Co-reporter:Omkar V. Zore, Patrick J. Lenehan, Challa V. Kumar, and Rajeswari M. Kasi
Langmuir May 13, 2014 Volume 30(Issue 18) pp:5176-5184
Publication Date(Web):May 13, 2014
DOI:10.1021/la501034b
We previously reported that the stability and aqueous catalytic activity of met-hemoglobin (Hb) was improved when covalently conjugated with poly(acrylic acid) (PAA). In the current study, the Hb–PAA–water interface was modified to improve Hb catalytic efficiency in organic solvents (0–80% v/v organic solvent; remainder is the conjugate, the substrate, and water). The protein–polymer–solvent interface modification was achieved by esterifying the carboxylic acid groups of Hb–PAA with ethanol (EtOH) or 1-propanol (1-prop) after activation with carbodiimide. The resulting esters (Hb–PAA–Eth and Hb–PAA–1-prop, respectively) showed high peroxidase-like catalytic activities in acetonitrile (ACN), dimethylformamide (DMF), EtOH, and methanol (MeOH). Catalytic activities depended on the log(P) values of the solvents, which is a measure of solvent lipophilicity. The highest weighted-average activities were noted in MeOH for all three conjugates, and the lowest average activities were noted in DMF for two of the conjugates. Interestingly, the average activities of the conjugates were higher than that of Hb in all solvents except in ACN. The ratio of the catalytic rate constant (kcat) to the Michaelis constant (KM), the catalytic efficiency, for Hb–PAA–Eth in MeOH was the highest noted, and it is ∼3-fold higher than that of Hb in buffer; conjugates offered higher efficiencies than Hb at most solvent compositions. This is the very first general, versatile, modular strategy of coupling the enhanced stability of Hb with improved activity in organic solvents via the chemical manipulation of the polymer shell around Hb and provides a robust approach to efficient biocatalysis in organic solvents.
Co-reporter:Omkar V. Zore;Paritosh Pande;Oghenenyerovwo Okifo;Ashis K. Basu;Rajeswari M. Kasi
RSC Advances (2011-Present) 2017 vol. 7(Issue 47) pp:29563-29574
Publication Date(Web):2017/06/05
DOI:10.1039/C7RA05666D
We report a general and modular approach for the synthesis of multi enzyme–polymer conjugates (MECs) consisting of five different enzymes of diverse isoelectric points and distinct catalytic properties conjugated within a single universal polymer scaffold. The five model enzymes chosen include glucose oxidase (GOx), acid phosphatase (AP), lactate dehydrogenase (LDH), horseradish peroxidase (HRP) and lipase (Lip). Poly(acrylic acid) (PAA) is used as the model synthetic polymer scaffold that will covalently conjugate and stabilize multiple enzymes concurrently. Parallel and sequential synthetic protocols are used to synthesise MECs, 5-P and 5-S, respectively. Also, five different single enzyme–PAA conjugates (SECs) including GOx–PAA, AP–PAA, LDH–PAA, HRP–PAA and Lip–PAA are synthesized. The composition, structure and morphology of MECs and SECs are confirmed by agarose gel electrophoresis, dynamic light scattering, circular dichroism spectroscopy and transmission electron microscopy. The bioreactor comprising MEC functions as a single biocatalyst can carry out at least five different or orthogonal catalytic reactions by virtue of the five stabilized enzymes, which has never been achieved to-date. Using activity assays relevant for each of the enzymes, for example AP, the specific activity of AP at room temperature and 7.4 pH in PB is determined and set at 100%. Interestingly, MECs 5-P and 5-S show specific activities of 1800% and 600%, respectively, compared to 100% specific activity of AP at room temperature (RT). The catalytic efficiencies of 5-P and 5-S are 1.55 × 10−3 and 1.68 × 10−3, respectively, compared to 9.11 × 10−5 for AP under similar RT conditions. Similarly, AP relevant catalytic activities of 5-P and 5-S at 65 °C show 100 and 300%, respectively, relative to native AP activity at RT as the native AP is catalytically inactive at 65 °C. The catalytic activity trends suggest: (1) MECs show enhanced catalytic activities compared to native enzymes under similar assay conditions and (2) 5-S is better suited for high temperature biocatalysis, while both 5-S and 5-P are suitable for room temperature biocatalysis. Initial cytotoxicity results show that these MECs are non-lethal to human cells including human embryonic kidney [HEK] cells when treated with doses of 0.01 mg mL−1 for 72 h. This cytotoxicity data is relevant for future biological applications.
Co-reporter:Islam M. Mosa;Ajith Pattammattel;Karteek Kadimisetty;Paritosh Pe;Maher F. El-Kady;Gregory W. Bishop;Marc Novak;Richard B. Kaner;Ashis K. Basu;James F. Rusling
Advanced Energy Materials 2017 Volume 7(Issue 17) pp:
Publication Date(Web):2017/09/01
DOI:10.1002/aenm.201700358
Nearly all implantable bioelectronics are powered by bulky batteries which limit device miniaturization and lifespan. Moreover, batteries contain toxic materials and electrolytes that can be dangerous if leakage occurs. Herein, an approach to fabricate implantable protein-based bioelectrochemical capacitors (bECs) employing new nanocomposite heterostructures in which 2D reduced graphene oxide sheets are interlayered with chemically modified mammalian proteins, while utilizing biological fluids as electrolytes is described. This protein-modified reduced graphene oxide nanocomposite material shows no toxicity to mouse embryo fibroblasts and COS-7 cell cultures at a high concentration of 1600 µg mL−1 which is 160 times higher than those used in bECs, unlike the unmodified graphene oxide which caused toxic cell damage even at low doses of 10 µg mL−1. The bEC devices are 1 µm thick, fully flexible, and have high energy density comparable to that of lithium thin film batteries. COS-7 cell culture is not affected by long-term exposure to encapsulated bECs over 4 d of continuous charge/discharge cycles. These bECs are unique, protein-based devices, use serum as electrolyte, and have the potential to power a new generation of long-life, miniaturized implantable devices.
Co-reporter:Bobbi Stromer, Melissa Limbacher, Dhanya T. Jayaram, Sudarat Yenjai, Ruma Chowdhury, Apinya Buranaprapuk, Danaboyina Ramaiah, Challa V. Kumar
Journal of Photochemistry and Photobiology A: Chemistry 2017 Volume 340(Volume 340) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.jphotochem.2017.01.027
•A powerful strategy to photocleave the protein backbone at a single site, developed over nearly two decades of our work, is described here.•The photocleavage has occurred at a single site, for most of the examples, although a minor product has been noted in smaller amounts in some cases.•The newly produced peptide fragments were sequenceable, a major advantage to identify the cleavage site.•Molecular modeling studies provided a strong predictive power to target given sites on a protein.A powerful strategy to photocleave the protein backbone at a single site, developed over nearly two decades, is described here. The remarkable ability to target one or even a couple of sites on a large protein with a small molecule, under photochemical control, is a considerable challenge. Systematic modification of the structure of the photoreagent provides valuable information on the binding site recognition as well as the mechanism of the photocleavage reaction. Some factors that impact the photocleavage reaction include the exact location of the probe binding site on the protein, conformations of the bound probe, protein size, functional groups present on the probe that interact with the protein surroundings either in a favorable or unfavorable manner, overall charge on the photoreagent, and photophysical/photochemical properties of the probe. The protein photocleavage studies were preceded by detailed binding studies by a variety of spectroscopic methods. Methods as simple as absorption and fluorescence spectroscopies or more sophisticated circular dichroism spectroscopy were used. Conclusions that are most consistent with the binding data indicated a single binding site on most proteins, irrespective of the probe or the protein, with only one exception noted so far. Such high selectivity for targeting a binding site on a biomolecule by a small molecule is remarkable. Photocleavage studies and laser flash photolysis studies were used to probe the photochemical peptide bond cleavage reactions, which indicated the important role of aromatic cation radicals in the mechanistic path. Our work focused mainly on few generic proteins serum albumin, lysozyme, and avidin; but may be extended to other proteins with appropriate modifications. This article strives to provide some guidance on such design strategies for other proteins or other photoreagents. Molecular modeling studies clearly supported our conclusions and the methods described here could be useful in a rational design of novel photoreagents to target desired sites on proteins in future studies.Download high-res image (143KB)Download full-size image
Co-reporter:Marc J. Novak, Ajith Pattammattel, Brianna Koshmerl, Megan Puglia, Christina Williams, and Challa V. Kumar
ACS Catalysis 2016 Volume 6(Issue 1) pp:339
Publication Date(Web):December 1, 2015
DOI:10.1021/acscatal.5b01968
A rational strategy is presented here to enhance enzyme stability by gaining control over the noncovalent interactions at the enzyme–graphene interface (EGI). The charge (n) on a model enzyme, glucose oxidase (GOx), was systematically varied from −67 to +78 via chemical modification of its COOH groups with polyamines, and chemically modified GOx(n) has been adsorbed onto graphene oxide (GO). Control of the net charge on GOx(n) provided an excellent handle to engineer the EGI to enhance the stability of the GOx(n)/GO biocatalyst while retaining full activity. Enzyme loading (w/w, GOx(n) to GO) increased with increased n, and a maximum enzyme loading of 420% (w/w) was noted when n = 0; a further increase in n decreased loading. Enzymatic activities of the GOx(n)/GO hybrids increased steadily with n, and the maximum specific activity was about 1.6 times greater than that of GOx. There has been a good correlation between the retention of enzyme secondary structure and the corresponding enzymatic activities. These indicated an unprecedented increase in kinetic stability as a function of n, measured at 40 °C, with a maximum half-life of 38 days (n = 0, +35), which is about 150 times higher than that of GOx, under the same conditions. The kinetic barrier to denaturation increased steadily from 58.7 kcal/mol for GOx to 294 kcal mol–1 for GOx(+35)/GO and then decreased slightly with increased n. Chemical denaturation studies showed that the improved stability is not due to changes in thermodynamic stability and that the increased kinetic stability is due to an increased barrier to denaturation. Adsorption onto GO played a key role in enhancing enzyme stability, and without GO, the stability enhancements were only marginal. Thus, engineering the EGI provided exciting opportunities to generate highly stable biocatalysts that can be stored under ambient conditions, while retaining high activities, with very long lifetimes, much needed for practical applications.Keywords: chemical modification; glucose oxidase; graphene oxide; half-life; stability
Co-reporter:C. M. Riccardi, D. Mistri, O. Hart, M. Anuganti, Y. Lin, R. M. Kasi and C. V. Kumar
Chemical Communications 2016 vol. 52(Issue 12) pp:2593-2596
Publication Date(Web):05 Jan 2016
DOI:10.1039/C6CC00037A
A modular, general method for trapping enzymes within the voids of paper, without chemical activation of cellulose, is reported. Glucose oxidase and peroxidase were crosslinked with poly(acrylic acid) via carbodiimide chemistry, producing 3-dimensional networks interlocked in cellulose fibers. Interlocking prevented enzyme activity loss and enhanced the washability and stability.
