. Michael M. Forde;Adam Thetford;Dr. Damien M. Murphy;Dr. Henk Hagen;Dr. Eric E. Stangl; Jacob M. Moulijn;Dr. Stuart H. Taylor;Dr. David J. Willock; Graham J. Hutchings
Chemistry - A European Journal 2012 Volume 18( Issue 49) pp:15735-15745
Publication Date(Web):
DOI:10.1002/chem.201202802
Abstract
The partial oxidation of methane to methanol presents one of the most challenging targets in catalysis. Although this is the focus of much research, until recently, approaches had proceeded at low catalytic rates (<10 h−1), not resulted in a closed catalytic cycle, or were unable to produce methanol with a reasonable selectivity. Recent research has demonstrated, however, that a system composed of an iron- and copper-containing zeolite is able to catalytically convert methane to methanol with turnover frequencies (TOFs) of over 14 000 h−1 by using H2O2 as terminal oxidant. However, the precise roles of the catalyst and the full mechanistic cycle remain unclear. We hereby report a systematic study of the kinetic parameters and mechanistic features of the process, and present a reaction network consisting of the activation of methane, the formation of an activated hydroperoxy species, and the by-production of hydroxyl radicals. The catalytic system in question results in a low-energy methane activation route, and allows selective C1-oxidation to proceed under intrinsically mild reaction conditions.
Co-reporter:Dr. Ceri Hammond;Dr. Robert L. Jenkins;Dr. Nikolaos Dimitratos;Dr. Jose Antonio Lopez-Sanchez;Dr. Mohd Hasbi abRahim;Dr. Michael M. Forde;Adam Thetford;Dr. Damien M. Murphy;Dr. Henk Hagen;Dr. Eric E. Stangl; Jacob M. Moulijn;Dr. Stuart H. Taylor;Dr. David J. Willock; Graham J. Hutchings
Chemistry - A European Journal 2012 Volume 18( Issue 49) pp:
Publication Date(Web):
DOI:10.1002/chem.201290208
Co-reporter:Dr. Ceri Hammond;Michael M. Forde;Dr. Mohd Hasbi AbRahim;Adam Thetford;Qian He;Dr. Robert L. Jenkins;Dr. Nikolaos Dimitratos;Dr. Jose A. Lopez-Sanchez;Dr. Nicholas F. Dummer;Dr. Damien M. Murphy;Dr. Albert F. Carley;Dr. Stuart H. Taylor;Dr. David J. Willock;Dr. Eric E. Stangl;Dr. Joo Kang;Dr. Henk Hagen; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie International Edition 2012 Volume 51( Issue 21) pp:5129-5133
Publication Date(Web):
DOI:10.1002/anie.201108706
Co-reporter:Dr. Ceri Hammond;Michael M. Forde;Dr. Mohd Hasbi AbRahim;Adam Thetford;Qian He;Dr. Robert L. Jenkins;Dr. Nikolaos Dimitratos;Dr. Jose A. Lopez-Sanchez;Dr. Nicholas F. Dummer;Dr. Damien M. Murphy;Dr. Albert F. Carley;Dr. Stuart H. Taylor;Dr. David J. Willock;Dr. Eric E. Stangl;Dr. Joo Kang;Dr. Henk Hagen; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie 2012 Volume 124( Issue 21) pp:5219-5223
Publication Date(Web):
DOI:10.1002/ange.201108706
Co-reporter:Mohd Izham binSaiman;Gemma L. Brett;Ramchra Tiruvalam;Michael M. Forde;Kate Sharples;Adam Thetford;Dr. Robert L. Jenkins;Dr. Nikolaos Dimitratos;Dr. Jose A. Lopez-Sanchez;Dr. Damien M. Murphy; Donald Bethell;Dr. David J. Willock;Dr. Stuart H. Taylor; David W. Knight; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie 2012 Volume 124( Issue 24) pp:6083-6087
Publication Date(Web):
DOI:10.1002/ange.201201059
Co-reporter:Mosaed Alhumaimess;Dr. Zhongjie Lin;Weihao Weng;Dr. Nikolaos Dimitratos;Dr. Nicholas F. Dummer;Dr. Stuart H. Taylor;Dr. Jonathan K. Bartley; Christopher J. Kiely; Graham J. Hutchings
ChemSusChem 2012 Volume 5( Issue 1) pp:125-131
Publication Date(Web):
DOI:10.1002/cssc.201100374
Abstract
The efficacy of using cerium oxide foams as a support for Au nanoparticles and subsequent use as oxidation catalysts have been investigated. These were synthesized using L-asparagine to produce a cerium coordination polymer foam, which was calcined to give the oxide foam. Au nanoparticles were supported on the CeO2 foams using a sol-immobilization method. The activity of the Au/foamCeO2 for solvent-free benzyl alcohol oxidation was superior to standard Au/CeO2 catalysts, and the activity was found to be dependent on the crystallization time of the precursor foam. A crystallization time of 4 h was found to produce the most active catalyst, which retained activity and a high selectivity to benzaldehyde (ca. 96 %) when re-used and this is related to the structure of the material. The high activity is attributed to the greater lability of surface oxygen in the support compared with commercial CeO2 materials.
Co-reporter:Mohd Izham binSaiman;Gemma L. Brett;Ramchra Tiruvalam;Michael M. Forde;Kate Sharples;Adam Thetford;Dr. Robert L. Jenkins;Dr. Nikolaos Dimitratos;Dr. Jose A. Lopez-Sanchez;Dr. Damien M. Murphy; Donald Bethell;Dr. David J. Willock;Dr. Stuart H. Taylor; David W. Knight; Christopher J. Kiely; Graham J. Hutchings
Angewandte Chemie International Edition 2012 Volume 51( Issue 24) pp:5981-5985
Publication Date(Web):
DOI:10.1002/anie.201201059
Co-reporter:Edwin N. Ntainjua;Simon J. Freakley
Topics in Catalysis 2012 Volume 55( Issue 11-13) pp:718-722
Publication Date(Web):2012 August
DOI:10.1007/s11244-012-9866-3
The direct synthesis of hydrogen peroxide is investigated using ruthenium containing catalysts. Ruthenium is not soluble in Au but forms alloys with palladium. We have therefore investigated Ru–Au, Ru–Pd as well as trimetallic formulations supported on titania. The addition of ruthenium enhances the direct synthesis activity for all the catalysts studied and the effect is dependent on the amount of ruthenium added. The calcination conditions are shown to affect both activity and reusability.
Co-reporter:Meenakshisundaram Sankar, Qian He, Moataz Morad, James Pritchard, Simon J. Freakley, Jennifer K. Edwards, Stuart H. Taylor, David J. Morgan, Albert F. Carley, David W. Knight, Christopher J. Kiely, and Graham J. Hutchings
ACS Nano 2012 Volume 6(Issue 8) pp:6600
Publication Date(Web):July 7, 2012
DOI:10.1021/nn302299e
We report a convenient excess anion modification and post-reduction step to the impregnation method which permits the reproducible preparation of supported bimetallic AuPd nanoparticles having a tight particle size distribution comparable to that found for sol-immobilization materials but without the complication of ligands adsorbed on the particle surface. The advantageous features of the modified impregnation materials compared to those made by conventional impregnation include a smaller average particle size, an optimized random alloy composition, and improved compositional uniformity from particle-to-particle resulting in higher activity and stability compared to the catalysts prepared using both conventional impregnation and sol immobilization methods. Detailed STEM combined with EDX analyses of individual particles have revealed that an increase in anion concentration increases the gold content of individual particles in the resultant catalyst, thus providing a method to control/tune the composition of the nanoalloy particles. The improved activity and stability characteristics of these new catalysts are demonstrated using (i) the direct synthesis of hydrogen peroxide and (ii) the solvent-free aerobic oxidation of benzyl alcohol as case studies.Keywords: anion-excess; direct synthesis of hydrogen peroxide; gold−palladium; ligand-free synthesis; modified impregnation
Co-reporter:Nikolaos Dimitratos;Jennifer K. Edwards
Applied Petrochemical Research 2012 Volume 2( Issue 1-2) pp:7-14
Publication Date(Web):2012 September
DOI:10.1007/s13203-012-0011-9
In recent years there has been a general realisation that supported gold and gold bimetallic nanoparticles can be very effective for a broad range of redox reactions. In this paper we review the preparation of gold palladium nanoparticles using a sol-immobilisation methodology and show their effectiveness for the oxidation of benzyl alcohol and the direct synthesis of hydrogen peroxide.
Co-reporter:Ren Su, Ramchandra Tiruvalam, Qian He, Nikolaos Dimitratos, Lokesh Kesavan, Ceri Hammond, Jose Antonio Lopez-Sanchez, Ralf Bechstein, Christopher J. Kiely, Graham J. Hutchings, and Flemming Besenbacher
ACS Nano 2012 Volume 6(Issue 7) pp:6284
Publication Date(Web):June 4, 2012
DOI:10.1021/nn301718v
Noble metal nanoparticles (Au, Pd, Au–Pd alloys) with a narrow size distribution supported on nanocrystalline TiO2 (M/TiO2) have been synthesized via a sol-immobilization route. The effect of metal identity and size on the photocatalytic performance of M/TiO2 has been systematically investigated using phenol as a probe molecule. A different phenol degradation pathway was observed when using M/TiO2 catalysts as compared to pristine TiO2. We propose a mechanism to illustrate how the noble metal nanoparticles enhance the efficiency of phenol decomposition based on photoreduction of p-benzoquinone under anaerobic conditions. Our results suggest that the metal nanoparticles not only play a role in capturing photogenerated electrons, but are strongly involved in the photocatalytic reaction mechanism. The analysis of the reaction intermediates allows us to conclude that on M/TiO2 undesired redox reactions that consume photogenerated radicals are effectively suppressed. The analysis of the final products shows that the reusability performance of the catalyst is largely dependent on the pretreatment of the catalyst and the identity of the metal nanoparticle. Interestingly, the as-prepared Pd and Au–Pd decorated TiO2 materials exhibit excellent long-term photoactivity, in which ∼90% of the phenol can be fully decomposed to CO2 in each cycle.Keywords: Au; Au−Pd alloys; CO2 conversion; Pd; phenol decomposition mechanism; photocatalysis; reusability; TiO2
Co-reporter:Salem Bawaked, Nicholas F. Dummer, Donald Bethell, David W. Knight and Graham J. Hutchings
Green Chemistry 2011 vol. 13(Issue 1) pp:127-134
Publication Date(Web):12 Nov 2010
DOI:10.1039/C0GC00550A
Selective oxidation is of immense importance in the synthesis of chemical intermediates and the epoxidation of alkenes by the electrophilic addition of oxygen to a carbon–carbon double bond remains one of the most significant challenges in oxidation catalysis. Although molecular oxygen is the most environmentally benign oxidant in many cases, far more reactive forms of oxygen are required to achieve reaction, and this can lead to by-products with a heavy environmental burden with respect to their disposal. We show that gold supported on graphite is a very effective catalyst for the epoxidation of cis-cyclooctene as long as catalytic amounts of a hydroperoxy species are present at the start of the reaction. Using mild solvent-free conditions the hydroperoxy initiator persists in solution for only a few minutes, being initially adsorbed on the catalyst surface. Subsequently, it decomposes to establish a reactive species that can propagate the selective oxidation process we observe. The observation of an induction period may in part be due to the adsorption of the radical initiator blocking surface sites as well as the establishment of the reactive species. We confirm that graphite is the best support and that tert-butyl hydroperoxide is the preferred initiator. We report extensive studies concerning the reusability of the gold/graphite catalyst as catalyst reusability is a key feature of green chemistry. The catalyst is found to be inhibited by the epoxide product but we demonstrate the effect of this is negligible for reused catalysts over a long reaction time.
Co-reporter:Salem Bawaked, Qian He, Nicholas F. Dummer, Albert F. Carley, David W. Knight, Donald Bethell, Christopher J. Kiely and Graham J. Hutchings
Catalysis Science & Technology 2011 vol. 1(Issue 5) pp:747-759
Publication Date(Web):03 Jun 2011
DOI:10.1039/C1CY00122A
Oxidation is an important route for the activation of chemical feedstocks for the synthesis of chemical intermediates. Alkene epoxidation by the electrophilic addition of oxygen to a carbon–carbon double bond is a major challenge in oxidation catalysis. In particular it is important to use molecular oxygen as the oxidant to avoid the formation of reagent by-products. We report the oxidation with air using graphite-supported gold-palladium catalysts of two alkenes, cis-cyclooctene, which gives mainly the epoxide, and crotyl alcohol (trans-but-2-en-1-ol). With cyclooctene, the reaction requires catalytic amounts of t-butyl hydroperoxide. The Au–Pd ratio has a major effect on the conversion with very low activities being associated with Au:Pd ratios of ca. 4:1 and 1:4 by weight. The selectivity to the epoxide is not affected by the Au:Pd ratio. With crotyl alcohol, t-butyl hydroperoxide was not required for activity. In the absence of Pd, crotonaldehyde was formed, but the introduction of Pd leads to an isomerisation pathway to 3-buten-1-ol being favoured over epoxidation and crotonaldehyde was a minor product.
Co-reporter:Charlotte L. Bracey, Albert F. Carley, Jennifer K. Edwards, Peter R. Ellis and Graham J. Hutchings
Catalysis Science & Technology 2011 vol. 1(Issue 1) pp:76-85
Publication Date(Web):31 Jan 2011
DOI:10.1039/C0CY00003E
The synthesis and catalytic application of supported CuAu is discussed. Different thermal treatments of a dried precursor of copper nitrate and tetrachloroauric acid on silica lead to catalysts with significantly different structures and properties. Direct calcination gives a catalyst which contains very large gold ensembles with minimal interaction with the copper present. Hydrogen reduction of the dried precursor leads to the formation of copper–gold alloy nanoparticles. Subsequent high temperature calcination de-alloys the copper from the gold to a significant extent. The presence of gold stabilises the formation of Cu+ on this catalyst. The activity and selectivity observed in the oxidation of propene with molecular oxygen, with or without co-fed hydrogen, depends on the pre-treatment, reaction conditions and the ratio of copper to gold in the catalyst. A number of different catalytic active sites are identified and discussed.
Co-reporter:Albert F. Carley, David J. Morgan, Nianxue Song, M. Wyn Roberts, Stuart H. Taylor, Jonathan K. Bartley, David J. Willock, Kara L. Howard and Graham J. Hutchings
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 7) pp:2528-2538
Publication Date(Web):10 Dec 2010
DOI:10.1039/C0CP01852J
The oxidation of CO by Au/Fe2O3 and Au/ZnO catalysts is compared in the very early stages of the reaction using a temporal analysis of products (TAP) reactor. For Au/Fe2O3 pre-dosing the catalyst with 18O labelled water gives an unexpected evolution order for the labelled CO2 product with the C18O2 emerging first, whereas no temporal differentiation is found for Au/ZnO. High pressure XPS experiments are then used to show that CO bond cleavage does occur for model catalysts consisting of Au particles deposited on iron oxide films but not when deposited on ZnO films. DFT calculations, show that this observation requires carbon monoxide to dissociate in such a way that cleavage of the CO bond occurs along with dynamically co-adsorbed oxygen so that the overall process of Au oxidation and CO dissociation is energetically favourable. Our results show that for Au/Fe2O3 there is a pathway for CO oxidation that involves atomic C and O surface species which operates along side the bicarbonate mechanism that is widely discussed in the literature. However, this minor pathway is absent for Au/ZnO.
Co-reporter:Ceri Hammond, Jose A. Lopez-Sanchez, Mohd Hasbi Ab Rahim, Nikolaos Dimitratos, Robert L. Jenkins, Albert F. Carley, Qian He, Christopher J. Kiely, David W. Knight and Graham J. Hutchings
Dalton Transactions 2011 vol. 40(Issue 15) pp:3927-3937
Publication Date(Web):21 Jan 2011
DOI:10.1039/C0DT01389G
The reaction of glycerol with urea to form glycerol carbonate is mostly reported in the patent literature and to date there have been very few fundamental studies of the reaction mechanism. Furthermore, most previous studies have involved homogeneous catalysts whereas the identification of heterogeneous catalysts for this reaction would be highly beneficial. This is a very attractive reaction that utilises two inexpensive and readily available raw materials in a chemical cycle that overall, results in the chemical fixation of CO2. This reaction also provides a route to up-grade waste glycerol produced in large quantities during the production of biodiesel. Previous reports are largely based on the utilisation of high concentrations of metal sulfates or oxides, which suffer from low intrinsic activity and selectivity. We have identified heterogeneous catalysts based on gallium, zinc, and gold supported on a range of oxides and the zeolite ZSM-5, which facilitate this reaction. The addition of each component to ZSM-5 leads to an increase in the reaction yield towards glycerol carbonate, but supported gold catalysts display the highest activity. For gold-based catalysts, MgO is the support of choice. Catalysts have been characterised by XRD, TEM, STEM and XPS, and the reaction has been studied with time-on-line analysis of products via a combination of FT-IR spectroscopy, HPLC, 13C NMR and GC-MS analysis to evaluate the reaction pathway. Our proposed mechanism suggests that glycerol carbonate forms via the cyclization of a 2,3-dihydroxypropyl carbamate and that a subsequent reaction of glycerol carbonate with urea yields the carbamate of glycerol carbonate. Stability and reactivity studies indicate that consecutive reactions of glycerol carbonate can limit the selectivity achieved and reaction conditions can be selected to avoid this. The effect of the catalyst in the proposed mechanism is discussed.
Co-reporter:Gemma L. Brett;Qian He;Ceri Hammond;Dr. Peter J. Miedziak;Dr. Nikolaos Dimitratos;Dr. Meenakshisundaram Sankar;Dr. Andrew A. Herzing;Dr. Marco Conte;Dr. Jose Antonio Lopez-Sanchez; Christopher J. Kiely; David W. Knight;Dr. Stuart H. Taylor; Graham J. Hutchings
Angewandte Chemie 2011 Volume 123( Issue 43) pp:10318-10321
Publication Date(Web):
DOI:10.1002/ange.201101772
Co-reporter:Dr. Meenakshisundaram Sankar;Ewa Nowicka;Ramchra Tiruvalam;Qian He;Dr. Stuart H. Taylor; Christopher J. Kiely; Donald Bethell; David W. Knight; Graham J. Hutchings
Chemistry - A European Journal 2011 Volume 17( Issue 23) pp:6524-6532
Publication Date(Web):
DOI:10.1002/chem.201003484
Abstract
In the solvent-free oxidation of benzyl alcohol to benzaldehyde using supported gold–palladium nanoparticles as catalysts, two pathways have been identified as the sources of the principal product, benzaldehyde. One is the direct catalytic oxidation of benzyl alcohol to benzaldehyde by O2, whereas the second is the disproportionation of two molecules of benzyl alcohol to give equal amounts of benzaldehyde and toluene. Herein we report that by changing the metal oxide used to support the metal–nanoparticles catalyst from titania or niobium oxide to magnesium oxide or zinc oxide, it is possible to switch off the disproportionation reaction and thereby completely stop the toluene formation. It has been observed that the presence of O2 increases the turnover number of this disproportionation reaction as compared to reactions in a helium atmosphere, implying that there are two catalytic pathways leading to toluene.
Co-reporter:Gemma L. Brett;Qian He;Ceri Hammond;Dr. Peter J. Miedziak;Dr. Nikolaos Dimitratos;Dr. Meenakshisundaram Sankar;Dr. Andrew A. Herzing;Dr. Marco Conte;Dr. Jose Antonio Lopez-Sanchez; Christopher J. Kiely; David W. Knight;Dr. Stuart H. Taylor; Graham J. Hutchings
Angewandte Chemie International Edition 2011 Volume 50( Issue 43) pp:10136-10139
Publication Date(Web):
DOI:10.1002/anie.201101772
Co-reporter:Mohd Izham bin Saiman;Lokesh Kesavan;Dan I. Enache;Ramchandra Tiruvalam;Robert L. Jenkins;Mohd Hasbi Ab Rahim;Nikolaos Dimitratos;Jose A. Lopez-Sanchez;Stuart H. Taylor;David W. Knight;Christopher J. Kiely
Science 2011 Volume 331(Issue 6014) pp:195-199
Publication Date(Web):14 Jan 2011
DOI:10.1126/science.1198458
A gold- and palladium-based catalyst can be used to oxidize toluene and form a commercially useful ester.
Co-reporter:James C. Pritchard, Qian He, Edwin N. Ntainjua, Marco Piccinini, Jennifer K. Edwards, Andrew. A. Herzing, Albert F. Carley, Jacob A. Moulijn, Christopher J. Kiely and Graham J. Hutchings
Green Chemistry 2010 vol. 12(Issue 5) pp:915-921
Publication Date(Web):01 Apr 2010
DOI:10.1039/B924472G
The direct synthesis of hydrogen peroxide from H2 and O2 offers the possibility of a new green production method for this important commodity chemical. Active catalysts for this reaction are typically prepared using an impregnation method and it is important to identify improvements in the preparation methodology that can result in more active catalysts that retain their stability. The effect of the precise procedure by which the metals are impregnated onto TiO2 and C supports during the preparation of supported Au–Pd catalysts has been investigated and it is shown that the two supports exhibit significant differences. The concentration of the solution of the mixed aqueous solution of HAuCl4 and PdCl2 immediately prior to the initial drying step has a profound effect on the structure and activity of the TiO2-supported catalysts. TiO2-supported catalysts prepared using impregnation with the minimal amount of added water whilst ensuring that the catalyst is not formed into a paste (i.e. still contains ca. 1.5–2 ml of H2O) prior to drying at 110 °C exhibit very high activity (ca. 120 mol H2O2 kgcat−1 h−1) which is equivalent to the corresponding carbon-supported catalyst. The presence of more water (ca. 2–28 ml) in the catalyst impregnation step prior to drying leads to a significant change in the particle size distribution and a bimodal distribution is observed for the TiO2-supported catalysts. These catalysts also show a change in the nature of the Au and Pd nanoparticles. Unfortunately, TiO2-supported catalysts prepared in this manner are not stable on re-use. However, catalysts prepared using a similar method, but with the removal of ca. 75% of the initial H2O ensuring that a paste is formed prior to drying, are found to be fully re-usable. In contrast, for carbon-supported catalysts dilution of the Au and Pd compounds during the initial impregnation step, coupled with subsequent removal of water to form paste with varying water content, did not affect the activity and these catalysts could be re-used without loss of catalyst performance. The effect of the catalyst structure on activity and re-usability is discussed.
Co-reporter:Marco Piccinini, Edwin Ntainjua N., Jennifer K. Edwards, Albert F. Carley, Jacob A. Moulijn and Graham J. Hutchings
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 10) pp:2488-2492
Publication Date(Web):21 Jan 2010
DOI:10.1039/B921815G
The direct synthesis of hydrogen peroxide from H2 and O2 has been studied using a high activity AuPd/TiO2 catalyst. In particular, the effect of variation in the reaction conditions on the productivity of hydrogen peroxide formation is investigated in detail. The effect of H2/O2 molar ratio, temperature, total pressure and solvent composition has been studied and optimised conditions identified. In addition, the effect of carrying out the synthesis reaction in the presence of hydrogen peroxide is investigated and the competing reactions of hydrogen peroxide formation, decomposition and hydrogenation are discussed and optimal operating conditions are identified.
Co-reporter:Raja AlOtaibi;Weihao Weng;JonathanK. Bartley Dr.;NicholasF. Dummer Dr.;ChristopherJ. Kiely ;GrahamJ. Hutchings
ChemCatChem 2010 Volume 2( Issue 4) pp:443-452
Publication Date(Web):
DOI:10.1002/cctc.200900274
Abstract
Vanadium phosphate materials, based on a VOHPO4⋅0.5 H2O precursor and with (VO)2P2O7 as an active high-temperature phase, are used as catalysts for the oxidation of alkanes. VOHPO4⋅0.5 H2O is prepared from VOPO4⋅2 H2O using 1-octanol, 3-octanol, 2-butanol, or 2-methyl-1-propanol as both solvent and reducing agent. With 1-octanol, the reaction temperature was found to be crucial in obtaining a high yield of the precursor phase. At temperatures of 160 °C or greater, a solution containing V4+ ions formed in preference to VOHPO4⋅0.5 H2O. However, VOHPO4⋅0.5 H2O formation can be achieved above 160 °C by carrying out the reduction process in the presence of a small amount of vanadium phosphate material, which effectively acts as a templating seed. The use of this seeding concept is shown to have a dramatic effect on the morphology of the final activated catalyst. In contrast, when 3-octanol is used, solely VO(H2PO4)2 is generated, except in the presence of a vanadium phosphate seed where significant amounts of VOHPO4⋅0.5 H2O can also be formed. Furthermore, VO(H2PO4)2 can be transformed to VOHPO4⋅0.5 H2O by heating at reflux with an alcohol in the presence of VOHPO4⋅0.5 H2O precursor seeds. The findings reported herein show that both the phase composition and morphology of vanadium phosphates can be influenced by the use of seeds during the preparation process.
Co-reporter:James Pritchard, Lokesh Kesavan, Marco Piccinini, Qian He, Ramchandra Tiruvalam, Nikolaos Dimitratos, Jose A. Lopez-Sanchez, Albert F. Carley, Jennifer K. Edwards, Christopher J. Kiely, and Graham J. Hutchings
Langmuir 2010 Volume 26(Issue 21) pp:16568-16577
Publication Date(Web):May 12, 2010
DOI:10.1021/la101597q
We report the preparation of Au−Pd nanocrystalline catalysts supported on activated carbon prepared via a sol-immobilization technique and explore their use for the direct synthesis of hydrogen peroxide and the oxidation of benzyl alcohol. In particular, we examine the synthesis of a systematic set of Au−Pd colloidal nanoparticles having a range of Au/Pd ratios. The catalysts have been structurally characterized using a combination of UV−visible spectroscopy, transmission electron microscopy, STEM HAADF/XEDS, and X-ray photoelectron spectroscopy. The Au−Pd nanoparticles are found in the majority of cases to be homogeneous alloys, although some variation is observed in the AuPd composition at high Pd/Au ratios. The optimum performance for the synthesis of hydrogen peroxide is observed for a catalyst having a Au/Pd 1:2 molar ratio. However, the competing hydrogenation reaction of hydrogen peroxide increases with increasing Pd content, although Pd alone is less effective than when Au is also present. Investigation of the oxidation of benzyl alcohol using these materials also shows that the optimum selective oxidation to the aldehyde occurs for the Au/Pd 1:2 molar ratio catalyst. These measured activity trends are discussed in terms of the structure and composition of the supported Au−Pd nanoparticles.
Co-reporter:Jennifer K. Edwards;Benjamin Solsona;Edwin Ntainjua N;Albert F. Carley;Andrew A. Herzing;Christopher J. Kiely
Science 2009 Vol 323(5917) pp:1037-1041
Publication Date(Web):20 Feb 2009
DOI:10.1126/science.1168980
Abstract
Hydrogen peroxide (H2O2) is an important disinfectant and bleach and is currently manufactured from an indirect process involving sequential hydrogenation/oxidation of anthaquinones. However, a direct process in which H2 and O2 are reacted would be preferable. Unfortunately, catalysts for the direct synthesis of H2O2 are also effective for its subsequent decomposition, and this has limited their development. We show that acid pretreatment of a carbon support for gold-palladium alloy catalysts switches off the decomposition of H2O2. This treatment decreases the size of the alloy nanoparticles, and these smaller nanoparticles presumably decorate and inhibit the sites for the decomposition reaction. Hence, when used in the direct synthesis of H2O2, the acid-pretreated catalysts give high yields of H2O2 with hydrogen selectivities greater than 95%.
Co-reporter:Charlotte L. Bracey, Peter R. Ellis and Graham J. Hutchings
Chemical Society Reviews 2009 vol. 38(Issue 8) pp:2231-2243
Publication Date(Web):01 Jun 2009
DOI:10.1039/B817729P
The use of nanoalloys in catalysis is a rapidly expanding field. There has been immense interest in the use of supported gold nanoparticles as catalysts, and bimetallic catalysts containing gold in combination with other metals represents an emerging field of research. While bulk copper–gold alloys are well-known and, indeed, are much studied systems, bimetallic copper–gold nanoalloys have received relatively little attention. In this tutorial review we review the literature on bimetallic CuAu catalysts and present some options for their future development.
Co-reporter:Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, Sankar Meenakshisundaram, Jinto Manjaly Anthonykutty, Gemma Brett, Albert F. Carley, Stuart H. Taylor, David W. Knight and Graham J. Hutchings
Green Chemistry 2009 vol. 11(Issue 8) pp:1209-1216
Publication Date(Web):28 May 2009
DOI:10.1039/B823285G
The use of bio-renewable resources, such as glycerol, a by-product from bio-diesel manufacture, can provide a viable way to make valuable products using greener technology. In particular, glycerol can be reduced to give 1,2-propanediol that can then be selectively oxidised to lactate, which has immense potential as a monomer for the synthesis of biodegradable polymers. We show that gold-palladium alloy catalysts can be very effective for the selective oxidation of 1,2-propanediol to lactate. Two supports, TiO2 and carbon, and two preparation methods, wet impregnation and sol-immobilisation, are contrasted. The addition of palladium to gold significantly enhances the activity and retains the high selectivity to lactate using O2 as oxidant (we observe 96% lactate selectivity at 94% conversion). Use of hydrogen peroxide is also possible but lower activities are observed as a result of the reaction conditions, but in this case no marked enhancement is observed on addition of palladium to gold. Comparison of the activity for C3 alcohols shows that the reactivity decreases in the order: glycerol > 1,2-propanediol > 1,3-propanediol ∼ 1-propanol > 2-propanol. The use of a sol-immobilisation preparation method as compared to impregnation leads to alloy catalysts with the highest activity for lactate formation from the oxidation of 1,2-propanediol; the origins of these activity trends are discussed.
Co-reporter:Salem Bawaked, Nicholas F. Dummer, Nikolaos Dimitratos, Donald Bethell, Qian He, Christopher J. Kiely and Graham J. Hutchings
Green Chemistry 2009 vol. 11(Issue 7) pp:1037-1044
Publication Date(Web):28 Apr 2009
DOI:10.1039/B823286P
Oxidation is one of the major pathways for the synthesis of chemical intermediates. The epoxidation of alkenes by the electrophilic addition of oxygen to a carbon–carbon double bond remains one of the most significant challenges in oxidation. Of key importance is the use of oxygen as the oxidant, but in many cases more reactive, and less green, sources of oxygen are used. We report the solvent-free epoxidation of cyclooctene with air using supported gold catalysts with small amounts of a hydroperoxide. We identify the appropriate reaction conditions to maximize the selectivity of the epoxide. In the absence of a hydroperoxide initiator, using air at atmospheric pressure, no reaction is observed. Choice of the peroxide initiator is crucial and in the absence of a catalyst or a support the reaction of the alkene can be observed with di-t-butyl peroxide and t-butyl hydroperoxide (TBHP) only when high concentrations are used at high temperatures ≥ 80 °C, and TBHP was found to be the more selective to epoxide formation. In contrast, cumene hydroperoxide was highly reactive under all conditions evaluated. TBHP was selected for more detailed study. Use of graphite as a support was found to give the best combination of selectivity and conversion. In general the selectivity to the epoxide increased with reaction temperature from 60–80 °C and was highest at 80 °C. Other carbon supports, e.g. activated carbon, were found to be less effective. TiO2- and SiO2-supported Au catalysts were also selective for the epoxidation reaction and the general order of activity was: graphite > SiO2 > TiO2. The major by-product is the allylic alcohol and the reaction pathways to the epoxides and the allylic alcohol are discussed. Preparation of catalysts using a sol-immobilisation method significantly enhanced catalyst activity with retention of selectivity to the epoxide.
Co-reporter:Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, Jinto Manjaly Anthonykutty, Gemma Brett, Albert F. Carley, Ram Chandra Tiruvalam, Andrew A. Herzing, Christopher J. Kiely, David W. Knight and Graham J. Hutchings
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 25) pp:4952-4961
Publication Date(Web):17 Apr 2009
DOI:10.1039/B904317A
The use of bio-renewable resources for the generation of materials and chemicals continues to attract significant research attention. Glycerol, a by-product from biodiesel manufacture, is a highly functionalised renewable raw material, and in this paper the oxidation of glycerol in the presence of base using supported gold, palladium and gold–palladium alloys is described and discussed. Two supports, TiO2 and carbon, and two preparation methods, wet impregnation and sol-immobilisation, are compared and contrasted. For the monometallic catalysts prepared by impregnation similar activities are observed for Au and Pd, but the carbon-supported monometallic catalysts are more active than those on TiO2. Glycerate is the major product and lesser amounts of tartronate, glycolate, oxalate and formate are observed, suggesting a sequential oxidation pathway. Combining the gold and palladium as supported alloy nanocrystals leads to a significant enhancement in catalyst activity and the TiO2-supported catalysts are significantly more active for the impregnated catalysts. The use of a sol-immobilisation preparation method as compared to impregnation leads to the highest activity alloy catalysts and the origins of these activity trends are discussed.
