Co-reporter:Benjamin J. Foust, Michael M. Poe, Nicholas A. Lentini, Chia-Hung Christine Hsiao, Andrew J. Wiemer, and David F. Wiemer
ACS Medicinal Chemistry Letters September 14, 2017 Volume 8(Issue 9) pp:914-914
Publication Date(Web):August 9, 2017
DOI:10.1021/acsmedchemlett.7b00245
Studies of aryl phosphonate derivatives of a butyrophilin 3A1 ligand have resulted in identification of a potent stimulant of Vγ9 Vδ2 T cells. This compound, a mixed ester bearing one pivaloyloxymethyl substituent and one 1-naphthyl ester displayed an EC50 of 0.79 nM as a stimulant of T cell proliferation, and a 9.0 nM EC50 in an assay designed to measure interferon gamma production. In both assays, this is the most potent butyrophilin ligand prodrug yet reported, and thus it should be a valuable tool for studies of T cell function. Furthermore, mixed aryl/acyloxyalkyl esters may represent a new class of phosphonate prodrugs with high efficacy.Keywords: Aryl phosphonates; BTN3A1; butyrophilin; ligand; phosphoantigen; prodrug;
Co-reporter:David P. Stockdale, John A. Beutler, David F. Wiemer
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.08.016
The schweinfurthins are plant-derived stilbenes with an intriguing profile of anti-cancer activity. To obtain analogues of the schweinfurthins that might preserve the biological activity but have greater water solubility, a formal replacement of the central olefin with an amide has been explored. Two pairs of amides have been prepared, each containing the same hexahydroxanthene “left half” joined through an amide linkage to two different “right halves.” In each series, the amide has been inserted in both possible orientations, placing the carbonyl group on the tricyclic ABC ring system and the amine on the D-ring, or placing the amine on the hexahydroxanthene and the carbonyl group on the D-ring. The four new schweinfurthin analogues have been tested in the NCI 60 cell line screen, and in both cases the more active isomer carried the carbonyl group on the C-ring.Download high-res image (109KB)Download full-size image
Co-reporter:Benjamin J. Foust, Cheryl Allen, Sarah A. Holstein, David F. Wiemer
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 16) pp:3734-3741
Publication Date(Web):15 August 2016
DOI:10.1016/j.bmc.2016.06.019
The enzyme geranylgeranyl diphosphate synthase (GGDPS) is believed to receive the substrate farnesyl diphosphate through one lipophilic channel and release the product geranylgeranyl diphosphate through another. Bisphosphonates with two isoprenoid chains positioned on the α-carbon have proven to be effective inhibitors of this enzyme. Now a new motif has been prepared with one isoprenoid chain on the α-carbon, a second included as a phosphonate ester, and the potential for a third at the α-carbon. The pivaloyloxymethyl prodrugs of several compounds based on this motif have been prepared and the resulting compounds have been tested for their ability to disrupt protein geranylgeranylation and induce cytotoxicity in myeloma cells. The initial biological studies reveal activity consistent with GGDPS inhibition, and demonstrate a structure–function relationship which is dependent on the nature of the alkyl group at the α-carbon.
Co-reporter: Andrew J. Wiemer;Rebekah R. Shippy;Ashley M. Kilcollins;Jin Li;Dr. Chia-Hung Christine Hsiao; Rocky J. Barney; M. Lei Geng; David F. Wiemer
ChemBioChem 2016 Volume 17( Issue 1) pp:52-55
Publication Date(Web):
DOI:10.1002/cbic.201500484
Abstract
Cell-cleavable protecting groups often enhance cellular delivery of species that are charged at physiological pH. Although several phosphonate protecting groups have achieved clinical success, it remains difficult to use these prodrugs in live cells to clarify biological mechanisms. Here, we present a strategy that uses a 7-methoxycoumarin-3-carboxylic acid ester as a fluorescent protecting group. This strategy was applied to synthesis of an (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP) analogue to assess cellular uptake and human Vγ9Vδ2 T cell activation. The fluorescent ester displayed low cellular toxicity (IC50>100 μm) and strong T cell activation (EC50=0.018 μm) relative to the unprotected anion (EC50=23 μm). The coumarin-derived analogue allowed no-wash analysis of biological deprotection, which revealed rapid internalization of the prodrug. These results demonstrate that fluorescent groups can be applied both as functional drug delivery tools and useful biological probes of drug uptake.
Co-reporter:Kevyn D. Gardner and David F. Wiemer
The Journal of Organic Chemistry 2016 Volume 81(Issue 4) pp:1585-1592
Publication Date(Web):January 15, 2016
DOI:10.1021/acs.joc.5b02756
Pawhuskin A is a prenylated stilbene that functions as an antagonist of the kappa opioid receptor. Analogues of this natural product bearing different placements of the prenyl group in the A-ring have shown selectivity for either the kappa or the delta receptors subtypes. This differential activity has drawn attention to regiospecific preparation of the C-2, C-5, and C-6 prenylated A-ring regioisomers. Through halogen metal exchange, advanced intermediates representing each of these regioisomers have been prepared, and the new C-6 intermediate has been converted to a new analogue of the natural stilbene.
