Co-reporter:Qingyuan Yang and Chongli Zhong
Langmuir February 17, 2009 Volume 25(Issue 4) pp:2302-2308
Publication Date(Web):January 20, 2009
DOI:10.1021/la8035902
In this work, grand canonical Monte Carlo simulations were performed to investigate the adsorption behaviors of three important gases (CO2, CH4 and H2) in two two-dimensional (2D) covalent organic frameworks (COFs) with different pore sizes. The simulation results show that stepped behavior is common in gas adsorption in 2D COFs, and multilayer formation is likely to be the underlying mechanism. For CO2 adsorption in 2D COFs, stepped phenomena easily occur, and the electrostatic interactions between CO2−CO2 molecules play a dominant role, while, within the temperature range studied, no stepped behaviors were found in isotherms for H2 adsorption in 2D COFs because of the too weak interactions in the systems. In addition, this work demonstrates that the stepped behaviors are highly affected by temperature, pore size, and the interaction strengths between adsorbates as well as those between adsorbates and adsorbents.
Co-reporter:Jing Ma, Xiangyu Guo, Yunpan Ying, Dahuan Liu, Chongli Zhong
Chemical Engineering Journal 2017 Volume 313(Volume 313) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.cej.2016.10.127
•The anchored UiO-66 on GO layers can avoid the layer stacking of GO.•UiO-66@GO composite combined the unique properties of UiO-66 and GO.•UiO-66@GO/PES membranes show high water purification performance.•UiO-66@GO/PES membranes exhibit impressive antifouling performance.Developing advanced filtration membrane with high flux, good solute rejection and excellent antifouling performance is highly demanded. Hydrophilic graphene oxide (GO) nanosheets are attractive fillers for the preparation of composite membranes for water purification. However, strategies that can fully exploit the advantages and remedy the drawbacks of GO nanosheets are still needed. In this work, UiO-66 was specifically anchored to the GO layers as a porous modifier. The incorporated UiO-66 can effectively prevent the GO layers from stacking and introduce unique properties into the composite (UiO-66@GO). A series of novel composite membranes were prepared with the obtained UiO-66@GO composite and polyethersulfone (PES). As a result, the prepared composite membranes (UiO-66@GO/PES) exhibit high hydrophilicity and water purification performance. Especially, the water flux of composite membrane with 3.0 wt% UiO-66@GO loading shows an increase of 351% and 78% respectively in comparison with that of the PES and GO/PES membranes. Moreover, the UiO-66@GO/PES membranes exhibit good solute rejection and impressive antifouling performance, which is appealing for the application of industrial water purification.Download high-res image (228KB)Download full-size image
Co-reporter:Keke Wang, Yuanzhe Tang, Qin Jiang, Youshi Lan, ... Chongli Zhong
Journal of Energy Chemistry 2017 Volume 26, Issue 5(Volume 26, Issue 5) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jechem.2017.07.007
In this work, a 2D covalent triazine-based framework was prepared by using 1,3-dicyanobenzo[c]thiophene (DCBT) as monomer to effectively capture CO2. The resulting CTF-DCBT was characterized by FT-IR, XPS, PXRD, elemental analysis, SEM, TEM, and N2 adsorption-desorption. The results indicate that CTF-DCBT is partially crystalline and has ultramicropore (6.5 Å) as well as high heteroatom contents (11.24 wt% and 12.61 wt% for N and S, respectively). In addition, the BET surface area and total pore volume of CTF-DCBT are 500 m2/g and 0.26 cm3/g, respectively. CTF-DCBT possesses excellent thermal stability (450 °C) and chemical stability towards boiling water, 4 M HCl, and 1 M NaOH. The CO2 adsorption capacity of CTF-DCBT is 37.8 cm3/g at 1 bar and 25 °C. After six adsorption–desorption cycles, there is no obvious loss of CO2 uptake observed. Due to the ultramicropore and high heteroatom contents, CTF-DCBT has high isosteric heats of adsorption for CO2 and high selectivities of CO2 over N2 and CH4. At 25 °C, the CO2/N2 and CO2/CH4 selectivities are 112.5 and 10.3, respectively, which are higher than those of most POFs. Breakthrough curves indicate that CTF-DCBT could effectively separate CO2/N2 and CO2/CH4 mixtures.Download high-res image (81KB)Download full-size imageA covalent triazine-based framework with both ultramicropore and high heteroatom contents is obtained to selectively capture CO2.