Co-reporter:Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, David Morgan, Albert F. Carley, Ramchandra Tiruvalam, Christopher J. Kiely, Donald Bethell and Graham J. Hutchings
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 25) pp:5142-5153
Publication Date(Web):03 Apr 2009
DOI:10.1039/B900151B
We report the preparation of Au–Pd nanocrystalline catalysts supported on TiO2 and carbon prepared via a sol-immobilisation technique using three different preparation strategies; namely, simultaneous formation of the sols for both metals or initial formation of a seed sol of one of the metals followed by a separate step in which a coating sol of the second metal is added. The catalysts have been structurally characterised using a combination of transmission electron microscopy and X-ray photoelectron spectroscopy. The catalysts have been evaluated for the oxidation of benzyl alcohol under solvent-free conditions. The catalysts prepared using the sol immobilisation technique show higher activity when compared with catalysts prepared by impregnation, particularly as lower metal concentrations can be used. The Au–Pd catalysts were all more active than the corresponding monometallic supported Au or Pd catalysts. For 1 wt% Au–Pd/TiO2 the order of metal addition in the preparation was not observed to be significant with respect to selectivity or activity. However, the 1 wt% Au–Pd/carbon catalysts are more active but less selective to benzaldehyde than the TiO2-supported catalysts when compared at iso-conversion. Furthermore, for the carbon-supported catalyst the order of metal addition has a very marked affect on activity. The carbon-supported catalysts are also more significantly affected by heat treatment, e.g.calcination at 400 °C leads to the activity being decreased by an order of magnitude, whereas the TiO2-supported catalysts show a 50% decrease in activity. Toluene is observed as a by-product of the reaction and conditions have been identified that minimise its formation. It is proposed that toluene and benzaldehyde are formed by competing parallel reactions of the initial benzyl intermediate via an adsorbed benzylidene species that can either be hydrogenated or oxidised. Hence, conditions that maximise the availability of oxygen on the catalyst surface favour the synthesis of benzaldehyde.
Co-reporter:Graham J. Hutchings
Topics in Catalysis 2009 Volume 52( Issue 8) pp:982-987
Publication Date(Web):2009 July
DOI:10.1007/s11244-009-9248-7
Catalysts are intrinsically considered to be a key part of green and sustainable technology. However, many catalysts require the use of non-green methodologies during their manufacture. With the increased focus on green issues that is now emerging, it is essential that some attention is focussed on synthesing catalysts using green approaches. However, it is critical that any material prepared using these new green methods has to exhibit key advantages over current technology, such as significantly enhanced performance, if they are o be adopted in this competitive industry. Here the use of supercritical carbon dioxide as an anti-solvent is discussed and two examples, namely the synthesis of vanadium phosphate and ceria-supported gold catalysts, are described using supercritical carbon dioxide in the preparation. Both of these examples demonstrate significant advantages with respect to higher activity when compared with catalysts prepared using standard methods.
Co-reporter:Nikolaos Dimitratos;Jose Antonio Lopez-Sanchez
Topics in Catalysis 2009 Volume 52( Issue 3) pp:258-268
Publication Date(Web):2009 April
DOI:10.1007/s11244-008-9162-4
The use of renewable feedstocks, derived from biomass, for the chemical industry is discussed. The modern chemical industry is based around platform chemicals, e.g. ethene, propene, benzene and xylenes, which are readily derived from oil, and using these intermediates a broad range of finished products can be derived. While it is feasible that biomass can be converted to syngas and hence to existing key platform chemicals, this loses all of the chemical complexity that is inherent in bio-derived molecules. In this paper some of the options are considered and, in particular, the oxidation of glucose and glycerol using gold nanoparticles supported on carbon is described. We also contrast the oxidation of glycerol using supported gold and gold–palladium alloys prepared using an impregnation technique, since the gold–palladium alloys have been shown to be highly effective for the oxidation of alcohols and the synthesis of hydrogen peroxide.
Co-reporter:Edwin NtainjuaN. Dr.;Marco Piccinini;JamesC. Pritchard;Qian He;JenniferK. Edwards Dr.;AlbertF. Carley Dr.;JacobA. Moulijn ;ChristopherJ. Kiely ;GrahamJ. Hutchings
ChemCatChem 2009 Volume 1( Issue 4) pp:479-484
Publication Date(Web):
DOI:10.1002/cctc.200900171
Abstract
The effect of pretreating Au–Pd catalysts on MgO and C supports with aqueous bromide solution, prior to using them for the direct synthesis of hydrogen peroxide, has been investigated. These two supports were selected since the parent materials exhibit contrasting microstructures and activities. The carbon-supported catalysts comprise homogeneous Au–Pd alloy nanoparticles, which give high activity, whereas the MgO-supported catalyst has Au–Pd alloys with a Pd-rich surface and a Au-rich core, which result in lower activity. Pretreatment of these catalysts with bromide was found to enhance H2O2 productivity and the degree of enhancement was largely dependent on the nature of the Au–Pd nanoparticles. Whereas bromide pretreatment significantly enhanced H2O2 productivity over the MgO-based catalysts, the carbon-based catalyst only showed a subtle promotional effect. Very low loadings of bromide (0.00034–0.044 wt %) were required to yield a significant positive effect. Higher bromide loadings (0.5–8.3 wt %) proved deleterious. The promotional effect has been correlated to selective poisoning of sites responsible for H2O2 hydrogenation and decomposition. In view of the limited effect of bromide pretreatment on the yield of H2O2 coupled with the effective performance of the carbon supported Au-Pd catalysts in the absence of halides, for practical processes the addition of halides is not considered advisable with this catalyst system.
Co-reporter:Meenakshisundaram Sankar Dr.;Nikolaos Dimitratos Dr.;DavidW. Knight ;AlbertF. Carley Dr.;Ramchra Tiruvalam;ChristopherJ. Kiely ;Damian Thomas Dr.;GrahamJ. Hutchings
ChemSusChem 2009 Volume 2( Issue 12) pp:1145-1151
Publication Date(Web):
DOI:10.1002/cssc.200900133
Abstract
Glycolic acid is an important chemical that has uses as a cleaning agent as well as a chemical intermediate. At present glycolic acid is manufactured from either chloroacetic acid or from formaldehyde hydrocyanation, both routes being nongreen and using nonsustainable resources. We investigate the possibility of producing glycolate from the oxidation of glycerol, a sustainable raw material. We show that by using 1 % wt Au/carbon catalysts prepared using a sol-immobilization method glycolate yields of ca. 60 % can be achieved, using hydrogen peroxide as oxidant in an autoclave reactor. We describe and discuss the reaction mechanism and consider the reaction conditions that maximize the formation of glycolate.
Co-reporter:Edwin Ntainjua N. Dr.;Marco Piccinini;JamesC. Pritchard;JenniferK. Edwards Dr.;AlbertF. Carley Dr.;JacobA. Moulijn ;GrahamJ. Hutchings
ChemSusChem 2009 Volume 2( Issue 6) pp:575-580
Publication Date(Web):
DOI:10.1002/cssc.200800257
Co-reporter:Edwin Ntainjua N., Jennifer K. Edwards, Albert F. Carley, Jose Antonio Lopez-Sanchez, Jacob A. Moulijn, Andrew A. Herzing, Christopher J. Kiely and Graham J. Hutchings
Green Chemistry 2008 vol. 10(Issue 11) pp:1162-1169
Publication Date(Web):26 Sep 2008
DOI:10.1039/B809881F
Pd-only, Au-only and bimetallic AuPd catalysts supported on a range of supports (Al2O3, TiO2, MgO, and C) have been prepared by impregnation and tested for the hydrogenation and decomposition of hydrogen peroxide under conditions similar to those used in direct synthesis of hydrogen peroxide. Hydrogenation and decomposition are the main pathways for loss of selectivity and yield in the direct synthesis reaction, and the support is found to be a crucial parameter with respect to hydrogenation and decomposition activity. We show that by making the right choice of support for both the monometallic and bimetallic Au and Pd catalysts, it is possible to achieve very low hydrogen peroxide hydrogenation and decomposition activity, thus enhancing hydrogen peroxide productivity during synthesis. Carbon is found to be the optimal support for both monometallic Au and Pd catalysts as well as Au–Pd alloys, since carbon-supported catalysts gave the lowest hydrogenation and decomposition activities. Au-only catalysts were generally less active than Pd-only catalysts when utilizing the same support and metal loading. The addition of Au to Pd catalysts supported on TiO2 and carbon resulted in a decrease in both H2O2 hydrogenation and decomposition while the reverse effect was observed for the Al2O3 and MgO-supported catalysts. These effects are discussed in terms of the basicity of the support, and in particular the isoelectronic point of the support, which is a major factor in controlling the stability of hydrogen peroxide under reaction conditions.
Co-reporter:Hongmei Huang, Nicola Young, B. Peter Williams, Stuart H. Taylor and Graham Hutchings
Green Chemistry 2008 vol. 10(Issue 5) pp:571-577
Publication Date(Web):11 Mar 2008
DOI:10.1039/B717031A
The effect of rare earth doping of alumina catalysts is investigated for the carbonyl sulfide (COS) hydrolysis reaction (COS + H2O = CO2 + H2S). The effect of the catalyst preparation method is described and discussed, and three methods are compared, namely: impregnation by incipient wetness, coprecipitation and deposition precipitation. The most effective catalysts are prepared using the incipient wetness impregnation method. The addition of rare earth oxides, namely Y2O3, Gd2O3, Nd2O3, La2O3, increases the basicity of the material as shown by pulsed CO2 chemisorption and the basicity increases with the amount of rare earth oxide added. CO2 TPD shows that the La2O3-doped alumina has the strongest basic sites. The promoted catalysts are all effective for the COS hydrolysis reaction and the best results are obtained with Y2O3-doped materials, as these have the most pronounced promotion of activity over the reaction timescale we have examined. The combination of the results for COS conversion with the H2S selectivity data and the effects of H2S pre-treatment shows that a highly active catalyst also has a high H2S selectivity. The La2O3-doped materials deactivate rapidly and have poor H2S selectivities, and we propose that the higher basicity of this material leads to reaction with the acidic COS and H2S leading to the formation of the less basic lanthanum sulfide. This study has presented results for the first time showing that an alumina catalyst for COS hydrolysis can be promoted by the addition of rare earth oxides, and this is related to the enhanced basicity of the promoted catalyst.
Co-reporter:Jennifer K. Edwards, Adrian Thomas, Albert F. Carley, Andrew A. Herzing, Christopher J. Kiely and Graham J. Hutchings
Green Chemistry 2008 vol. 10(Issue 4) pp:388-394
Publication Date(Web):20 Nov 2007
DOI:10.1039/B714553P
The direct synthesis of hydrogen peroxide with Au–Pd catalysts is described and discussed: in particular, the roles of the support and promoters. Catalysts prepared by co-impregnation on various supports with calcination at 400 °C were stable and could be re-used several times without loss of metal. Catalysts calcined at lower temperatures were found to be unstable and could not be successfully re-used. Au–Pd/carbon and Au–Pd/silica catalysts gave the highest rate of H2O2 production, and the order of reactivity observed for the support materials investigated is: carbon ∼ silica > TiO2 > Al2O3. Bimetallic Au–Pd particles on TiO2 and Al2O3 were found to exhibit a core–shell structure, Pd being concentrated on the surface. It is considered that the Au–Pd/silica catalysts have a similar core–shell morphology based on X-ray photoelectron spectroscopy studies, whereas, in contrast, the calcined Au–Pd/carbon catalysts are observed to be homogeneous alloys. TEM studies showed that the silica contains impurities of carbon and that the Au–Pd alloys all preferentially interact with these carbonaceous impurities, hence resulting in a rather similar catalytic performance to that of the carbon supported Au–Pd catalysts. The origin of the enhanced activity for the silica and carbon supported catalysts is a result of higher H2 selectivity for the formation of hydrogen peroxide, which is due to the surface composition and size distribution of the nanoparticles. The effect of promoters is investigated, and it is shown that addition of Br− and PO43− is deleterious under our conditions, which contrasts markedly with Pd catalysts for which such species are essential. Furthermore, we show the acid solution formed by CO2 in water increases the rate of H2O2 synthesis, and thereby the CO2 diluent in our experiments acts as green in situ acid promoter.
Co-reporter:Jose Antonio Lopez-Sanchez, Nikolaos Dimitratos, Peter Miedziak, Edwin Ntainjua, Jennifer K. Edwards, David Morgan, Albert F. Carley, Ramchandra Tiruvalam, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 14) pp:1921-1930
Publication Date(Web):18 Feb 2008
DOI:10.1039/B719345A
Catalysis by gold and gold–palladium nanoparticles has attracted significant research attention in recent years. These nanocrystalline materials have been found to be highly effective for selective and total oxidation, but in most cases the catalysts are prepared using precipitation or impregnation. We report the preparation of Au–Pd nanocrystalline catalysts supported on carbon prepared via a sol-immobilisation technique and these have been compared with Au–Pd catalysts prepared via impregnation. The catalysts have been evaluated for two selective chemical syntheses, namely, oxidation of benzyl alcohol and the direct synthesis of hydrogen peroxide. The catalysts have been structurally characterised using a combination of scanning transmission electron microscopy and X-ray photoelectron spectroscopy. The catalysts prepared using the sol immobilisation technique show higher activity when compared with catalysts prepared by impregnation as they are more active for both hydrogen peroxide synthesis and hydrogenation, and also for benzyl alcohol oxidation. The method facilitates the use of much lower metal concentrations which is a key feature in catalyst design, particularly for the synthesis of hydrogen peroxide.
Co-reporter:Marco Conte;Albert F. Carley
Catalysis Letters 2008 Volume 124( Issue 3-4) pp:165-167
Publication Date(Web):2008 August
DOI:10.1007/s10562-008-9583-5
Acetylene hydrochlorination using a carbon-supported gold catalyst is studied. Reactivation of the catalyst is demonstrated using a brief treatment of the spent catalyst with boiling aqua regia and the process of reactivation and deactivation is characterised using X-ray photoelectron spectroscopy. Deactivation is considered to be due to loss of Au3+ which is restored by the aqua regia treatment.
Co-reporter:Graham J. Hutchings
Topics in Catalysis 2008 Volume 48( Issue 1-4) pp:55-59
Publication Date(Web):2008 May
DOI:10.1007/s11244-008-9048-5
The use of both heterogeneous and homogeneous gold catalysts for the reactions of alkynes is described. The reaction of ethyne with hydrogen chloride to make vinyl chloride monomer is initially described using gold supported on activated carbon. This reaction involves Au3+ as the reactive species and it is now known that higher alkynes can also be hydrochlorinated with the products of Markovnikov addition being formed. Subsequently, there is now a rich chemistry involving cationic gold or gold complexes as homogeneous catalysts for a very broad range of reactions involving alkynes, and some examples are discussed, including the nucleophilic addition of amines and alcohols to alkynes. Given the range of reactions that gold catalyses as a homogeneous catalysts this may be indicative that many more heterogeneously catalysed reactions using gold can be expected to be discovered.
Co-reporter:Albert F. Carley;Christopher J. Kiely;Philip Landon;Andrew A. Herzing
Science 2008 Volume 321(Issue 5894) pp:1331-1335
Publication Date(Web):05 Sep 2008
DOI:10.1126/science.1159639
Abstract
Gold nanocrystals absorbed on metal oxides have exceptional properties in oxidation catalysis, including the oxidation of carbon monoxide at ambient temperatures, but the identification of the active catalytic gold species among the many present on real catalysts is challenging. We have used aberration-corrected scanning transmission electron microscopy to analyze several iron oxide–supported catalyst samples, ranging from those with little or no activity to others with high activities. High catalytic activity for carbon monoxide oxidation is correlated with the presence of bilayer clusters that are ∼0.5 nanometer in diameter and contain only ∼10 gold atoms. The activity of these bilayer clusters is consistent with that demonstrated previously with the use of model catalyst systems.
Co-reporter:Kalala Jalama;Neil J. Coville;Diane Hildebrandt;David Glasser
Topics in Catalysis 2007 Volume 44( Issue 1-2) pp:129-136
Publication Date(Web):2007 June
DOI:10.1007/s11244-007-0286-8
The effect of Au doping on a 10%Co/TiO2 Fischer–Tropsch catalyst has been investigated by varying the amount of Au (0.2–5 wt.%) added to the catalyst. Addition of Au to the 10%Co/TiO2 system improved the cobalt dispersion on the catalyst surface and shifted the reduction temperature for the cobalt oxides in interaction with the support to lower temperatures. The catalyst activity for FT reaction increased with an increase in Au loading and passed through a maximum in activity at 1 wt.% Au while the methane and light product selectivity monotonically increased with Au loading.
Co-reporter:Saleh Al-Sayari;Albert F. Carley;Stuart H. Taylor
Topics in Catalysis 2007 Volume 44( Issue 1-2) pp:123-128
Publication Date(Web):2007 June
DOI:10.1007/s11244-007-0285-9
The preparation of Au/ZnO and Au/Fe2O3 catalysts using two coprecipitation methods is investigated to determine the important factors that control the synthesis of high activity catalysts for the oxidation of carbon monoxide at ambient temperature. In particular, the factors involved in the preparation of catalysts that are active without the need for a calcination step are evaluated. The two preparation methods differ in the manner in which the pH is controlled during the precipitation, either constant pH throughout or variable pH in which the pH is raised from an initial low value to a defined end point. Non-calcined Au/ZnO catalysts prepared using both methods are very sensitive to pH and ageing time, and catalysts prepared at a maximum pH = 5 with a short ageing time (ca. 0–3 h) exhibit high activity. Catalysts prepared at higher pH give lower activity. However, all catalysts require a short operation period during which the oxidation activity increases. In contrast, the calcined catalysts are not particularly sensitive to the preparation conditions. Non-calcined Au/Fe2O3 catalysts exhibit high activity when prepared at pH ≥ 5. Calcined Au/Fe2O3 prepared using the controlled pH method retain high activity, whereas calcined catalysts prepared using the variable pH method are inactive. The study shows the immense sensitivity of the catalyst performance to the preparation methods. It is therefore not surprising that marked differences in the performance of supported Au catalysts for CO oxidation that are apparent in the extensive literature on this subject, particularly the effect of calcination, can be expected if the preparation parameters are not carefully controlled and reported.
Co-reporter:Luisa Sartoni, Andréas Delimitis, Jonathan K. Bartley, Andrew Burrows, Hervé Roussel, Jean-Marie Herrmann, Jean-Claude Volta, Christopher J. Kiely and Graham J. Hutchings
Journal of Materials Chemistry A 2006 vol. 16(Issue 44) pp:4348-4360
Publication Date(Web):20 Sep 2006
DOI:10.1039/B610233F
Vanadium phosphate (VPO) catalyst materials have been doped with gallium and subsequently tested for the mild oxidation of n-butane to maleic anhydride. At low Ga concentrations, this impurity is shown to be beneficial for n-butane conversion. For low dopant levels (Ga/V ⩽ 1 at%), the crystallinity of the hemihydrate (VOHPO4·0.5H2O) precursor phase is improved and its specific area is increased relative to the undoped material as a result of decreased platelet thickness. Electron diffraction and energy dispersive X-ray analysis (XEDS) revealed that the Ga is uniformly distributed in a substitutional manner throughout the hemihydrate structure. The presence of Ga also significantly shortens the activation time required to convert the hemihydrate precursor into a well crystallized vanadyl pyrophosphate (VO)2P2O7 phase under an n-butane/air gas flow at 400 °C. The intimate presence of Ga distributed within the VOHPO4·0.5H2O unit cell has also been confirmed by XANES and EXAFS. Such studies also show that the Ga is partially redistributed within the (VO)2P2O7 structure after catalyst activation. A complementary electrical conductivity study on these materials revealed that Ga3+ substitutes for (VO)2+ species in (VO)2P2O7 giving rise to an n-type semiconductivity which partially compensates the natural p-type conductivity character of the (VO)2P2O7 phase. For higher Ga doping levels (Ga/V ≈ 5 at%), the excess of Ga concentrates as a GaPO4 impurity phase, which is shown to have a detrimental effect on the catalytic performance of the Ga-doped VPO catalyst.
Co-reporter:F. Girgsdies, W.-S. Dong, J.K. Bartley, G.J. Hutchings, R. Schlögl, T. Ressler
Solid State Sciences 2006 Volume 8(Issue 7) pp:807-812
Publication Date(Web):July 2006
DOI:10.1016/j.solidstatesciences.2006.04.008
The crystal structure of ε-VOPO4 was determined in the space group Cc from X-ray powder diffraction data using a rigid body approach. The resulting structure is compared to a recently published, slightly different structure model (space group P21/nP21/n) using Rietveld refinement. It was found that the new Cc model consistently yields a better fit to the observed data and exhibits a less distorted, more stable geometry. The crystal structure of ε-VOPO4 is discussed in comparison to β-VOPO4, monoclinic VPO4⋅H2O, and other related structures.
Co-reporter:Wen-Sheng Dong, Jonathan K. Bartley, Nicholas F. Dummer, Frank Girgsdies, Dansheng Su, Robert Schlögl, Jean-Claude Volta and Graham J. Hutchings
Journal of Materials Chemistry A 2005 vol. 15(Issue 31) pp:3214-3220
Publication Date(Web):30 Jun 2005
DOI:10.1039/B505586P
The reaction of VOPO4·2H2O, β-VOPO4 and VOHPO4·0.5H2O with alcohols in an autoclave at elevated temperatures (100–400 °C) and pressures (1–150 bar) is described and discussed. The reduction of VOPO4·2H2O with alcohols at ambient pressure is a standard method of preparation for VOHPO4·0.5H2O which is a valuable commercial catalyst precursor for the oxidation of butane to maleic anhydride. Surprisingly, the use of higher reaction pressures presents an unexplored region, and we show that primary alcohols at high temperature and pressure reduce both VOPO4·2H2O and β-VOPO4 to tetragonal VPO4·H2O, whereas VOHPO4·0.5H2O is reduced to form monoclinic VPO4·H2O. Previously these materials have been prepared by slow hydrothermal synthesis requiring the presence of templates, and hence we present new more facile synthetic pathways to these V(III) compounds. The catalytic performance for the selective oxidation of butane to maleic anhydride of these materials pretreated in situ with butane/air is also described and discussed in terms of the structures of the materials formed under the reaction conditions.
Co-reporter:Jennifer K. Edwards, Benjamin Solsona, Philip Landon, Albert F. Carley, Andrew Herzing, Masashi Watanabe, Christopher J. Kiely and Graham J. Hutchings
Journal of Materials Chemistry A 2005 vol. 15(Issue 43) pp:4595-4600
Publication Date(Web):23 Sep 2005
DOI:10.1039/B509542E
The direct synthesis of hydrogen peroxide from H2 and O2 using a range of Au, Pd and Au–Pd metal nanoparticles supported on iron oxide is described and discussed, and in particular the microstructure of the catalysts are investigated using a detailed electron microscopy study. Iron oxide was selected as a support because Au/Fe2O3 catalysts are known to be very active for low temperature CO oxidation. Hydrogen peroxide synthesis was investigated at low temperatures (2 °C) and short reaction (residence) time, and the addition of Pd to the Au catalyst was found to increase the rate of hydrogen peroxide synthesis as well as the concentration of hydrogen peroxide formed. Indeed the rates of hydrogen peroxide synthesis are higher for the Au–Pd alloy catalysts as compared to the Au or Pd only catalysts. These catalyst materials were also investigated for CO oxidation at 25 °C and all were found to be almost inactive. In contrast, Au-based catalysts that are very effective for low temperature CO oxidation were found to be totally inactive for H2 oxidation to H2O2. This suggests an inverse correlation between catalysts that are active for either CO or H2 activation. The microstructure of the Au–Pd/Fe2O3 catalysts was studied using scanning transmission electron microscopy and the metal alloy nanoparticles were found to have a core–shell morphology with Pd concentrated on the catalyst surface.
Co-reporter:Wen-Sheng Dong, Jonathan K. Bartley, Frank Girgsdies, Robert Schlögl and Graham J. Hutchings
Journal of Materials Chemistry A 2005 vol. 15(Issue 38) pp:4147-4153
Publication Date(Web):18 Aug 2005
DOI:10.1039/B509296E
The reaction of vanadyl pyrophosphate, (VO)2P2O7, with water was carried out at various temperatures and pressures in an autoclave and the resulting hydration products characterized using elemental analysis, powder X-ray diffraction, laser Raman spectroscopy and infrared spectroscopy. The hydration products were strongly dependent on the reaction conditions. At 150 °C (VO)3(PO4)2·6H2O was formed; at 200 °C VOHPO4·0.5H2O; and at ≥250 °C a phosphorus deficient, vanadium(III) phosphate (V1.23(PO4)(OH)0.69(H2O)0.31·0.33H2O or V5.12(PO4)4(OH)3.36(H2O)0.64·0.84H2O) was found to be the main product. The addition of 1-propanol to the reaction promotes the transformation of (VO)2P2O7 to the phosphorus deficient, vanadium(III) phosphate and the transformation to this phase is described in terms of the formation of extended glide shear defects due to the loss of lattice oxygen as well as the removal of phosphorus species from the catalyst lattice.
Co-reporter:Philip Landon, Jonathan Ferguson, Benjamin E. Solsona, Tomas Garcia, Albert F. Carley, Andrew A. Herzing, Christopher J. Kiely, Stanislaw E. Golunski and Graham J. Hutchings
Chemical Communications 2005 (Issue 27) pp:3385-3387
Publication Date(Web):17 May 2005
DOI:10.1039/B505295P
An Au/Fe2O3 catalyst prepared using a two-stage calcination procedure achieves target conversion and selectivity for the competitive oxidation of dilute CO in the presence of moist excess H2 and CO2.
Co-reporter:William P. Hems, Paul McMorn, Stewart Riddel, Simon Watson, Frederich E. Hancock and Graham J. Hutchings
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 8) pp:1547-1550
Publication Date(Web):18 Mar 2005
DOI:10.1039/B501359C
Rh diphosphine complexes using DuPhos and JosiPhos as chiral ligands have been immobilised by ion exchange into the mesoporous material MCM-41. When used as catalysts for the enantioselective hydrogenation of dimethyl itaconate and methyl-2-acetamidoacrylate, these heterogeneous catalysts give catalytic performance in terms of yield and enantioselection that are comparable to the corresponding homogeneous catalysts. Furthermore, the heterogeneous catalysts can be readily recovered and reused without loss of catalyst performance. A second immobilisation strategy is described in which [Rh(COD)2]+BF4− is initially immobilised by ion exchange and subsequently modified by the chiral diphosphine and this give comparable catalyst performance. This immobilisation strategy opens up the possibility of easy ligand-screening for parallel synthesis and libraries.
Co-reporter:Mathew D. Hughes, Yi-Jun Xu, Patrick Jenkins, Paul McMorn, Philip Landon, Dan I. Enache, Albert F. Carley, Gary A. Attard, Graham J. Hutchings, Frank King, E. Hugh Stitt, Peter Johnston, Ken Griffin
and Christopher J. Kiely
Nature 2005 437(7062) pp:1132
Publication Date(Web):
DOI:10.1038/nature04190
Co-reporter:Baojian Zhang, Stuart H. Taylor and Graham J. Hutchings
New Journal of Chemistry 2004 vol. 28(Issue 4) pp:471-476
Publication Date(Web):26 Feb 2004
DOI:10.1039/B312340P
Sustained synthesis of methanethiol from the reaction of CO/H2/H2S mixtures is reported and discussed. Surprisingly, unmodified α-Al2O3 gives the best results for this reaction and methanethiol selectivities of >98% at CO conversions of ca. 6% can be readily obtained (CO∶H2∶H2S=4∶5∶1, 340°C, total pressure=20 bar, 200 h−1). Reaction of CO+H2
(CO∶H2=1∶1) in the absence of H2S using α-Al2O3 under comparable conditions gives a lower CO conversion (ca. 1.3%) with significant selectivities to methane (20%), methanol (28.5%) and ethanol (21.1%). When H2S is added to the synthesis gas feedstock, the product selectivity switches to sulfur-containing products, almost exclusively methanethiol, but some by-product thiophene is also observed. A range of other catalysts were also investigated (e.g., γ-Al2O3, Cr2O3, Cr2O3/Al2O3, Cu/Cr2O3) but all give inferior catalytic performance when compared with α-Al2O3. The mechanism of the synthesis of methanethiol is discussed, based on a modification of chain propagation in the Fischer–Tropsch synthesis reaction.
Co-reporter:Luisa Sartoni, Jonathan K Bartley, Richard P.K Wells, Christopher J Kiely, Jean Claude Volta, Graham J Hutchings
Journal of Molecular Catalysis A: Chemical 2004 Volume 220(Issue 1) pp:85-92
Publication Date(Web):27 September 2004
DOI:10.1016/j.molcata.2004.03.052
The doping of vanadium phosphate catalysts by low levels of gallium is described an discussed. VOHPO4·0.5H2O precursors doped with Ga were prepared using a two stage method in which V2O5 is initially reacted with isobutanol before reaction with H3PO4 and Ga(acac)3. These were transformed to (VO)2P2O7 by reaction with 1.7% n-butane in air at 400 °C for 72 h. The materials were characterised using a combination of powder XRD, BET surface area measurement, laser Raman spectroscopy, X-ray photoelectron spectroscopy and scanning electron microscopy. The addition of 0.1 mol% Ga significantly enhances the activity of the catalyst. The effect is due in part to a structural effect as the surface area of the catalyst is increased by ca. 50–100% when up to 1 mol% Ga is added. However, the intrinsic activity (mol maleic anhydride produced/m2/h) is also significantly enhanced at these low doping levels. Hence, the promotional effect is considered to be due to both structural and electronic effects. The source of Ga was found to be and experiments were carried out with Ga2O3 and GaPO4 in place of Ga(acac)3. GaPO4 was found to give some enhancement in activity, but neither Ga2O3 nor GaPO4 gave an enhancement in surface area.The doping of vanadium phosphate catalysts by low levels of gallium significantly enhances the activity of the catalyst. The promotional effect is considered to be due to both structural and electronic effects.
Co-reporter:Louisa Griesel, Jonathan K Bartley, Richard P.K Wells, Graham J Hutchings
Journal of Molecular Catalysis A: Chemical 2004 Volume 220(Issue 1) pp:113-119
Publication Date(Web):27 September 2004
DOI:10.1016/j.molcata.2004.02.027
The preparation of VOPO4·2H2O is described and discussed. Three samples of the dihydrate are prepared with different ageing times following the initial reflux of V2O5 with H3PO4 in water for 24 h. The materials were characterised using a combination of powder XRD, BET surface area measurement, laser Raman spectroscopy and scanning electron microscopy. A sample of VOPO4·2H2O was isolated by immediate filtration of the reaction mixture and comprised flat oval crystallites with a broad size distribution between ca. 2 and 20 μm in diameter. Materials isolated following ageing of the initial reaction mixture (20 °C, 24 h) comprise square platelets again with a very broad size distribution. Using pyrophosphoric acid as the phosphorus source in place of phosphoric acid also affected the morphology of the VOPO4·2H2O. The dihydrates were reacted with isobutanol to form VOHPO4·0.5H2O and these were transformed to (VO)2P2O7 by reaction with 1.7% n-butane in air at 400 °C. The most active catalyst was derived from VOPO4·2H2O prepared from ageing a reaction mixture following the removal of the first crop of crystals. The study shows that the method of preparation of VOPO4·2H2O and, in particular, its morphology is of importance in the preparation of vanadium phosphate catalysts using the two stage method based on the reaction of the dihydrate with an alcohol to form the hemihydrate precursor.Graphic