Co-reporter:Robert A. Matthiesen, Veronica S. Wills, Joseph I. Metzger, Sarah A. Holstein, and David F. Wiemer
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:9438-9442
Publication Date(Web):September 20, 2016
DOI:10.1021/acs.joc.6b01693
Isoprenoid-substituted bisphosphonates are known to serve as inhibitors of the enzyme geranylgeranyl diphosphate synthase, and their activity can be highly sensitive to olefin stereochemistry. A mixture of homogeranyl and homoneryl triazole bisphosphonates has previously demonstrated potent activity, and thus stereocontrolled syntheses of the individual isomers have been developed.
Co-reporter:Veronica S. Wills, Cheryl Allen, Sarah A. Holstein, and David F. Wiemer
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 12) pp:1195
Publication Date(Web):October 28, 2015
DOI:10.1021/acsmedchemlett.5b00334
Studies of triazole bisphosphonates have resulted in identification of a potent inhibitor of geranylgeranyl diphosphate synthase (IC50 = 45 nM) with very good selectivity for this enzyme over farnesyl diphosphate synthase (IC50 = 28 μM). This compound also potently disrupts geranylgeranylation and induces cytotoxicity in human myeloma cells at submicromolar levels, suggesting that it may serve as a lead compound for treatment of malignancies characterized by excessive protein secretion.Keywords: bisphosphonate; farnesyl diphosphate synthase; Geranylgeranyl diphosphate synthase; GGDPS; myeloma
Co-reporter:Xiang Zhou, Ella J. Born, Cheryl Allen, Sarah A. Holstein, David F. Wiemer
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 11) pp:2331-2334
Publication Date(Web):1 June 2015
DOI:10.1016/j.bmcl.2015.04.021
The N-oxide derivatives of [2-(3-pyridinyl)-1-hydroxyethylidene-1,1-phosphonocarboxylic acid (or PEHPC) and [2-(3-pyridinyl)-1-ethylidene-1,1-phosphonocarboxylic acid (or PEPC) have been prepared and evaluated for their activity against several enzymes which utilize isoprenoids. The parent pyridines are known inhibitors of GGTase II, but the N-oxide derivatives show no improvement in biological activity in assays with the isolated enzyme. However, the PEHPC N-oxide did induce significant accumulation of intracellular light chain in myeloma cells, consistent with inhibition of Rab geranylgeranylation.
Co-reporter:Xiang Zhou, Sarah D. Ferree, Veronica S. Wills, Ella J. Born, Huaxiang Tong, David F. Wiemer, Sarah A. Holstein
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 9) pp:2791-2798
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmc.2014.03.014
When inhibitors of enzymes that utilize isoprenoid pyrophosphates are based on the natural substrates, a significant challenge can be to achieve selective inhibition of a specific enzyme. One element in the design process is the stereochemistry of the isoprenoid olefins. We recently reported preparation of a series of isoprenoid triazoles as potential inhibitors of geranylgeranyl transferase II but these compounds were obtained as a mixture of olefin isomers. We now have accomplished the stereoselective synthesis of these triazoles through the use of epoxy azides for the cycloaddition reaction followed by regeneration of the desired olefin. Both geranyl and neryl derivatives have been prepared as single olefin isomers through parallel reaction sequences. The products were assayed against multiple enzymes as well as in cell culture studies and surprisingly a Z-olefin isomer was found to be a potent and selective inhibitor of geranylgeranyl diphosphate synthase.
Co-reporter:John G. Kodet, John A. Beutler, David F. Wiemer
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 8) pp:2542-2552
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmc.2014.02.043
Co-reporter:Xiang Zhou, Sara V. Hartman, Ella J. Born, Jacqueline P. Smits, Sarah A. Holstein, David F. Wiemer
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:764-766
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.11.089
A small set of triazole bisphosphonates has been prepared and tested for the ability to inhibit geranylgeranyltransferase II (GGTase II). The compounds were prepared through use of click chemistry to assemble a central triazole that links a polar head group to a hydrophobic tail. The resulting compounds were tested for their ability to inhibit GGTase II in an in vitro enzyme assay and also were tested for cytotoxic activity in an MTT assay with the human myeloma RPMI-8226 cell line. The most potent enzyme inhibitor was the triazole with a geranylgeranyl tail, which suggests that inhibitors that can access the enzyme region that holds the isoprenoid tail will display greater activity.
Co-reporter:John G. Kodet and David F. Wiemer
The Journal of Organic Chemistry 2013 Volume 78(Issue 18) pp:9291-9302
Publication Date(Web):August 19, 2013
DOI:10.1021/jo4014244
An interest in the schweinfurthins, natural stilbenes with significant antiproliferative activity, has prompted efforts to prepare a set of indole analogues. To approach the desired compounds through a Horner–Wadsworth–Emmons condensation, new indole derivatives bearing a phosphonomethyl substituent in the B-ring were required. The parent indole system with the necessary substitution pattern was obtained through Stobbe condensation and cyclization. A prenyl substituent was incorporated at the C3 position of a 4,6-disubstituted indole through a highly regioselective electrophilic aromatic substitution reaction, while metalation and alkylation provided the C2-prenylated indole. After introduction of the phosphonate group through classical reactions, the new indole phosphonates were found to undergo the desired condensation with nonracemic aldehydes representing the schweinfurthin left half. This approach provides facile access to new heteroaromatic analogues of the natural schweinfurthins and should be applicable to many other natural stilbenes as well.