Co-reporter:Ranjan K. Kamat, Yuting Zhang, Murali Anuganti, Wanfu Ma, Iman Noshadi, Hailin Fu, Stephen Ekatan, Richard Parnas, Changchun Wang, Challa V. Kumar, and Yao Lin
Langmuir 2016 Volume 32(Issue 44) pp:11573-11579
Publication Date(Web):October 24, 2016
DOI:10.1021/acs.langmuir.6b02573
Polycatalytic enzyme complexes made by immobilization of industrial enzymes on polymer- or nanoparticle-based scaffolds are technologically attractive due to their recyclability and their improved substrate binding and catalytic activities. Herein, we report the synthesis of polycatalytic complexes by the immobilization of nonprocessive cellulases on the surface of colloidal polymers with a magnetic nanoparticle core and the study of their binding and catalytic activities. These polycatalytic cellulase complexes have increased binding affinity for the substrate. But due to their larger size, these complexes were unable to access to the internal surfaces of cellulose and have significantly lower binding capacity when compared to those of the corresponding free enzymes. Analysis of released soluble sugars indicated that the formation of complexes may promote the prospect of having consistent, multiple attacks on cellulose substrate. Once bound to the substrate, polycatalytic complexes tend to remain on the surface with very limited mobility due to their strong, multivalent binding to cellulose. Hence, the overall performance of polycatalytic complexes is limited by its substrate accessibility as well as mobility on the substrate surface.
Co-reporter:Clive L. Baveghems, Ajith Pattammattel, and Challa V. Kumar
The Journal of Physical Chemistry B 2016 Volume 120(Issue 46) pp:11880-11887
Publication Date(Web):October 28, 2016
DOI:10.1021/acs.jpcb.6b08651
An artificial histone is synthesized that functions as a DNA–protein digital switch, where DNA binding is all or none, controlled by a sharp threshold of protein charge. A non-DNA-binding protein, glucose oxidase (GOx), was chemically modified by attaching an increasing number of triethylenetetramine (TETA) side chains to its glutamate/aspartate groups to obtain a small library of covalently modified GOx(n) derivatives. The parameter n denotes the net charge on the protein at pH 7, which was increased from −62 (pristine GOx) to +75 by attaching an increasing number of TETA residues to the protein. All GOx(n) derivatives retained their secondary structure to a good extent, as monitored by UV circular dichroism (CD) spectroscopy, and they also retained oxidase activities to a significant extent. The interaction of the GOx(n) with calf thymus DNA was examined by isothermal titration calorimetry (ITC). Pristine GOx of −62 charge at pH 7 in 10 mM Tris–HCl and 50 mM NaCl buffer had no affinity for the negatively charged DNA helix, and GOx(n) with n < +30 had no affinity for DNA either. However, binding has been turned on abruptly when n ≥ +30 with binding constants (Kb) ranging from (1.5 ± 0.7) × 107 to (7.3 ± 2.8) × 107 M–1 for n values of +30 and +75, respectively, and this type of “all-or-none” binding based on protein charge is intriguing. Furthermore, thermodynamic analysis of the titration data revealed that binding is entirely entropy-driven with ΔS ranging from 0.09 ± 0.007 to 0.19 ± 0.008 kcal/mol K with enthalpic penalties of 17.0 ± 2.3 and 46.1 ± 2.1 kcal/mol, respectively. The binding had intrinsic propensities (ΔG) ranging from −9.8 ± 0.14 to −10.7 ± 0.25 kcal/mol, independent of n. DNA binding distorted protein–DNA secondary structure, as evidenced by CD spectroscopy, but oxidase activity of GOx(n)/DNA complexes has been unaffected. This is the very first example of an artificial histone (GOx(n)) where the protein charge functioned as a DNA-binding switch; protein charge is in turn under complete chemical control while preserving the biological activity of the protein. The new insight gained here could be useful in the design of novel “on–off” protein switches.
Co-reporter:Ajith Pattammattel ;Challa Vijaya Kumar
Advanced Functional Materials 2015 Volume 25( Issue 45) pp:7088-7098
Publication Date(Web):
DOI:10.1002/adfm.201503247
A high yielding aqueous phase exfoliation of graphite to high quality graphene using edible proteins and kitchen chemistry is reported here. Bovine serum albumin (BSA), β-lactoglobulin, ovalbumin, lysozyme, and hemoglobin are used to exfoliate graphite and the exfoliation efficiency depended on the sign and magnitude of the protein charge. BSA showed maximum exfoliation rate, facilitated graphite exfoliation in water, at room temperature, by turbulence/shear force generated in a kitchen blender at exfoliation efficiencies exceeding 4 mg mL−1 h−1. Raman spectroscopy and transmission electron microscopy indicated 3–5 layer, defect-free graphene of 0.5 μm size. Graphene dispersions loaded on a cellulose paper (650 μg cm−2) showed the film conductivity of 32 000 S m−1, which is much higher than graphene/polymer composites. Our method yielded ≈7 mg mL−1, BSA-coated graphene with controllable surface charge, which is stable under wide ranges of pH (3.0–11) and temperature (5.0–50 °C), and in fetal bovine serum, for more than two months.These findings may lead to the large scale production of graphene for biological applications.
