Co-reporter:Fuzhu Liu, Chao Wu, and Shengchun Yang
The Journal of Physical Chemistry C October 12, 2017 Volume 121(Issue 40) pp:22139-22139
Publication Date(Web):September 20, 2017
DOI:10.1021/acs.jpcc.7b07081
The strain and ligand effects on the adsorption energies of key intermediates (*COOH, *CO, *CHO, and *COH) in CO2 reduction reactions on the Cu–M(111) (M = Ni, Co, Cu, Rh, Ir, Pd, Pt) heterolayered catalysts have been quantitatively separated using first-principles calculations. Contrary to the common belief that strain is always the leading factor influencing catalytic performance of the core–shell type heterostructure catalysts, the ligand effect due to the underlying hetero elements should not be ignored and may become dominant for strain-insensitive adsorbates (*CO and *COH). Moreover, the models of Cu(2 ML/3 ML)–M(111) (M = Ir, Rh, Pt, Pd) have been shown to be better catalysts for CO2 reduction, as they require lower overpotential to drive the reaction than the Cu(111) slab. Particularly, the overpotential is predicted to be lowered by 0.17 V for Cu(3 ML)–Ir(111) model catalyst. Thus, both effects should be considered in heterostructure catalyst design.
Co-reporter:Y. Zhang, D. Lu, J.-J. Zhang and C. Wu
RSC Advances 2016 vol. 6(Issue 70) pp:66078-66086
Publication Date(Web):06 Jul 2016
DOI:10.1039/C6RA10356A
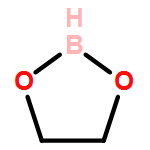
Thirteen new 1,3-substituted imidazolium poly(azolyl)borate salts with the general formula [R1R2im][B(H)4−n(azolyl)n] (R1 = methyl, n-butyl, 2-(diethylamino)ethyl; R2 = methyl; azolyl = pyrazolyl, imidazolyl, 1,2,4-triazolyl, tetrazolyl; n = 2, 3) have been synthesized through salt metathesis of the corresponding imidazolium chloride and potassium poly(azolyl)borates. The newly synthesized dihydrobis(azolyl)borate and hydrotris(azolyl)borate salts are liquids at room temperature except two of the 1,3-dimethylimidazolium derivatives, [mmim][H2B(pz)2] and [mmim][HB(pz)3]. Conductivity, thermal and physicochemical properties of the new borate ionic liquids were systematically investigated. With multiple azolyl groups serving as binding sites, these ionic liquids (ILs) generally exhibit high SO2 absorption capacity (up to 5.8 mol mol−1 of IL or 1.05 g g−1 of IL for 1-methyl-3-n-butylimidazolium hydrotris(imidazolyl)borate, compound 13).
Co-reporter:Huarong Tang, Dongmei Lu and Chao Wu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 24) pp:15725-15731
Publication Date(Web):12 May 2015
DOI:10.1039/C5CP01793A
Cation-assisted interactions between N-containing heterocycles (NHCs) and CO2 have been systematically studied by using density functional theory (DFT). For neutral and anionic (non-carbenoid) NHCs, the effects of monovalent cations (i.e., alkali metal ions) are moderate to small (the NHC–CO2 binding energy change, ΔBE usually < 25 kJ mol−1). However, for NHC carbenes, due to their strong basicity, the effects are strong (ΔBE > 60 kJ mol−1) and the monovalent cations play a critical role in the single carboxylation of dicarbenes with CO2. In comparison, divalent alkali earth metal cations, due to both their smaller sizes and higher formal charges, exhibit a much stronger influence (ΔBE > 100 kJ mol−1). Divalent cations should be incorporated into next generation CO2 capture reagents. Other aspects including the reaction potential energy surface (PES), orbital-based analyses of interactions, substitution effects, and the reactivity descriptors (cation size, reacting N lone pair orbital energy, etc.) have been discussed in detail as well.
Co-reporter:Zhengzheng Chen, Pengfei Li and Chao Wu
RSC Advances 2015 vol. 5(Issue 16) pp:11791-11796
Publication Date(Web):14 Jan 2015
DOI:10.1039/C4RA15322G
A novel 2D structure composed only of carbon and nitrogen elements at the molar ratio of 1:1 is predicted by first-principles calculations. The basic structural units are 1,3,5-triazine molecules which trimerize and polymerize into a 2D network of semiconductor nature. Six triazine units comprise a hollow hexagon with a van der Waals hole diameter of about 2.4 Å, which is suitable for H2 separation from larger molecules. Metal atoms of various sizes can strongly bind over the polynitrogen pore, which suggests that the 2D network is an ideal support for single-atom catalysis.