Co-reporter:Weixin Zhang, Yunpan Ying, Jing Ma, Xiangyu Guo, Hongliang Huang, Dahuan Liu, Chongli Zhong
Journal of Membrane Science 2017 Volume 527(Volume 527) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.memsci.2017.01.001
•Interfacial modification was used to solve the compatibility issue in MMMs.•MOF fillers were coated by a thin and uniform PD layer to improve compatibility.•The record water flux and selectivity were obtained for dehydration of EG.In order to solve the issue of incompatibility between metal-organic frameworks (MOFs) and polymer matrix, mixed-matrix membranes (MMMs) on the basis of a hydrophilic polymer poly(vinyl alcohol) (PVA) and a MOF with hydrophilic sulfonic acid group (SO3H-MIL-101-Cr) coated by a thin and uniform polydopamine (PD) layer were prepared for separation of organic solvent from aqueous solutions by taking ethylene glycol (EG) as an example. The PD layer can control the thickness effectively on the surface of SO3H-MIL-101-Cr through a self-polymerization of dopamine and enhance the compatibility between MOF and PVA due to the formation of hydrogen bond between the abundant amine groups in PD and the hydroxyl in PVA matrix. As a result, the prepared SO3H-MIL-101-Cr@PD-PVA MMM exhibits a largely improved water permeability of 7.05×10−5 g m−1 h−1 kPa−1 (the water flux is 540 g m−2 h−1) and the selectivity is as high as 68.1 (the separation factor is 2864) for EG aqueous solution (water content: 10 wt%) separation at 343 K. Compared with those in pure PVA membrane, the water permeability and selectivity were increased by 483% and 567% respectively. To the best of our knowledge, this separation performance is superior to those in all of the reported membranes so far. Therefore, this interfacial modification of MOF fillers may be an effective way to enhance the PV separation performance of MMMs, which can be conveniently extended to prepare other kinds of membranes with advanced properties.Download high-res image (296KB)Download full-size image
Co-reporter:Yunpan Ying, Dahuan LiuWeixin Zhang, Jing Ma, Hongliang Huang, Qingyuan Yang, Chongli Zhong
ACS Applied Materials & Interfaces 2017 Volume 9(Issue 2) pp:
Publication Date(Web):December 21, 2016
DOI:10.1021/acsami.6b14371
Graphene oxide (GO) membranes assembled by single-atom thick GO nanosheets have displayed huge potential application both in gas and liquid separation processes due to its facile and large-scale preparation resulting from various functional groups, such as hydroxyl, carboxyl, and epoxide groups. Taking advantage of these characters, GO membranes intercalated by superhydrophilic metal–organic frameworks (MOFs) as strengthening separation fillers were prepared on modified polyacrylonitrile (PAN) support by a novel pressure-assisted self-assembly (PASA) filtration technique instead of traditional vacuum filtration method for the first time. The synthesized MOF@GO membranes were characterized with several spectroscopic techniques including X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS), as well as scanning electron microscopy (SEM). Compared with GO membrane, these MOF@GO membranes combine the unique properties of MOF and GO and thus have significant enhancements of pervaporation (PV) permeation flux and separation factor simultaneously for ethyl acetate/water mixtures (98/2, w/w) through the PV process, which are also superior to the reported other kinds of membranes. Especially, for MOF@GO-0.3 membrane (corresponding MOF loading: 23.08 wt %), the increments are 159% and 244%, respectively, at 303 K, and the permeate water content can reach as high as 99.5 wt % (corresponding separation factor, 9751) with a high permeation flux of 2423 g m–2 h–1. Moreover, the procedures of both the synthesis of MOF and membranes preparation are environmentally friendly that only water was used as solvent. Such a nanosized MOF-intercalating approach may be also extended to other laminated membranes, providing valuable insights in designing and developing of advanced membranes for effective separation of aqueous organic solution through nanostructure manipulation of the nanomaterials.Keywords: dehydration; graphene oxide; membrane separations; metal−organic frameworks; pervaporation;
Co-reporter:Jing Ma, Yunpan Ying, Xiangyu Guo, Hongliang Huang, Dahuan Liu and Chongli Zhong
Journal of Materials Chemistry A 2016 vol. 4(Issue 19) pp:7281-7288
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6TA02611G
Mixed-matrix membranes (MMMs) have exhibited advantages in membrane-based gas separation in recent years, however, there is still intensive demand for the development of a proper method to design effective fillers to further enhance the gas separation performance of MMMs. In this work, a nanoporous material to selectively facilitate CO2 transport was proposed through the loading of a task-specific ionic liquid (TSIL) into a metal–organic framework (MOF). [C3NH2bim][Tf2N] and NH2-MIL-101(Cr) were selected as a demonstrative TSIL and MOF, respectively. The amine-containing TSIL worked as a selective CO2 transport carrier, which can be beneficial for the improvement of CO2 permeability and CO2/N2 selectivity. Simultaneously, NH2-MIL-101(Cr) is an appropriate porous host material that can control the good dispersion of TSIL and can effectively expose more active adsorption sites of the TSIL. Meanwhile, the amine-containing porous MOF is helpful for rapid CO2 transport and further increases the CO2 permeability. We further incorporated the porous composite into PIM-1 to fabricate MMMs with different loadings. The prepared TSIL@NH2-MIL-101(Cr)/PIM-1 membrane exhibits largely improved gas permeability and selectivity for CO2/N2 separation, with CO2 permeation values of 2979 Barrer and a CO2/N2 separation selectivity of 37 at 5 wt% loading. Compared with NH2-MIL-101(Cr)/PIM-1 and PIM-1 membranes, the CO2/N2 separation selectivity was increased by 116% and 119%, respectively, at the same loading.