Co-reporter:Omkar V. Zore, Ajith Pattammattel, Shailaja Gnanaguru, Challa V. Kumar, and Rajeswari M. Kasi
ACS Catalysis 2015 Volume 5(Issue 9) pp:4979
Publication Date(Web):July 8, 2015
DOI:10.1021/acscatal.5b00958
An example of a highly stable and functional bienzyme–polymer conjugate triad assembled on a topologically orthogonal support, layered graphene oxide (GO), is reported here. Glucose oxidase (GOx) and horseradish peroxidase (HRP) catalytic dyad were used as the model system for cascade biocatalysis. Poly(acrylic acid) (PAA) was used to covalently conjugate these enzymes, and then the conjugate has been subsequently adsorbed onto GO. The resultant nanobiocatalysts are represented as GOx-HRP-PAA/GO. Their morphology and structural characteristics were examined by transmission electron microscopy (TEM), agarose gel electrophoresis, circular dichroism (CD), and zeta potential. These robust conjugates remarkably functioned as active catalysts under biologically challenging conditions such as extreme pHs, high temperature (65 °C), and in the presence of a chemical denaturant. In one example, GOx-HRP-PAA/GO presented doubling of the Kcat (TON) (68 × 10–2 s–1) at pH 7.0 and room temperature, when compared to the corresponding physical mixture of GOx/HRP (32 × 10–2 s–1) under similar conditions. In another case, at 65 °C, GOx-HRP-PAA/GO displayed ∼120% specific activity, whereas GOx/HRP showed only 16% of its original activity. At pH 2.0 and in the presence of 4.0 mM SDS as the denaturant, GOx-HRP-PAA/GO presented greater than 100% specific activity, whereas GOx/HRP was completely deactivated under these conditions. Thus, we combined two concepts, enzyme–polymer conjugation followed by adsorption onto a 2D nanolayered material to obtain enhanced substrate channeling and excellent enzyme stability under challenging conditions. These features have never been attained by either traditional enzyme–polymer conjugates or enzyme–GO hybrids. This general, modular, and powerful approach may also be used to produce environmentally benign, biologically compatible (edible), and efficient cascade biocatalysts.Keywords: enzyme−polymer conjugate; graphene oxide; high temperature activity; multienzyme; substrate channeling
Co-reporter:Inoka K. Deshapriya, Bobbi S. Stromer, Ajith Pattammattel, Christina S. Kim, Ramiro Iglesias-Bartolome, Laura Gonzalez-Fajardo, Vyomesh Patel, J. Silvio Gutkind, Xiuling Lu, and Challa V. Kumar
Bioconjugate Chemistry 2015 Volume 26(Issue 3) pp:396
Publication Date(Web):January 21, 2015
DOI:10.1021/bc500621h
A simple and effective method for synthesizing highly fluorescent, protein-based nanoparticles (Prodots) and their facile uptake into the cytoplasm of cells is described here. Prodots made from bovine serum albumin (nBSA), glucose oxidase (nGO), horseradish peroxidase (nHRP), catalase (nCatalase), and lipase (nLipase) were found to be 15–50 nm wide and have been characterized by gel electrophoresis, transmission electron microscopy (TEM), circular dichroism (CD), fluorescence spectroscopy, dynamic light scattering (DLS), and optical microscopic methods. Data showed that the secondary structure of the protein in Prodots is retained to a significant extent and specific activities of nGO, nHRP, nCatalase, and nLipase were 80%, 70%, 65%, and 50% of their respective unmodified enzyme activities. Calorimetric studies indicated that the denaturation temperatures of nGO and nBSA increased while those of other Prodots remained nearly unchanged, and accelerated storage half-lives of Prodots at 60 °C increased by 4- to 8-fold. Exposure of nGO and nBSA+ nGO to cells indicated rapid uptake within 1–3 h, accompanied by significant blebbing of the plasma membrane, but no uptake has been noted in the absence of nGO. The presence of nGO/glucose in the media facilitated the uptake, and hydrogen peroxide induced membrane permeability could be responsible for this rapid uptake of Prodots. In control studies, FITC alone did not enter the cell, BSA-FITC was not internalized even in the presence of nGO, and there has been no uptake of nBSA-FITC in the absence of nGO. These are the very first examples of very rapid cellular uptake of fluorescent nanoparticles into cells, particularly nanoparticles made from pure proteins. The current approach is a simple and efficient method for the preparation of bioactive, fluorescent protein nanoparticles of controllable size for cellular imaging, and cell uptake is under the control of two separate chemical triggers.
Co-reporter:Ajith Pattammattel, Christina L. Williams, Paritosh Pande, William G. Tsui, Ashis K. Basu and Challa Vijaya Kumar
RSC Advances 2015 vol. 5(Issue 73) pp:59364-59372
Publication Date(Web):01 Jul 2015
DOI:10.1039/C5RA10306A
The influence of oxidative debris (OD) present in as-prepared graphene oxide (GO) suspensions on proteins and its toxicity to human embryonic kidney cells (HEK-293T) are reported here. The OD was removed by repeated washing with aqueous ammonia to produce the corresponding base-washed GO (bwGO). The loading (w/w) of bovine serum albumin (BSA) was increased by 85% after base washing, whereas the loading of hemoglobin (Hb) and lysozyme (Lyz), respectively, was decreased by 160% and 100%. The secondary structures of 13 different proteins bound to bwGO were compared with the corresponding proteins bound to GO using the UV circular dichroism spectroscopy. There was a consistent loss of protein secondary structure with bwGO when compared with proteins bound to GO, but no correlation between either the isoelectric point or hydrophobicity of the protein and the extent of structure loss was observed. All enzymes bound to bwGO and GO indicated significant activities, and a strong correlation between the enzymatic activity and the extent of structure retention was noted, regardless of the presence or absence of OD. At low loadings (<100 μg mL−1) both GO and bwGO showed excellent cell viability but substantial cytotoxicity (∼40% cell death) was observed at high loadings (>100 μg mL−1). In control studies, OD by itself did not alter the growth rate even after a 48 h incubation. Thus, the presence of OD in GO played a very important role in controlling the chemical and biological nature of the protein–GO interface and the presence of OD in GO improved its biological compatibility when compared to bwGO.
Co-reporter:Challa V. Kumar, Marc J. Novak, Kyle R. Benson, Clive Baveghems, Vindya K. Thilakarathne, Bobbi S. Stromer and Filicia M. Ross
RSC Advances 2015 vol. 5(Issue 88) pp:72416-72422
Publication Date(Web):19 Aug 2015
DOI:10.1039/C5RA14208C
Artificial antenna complexes built via self-assembly are reported here, which indicated excellent energy transfer efficiency, macroscopic organization, unprecedented thermal stability, and ease of formation. Our system consists of four fluorescent donor–acceptor dyes, double-helical DNA and cationized bovine serum albumin, all self-assembled on cover glass slips to form functional materials. These captured radiation in the range of 330–590 nm, and excitation of any of the donor dyes resulted in efficient emission from the terminal acceptor. Excitation spectra provided unequivocal proof of energy transfer via jumper dyes, and transfer was interrupted when one of the jumper dyes was omitted, another direct evidence for cascade energy transfer. The entire assembly indicated unusually high thermal stability and continued to function efficiently even after exposure to 80 °C for >169 days, an important consideration for field applications. These unusually stable, high efficiency, multi-chromophoric, artificial antennas are the first of their kind to demonstrate self-assembled 4-dye energy cascade, converting blue photons to red photons.
Co-reporter:Inoka K. Deshapriya, Christina S. Kim, Marc J. Novak, and Challa V. Kumar
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 12) pp:9643
Publication Date(Web):May 22, 2014
DOI:10.1021/am502070w
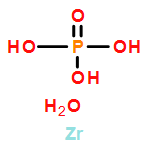
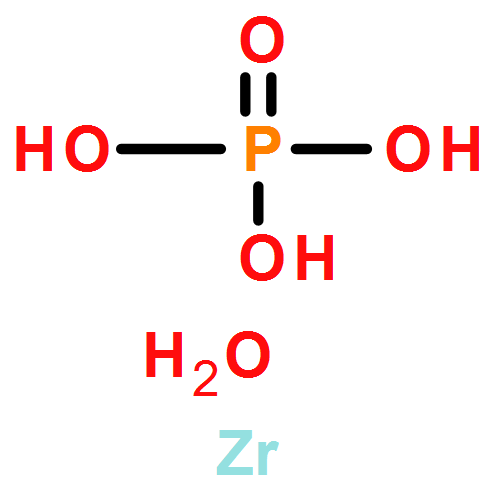
Controlling the properties of enzymes bound to solid surfaces in a rational manner is a grand challenge. Here we show that preadsorption of cationized bovine serum albumin (cBSA) to α-Zr(IV) phosphate (α-ZrP) nanosheets promotes enzyme binding in a predictable manner, and surprisingly, the enzyme binding is linearly proportional to the number of residues present in the enzyme or its volume, providing a powerful, new predictable tool. The cBSA loaded α-ZrP (denoted as bZrP) was tested for the binding of pepsin, glucose oxidase (GOX), tyrosinase, catalase, myoglobin and laccase where the number of residues increased from the lowest value of ∼153 to the highest value of 2024. Loading depended linearly on the number of residues, rather than enzyme charge or its isoelectric point. No such correlation was seen for the binding of these enzymes to α-ZrP nanosheets without the preadsorption of cBSA, under similar conditions of pH and buffer. Enzyme binding to bZrP was supported by centrifugation studies, powder X-ray diffraction and scanning electron microscopy/energy-dispersive X-ray spectroscopy. All the bound enzymes retained their secondary structure and the extent of structure retention depended directly on the amount of cBSA preadsorbed on α-ZrP, prior to enzyme loading. Except for tyrosinase, all enzyme/bZrP biocatalysts retained their enzymatic activities nearly 90–100%, and biofunctionalization enhanced the loading, improved structure retention and supported higher enzymatic activities. This approach of using a chemically modified protein to serve as a glue, with a predictable affinity/loading of the enzymes, could be useful to rationally control enzyme binding for applications in advanced biocatalysis and biomedical applications.Keywords: bZrP; cationization; intercalation; nanomaterials; protein-o-philic; α-ZrP;
Co-reporter:Caterina M. Riccardi, Kyle S. Cole, Kyle R. Benson, Jessamyn R. Ward, Kayla M. Bassett, Yiren Zhang, Omkar V. Zore, Bobbi Stromer, Rajeswari M. Kasi, and Challa V. Kumar
Bioconjugate Chemistry 2014 Volume 25(Issue 8) pp:1501