Co-reporter:Silvio Carrettin, Paul McMorn, Peter Johnston, Ken Griffin, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 6) pp:1329-1336
Publication Date(Web):10 Feb 2003
DOI:10.1039/B212047J
The oxidation of aqueous solutions of glycerol is described and discussed for Pd, Pt and Au nanoparticles supported on graphite and activated carbon. The oxidation in a batch reactor at 60°C and 1 bar pressure using air as oxidant was initially investigated. Under these conditions, supported Pd and Pt catalysts give some selectivity to glyceric acid, but the main reaction products are considered to be non-desired C1 by-products, e.g. CO2, HCHO and HCOOH. In addition, under these conditions, supported Au catalysts were totally inactive. Using an autoclave with pure oxygen at 3 bar pressure gave a significant improvement in reactivity and, for Pt and Au catalysts, the formation of C1 by-products was eliminated when NaOH was added. In particular, it was noted that, in the absence of NaOH, the Au/C catalyst was inactive. For 1 wt.% Au/graphite or activated carbon, 100% selectivity to glyceric acid at high conversion was readily achieved. The role of the base is discussed and it is proposed that the base aids the initial dehydrogenation via H-abstraction of one of the primary OH groups of glycerol and, in this way, the rate limiting step in the oxidation process is overcome.
Co-reporter:Philip Landon, Paul J. Collier, Albert F. Carley, David Chadwick, Adam J. Papworth, Andrew Burrows, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 9) pp:1917-1923
Publication Date(Web):03 Apr 2003
DOI:10.1039/B211338B
The direct synthesis of hydrogen peroxide from H2 and O2 using a range of supported metal catalysts is described and discussed. A detailed study of the factors influencing the formation and decomposition of hydrogen peroxide is presented for a Pd/sulfonated carbon catalyst in a methanol/water solvent. The use of low temperatures (1–2°C) and short reaction (residence) time are identified as the key factors that favour high selectivity to hydrogen peroxide. Decomposition of hydrogen peroxide, mainly via further hydrogenation, prevents the formation of high concentrations of hydrogen peroxide. Combustion of hydrogen to water is a competing reaction that becomes significant at higher temperatures, but this can be partially inhibited by the addition of HBr. A second set of supported Pd and Au catalysts are evaluated for the direct synthesis of hydrogen peroxide using supercritical CO2 as a solvent. The use of supercritical CO2 is shown to be beneficial when compared with hydrogen peroxide formation at a temperature just below the critical temperature for CO2. However, at the critical temperature of CO2
(31.1°C), the decomposition of hydrogen peroxide is rapid and only low rates of hydrogen peroxide formation are observed. At low temperature (2°C) supported Au catalysts are shown to be very selective for the synthesis of hydrogen peroxide. The rate of hydrogen peroxide synthesis is enhanced markedly when Pd is present with Au and a detailed scanning transmission electron microscopy study shows that the 2–9 nm metal nanoparticles present in this supported catalyst are a Au∶Pd alloy.
Co-reporter:Jose Antonio Lopez-Sanchez, Louisa Griesel, Jonathan K. Bartley, Richard P. K. Wells, Andrzej Liskowski, Dangsheng Su, Robert Schlögl, Jean-Claude Volta and Graham J. Hutchings
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 16) pp:3525-3533
Publication Date(Web):14 Jul 2003
DOI:10.1039/B305437N
Vanadium phosphate catalysts prepared in aqueous solution at elevated temperature (145°C) using either H3PO3 or V2O4 as reactants are described and discussed. This methodology produces catalysts with a much higher surface area (ca. 20 m2g−1) compared with those prepared using aqueous routes using HCl as reducing agent (ca. 4 m2g−1). The materials were characterised using a combination of powder XRD, BET surface area measurement, laser Raman spectroscopy, TGA, electron microscopy and 31P spin echo mapping NMR spectroscopy. Refluxing the precursors in water prior to activation was crucial in obtaining high surface area materials, and 31P spin echo mapping NMR together with electron microscopy data indicate that the water reflux step influences the relative amounts of V4+ and V5+ phases present in the catalyst, as well as reducing the size of the crystallites. A correlation between the activity of the catalyst and the surface area is observed. However, a small group of catalysts display a higher activity than that expected from this correlation, and this increased activity is discussed in terms of the interaction of V4+ and V5+ phases.
Co-reporter:Silvio Carrettin, Paul McMorn, Peter Johnston, Ken Griffin and Graham J. Hutchings
Chemical Communications 2002 (Issue 7) pp:696-697
Publication Date(Web):05 Mar 2002
DOI:10.1039/B201112N
Glycerol is oxidised to glyceric acid with 100% selectivity using either 1% Au/charcoal or 1% Au/graphite catalyst under mild reaction conditions (60 °C, 3 h, water as solvent).
Co-reporter:Philip Landon, Paul J. Collier, Adam J. Papworth, Christopher J. Kiely and Graham J. Hutchings
Chemical Communications 2002 (Issue 18) pp:2058-2059
Publication Date(Web):14 Aug 2002
DOI:10.1039/B205248M
Supported Au catalysts are very selective for the direct formation of hydrogen peroxide from H2/O2 mixtures at 2 °C; the rate of H2O2 synthesis is markedly increased if Au–Pd alloy nanoparticles are generated by the addition of Pd.
Co-reporter:Richard Tanner, Philip Gill, Richard Wells, Jillian E. Bailie, Gordon Kelly, S. David Jackson and Graham J. Hutchings
Physical Chemistry Chemical Physics 2002 vol. 4(Issue 4) pp:688-695
Publication Date(Web):09 Jan 2002
DOI:10.1039/B108794K
The acid–base properties of vanadium phosphate catalysts are investigated using the aldol condensation of acetone and the reactions of 2-methylbut-3-yn-2-ol (MBOH). Three well characterised samples of VOHPO4·0.5H2O were prepared using the reaction of V2O5 and H3PO4 with aqueous hydrochloric acid or isobutanol as reducing agents, or from the reaction of VOPO4.2H2O with isobutanol. (VO)2P2O7, prepared by heating VOHPO4·0.5H2O in He (8 h, 750°C), before and following partial oxidation in air or butane/air, and αI-VOPO4 were also investigated. The reaction of MBOH was used to probe the nature of the acid–base properties of the vanadium phosphates. The V4+ phases (VOHPO4·0.5H2O and (VO)2P2O7)
exhibited only acidic active sites, whereas the V5+ phases (αI-VOPO4 and oxidised (VO)2P2O7) exhibited some basic sites in addition to the acid sites. For the aldol condensation reactions of acetone, the V4+ phases were found to be selective for the formation of isophorone from acetone alone and methyl vinyl ketone from the reaction of acetone and formaldehyde. In contrast, vanadium phosphate catalysts containing V5+ phases are not selective to these products and only form hydrocarbons (typically isobutane and isobutene). For all these reactions, the catalyst activity is short lived and the deactivation that is observed is due to the surface becoming fouled by the adsorption of products of polymerisation of the reaction products. However, the catalyst reactivity can be restored by a simple oxidation treatment. The nature of active sites in n-butane
oxidation to maleic anhydride is also discussed and it is concluded that basic sites are required in addition to acidic surface sites for the selective formation of maleic anhydride. For the reaction of MBOH, the data are found to give a linear relationship for a Cremer–Constable plot and this is discussed in terms of the enthalpy of adsorption of MBOH.
Co-reporter:Lee J. Schofield, Owain J. Kerton, Paul McMorn, Donald Bethell, Simon Ellwood and Graham J. Hutchings
Organic & Biomolecular Chemistry 2002 (Issue 8) pp:1475-1481
Publication Date(Web):26 Jun 2002
DOI:10.1039/B201724P
The regioselective epoxidation of monoterpenes in the liquid phase has been studied using the titanosilicates TS-1 and TiAlβ. A range of oxidants (hydrogen peroxide, tert-butyl hydroperoxide and urea–hydrogen peroxide complex) have been studied in detail. The allylic alcohols linalool and geraniol have been studied alongside the non-allylic alcohol citronellol and the diene dihydromyrcene to help determine the role of the hydroxy group in these reactions. Dihydromyrcene is selectively epoxidised at the more electron rich double bond regardless of the catalyst–oxidant–solvent system used. Geraniol can undergo allylic assisted epoxidation with TS-1–acetone–hydrogen peroxide and TiAlβ–acetonitrile–urea–hydrogen peroxide. With TiAlβ–hydrogen peroxide–methanol, the reaction shows an induction period in the conversion of geraniol which is considered to be characteristic of the autocatalytic removal
of titanium from the catalyst framework. Reactions with citronellol show this titanium removal is entirely due to the presence of the allylic alcohol moiety. Finally, epoxidation of linalool and the subsequent in situ conversion of the epoxide to the furano- and pyrano-oxides were studied. The ratio of furano- and pyrano-oxides formed was considered to be due, in part, to the pore geometry and the Brønsted acidity of the catalyst.
Co-reporter:Lee J. Schofield, Owain J. Kerton, Paul McMorn, Donald Bethell, Simon Ellwood and Graham J. Hutchings
Organic & Biomolecular Chemistry 2002 (Issue 12) pp:2064-2071
Publication Date(Web):2002/10/23
DOI:10.1039/B207425G
The regioselective epoxidation of dihydromyrcene has been studied in the presence of the titanium-containing silicates TS-1 and TiAlβ using aqueous hydrogen peroxide, tert-butyl hydroperoxide and urea–hydrogen peroxide as oxidants. Epoxides were observed with TS-1 and aqueous hydrogen peroxide, and with TiAlβ when used in conjunction with the urea–hydrogen peroxide complex and tert-butyl hydroperoxide. Epoxidation occurs exclusively at the more electron-rich double bond in the presence of both catalysts. The epoxidation of dihydromyrcene has also been studied under triphasic conditions (two immiscible liquid phases and one catalyst phase) rather than biphasic conditions (one liquid phase and one catalyst phase). The alcoholysis reaction of the resulting epoxide was found to proceed via the more stabilised cation intermediate under biphasic conditions. In contrast, alcoholysis under triphasic conditions proceeded to form both the favoured (major) and unfavoured (minor) ether alcohols in ratios up to 2 ∶ 1. Model compounds, (2-methylpent-2-ene and 3-methylpent-1-ene) which simulate the electronic environment around each of the double bonds in dihydromyrcene, have been used to study the degree of epoxidation of each double bond separately and under competitive conditions. When the model substrates are studied separately, the rate of epoxidation of the two double bonds are comparable. When the model substrates are epoxidised in a competitive manner, the electron-deficient double bond is oxidised in preference which is different to that observed for dihydromyrcene.
Co-reporter:Jose Antonio Lopez-Sanchez, Jonathan K. Bartley, Richard P. K. Wells, Colin Rhodes and Graham J. Hutchings
New Journal of Chemistry 2002 vol. 26(Issue 11) pp:1613-1618
Publication Date(Web):17 Oct 2002
DOI:10.1039/B203192M
The synthesis of vanadium phosphorus oxide catalysts using water as solvent is described and discussed. The use of H3PO3 as a reducing agent is contrasted with aqueous hydrochloric acid. Using H3PO3 as a reducing agent for V2O5 at 145°C for 72 h, together with H3PO4 or H4P2O7 as the additional phosphorus source, is found to produce VOHPO4·0.5H2O. Following activation in n-butane–air for 72 h, the catalysts derived from this method have surface areas (17–23 m2 g−1) that are comparable to those prepared using a standard non-aqueous solvent method. The specific (mol maleic anhydride per g catalyst per h) and intrinsic (mol maleic anhydride m−2 h−1) activities of the catalysts derived from using H3PO3 with water as solvent are also comparable to those for catalysts prepared using non-aqueous solvents. The activated catalysts comprise (VO)2P2O7, together with αII- and δ-VOPO4. Refluxing the VOHPO4·0.5H2O catalyst precursors in water is found to decrease the crystallite size of the precursor, and this leads to higher area activated catalysts, but does not affect the specific or intrinsic activity significantly.
Co-reporter:Paul Laidlaw, Donald Bethell, Stephen M Brown, Graeme Watson, David J Willock, Graham J Hutchings
Journal of Molecular Catalysis A: Chemical 2002 Volume 178(1–2) pp:205-209
Publication Date(Web):23 January 2002
DOI:10.1016/S1381-1169(01)00321-1
The sulfonylation of substituted benzenes is investigated for Zn-exchanged zeolites (ZSM-5, Y, β) using two sulfonating agents (methanesulfonyl chloride and benzenesulfonyl chloride). Zinc-exchanged zeolites are significantly more active than their corresponding proton form. Zinc-exchanged zeolites prepared using zinc acetate give higher level of Zn2+-exchange acid and are more active than those prepared from zinc nitrate. Methanesulfonyl chloride gives similar selectivities for the 2- and 4-substituted products, but benzenesulfonyl chloride gives enhanced selectivity to the 4-product. The highest yields of the 4-product are observed with Zn-H-β as catalyst. For most zinc-exchanged zeolites no leaching of Zn2+ is observed and furthermore, the Zn2+ that is leached into solution is found to be inactive as a homogeneous catalyst for the reaction. This indicates that the observed reactions are wholly heterogeneously catalysed. The catalytic data are discussed in relation to pore access calculations for the arenium ion intermediates formed during the sulfonylation reaction.
Co-reporter:John Gullick, Sophia Taylor, Paul McMorn, Donald Bethell, Philip C. Bulman-Page, Frederick E. Hancock, Frank King, Graham J. Hutchings
Journal of Molecular Catalysis A: Chemical 2002 Volumes 182–183() pp:571-575
Publication Date(Web):31 May 2002
DOI:10.1016/S1381-1169(01)00497-6
The copper-catalysed aziridination of styrene with copper-exchanged zeolite Y (CuHY) and copper(II) triflate (Cu(OTf)2) as catalysts is described and discussed. In particular, the effects of reaction conditions on the yield and enantiomeric excess of the aziridine product are described using [N-(p-nitrophenylsulfonyl)imino]phenyliodinane (PhINNs) as nitrene donor. By careful control of the styrene:nitrene donor molar ratio and the solvent, an ee of 95% can be obtained for the heterogeneously catalysed bis(oxazoline)-modified zeolite CuHY. The ee achieved with the zeolite immobilised catalyst is significantly higher than that achieved for the non-immobilised homogeneous catalyst under comparable reaction conditions.
Co-reporter:Meleri Johns, Philip Landon, Tony Alderson and Graham J. Hutchings
Chemical Communications 2001 (Issue 23) pp:2454-2455
Publication Date(Web):13 Nov 2001
DOI:10.1039/B108353H
A composite catalyst comprising a physical mixture of a zeolite and a cobalt/manganese oxide Fischer–Tropsch catalyst decreases the formation of methane in the hydrogenation of carbon monoxide without significantly affecting conversion.
Co-reporter:Sophia Taylor, John Gullick, Paul McMorn, Donald Bethell, Philip C. Bulman Page, Frederick E. Hancock, Frank King and Graham J. Hutchings
Organic & Biomolecular Chemistry 2001 (Issue 9) pp:1724-1728
Publication Date(Web):09 Aug 2001
DOI:10.1039/B104526C
The stability of the heterogeneous CuHY catalyst for the aziridination of styrene using nitrene donors, with and without the presence of chiral bis(oxazoline) modifiers, is described in detail. Cu2+ is found to leach from the CuHY and the rate of Cu-leaching is dependent on the reaction time, the nature of the nitrene donor, the structure of the bis(oxazoline), the presence of solvent and the breakdown products of the nitrene donor. It is found that between 0.08–6.8% by weight of the Cu present in CuHY can be leached during standard reaction conditions. However, using short reaction times it is shown that the amount of Cu removed can be limited readily to the lower value. Detailed studies show that for this reaction system the leached Cu2+ plays no significant role in the formation of the aziridine, and that the high enantioselectivities observed with CuHY are due to Cu2+ which is electrostatically bound within the pores of the zeolite and modified by a chiral bis(oxazoline) ligand.
Co-reporter:Sophia Taylor, John Gullick, Paul McMorn, Donald Bethell, Philip C. Bulman Page, Frederick E. Hancock, Frank King and Graham J. Hutchings
Organic & Biomolecular Chemistry 2001 (Issue 9) pp:1714-1723
Publication Date(Web):09 Aug 2001
DOI:10.1039/B104522A
The copper-catalysed aziridination of styrene with copper-exchanged zeolite Y (CuHY) and copper(II) triflate (trifluoromethanesulfonate) (Cu(OTf)2) as catalysts is described in detail. Two nitrene donors, [N-(p-tolylsulfonyl)imino)]phenyliodinane (PhINTs) and [N-(p-nitrophenylsulfonyl)imino]phenyliodinane (PhINNs) are compared. Modification of the catalyst with bis(oxazolines) affords enantioselective catalysts and a range of chiral bis(oxazolines) has been studied. The ratio of nitrene donor to styrene is shown to be an important factor controlling both the yield and ee of aziridine formed. The best results are obtained with PhINNs; ee, ≥ 90%, together with high yields (≥ 85%), can readily be achieved with this nitrene donor using acetonitrile as solvent. Addition of the nitrene donor over a period of time, rather than all at the start of the reaction, is shown to enhance the yield of the aziridine but the ee is significantly decreased for both the homogeneous and the heterogeneous catalysts. Experiments in which the breakdown products of the nitrene donor, iodobenzene and the corresponding sulfonamide, are added at the start of the reaction show that a complex interplay exists at the copper active site between the reactants, products, chiral modifier and the solvent. However, the heterogeneous catalyst, CuHY, is found to give enhanced enantioselection for a range of bis(oxazolines) compared to the homogeneous catalyst, and the effect is considered to be due to the confinement of the catalyst within the micropores of the zeolite.
Co-reporter:Jillian E. Bailie, Halim A. Abdullah, James A. Anderson, Colin H. Rochester, Neville V. Richardson, Nicholas Hodge, Jian-Guo Zhang, Andy Burrows, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2001 vol. 3(Issue 18) pp:4113-4121
Publication Date(Web):31 Aug 2001
DOI:10.1039/B103880J
The hydrogenation of but-2-enal over supported Au catalysts is discussed, together with a detailed characterisation study using X-ray diffraction, infrared spectroscopy and transmission electron microscopy. Au/ZnO catalysts are found to be selective for the formation of the unsaturated alcohol, but-2-en-1-ol rather than the saturated aldehyde, butanal. In general, the addition of thiophene is found to enhance the yield of the unsaturated alcohol. Detailed transmission electron microscopy and infrared spectroscopy studies show that thiophene modification of Au/ZnO catalysts does not affect the Au-particle size or morphology; rather, thiophene undergoes irreversible dissociative adsorption giving a surface in which the Au sites are electronically promoted by sulfur. It is observed that thiophene modification does not give any marked effect on catalyst performance for the catalysts that contain large Au-particles
(10 nm) and, hence, it is considered that the sulfur promotion observed is associated with smaller Au nanoparticles. The highest but-2-en-1-ol selectivities (∽80%) are observed for 5 wt.% Au/ZnO catalysts reduced at 400°C prior to reaction. It is proposed that the origin of high selectivity is associated with large Au particles (10–20 nm in diameter) that are present in this catalyst.
Co-reporter:Jonathan K. Bartley, Ian J. Ellison, Andreas Delimitis, Christopher J. Kiely, Asghar-Zeini Isfahani, Colin Rhodes and Graham J. Hutchings
Physical Chemistry Chemical Physics 2001 vol. 3(Issue 20) pp:4606-4613
Publication Date(Web):25 Sep 2001
DOI:10.1039/B105304N
Vanadium phosphate catalysts prepared from VOPO4
·2H2O are described and discussed. The use of H4P2O7 as a phosphorus source, as compared with H3PO4, is shown to give an improved synthesis of VOPO4·2H2O. Reduction of VOPO4·2H2O with alcohols from both the materials leads to the formation of VOHPO4·0.5H2O. However, the material derived from H4P2O7 has a higher intrinsic activity (mol maleic anhydride formed per unit surface area) than that derived from H3PO4. The final catalysts are shown to comprise (VO)2P2O7 together with a range of V(V) VOPO4 phases and the origin of the enhanced activity is discussed in terms of the catalyst structure.
Co-reporter:Paul Laidlaw, Donald Bethell, Stephen M. Brown, Graham J. Hutchings
Journal of Molecular Catalysis A: Chemical 2001 Volume 174(1–2) pp:187-191
Publication Date(Web):1 October 2001
DOI:10.1016/S1381-1169(01)00164-9
The benzoylation of substituted arenes using benzoyl chloride has been investigated for Zn- and Fe- exchanged zeolites (H-ZSM-5, mordenite and zeolite Y). Toluene benzoylation preferentially gave 4-methylbenzophenone for all zeolites. The Zn-exchanged zeolites were more active than the proton forms of zeolites, but gave the same product selectivity, and it has been shown that extensive leaching of Zn2+ cations into solution occurs, particularly when zinc acetate is used to prepare the catalyst. For the Zn-exchanged zeolites it is concluded that a significant proportion of the reaction occurs in solution. However, Fe-exchanged zeolites gave only limited leaching of Fe cations into solution and in this case the cation-exchanged zeolite acts as a heterogeneous catalyst. The Fe-Na-Y has been shown to give high yields of substituted benzophenones for a range of substrates. The form of conversion/time curves is consistent with the existence of two types of catalytic site. One type appears to be inhibited by the product benzophenone and is responsible for the initial rapid rate of conversion. The other is not inhibited, giving rise to an approximately constant, slower reaction rate in the later stages.
Co-reporter:Lucinda J Davies, Paul McMorn, Donald Bethell, Philip C Bulman Page, Frank King, Frederick E Hancock, Graham J Hutchings
Journal of Molecular Catalysis A: Chemical 2001 Volume 165(1–2) pp:243-247
Publication Date(Web):8 January 2001
DOI:10.1016/S1381-1169(00)00430-1
A comparative study of the oxidation of the crotyl alcohol using hydrogen peroxide and tert-butyl hydroperoxide as oxidants with TS-1, Ti-β, Ti-Alβ, Ti-MCM-41, Ti-Al-MCM-41 and Ti-grafted-MCM-41 as catalysts is described and discussed. With hydrogen peroxide as oxidant, significant Ti-leaching is observed with all the catalysts except TS-1 (Ti-Alβ>Ti-grafted- MCM-41>Ti-MCM-41>Tiβ>Ti-Al-MCM-41⪢TS-1). For Ti-Alβ, Ti-grafted- MCM-41 and Ti-Al-MCM-41, initial heterogeneously catalysed formation of the epoxide was observed. However, the formation of a Ti-species in solution is shown to contribute to competing homogeneously catalysed formation of ether diols and triol. Using tert-butyl hydroperoxide as oxidant the Ti-leaching was minimised and selective epoxide formation was observed with Ti-β, Ti-Alβ and Ti-MCM-41 as heterogeneous catalysts, although, with Ti-Alβ, the ether diols and triol products dominated due to acid catalysed solvolysis of the epoxide.
Co-reporter:Paola Piaggio, Paul McMorn, Damien Murphy, Donald Bethell, Philip C. Bulman Page, Frederick E. Hancock, Christopher Sly, Owain J. Kerton and Graham J. Hutchings
Organic & Biomolecular Chemistry 2000 (Issue 10) pp:2008-2015
Publication Date(Web):18 Sep 2000
DOI:10.1039/B005752P
Manganese-exchanged Al-MCM-41 modified by the chiral salen ligand [(R,R)-(−)-N,N′-bis(3,5-di-tert-butylsalicylidene)cyclohexane-1,2-diamine] has been investigated as a heterogeneous catalyst for the enantioselective epoxidation of (Z)-stilbene using iodosylbenzene as oxygen donor, with particular interest in the effect of reaction conditions on the cis∶trans ratio of the epoxide product. Immobilisation of the chiral Mn–salen complex in Al-MCM-41 increases the cis∶trans ratio of the epoxide product when compared to the non-immobilised complex under the same conditions. Increasing the level of Mn-exchange in the Al-MCM-41 increases the amount of trans-epoxide, whereas increasing the iodosylbenzene∶substrate ratio increases the amount of cis product formed. Increasing the reaction temperature also increases the amount of trans-epoxide for the homogeneous Mn-complex under the same conditions. A series of experiments is described in which the external ion-exchange sites on Al-MCM-41 are preferentially silanised, which enables the cis/trans selectivity for external and internal sites to be determined. Mn–salen immobilised on the external surface of Al-MCM-41 gives the same cis∶trans ratio as that observed with the non-immobilised Mn–salen complex in solution, whereas Mn–salen immobilised within the pores gives the cis-epoxide preferentially.The enantioselection of the immobilised chiral Mn–salen complex is shown to decrease with reaction time at −10 °C, but the cis∶trans epoxide ratio remains unchanged; whereas for the non-immobilised complex in solution the enantioselection is independent of reaction time. Iodobenzene, a decomposition product formed from iodosylbenzene, is found to act as a poison for the immobilised catalyst, leading to a slower reaction and lower enantioselection.
Co-reporter:Michael M. Forde, Bezzu C. Grazia, Robert Armstrong, Robert L. Jenkins, Mohammed Hasbi Ab Rahim, Albert F. Carley, Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, Stuart H. Taylor, Neil B. McKeown, Graham John Hutchings
Journal of Catalysis (June 2012) Volume 290() pp:177-185
Publication Date(Web):1 June 2012
DOI:10.1016/j.jcat.2012.03.013
Methane oxidation in water using hydrogen peroxide with a μ-nitrido iron phthalocyanine complex grafted on silica has been investigated in detail. Methyl hydroperoxide is identified as the main primary reaction product from methane oxidation. The catalyst is unstable under reaction conditions and this is discussed. However, the unmodified silica support is also found to be active for this reaction, and in particular, a Fe/SiO2 catalyst prepared by wet impregnation is also found to be as effective as the μ-nitrido iron phthalocyanine complex grafted on silica, with higher selectivity to useful oxygenates (>80%) and displays minimal leaching or instability. A method for the reliable quantification of CO2 in both the gas and aqueous phase is reported as this presents a key experimental difficulty in the oxidation of methane in aqueous media.Graphical abstractMethane oxidation in water using H2O2 with a μ-nitrido iron phthalocyanine complex grafted on silica gives a number of oxygenated products derived from both the oxidation of methane and the decomposition of the catalyst. However, a much simpler catalyst Fe/SiO2 is equally active and is stable.Download high-res image (110KB)Download full-size imageHighlights► Methane oxidation with a μ-nitrido iron phthalocyanine complex is investigated. ► Methyl hydroperoxide is identified as the primary reaction product of methane oxidation. ► The μ-nitrido iron phthalocyanine complex grafted on silica catalyst is highly unstable. ► Fe/SiO2 prepared by wet impregnation is as effective as the bio-mimetic catalyst studied. ► A reliable method CO2 analysis in both the gas and the aqueous phase is described.
Co-reporter:Marco Conte, Catherine J. Davies, David J. Morgan, Thomas E. Davies, David J. Elias, Albert F. Carley, Peter Johnston, Graham J. Hutchings
Journal of Catalysis (January 2013) Volume 297() pp:128-136
Publication Date(Web):1 January 2013
DOI:10.1016/j.jcat.2012.10.002
Au/C catalysts are effective materials for the gas phase hydrochlorination of acetylene to vinyl chloride monomer, and to date, the most effective catalyst preparation protocol makes use of impregnation using aqua regia. In the present study, the effect of this solvent is evaluated and discussed in detail by modifying the ratio of HCl and HNO3 and the temperature of the impregnation step. These factors are observed to affect the Au3+/Au0 ratio of the final catalyst, in addition to the modification of the functional groups of the carbon used as support. The results can be rationalised by the oxidation effect of HNO3 on both the gold nanoparticles and the functional groups on the carbon surface, as well as a nucleation effect of HCl towards gold over the carbon support.Graphical abstractAu/C catalysts prepared using aqua regia as a solvent display superior activity in the hydrochlorination reaction of acetylene to vinyl chloride monomer. A synergistic effect between HCl and HNO3 is observed, which is linked to the nucleation of Au nanoparticles.Download high-res image (102KB)Download full-size imageHighlights► Impregnated Au/C catalyst with aqua regia as a solvent displayed superior activity in the hydrochlorination reaction of acetylene to vinyl chloride monomer. ► A synergistic effect between HCl and HNO3 is present, driving the Au nanoparticles nucleation process over the carbon support. ► The impregnating acid mixture can affect the carbon support enriching the presence of oxygen functional groups. ► The reaction occurs over Au3+ centres at the Au/C interface.
Co-reporter:Muhammad H. Haider, Nicholas F. Dummer, Dazhi Zhang, Peter Miedziak, Thomas E. Davies, Stuart H. Taylor, David J. Willock, David W. Knight, David Chadwick, Graham J. Hutchings
Journal of Catalysis (February 2012) Volume 286() pp:206-213
Publication Date(Web):1 February 2012
DOI:10.1016/j.jcat.2011.11.004
Dehydration of glycerol was carried out using rubidium- and caesium-doped silicotungstic acid catalysts. These catalysts were prepared by varying concentration of the dopant metal cations while keeping the concentration of heteropoly acid unchanged. High acrolein selectivity (94–96%) was observed with unsupported caesium-doped silicotungstic acid and rubidium-doped silicotungstic acid with a dilute glycerol feed (0.5 wt.% in water). These catalysts were then supported on alpha-alumina and an alumina comprising a theta-delta mixture. Caesium-doped silicotungstic acid supported on theta-delta alumina gave a maximum selectivity of ca. 90% at 100% glycerol conversion for 90-h time online, with a 10 wt.% glycerol solution. With a more concentrated glycerol feed (20 wt.%), this catalyst achieved a space time yield of 210 g(acrolein)kg(cat)-1h-1. The catalyst was investigated further to determine the origin of the long-term stability. The binding strength of the partially doped silicotungstic acid on the alumina was found to be crucial to sustain the supported Keggin structure and hence the acidity of the active sites resulting in a high acrolein yield.Graphical abstractDehydration of glycerol to acrolein was carried out using rubidium- and caesium-doped silicotungstic acid catalysts supported on alumina. Cs-doped silicotungstic acid supported on theta-delta alumina gave a maximum selectivity of ca. 90% at 100% glycerol conversion and with a more concentrated glycerol feed (20 wt.%) achieved an space time yield of 210 g(acrolein)kg(cat)-1h-1. The catalysts were tested for up to 200 h on-stream and found to be stable for 90 h (10 wt.% glycerol).Download high-res image (72KB)Download full-size imageHighlights► Caesium-doped silicotungstic acid catalysts, CsSTA/Al2O3, are selective for glycerol dehydration to acrolein. ► Catalysts are stable for up to 90-h time-on-stream without co-fed O2. ► Catalysts characterisation indicates that high acrolein yield can be correlated with an increased interaction of the active silicotungstic acid with the support.
Co-reporter:Jennifer K. Edwards, James Pritchard, Marco Piccinini, Greg Shaw, Qian He, Albert F. Carley, Christopher J. Kiely, Graham J. Hutchings
Journal of Catalysis (August 2012) Volume 292() pp:227-238
Publication Date(Web):1 August 2012
DOI:10.1016/j.jcat.2012.05.018
The direct synthesis of hydrogen peroxide using supported gold palladium catalysts prepared by incipient wetness impregnation is described and discussed. The effect of an acid pre-treatment step on the activated carbon support prior to the deposition of the metals, together with the effect of the calcination temperature, has been investigated. The acid pre-treated samples all show superior activity to those materials prepared with the omission of this acid pre-treatment stage. The calcination temperature affects both the re-usability and hydrogenation activity of the catalysts. Detailed characterisation using X-ray photoelectron spectroscopy and aberration-corrected scanning transmission electron microscopy is described. The enhanced activity is associated with a higher surface concentration of palladium in the acid pre-treated samples which is principally present as Pd2+. Calcination of the catalysts at 400 °C is required to achieve re-usable and stable catalysts, and this is associated with the morphology and dispersion of the metal nanoparticles. The surface ratio of Pd0/Pd2+ is found to be an important factor controlling the hydrogenation of hydrogen peroxide, and a series of controlled reduction and re-oxidation of a sample show how the Pd0/Pd2+ surface ratio can influence the relative rates of hydrogen peroxide synthesis and hydrogenation.Graphical abstractCalcination of AuPd/carbon catalysts controls the Pd2+/Pd0 ratio and permits the direct synthesis of H2O2 without sequential decomposition or hydrogenationDownload high-res image (88KB)Download full-size imageHighlights► Acid pre-treatment of carbon-supported AuPd catalysts switches off the non-selective reactions in direct synthesis of H2O2. ► The acid pre-treated samples all show superior activity to those prepared with the omission of this acid pre-treating stage. ► X-ray photoelectron spectroscopy and aberration-corrected scanning transmission electron microscopy show the effect of calcination on activity. ► The enhanced activity is associated with a higher surface concentration of palladium in the acid pre-treated samples which is principally present as Pd2+.