Co-reporter:Minman Tong, Qingyuan Yang, Qintian Ma, Dahuan Liu and Chongli Zhong
Journal of Materials Chemistry A 2016 vol. 4(Issue 1) pp:124-131
Publication Date(Web):12 Nov 2015
DOI:10.1039/C5TA06707C
Ultrathin films have the intrinsic feature to show high flux, which may become ideal membranes if they are fabricated to also have high selectivity. 2D covalent organic frameworks (COFs) are a class of crystalline materials with well-defined layered porous structures. Utilizing this feature, 2D-COF nanosheets can be stacked into few-layered ultrathin membranes, and thus the newly formed interlayer flow passages can be regulated to tune their separation properties, making them potential candidates for high-performance membranes. In this work, a series of few-layered 2D-COF membranes were constructed to explore their capability for gas separation as well as the underlying gas transport mechanisms by taking CO2/N2 separation as an example. The results showed that various few-layered 2D-COF membranes can be fabricated to show very different separation performances, from nonselective to highly selective, and even to the molecular sieving level. Furthermore, it was revealed that the energetic microenvironment around the narrow interlayer passages plays a crucial role, and introduction of interacting surfaces to generate van der Waals potential sites near these passages by tuning stacking modes can achieve few-layered membranes with both high CO2 flux and high CO2/N2 selectivity, leading to their separation performance far above the Robeson's upper bound. The mechanisms revealed and the design strategies proposed in this work may provide useful guidance for preparing ultrathin membranes with outstanding performance for gas separation.
Co-reporter:Yaguang Peng, Hongliang Huang, Dahuan Liu, and Chongli Zhong
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 13) pp:8527
Publication Date(Web):March 21, 2016
DOI:10.1021/acsami.6b00900
Highly efficient and irreversible capture of radioactive barium from aqueous media remains a serious task for nuclear waste disposal and environmental protection. To address this task, here we propose a concept of barium ion trap based on metal–organic framework (MOF) with a strong barium-chelating group (sulfate and sulfonic acid group) in the pore structures of MOFs. The functionalized MOF-based ion traps can remove >90% of the barium within the first 5 min, and the removal efficiency reaches 99% after equilibrium. Remarkably, the sulfate-group-functionalized ion trap demonstrates a high barium uptake capacity of 131.1 mg g–1, which surpasses most of the reported sorbents and can selectively capture barium from nuclear wastewater, whereas the sulfonic-acid-group-functionalized ion trap exhibits ultrafast kinetics with a kinetic rate constant k2 of 27.77 g mg–1 min–1, which is 1–3 orders of magnitude higher than existing sorbents. Both of the two MOF-based ion traps can capture barium irreversibly. Our work proposes a new strategy to design barium adsorbent materials and provides a new perspective for removing radioactive barium and other radionuclides from nuclear wastewater for environment remediation. Besides, the concrete mechanisms of barium–sorbent interactions are also demonstrated in this contribution.Keywords: adsorption; functionalization; metal−organic framework; nuclear wastewater; porous material; radioactive barium ion trap
Co-reporter:Keke Wang, Hongliang Huang, Dahuan Liu, Chang Wang, Jinping Li, and Chongli Zhong
Environmental Science & Technology 2016 Volume 50(Issue 9) pp:4869-4876
Publication Date(Web):April 15, 2016
DOI:10.1021/acs.est.6b00425