Publication Date(Web):July 21, 2014
DOI:10.1021/bc500233u
Several key properties of catalase such as thermal stability, resistance to protease degradation, and resistance to ascorbate inhibition were improved, while retaining its structure and activity, by conjugation to poly(acrylic acid) (PAA, Mw 8000) via carbodiimide chemistry where the amine groups on the protein are appended to the carboxyl groups of the polymer. Catalase conjugation was examined at three different pH values (pH 5.0, 6.0, and 7.0) and at three distinct mole ratios (1:100, 1:500, and 1:1000) of catalase to PAA at each reaction pH. The corresponding products are labeled as Cat-PAA(x)-y, where x is the protein to polymer mole ratio and y is the pH used for the synthesis. The coupling reaction consumed about 60–70% of the primary amines on the catalase; all samples were completely water-soluble and formed nanogels, as evidenced by gel electrophoresis and electron microscopy. The UV circular dichroism (CD) spectra indicated substantial retention of protein secondary structure for all samples, which increased to 100% with increasing pH of the synthesis and polymer mole fraction. Soret CD bands of all samples indicated loss of ∼50% of band intensities, independent of the reaction pH. Catalytic activities of the conjugates increased with increasing synthesis pH, where 55–80% and 90–100% activity was retained for all samples synthesized at pH 5.0 and pH 7.0, respectively, and the Km or Vmax values of Cat-PAA(100)-7 did not differ significantly from those of the free enzyme. All conjugates synthesized at pH 7.0 were thermally stable even when heated to ∼85–90 °C, while native catalase denatured between 55 and 65 °C. All conjugates retained 40–90% of their original activities even after storing for 10 weeks at 8 °C, while unmodified catalase lost all of its activity within 2 weeks, under similar storage conditions. Interestingly, PAA surrounding catalase limited access to the enzyme from large molecules like proteases and significantly increased resistance to trypsin digestion compared to unmodified catalase. Similarly, negatively charged PAA surrounding the catalase in these conjugates protected the enzyme against inhibition by negatively charged inhibitors such as ascorbate. While Cat-PAA(100)-7 did not show any inhibition by ascorbate in the presence of 270 μM ascorbate, unmodified catalase lost ∼70% of its activity under similar conditions. This simple, facile, and rational methodology produced thermostable, storable catalase that is also protected from protease digestion and ascorbate inhibition and most likely prevented the dissociation of the multimer. Using synthetic polymers to protect and improve enzyme properties could be an attractive approach for making “Stable-on-the-Table” enzymes, as a viable alternative to protein engineering.
Co-reporter:Omkar V. Zore, Patrick J. Lenehan, Challa V. Kumar, and Rajeswari M. Kasi
Langmuir 2014 Volume 30(Issue 18) pp:5176-5184
Publication Date(Web):2017-2-22
DOI:10.1021/la501034b
We previously reported that the stability and aqueous catalytic activity of met-hemoglobin (Hb) was improved when covalently conjugated with poly(acrylic acid) (PAA). In the current study, the Hb–PAA–water interface was modified to improve Hb catalytic efficiency in organic solvents (0–80% v/v organic solvent; remainder is the conjugate, the substrate, and water). The protein–polymer–solvent interface modification was achieved by esterifying the carboxylic acid groups of Hb–PAA with ethanol (EtOH) or 1-propanol (1-prop) after activation with carbodiimide. The resulting esters (Hb–PAA–Eth and Hb–PAA–1-prop, respectively) showed high peroxidase-like catalytic activities in acetonitrile (ACN), dimethylformamide (DMF), EtOH, and methanol (MeOH). Catalytic activities depended on the log(P) values of the solvents, which is a measure of solvent lipophilicity. The highest weighted-average activities were noted in MeOH for all three conjugates, and the lowest average activities were noted in DMF for two of the conjugates. Interestingly, the average activities of the conjugates were higher than that of Hb in all solvents except in ACN. The ratio of the catalytic rate constant (kcat) to the Michaelis constant (KM), the catalytic efficiency, for Hb–PAA–Eth in MeOH was the highest noted, and it is ∼3-fold higher than that of Hb in buffer; conjugates offered higher efficiencies than Hb at most solvent compositions. This is the very first general, versatile, modular strategy of coupling the enhanced stability of Hb with improved activity in organic solvents via the chemical manipulation of the polymer shell around Hb and provides a robust approach to efficient biocatalysis in organic solvents.
Co-reporter:Ananta Ghimire, Rajeswari M. Kasi, and Challa V. Kumar
The Journal of Physical Chemistry B 2014 Volume 118(Issue 19) pp:5026-5033
Publication Date(Web):April 16, 2014
DOI:10.1021/jp500310w
Rational design of protein–polymer composites and their use, under the influence of the stimulus, for numerous applications requires a clear understanding of protein–polymer interfaces. Here, using poly(acrylic acid) (PAA) and lysozyme as model systems, the binding interactions between these macromolecules were investigated by isothermal titration calorimetry. The binding is proposed to require and be governed by “charge neutralization of the protein/polymer interface” and predicted to depend on solution pH. Calorimetric data show strong exothermic binding of lysozyme to PAA with a molar ΔH and TΔS values of −107 and −95 kcal/mol, respectively, at pH 7 and room temperature. Both ΔH and TΔS decreased linearly with increasing pH from 3 to 8, and these plots had slopes of −17.7 and −17.5 kcal/mol per pH unit, respectively. The net result was that the binding propensity (ΔG) was nearly independent of pH but the binding stoichiometry, surprisingly, increased rapidly with increasing pH from 1 lysozyme binding per PAA molecule at pH 3 to 16 lysozyme molecules binding per PAA molecule at pH 8. A plot of stoichiometry vs pH was linear, and consistent with this result, a plot of ln(average size of the protein/polymer complex) vs pH was also linear. Thus, protonation–deprotonation plays a major role in the binding mechanism. “Charge neutralization” of the lysozyme/PAA interface controls the binding stoichiometry as well as the binding enthalpies/entropies in a predictable fashion, but it did not control the binding affinity (ΔG). The pH dependence of lysozyme binding to PAA, demonstrated here, provides a stimuli-responsive system for protein binding and release from the polymer surface.
Co-reporter:Ajith Pattammattel, Megan Puglia, Subhrakanti Chakraborty, Inoka K. Deshapriya, Prabir K. Dutta, and Challa V. Kumar
Langmuir 2013 Volume 29(Issue 50) pp:15643-15654
Publication Date(Web):2017-2-22
DOI:10.1021/la404051c
Graphene oxide (GO) is being investigated extensively for enzyme and protein binding, but many enzymes bound to GO denature considerably and lose most of their activities. A simple, novel, and efficient approach is described here for improving the structures and activities of enzymes bound to GO such that bound enzymes are nearly as active as those of the corresponding unbound enzymes. Our strategy is to preadsorb highly cationized bovine serum albumin (cBSA) to passivate GO, and cBSA/GO (bGO) served as an excellent platform for enzyme binding. The binding of met-hemoglobin, glucose oxidase, horseradish peroxidase, BSA, catalase, lysozyme, and cytochrome c indicated improved binding, structure retention, and activities. Nearly 100% of native-like structures of all the seven proteins/enzymes were noted at near monolayer formation of cBSA on GO (400% w/w), and all bound enzymes indicated 100% retention of their activities. A facile, benign, simple, and general method has been developed for the biofunctionalization of GO, and this approach of coating with suitable protein glues expands the utility of GO as an advanced biophilic nanomaterial for applications in catalysis, sensing, and biomedicine.
Co-reporter:Inoka K. Deshapriya and Challa V. Kumar
Langmuir 2013 Volume 29(Issue 46) pp:14001-14016
Publication Date(Web):October 8, 2013
DOI:10.1021/la403165y
Specific approaches to the rational design of nanobio interfaces for enzyme and protein binding to nanomaterials are vital for engineering advanced, functional nanobiomaterials for biocatalysis, sensing, and biomedical applications. This feature article presents an overview of our recent discoveries on structural, functional, and mechanistic details of how enzymes interact with inorganic nanomaterials and how they can be controlled in a systematic manner using α-Zr(IV)phosphate (α-ZrP) as a model system. The interactions of a number of enzymes having a wide array of surface charges, sizes, and functional groups are investigated. Interactions are carefully controlled to screen unfavorable repulsions and enhance favorable interactions for high affinity, structure retention, and activity preservation. In specific cases, catalytic activities and substrate selectivities are improved over those of the pristine enzymes, and two examples of high activity near the boiling point of water have been demonstrated. Isothermal titration calorimetric studies indicated that enzyme binding is coupled to ion sequestration or release to or from the nanobio interface, and binding is controlled in a rational manner. We learned that (1) bound enzyme stabilities are improved by lowering the entropy of the denatured state; (2) maximal loadings are obtained by matching charge footprints of the enzyme and the nanomaterial surface; (3) binding affinities are improved by ion sequestration at the nanobio interface; and (4) maximal enzyme structure retention is obtained by biophilizing the nanobio interface with protein glues. The chemical and physical manipulations of the nanobio interface are significant not only for understanding the complex behaviors of enzymes at biological interfaces but also for desiging better functional nanobiomaterials for a wide variety of practical applications.