Co-reporter:John G. Kodet, Joseph J. Topczewski, Kevyn D. Gardner, David F. Wiemer
Tetrahedron 2013 69(44) pp: 9212-9218
Publication Date(Web):
DOI:10.1016/j.tet.2013.08.056
Co-reporter:Rebekah M. Richardson, Rocky J. Barney, David F. Wiemer
Tetrahedron Letters 2012 Volume 53(Issue 49) pp:6682-6684
Publication Date(Web):5 December 2012
DOI:10.1016/j.tetlet.2012.09.114
Several trialkyl and two triaryl phosphites have been tested for their reactivity in the zinc iodide mediated conversion of benzyl alcohol to the corresponding phosphonates. Most react smoothly to afford the desired phosphonate diesters, including hindered and nonracemic phosphites. The implications of this reactivity on the reactions involved in the transformation are discussed.
Co-reporter:Joseph J. Topczewski, Michael P. Callahan, John G. Kodet, Jery D. Inbarasu, Nolan R. Mente, John A. Beutler, David F. Wiemer
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 24) pp:7570-7581
Publication Date(Web):15 December 2011
DOI:10.1016/j.bmc.2011.10.034
The schweinfurthins are an intriguing group of anti-proliferative agents that display low nanomolar activities against several cell types, including the human-derived glioblastoma cell line SF-295, but have little impact on other cell lines even at micromolar concentrations. This activity has inspired the synthesis of seven of the natural schweinfurthins, all with the correct absolute stereochemistry, and a variety of analogues designed to probe different facets of the pharmacophore. Reported herein is the synthesis of several new schweinfurthin analogues varied at the C-5 position along with data on their biological activity in the NCI 60 cell-line assay.
Co-reporter:Joseph J. Topczewski, David F. Wiemer
Tetrahedron Letters 2011 Volume 52(Issue 14) pp:1628-1630
Publication Date(Web):6 April 2011
DOI:10.1016/j.tetlet.2011.01.137
The total synthesis of (+)-vedelianin has been accomplished in 18 steps from vanillin. Preparation of a key intermediate in nonracemic form through a Shi epoxidation has allowed determination of the absolute stereochemistry of the natural product as the (2S,3R,4aR,9aR)-isomer.
Co-reporter:Jacqueline P. Smits and David F. Wiemer
The Journal of Organic Chemistry 2011 Volume 76(Issue 21) pp:8807-8813
Publication Date(Web):September 14, 2011
DOI:10.1021/jo201523w
The α-trisphosphonic acid esters provide a unique spatial arrangement of three phosphonate groups and may represent an attractive motif for inhibitors of enzymes that utilize di- or triphosphate substrates. To advance studies of this unique functionality, a general route to alkyl derivatives of the parent system (R = H) has been developed. A set of new α-alkyl-1,1,1-trisphosphonate esters has been prepared through phosphinylation and subsequent oxidation of tetraethyl alkylbisphosphonates, and the reactivity of these new compounds has been studied in representative reactions that afford additional examples of this functionality.
Co-reporter:Rocky J. Barney, Rebekah M. Richardson, and David F. Wiemer
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2875-2879
Publication Date(Web):March 15, 2011
DOI:10.1021/jo200137k
Benzyl phosphonate esters often serve as reagents in Horner−Wadsworth−Emmons reactions. In most cases, they can be prepared from benzylic alcohols via formation of the corresponding halide followed by an Arbuzov reaction. To identify a more direct synthesis of phosphonate esters, we have developed a one-flask procedure for conversion of benzylic and allylic alcohols to the corresponding phosphonates through treatment with triethyl phosphite and ZnI2.
Co-reporter:Rocky J. Barney, Brian M. Wasko, Amel Dudakovic, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 20) pp:7212-7220
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmc.2010.08.036
Geminal bisphosphonates display varied biological activity depending on the nature of the substituents on the central carbon atom. For example, the nitrogenous bisphosphonates zoledronate and risedronate inhibit the enzyme farnesyl diphosphate synthase while digeranyl bisphosphonate has been shown to inhibit the enzyme geranylgeranyl diphosphate synthase. We now have synthesized isoprenoid bisphosphonates where an aromatic ring has been used to replace one of the isoprenoid olefins in an isoprenoid bisphosphonate and investigated the ability of these new compounds to impair protein geranylgeranylation within cells. Several of these new compounds are potent inhibitors of the enzyme geranylgeranyl diphosphate synthase.A series of isoprenoid bisphosphonates has been prepared where an aromatic ring has been used to replace one of the isoprenoid olefins. These compounds have been investigated for their ability to inhibit protein geranylgeranylation in enzyme assays and within cells.
