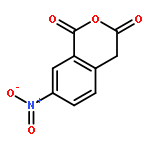
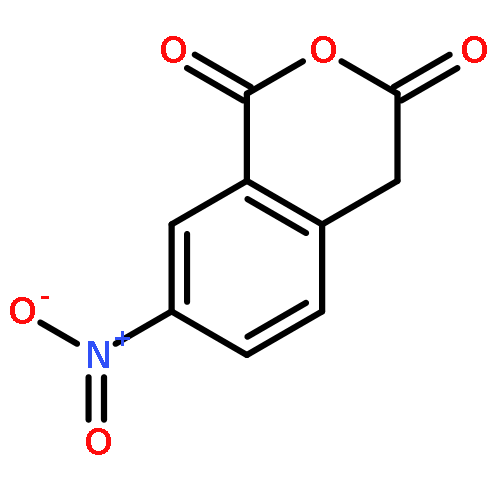
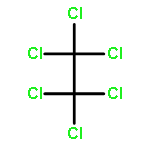
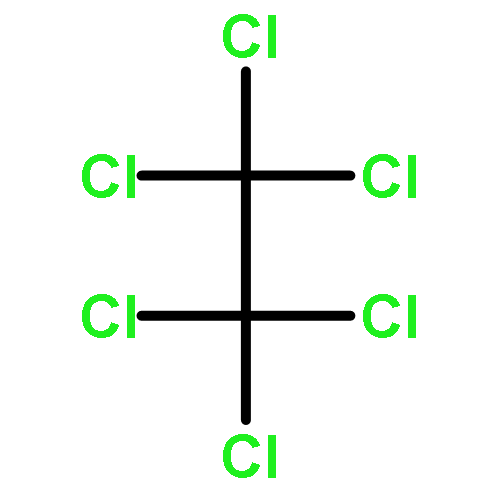
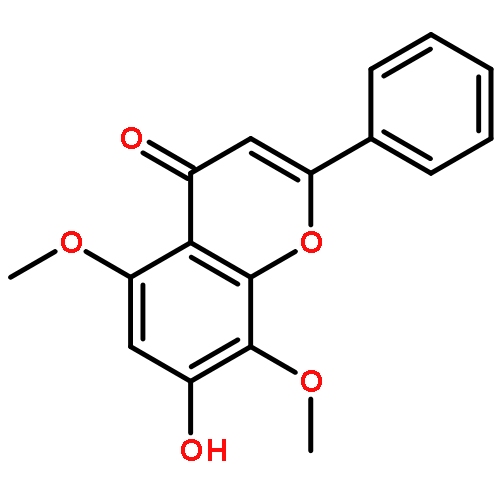
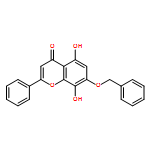
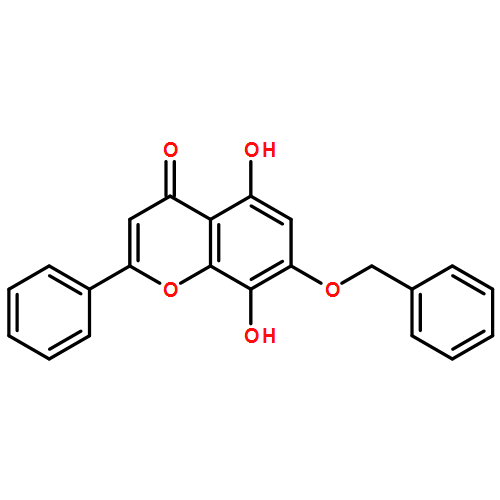
Co-reporter:Zhouling Xie, Benimana Oscar, Lulu Zhao, Xue Ding, Chen Cao, Sen Feng, Han Li, Chen Pan, Ziyi Bian, Yang Li, Weijun Wang, Yi Kong, Zhiyu Li
European Journal of Medicinal Chemistry 2017 Volume 125() pp:197-209
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.09.032
•LX5 was identified based on an identified antiplatelet aggregation agent LX2421.•LX14 shows comparable ability as Tirofiban in inhibiting platelet aggregation and GPIIb/IIIa receptor.•LX14 has less bleeding risk in comparison with Tirofiban.Based upon LX2421, a previously identified antiplatelet aggregation agent, a series of novel 2-amino-3-(naphth-2-yl)propanoic acid derivatives were designed, synthesized and evaluated. Among them, compounds LX14 and LX25 were identified as promising antiplatelet aggregation agents. The in vitro biologic study demonstrated that LX14 can block platelet aggregation induced by four different inducers and displays comparable potency in inhibiting GPIIb/IIIa receptor in comparison with Tirofiban. In addition, LX14 has much lower risk of bleeding than Tirofiban and shows significant antithrombotic activity in vivo. Taking together, the results indicated that LX14 is a promising GPIIb/IIIa receptor antagonist against platelet aggregation worthy of further evaluation.
Co-reporter:Minzan Zuo, Xi Xu, Zhouling Xie, Raoling Ge, Ziyu Zhang, Zhiyu Li, Jinlei Bian
European Journal of Medicinal Chemistry 2017 Volume 125() pp:1002-1022
Publication Date(Web):5 January 2017
DOI:10.1016/j.ejmech.2016.10.049
•A series of indoline thiohydantoin derivatives were synthesized and evaluated in vitro.•The most potent compound 48c shows comparable ability with enzalutamide in proliferation inhibition of LNCaP cells.•Compound 48c has less cytotoxic to AR-negative cells compared with Enzalutamide.•The bicalutamide-resistant mechanism was clarified and overcome by compound 48c.A novel scaffold of indoline thiohydantoin was discovered as potent androgen receptor (AR) antagonist through rational drug designation. Several compounds showed good biological profiles in AR binding and higher selective toxicity than enzalutamide toward LNCaP cells (AR-rich) versus DU145 cells (AR-deficient). In addition, the docking studies supported the rationalization of the biological evaluation. Among these compounds, the representative compound 48c exhibited the strongest inhibitory effect on LNCaP growth and also acted as a competitive AR antagonist. Further preliminary mechanism study confirmed that 48c exerted its AR antagonistic activity through impairing AR nuclear translocation. All these results indicated that the novel scaffold compounds demonstrated AR antagonistic behavior and promising candidates for future development were identified.
Co-reporter:Jubo Wang, Raoling Ge, Xiaqiu Qiu, Xi Xu, Libin Wei, Zhiyu Li, Jinlei Bian
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.09.046
•A series of novel flavonoids were synthesized and evaluated as potent antitumor compounds.•Compounds 18n and 20b exhibited potent antitumor activity against several cancer cells.•Compounds 18n and 20b could significantly enhance the intracellular ROS level and induce the cell apoptosis.•Through similarity searching, CDK9 was identified as the potential target for 20b, which could be a start point for next drug design.Phenotypic screening of high quality compound library is one of the most effective strategy to obtain novel bioactive compounds. Recently, our group have constructed a Wogonin-scaffold library with substituents diversity and successfully obtained a series of potent compounds. Herein, we reported the synthesis of these compounds and evaluated the in vitro antitumor activity against a panel of human tumor cell lines. Most of them showed good activity with a broad spectrum and preliminary structure-activity relationship for the substitutions were obtained. Further biological assays showed that the most potent compounds 18n and 20b could significantly enhance the intracellular ROS level and induce the cell apoptosis at low micromole level. Through similarity searching, CDK9 was identified as the potential target for 20b, which could be a start point for next structure-based drug design.Download high-res image (208KB)Download full-size image
Co-reporter:Dr. Fei Ji;Yun Fan;Run Yang;Yueyan Yang;Dianwen Yu;Minyu Wang; Dr. Zhiyu Li
Asian Journal of Organic Chemistry 2017 Volume 6(Issue 6) pp:682-685
Publication Date(Web):2017/06/01
DOI:10.1002/ajoc.201700050
AbstractA simple approach for the synthesis of thiocyanate-containing isoxazolines by FeIII/K2S2O8 mediated radical thiocyanation/cyclization cascade reaction of accessible β,γ-unsaturated oximes has been developed. The high selectivity, mild reaction conditions and absence of harmful heavy-metal reagents render this reaction attractive. Additionally, the thiocyanate group from isoxazolines could be easily converted to various important functional groups, which could make these compounds important building blocks in organic synthesis.
Co-reporter:Tinghan Li, Tianwei Weng, Jubo Wang, Zhihui Wei, Lu Zhao, and Zhiyu Li
Organic Process Research & Development 2017 Volume 21(Issue 2) pp:
Publication Date(Web):January 10, 2017
DOI:10.1021/acs.oprd.6b00249
A scalable and practical route to wogonin, a bioactive natural product with multiple pharmacological activities which is currently under phase I/II clinical studies, is described. Wogonin was obtained via a four-step process starting from commercially available chrysin and including the Elbs oxidation, benzylation, methylation, and the final debenzylation. The whole procedure gives the target product in a 38% overall yield with >99% purity. Key steps in this process including oxidation and methylation are discussed in detail. The optimized process has been successfully demonstrated on the kilogram scale to support ongoing clinical development of wogonin.Keywords: Elbs oxidation; large-scale synthesis; natural product; reaction condition optimization;
Co-reporter:Raoling Ge, Qian Zhao, Zhouling Xie, Lu Lu, Qinglong Guo, Zhiyu Li, Li Zhao
European Journal of Medicinal Chemistry 2016 Volume 122() pp:465-474
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.054
•Some of our compounds demonstrate potent Top I inhibitory activity as Camptothecin.•18f and 18g show comparable ability as Camptothecin in Top I inhibitory activity and cellular inhibitory activity.•18f and 18g impair the cell cycle progression in the s phase.•18f and 18g represent more safety compared with hydroxycamptothecin.The design and synthesis of a new series of 6-fluoro-3-phenyl-7-piperazinyl quinolone derivatives, built on the structure of 1-ethyl-3-(6-nitrobenzoxazol-2-yl)-6,8-difluoro-7-(3-methylpiperazin-1-yl)-4(1H)-quinolone, are described. These compounds provide new scaffold for the discovery of Topoisomerase I (Top I) inhibitors and target based assay showed that they can obviously inhibited Top I at 100 μM. The in vitro anti-proliferative activity of these new compounds was evaluated against A549, Hela, BGC-823, and HepG2 cell lines. Compounds 18a-g showed potent inhibitory activity against the growth of those cancer cell lines. The most positive compounds 18f and 18g demonstrated as potent as camptothecin in Top I inhibition assay and MTT assay. Compounds 18f and 18g led to an obvious increase in the percentage of S phase of the cells in 24 h. The in vivo data showed that 18f and 18g inhibited tumor growth with the inhibitory rate of 29.25% and 42.75% at 20 mg/kg, respectively. The data suggested the therapeutic potential for further development.