Co-reporter:Vamsi K. Mudhivarthi, Kyle S. Cole, Marc J. Novak, Westley Kipphut, Inoka K. Deshapriya, Yuxiang Zhou, Rajeswari M. Kasi and Challa V. Kumar
Journal of Materials Chemistry A 2012 vol. 22(Issue 38) pp:20423-20433
Publication Date(Web):08 Aug 2012
DOI:10.1039/C2JM34434C
Stabilization of proteins against thermal deactivation is a major challenge, and a simple, facile, novel chemical approach is described here to overcome this hurdle. We report here, for the first time, the successful synthesis of ultrastable protein nanoparticles consisting of met-hemoglobin (Hb) conjugated with low molecular weight polyacrylic acid (PAA, Mw 8000) to form discrete nanoparticles. Hb–PAA nanoparticles were not deactivated when subjected to prolonged thermal treatment such as steam sterilization conditions (122 °C, 40 minutes, 17–20 psi), while the unprotected Hb lost most of its activity when subjected to the same heating conditions. Several Hb–PAA derivatives which resist thermal inactivation, in a similar manner, are produced and characterized. Interestingly, the highest activity retention, after the above thermal treatment, was ∼100% for the untreated samples. This resistance to heat is attributed to the enhanced thermodynamic stability of the Hb–PAA conjugate and improved re-folding of the denatured state to the native form, facilitated by PAA conjugation to Hb. This is a unique approach to stabilize Hb against thermal inactivation, and it is a major breakthrough in the production of stable Hb-based nanomaterials that can be safely sterilized in an autoclave for biomedical/in vivo applications.
Co-reporter:Ruma Chowdhury, Bobbi Stromer, Binod Pokharel, and Challa V. Kumar
Langmuir 2012 Volume 28(Issue 32) pp:11881-11889
Publication Date(Web):July 23, 2012
DOI:10.1021/la3022003
Electrostatic forces could contribute significantly toward enzyme–solid interactions, and controlling these charge–charge interactions while maintaining high affinity, benign adsorption of enzymes on solids is a challenge. Here, we demonstrate that chemical modification of the surface carboxyl groups of enzymes can be used to adjust the net charge of the enzyme and control binding affinities to solid surfaces. Negatively charged nanosolid, α-Zr(HPO4)2·H2O (abbreviated as α-ZrP) and two negatively charged proteins, glucose oxidase (GO) and methemoglobin (Hb), have been chosen as model systems. A limited number of the aspartate and glutamate side chains of these proteins are covalently modified with tetraethylenepentamine (TEPA) to convert these negatively charged proteins into the corresponding positively charged ones (cationized). Cationized proteins retained their structure and activities to a significant extent, and the influence of cationization on binding affinities has been tested. Cationized GO, for example, showed 250-fold increase in affinity for the negatively charged α-ZrP, when compared to that of the unmodified GO, and cationized Hb, similarly, indicated 26-fold increase in affinity. Circular dichroism spectra showed that α-ZrP-bound cationized GO retained native-like structure to a significant extent, and activity studies showed that cationized GO/α-ZrP complex is ∼2.5-fold more active than GO/α-ZrP. Cationized Hb/α-ZrP retained ∼75% of activity of Hb/α-ZrP. Therefore, enzyme cationization enhanced affinities by 1–2 orders of magnitude, while retaining considerable activity for the bound biocatalyst. This benign, chemical control over enzyme charge provided a powerful new strategy to rationally modulate enzyme–solid interactions while retaining their biocatalytic properties.
Co-reporter:Vindya K. Thilakarathne, Victoria A. Briand, Rajeswari M. Kasi, and Challa V. Kumar
The Journal of Physical Chemistry B 2012 Volume 116(Issue 42) pp:12783-12792
Publication Date(Web):October 2, 2012
DOI:10.1021/jp307206h
Protein–polymer interactions play a very important role in a number of applications, but details of these interactions are not fully understood. Chemical modification was introduced here to tune protein–polymer interactions in a systematic manner, where methemoglobin (Hb) and poly(acrylic acid) (PAA) served as a model system. Under similar conditions of pH and ionic strength, the influence of protein charge on Hb/PAA interaction was studied using chemically modified Hb by isothermal titration calorimetry (ITC). A small fraction of COOH groups of Hb were amidated with triethylenetetramine (TETA) or ammonium chloride to produce the corresponding charge ladders of Hb-TETA and Hb-ammonia derivatives, respectively. All the Hb/PAA complexes produced here are bioactive, entirely soluble in water, and indicated the retention of Hb structure to a significant extent. Binding of Hb to PAA was exothermic (ΔH < 0). The binding of Hb-TETA charge ladder to PAA indicated decrease of ΔH from −8 ± 0.2 to −89 ± 4 kcal/mol, at a rate of −3.8 kcal/mol per unit charge introduced via modification. The Hb-ammonia charge ladder, in contrast, showed a decrease of ΔH from −8 ± 0.2 to −17 ± 1.5 kcal/mol, at much slower rate of −1.0 kcal/mol per unit charge. Thus, the amine used for the modification played a strong role in tuning Hb/PAA interactions, even after correcting for the charge, synergistically. Charge clustering may be responsible for this synergy, and this interesting observation may be exploited to construct protein/polymer platforms for advanced biomacromolecular applications.
Co-reporter:Victoria A. Briand, Vindya Thilakarathne, Rajeswari M. Kasi, Challa V. Kumar
Talanta 2012 Volume 99() pp:113-118
Publication Date(Web):15 September 2012
DOI:10.1016/j.talanta.2012.05.026
We have developed a novel surface plasmon resonance (SPR) biosensor for heme detection that utilizes the reconstitution of the heme cofactor with apohemoglobin (apoHb), hemoglobin from which the heme has been removed, as the sensing mechanism. The binding is highly specific, efficient and generated very strong SPR signals. This is the first report that uses immobilization of the apoprotein in a hydrophilic polymer matrix and senses the corresponding cofactor by SPR. This is also the first report of high sensitivity heme detection in real time by SPR and the sensing surface is re-generated many times without loss of sensitivity or selectivity. The sensing surface was fabricated by covalent immobilization of hemoglobin in a polyacrylic acid matrix in situ, which allowed for a high concentration of protein to be located in the plasmon detection range on the Au chip. Removal of the heme from the hemoglobin–polymer conjugate (Hb–PAA) resulted in a surface anchored apoHb–polymer conjugate. The limit of detection was approximately 2 μM or 1.30 μg/mL, which is relevant for biological heme levels (1–50 μM for hemolytic pathological conditions). This apoHb–polyacrylic acid system demonstrates a new concept in SPR detection with the use of protein cofactor binding pockets for analyte detection. The methodology that we developed here may be extended for the detection of a number of other cofactor molecules with high sensitivity, selectivity and low detection limits. In future, such sensors could be useful for the development of point-of-care devices to detect biologically important small molecules.Highlights► Novel sensing surface with apoHb immobilized in polyacrylic acid. ► Detection of heme by cofactor reconstitution on SPR surface. ► Biologically relevant detection of heme with a detection limit of 2 μM. ► SPR sensor that can be re-used for up to 12 cycles without significant loss of sensitivity or selectivity.
Co-reporter:Vindya Thilakarathne, Victoria A. Briand, Yuxiang Zhou, Rajeswari M. Kasi, and Challa V. Kumar
Langmuir 2011 Volume 27(Issue 12) pp:7663-7671
Publication Date(Web):May 18, 2011
DOI:10.1021/la2015034
The synthesis, characterization, and evaluation of a novel polymer–protein conjugate are reported here. The covalent conjugation of high-molecular weight poly(acrylic acid) (PAA) to the lysine amino groups of met-hemoglobin (Hb) resulted in the covalent conjugation of Hb to PAA (Hb-PAA conjugate), as confirmed by dialysis and electrophoresis studies. The retention of native-like structure of Hb in Hb-PAA was established from Soret absorption, circular dichroism studies, and the redox activity of the iron center in Hb-PAA. The peroxidase-like activities of the Hb-PAA conjugate further confirmed the retention of Hb structure and biological activity. Thermal denaturation of the conjugate was investigated by differential scanning calorimetry and steam sterilization studies. The Hb-PAA conjugate indicated an improved denaturation temperature (Td) when compared to that of the unmodified Hb. One astonishing observation was that polymer conjugation significantly enhanced the Hb-PAA storage stability at room temperature. After 120 h of storage at room temperature in phosphate-buffered saline (PBS) at pH 7.4, for example, Hb-PAA retained 90% of its initial activity and unmodified Hb retained <60% of its original activity under identical conditions of buffer, pH, and temperature. Our conjugate demonstrates the key role of polymers in enhancing Hb stability via a very simple, efficient, general route. Water-swollen, lightly cross-linked, stable Hb-polymer nanogels of 100–200 nm were produced quickly and economically by this approach for a wide variety of applications.
