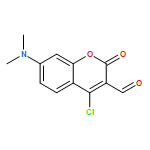
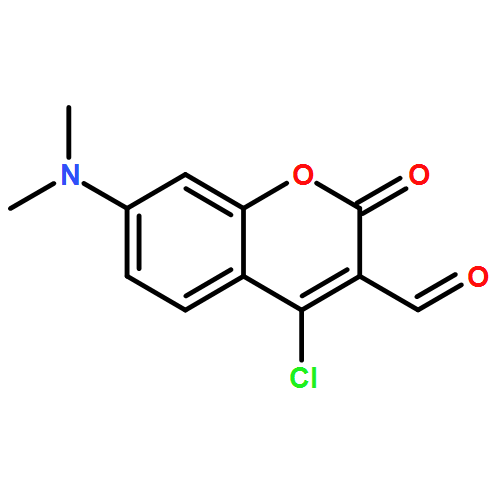
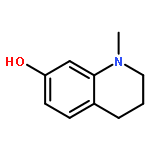
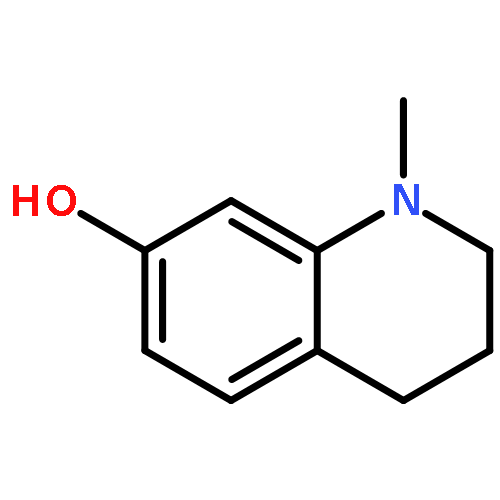
Co-reporter:Richard Wolfenden;Charles A. Lewis, Jr.;Ronald Swanstrom;Jesse Crayle;Shuntai Zhou
PNAS 2016 Volume 113 (Issue 29 ) pp:8194-8199
Publication Date(Web):2016-07-19
DOI:10.1073/pnas.1607580113
The hydrolytic deamination of cytosine and 5-methylcytosine residues in DNA appears to contribute significantly to the appearance
of spontaneous mutations in microorganisms and in human disease. In the present work, we examined the mechanism of cytosine
deamination and the response of the uncatalyzed reaction to changing temperature. The positively charged 1,3-dimethylcytosinium
ion was hydrolyzed at a rate similar to the rate of acid-catalyzed hydrolysis of 1-methylcytosine, for which it furnishes
a satisfactory kinetic model and a probable mechanism. In agreement with earlier reports, uncatalyzed deamination was found
to proceed at very similar rates for cytosine, 1-methylcytosine, cytidine, and cytidine 5′-phosphate, and also for cytosine
residues in single-stranded DNA generated from a phagemid, in which we sequenced an insert representing the gene of the HIV-1
protease. Arrhenius plots for the uncatalyzed deamination of cytosine were linear over the temperature range from 90 °C to
200 °C and indicated a heat of activation (ΔH‡) of 23.4 ± 0.5 kcal/mol at pH 7. Recent evidence indicates that the surface of the earth has been cool enough to support
life for more than 4 billion years and that life has been present for almost as long. If the temperature at Earth's surface
is assumed to have followed Newton's law of cooling, declining exponentially from 100 °C to 25 °C during that period, then
half of the cytosine-deaminating events per unit biomass would have taken place during the first 0.2 billion years, and <99.4%
would have occurred during the first 2 billion years.
Co-reporter:Richard Wolfenden;Charles A. Lewis Jr.;Yang Yuan;Charles W. Carter Jr.