Co-reporter:Qian Zhao, Xi Xu, Zhouling Xie, Xiao Liu, Qidong You, Qinglong Guo, Yi Zhong, Zhiyu Li
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 3) pp:1068-1072
Publication Date(Web):1 February 2016
DOI:10.1016/j.bmcl.2015.12.014
A new series of indenoisoquinoline derivatives was designed and synthesized. The in vitro anti-proliferative activity of these novel compounds was evaluated in HepG2, A549 and HCT-116 cell lines. Compounds 9a, 9b, 10a, 10c, 10e, 18a and 18b manifested potent inhibitory activity against the three tested cancer cell lines. Nineteen compounds were also tested for Top I inhibition at 50 μM. Almost all the tested compounds showed potent Top I inhibition activity at this concentration. The most potent compounds 9a and 10a demonstrated more cytotoxicity than HCPT and TPT and was comparable to CPT in inhibitory activities on Top I in our biological assay.
Co-reporter:Mingping Wang, Wei Tian, Chongqing Wang, Shihai Lu, Chao Yang, Juan Wang, Yunlong Song, Youjun Zhou, Ju Zhu, Zhiyu Li, Canhui Zheng
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 21) pp:5207-5211
Publication Date(Web):1 November 2016
DOI:10.1016/j.bmcl.2016.09.061
The anti-apoptotic Bcl-2 proteins are attractive targets for anti-cancer drug development, and the discovery of their selective inhibitors has become a research focus. In this Letter, obvious differences in the P1 pocket of the active site between Bcl-2, Bcl-xL, and Mcl-1 proteins were proposed by the structural comparison of these proteins. As a result, the groups in their inhibitors binding to the P1 pockets may have significant effect on the selectivity for these proteins. Based on this hypothesis, five types of derivatives of the lead compound B-1 were designed, and several highly selective inhibitors of Bcl-xL (E-1) or Mcl-1 proteins (G) were found. The selective inhibitors of Mcl-1 protein found in this Letter provide new structural types for the development of novel antitumor agents.
Co-reporter:Zhouling Xie, Sen Feng, Ying Wang, Chen Cao, Jing Huang, Yahui Chen, Yi Kong, Zhiyu Li
European Journal of Medicinal Chemistry 2015 Volume 102() pp:363-374
Publication Date(Web):18 September 2015
DOI:10.1016/j.ejmech.2015.07.016
•The small molecule compound 70 was identified based on the three mer peptide pENW (pGlu-Asn-Trp).•A series of Trp derivatives were synthesized and evaluated for antiplatelet aggregation activity.•The most potent compound 87 in our paper shows comparable ability as Tirofiban in inhibiting platelet aggregation.•Compound 87 has less bleeding risk compared with Tirofiban.pENW, a three mer peptide derived from Agkistrodon acutus Guenther venom, has been found to be an antagonist of the GPIIb/IIIa receptor and shows antiplatelet aggregation activity. Based on pENW and a GPIIb/IIIa inhibitor Tirofiban, a series of tryptophan derivatives were designed, synthesized and evaluated for their antiplatelet aggregation activity induced by ADP. The most potent compound 87 was also tested for the bleeding time and antithrombotic activity in vivo in comparison with Tirofiban. The results indicated that 87 shows similar antiplatelet aggregation activity as Tirofiban to the aggregation of platelet induced by all of the four agonists, but has lower bleeding risk than Tirofiban, representing a promising lead compound for further study.
Co-reporter:Zhouling Xie, Youli Zhou, Wei Zhao, He Jiao, Yu Chen, Yong Yang, Zhiyu Li
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 20) pp:4557-4561
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmcl.2015.08.060
A series of AG014699 derivatives containing a novel scaffold of 2,3-dihydro-1H-[1,2]diazepino[4,5,6-cd]indole-1,4(6H)-dione were synthesized and evaluated for their inhibitory activities toward PARP-1 enzyme and two cell lines, MCF-7 cells and the BRCA1-deficient MDA-MB-436 cells. Our results demonstrated that of all AG014699 derivatives synthesized in this work, compounds 6 and 7 showed strong PARP-1 inhibitory activity (IC50 = 3.5 nM and 2.4 nM, respectively), only four and three times less potent than AG014699. Compound 6 also had significantly cell inhibitory activity against both MCF-7 cells (CC50 = 25.8 μM) and the BRCA1-deficient MDA-MB-436 cells (CC50 = 5.4 μM), nearly as good as AG014699, indicating that it can be a promising compound for further evaluation.
Co-reporter:Rong Luo, Jubo Wang, Li Zhao, Na Lu, Qidong You, Qinglong Guo, Zhiyu Li
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 5) pp:1334-1338
Publication Date(Web):1 March 2014
DOI:10.1016/j.bmcl.2014.01.053
Co-reporter:Jun-Peng Zhang, Jie Huang, Chao Liu, Xu-Fang Lu, Bao-Xiang Wu, Li Zhao, Na Lu, Qing-Long Guo, Zhi-Yu Li, Cheng Jiang
Chinese Chemical Letters 2014 Volume 25(Issue 7) pp:1025-1028
Publication Date(Web):July 2014
DOI:10.1016/j.cclet.2014.05.048
A series of new 3-benzoheterocyclic substituted pyridopyrimidines were designed and synthesized. Structures of the compounds were determined by IR, 1H NMR, and elemental analyses. The anti-proliferation activity of 13 novel compounds was evaluated in A549, HL-60, BGC-823 and SMMC-7721 cell lines. Compounds 3, 5, 7, 8, 9, 10 showed potent inhibitory activity against the four tested cancer cell lines. These six compounds were examined for Top I inhibition at 100 μmol/L by measuring the relaxation of supercoiled DNA in plasmid pBR322. Most of the tested compounds inhibited the enzyme at this concentration. The most potent compound 9 was as potent as camptothecin.Using a scaffold modification strategy, we identified a series of new 3-benzoheterocyclic substituted pyridopyrimidines as potential topoisomerase I inhibitors.
Co-reporter:Jinlei Bian, Tinghan Li, Tianwei Weng, Jubo Wang, Yu Chen, Zhiyu Li
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2016.12.076
A novel series of 49 wogonin derivatives were synthesized by introducing group at 7-, 8- or B ring of wogonin. The cytotoxic activities against HepG2, A549 and BCG-823 cancer cell lines were also investigated in vitro. Several of them showed obvious cytotoxic activities and compound 3h possessed the highest potency against HepG2, A549, and BCG-823 with IC50 values of 1.07 μM, 1.74 μM and 0.98 μM, respectively. A quantitative structure-activity relationship (QSAR) study of these synthetic derivatives as well as wogonin indicated that high solubility and low octanol/water partition coefficient are favorable, and excessive electrostatic properties and refractivity are unfavorable for the cytotoxic activities of these wogonin derivatives. These findings and results provide a base for further investigations.