Co-reporter:Thota Jyotsna and Challa V. Kumar, Steffen Jockusch and Nicholas J. Turro
Langmuir 2010 Volume 26(Issue 3) pp:1966-1972
Publication Date(Web):October 7, 2009
DOI:10.1021/la902611j
Steady-state and time-resolved studies of site-selective photocleavage of lysozyme by cobalt(III) complexes [Co(NH3)5Br]2+ and ([Co(NH3)4CO3]+ are reported. Photocleavage resulted in two fragments of molecular masses ∼10.5 kDa and ∼3.5 kDa, and the yield increased (8−33%) with irradiation time (0.16−0.8 h) as well as with the metal complex concentration (0.1−5 mM). The reaction proceeded to a significant extent even when nearly stoichiometric amounts of the reagents were used. Photocleavage was effective at wavelengths ranging from 310 to 390 nm, and cleavage was inhibited by the addition of selected metal ions such as Gd(III) at moderate concentrations (2 mM). Gd(III) is known to bind at Asp52/Glu35 residues on lysozyme, and these residues are located at the enzyme active site. Current and previous studies suggest that Co(III) metal complexes bind at this site on lysozyme. Consistent with this hypothesis, [Co(NH3)4CO3]+ (8 mM) inhibited lysozyme activity by 67%. Laser flash photolysis studies show that excitation of the metal complexes [Co(NH3)5Br]2+ and ([Co(NH3)4CO3]+ (308 nm, 20 ns pulse width) resulted in the corresponding ligand-derived radical intermediates. For example, photoexcitation of an aqueous solution of [Co(NH3)5Br]2+ at 308 nm resulted in the formation of Br2−•. When the excitation was carried out in the presence of lysozyme, Br2−• was quenched with a bimolecular rate constant of 1.4 × 109 M−1 s−1. Quenching resulted in protein-derived radicals (Trp+• and Tyr+•), as identified by their characteristic known transient absorption bands. Steady-state studies correlated with the time-resolved data, and taken together, these illustrated the reactivities of Co(III) metal complexes to direct protein photocleavage with high selectivity.
Co-reporter:Challa V. Kumar ;Michael. R. Duff ; Jr.
Journal of the American Chemical Society 2009 Volume 131(Issue 44) pp:16024-16026
Publication Date(Web):October 21, 2009
DOI:10.1021/ja904551n
Solar radiation reaching this planet is distributed over a wide range of wavelengths, and efficient collection and conversion of solar energy requires light harvesting over multiple wavelengths. Yet, the design, synthesis, and testing of novel, efficient, inexpensive light harvesting complexes are lacking. Engineered protein−DNA complexes are used here to self-assemble donor and acceptor molecules into artificial light harvesting units with an association constant of 3.3 ± 1.2 μM−1. Excitation of the DNA-bound donors resulted in a 540% increase in emission from the protein-bound acceptors, and the presence of one acceptor for each pair of donors was sufficient to quench ∼50% of donor emission. Successful self-assembly of DNA-based light harvesting units is expected to facilitate economic/efficient conversion of solar energy, and model systems to achieve this goal are demonstrated here. We anticipate that success along these lines would facilitate more efficient approaches for solar energy capture.
Co-reporter:Michael R. Duff, Jr. and Challa V. Kumar
Metallomics 2009 vol. 1(Issue 6) pp:518-523
Publication Date(Web):02 Sep 2009
DOI:10.1039/B910253A
Metal binding to serum albumins is examined by oxidative protein-cleavage chemistry, and relative affinities of multiple metal ions to particular sites on these proteins were identified using a fast and reliable chemical footprinting approach. Fe(II) and Cu(II), for example, mediate protein cleavage at their respective binding sites on serum albumins, in the presence of hydrogen peroxide and ascorbate. This metal-mediated protein-cleavge reaction is used to evaluate the binding of metal ions, Na+, Mg2+, Ca2+, Al3+, Cr3+, Mn2+, Co2+, Ni2+, Zn2+, Cd2+, Hg2+, Pb2+, and Ce3+ to albumins, and the relative affinities (selectivities) of the metal ions are rapidly evaluated by examining the extent of inhibition of protein cleavage. Four distinct systems Fe(II)/BSA, Cu(II)/BSA, Fe(II)/HSA and Cu(II)/HSA are examined using the above strategy. This metallomics approach is novel, even though the cleavage of serum albumins by Fe(II)/Cu(II) has been reported previously by this laboratory and many others. The protein cleavage products were analyzed by SDS PAGE, and the intensities of the product bands quantified to evaluate the extent of inhibition of the cleavage and thereby evaluate the relative binding affinities of specific metal ions to particular sites on albumins. The data show that Co(II) and Cr(III) showed the highest degree of inhibition, across the table, followed by Mn(II) and Ce(III). Alakali metal ions and alkaline earth metal ions showed very poor affinity for these metal sites on albumins. Thus, metal binding profiles for particular sites on proteins can be obtained quickly and accurately, using the metallomics approach.
Co-reporter:Michael R. Duff Jr., Vamsi K. Mudhivarthi and Challa V. Kumar
The Journal of Physical Chemistry B 2009 Volume 113(Issue 6) pp:1710-1721
Publication Date(Web):January 21, 2009
DOI:10.1021/jp807164f
Achieving the goal of rational design of DNA-binding ligands is important, and many inroads have been made in this direction. Toward that goal, we report a simple, systematic, and quantitative approach to design DNA-binding anthracene derivatives. Current data show that the binding free energies (ΔG°) as well as enthalpies (ΔH°) are related to specific structural features of the binders. Systematic design of anthracene probes, for example, indicated that the affinity can be enhanced via the introduction of methylene groups. Each methylene group contributed, on an average, −0.08 ± 0.002 kcal/mol (at 1 M ionic strength, 293 K) toward the total binding free energy. Binding of the probes to DNA depended on ionic strength, and ionic strength studies were used to factor out to parse free-energy contributions due to specific interactions. The intrinsic free-energy contributions (ΔGMol) of the probes are obtained by factoring out contributions from ionic interactions, hydration, conformational changes, polyelectrolyte effect, and the loss of rotational/translational motion. A strong, linear correlation was noted between ΔGMol and the number of methylene groups present in the probe, and the correlation indicated free-energy contributions of −1.49 kcal/mol per methylene (at 50 mM NaCl, 293 K). This important observation provides a convenient handle to systematically fine-tune the intrinsic affinities of DNA binders. ΔH values also showed clear trends, and each methylene contributed +0.28 kcal/mol toward the overall binding enthalpy (at 50 mM NaCl, 293 K), and this aspect is useful to fine-tune ΔH contributions to binding. These important physical insights, derived from systematic modifications of the side chains of the DNA binders, are useful in the rational design of novel DNA binders.
Co-reporter:Akhilesh Bhambhani, Challa V. Kumar
Microporous and Mesoporous Materials 2008 Volume 109(1–3) pp:223-232
Publication Date(Web):1 March 2008
DOI:10.1016/j.micromeso.2007.04.048
Direct assessment of the thermodynamic stabilities of enzymes bound to solids is essential to understand the factors that control bound enzyme stability. Here, the first reports of the thermodynamic stabilities of enzymes/proteins which are bound to an inorganic solid [α-Zr(HPO4)2 · H2O, abbreviated as α-ZrP] are described. The thermal denaturation of hen egg white lysozyme (Lys), met-myoglobin (Mb) and met-hemoglobin (Hb) bound to α-ZrP occurs over a wide range of temperatures (50–100 °C). This is in contrast to the behavior of the free enzyme/proteins in the solution, which indicated sharp transitions at their respective denaturation temperatures. Denaturation of the bound protein depended on the scan rate and the denaturation process was kinetically controlled. At rapid scan rates (2 °C/min), for example, the thermal profiles of the intercalated proteins became sharper while the free proteins indicated little or no changes. Careful analysis of the calorimetric data provided a clear distinction between the moving-boundary model and the uniform distribution model for protein binding. Calorimetric data also revealed that a distribution of thermodynamic states or kinetically-slow forming states are important in the denaturation. While the thermal denaturation of Mb bound to α-ZrP indicated a significant extent of reversibility, Lys and Hb did not. The solid stabilized a fraction of the intercalated protein, and efforts will be focused to maximize this portion. Improved thermal stabilities are important for biosensor or biocatalysis applications of enzyme–inorganic materials.