PNAS 2015 112 (24 ) pp:7484-7488
Publication Date(Web):2015-06-16
DOI:10.1073/pnas.1507565112
The hydrophobicities of the 20 common amino acids are reflected in their tendencies to appear in interior positions in globular
proteins and in deeply buried positions of membrane proteins. To determine whether these relationships might also have been
valid in the warm surroundings where life may have originated, we examined the effect of temperature on the hydrophobicities
of the amino acids as measured by the equilibrium constants for transfer of their side-chains from neutral solution to cyclohexane
(Kw>c). The hydrophobicities of most amino acids were found to increase with increasing temperature. Because that effect is more
pronounced for the more polar amino acids, the numerical range of Kw>c values decreases with increasing temperature. There are also modest changes in the ordering of the more polar amino acids.
However, those changes are such that they would have tended to minimize the otherwise disruptive effects of a changing thermal
environment on the evolution of protein structure. Earlier, the genetic code was found to be organized in such a way that—with
a single exception (threonine)—the side-chain dichotomy polar/nonpolar matches the nucleic acid base dichotomy purine/pyrimidine
at the second position of each coding triplet at 25 °C. That dichotomy is preserved at 100 °C. The accessible surface areas
of amino acid side-chains in folded proteins are moderately correlated with hydrophobicity, but when free energies of vapor-to-cyclohexane
transfer (corresponding to size) are taken into consideration, a closer relationship becomes apparent.
Co-reporter:Richard Wolfenden;Charles A. Lewis Jr.;Yang Yuan;Charles W. Carter Jr.
PNAS 2015 112 (24 ) pp:7484-7488
Publication Date(Web):2015-06-16
DOI:10.1073/pnas.1507565112
The hydrophobicities of the 20 common amino acids are reflected in their tendencies to appear in interior positions in globular
proteins and in deeply buried positions of membrane proteins. To determine whether these relationships might also have been
valid in the warm surroundings where life may have originated, we examined the effect of temperature on the hydrophobicities
of the amino acids as measured by the equilibrium constants for transfer of their side-chains from neutral solution to cyclohexane
(Kw>c). The hydrophobicities of most amino acids were found to increase with increasing temperature. Because that effect is more
pronounced for the more polar amino acids, the numerical range of Kw>c values decreases with increasing temperature. There are also modest changes in the ordering of the more polar amino acids.
However, those changes are such that they would have tended to minimize the otherwise disruptive effects of a changing thermal
environment on the evolution of protein structure. Earlier, the genetic code was found to be organized in such a way that—with
a single exception (threonine)—the side-chain dichotomy polar/nonpolar matches the nucleic acid base dichotomy purine/pyrimidine
at the second position of each coding triplet at 25 °C. That dichotomy is preserved at 100 °C. The accessible surface areas
of amino acid side-chains in folded proteins are moderately correlated with hydrophobicity, but when free energies of vapor-to-cyclohexane
transfer (corresponding to size) are taken into consideration, a closer relationship becomes apparent.
Co-reporter:Richard Wolfenden
Cellular and Molecular Life Sciences 2014 Volume 71( Issue 15) pp:2909-2915
Publication Date(Web):2014 August
DOI:10.1007/s00018-014-1587-2
Ever since the publication of Darwin’s Origin of Species, questions have been raised about whether enough time has elapsed for living organisms to have reached their present level of complexity by mutation and natural selection. More recently, it has become apparent that life originated very early in Earth’s history, and there has been controversy as to whether life originated in a hot or cold environment. This review describes evidence that rising temperature accelerates slow reactions disproportionately, and to a much greater extent than has been generally recognized. Thus, the time that would have been required for primordial chemistry to become established would have been abbreviated profoundly at high temperatures. Moreover, if the catalytic effect of a primitive enzyme (like that of modern enzymes) were to reduce a reaction’s heat of activation, then the rate enhancement that it produced would have increased as the surroundings cooled, quite aside from changes arising from mutation (which is itself highly sensitive to temperature). Some nonenzymatic catalysts of slow reactions, including PLP as a catalyst of amino acid decarboxylation, and the CeIV ion as a catalyst of phosphate ester hydrolysis, have been shown to meet that criterion. The work reviewed here suggests that elevated temperatures collapsed the time required for early evolution on Earth, furnishing an appropriate setting for exploring the vast range of chemical possibilities and for the rapid evolution of enzymes from primitive catalysts.