![N-METHYL-2-[[3-[(E)-2-PYRIDIN-2-YLETHENYL]-2,3,3A,4,5,6,7,7A-OCTAHYDRO-1H-INDAZOL-6-YL]SULFANYL]BENZAMIDE](http://img.cochemist.com/ccimg/1096200/1096120-30-7.png)
![N-METHYL-2-[[3-[(E)-2-PYRIDIN-2-YLETHENYL]-2,3,3A,4,5,6,7,7A-OCTAHYDRO-1H-INDAZOL-6-YL]SULFANYL]BENZAMIDE](http://img.cochemist.com/ccimg/1096200/1096120-30-7_b.png)
![1-[4-(3-FLUOROANILINO)PIPERIDIN-1-YL]-2-[4-[[(3S)-3-METHYLPIPERAZIN-1-YL]METHYL]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/1096200/1096120-23-8.png)
![1-[4-(3-FLUOROANILINO)PIPERIDIN-1-YL]-2-[4-[[(3S)-3-METHYLPIPERAZIN-1-YL]METHYL]PHENYL]ETHANONE](http://img.cochemist.com/ccimg/1096200/1096120-23-8_b.png)












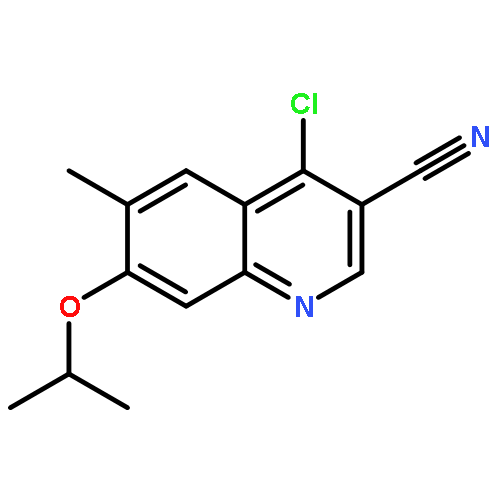
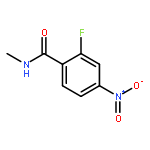