Co-reporter:Thota Jyotsna, Kalpanie Bandara and Challa V. Kumar
Photochemical & Photobiological Sciences 2008 vol. 7(Issue 12) pp:1531-1539
Publication Date(Web):28 Oct 2008
DOI:10.1039/B810422K
Photocleavage of chicken hen egg lysozyme by three Co(III)ammine complexes, hexamminecobalt(III) chloride ([Co(NH3)6]+3), pentamminechloro cobalt(III)chloride ([Co(NH3)5Cl]+2), and tetramminecarbonato cobalt(III) nitrate ([Co(NH3)4CO3]+), is reported here. Photocleavage resulted in two fragments of molecular masses of ∼10.5 kDa and ∼3.5 kDa which add-up to that of the parent molar mass. Detailed studies on the influence of irradiation time, excitation wavelength, the type of ligand coordinated to Co(III), concentration of the metal complex, the addition of competing metal ions, and quenchers on the protein photocleavage are reported. The Co(III) complexes also photocleaved apotransferrin, bovine serum albumin, and yeast enolase. Near-equimolar concentrations of Ni(II), Co(II) or Gd(III) inhibited the photocleavage, and therefore, binding of Co(III) metal complexes to Ni(II)/Co(II)/Gd(III) binding sites on lysozyme is necessary for the observed photocleavage. Since these ions are known to bind to Asp52 on lysozyme, we suspect that the above Co(III) complexes bind at this site, and initiate the protein cleavage. The Co(III) complexes have appropriate photochemical reactivities to cleave the peptide backbone, and they may be useful in the design of novel photochemical approaches to cleave the protein backbone.
Co-reporter:Challa V. Kumar and Michael R. Duff, Jr.
Photochemical & Photobiological Sciences 2008 vol. 7(Issue 12) pp:1522-1530
Publication Date(Web):16 Oct 2008
DOI:10.1039/B811091C
Specific donor and acceptor pairs have been assembled in bovine serum albumin (BSA), at neutral pH and room temperature, and these dye–protein complexes indicated efficient donor to acceptor singlet–singlet energy transfer. For example, pyrene-1-butyric acid served as the donor and Coumarin 540A served as the acceptor. Both the donor and the acceptor bind to BSA with affinity constants in excess of 2 × 105 M−1, as measured in absorption and circular dichroism (CD) spectral titrations. Simultaneous binding of both the donor and the acceptor chromophores was supported by CD spectra and one chromophore did not displace the other from the protein host, even when limited concentrations of the host were used. For example, a 1:1:1 complex between the donor, acceptor and the host can be readily formed, and spectral data clearly show that the binding sites are mutually exclusive. The ternary complexes (two different ligands bound to the same protein molecule) provided opportunities to examine singlet–singlet energy transfer between the protein-bound chromophores. Donor emission was quenched by the addition of the acceptor, in the presence of limited amounts of BSA, while no energy transfer was observed in the absence of the protein host, under the same conditions. The excitation spectra of the donor–acceptor-host complexes clearly show the sensitization of acceptor emission by the donor. Protein denaturation, as induced by the addition of urea or increasing the temperature to 360 K, inhibited energy transfer, which indicate that protein structure plays an important role. Sensitization also proceeded at low temperature (77 K) and diffusion of the donor or the acceptor is not required for energy transfer. Stern–Volmer quenching plots show that the quenching constant is (3.1 ± 0.2) × 104 M−1, at low acceptor concentrations (<35 μM). Other albumins such as human and porcine proteins also served as good hosts for the above experiments. For the first time, non-natural systems have been self-assembled which can capture donor–acceptor pairs and facilitate singlet–singlet energy transfer. Such systems may form a basis for the design and construction of protein-based multi-chromophore self-assemblies for solar light harvesting, conversion and storage.
Co-reporter:Akhilesh Bhambhani, Soonwoo Chah, Eli G. Hvastkovs, Gary C. Jensen, James F. Rusling, Richard N. Zare and Challa V. Kumar
The Journal of Physical Chemistry B 2008 Volume 112(Issue 30) pp:9201-9208
Publication Date(Web):July 4, 2008
DOI:10.1021/jp7121642
The free energy change (ΔG°) for the unfolding of immobilized yeast iso-1-cytochrome c (Cyt c) at nanoassemblies was measured by surface plasmon resonance (SPR) spectroscopy. Data show that SPR is sensitive to protein conformational changes, and protein solid interface exerts a major influence on bound protein stability. First, Cyt c was self-assembled on the Au film via the single thiol of Cys-102. Then, crystalline sheets of layered α-Zr(O3POH)2·H2O (α-ZrP) or Zr(O3PCH2CH2COOH)2·xH2O (α-ZrCEP) were adsorbed to construct α-ZrP/Cyt c/Au or α-ZrCEP/Cyt c/Au nanoassemblies. The construction of each layer was monitored by SPR, in real time, and the assemblies were further characterized by atomic force microscopy and electrochemical studies. Thermodynamic stability of the protein nanoassembly was assessed by urea-induced unfolding. Surprisingly, unfolding is reversible in all cases studied here. Stability of Cyt c in α-ZrP/Cyt c/Au increased by ∼4.3 kJ/mol when compared to the unfolding free energy of Cyt c/Au assembly. In contrast, the protein stability decreased by ∼1.5 kJ/mol for α-ZrCEP/Cyt c/Au layer. Thus, OH-decorated surfaces stabilized the protein whereas COOH-decorated surfaces destabilized it. These data quantitate the role of specific functional groups of the inorganic layers in controlling bound protein stability.
Co-reporter:Apinya Buranaprapuk, Yaowaluk Malaikaew, Jisnuson Svasti and Challa V. Kumar
The Journal of Physical Chemistry B 2008 Volume 112(Issue 30) pp:9258-9265
Publication Date(Web):July 4, 2008
DOI:10.1021/jp802791c
Strong chiral discrimination and site-selective photocleavage of two model proteins, lysozyme and bovine serum albumin (BSA), by new pyrenyl probes are reported here. The enantiomeric pyrenyl probes d-phenylalanine-1(1-pyrene)methylamide (PMA-d-Phe) and l-phenylalanine-1(1-pyrene)methylamide (PMA-l-Phe) were synthesized by coupling the carboxyl function of d-phenylalanine or l-phenylalanine with the amino group of 1(1-pyrene)methylamine. Binding affinities of the two enantiomers with the proteins were quantitated in absorption titrations. BSA indicated 10-fold selectivity for PMA-d-Phe, and the binding constants for the l- and d-enantiomers were 3.8 × 105 and 4.0 × 106 M−1, respectively. Lysozyme, similarly, indicated a 6-fold preference for PMA-d-Phe with binding constants of 3.3 × 105 and 2.0 × 106 M−1 for the l- and d-isomers, respectively. Such strong chiral discrimination illustrates the key role of the chiral center of the probe (Phe) in the binding interactions. The enantiomers were tested to examine how the chiral discrimination for their binding influences reactivity toward protein photocleavage. Irradiation of the probe−protein complexes, at 342 nm in the presence of hexammine cobalt(III) chloride, resulted in the cleavage of the protein backbone. Photocleavage did not proceed in the dark or in the absence of the pyrenyl probes. Both enantiomers indicated low reactivity with BSA (<5% yield), while large photocleavage yields (∼57%) have been noted with lysozyme. This lysozyme photocleavage yield is a significant improvement over previous reports. However, both enantiomers cleaved lysozyme at the same location between Trp108-Val109, despite the strong chiral selectivity for binding. H-atom abstraction from Trp 108, accessible from the active site cleft, could initiate the observed peptide bond cleavage.
Co-reporter:A. Bhambhani;C. V. Kumar
Advanced Materials 2006 Volume 18(Issue 7) pp:939-942
Publication Date(Web):24 MAR 2006
DOI:10.1002/adma.200502230
Successful intercalation of calf thymus DNA into the galleries of layered alpha zirconium phosphate (α-Zr(HPO4)2· H2O, α-ZrP) was facilitated by met-hemoglobin (Hb) and tetrabutylammonium (TBA) hydroxide. Intercalation of DNA, in turn, improved the properties of the co-intercalated protein. The protein structure, for example, was improved significantly and activities were enhanced by more than fivefold.