Co-reporter:Qian Yang, Wilm Jones, Peter P. Wells, David Morgan, Lichun Dong, Baoshan Hu, Nikolaos Dimitratos, Mingdong Dong, Mike Bowker, Flemming Besenbacher, Ren Su, Graham Hutchings
Applied Catalysis A: General (25 May 2016) Volume 518() pp:213-220
Publication Date(Web):25 May 2016
DOI:10.1016/j.apcata.2015.10.023
Co-reporter:Simon A. Kondrat, Thomas E. Davies, Zhongling Zu, Paul Boldrin, Jonathan K. Bartley, Albert F. Carley, Stuart H. Taylor, Matthew J. Rosseinsky, Graham J. Hutchings
Journal of Catalysis (25 July 2011) Volume 281(Issue 2) pp:279-289
Publication Date(Web):25 July 2011
DOI:10.1016/j.jcat.2011.05.012
The auto-reduction of copper and manganese acetates has been investigated using in situ X-ray diffraction and thermogravimetric analysis, with the intention of manipulating the phenomena to tailor specific phase formation for synthesising catalysts. Subsequently catalysts prepared in this controlled manner were evaluated for ambient temperature CO oxidation. The decomposition of mixed copper and manganese acetate systems was controlled to form MnOx-supported Cu or CuMnOx spinel structures, depending on the oxygen concentration and flow conditions during the heat treatment. Catalyst precursors were prepared by physical grinding and by a supercritical CO2 anti-solvent precipitation process. The use of supercritical anti-solvent precipitation allows for the formation of well-mixed metal acetates that decompose to form active spinel CO-oxidation catalysts or small copper nano-particles supported on MnOx, depending on the oxygen content of the heat treatment atmosphere. The ability to tune oxidation state and phase composition of catalysts is a key preparation parameter for controlling the activity and provides insight into the active sites for CO oxidation.Graphical abstractThe auto-reduction of copper and manganese acetates has been manipulated, to tailor specific Cu/Mn/O phases for the purpose of investigating their relation to activity for CO oxidation. A range of phases were produced from Hopcalite to discrete metallic copper particles supported by manganese oxide. The Hopcalite spinel phase was found to be required for the activity, while copper and manganese oxides were found to be inactive.Download high-res image (50KB)Download full-size imageHighlights► The use of supercritical anti-solvent precipitation allows for the formation of well-mixed metal acetates. ► The auto-reduction of Cu and Mn acetates has been controlled to tailor specific phase formation for synthesising catalysts. ► MnOx-supported Cu nanoparticles or CuMnOx spinel structures were formed, depending on the heat treatment conditions. ► The ability to tune oxidation state and phase composition of catalysts is a key preparation parameter for controlling the activity.
Co-reporter:Nicholas F. Dummer, Robert Jenkins, Xiabao Li, Salem M. Bawaked, Paul McMorn, Andrew Burrows, Christopher J. Kiely, Richard P.K. Wells, David J. Willock, Graham J. Hutchings
Journal of Catalysis (25 January 2007) Volume 245(Issue 2) pp:
Publication Date(Web):25 January 2007
DOI:10.1016/j.jcat.2006.11.010
Co-reporter:Graham J. Hutchings
Catalysis Today (15 October 2008) Volume 138(Issues 1–2) pp:9-14
Publication Date(Web):15 October 2008
DOI:10.1016/j.cattod.2008.04.029
Catalysts based on gold are now well established as very active and selective for broad ranges of redox reactions. Although primarily known for selective and preferential oxidation reactions, gold catalysts are also highly effective for selective hydrogenation. Hydrogenation reactions provide the focus for this perspective paper that is based on a François Gault lecture given at the Sabatier Conference in 2007. In particular, two reactions will be discussed; namely, the use of supported gold catalysts for selective hydrogenation of α,β-unsaturated aldehydes to unsaturated alcohols, and the use of supported gold palladium alloys for the direct hydrogenation of molecular oxygen to form hydrogen peroxide in preference to water.
Co-reporter:Edwin N. Ntainjua, Marco Piccinini, James C. Pritchard, Jennifer K. Edwards, Albert F. Carley, Christopher J. Kiely, Graham J. Hutchings
Catalysis Today (15 December 2011) Volume 178(Issue 1) pp:47-50
Publication Date(Web):15 December 2011
DOI:10.1016/j.cattod.2011.06.024
A key discovery in the last two decades has been the realisation that gold, when prepared as supported nanoparticles, is exceptionally effective as a redox catalyst. The catalytic efficacy is enhanced further by the alloying of gold with palladium and this is particularly exemplified for the direct synthesis of hydrogen peroxide where supported gold palladium alloy nanoparticles are found to be highly active. In this paper we report a study of ceria-supported gold, palladium and gold–palladium nanoparticles for the direct synthesis of hydrogen peroxide and show that ceria can be a potentially interesting support for this reaction. However, with current methods of catalyst fabrication ceria-supported monometallic palladium catalysts have a superior performance to bimetallic gold–palladium catalysts.Graphical abstractPalladium supported on ceria is a very active and reusable catalyst for the direct synthesis of hydrogen peroxide.Download high-res image (107KB)Download full-size imageHighlights► Observation of direct synthesis of hydrogen peroxide using ceria as a support with Pd nanoparticles gives enhanced activity. ► Addition of Au does not lead to a synergistic enhancement in contrast to all previous AuPd supported catalysts. ► Ceria supported AuPd and Pd catalysts are stable and reusable for this reaction.
Co-reporter:David Freeman, Richard P. K. Wells and Graham J. Hutchings
Chemical Communications 2001(Issue 18) pp:NaN1755-1755
Publication Date(Web):2001/08/28
DOI:10.1039/B104844A
Addition of β-Ga2O3 to H-ZSM-5, as
a physical mixture, enhances the formation of aromatic hydrocarbons in the
methanol to hydrocarbons reaction.
Co-reporter:Moataz Morad, Ewa Nowicka, Mark Douthwaite, Sarwat Iqbal, Peter Miedziak, Jennifer K. Edwards, Gemma L. Brett, Qian He, David Morgan, Hamed Alshammari, Donald Bethell, David W. Knight, Meenakshisundaram Sankar and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 9) pp:NaN1936-1936
Publication Date(Web):2017/03/28
DOI:10.1039/C7CY00184C
We report the one-pot tandem synthesis of 4-(4-methoxyphenyl)butan-2-one directly from 4-methoxybenzyl alcohol and acetone using a multifunctional supported AuPd nanoalloy catalyst. This one-pot synthesis involves dehydrogenation, aldol condensation and hydrogenation of CC. In this supported AuPd catalyst, the bimetallic sites catalyse the dehydrogenation and hydrogenation steps and, in combination with the support, catalyse the C–C coupling (aldol) process. This supported bimetallic catalyst is also effective in utilizing hydrogen from the dehydrogenation reaction for the hydrogenation of 4-(4-methoxyphenyl)but-3-en-2-one to 4-(4-methoxyphenyl)butane-2-one via a hydrogen auto transfer route. These multifunctional catalysts were characterised using transmission electron microscopy, X-ray diffraction and X-ray photoelectron spectroscopy.
Co-reporter:Charlotte L. Bracey, Peter R. Ellis and Graham J. Hutchings
Chemical Society Reviews 2009 - vol. 38(Issue 8) pp:NaN2243-2243
Publication Date(Web):2009/06/01
DOI:10.1039/B817729P
The use of nanoalloys in catalysis is a rapidly expanding field. There has been immense interest in the use of supported gold nanoparticles as catalysts, and bimetallic catalysts containing gold in combination with other metals represents an emerging field of research. While bulk copper–gold alloys are well-known and, indeed, are much studied systems, bimetallic copper–gold nanoalloys have received relatively little attention. In this tutorial review we review the literature on bimetallic CuAu catalysts and present some options for their future development.
Co-reporter:Raiedhah Alsaiari, Luke T. Perrott, Ewa Nowicka, Rebecca V. Engel, Peter J. Miedziak, Simon A. Kondrat, Jennifer K. Edwards, David J. Willock and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 6) pp:NaN1439-1439
Publication Date(Web):2017/03/07
DOI:10.1039/C6CY02448C
Carbon dioxide utilisation technology can contribute to the reduction of atmospheric CO2 levels both through its sequestration from flue gases and indirectly by relieving pressure on conventional feedstocks in chemical manufacturing. A promising approach is to employ CO2 to produce valuable cyclic carbonates (CCs) in reaction with suitable epoxides. This also has the advantage that carbon dioxide replaces toxic and hazardous reactants such as phosgene. In earlier work we have investigated the synthesis of epoxides from cycloalkenes using supported gold and gold–palladium nanoparticles as catalysts and oxygen from air as the oxidant under solvent free conditions. A strong dependence of epoxide selectivity on ring size was observed with C5 < C6 < C7 ≪ C8. In this study we extend this work to the investigation of cycloaddition of CO2 to different cycloalkene oxides with the ultimate aim of designing a process in which both epoxidation of an alkene and incorporation of CO2 could be achieved in a single process. However, we have found the opposite trend for the selectivity to carbonates: smaller ring cycloalkene oxides giving the highest carbonate selectivities while large rings do not yield CCs at all. The product distributions suggest that an alternative ring opening of the epoxides to yield alcohols and ketones is preferred under all the experimental conditions explored for larger ring systems. Additionally, the mechanism of the CC synthesis using a quaternary ammonium salt and ZnBr2 as the catalyst system was investigated using DFT methods. The results of the calculations support the experimental findings.
Co-reporter:Alberto Villa, Nikolaos Dimitratos, Carine E. Chan-Thaw, Ceri Hammond, Gabriel M. Veith, Di Wang, Maela Manzoli, Laura Prati and Graham J. Hutchings
Chemical Society Reviews 2016 - vol. 45(Issue 18) pp:NaN4994-4994
Publication Date(Web):2016/05/20
DOI:10.1039/C5CS00350D
Au-based catalysts have established a new important field of catalysis, revealing specific properties in terms of both high activity and selectivity for many reactions. However, the correlation between the morphology and the activity of the catalyst is not always clear although much effort has been addressed to this task. To some extent the problem relates to the complexity of the characterisation techniques that can be applied to Au catalyst and the broad range of ways in which they can be prepared. Indeed, in many reports only a few characterization techniques have been used to investigate the potential nature of the active sites. The aim of this review is to provide a critical description of the techniques that are most commonly used as well as the more advanced characterization techniques available for this task. The techniques that we discuss are (i) transmission electron microscopy methods, (ii) X-ray spectroscopy techniques, (iii) vibrational spectroscopy techniques and (iv) chemisorption methods. The description is coupled with developing an understanding of a number of preparation methods. In the final section the example of the supported AuPd alloy catalyst is discussed to show how the techniques can gain an understanding of an active oxidation catalyst.
Co-reporter:Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, David Morgan, Albert F. Carley, Ramchandra Tiruvalam, Christopher J. Kiely, Donald Bethell and Graham J. Hutchings
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 25) pp:NaN5153-5153
Publication Date(Web):2009/04/03
DOI:10.1039/B900151B
We report the preparation of Au–Pd nanocrystalline catalysts supported on TiO2 and carbon prepared via a sol-immobilisation technique using three different preparation strategies; namely, simultaneous formation of the sols for both metals or initial formation of a seed sol of one of the metals followed by a separate step in which a coating sol of the second metal is added. The catalysts have been structurally characterised using a combination of transmission electron microscopy and X-ray photoelectron spectroscopy. The catalysts have been evaluated for the oxidation of benzyl alcohol under solvent-free conditions. The catalysts prepared using the sol immobilisation technique show higher activity when compared with catalysts prepared by impregnation, particularly as lower metal concentrations can be used. The Au–Pd catalysts were all more active than the corresponding monometallic supported Au or Pd catalysts. For 1 wt% Au–Pd/TiO2 the order of metal addition in the preparation was not observed to be significant with respect to selectivity or activity. However, the 1 wt% Au–Pd/carbon catalysts are more active but less selective to benzaldehyde than the TiO2-supported catalysts when compared at iso-conversion. Furthermore, for the carbon-supported catalyst the order of metal addition has a very marked affect on activity. The carbon-supported catalysts are also more significantly affected by heat treatment, e.g.calcination at 400 °C leads to the activity being decreased by an order of magnitude, whereas the TiO2-supported catalysts show a 50% decrease in activity. Toluene is observed as a by-product of the reaction and conditions have been identified that minimise its formation. It is proposed that toluene and benzaldehyde are formed by competing parallel reactions of the initial benzyl intermediate via an adsorbed benzylidene species that can either be hydrogenated or oxidised. Hence, conditions that maximise the availability of oxygen on the catalyst surface favour the synthesis of benzaldehyde.
Co-reporter:Inés Moreno, Nicholas F. Dummer, Jennifer K. Edwards, Mosaed Alhumaimess, Meenakshisundaram Sankar, Raul Sanz, Patricia Pizarro, David P. Serrano and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 9) pp:NaN2434-2434
Publication Date(Web):2013/07/16
DOI:10.1039/C3CY00493G
Benzyl alcohol was oxidized by an “in situ generated” hydrogen peroxy species, formed from a dilute mixture of hydrogen and oxygen, under mild conditions at a high rate over gold, palladium and gold–palladium nanoparticles supported on hierarchical titanium silicate materials. Hierarchical TS-1 supports were obtained from the crystallization of silanized protozeolitic units, being characterized by having a secondary porous system within supermicro/mesopore range and an enhanced surface area over a standard reference TS-1 material. The presence of the secondary porosity not only improves the accessibility to the active sites of the relatively large reactant molecules but also enhances the metal dispersion, leading to an improved catalytic performance for alcohol oxidation. The catalytic activity of metal loaded hierarchical TS-1 materials was found to be higher in reactions conducted in the presence of diluted hydrogen and oxygen, resulting in a 5-fold increase in the yield of benzaldehyde at 30 °C with an AuPd catalyst with secondary porosity. The improvement in rate observed is due to the oxidizing efficacy of in situ generated hydroperoxy species as compared to molecular oxygen alone as the terminal oxidant.
Co-reporter:Gerolamo Budroni, Simon A. Kondrat, Stuart H. Taylor, David J. Morgan, Albert F. Carley, Peter B. Williams and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 10) pp:NaN2754-2754
Publication Date(Web):2013/08/01
DOI:10.1039/C3CY00449J
Alumina supported nickel palladium bimetallic catalysts were prepared by a selective redox deposition method and used for the hydrogenation of crotonaldehyde. Supported nickel catalysts, prepared by incipient wetness, were reduced to form a nickel hydride surface prior to contact with palladium(II) salt solutions. Palladium was successfully deposited selectively onto the nickel hydride surfaces, by a redox reaction. Catalysts were prepared using two different palladium(II) salt solutions; (a) pH 1 palladium chloride in hydrochloric acid solution or (b) pH 3 palladium nitrate in nitric acid. The deposition of palladium onto the supported nickel nanoparticles was strikingly different when using the two palladium solutions, with strong alloy formation with the pH 1 solution and a weaker segregated nickel palladium catalyst with the pH 3 solution. Both catalysts were compared with monometallic palladium and nickel supported catalysts for the hydrogenation of crotonaldehyde with the sample prepared at pH 1 being more active.
Co-reporter:Upendra Nath Gupta, Hamed Alshammari, Nicholas F. Dummer, Robert L. Jenkins, Donald Bethell and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 2) pp:NaN1313-1313
Publication Date(Web):2014/11/12
DOI:10.1039/C4CY01355G
The oxidation of dec-1-ene is investigated under solvent-free conditions using gold nanoparticles supported on graphite and in a batch reactor in the presence of a radical initiator using oxygen from air as the terminal oxidant. The evolution of the products with reaction time shows that there is an initial induction period and during this time very little epoxide is fomed and the products of allylic oxidation are dominant. Subsequently the epoxide becomes the major product prior to the diol being formed from hydrolysis due to the presence of by-product water formed from the selective oxidation reaction. It is considered that the allylic oxidation products are in part converted in situ into aldehydes which form peracids during the induction period; the peracid leads to epoxide formation as the major product as the conversion is increased. The effect of addition of a number of aldehydes is investigated, all leading to enhanced epoxide formation when added in small amounts. Molar enhancements of epoxide yield can approach twice the amount of aldehyde initially added. This behaviour is in contrast to earlier studies which utilise aldehydes in greater than stoichiometric amounts as sacrificial reactants. The importance of in situ aldehyde formation is also demonstrated by the addition of benzyl alcohol which under the reaction conditions rapidly gives benzaldehyde and enhanced epoxide formation. Possible mechanistic interpretations of the observations are discussed.
Co-reporter:Albert F. Carley, David J. Morgan, Nianxue Song, M. Wyn Roberts, Stuart H. Taylor, Jonathan K. Bartley, David J. Willock, Kara L. Howard and Graham J. Hutchings
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 7) pp:NaN2538-2538
Publication Date(Web):2010/12/10
DOI:10.1039/C0CP01852J
The oxidation of CO by Au/Fe2O3 and Au/ZnO catalysts is compared in the very early stages of the reaction using a temporal analysis of products (TAP) reactor. For Au/Fe2O3 pre-dosing the catalyst with 18O labelled water gives an unexpected evolution order for the labelled CO2 product with the C18O2 emerging first, whereas no temporal differentiation is found for Au/ZnO. High pressure XPS experiments are then used to show that CO bond cleavage does occur for model catalysts consisting of Au particles deposited on iron oxide films but not when deposited on ZnO films. DFT calculations, show that this observation requires carbon monoxide to dissociate in such a way that cleavage of the CO bond occurs along with dynamically co-adsorbed oxygen so that the overall process of Au oxidation and CO dissociation is energetically favourable. Our results show that for Au/Fe2O3 there is a pathway for CO oxidation that involves atomic C and O surface species which operates along side the bicarbonate mechanism that is widely discussed in the literature. However, this minor pathway is absent for Au/ZnO.
Co-reporter:Ren Su, Michael M. Forde, Qian He, Yanbin Shen, Xueqin Wang, Nikolaos Dimitratos, Stefan Wendt, Yudong Huang, Bo B. Iversen, Christopher J. Kiely, Flemming Besenbacher and Graham J. Hutchings
Dalton Transactions 2014 - vol. 43(Issue 40) pp:NaN14982-14982
Publication Date(Web):2014/06/19
DOI:10.1039/C4DT01309C
As co-catalyst materials, metal nanoparticles (NPs) play crucial roles in heterogeneous photocatalysis. The photocatalytic performance strongly relies on the physical properties (i.e., composition, microstructure, and surface impurities) of the metal NPs. Here we report a convenient chemical vapour impregnation (CVI) approach for the deposition of monometallic-, alloyed, and core–shell structured metal co-catalysts onto the TiO2 photocatalyst. The as-synthesised metal NPs are highly dispersed on the support and show narrow size distributions, which suit photocatalysis applications. More importantly, the surfaces of the as-synthesised metal NPs are free of protecting ligands, enabling the photocatalysts to be ready to use without further treatment. The effect of the metal identity, the alloy chemical composition, and the microstructure on the photocatalytic performance has been investigated for hydrogen production and phenol decomposition. Whilst the photocatalytic H2 production performance can be greatly enhanced by using the core–shell structured co-catalyst (Pdshell–Aucore and Ptshell–Aucore), the Ptshell–Aucore modified TiO2 yields enhanced quantum efficiency but a reduced effective decomposition of phenol to CO2 compared to that of the monometallic counterparts. We consider the CVI approach provides a feasible and elegant process for the decoration of photocatalyst materials.
Co-reporter:Marco Conte, Catherine J. Davies, David J. Morgan, Thomas E. Davies, Albert F. Carley, Peter Johnston and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 1) pp:NaN134-134
Publication Date(Web):2012/08/07
DOI:10.1039/C2CY20478A
The effect of the gold oxidation state and carbon structure on the activity of Au/C catalysts for the hydrochlorination of acetylene was investigated by a combined approach using TPR, XPS and porosimetry determinations. The activity of the catalyst in the synthesis of vinyl chloride monomer was found to be dependent on the presence of Au3+ species in the catalyst. However, by preparing catalysts with different Au3+ content it was possible to determine the existence of a threshold Au3+ amount, beyond which the excess of Au3+ was not active for the reaction. This was explained by the existence of active sites at the Au/C interface, and not just by the presence of Au3+ species on top of Au nanoparticles, as explained by current models for these catalysts. It was also possible to determine the existence of a subset of Au nanoclusters which do not take part in the reaction, as well as changes in the textural properties of the carbon that can affect its long term reusability.
Co-reporter:Hamed Alshammari, Peter J. Miedziak, David W. Knight, David J. Willock and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 6) pp:NaN1539-1539
Publication Date(Web):2013/03/04
DOI:10.1039/C3CY20864H
In this work we expand on our previous studies on the oxidation of cyclic alkenes using supported gold nanoparticles together with catalytic amounts of peroxides. We demonstrate that various sized cyclic alkenes can be oxidised by this catalyst, under green conditions, without solvent and using air as the oxidant gas. The effect of support, preparation method and choice of metal have been investigated, we demonstrate that supported gold is superior to palladium or a gold palladium alloy, we show that oxides provide the best support for these gold catalysts and the preparation methods that afford the smallest particles are the most active. We show that the reactivity of the cyclic alkenes is related to the ring size with the smaller rings more reactive than the larger rings at the same temperature. The selectivity to the epoxide is dependent on the size of the cyclic alkene ring. In particular, the epoxide selectivity is very low for rings containing fewer than seven carbon atoms. We discuss the origins of this selectivity effect, using DFT calculations to investigate the effect of ring strain on the intermediates and reaction products.
Co-reporter:Paul J. Smith, Simon A. Kondrat, Philip A. Chater, Benjamin R. Yeo, Greg M. Shaw, Li Lu, Jonathan K. Bartley, Stuart H. Taylor, Michael S. Spencer, Christopher J. Kiely, Gordon J. Kelly, Colin W. Park and Graham J. Hutchings
Chemical Science (2010-Present) 2017 - vol. 8(Issue 3) pp:NaN2447-2447
Publication Date(Web):2017/01/03
DOI:10.1039/C6SC04130B
Zincian georgeite, an amorphous copper–zinc hydroxycarbonate, has been prepared by co-precipitation using acetate salts and ammonium carbonate. Incorporation of zinc into the georgeite phase and mild ageing conditions inhibits crystallisation into zincian malachite or aurichalcite. This zincian georgeite precursor was used to prepare a Cu/ZnO catalyst, which exhibits a superior performance to a zincian malachite derived catalyst for methanol synthesis and the low temperature water–gas shift (LTS) reaction. Furthermore, the enhanced LTS activity and stability in comparison to that of a commercial Cu/ZnO/Al2O3 catalyst, indicates that the addition of alumina as a stabiliser may not be required for the zincian georgeite derived Cu/ZnO catalyst. The enhanced performance is partly attributed to the exclusion of alkali metals from the synthesis procedure, which are known to act as catalyst poisons. The effect of residual sodium on the microstructural properties of the catalyst precursor was investigated further from preparations using sodium carbonate.
Co-reporter:Adeeba Akram, Simon J. Freakley, Christian Reece, Marco Piccinini, Greg Shaw, Jennifer K. Edwards, Frédérique Desmedt, Pierre Miquel, Eero Seuna, David. J. Willock, Jacob A. Moulijn and Graham J. Hutchings
Chemical Science (2010-Present) 2016 - vol. 7(Issue 9) pp:NaN5837-5837
Publication Date(Web):2016/05/11
DOI:10.1039/C6SC01332E
Hydrogen peroxide synthesis from hydrogen and oxygen in the gas phase is postulated to be a key reaction step in the gas phase epoxidation of propene using gold–titanium silicate catalysts. During this process H2O2 is consumed in a secondary step to oxidise an organic molecule so is typically not observed as a reaction product. We demonstrate that using AuPd nanoparticles, which are known to have high H2O2 synthesis rates in the liquid phase, it is possible to not only oxidise organic molecules in the gas phase but to detect H2O2 for the first time as a reaction product in both a fixed bed reactor and a pulsed Temporal Analysis of Products (TAP) reactor without stabilisers present in the gas feed. This observation opens up possibility of synthesising H2O2 directly using a gas phase reaction.
Co-reporter:Nikolaos Dimitratos, Jose Antonio Lopez-Sanchez, Jinto Manjaly Anthonykutty, Gemma Brett, Albert F. Carley, Ram Chandra Tiruvalam, Andrew A. Herzing, Christopher J. Kiely, David W. Knight and Graham J. Hutchings
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 25) pp:NaN4961-4961
Publication Date(Web):2009/04/17
DOI:10.1039/B904317A
The use of bio-renewable resources for the generation of materials and chemicals continues to attract significant research attention. Glycerol, a by-product from biodiesel manufacture, is a highly functionalised renewable raw material, and in this paper the oxidation of glycerol in the presence of base using supported gold, palladium and gold–palladium alloys is described and discussed. Two supports, TiO2 and carbon, and two preparation methods, wet impregnation and sol-immobilisation, are compared and contrasted. For the monometallic catalysts prepared by impregnation similar activities are observed for Au and Pd, but the carbon-supported monometallic catalysts are more active than those on TiO2. Glycerate is the major product and lesser amounts of tartronate, glycolate, oxalate and formate are observed, suggesting a sequential oxidation pathway. Combining the gold and palladium as supported alloy nanocrystals leads to a significant enhancement in catalyst activity and the TiO2-supported catalysts are significantly more active for the impregnated catalysts. The use of a sol-immobilisation preparation method as compared to impregnation leads to the highest activity alloy catalysts and the origins of these activity trends are discussed.
Co-reporter:Jose Antonio Lopez-Sanchez, Nikolaos Dimitratos, Peter Miedziak, Edwin Ntainjua, Jennifer K. Edwards, David Morgan, Albert F. Carley, Ramchandra Tiruvalam, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 14) pp:NaN1930-1930
Publication Date(Web):2008/02/18
DOI:10.1039/B719345A
Catalysis by gold and gold–palladium nanoparticles has attracted significant research attention in recent years. These nanocrystalline materials have been found to be highly effective for selective and total oxidation, but in most cases the catalysts are prepared using precipitation or impregnation. We report the preparation of Au–Pd nanocrystalline catalysts supported on carbon prepared via a sol-immobilisation technique and these have been compared with Au–Pd catalysts prepared via impregnation. The catalysts have been evaluated for two selective chemical syntheses, namely, oxidation of benzyl alcohol and the direct synthesis of hydrogen peroxide. The catalysts have been structurally characterised using a combination of scanning transmission electron microscopy and X-ray photoelectron spectroscopy. The catalysts prepared using the sol immobilisation technique show higher activity when compared with catalysts prepared by impregnation as they are more active for both hydrogen peroxide synthesis and hydrogenation, and also for benzyl alcohol oxidation. The method facilitates the use of much lower metal concentrations which is a key feature in catalyst design, particularly for the synthesis of hydrogen peroxide.
Co-reporter:Raimon P. Marin, Simon A. Kondrat, Thomas E. Davies, David J. Morgan, Dan I. Enache, Gary B. Combes, Stuart H. Taylor, Jonathan K. Bartley and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 7) pp:NaN1978-1978
Publication Date(Web):2014/04/08
DOI:10.1039/C4CY00044G
Cobalt zinc oxide catalysts have been prepared by anti-solvent precipitation in supercritical CO2 and investigated for CO hydrogenation. Here we show how the textural and catalytic properties of the catalyst can be tailored by the addition of water to the initial solution of cobalt and zinc acetates in methanol. Characterization of the catalysts by powder X-ray diffraction, infra-red and Raman spectroscopy showed that in the absence of water a high surface area mixed acetate was produced which upon calcination formed wurtzite type Zn1−xCoxO and spinel type ZnxCo3−xO4. The addition of 5 vol.% water resulted in a phase separated Co3O4/ZnO catalyst and enhanced active cobalt surface area as a result of disruption of the solvent/CO2 phase equilibrium during precipitation.
Co-reporter:Meenakshisundaram Sankar, Nikolaos Dimitratos, Peter J. Miedziak, Peter P. Wells, Christopher J. Kiely and Graham J. Hutchings
Chemical Society Reviews 2012 - vol. 41(Issue 24) pp:NaN8139-8139
Publication Date(Web):2012/10/23
DOI:10.1039/C2CS35296F
This Critical Review provides an overview of the recent developments in the synthesis and characterization of bimetallic nanoparticles. Initially the review follows a materials science perspective on preparing bimetallic nanoparticles with designer morphologies, after which the emphasis shifts towards recent developments in using these bimetallic particles for catalysing either oxidation or reduction. In the final part of this review we present an overview of the utilization of bimetallic catalyst systems for the transformation of bio-renewable substrates and reactions related to the realization of a bio-refinery. Because of the sheer number of examples of transformations in this area, a few key examples, namely selective oxidation, hydrogenation/hydrogenolysis and reforming of biomass derived molecules, have been chosen for this review. Reports of bimetallic catalysts being used for the aforementioned transformations are critically analysed and the potential for exploiting such bimetallic catalysts have also been highlighted. A specific objective of this review article is to motivate researchers to synthesize some of the “designer” bimetallic catalysts with specific nanostructures, inspired from recent advances in the area of materials chemistry, and to utilize them for the transformation of biomass derived materials that are very complex and pose different challenges compared to those of simple organic molecules. We consider that supported bimetallic nanoparticles have an important role to play as catalysts in our quest for a more green and sustainable society.