Porous organic frameworks (POFs) are a class of porous materials composed of organic precursors linked by covalent bonds. The objective of this work is to develop POFs with both ultramicropores and high nitrogen contents for CO2 capture. Specifically, two covalent triazine-based frameworks (CTFs) with ultramicropores (pores of width <7 Å) based on short (fumaronitrile, FUM) and wide monomers (1,4-dicyanonaphthalene, DCN) were synthesized. The obtained CTF-FUM and CTF-DCN possess excellent chemical and thermal stability with ultramicropores of 5.2 and 5.4 Å, respectively. In addition, they exhibit excellent ability to selectively capture CO2 due to ultramicroporous nature. Especially, CTF-FUM-350 has the highest nitrogen content (27.64%) and thus the highest CO2 adsorption capacity (57.2 cc/g at 298 K) and selectivities for CO2 over N2 and CH4 (102.4 and 20.5 at 298 K, respectively) among all CTF-FUM and CTF-DCN. More impressively, as far as we know, the CO2/CH4 selectivity is larger than that of all reported CTFs and ranks in top 10 among all reported POFs. Dynamic breakthrough curves indicate that both CTFs could indeed separate gas mixtures of CO2/N2 and CO2/CH4 completely.
Co-reporter:Xudong Zhao, Dahuan Liu, Hongliang Huang, Chongli Zhong
Microporous and Mesoporous Materials 2016 Volume 224() pp:149-154
Publication Date(Web):April 2016
DOI:10.1016/j.micromeso.2015.11.042
•Eu3+ was successfully incorporated into two water-stable UiO-MOFs.•The specific binding site between two free –COOH is the key of immobilization of Cu2+.•Eu3+@UiO-66-2COOH proves to be a highly selective and sensitive sensor for Cu2+.•Eu3+@UiO-66-2COOH is a fast-response fluorescence probe.Eu3+ was successfully incorporated into two isostructural MOFs with different distributions of carboxyl groups, UiO-66-COOH and UiO-66-2COOH. Utilizing the specific binding site between two free carboxyl groups for efficiently capturing Cu2+, Eu3+@UiO-66-2COOH shows highly selective and sensitive luminescence quenching effect for Cu2+ in aqueous solution, while Eu3+@UiO-66-COOH with single free carboxyl group on the ligand possesses relatively poor performance. Meanwhile, such immobilization behavior of Cu2+ makes Eu3+@UiO-66-2COOH remarkably sensitive for Cu2+ at nano-molar concentration, superior to the majority of reported MOF sensors.
Co-reporter:Imteaz Ahmed
The Journal of Physical Chemistry C 2016 Volume 120(Issue 1) pp:407-415
Publication Date(Web):December 15, 2015
DOI:10.1021/acs.jpcc.5b10578
Adsorptive denitrogenation (ADN) was carried out by adsorption of indole (IND) and quinoline (QUI) over metal–organic frameworks (MOFs) including acidic UiO-66—SO3H for the first time. The adsorbed amount of IND increased with increasing content of —SO3H in UiO-66. The favorable effect of the —SO3H group on the adsorptive removal of IND could be explained by hydrogen bonding between the O of —SO3H and the H of IND, which was firmly supported by the adsorption of pyrrole and methylpyrrole and by theoretical calculations. The application of an —SO3H group in the adsorptive removal of neutral IND is meaningful since neutral nitrogen-containing compounds are not easy to remove and since UiO-66—SO3H is reusable after simple washing with ethanol. The expected increase in QUI adsorption (due to acid–base interaction) with acidic —SO3H was observed when QUI was present at low concentrations (<∼400 ppmw). This favorable contribution of acidic —SO3H to the adsorption of basic QUI was also supported by calculations for the adsorption of one QUI molecule on the —SO3H group of UiO-66. Interestingly, the adsorbed amount of QUI decreased with increasing content of —SO3H in UiO-66 when the QUI concentration was high (initial concentration of 1000 ppmw). One of the reasons for the negative effect of acidic —SO3H on QUI adsorption might be the presence of only one H atom in —SO3H or steric hindrance (due to decreased pore space), although detailed works are needed to support this.