Co-reporter:Joseph J. Topczewski, Craig H. Kuder, Jeffrey D. Neighbors, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 18) pp:6734-6741
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmc.2010.07.056
The natural tetracyclic schweinfurthins are potent and selective inhibitors of cell growth in the National Cancer Institute’s 60 cell-line screen. At this time, the mechanism or cellular target that underlies this activity has not yet been identified, and efforts to illuminate the schweinfurthins’ mode of action would benefit from development of potent fluorescent analogs that could be readily visualized within cells. This report describes the synthesis of fluorescent analogs of schweinfurthins B and F, and demonstrates that these compounds retain the potent and differentially toxic activities against select human cancer cells that are characteristic of the natural schweinfurthins. In addition, the synthesis of control compounds that maintain parallel fluorescent properties, but lack the potent activity of the natural schweinfurthin is described. Use of fluorescence microscopy shows differences between the localization of the active and relatively inactive schweinfurthin analogs. The active compounds localize in peripheral puncta which may identify the site(s) of activity.The synthesis of several new fluorescent analogs of schweinfurthins B and F is described, along with assays that demonstrates that these compounds retain potent and differential activities against select human cancer cell lines. Use of fluorescence microscopy shows differences between the localization of the active and relatively inactive schweinfurthin analogs.
Co-reporter:Natalie C. Ulrich, John G. Kodet, Nolan R. Mente, Craig H. Kuder, John A. Beutler, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 4) pp:1676-1683
Publication Date(Web):15 February 2010
DOI:10.1016/j.bmc.2009.12.063
The natural tetracyclic schweinfurthins are potent and selective inhibitors of cell growth in the National Cancer Institute’s 60-cell line screen. An interest in determination of their cellular or molecular target has inspired our efforts to prepare both the natural products and analogues. In this paper, chemical synthesis of analogues modified in different olefinic positions, and preliminary results from studies of their biological activity, are reported.Synthesis of a set of schweinfurthin analogues varied in the D-ring alkyl substituent and stilbene moiety has been accomplished, and the activity of these compounds has been measured in a two-cell line screen. The most potent analogue and a much less active compound also were tested in the NCI 60-cell line assay, and showed comparable potency in that more extensive screen.
Co-reporter:Natalie C. Ulrich, Craig H. Kuder, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 22) pp:6716-6720
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmcl.2010.08.143
As a prelude to efforts to identify schweinfurthin binding proteins, an ester conjugate and an amide conjugate of schweinfurthin F and biotin have been prepared by chemical synthesis. These compounds maintain activity in SF-295 cells comparable to the parent system, and display the lower potency in A549 cells that is a characteristic of the schweinfurthin pattern of activity.
Co-reporter:Joseph J. Topczewski ; Michael P. Callahan ; Jeffrey D. Neighbors
Journal of the American Chemical Society 2009 Volume 131(Issue 41) pp:14630-14631
Publication Date(Web):September 25, 2009
DOI:10.1021/ja906468v
When cascade cyclizations initiated by Lewis acid-mediated opening of an epoxide are terminated through reaction with a MOM-protected phenol, a tandem electrophilic aromatic substitution can be obtained. This highly regioselective tandem process has been employed in the first synthesis of (+)-angelichalcone.
Co-reporter:Craig H. Kuder, Jeffrey D. Neighbors, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 13) pp:4718-4723
Publication Date(Web):1 July 2009
DOI:10.1016/j.bmc.2009.04.071
Most of the natural schweinfurthins are potent and selective inhibitors of cell growth as measured by the National Cancer Institute’s 60-cell line screen. Due to the limited supply of these natural products, we have initiated a program aimed at their synthesis. To date, this effort has led to the preparation of three natural schweinfurthins and more than 40 analogues, and assays on these compounds have afforded some understanding of structure–activity relationships in this family. Further development of schweinfurthins as chemotherapeutic agents would benefit from characterization of their mechanism(s) of action. This perspective led to development of a fluorescent schweinfurthin analogue that retains the differential activity of the natural products, and yet has properties that facilitate its visualization within cells.Synthesis of a fluorescent schweinfurthin analogue has been accomplished, and its biological activity and sub-cellular localization have been studied.
Co-reporter:Jeffrey D. Neighbors, Joseph J. Topczewski, Dale C. Swenson, David F. Wiemer
Tetrahedron Letters 2009 50(27) pp: 3881-3884
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.052
Co-reporter:Jeffrey D. Neighbors, Matthew J. Buller, Kelly D. Boss and David F. Wiemer
Journal of Natural Products 2008 Volume 71(Issue 11) pp:1949-1952
Publication Date(Web):October 15, 2008
DOI:10.1021/np800351c
Pawhuskin A is an isoprenylated stilbene that was isolated from Dalea purpurea and reported to have affinity for the opioid receptor in vitro. It has been synthesized through a convergent sequence that joins a prenylated aldehyde with a geranylated phosphonate in a stereoselective Horner−Wadsworth−Emmons condensation to afford the target E olefin isomer. This synthesis confirms the structure assigned to the natural product and establishes a route that may be used to explore its biological activity and to prepare more active analogues.