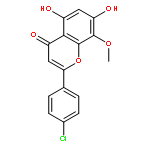
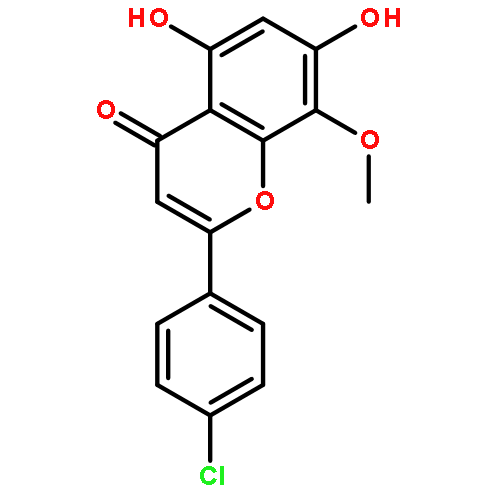
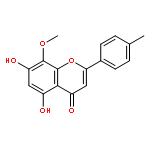
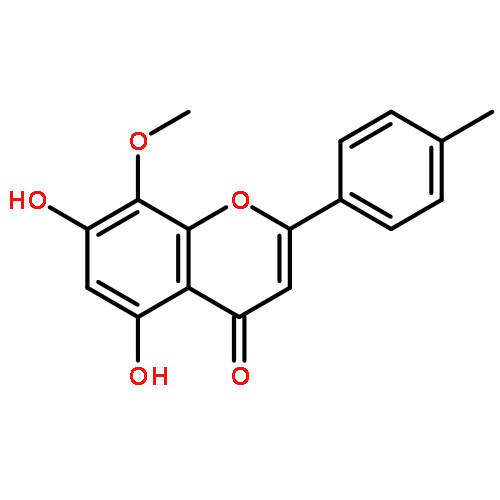

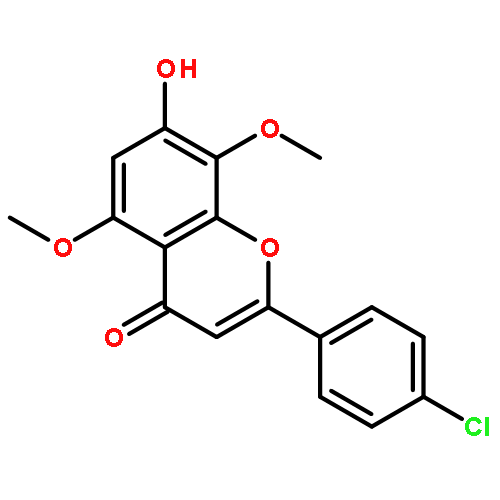

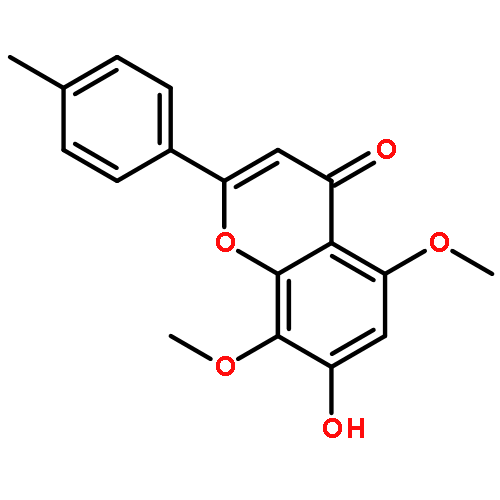

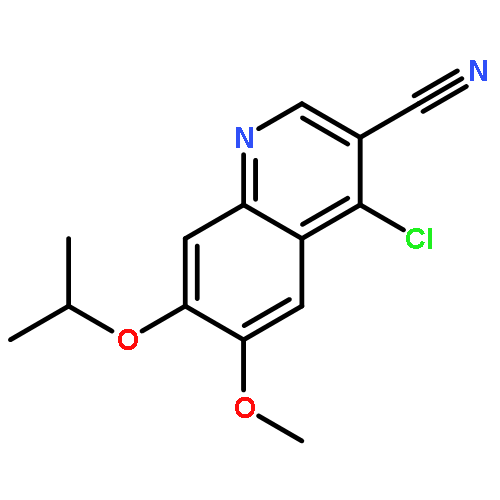

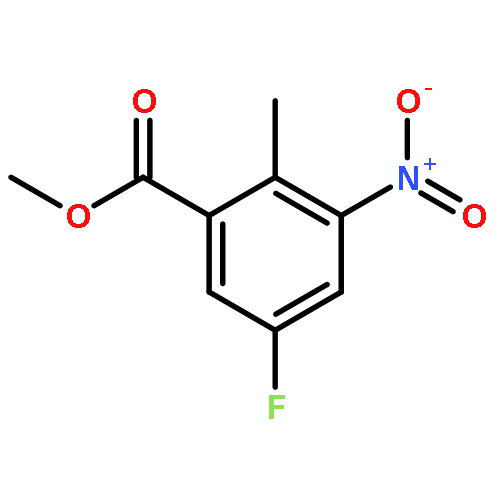

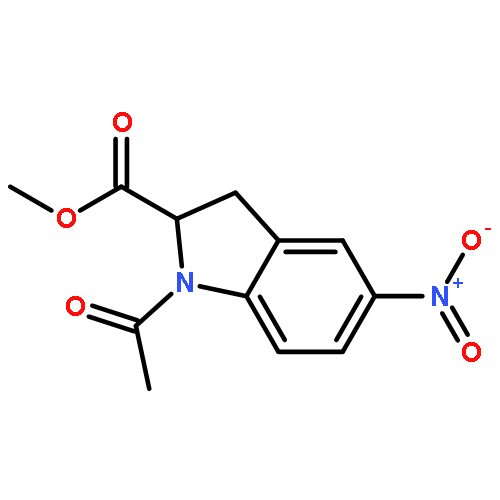
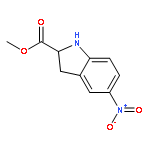
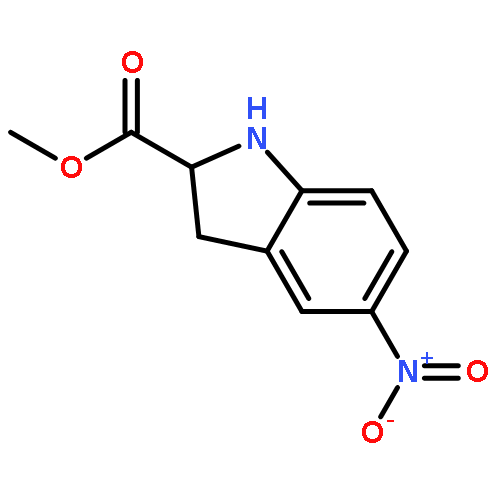