Co-reporter:Zhenni Wang, Zhengzheng Chen, Hui Zhang, Zhaorui Zhang, Haijun Wu, Mingshang Jin, Chao Wu, Deren Yang, and Yadong Yin
ACS Nano 2015 Volume 9(Issue 3) pp:3307
Publication Date(Web):March 6, 2015
DOI:10.1021/acsnano.5b00475
Synthesis of anisotropic nanostructures from materials with isotropic crystal structures often requires the use of seeds containing twin planes to break the crystalline symmetry and promote the preferential anisotropic growth. Controlling twinning in seeds is therefore critically important for high-yield synthesis of many anisotropic nanostructures. Here, we demonstrate a unique strategy to induce twinning in metal nanostructures for anisotropic growth by taking advantage of the large lattice mismatch between two metals. By using Au–Cu as an example, we show, both theoretically and experimentally, that deposition of Cu to the surface of single-crystalline Au seeds can build up strain energy, which effectively induces the formation of twin planes. Subsequent seeded growth allows the production of Cu nanorods with high shape anisotropy that is unachievable without the use of Au seeds. This work provides an effective strategy for the preparation of anisotropic metal nanostructures.Keywords: copper; core−shell; epitaxial growth; gold; lattice mismatch; twinned structure;
Co-reporter:Fuzhu Liu
The Journal of Physical Chemistry C 2015 Volume 119(Issue 27) pp:15500-15505
Publication Date(Web):June 8, 2015
DOI:10.1021/acs.jpcc.5b04511
The oxidation of CO on strained Pt(100) surface was studied using periodic density functional theory (DFT). Unlike the uniform response of global properties (e.g., d-band center) to strain, the localized nature of adsorption leads to complex site-dependent and adsorbate-dependent responses, invalidating the generally believed statement of “tension strengthens binding”. Moreover, the complex responses of reaction energetics to strain require direct study of the reaction under strain rather than extrapolating the known behaviors of individual adsorbates under strain or reaction energetics on unstrained surfaces. We show that the tensile strain lowers the reaction barrier of CO oxidation over the Pt(100) surface. This work provides a theoretical basis of utilizing strain to improve the Pt catalysts with a higher tolerance toward CO poisoning.
Co-reporter:Yu Wei, Huarong Tang, Xuefeng Cong, Bin Rao, Chao Wu, and Xiaoming Zeng
Organic Letters 2014 Volume 16(Issue 8) pp:2248-2251
Publication Date(Web):March 31, 2014
DOI:10.1021/ol500745t
An example of using readily available, less reactive aryl bromides as arylating reagents in the Pd(II)-catalyzed intermolecular arylation of unactivated C(sp3)–H bonds is described. This reaction was promoted by a crucial 8-aminoquinolinyl directing group and a K2CO3 base, enabling regiospecific installation of an aryl scaffold at the β-position of carboxamides. A mechanistic study by DFT calculations reveals a C(sp3)–H activation-led pathway featuring the oxidative addition as the highest energy transition state.
Co-reporter:Yuan-Chao Pang, Xiufang Hou, Lei Qin, Chao Wu, Wei Xue, Yan-Zhen Zheng, Zhiping Zheng and Xiao-Ming Chen
Chemical Communications 2014 vol. 50(Issue 22) pp:2910-2912
Publication Date(Web):24 Jan 2014
DOI:10.1039/C3CC49287G
Allylic rearrangement or the migration of a double bond from its original position in the carbon skeleton to an adjacent site was observed when 3,4,5,6-tetrahydrophthalate was hydrolyzed in a basic solution and in the presence of Co(II) and Mn(II) under hydrothermal conditions.
Co-reporter:Ran Li, Huarong Tang, Haixing Fu, Hailong Ren, Xuemei Wang, Chunrui Wu, Chao Wu, and Feng Shi
The Journal of Organic Chemistry 2014 Volume 79(Issue 3) pp:1344-1355
Publication Date(Web):January 16, 2014
DOI:10.1021/jo402754d
Arynes are shown to insert into some C═X double bonds, leading to benzannulated four-membered rings. The strain of these rings allow for a ready, spontaneous opening to afford o-quinomethide analogues. Subsequent nucleophilic addition re-aromatizes the intermediates to achieve ortho-difunctionalization of arynes. In this report, we describe the aryne insertion into the C═C double bonds of vinylogous amides and the C═N double bonds of carbodiimides. The correlation and comparison with aryne single bond insertion chemistry will be discussed. Computational studies for the ring-opening step, as well as the nature of the o-quinomethide intermediates, will also be discussed.