Co-reporter:Andrew J. Wiemer, Jose S. Yu, Kimberly M. Lamb, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 1) pp:390-399
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmc.2007.09.029
Nitrogenous bisphosphonates are used clinically to reduce bone resorption associated with osteoporosis or metastatic bone disease, and are recognized as inhibitors of farnesyl diphosphate synthase. Inhibition of this enzyme decreases cellular levels of both farnesyl diphosphate and geranylgeranyl diphosphate which results in a variety of downstream biological effects including inhibition of protein geranylgeranylation. Our lab recently has prepared several isoprenoid bisphosphonates that inhibit protein geranylgeranylation and showed that one selectively inhibits geranylgeranyl diphosphate synthase. This results in depletion of intracellular geranylgeranyl diphosphate and impacts protein geranylgeranylation but does not affect protein farnesylation. To clarify the structural features of isoprenoid bisphosphonates that account for their geranylgeranyl diphosphate synthase inhibition, we have prepared a new group of isoprenoid bisphosphonates. The complete set of compounds has been tested for in vitro inhibition of human recombinant geranylgeranyl diphosphate synthase and cellular inhibition of protein geranylgeranylation. These results show some surprising relationships between in vitro and cellular activity, and will guide development of clinical agents directed at geranylgeranyl diphosphate synthase.
Co-reporter:Andrew J. Wiemer, Jose S. Yu, Larry W. Shull, Rocky J. Barney, Brian M. Wasko, Kimberly M. Lamb, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 7) pp:3652-3660
Publication Date(Web):1 April 2008
DOI:10.1016/j.bmc.2008.02.016
Nitrogenous bisphosphonate inhibitors of farnesyl disphosphate synthase have been used clinically for treatment of bone disease. Because many of their effects may be mediated by depletion of geranylgeranyl diphosphate, our group has sought compounds that do this more directly through inhibition of geranylgeranyl diphosphate synthase and we have discovered a number of isoprenoid-containing bisphosphonates that selectively inhibit this enzyme. These compounds have a high negative charge at physiological pH which is necessary for inhibition of the enzyme but may limit their ability to enter cells. Therefore, chemical modifications that mask this charge may enhance their cellular potency. We now have synthesized novel pivaloyloxymethyl-modified isoprenoid bisphosphonates and investigated their ability to inhibit protein geranylgeranylation within cells. We have found that addition of pivaloyloxymethyl moieties to isoprenoid bisphosphonates increases their potency towards cellular geranylgeranylation even though this modification decreases their in vitro inhibition of geranylgeranyl diphosphate synthase. Pivaloyloxymethyl modifications more effectively increase the cellular activity of the more polar isoprenoid bisphosphonates. These results reveal structural relationships between in vitro and cellular activity which may serve as the basis for future development of more potent and/or drug-like inhibitors of geranylgeranyl diphosphate synthase.
Co-reporter:Nolan R. Mente, Andrew J. Wiemer, Jeffrey D. Neighbors, John A. Beutler, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 4) pp:911-915
Publication Date(Web):15 February 2007
DOI:10.1016/j.bmcl.2006.11.096
Total synthesis of the (R,R,R)- and (S,S,S)-enantiomers of the natural product schweinfurthin F has been completed. Comparisons of spectral data and optical rotations with those reported for the natural product, as well as a variety of bioassay data, allow assignment of the natural material as the (R,R,R)-isomer.
Co-reporter:Larry W. Shull, Andrew J. Wiemer, Raymond J. Hohl, David F. Wiemer
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 12) pp:4130-4136
Publication Date(Web):15 June 2006
DOI:10.1016/j.bmc.2006.02.010
Bisphosphonates have been used in the clinic to treat osteoporosis and to reduce bone resorption and the accompanying pathological bone fractures that attend a number of malignancies including multiple myeloma and cancers of the prostate, breast, and lung. There is also evidence that some bisphosphonates have direct anticancer activity. Expansion of the current class of bisphosphonates may lead to compounds that more selectively and potently target these cancers through inhibition of the mevalonate pathway. To this end, a set of dialkyl bisphosphonates bearing isoprenoid chains of varying lengths has been synthesized. Some of these compounds were found to have biological activity on post-translational processing of the oncogenic small GTPases, Ras and Rap1a, in human-derived K562 leukemia cells. Most importantly, these compounds impair protein geranylgeranylation and not protein farnesylation.
Co-reporter:Jeffrey D. Neighbors, Maya S. Salnikova, John A. Beutler, David F. Wiemer
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 6) pp:1771-1784
Publication Date(Web):15 March 2006
DOI:10.1016/j.bmc.2005.10.025
The synthesis and biological evaluation of several enantioenriched schweinfurthin B analogs were undertaken to develop structure–activity relationships and guide design of probes for their putative molecular target. The desired stilbenes contain a common left-half hexahydroxanthene ring system and an aromatic right-half with varied substituents. The synthesis involves penultimate Horner–Wadsworth–Emmons coupling of one of several right-half phosphonates with the aldehyde comprising the left-half of 3-deoxyschweinfurthin B. Preparation of the requisite phosphonates, and the respective stilbenes, as well as the cytotoxicity profiles of these new compounds in the National Cancer Institute’s 60 cell-line anticancer screen is described. Several of these analogs displayed cytotoxicity patterns well-correlated with the natural product and differences in activity of ∼103 across the various cell lines. Together, these assay results indicate the importance of at least one free phenol group on the aromatic D-ring of this system for differential cytotoxicity.