Co-reporter:Danielle C. Lohman ; David R. Edwards
Journal of the American Chemical Society 2013 Volume 135(Issue 39) pp:14473-14475
Publication Date(Web):September 16, 2013
DOI:10.1021/ja406381b
In the biological fixation of halide ions, several enzymes have been found to catalyze alkyl transfer from S-adenosylmethionine to halide ions. It proves possible to measure the rates of reaction of the trimethylsulfonium ion with I–, Br–, Cl–, F–, HO–, and H2O in water at elevated temperatures. Comparison of the resulting second-order rate constants, extrapolated to 25 °C, with the values of kcat/Km reported for fluorinase and chlorinase indicates that these enzymes enhance the rates of alkyl halide formation by factors of 2 × 1015- and 1 × 1017-fold, respectively. These rate enhancements, achieved without the assistance of cofactors, metal ions, or general acid–base catalysis, are the largest that have been reported for an enzyme that acts on two substrates.
Co-reporter:David R. Edwards and Richard Wolfenden
The Journal of Organic Chemistry 2012 Volume 77(Issue 9) pp:4450-4453
Publication Date(Web):April 9, 2012
DOI:10.1021/jo300386u
The hydrolysis of N-methyl O-phenyl sulfamate (1) has been studied as a model for steroid sulfatase inhibitors such as Coumate, 667 Coumate, and EMATE. At neutral pH, simulating physiological conditions, hydrolysis of 1 involves an intramolecular proton transfer from nitrogen to the bridging oxygen atom of the leaving group. Remarkably, this proton transfer is estimated to accelerate the decomposition of 1 by a factor of 1011. Examination of existing kinetic data reveals that the sulfatase PaAstA catalyzes the hydrolysis of sulfamate esters with catalytic rate accelerations of ∼104, whereas the catalytic rate acceleration generated by the enzyme for its cognate substrate is on the order of ∼1015. Rate constants for hydrolysis of a wide range of sulfuryl esters, ArOSO2X–, are shown to be correlated by a two-parameter equation based on pKaArOH and pKaArOSO2XH.
Co-reporter:Richard Wolfenden ; Charles A. Lewis ; Jr.;Yang Yuan
Journal of the American Chemical Society 2011 Volume 133(Issue 15) pp:5683-5685
Publication Date(Web):March 24, 2011
DOI:10.1021/ja111457h
To compare the powers of the corresponding enzymes as catalysts, the rates of uncatalyzed decarboxylation of several aliphatic acids (oxalate, malonate, acetoacetate, and oxaloacetate) were determined at elevated temperatures and extrapolated to 25 °C. In the extreme case of oxalate, the rate of the uncatalyzed reaction at pH 4.2 was 1.1 × 10−12 s−1, implying a 2.5 × 1013-fold rate enhancement by oxalate decarboxylase. Whereas the enzymatic decarboxylation of oxalate requires O2 and MnII, the uncatalyzed reaction is unaffected by the presence of these cofactors and appears to proceed by heterolytic elimination of CO2.
Co-reporter:Richard Wolfenden ;Yang Yuan
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:13821-13823
Publication Date(Web):July 27, 2011
DOI:10.1021/ja204116a
Experiments at elevated temperatures permit the determination of rate constant and thermodynamic activation parameters for the neutral hydrolysis of the neurotransmitter acetylcholine in water. At 25 °C, the extrapolated rate constant for the uncatalyzed (or neutral) hydrolysis of acetylcholine is 3.9 × 10–7 s–1 at 25 °C (ΔH‡ = 20.0 kcal/mol; TΔS‡ = −6.1 kcal/mol). Acetylcholine is more susceptible to neutral and base-catalyzed hydrolysis than ethyl acetate but less susceptible to acid-catalyzed hydrolysis. For acetylcholinesterase from the electric eel, the catalytic proficiency [(kcat/Km)/kneutral] is 2 × 1016 M–1, comparable in magnitude with the catalytic proficiencies of aminohydrolases that act on peptides and nucleosides.