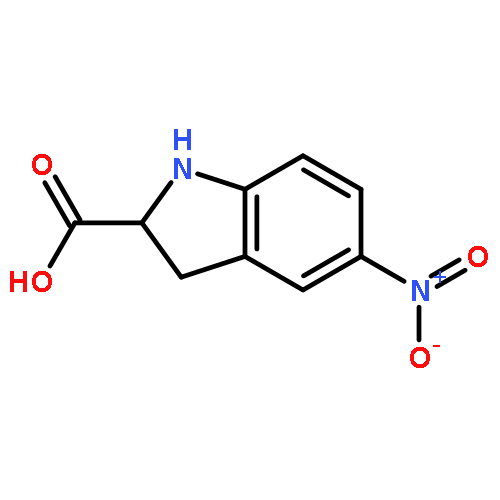
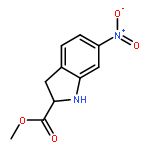
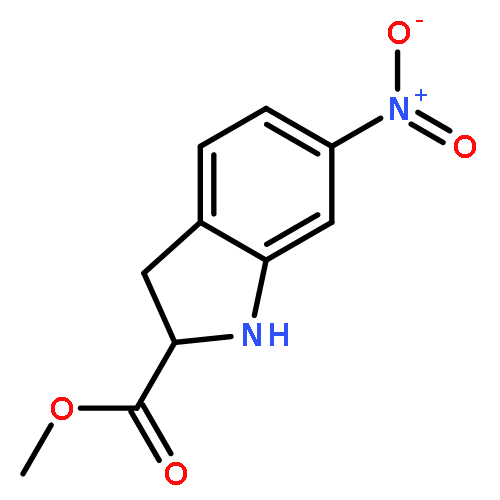
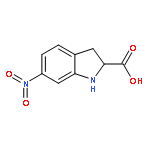
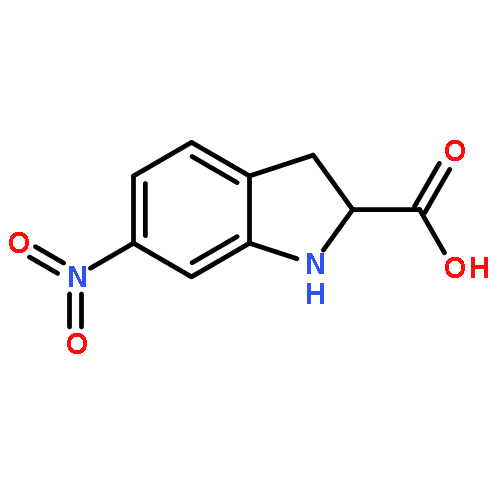
![Benzenesulfonamide, 3-nitro-4-[[2-(phenylthio)ethyl]amino]-](http://img.cochemist.com/ccimg/406300/406233-13-4.png)
![Benzenesulfonamide, 3-nitro-4-[[2-(phenylthio)ethyl]amino]-](http://img.cochemist.com/ccimg/406300/406233-13-4_b.png)




![1-Propanamine, 3-chloro-N-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/367300/367280-93-1.png)
![1-Propanamine, 3-chloro-N-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/367300/367280-93-1_b.png)



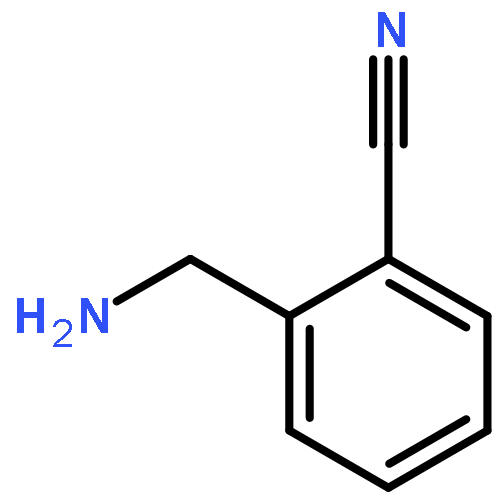





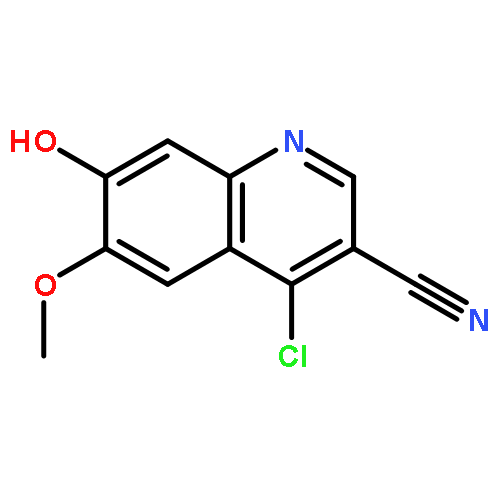
![4-Imidazolidinone, 5-[2-(methylthio)ethyl]-3-(phenylmethyl)-2-thioxo-](http://img.cochemist.com/ccimg/256400/256377-35-2.png)
![4-Imidazolidinone, 5-[2-(methylthio)ethyl]-3-(phenylmethyl)-2-thioxo-](http://img.cochemist.com/ccimg/256400/256377-35-2_b.png)






![BENZOIC ACID, 4-[[1-OXO-3-[4-(TRIFLUOROMETHYL)PHENYL]-2-PROPENYL]AMINO]-](http://img.cochemist.com/ccimg/184000/183991-47-1.png)
![BENZOIC ACID, 4-[[1-OXO-3-[4-(TRIFLUOROMETHYL)PHENYL]-2-PROPENYL]AMINO]-](http://img.cochemist.com/ccimg/184000/183991-47-1_b.png)
![Benzoic acid, 4-[[3-(3-methoxyphenyl)-1-oxo-2-propenyl]amino]-](http://img.cochemist.com/ccimg/184000/183991-30-2.png)
![Benzoic acid, 4-[[3-(3-methoxyphenyl)-1-oxo-2-propenyl]amino]-](http://img.cochemist.com/ccimg/184000/183991-30-2_b.png)


![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5.png)
![1,1'-[1,4-Phenylenebis(methylene)]bis[1,4,8,11-tetraazacyclotetradecane]](http://img.cochemist.com/ccimg/155200/155148-31-5_b.png)

















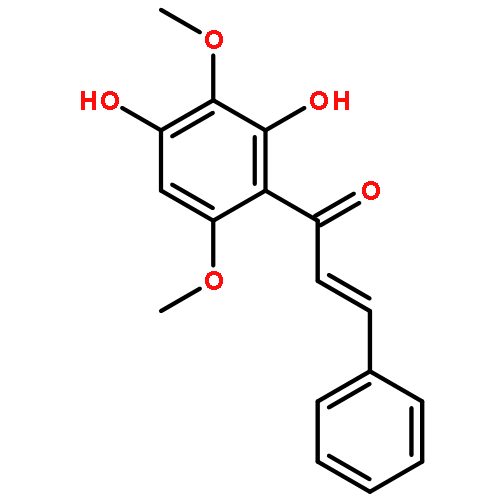


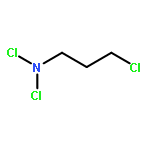
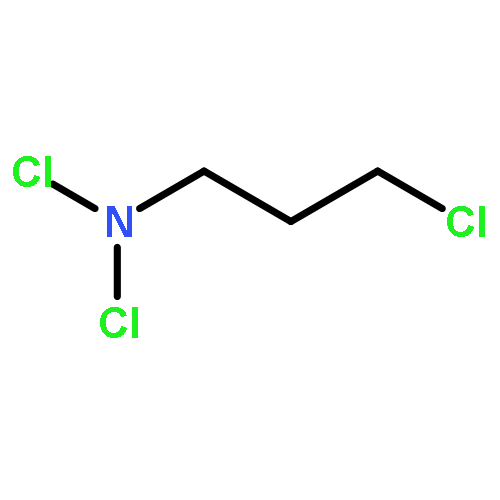