Co-reporter:V. Jagannadham, Akhilesh Bhambhani, Challa V. Kumar
Microporous and Mesoporous Materials 2006 Volume 88(1–3) pp:275-282
Publication Date(Web):21 January 2006
DOI:10.1016/j.micromeso.2005.09.019
Thermal stability of met-hemoglobin (Hb) intercalated in three layered solid materials was investigated at 60 °C. At this temperature, free Hb undergoes rapid thermal denaturation, which is accompanied by the loss of its peroxidase-like activity and protein secondary structure. Heating of Hb intercalated in the galleries of α-zirconium(IV) phosphate (at 60 °C) also indicated initial loss of peroxidase-like activity of bound Hb, within 2 h. Continued equilibration of Hb bound to this solid (at 60 °C) indicated steady increase in its activity (>50%), as a function of continued equilibration at this temperature. The extent of this recovery also depended on the type surface functions present on the solid, and these are in the order α-Zr(PO3CH2CH2COOH)2 > α-Zr(PO3CH2COOH)2 > α-Zr(HPO4)2. In the case of α-Zr(PO3CH2CH2COOH)2, the recovered activity was nearly twice as much as the activity, before heating. The circular dichroism spectra of the samples, heated at 60 °C for various time intervals, also indicated the initial loss of structure followed by significant recovery. This behavior is highly unusual, and these are the very first examples of activity recovery of proteins bound to solids, upon thermal treatment at moderate temperatures. These studies clearly indicate the important role of layered solids and their surface functions in the recovery of Hb structure. Annealing could be a useful method in recovering or improving the activities of specific proteins bound to selected solid surfaces.
Co-reporter:Alison Rodger;Willy B. Tan;Akhilesh Bhambhani;Michael R. Duff
Photochemistry and Photobiology 2006 Volume 82(Issue 1) pp:20-30
Publication Date(Web):30 APR 2007
DOI:10.1562/2005-05-24-RA-539
The binding properties of two anthracene derivatives with calf thymus DNA (CT DNA), poly(dA-dT), and poly(dG).poly(dC) are reported. One contained bulky, cyclic cationic substituents at the 9 and 10 positions, and the other carried acylic, branched, cationic substituents. Binding of the probes to the DNA was examined by calorimetry, spectroscopy and helix melting studies. The cyclic derivative indicated exothermic binding, strong hypochromism, bathochromism, positive induced cir-cular dichroism (CD, 300–400 nm), significant unwinding of the helix, large increases in the helix melting temperature, strong but negative linear dichroism (LD, 300–400 nm) and consider-able stabilization of the helix. In contrast, the acyclic analog indicated thermoneutral binding, smaller hypochromism, no bathochromism, very weak induced CD, and no change in the helix melting temperature with any of the DNA polymers. A sharp distinction between the binding properties of the two probes is indicated, and both have intrinsic binding constants of ∼106M-1 for the three polymers. However, when the ionic strength of the medium was lowered (10 mM NaCl), the absorption as well as CD spectral changes associated with the binding of the acyclic derivative corresponded with those of the cyclic derivative. The acyclic derivative showed large preference (10-fold) for poly(dG)·poly(dC) over poly(dA-dT), whereas the cyclic analog showed no preference. The characteristic spectroscopic signatures of the two distinct binding modes of these probes will be helpful in deciphering the interaction of other anthracene derivatives with DNA.
Co-reporter:N.K. Modukuru, K.J. Snow, B. Scott Perrin Jr., A. Bhambhani, M. Duff, Challa V. Kumar
Journal of Photochemistry and Photobiology A: Chemistry 2006 Volume 177(Issue 1) pp:43-54
Publication Date(Web):1 January 2006
DOI:10.1016/j.jphotochem.2005.05.010
The DNA binding properties of an anthracene derivative with substituents at the 9 and 10 positions, carrying four positive charges, are examined in calorimetric, spectroscopic and photocleavage studies. Isothermal titration calorimetric data indicated exothermic binding of the ligand to calf thymus DNA with a binding constant of (1.4 ± 0.5) × 105 M−1 and this value is much greater than binding of similar monocationic derivatives. The values for the other binding parameters were, ΔH = −3.5 ± 0.4 kcal/mol; ΔS = 11.6 ± 1.6 cal/mol K, and a binding site size of ∼4 base pairs. Absorption spectral studies indicated small, but significant red shifts in the vibronic bands, and ∼70% of hypochromism. The binding plots indicated bi-phasic binding of the ligand. At higher ionic strengths, the red shifts in the absorption spectra were abolished but significant hypochromism persisted.Excitation and sensitized fluorescence spectral studies indicated weak energy transfer from the DNA bases to the ligand. Further more, energy transfer was reduced substantially at higher ionic strengths. Strong induced circular dichroism bands are noted, in the 300–400 nm region, and these are most likely dominated by the contributions from the groove bound form as well as the intercalated chromophore. Helix melting studies indicated improvement in the helix stability, and substantial increase in the melting temperature (ΔTm > 17 °C). Differential scanning calorimetric data, on the other hand, indicated only minor improvements in the thermodynamic parameters. Irradiation of a mixture of the ligand (2 μM) and supercoiled pUC18 DNA (20 μM, @374 nm) resulted in the efficient formation of nicked circular DNA (>90%) in an hour. The data indicated at least two distinct binding modes, and one of these persisted at high ionic strengths (375 mM NaCl). Substitution at 9 and 10 positions of the anthracene ring system with positively charged residues resulted in multiple binding modes, and these are resolvable in ionic strength studies.
Co-reporter:Michael R. Duff Jr.,Challa V. Kumar
Angewandte Chemie International Edition 2006 45(1) pp:137-139
Publication Date(Web):
DOI:10.1002/anie.200502344
Co-reporter:Michael R. Duff Jr.
Angewandte Chemie 2006 Volume 118(Issue 1) pp:
Publication Date(Web):21 NOV 2005
DOI:10.1002/ange.200502344
Fußspuren eines Metalls: Das Uranyl-Ion (UO22+) bindet mit hoher Affinität und spezifisch an ausgewählte Proteine. Diese Eigenschaft wurde genutzt, um Proteine an der UO22+-Bindungsstelle photolytisch zu spalten. Abfangstudien ergaben, dass freies UO22+ für die Spaltung nicht verantwortlich ist. Dies ist der erste Bericht über die durch Anregung des Uranyl-Ions mit sichtbarem Licht ausgelöste hoch selektive Photospaltung eines Proteins.
Co-reporter:C. M. Riccardi, D. Mistri, O. Hart, M. Anuganti, Y. Lin, R. M. Kasi and C. V. Kumar
Chemical Communications 2016 - vol. 52(Issue 12) pp:NaN2596-2596
Publication Date(Web):2016/01/05
DOI:10.1039/C6CC00037A
A modular, general method for trapping enzymes within the voids of paper, without chemical activation of cellulose, is reported. Glucose oxidase and peroxidase were crosslinked with poly(acrylic acid) via carbodiimide chemistry, producing 3-dimensional networks interlocked in cellulose fibers. Interlocking prevented enzyme activity loss and enhanced the washability and stability.
Co-reporter:Vamsi K. Mudhivarthi, Kyle S. Cole, Marc J. Novak, Westley Kipphut, Inoka K. Deshapriya, Yuxiang Zhou, Rajeswari M. Kasi and Challa V. Kumar
Journal of Materials Chemistry A 2012 - vol. 22(Issue 38) pp:NaN20433-20433
Publication Date(Web):2012/08/08
DOI:10.1039/C2JM34434C
Stabilization of proteins against thermal deactivation is a major challenge, and a simple, facile, novel chemical approach is described here to overcome this hurdle. We report here, for the first time, the successful synthesis of ultrastable protein nanoparticles consisting of met-hemoglobin (Hb) conjugated with low molecular weight polyacrylic acid (PAA, Mw 8000) to form discrete nanoparticles. Hb–PAA nanoparticles were not deactivated when subjected to prolonged thermal treatment such as steam sterilization conditions (122 °C, 40 minutes, 17–20 psi), while the unprotected Hb lost most of its activity when subjected to the same heating conditions. Several Hb–PAA derivatives which resist thermal inactivation, in a similar manner, are produced and characterized. Interestingly, the highest activity retention, after the above thermal treatment, was ∼100% for the untreated samples. This resistance to heat is attributed to the enhanced thermodynamic stability of the Hb–PAA conjugate and improved re-folding of the denatured state to the native form, facilitated by PAA conjugation to Hb. This is a unique approach to stabilize Hb against thermal inactivation, and it is a major breakthrough in the production of stable Hb-based nanomaterials that can be safely sterilized in an autoclave for biomedical/in vivo applications.
.jpg)


![1H,5H,11H-[1]Benzopyrano[6,7,8-ij]quinolizin-11-one, 2,3,6,7-tetrahydro-9-(trifluoromethyl)-](/data/chemimg/905400/53518-18-6.png)
![1H,5H,11H-[1]Benzopyrano[6,7,8-ij]quinolizin-11-one, 2,3,6,7-tetrahydro-9-(trifluoromethyl)-](/data/chemimg/905400/53518-18-6_b.png)













![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)