Co-reporter:Randy B. Stockbridge and Richard Wolfenden
Chemical Communications 2010 vol. 46(Issue 24) pp:4306-4308
Publication Date(Web):06 May 2010
DOI:10.1039/C0CC00229A
The hydrolysis of phosphate diesters is one of the most difficult reactions known. Here we show that in acetone or cyclohexane, at 25 °C, phosphodiesters undergo hydrolysis 5 × 105 and 2 × 109-fold more rapidly than in water, respectively, and that this rate enhancement is achieved by lowering the enthalpy of activation.
Co-reporter:Randy B. Stockbridge, Gottfried K. Schroeder, Richard Wolfenden
Bioorganic Chemistry 2010 Volume 38(Issue 5) pp:224-228
Publication Date(Web):October 2010
DOI:10.1016/j.bioorg.2010.05.003
Previous estimates of the rate of spontaneous cleavage of the glycosidic bond of adenosine were determined by extrapolating the rates of the acid- and base-catalyzed reactions to neutral pH. Here we show that cleavage also proceeds through a pH-independent mechanism. Rate constants were determined as a function of temperature at pH 7 and a linear Arrhenius plot was constructed. Uncatalyzed cleavage occurs with a rate constant of 3.7 × 10−12 s−1 at 25 °C, and the rate enhancement generated by the corresponding glycoside hydrolase is ∼5 × 1012-fold.We show that hydrolysis of the glycosidic CN bond of adenosine, previously known to be subject to hydrolysis in acid and base, also proceeds through a pH-independent mechanism with a rate constant of 4 × 10−12 s−1 at 25 °C.
Co-reporter:Randy B. Stockbridge;Charles A. Lewis, Jr.;Yang Yuan;Richard Wolfenden
PNAS 2010 Volume 107 (Issue 51 ) pp:22102-22105
Publication Date(Web):2010-12-21
DOI:10.1073/pnas.1013647107
All reactions are accelerated by an increase in temperature, but the magnitude of that effect on very slow reactions does
not seem to have been fully appreciated. The hydrolysis of polysaccharides, for example, is accelerated 190,000-fold when
the temperature is raised from 25 to 100 °C, while the rate of hydrolysis of phosphate monoester dianions increases 10,300,000-fold.
Moreover, the slowest reactions tend to be the most heat-sensitive. These tendencies collapse, by as many as five orders of
magnitude, the time that would have been required for early chemical evolution in a warm environment. We propose, further,
that if the catalytic effect of a “proto-enzyme”—like that of modern enzymes—were mainly enthalpic, then the resulting rate
enhancement would have increased automatically as the environment became cooler. Several powerful nonenzymatic catalysts of
very slow biological reactions, notably pyridoxal phosphate and the ceric ion, are shown to meet that criterion. Taken together,
these findings greatly reduce the time that would have been required for early chemical evolution, countering the view that
not enough time has passed for life to have evolved to its present level of complexity.
Co-reporter:Randy B. Stockbridge
Journal of the American Chemical Society 2009 Volume 131(Issue 51) pp:18248-18249
Publication Date(Web):December 3, 2009
DOI:10.1021/ja907967y
The hydrolysis of simple phosphate monoesters is among the most difficult reactions that are subject to catalysis by enzymes, and it has been suggested that extraction of the substrates from solvent water may contribute to the catalytic effects of phosphohydrolases. Here, we show that the tetrabutylammonium salt of neopentyl phosphate enters wet cyclohexane at concentrations sufficient to allow determination of its rate of hydrolysis. The second-order rate constant for hydrolysis of the phosphomonoester dianion is enhanced ∼2 × 1012-fold by transfer from water to cyclohexane. That rate enhancement arises from an increase in the entropy of activation.