Co-reporter:Alberto Villa, Simon J. Freakley, Marco Schiavoni, Jennifer K. Edwards, Ceri Hammond, Gabriel M. Veith, Wu Wang, Di Wang, Laura Prati, Nikolaos Dimitratos and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 3) pp:NaN697-697
Publication Date(Web):2015/12/03
DOI:10.1039/C5CY01880C
In this work, we show that the introduction of acidic oxygen functionalities to the surface of carbon nanofibers serves to depress the hydrogenation and the decomposition of hydrogen peroxide during the direct synthesis of H2O2. Moreover, the presence of acidic groups enhances the H2O2 productivity in the case of supported AuPd nanoparticles.
Co-reporter:Marco Conte, Jose A. Lopez-Sanchez, Qian He, David J. Morgan, Yulia Ryabenkova, Jonathan K. Bartley, Albert F. Carley, Stuart H. Taylor, Christopher J. Kiely, Karim Khalid and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 1) pp:NaN112-112
Publication Date(Web):2011/10/27
DOI:10.1039/C1CY00299F
Catalysts comprising zeolite ZSM-5 impregnated with precious metals including Ag, Cu, Ni, Pd, Ir and Ru, have been tested for the methanol to hydrocarbons reaction in a continuous flow fixed bed reactor. Comparison with the activity of unmodified ZSM-5 showed that Ag, Cu and Ni enhanced the selectivity to C6–C11 aromatic products by a factor of two or higher. Moreover, Ag/ZSM-5 showed improved selectivity for the C6–C7 fraction of aromatic products. Ni/ZSM-5 was found to be selective to naphthalene, while Cu/ZSM-5 was selective for C9–C11 aromatic products. It was ascertained that all the impregnated metals were present as metal oxides in the starting materials. It is therefore proposed that the enhanced selectivity to aromatic products is due to the interaction of the acid sites of the zeolite with the basic sites of the metal oxide at the edge of the zeolite crystals, as well as the possible coordination of propene molecules formed during the reaction, that are likely to be the building blocks for the formation of aromatics.
Co-reporter:Moataz Morad, Meenakshisundaram Sankar, Enhong Cao, Ewa Nowicka, Thomas E. Davies, Peter J. Miedziak, David J. Morgan, David W. Knight, Donald Bethell, Asterios Gavriilidis and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 9) pp:NaN3128-3128
Publication Date(Web):2014/05/22
DOI:10.1039/C4CY00387J
The synthesis of stable, supported, bimetallic nanoalloys with controlled size, morphology and composition is of great practical importance. Compared to their monometallic analogues, such materials exhibit remarkable enhancement in functional properties, which can be exploited in various fields including catalysis. Recently, we have reported a simple excess anion modification of the impregnation method to prepare supported gold–palladium catalysts which gives very good control over the particle sizes and the composition without using any stabilizer ligands in the preparation. Here, we report the results from a comparative study of using this modified impregnation catalyst for the solvent-free aerobic oxidation of alcohols in two different reactors: a glass stirred reactor and a micro packed bed reactor under batch and continuous mode respectively. These modified impregnation catalysts are exceptionally active and more importantly, when tested in a micro packed bed reactor under flow conditions, are observed to be stable for several days without any sign of deactivation in contrast to the same catalyst prepared by the sol immobilization method in the presence of stabilizer ligands which showed a 3–5% decrease in conversion over 10–12 h.
Co-reporter:Obaid F. Aldosari, Sarwat Iqbal, Peter J. Miedziak, Gemma L. Brett, Daniel R. Jones, Xi Liu, Jennifer K. Edwards, David J. Morgan, David K. Knight and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 1) pp:NaN242-242
Publication Date(Web):2015/11/03
DOI:10.1039/C5CY01650A
The selective hydrogenation of furfural at ambient temperature has been investigated using a Pd/TiO2 catalyst. The effect of the solvent was studied and high activity and selectivity to 2-methylfuran and furfuryl alcohol was observed using octane as solvent but a number of byproducts were observed. The addition of Ru to the PdTiO2 catalyst decreased the catalytic activity but improved the selectivity towards 2-methylfuran and furfuryl alcohol with decreased byproduct formation. Variation of the Ru/Pd ratio has shown an interesting effect on the selectivity. The addition of a small amount of Ru (1 wt%) shifted the selectivity towards furfuryl alcohol and 2-methylrofuran. Further increasing the Ru ratio decreased the catalytic activity and also showed a very poor selectivity to 2-methylfuran.
Co-reporter:Marco Conte, Xi Liu, Damien M. Murphy, Keith Whiston and Graham J. Hutchings
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 47) pp:NaN16285-16285
Publication Date(Web):2012/10/29
DOI:10.1039/C2CP43363J
The liquid phase oxidation of cyclohexane was undertaken using Au/MgO and the reaction mechanism was investigated by means of continuous wave (CW) EPR spectroscopy employing the spin trapping technique. Activity tests aimed to determine the conversion and selectivity of Au/MgO catalyst showed that Au was capable of selectivity control to cyclohexanol formation up to 70%, but this was accompanied by a limited enhancement in conversion when compared with the reaction in the absence of catalyst. In contrast, when radical initiators were used, in combination with Au/MgO, an activity comparable to that observed in industrial processes at ca. 5% conversion was found, with retained high selectivity. By studying the free radical autoxidation of cyclohexane and the cyclohexyl hydroperoxide decomposition in the presence of spin traps, we show that Au nanoparticles are capable of an enhanced generation of cyclohexyl alkoxy radicals, and the role of Au is identified as a promoter of the catalytic autoxidation processes, therefore demonstrating that the reaction proceeds via a radical chain mechanism.
Co-reporter:Virginie Peneau, Qian He, Gregory Shaw, Simon A. Kondrat, Thomas E. Davies, Peter Miedziak, Michael Forde, Nikolaos Dimitratos, Christopher J. Kiely and Graham J. Hutchings
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 26) pp:NaN10644-10644
Publication Date(Web):2013/04/02
DOI:10.1039/C3CP50361E
Supported nano-alloys have been prepared using the sol-immobilisation method for two bimetallic combinations, namely gold–platinum and palladium–platinum, using activated carbon and titania as supports. Some of the materials were prepared using a method where both metals are simultaneously reduced, thereby leading to homogeneous alloys being formed. In addition, sequential reduction of the metal combinations has also been investigated to facilitate the formation of core–shell structures. The materials have been characterized using X-ray photoelectron spectroscopy and aberration-corrected scanning transmission electron microscopy. The supported nanoparticles have been tested for a two selective oxidation reactions, namely the oxidation of toluene and benzyl alcohol using tertiary butyl hydroperoxide at 80 °C, in order to elucidate any potential structure–activity relationships.
Co-reporter:Mohd Hasbi Ab Rahim, Robert D. Armstrong, Ceri Hammond, Nikolaos Dimitratos, Simon J. Freakley, Michael M. Forde, David J. Morgan, Georgi Lalev, Robert L. Jenkins, Jose Antonio Lopez-Sanchez, Stuart H. Taylor and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 10) pp:NaN3418-3418
Publication Date(Web):2015/12/15
DOI:10.1039/C5CY01586C
The selective oxidation of methane to methanol has been studied using trimetallic AuPdCu/TiO2 catalysts prepared by incipient wetness impregnation. They are able to catalyse the selective oxidation of methane to methanol under mild aqueous reaction conditions using H2O2 as the oxidant. When compared with bimetallic, Au–Pd/TiO2 analogues, the new trimetallic catalysts present productivities which are up to 5 times greater under the same test conditions, and this is coupled with methanol selectivity of up to 83%. Characterisation shows that whilst Au–Pd is present as Au-core–Pd-shell nanoparticles, copper is present as either Cu or Cu2O in <5 nm particles.
Co-reporter:Marco Piccinini, Edwin Ntainjua N., Jennifer K. Edwards, Albert F. Carley, Jacob A. Moulijn and Graham J. Hutchings
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 10) pp:NaN2492-2492
Publication Date(Web):2010/01/21
DOI:10.1039/B921815G
The direct synthesis of hydrogen peroxide from H2 and O2 has been studied using a high activity AuPd/TiO2 catalyst. In particular, the effect of variation in the reaction conditions on the productivity of hydrogen peroxide formation is investigated in detail. The effect of H2/O2 molar ratio, temperature, total pressure and solvent composition has been studied and optimised conditions identified. In addition, the effect of carrying out the synthesis reaction in the presence of hydrogen peroxide is investigated and the competing reactions of hydrogen peroxide formation, decomposition and hydrogenation are discussed and optimal operating conditions are identified.
Co-reporter:Wilm Jones, Ren Su, Peter P. Wells, Yanbin Shen, Nikolaos Dimitratos, Michael Bowker, David Morgan, Bo B. Iversen, Arunabhiram Chutia, Flemming Besenbacher and Graham Hutchings
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 48) pp:NaN26644-26644
Publication Date(Web):2014/10/27
DOI:10.1039/C4CP04693E
The development of efficient photocatalytic routines for producing hydrogen is of great importance as society moves away from energy sources derived from fossil fuels. Recent studies have identified that the addition of metal nanoparticles to TiO2 greatly enhances the photocatalytic performance of these materials towards the reforming of alcohols for hydrogen production. The core–shell structured Au–Pd bimetallic nanoparticle supported on TiO2 has being of interest as it exhibited extremely high quantum efficiencies for hydrogen production. However, the effect of shell composition and thickness on photocatalytic performance remains unclear. Here we report the synthesis of core–shell structured AuPd NPs with the controlled deposition of one and two monolayers (ML) equivalent of Pd onto Au NPs by colloidal and photodeposition methods. We have determined the shell composition and thickness of the nanoparticles by a combination of X-ray absorption fine structure and X-ray photoelectron spectroscopy. Photocatalytic ethanol reforming showed that the core–shell structured Au–Pd promoters supported on TiO2 exhibit enhanced activity compared to that of monometallic Au and Pd as promoters, whilst the core–shell Au–Pd promoters containing one ML equivalent Pd provide the optimum reactivity.
Co-reporter:Ewa Nowicka, Jan P. Hofmann, Stewart F. Parker, Meenakshisundaram Sankar, Giacomo M. Lari, Simon A. Kondrat, David W. Knight, Donald Bethell, Bert M. Weckhuysen and Graham J. Hutchings
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 29) pp:NaN12155-12155
Publication Date(Web):2013/05/02
DOI:10.1039/C3CP50710F
In the solvent free oxidation of benzyl alcohol, using supported gold–palladium nanoalloys, toluene is often one of major by-products and it is formed by the disproportionation of benzyl alcohol. Gold–palladium catalysts on acidic supports promote both the disproportionation of benzyl alcohol and oxidative dehydrogenation to form benzaldehyde. Basic supports completely switch off disproportionation and the gold–palladium nanoparticles catalyse the oxidative dehydrogenation reaction exclusively. In an attempt to provide further details on the course of these reactions, we have utilized in situ ATR-IR, in situ DRIFT and inelastic neutron scattering spectroscopic methods, and in this article we present the results of these in situ spectroscopic studies.
Co-reporter:Giacomo M. Lari, Ewa Nowicka, David J. Morgan, Simon A. Kondrat and Graham J. Hutchings
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 35) pp:NaN23244-23244
Publication Date(Web):2015/08/05
DOI:10.1039/C5CP02512E
The sol immobilisation technique, in which a stabilising ligand (such as polyvinyl alcohol or polyvinyl pyridine) can be used to tune metal particle size and composition, has become a valuable method of making supported nanoparticle catalysts. An unfortunate consequence of the stabilising ligand is that often access of reactant molecules to the metal nanoparticle surface is impeded. Several methods have been proposed for the removal of these ligands, though determination of the degree of their success is difficult. Here, we demonstrate the use of in situ infrared and UV-Vis spectroscopy to elucidate the access of carbon monoxide to the surface of Au/TiO2 catalysts before and after various ligand removal treatments. These were contrasted with a catalyst prepared by deposition precipitation prepared in the absence of stabilising ligand as a control. Changes were observed in the infrared spectrum, with the wavenumber of carbon monoxide linearly bonded to Au for catalysts shifting before and after ligand removal, which correlated well with the activity of the catalyst for carbon monoxide oxidation. Also the extent of shifting of the Au surface resonance plasmon band on the addition of carbon monoxide, observed by UV-Vis, also correlated well with catalyst activity. These simple methods can be used to determine the quantity of exposed metal sites after a ligand removal treatment and so determine the treatments effectiveness.
Co-reporter:Daniel R. Jones, Sarwat Iqbal, Simon A. Kondrat, Giacomo M. Lari, Peter J. Miedziak, David J. Morgan, Stewart F. Parker and Graham J. Hutchings
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 26) pp:NaN17264-17264
Publication Date(Web):2016/04/01
DOI:10.1039/C6CP01311B
A series of ruthenium catalysts supported on two different carbons were tested for the hydrogenation of lactic acid to 1,2-propanediol and butanone to 2-butanol. The properties of the carbon supports were investigated by inelastic neutron scattering and correlated with the properties of the ruthenium deposited onto the carbons by wet impregnation or sol-immobilisation. It was noted that the rate of butanone hydrogenation was highly dependent on the carbon support, while no noticeable difference in rates was observed between different catalysts for the hydrogenation of lactic acid.
Co-reporter:Xi Liu, Marco Conte, Weihao Weng, Qian He, Robert L. Jenkins, Masashi Watanabe, David J. Morgan, David W. Knight, Damien M. Murphy, Keith Whiston, Christopher J. Kiely and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 1) pp:NaN227-227
Publication Date(Web):2014/10/23
DOI:10.1039/C4CY01213E
Molybdenum blue (MB), a multivalent molybdenum oxide with a nano-ring morphology is well-known in analytical chemistry but, to date it has been largely ignored in other applications. In the present work, MB has been characterized by STEM-HAADF imaging for the first time, showing the nano-ring morphology of this complex molybdenum oxide and the ordered super-molecular framework crystals that can result from the self-assembly of these MB nano-ring units. The potential of MB as an oxidation catalyst has also been investigated, where it is shown to have excellent catalytic activity and stability in the selective oxidation of cyclohexane to cyclohexanol and cyclohexanone which are important intermediates in the production of nylon.
Co-reporter:Gemma L. Brett, Peter J. Miedziak, Nikolaos Dimitratos, Jose A. Lopez-Sanchez, Nicholas F. Dummer, Ramchandra Tiruvalam, Christopher J. Kiely, David W. Knight, Stuart H. Taylor, David J. Morgan, Albert F. Carley and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 1) pp:NaN104-104
Publication Date(Web):2011/08/31
DOI:10.1039/C1CY00254F
The oxidative esterification of 1,2-propanediol to methyl lactate and methyl pyruvate was investigated using gold and gold palladium nanoparticles supported on a variety of supports. Methyl lactate can be used in cosmetics and personal care products, whereas methyl pyruvate is useful in the treatment of diseases of the nervous system. We show that gold-palladium alloy catalysts can be very effective for the oxidative esterification of 1,2-propanediol to methyl lactate and methyl pyruvate. Five supports, titania, carbon, silica, iron oxide and ceria are contrasted. The addition of palladium to gold significantly enhances the activity and retains the high selectivity to methyl lactate using O2 as oxidant. Using ceria as support a significant improvement in the selectivity to methyl lactate was observed, whereas using silica as support high selectivity to methyl pyruvate was achieved. The use of colloidal methods and the effect of support and Au/Pd molar ratio are discussed.
Co-reporter:Daniel R. Jones, Sarwat Iqbal, Satoshi Ishikawa, Christian Reece, Liam M. Thomas, Peter J. Miedziak, David J. Morgan, Jennifer K. Edwards, Jonathon K. Bartley, David J. Willock and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 15) pp:NaN6030-6030
Publication Date(Web):2016/05/03
DOI:10.1039/C6CY00382F
A series of Cu–ZrO2 catalysts prepared by a co-precipitation method were studied for the hydrogenation of levulinic acid to give γ-valerolactone (GVL). The effects of a range of catalyst preparation parameters, namely molar Cu/Zr ratio, calcination temperature and the ageing time of the precipitates, were systematically investigated. The molar Cu/Zr ratio was found to have a strong influence on the BET surface area of the material leading to a high activity for catalysts prepared with a Cu/Zr molar ratio of unity. Using this molar ratio the calcination temperature was varied from 300 °C to 800 °C, the material calcined at 400 °C showed the highest activity. Increasing the ageing time used in the catalyst preparation identified 6 h as the optimum to achieve the highest activity for LA conversion. Based on characterisation of all materials we conclude that the active Cu species is present in only low concentration suggesting that it should be possible to produce a catalyst of high activity with much lower Cu content.
Co-reporter:Salem Bawaked, Qian He, Nicholas F. Dummer, Albert F. Carley, David W. Knight, Donald Bethell, Christopher J. Kiely and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2011 - vol. 1(Issue 5) pp:NaN759-759
Publication Date(Web):2011/06/03
DOI:10.1039/C1CY00122A
Oxidation is an important route for the activation of chemical feedstocks for the synthesis of chemical intermediates. Alkene epoxidation by the electrophilic addition of oxygen to a carbon–carbon double bond is a major challenge in oxidation catalysis. In particular it is important to use molecular oxygen as the oxidant to avoid the formation of reagent by-products. We report the oxidation with air using graphite-supported gold-palladium catalysts of two alkenes, cis-cyclooctene, which gives mainly the epoxide, and crotyl alcohol (trans-but-2-en-1-ol). With cyclooctene, the reaction requires catalytic amounts of t-butyl hydroperoxide. The Au–Pd ratio has a major effect on the conversion with very low activities being associated with Au:Pd ratios of ca. 4:1 and 1:4 by weight. The selectivity to the epoxide is not affected by the Au:Pd ratio. With crotyl alcohol, t-butyl hydroperoxide was not required for activity. In the absence of Pd, crotonaldehyde was formed, but the introduction of Pd leads to an isomerisation pathway to 3-buten-1-ol being favoured over epoxidation and crotonaldehyde was a minor product.
Co-reporter:Charlotte L. Bracey, Albert F. Carley, Jennifer K. Edwards, Peter R. Ellis and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2011 - vol. 1(Issue 1) pp:NaN85-85
Publication Date(Web):2011/01/31
DOI:10.1039/C0CY00003E
The synthesis and catalytic application of supported CuAu is discussed. Different thermal treatments of a dried precursor of copper nitrate and tetrachloroauric acid on silica lead to catalysts with significantly different structures and properties. Direct calcination gives a catalyst which contains very large gold ensembles with minimal interaction with the copper present. Hydrogen reduction of the dried precursor leads to the formation of copper–gold alloy nanoparticles. Subsequent high temperature calcination de-alloys the copper from the gold to a significant extent. The presence of gold stabilises the formation of Cu+ on this catalyst. The activity and selectivity observed in the oxidation of propene with molecular oxygen, with or without co-fed hydrogen, depends on the pre-treatment, reaction conditions and the ratio of copper to gold in the catalyst. A number of different catalytic active sites are identified and discussed.
Co-reporter:Sivaram Pradhan, Rhys Lloyd, Jonathan K. Bartley, Donald Bethell, Stan Golunski, Robert L. Jenkins and Graham J. Hutchings
Chemical Science (2010-Present) 2012 - vol. 3(Issue 10) pp:NaN2964-2964
Publication Date(Web):2012/07/02
DOI:10.1039/C2SC20683H
Zeolite catalysts have been evaluated for the gas-phase conversion of decane, to study new routes for upgrading intermediate-length straight chain hydrocarbons. For a gas-feed of dilute n-decane in an inert carrier, at a contact time of 4 s, the initial activity of ZSM-5 and Ga/ZSM-5 was consistently high (>95% conversion) over the temperature range 300–460 °C. The parent zeolite produced almost equal yields of cracked hydrocarbons and aromatics, while the Ga-modified zeolite produced predominantly BTX and other heavier aromatics. This difference in product distribution is consistent with the short-chain alkanes formed within the internal pore structure of the zeolite being intermediates in a Cyclar-type aromatisation mechanism, while the direct conversion of decane to heavy (C10–C15) aromatics occurs at the unconstrained external acid sites. Under aerobic conditions, the rate of CO formation was negligible and CO2 was barely detectable over either the parent or the Ga-modified zeolite, even though all the O2 was consumed. The ability of Ga/ZSM-5 to catalyse selective oxidation, of the H2 released during the dehydrogenation steps, thus provides the prospect of the aromatisation reaction being operated autothermally. Although the external sites are preferentially blocked by carbon retention, rapid deactivation did not occur until after 65 h on line (under either anaerobic or aerobic conditions) when blocking of the internal pore structure became limiting.
Co-reporter:Yueling Cao, Xi Liu, Sarwat Iqbal, Peter J. Miedziak, Jennifer K. Edwards, Robert D. Armstrong, David J. Morgan, Junwei Wang and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 1) pp:NaN117-117
Publication Date(Web):2015/08/25
DOI:10.1039/C5CY00732A
1% Au/TiO2 catalysts prepared by a range of preparation methods were studied for the base-free oxidation of glucose. The highest catalytic activity was observed with the catalyst prepared by the sol-immobilization method. Furthermore we have studied the effect of the post-synthesis treatments of treatment with water, or heating in air on the activity. The catalyst calcined at 250 °C showed optimal activity and selectivity. Additionally, we studied the effect of the amount of stabilising ligand in the sol-immobilisation method and observed that this is a key parameter with respect to determining the catalyst's activity.
Co-reporter:Simon A. Kondrat, Greg Shaw, Simon J. Freakley, Qian He, Joanna Hampton, Jennifer K. Edwards, Peter J. Miedziak, Thomas E. Davies, Albert F. Carley, Stuart H. Taylor, Christopher. J. Kiely and Graham J. Hutchings
Chemical Science (2010-Present) 2012 - vol. 3(Issue 10) pp:NaN2971-2971
Publication Date(Web):2012/07/16
DOI:10.1039/C2SC20450A
We have prepared supported gold, palladium and gold–palladium bimetallic catalysts by the physical mixing of the acetate salts of the metals followed by a simple heat treatment. The use of the acetates as the metal precursor eliminates chloride from the catalyst preparation step. Extensive characterisation shows the formation of bimetallic alloy particles. These catalysts are extremely active for alcohol oxidations and the direct formation of hydrogen peroxide.
Co-reporter:Sarwat Iqbal, Xi Liu, Obaid F. Aldosari, Peter J. Miedziak, Jennifer K. Edwards, Gemma L. Brett, Adeeba Akram, Gavin M. King, Thomas E. Davies, David J. Morgan, David K. Knight and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2014 - vol. 4(Issue 8) pp:NaN2286-2286
Publication Date(Web):2014/04/14
DOI:10.1039/C4CY00184B
The selective hydrogenation of furfuryl alcohol into 2-methylfuran was investigated at room temperature using palladium supported catalysts. We have shown that Pd–TiO2 catalysts can be very effective for the synthesis of 2-methylfuran at room temperature and low pressure of hydrogen (1–3 bar). The effect of various reaction conditions (pressure, catalyst amount, and solvent) was studied.
Co-reporter:Peter J. Miedziak, Simon A. Kondrat, Noreen Sajjad, Gavin M. King, Mark Douthwaite, Greg Shaw, Gemma L. Brett, Jennifer K. Edwards, David J. Morgan, Ghulam Hussain and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 11) pp:NaN2917-2917
Publication Date(Web):2013/05/09
DOI:10.1039/C3CY00263B
We have investigated the optimisation of the catalytic parameters for the preparation of catalysts by the simple mixing and thermal treatment of a support and metal acetate precursors. We have studied the effect of metal ratio and metal loading to produce a catalyst which has the optimum activity for a variety of reactions including benzyl alcohol oxidation, glycerol oxidation and the direct synthesis of hydrogen peroxide. We have demonstrated the high activity of these catalyst's for a variety of substrates and performed XPS and XRD studies on the catalysts to help elucidate the origin of the catalysts improved activity.
Co-reporter:James Pritchard, Marco Piccinini, Ramchandra Tiruvalam, Quian He, Nikolaos Dimitratos, Jose A. Lopez-Sanchez, David J. Morgan, Albert F. Carley, Jennifer K. Edwards, Christopher J. Kiely and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2013 - vol. 3(Issue 2) pp:NaN317-317
Publication Date(Web):2012/05/16
DOI:10.1039/C2CY20234D
We have investigated the effect of heat treatment in air on Au–Pd nanoparticles supported on titania and activated carbon prepared via the immobilisation of PVA-stabilised alloy nanoparticles. The catalytic activity of the gold–palladium nanoparticles was affected by both metal and PVA loading, as well as the degree of interaction of the nanoparticles with the support. The turnover frequency numbers for benzyl alcohol and hydrogen peroxide synthesis were also sensitive to the calcination procedure employed and find a doubling of catalytic activity when using activated carbon as opposed to TiO2 as the support material. These results illustrate the importance of understanding the precise metal–support interaction of catalyst systems designed for benzyl alcohol oxidation and hydrogen peroxide synthesis.
Co-reporter:Junting Feng, Chao Ma, Peter J. Miedziak, Jennifer K. Edwards, Gemma L. Brett, Dianqing Li, Yiyun Du, David J. Morgan and Graham J. Hutchings
Dalton Transactions 2013 - vol. 42(Issue 40) pp:NaN14508-14508
Publication Date(Web):2013/08/19
DOI:10.1039/C3DT51855H
Au–Pd nanoalloys supported on Mg–Al mixed metal oxides prepared using sol-immobilisation are found to be highly efficient and reusable catalysts for the solvent-free oxidation of benzyl alcohol using molecular oxygen under low pressure. When using this support alloying Pd with Au resulted in an increase in both activity and selectivity to benzaldehyde and moreover an improved resistance to catalyst deactivation compared with the monometallic Pd and Au catalysts. The turnover number for the Au/Pd 1:1 molar ratio catalyst achieved 13000 after 240 min and the selectivity to benzaldehyde was maintained at 93%; this high catalytic activity can be retained in full after three successive uses. The ensemble and electronic effect of Au–Pd nanoalloys were studied by IR spectroscopy using CO chemisorption, XPS and HRTEM. Moreover, the bifunctional nature of the acid–base MgAl-MMO support was found to be important as the acid sites are considered to be responsible for the improvement of catalytic activity; while, the basic sites gave rise to high selectivity. A possible mechanism with Au–Pd nanoparticles as the active sites has been proposed, illustrating that the oxidation of benzyl alcohol can proceed through the cooperation between the Au–Pd nanoalloys and the base/acid sites on the surface of the support.
Co-reporter:Ceri Hammond, Jose A. Lopez-Sanchez, Mohd Hasbi Ab Rahim, Nikolaos Dimitratos, Robert L. Jenkins, Albert F. Carley, Qian He, Christopher J. Kiely, David W. Knight and Graham J. Hutchings
Dalton Transactions 2011 - vol. 40(Issue 15) pp:NaN3937-3937
Publication Date(Web):2011/01/21
DOI:10.1039/C0DT01389G
The reaction of glycerol with urea to form glycerol carbonate is mostly reported in the patent literature and to date there have been very few fundamental studies of the reaction mechanism. Furthermore, most previous studies have involved homogeneous catalysts whereas the identification of heterogeneous catalysts for this reaction would be highly beneficial. This is a very attractive reaction that utilises two inexpensive and readily available raw materials in a chemical cycle that overall, results in the chemical fixation of CO2. This reaction also provides a route to up-grade waste glycerol produced in large quantities during the production of biodiesel. Previous reports are largely based on the utilisation of high concentrations of metal sulfates or oxides, which suffer from low intrinsic activity and selectivity. We have identified heterogeneous catalysts based on gallium, zinc, and gold supported on a range of oxides and the zeolite ZSM-5, which facilitate this reaction. The addition of each component to ZSM-5 leads to an increase in the reaction yield towards glycerol carbonate, but supported gold catalysts display the highest activity. For gold-based catalysts, MgO is the support of choice. Catalysts have been characterised by XRD, TEM, STEM and XPS, and the reaction has been studied with time-on-line analysis of products via a combination of FT-IR spectroscopy, HPLC, 13C NMR and GC-MS analysis to evaluate the reaction pathway. Our proposed mechanism suggests that glycerol carbonate forms via the cyclization of a 2,3-dihydroxypropyl carbamate and that a subsequent reaction of glycerol carbonate with urea yields the carbamate of glycerol carbonate. Stability and reactivity studies indicate that consecutive reactions of glycerol carbonate can limit the selectivity achieved and reaction conditions can be selected to avoid this. The effect of the catalyst in the proposed mechanism is discussed.
Co-reporter:Ren Su, Lokesh Kesavan, Mads M. Jensen, Ramchandra Tiruvalam, Qian He, Nikolaos Dimitratos, Stefan Wendt, Marianne Glasius, Christopher J. Kiely, Graham J. Hutchings and Flemming Besenbacher
Chemical Communications 2014 - vol. 50(Issue 84) pp:NaN12614-12614
Publication Date(Web):2014/07/11
DOI:10.1039/C4CC04024D
The selectivity of photocatalytic phenol production from the direct oxidation of benzene can be enhanced by fine adjustment of the morphology and composition of Au–Pd metal nanoparticles supported on titanium dioxide thereby suppressing the decomposition of benzene and evolution of phenolic compounds.
Co-reporter:Mohd Hasbi Ab Rahim, Qian He, Jose A. Lopez-Sanchez, Ceri Hammond, Nikolaos Dimitratos, Meenakshisundaram Sankar, Albert F. Carley, Christopher J. Kiely, David W. Knight and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 9) pp:NaN1924-1924
Publication Date(Web):2012/05/23
DOI:10.1039/C2CY20288C
Supported Au–Pd nanoparticles are shown to be effective catalysts for the transformation of glycerol into glycerol carbonate. The reaction of glycerol with urea to form glycerol carbonate is a very attractive reaction that utilises two inexpensive and readily available raw materials in a chemical cycle that, overall, results in the chemical fixation of carbon dioxide. Previous reports are largely based on the utilisation of high concentrations of metal sulphates or oxides, which suffer from low intrinsic activity and selectivity and limited recoverability due to the dissolution of the catalyst in the reaction media. We now report that magnesium oxide is an excellent support for gold and bimetallic gold–palladium nanoparticles for this reaction. The preparation method and pre-treatment affect the catalytic performance and a colloidal preparation route produces the most active catalysts.
Co-reporter:Marco Piccinini, Jennifer K. Edwards, Jacob A. Moulijn and Graham J. Hutchings
Catalysis Science & Technology (2011-Present) 2012 - vol. 2(Issue 9) pp:NaN1913-1913
Publication Date(Web):2012/06/13
DOI:10.1039/C2CY20282D
The direct synthesis of hydrogen peroxide has been studied using a highly active AuPd/C catalyst, where the activated carbon support has been pretreated with dilute HNO3 prior to metal deposition and consequently using standard reaction conditions this catalyst does not hydrogenate H2O2. The effect of reaction variables has been investigated on the synthesis and hydrogenation activity over this catalyst. The effect of H2/O2 molar ratio, temperature, total pressure and solvent composition has been studied and optimised conditions identified. The effect of these conditions on the hydrogenation activity was also evaluated; thereby permitting an optimal set of reaction conditions to be identified for both the synthesis of H2O2 and its hydrogenation/decomposition.