Co-reporter:Larry W. Shull, David F. Wiemer
Journal of Organometallic Chemistry 2005 Volume 690(Issue 10) pp:2521-2530
Publication Date(Web):16 May 2005
DOI:10.1016/j.jorganchem.2004.10.013
The copper-mediated displacement of allylic THP ethers by Grignard reagents has been examined in a system that contains a geminal bisphosphonate ester. With Grignard reagents derived from several aromatic halides or benzyl bromide the displacement proceeds in attractive yields, but more mixed results were obtained from reactions with alkyl halides. In addition to its role as a nucleophile, the Grignard reagent also appears to deprotonate the bisphosphonate to generate an anionic intermediate. Formation of this anion appears to limit competitive nucleophilic attack at the phosphonate group and provides an intermediate that can be trapped by reaction with an electrophilic reagent such as methyl iodide to access a more substituted system.The copper-mediated displacement of an allylic THP ether by Grignard reagents proceeds in a system that contains a geminal bisphosphonate ester.
Co-reporter:Diana M. Cermak, David F. Wiemer, Kriste Lewis, Raymond J. Hohl
Bioorganic & Medicinal Chemistry 2000 Volume 8(Issue 12) pp:2729-2737
Publication Date(Web):December 2000
DOI:10.1016/S0968-0896(00)00212-1
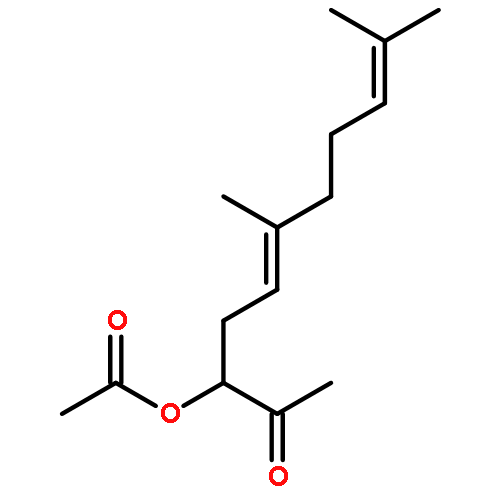
2-(Acyloxy)ethylphosphonate analogues of geranyl, farnesyl, and geranylgeranyl pyrophosphate have been prepared. Horner–Wadsworth–Emmons condensation of different terpene aldehydes with an unsymmetrical bisphosphonate was the key step in syntheses of the phosphonates bearing α,β-unsaturated acyloxy groups. After preparation of the respective phosphonic acids through reaction with TMSBr, both acids and esters were tested for their effects on DNA synthesis in human-derived myeloid and lymphoid leukemia cell lines. The phosphonate esters varied substantially in their ability to impair proliferation of the different cell lines, but testing against one possible target, farnesyl protein transferase (FPTase), revealed little impact at concentrations ranging up to 10 μM. Because the corresponding 2,3-dihydro compounds showed similar biological activity, conjugate addition would not appear to be involved in the toxicity.
Co-reporter:Kang-Yeoun Jung, Raymond J. Hohl, Andrew J. Wiemer, David F. Wiemer
Bioorganic & Medicinal Chemistry 2000 Volume 8(Issue 10) pp:2501-2509
Publication Date(Web):October 2000
DOI:10.1016/S0968-0896(00)00183-8
The vinyl phosphonate derivatives of uridine, cytidine, and cytosine arabinoside (ara-C) have been prepared through oxidation of appropriately protected nucleosides to the 5′ aldehydes and Wittig condensation with [(diethoxyphosphinyl)methylidine]triphenylphosphorane. Dihydroxylation of these vinyl phosphonates with an AD-mix reagent generated the new 5′,6′-dihydroxy-6′-phosphonates. After hydrolysis of the phosphonate esters and the various protecting groups, the six phosphonic acids were tested for their ability to serve as substrates for the enzyme nucleotide monophosphate kinase and for their toxicity to K562 cells.