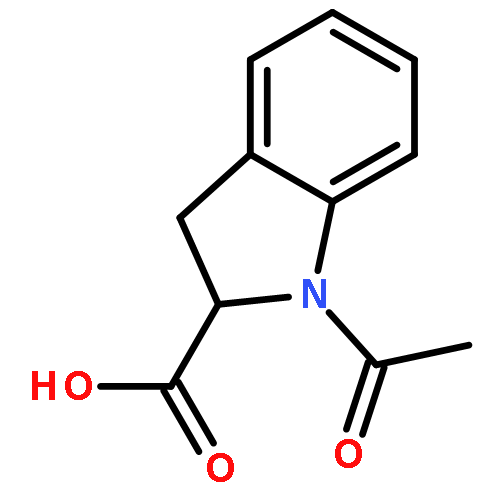
![5,12-Naphthacenedione,9-acetyl-9-amino-7-[(2-deoxy-b-D-erythro-pentopyranosyl)oxy]-7,8,9,10-tetrahydro-6,11-dihydroxy-,(7S,9S)-](http://img.cochemist.com/ccimg/110300/110267-81-7.png)
![5,12-Naphthacenedione,9-acetyl-9-amino-7-[(2-deoxy-b-D-erythro-pentopyranosyl)oxy]-7,8,9,10-tetrahydro-6,11-dihydroxy-,(7S,9S)-](http://img.cochemist.com/ccimg/110300/110267-81-7_b.png)




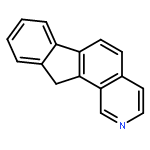



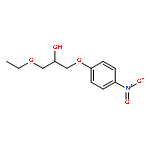
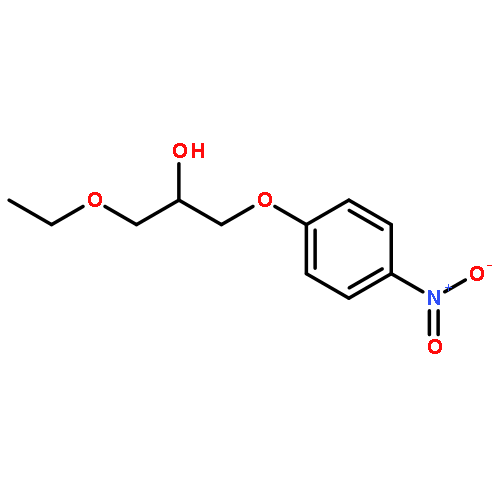
![N-[4-(3-Ethoxy-2-hydroxypropoxy)phenyl]-3-(methylthio)propanamide](http://img.cochemist.com/ccimg/94100/94057-02-0.png)
![N-[4-(3-Ethoxy-2-hydroxypropoxy)phenyl]-3-(methylthio)propanamide](http://img.cochemist.com/ccimg/94100/94057-02-0_b.png)
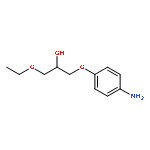



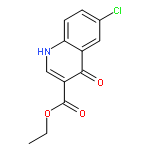

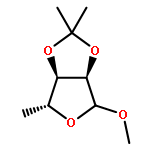

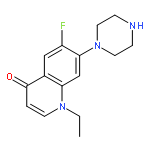

![4-[(PHENYLACETYL)AMINO]BENZOIC ACID](http://img.cochemist.com/ccimg/73600/73548-12-6.png)
![4-[(PHENYLACETYL)AMINO]BENZOIC ACID](http://img.cochemist.com/ccimg/73600/73548-12-6_b.png)


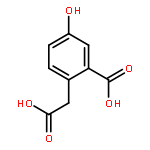




![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)
![Naphth[2,3-b]oxirene, 1a,2,7,7a-tetrahydro-3,6-dimethoxy-](http://img.cochemist.com/ccimg/58900/58851-64-2.png)
![Naphth[2,3-b]oxirene, 1a,2,7,7a-tetrahydro-3,6-dimethoxy-](http://img.cochemist.com/ccimg/58900/58851-64-2_b.png)
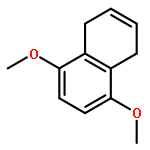
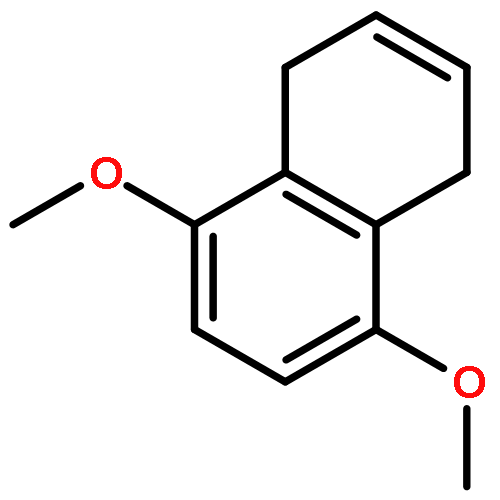
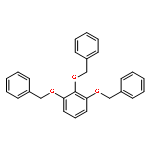
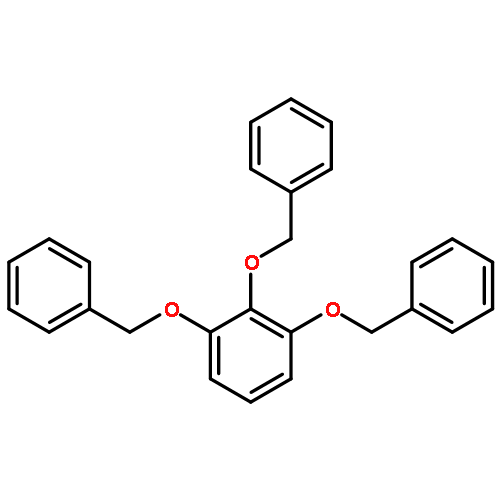
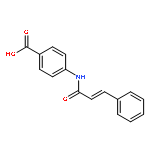