![Naphth[1,2-b]oxirene, 1a,2,3,7b-tetrahydro-, (1aS,7bR)-](http://img.cochemist.com/ccimg/58900/58800-12-7.png)
![Naphth[1,2-b]oxirene, 1a,2,3,7b-tetrahydro-, (1aS,7bR)-](http://img.cochemist.com/ccimg/58900/58800-12-7_b.png)
![Aziridine, 2-(4-methoxyphenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-05-5.png)
![Aziridine, 2-(4-methoxyphenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-05-5_b.png)
![Aziridine, 2-(4-bromophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-01-1.png)
![Aziridine, 2-(4-bromophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-01-1_b.png)


![Aziridine, 2-methyl-1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-03-3.png)
![Aziridine, 2-methyl-1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-03-3_b.png)
![Aziridine, 2-(4-chlorophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-95-0.png)
![Aziridine, 2-(4-chlorophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-95-0_b.png)
![9-Oxabicyclo[6.1.0]nonane, (1R,8S)-rel-](http://img.cochemist.com/ccimg/5000/4925-71-7.png)
![9-Oxabicyclo[6.1.0]nonane, (1R,8S)-rel-](http://img.cochemist.com/ccimg/5000/4925-71-7_b.png)

![Aziridine, 2-(3-methylphenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-46-7.png)
![Aziridine, 2-(3-methylphenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-46-7_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, (2S-trans)-](http://img.cochemist.com/ccimg/147200/147127-07-9.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, (2S-trans)-](http://img.cochemist.com/ccimg/147200/147127-07-9_b.png)