Co-reporter:Rebekah M. Richardson, Rocky J. Barney, David F. Wiemer
Tetrahedron Letters (5 December 2012) Volume 53(Issue 49) pp:6682-6684
Publication Date(Web):5 December 2012
DOI:10.1016/j.tetlet.2012.09.114
Several trialkyl and two triaryl phosphites have been tested for their reactivity in the zinc iodide mediated conversion of benzyl alcohol to the corresponding phosphonates. Most react smoothly to afford the desired phosphonate diesters, including hindered and nonracemic phosphites. The implications of this reactivity on the reactions involved in the transformation are discussed.Download full-size image
Co-reporter:Jacqueline E. Reilly, Xiang Zhou, Huaxiang Tong, Craig H. Kuder, David F. Wiemer, Raymond J. Hohl
Biochemical Pharmacology (15 July 2015) Volume 96(Issue 2) pp:83-92
Publication Date(Web):15 July 2015
DOI:10.1016/j.bcp.2015.04.009
.jpg)






![Bicyclo[4.1.0]heptane-3,4-diol,3,7,7-trimethyl-, (1S,3S,4S,6R)-](http://img.cochemist.com/ccimg/2200/2140-46-7.png)
![Bicyclo[4.1.0]heptane-3,4-diol,3,7,7-trimethyl-, (1S,3S,4S,6R)-](http://img.cochemist.com/ccimg/2200/2140-46-7_b.png)

![5-[(E)-4-hydroxystyryl]-2-[(2E,6E)-3,7,11-trimethyldodeca-2,6,10-trienyl]benzene-1,3-diol](http://img.cochemist.com/ccimg/1203500/1203460-65-4.png)
![5-[(E)-4-hydroxystyryl]-2-[(2E,6E)-3,7,11-trimethyldodeca-2,6,10-trienyl]benzene-1,3-diol](http://img.cochemist.com/ccimg/1203500/1203460-65-4_b.png)

![[3,4-BIS(METHOXYMETHOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/475300/475294-53-2.png)
![[3,4-BIS(METHOXYMETHOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/475300/475294-53-2_b.png)
![2H-Pyran, 2-[(2-bromo-3,5-dimethoxyphenyl)methoxy]tetrahydro-](http://img.cochemist.com/ccimg/409400/409360-06-1.png)
![2H-Pyran, 2-[(2-bromo-3,5-dimethoxyphenyl)methoxy]tetrahydro-](http://img.cochemist.com/ccimg/409400/409360-06-1_b.png)
![(2S,3R,4aR,9aR)-7-[(E)-2-{4-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-3,5-dihydroxyphenyl}ethenyl]-5-methoxy-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-2,3-diol](http://img.cochemist.com/ccimg/217500/217476-93-2.png)
![(2S,3R,4aR,9aR)-7-[(E)-2-{4-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-3,5-dihydroxyphenyl}ethenyl]-5-methoxy-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-2,3-diol](http://img.cochemist.com/ccimg/217500/217476-93-2_b.png)