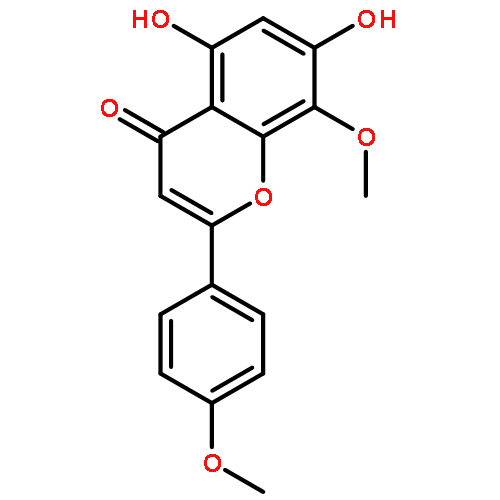
![Benzoic acid, 4-[[3-(4-chlorophenyl)-1-oxo-2-propenyl]amino]-](http://img.cochemist.com/ccimg/54000/53901-68-1.png)
![Benzoic acid, 4-[[3-(4-chlorophenyl)-1-oxo-2-propenyl]amino]-](http://img.cochemist.com/ccimg/54000/53901-68-1_b.png)
![Benzoic acid, 4-[[3-(4-methoxyphenyl)-1-oxo-2-propenyl]amino]-](http://img.cochemist.com/ccimg/54000/53901-60-3.png)
![Benzoic acid, 4-[[3-(4-methoxyphenyl)-1-oxo-2-propenyl]amino]-](http://img.cochemist.com/ccimg/54000/53901-60-3_b.png)
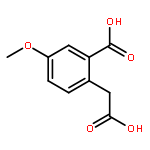
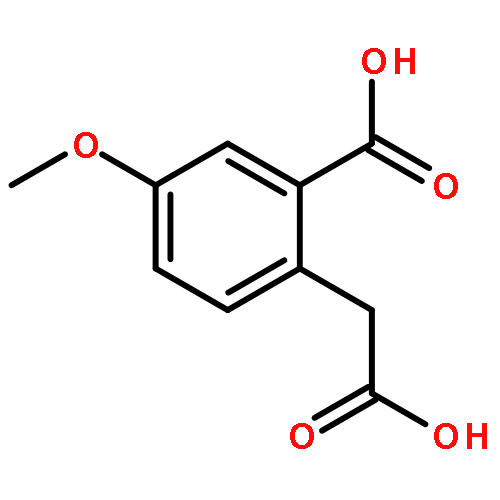


![2-ETHYL-8-METHOXY-2,3,4,5-TETRAHYDRO-1H-PYRIDO[4,3-B]INDOLE](http://img.cochemist.com/ccimg/39200/39123-65-4.png)
![2-ETHYL-8-METHOXY-2,3,4,5-TETRAHYDRO-1H-PYRIDO[4,3-B]INDOLE](http://img.cochemist.com/ccimg/39200/39123-65-4_b.png)

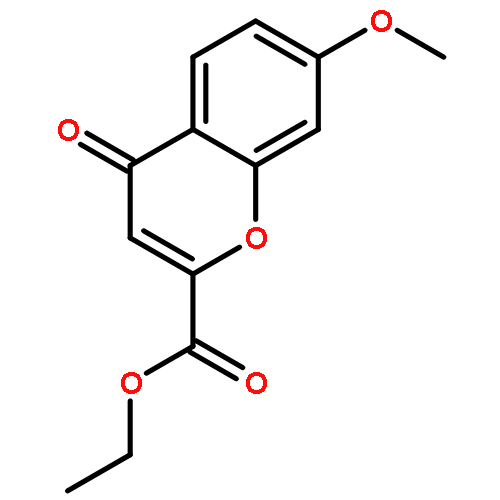

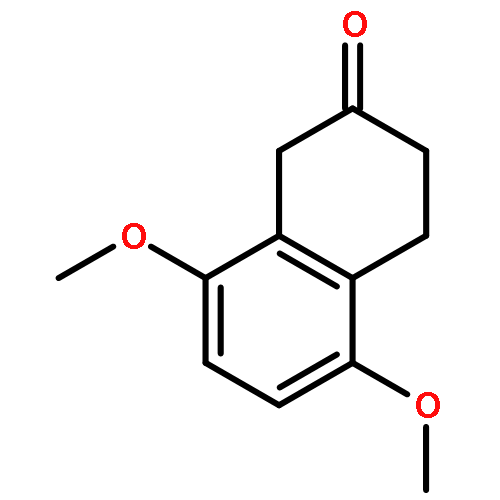
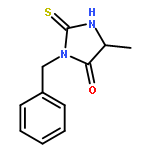


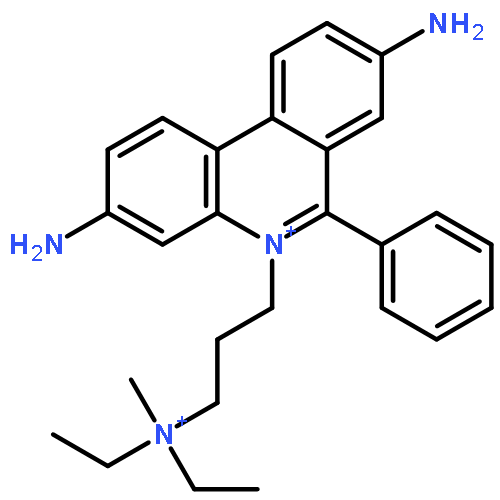
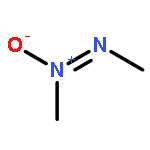
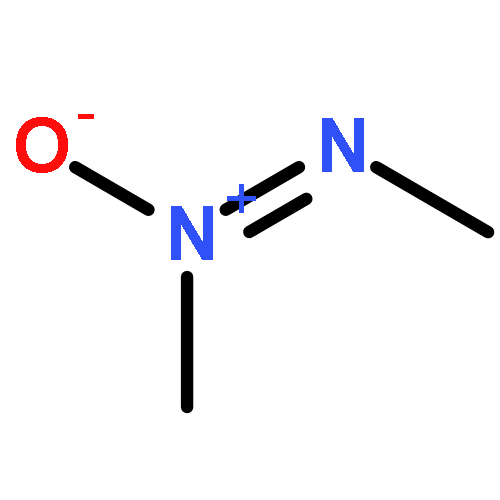
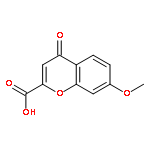