Co-reporter:Charles A. Lewis Jr. and Richard Wolfenden
Biochemistry 2009 Volume 48(Issue 36) pp:
Publication Date(Web):August 13, 2009
DOI:10.1021/bi901085m
OMP decarboxylase (ODCase) generates a very large rate enhancement without the assistance of metals or other cofactors. The uncatalyzed decarboxylation of 1-methylorotate in water is shown to involve the monoanion, although uncharged 1-methylorotic acid is decarboxylated at a similar rate. To measure the extent to which the rate of the nonenzymatic decarboxylation of orotate derivatives might be enhanced by their removal from solvent water, the 1-phosphoribosyl moiety of OMP was replaced with 1-substituents that would allow it to enter less polar solvents. When the tetrabutylammonium salt of 1-cyclohexylorotate was transferred from water to a series of dipolar aprotic solvents, its rate of decarboxylation increased markedly, varying with the relative ability of each solvent to release the substrate in the ground state from stabilization by solvent water acting as a proton donor. These findings are consistent with the view that separation of the substrate from solvent water may contribute, at least to a limited extent, to the rate enhancement produced by ODCase. This enzyme’s active site, like that of another cofactorless enzyme recently shown to produce a rate enhancement similar in magnitude (uroporphyrinogen decarboxylase), is equipped with an ammonium group positioned in such a way as to balance the electrostatic charge of the carboxylate group of the substrate and later supply a proton to the incipient carbanion in a relatively waterless environment.
Co-reporter:Richard Wolfenden;Yang Yuan;
Proceedings of the National Academy of Sciences 2007 104(1) pp:83-86
Publication Date(Web):December 20, 2006
DOI:10.1073/pnas.0609644104
Alkyl sulfate monoesters are involved in cell signaling and structure. Alkyl sulfates are also present in many commercial
detergents. Here, we show that monomethyl sulfate acts as an efficient alkylating agent in water, reacting spontaneously with
oxygen nucleophiles >100-fold more rapidly than do alkylsulfonium ions, the usual methyl donors in living organisms. These
reactions of methyl sulfate, which are much more rapid than its hydrolysis, are insensitive to the nature of the attacking
nucleophile, with a Brønsted βnuc value of −0.01. Experiments at elevated temperatures indicate a rate constant of 2 × 10−11 s−1 for the uncatalyzed hydrolysis of methyl sulfate at 25°C (t
1/2 = 1,100 y), corresponding to a rate enhancement of ≈1011-fold by a human alkylsulfatase. Equilibria of formation of methyl sulfate from methanol and sodium hydrogen sulfate indicate
a group transfer potential (ΔG′
pH7) of −8.9 kcal/mol for sulfate ester hydrolysis. The magnitude of that value, involving release of the strong acid HSO4
−, helps to explain the need for harnessing the free energy of hydrolysis of two ATP molecules in activating sulfate for the
biosynthesis of sulfate monoesters. The “energy-rich” nature of monoalkyl sulfate esters, coupled with their marked resistance
to hydrolysis, renders them capable of acting as sulfating or alkylating agents under relatively mild conditions. These findings
raise the possibility that, under appropriate circumstances, alkyl groups may undergo transfer from alkyl sulfate monoesters
to biological target molecules.
Co-reporter:Gottfried K. Schroeder;Chetan Lad;Paul Wyman;Nicholas H. Williams;Richard Wolfenden
PNAS 2006 Volume 103 (Issue 11 ) pp:4052-4055
Publication Date(Web):2006-03-14
DOI:10.1073/pnas.0510879103
Phosphodiester linkages, including those that join the nucleotides of DNA, are highly resistant to spontaneous hydrolysis.
The rate of water attack at the phosphorus atom of phosphodiesters is known only as an upper limit, based on the hydrolysis
of the dimethyl phosphate anion. That reaction was found to proceed at least 99% by C–O cleavage, at a rate suggesting an
upper limit of 10−15 s−1 for P–O cleavage of phosphodiester anions at 25°C. To evaluate the rate enhancement produced by P–O cleaving phosphodiesterases
such as staphylococcal nuclease, we decided to establish the actual value of the rate constant for P–O cleavage of a simple
phosphodiester anion. In dineopentyl phosphate, C–O cleavage is sterically precluded so that hydrolysis occurs only by P–O
cleavage. Measurements at elevated temperatures indicate that the dineopentyl phosphate anion undergoes hydrolysis in water
with a t
1/2 of 30,000,000 years at 25°C, furnishing an indication of the resistance of the internucleotide linkages of DNA to water attack
at phosphorus. These results imply that staphylococcal nuclease (k
cat = 95 s−1) enhances the rate of phosphodiester hydrolysis by a factor of ≈1017. In alkaline solution, thymidylyl-3′-5′-thymidine (TpT) has been reported to decompose 105-fold more rapidly than does dineopentyl phosphate. We find however that TpT and thymidine decompose at similar rates and
with similar activation parameters, to a similar set of products, at pH 7 and in 1 M KOH. We infer that the decomposition
of TpT is initiated by the breakdown of thymidine, not by phosphodiester hydrolysis.