![AZIRIDINE, 2-(3-FLUOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-98-3.png)
![AZIRIDINE, 2-(3-FLUOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-98-3_b.png)
![AZIRIDINE, 2-(3-BROMOPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-00-0.png)
![AZIRIDINE, 2-(3-BROMOPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-00-0_b.png)




![AZIRIDINE, 2-(2-METHYLPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-02-2.png)
![AZIRIDINE, 2-(2-METHYLPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832118-02-2_b.png)
![AZIRIDINE, 2-(2-FLUOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-96-1.png)
![AZIRIDINE, 2-(2-FLUOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-96-1_b.png)



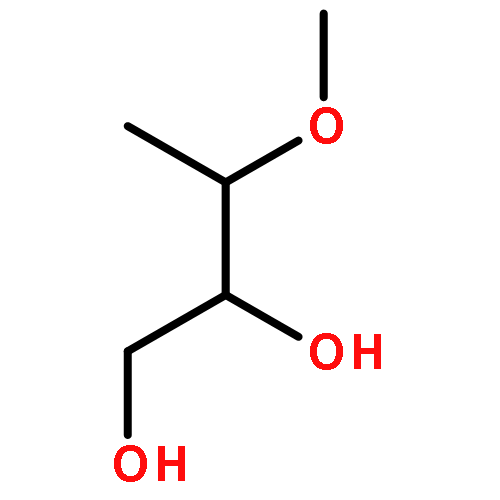
![AZIRIDINE, 2-METHYL-1-[(4-NITROPHENYL)SULFONYL]-3-PHENYL-, (3S)-](http://img.cochemist.com/ccimg/832200/832118-04-4.png)
![AZIRIDINE, 2-METHYL-1-[(4-NITROPHENYL)SULFONYL]-3-PHENYL-, (3S)-](http://img.cochemist.com/ccimg/832200/832118-04-4_b.png)
![AZIRIDINE, 2-(4-FLUOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-36-5.png)
![AZIRIDINE, 2-(4-FLUOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-36-5_b.png)
![AZIRIDINE, 2-(2-CHLOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-92-7.png)
![AZIRIDINE, 2-(2-CHLOROPHENYL)-1-[(4-NITROPHENYL)SULFONYL]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-92-7_b.png)