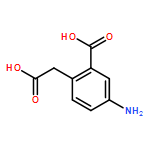
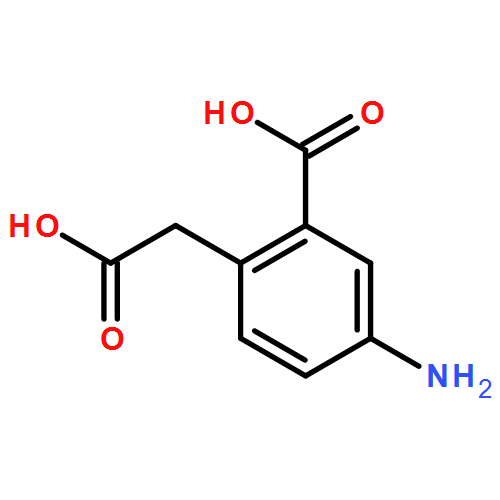
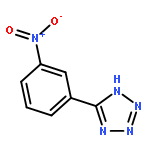
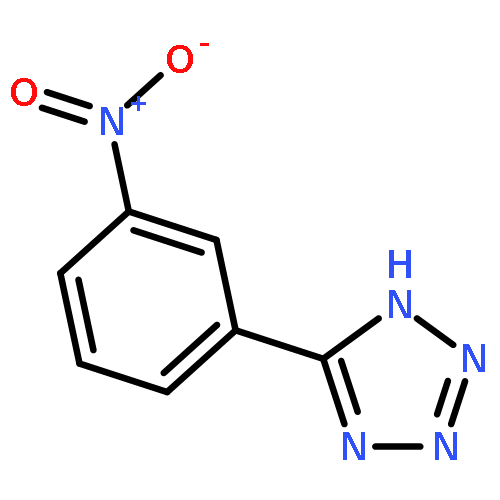
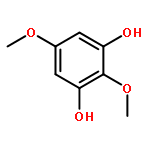
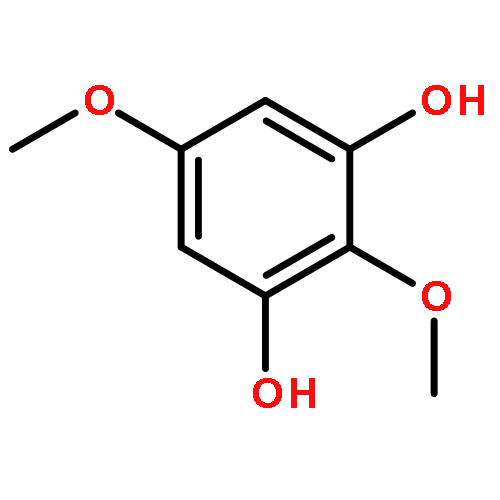





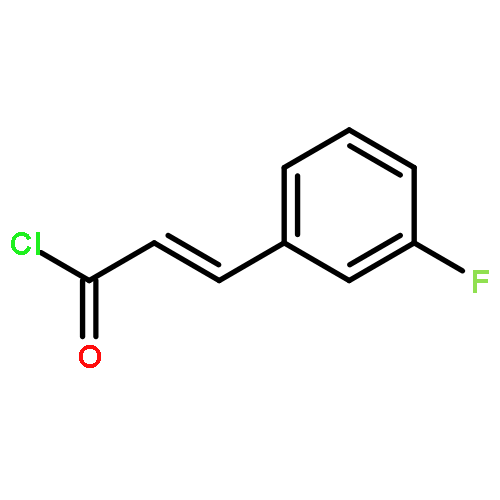


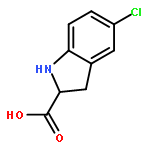
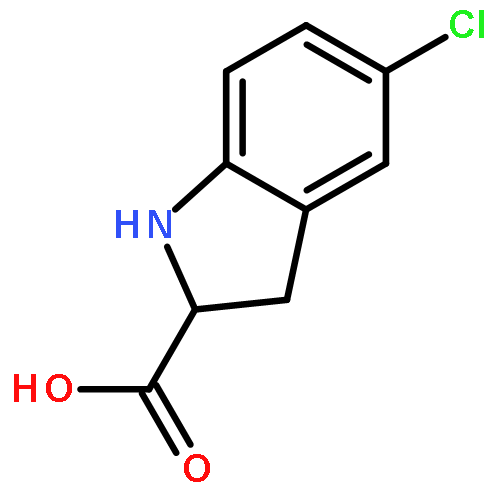
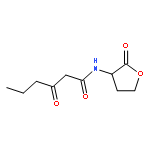
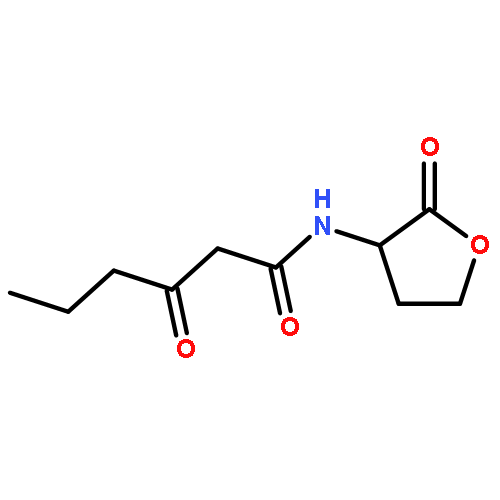


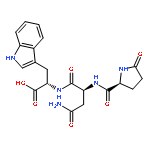


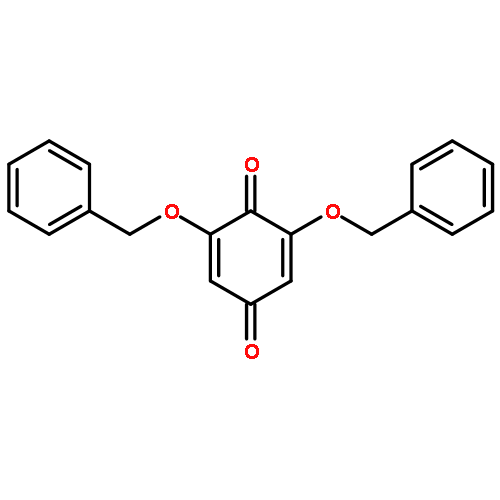
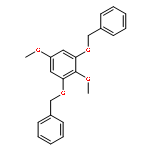
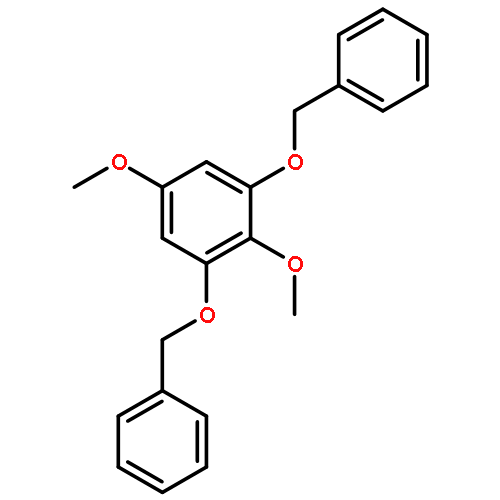




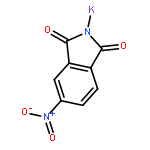

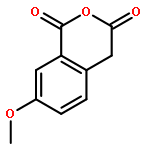



![N-[(2,6-DICHLOROPHENYL)SULFONYL]ALANINE](http://img.cochemist.com/ccimg/3900/3898-66-6.png)
![N-[(2,6-DICHLOROPHENYL)SULFONYL]ALANINE](http://img.cochemist.com/ccimg/3900/3898-66-6_b.png)




![L-Homoserine,O-[(aminoiminomethyl)amino]-](http://img.cochemist.com/ccimg/600/543-38-4.png)
![L-Homoserine,O-[(aminoiminomethyl)amino]-](http://img.cochemist.com/ccimg/600/543-38-4_b.png)