Co-reporter:Christopher M. Horvat;Richard V. Wolfenden
PNAS 2005 102 (45 ) pp:16199-16202
Publication Date(Web):2005-11-08
DOI:10.1073/pnas.0508176102
The soil of potato fields in The Netherlands harbors bacteria with the ability to metabolize 3-chloroacrylic acid, generated
by the degradation of a pesticide (1,3-dichloropropene) that entered the environment in 1946. From examination of rate constants
at elevated temperatures, we infer that the half-time at 25°C for spontaneous hydrolytic dechlorination of trans-3-chloroacrylic acid is 10,000 years, several orders of magnitude longer than half-times for spontaneous decomposition of
other environmental pollutants such as 1,2-dichloroethane (72 years), paraoxon (13 months), atrazine (5 months), and aziridine
(52 h). With thermodynamic parameters for activation similar to those for the spontaneous hydration of fumarate at pH 6.8,
this slow reaction proceeds at a constant rate through the pH range between 2 and 12. However, at the active site of the enzyme
3-chloroacrylate dehalogenase (CaaD), isolated from a pseudomonad growing in these soils, hydrolytic dechlorination proceeds
with a half-time of 0.18 s. Neither k
cat nor k
cat/K
m is reduced by increasing solvent viscosity with trehalose, implying that the rate of enzymatic dechlorination is controlled
by chemical events in catalysis rather than by diffusion-limited substrate binding or product release. CaaD achieves an ≈1012-fold rate enhancement, matching or surpassing the rate enhancements produced by many enzymes that act on more conventional
biological substrates. One of those enzymes is 4-oxalocrotonate tautomerase, with which CaaD seems to share a common evolutionary
origin.
Co-reporter:Christopher M. Horvat;Richard V. Wolfenden
PNAS 2005 102 (45 ) pp:16199-16202
Publication Date(Web):2005-11-08
DOI:10.1073/pnas.0508176102
The soil of potato fields in The Netherlands harbors bacteria with the ability to metabolize 3-chloroacrylic acid, generated
by the degradation of a pesticide (1,3-dichloropropene) that entered the environment in 1946. From examination of rate constants
at elevated temperatures, we infer that the half-time at 25°C for spontaneous hydrolytic dechlorination of trans-3-chloroacrylic acid is 10,000 years, several orders of magnitude longer than half-times for spontaneous decomposition of
other environmental pollutants such as 1,2-dichloroethane (72 years), paraoxon (13 months), atrazine (5 months), and aziridine
(52 h). With thermodynamic parameters for activation similar to those for the spontaneous hydration of fumarate at pH 6.8,
this slow reaction proceeds at a constant rate through the pH range between 2 and 12. However, at the active site of the enzyme
3-chloroacrylate dehalogenase (CaaD), isolated from a pseudomonad growing in these soils, hydrolytic dechlorination proceeds
with a half-time of 0.18 s. Neither k
cat nor k
cat/K
m is reduced by increasing solvent viscosity with trehalose, implying that the rate of enzymatic dechlorination is controlled
by chemical events in catalysis rather than by diffusion-limited substrate binding or product release. CaaD achieves an ≈1012-fold rate enhancement, matching or surpassing the rate enhancements produced by many enzymes that act on more conventional
biological substrates. One of those enzymes is 4-oxalocrotonate tautomerase, with which CaaD seems to share a common evolutionary
origin.