![1-Propanone, 3-[(4-methylphenyl)thio]-1,3-diphenyl-](http://img.cochemist.com/ccimg/95700/95625-60-8.png)
![1-Propanone, 3-[(4-methylphenyl)thio]-1,3-diphenyl-](http://img.cochemist.com/ccimg/95700/95625-60-8_b.png)
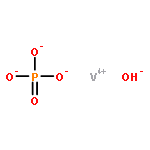
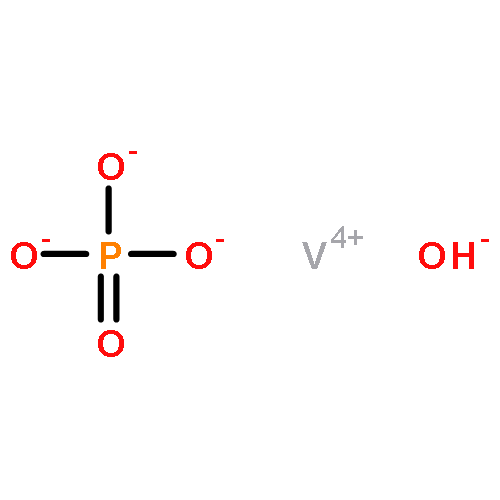


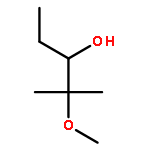


![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2R)-](http://img.cochemist.com/ccimg/194200/194156-25-7.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2R)-](http://img.cochemist.com/ccimg/194200/194156-25-7_b.png)






![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, cis-](http://img.cochemist.com/ccimg/17900/17879-98-0.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, cis-](http://img.cochemist.com/ccimg/17900/17879-98-0_b.png)


















![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, trans-](http://img.cochemist.com/ccimg/17900/17879-99-1.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2,3-diphenyl-, trans-](http://img.cochemist.com/ccimg/17900/17879-99-1_b.png)







![Aziridine, 1-[(2-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/392300/392274-55-4.png)
![Aziridine, 1-[(2-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/392300/392274-55-4_b.png)
![Acetic acid, [(diphenylmethyl)imino]-, ethyl ester](http://img.cochemist.com/ccimg/114400/114341-76-3.png)
![Acetic acid, [(diphenylmethyl)imino]-, ethyl ester](http://img.cochemist.com/ccimg/114400/114341-76-3_b.png)









![(r)-[(2s,5s)-5-ethenyl-1-azabicyclo[2.2.2]octan-2-yl]-quinolin-4-ylmethanol;hydrochloride](http://img.cochemist.com/ccimg/600/524-57-2.png)
![(r)-[(2s,5s)-5-ethenyl-1-azabicyclo[2.2.2]octan-2-yl]-quinolin-4-ylmethanol;hydrochloride](http://img.cochemist.com/ccimg/600/524-57-2_b.png)
![(2R)-1-[(4-chlorophenyl)sulfonyl]-2-phenylaziridine](http://img.cochemist.com/ccimg/50800/50707-39-6.png)
![(2R)-1-[(4-chlorophenyl)sulfonyl]-2-phenylaziridine](http://img.cochemist.com/ccimg/50800/50707-39-6_b.png)


![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/62600/62596-62-7.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (R)-](http://img.cochemist.com/ccimg/62600/62596-62-7_b.png)
![Aziridine, 2-(4-methylphenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-40-1.png)
![Aziridine, 2-(4-methylphenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-40-1_b.png)


![7-Azabicyclo[4.1.0]heptane, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/68900/68820-12-2.png)
![7-Azabicyclo[4.1.0]heptane, 7-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/68900/68820-12-2_b.png)




















![Aziridine, 2-(2-bromophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-99-4.png)
![Aziridine, 2-(2-bromophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-99-4_b.png)
![Aziridine, 2-(3-nitrophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-43-4.png)
![Aziridine, 2-(3-nitrophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/718700/718604-43-4_b.png)










![Aziridine, 2-(3-chlorophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-93-8.png)
![Aziridine, 2-(3-chlorophenyl)-1-[(4-nitrophenyl)sulfonyl]-, (2S)-](http://img.cochemist.com/ccimg/832200/832117-93-8_b.png)









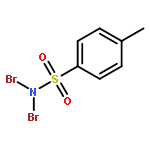








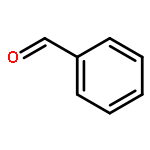
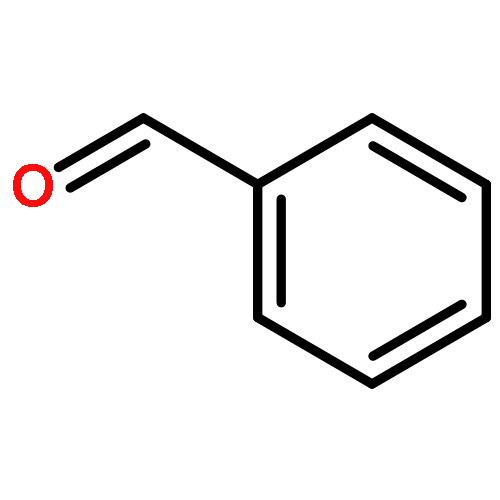
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0.png)
![Aziridine,1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/24400/24395-14-0_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-3-phenyl-, trans-](http://img.cochemist.com/ccimg/137600/137595-21-2_b.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/137600/137595-22-3.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/137600/137595-22-3_b.png)











![Aziridine, 2-(4-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-93-9.png)
![Aziridine, 2-(4-chlorophenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-93-9_b.png)

















![Aziridine, 2-(4-methylphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-87-1.png)
![Aziridine, 2-(4-methylphenyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/97500/97401-87-1_b.png)
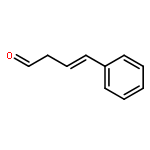
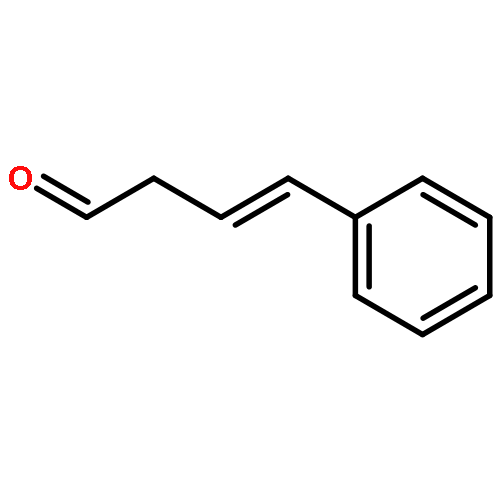


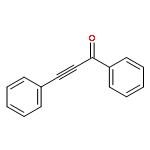
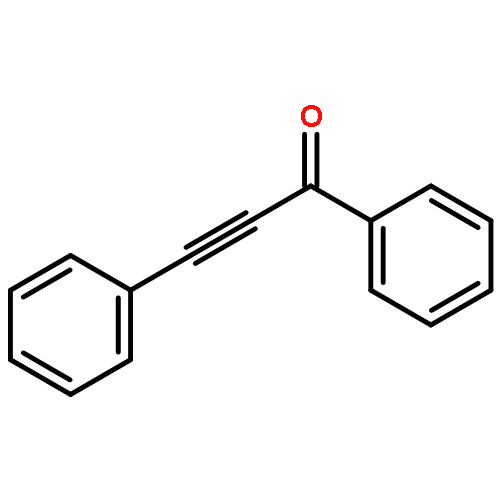


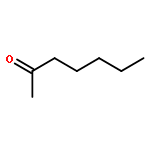
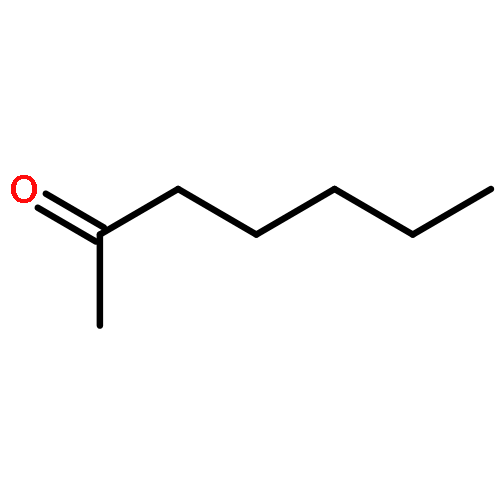
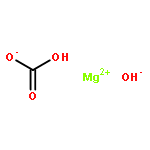
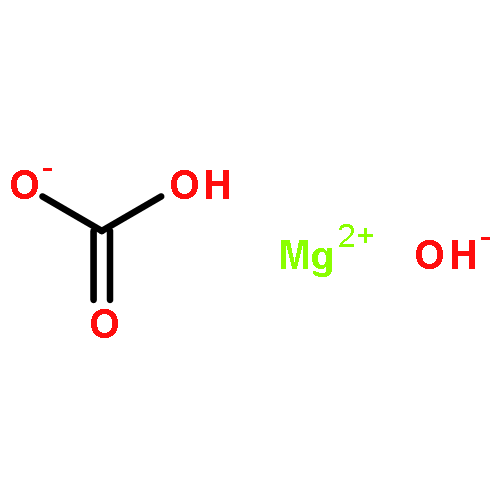




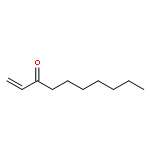
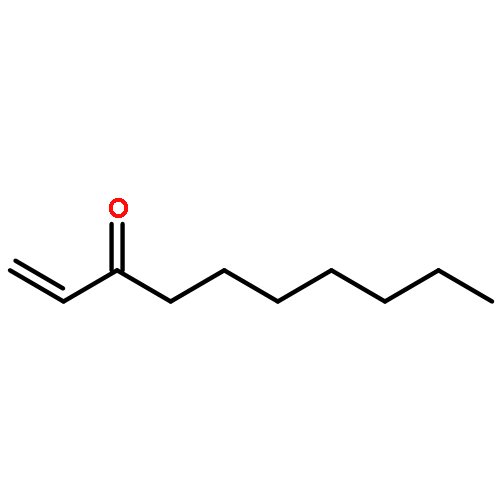







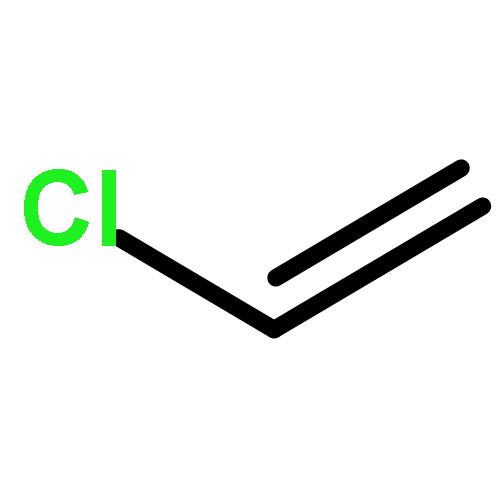





















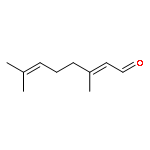
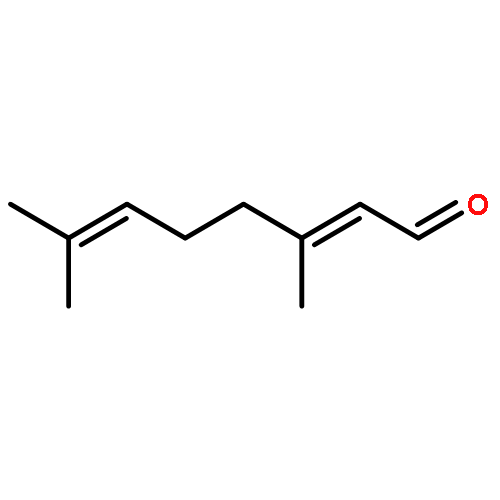
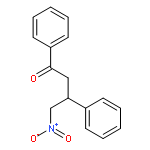
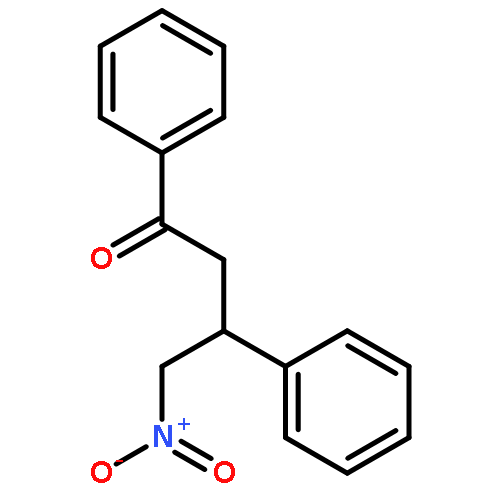






![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/194200/194156-28-0.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/194200/194156-28-0_b.png)
![Aziridine, 1-[(4-methoxyphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/181400/181306-57-0.png)
![Aziridine, 1-[(4-methoxyphenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/181400/181306-57-0_b.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/175300/175222-83-0.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-phenyl-](http://img.cochemist.com/ccimg/175300/175222-83-0_b.png)




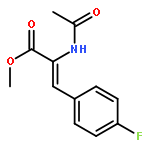

![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/149800/149769-84-6.png)
![Aziridine, 1-[(4-methylphenyl)sulfonyl]-2-phenyl-, (2S)-](http://img.cochemist.com/ccimg/149800/149769-84-6_b.png)

















![2,2′-Methylenebis[(4S)-4-tert-butyl-2-oxazoline]](/data/chemimg/106700/132098-54-5.png)
![2,2′-Methylenebis[(4S)-4-tert-butyl-2-oxazoline]](/data/chemimg/106700/132098-54-5_b.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4_b.png)