![(2S,3R,4aR,9aR)-7-[(E)-2-{4-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-3,5-dihydroxyphenyl}ethenyl]-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-2,3,5-triol](http://img.cochemist.com/ccimg/217500/217476-92-1.png)
![(2S,3R,4aR,9aR)-7-[(E)-2-{4-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-3,5-dihydroxyphenyl}ethenyl]-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-2,3,5-triol](http://img.cochemist.com/ccimg/217500/217476-92-1_b.png)
![Phosphonic acid, [(3,4-dimethylphenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/206100/206008-35-7.png)
![Phosphonic acid, [(3,4-dimethylphenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/206100/206008-35-7_b.png)






![Silane,[[3,5-bis(methoxymethoxy)phenyl]methoxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/144400/144337-52-0.png)
![Silane,[[3,5-bis(methoxymethoxy)phenyl]methoxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/144400/144337-52-0_b.png)




![3,5-Bis[[(tert-Butyl)dimethylsilyl]oxy]benzenemethanol](http://img.cochemist.com/ccimg/104000/103929-84-6.png)
![3,5-Bis[[(tert-Butyl)dimethylsilyl]oxy]benzenemethanol](http://img.cochemist.com/ccimg/104000/103929-84-6_b.png)






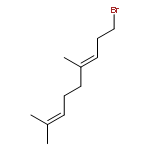
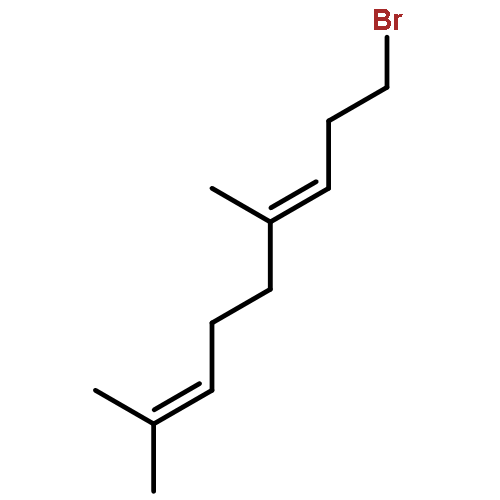


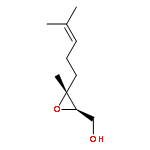
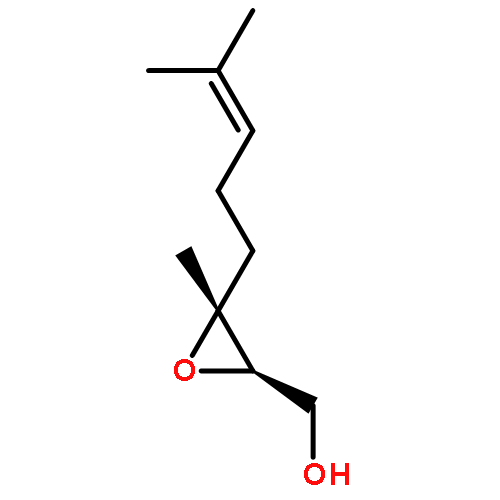
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)
![5-[(E)-2-(4-hydroxyphenyl)ethenyl]-2-(3-methylbut-2-en-1-yl)benzene-1,3-diol](http://img.cochemist.com/ccimg/61600/61517-87-1.png)
![5-[(E)-2-(4-hydroxyphenyl)ethenyl]-2-(3-methylbut-2-en-1-yl)benzene-1,3-diol](http://img.cochemist.com/ccimg/61600/61517-87-1_b.png)
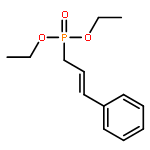
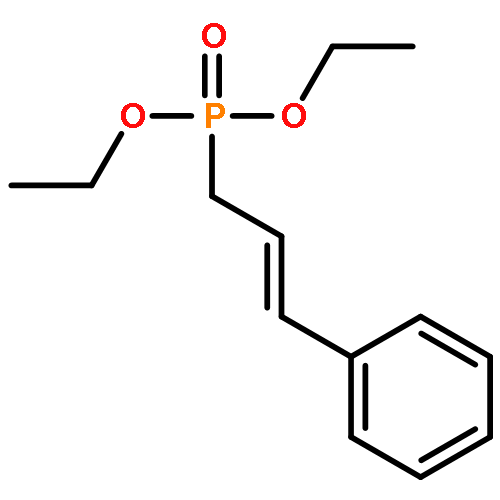
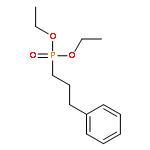
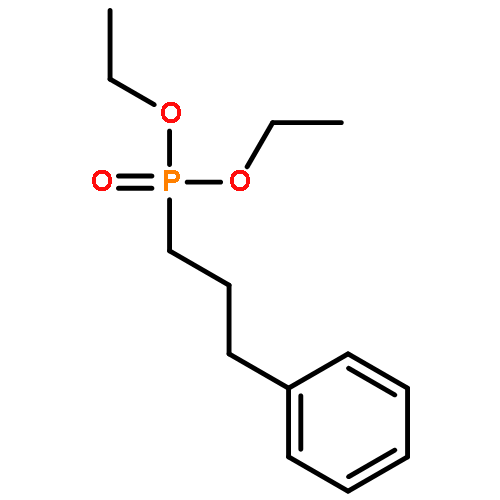














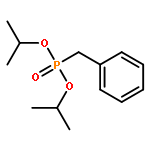
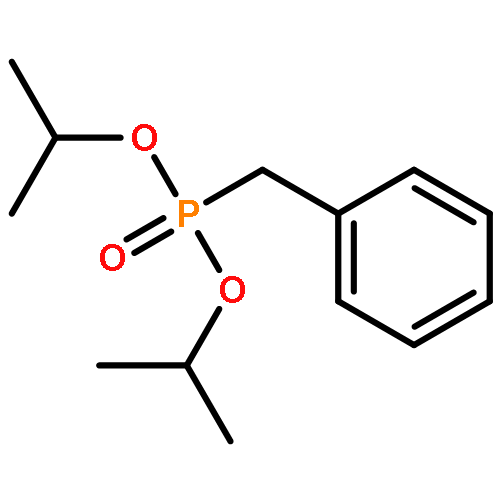
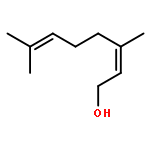
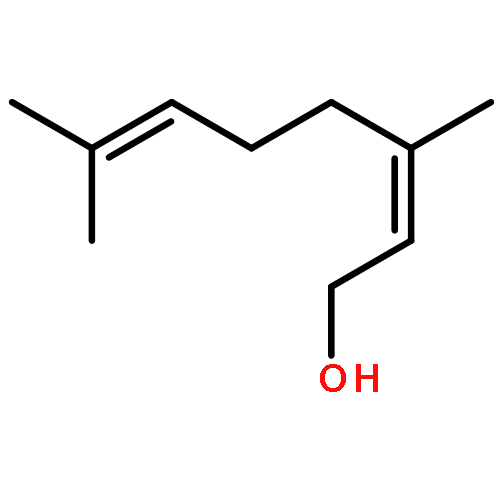




![[3,5-BIS(METHOXYMETHOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/76300/76280-60-9.png)
![[3,5-BIS(METHOXYMETHOXY)PHENYL]METHANOL](http://img.cochemist.com/ccimg/76300/76280-60-9_b.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3.png)
![Diphosphoric acid,P-[(2E,6E,10E)-3,7,11,15-tetramethyl-2,6,10,14-hexadecatetraen-1-yl] ester](http://img.cochemist.com/ccimg/6700/6699-20-3_b.png)