Co-reporter:Mark G. Snider;Brenda S. Temple;Richard Wolfenden
Journal of Physical Organic Chemistry 2004 Volume 17(Issue 6‐7) pp:586-591
Publication Date(Web):25 MAY 2004
DOI:10.1002/poc.761
A survey of kcat/Km values supports the view that most enzymes combine with substrates at rates that approach the limits imposed by diffusional encounter. The closeness of that approach precludes the obligate participation of any rare species of the substrate, or any rare species of the enzyme, in productive binding. Proceeding from the ground state through the transition state, the enzyme–substrate complex is assumed to remain in a state of quasi-equilibrium with the free enzyme and the unbound substrate in solution. Because the forces of attraction between an enzyme and substrate approach a maximum in the transition state for substrate transformation, the enzyme and substrate are also likely to approach a maximal state of distortion from their native ground-state structures at that stage in the reaction, furnishing a natural explanation of induced fit. There is now abundant evidence, from mutations of enzymes and substrates, that interactions between the enzyme and substrate, using ordinary chemical forces of attraction, are so strongly synergistic that they may suffice to explain the very high affinities that are achieved in the transition state. Copyright © 2004 John Wiley & Sons, Ltd.
Co-reporter:Victor E. Marquez;Steven A. Short;Gottfried K. Schroeder;Christoph H. Borchers;Mark J. Snider;Richard Wolfenden;J. Paul Speir
PNAS 2004 Volume 101 (Issue 43 ) pp:15341-15345
Publication Date(Web):2004-10-26
DOI:10.1073/pnas.0406781101
The structures of several powerful inhibitors of hydrolytic enzymes resemble that of the altered substrate in the transition
state, except that a hydrogen atom replaces one substituent (typically the leaving group). To test the hypothesis that a water
molecule might be present in the gap resulting from this replacement, we examined a transition-state analogue complex formed
by Escherichia coli cytidine deaminase by Fourier transform ion cyclotron resonance MS in electrospray mode. Upon nebularization from aqueous
solution under conditions (pH 5.6) where the enzyme is active, cytidine deaminase remains dimeric in the vapor phase. In the
presence of inhibitor, the enzyme's exact mass can be used to infer the presence at each active site of zinc, 5-fluoro-3,4-dihydrouridine,
and a single water molecule.
Co-reporter:Annette Sievers;Richard Wolfenden;Marina V. Rodnina;Malte Beringer
PNAS 2004 Volume 101 (Issue 21 ) pp:7897-7901
Publication Date(Web):2004-05-25
DOI:10.1073/pnas.0402488101
To determine the effectiveness of the ribosome as a catalyst, we compared the rate of uncatalyzed peptide bond formation,
by the reaction of the ethylene glycol ester of N-formylglycine with Tris(hydroxymethyl)aminomethane, with the rate of peptidyl transfer by the ribosome. Activation parameters
were also determined for both reactions, from the temperature dependence of their second-order rate constants. In contrast
with most protein enzymes, the enthalpy of activation is slightly less favorable on the ribosome than in solution. The 2 ×
107-fold rate enhancement produced by the ribosome is achieved entirely by lowering the entropy of activation. These results
are consistent with the view that the ribosome enhances the rate of peptide bond formation mainly by positioning the substrates
and/or water exclusion within the active site, rather than by conventional chemical catalysis.
Co-reporter:Richard V. Wolfenden
Biochemistry and Molecular Biology Education 2002 Volume 30(Issue 1) pp:
Publication Date(Web):3 NOV 2006
DOI:10.1002/bmb.2002.494030010003
Co-reporter:Randy B. Stockbridge and Richard Wolfenden
Chemical Communications 2010 - vol. 46(Issue 24) pp:NaN4308-4308
Publication Date(Web):2010/05/06
DOI:10.1039/C0CC00229A
The hydrolysis of phosphate diesters is one of the most difficult reactions known. Here we show that in acetone or cyclohexane, at 25 °C, phosphodiesters undergo hydrolysis 5 × 105 and 2 × 109-fold more rapidly than in water, respectively, and that this rate enhancement is achieved by lowering the enthalpy of activation.











![N-[(4-CYANOPHENYL)CARBAMOTHIOYL]BENZAMIDE](http://img.cochemist.com/ccimg/333400/333303-11-0.png)
![N-[(4-CYANOPHENYL)CARBAMOTHIOYL]BENZAMIDE](http://img.cochemist.com/ccimg/333400/333303-11-0_b.png)