Co-reporter:Huarong Tang ;Dr. Chao Wu
ChemSusChem 2013 Volume 6( Issue 6) pp:1050-1056
Publication Date(Web):
DOI:10.1002/cssc.201200986
Abstract
Azole anions are key components in CO2 capture materials that include ionic liquids and porous solids. Herein, we use density functional theory (DFT) and a Langmuir-type adsorption model to study azole anion–CO2 interactions. Linear CO2 has to be bent by approximately 45° to form an NC bond within the azole ring. The energy cost of bending renders CO2 absorption much more difficult compared to SO2 absorption. For different azole anions, the number of nitrogen atoms in the ring and the natural bond orbital energy of the reacting nitrogen lone pair, both linearly correlate with the calculated reaction enthalpy and are useful handles for new sorbent designs. Unlike for SO2, the azole parent architecture (unsubstituted) does not allow successive CO2 absorption under mild conditions (<0.12 MPa and at room temperature). Experimental CO2 and SO2 absorption isotherms are reproduced by using the Langmuir model parameterized with the calibrated DFT reaction enthalpies. This study provides insight for designing azole-based CO2-capture materials.
Co-reporter:Xuefeng Cong, Huarong Tang, Chao Wu, and Xiaoming Zeng
Organometallics 2013 Volume 32(Issue 21) pp:6565-6575
Publication Date(Web):October 9, 2013
DOI:10.1021/om400890p
We report here that mono-N-protected amino acids (MPAAs), an important environmentally compatible structural motif, enable acceleration of Pd(II)-catalyzed dehydrogenative Heck reactions between pyridines and electron-deficient arenes with simple alkenes, leading to diversely functionalized C3- or meta-selective alkenylated pyridines and benzenes via non-chelate-assisted C–H activation. A comprehensive theoretical study by DFT calculations discloses that the amino scaffold of the MPAA ligand facilely converts to an X-type ligand by an initial N–H activation, resulting in a relatively low activation barrier for the C–H cleavage of pyridine. Then a property reversal of the amino group from X-type to L-type ligand allows the alkene substitution to take place smoothly, while the carboxyl group enables the formation of an intramolecular hydrogen bond, significantly decreasing the activation barrier for the carbopalladation. The results of calculations and the kinetic isotopic effect measurement support a rate-limiting C–H activation by a mechanism involving a concerted metalation/deprotonation pathway, with an endothermicity of 31.0 kcal/mol in the process.
Co-reporter:Huarong Tang, Dongmei Lu and Chao Wu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 24) pp:NaN15731-15731
Publication Date(Web):2015/05/12
DOI:10.1039/C5CP01793A
Cation-assisted interactions between N-containing heterocycles (NHCs) and CO2 have been systematically studied by using density functional theory (DFT). For neutral and anionic (non-carbenoid) NHCs, the effects of monovalent cations (i.e., alkali metal ions) are moderate to small (the NHC–CO2 binding energy change, ΔBE usually < 25 kJ mol−1). However, for NHC carbenes, due to their strong basicity, the effects are strong (ΔBE > 60 kJ mol−1) and the monovalent cations play a critical role in the single carboxylation of dicarbenes with CO2. In comparison, divalent alkali earth metal cations, due to both their smaller sizes and higher formal charges, exhibit a much stronger influence (ΔBE > 100 kJ mol−1). Divalent cations should be incorporated into next generation CO2 capture reagents. Other aspects including the reaction potential energy surface (PES), orbital-based analyses of interactions, substitution effects, and the reactivity descriptors (cation size, reacting N lone pair orbital energy, etc.) have been discussed in detail as well.
Co-reporter:Yuan-Chao Pang, Xiufang Hou, Lei Qin, Chao Wu, Wei Xue, Yan-Zhen Zheng, Zhiping Zheng and Xiao-Ming Chen
Chemical Communications 2014 - vol. 50(Issue 22) pp:NaN2912-2912
Publication Date(Web):2014/01/24
DOI:10.1039/C3CC49287G
Allylic rearrangement or the migration of a double bond from its original position in the carbon skeleton to an adjacent site was observed when 3,4,5,6-tetrahydrophthalate was hydrolyzed in a basic solution and in the presence of Co(II) and Mn(II) under hydrothermal conditions.
























![3H-Imidazo[4,5-b]pyridin-7-amine](http://img.cochemist.com/ccimg/6800/6703-44-2.png)
![3H-Imidazo[4,5-b]pyridin-7-amine](http://img.cochemist.com/ccimg/6800/6703-44-2_b.png)