Co-reporter:Jing Ma, Yunpan Ying, Qingyuan Yang, Yujie Ban, Hongliang Huang, Xiangyu Guo, Yuanlong Xiao, Dahuan Liu, Yanshuo Li, Weishen Yang and Chongli Zhong
Chemical Communications 2015 vol. 51(Issue 20) pp:4249-4251
Publication Date(Web):27 Jan 2015
DOI:10.1039/C5CC00384A
To enhance dispersion and adhesion, functionalized porous metal–organic polyhedrons were incorporated into polysulfone as a filler to obtain mixed-matrix membranes, which exhibit largely improved gas permeability and separation factor simultaneously for CO2–CH4 separation.
Co-reporter:Yuyao Huang, Yuanlong Xiao, Hongliang Huang, Ziping Liu, Dahuan Liu, Qingyuan Yang and Chongli Zhong
Chemical Communications 2015 vol. 51(Issue 97) pp:17281-17284
Publication Date(Web):06 Oct 2015
DOI:10.1039/C5CC05061H
A ZIF-9 membrane covered by ionic liquid (ILs) functionalized carbon nanotubes (CNTs) was grown using heat treatment of the layer-by-layer deposition method. This hybrid membrane exhibits a high selectivity for H2/CO2 due to the cooperative effect of ZIFs, CNTs and ILs.
Co-reporter:Yunpan Ying, Yuanlong Xiao, Jing Ma, Xiangyu Guo, Hongliang Huang, Qingyuan Yang, Dahuan Liu and Chongli Zhong
RSC Advances 2015 vol. 5(Issue 36) pp:28394-28400
Publication Date(Web):16 Mar 2015
DOI:10.1039/C4RA15771K
Metal–organic frameworks (MOFs) have exhibited promising applications in gas and liquid separation. As a subclass of MOFs, zeolitic imidazolate frameworks (ZIFs) are attracting more and more interest because of their unique thermal and chemical stability. One of the representative ZIFs, ZIF-7, has super-hydrophobic pores, making it a perfect filler in polymer membranes for recovering acetone from fermentation broths. In this study, mixed matrix membranes (MMMs) based on ZIF-7 and polydimethylsiloxane (PDMS) were prepared, which display improved acetone–water total flux and separation factors simultaneously compared with a pure PDMS membrane. The separation factor can reach up to 39.1 with a high flux of 1236.8 g m−2 h−1 at 333 K, which is the highest value among those reported up to now to the best of our knowledge.
Co-reporter:Yuanlong Xiao, Xiangyu Guo, Hongliang Huang, Qingyuan Yang, Aisheng Huang and Chongli Zhong
RSC Advances 2015 vol. 5(Issue 10) pp:7253-7259
Publication Date(Web):19 Dec 2014
DOI:10.1039/C4RA13727B
A cheap and biocompatible metal–organic framework MIL-88B(Fe) (MIL = Material Institut Lavoisier) was synthesized and incorporated into Matrimid® 5218 to fabricate mixed-matrix membranes (MMMs) for gas separation. Separation performances of the MIL-88B(Fe)/Matrimid MMMs were tested for the single gas permeation of H2 and CH4 as well as the mixture gas separation of an equimolar binary H2–CH4 mixture. Due to the molecular sieving effect, the incorporation of MIL-88B(Fe) can enhance H2 permeability but hinder the transport of CH4 in MIL-88B(Fe)/Matrimid MMMs, thus resulting in enhanced separation selectivity of H2–CH4. At 298 K and ΔP = 3.0 bar, compared with those of a pure polymeric membrane, the H2 permeability and H2–CH4 mixture separation factor of MMMs with a MIL-88B(Fe) loading of 10% increased by 16% and 66%, respectively. In addition, the operation temperature has a significantly positive effect on the separation of a H2–CH4 mixture.
Co-reporter:Xudong Zhao, Xiao Han, Zhengjie Li, Hongliang Huang, Dahuan Liu, Chongli Zhong
Applied Surface Science 2015 Volume 351() pp:760-764
Publication Date(Web):1 October 2015
DOI:10.1016/j.apsusc.2015.05.186
Highlights
- •
Ag(I) was incorporated into a water-stable MOF via ion exchange process.
- •
This approach can prompt the distribution of Ag(I) homogeneous.
- •
It is the first time to remove iodide in aqueous solution using MOFs.
- •
MIL-101(Cr)-SO3Ag has an extremely high adsorption capacity of 244.2 mg g−1.
- •
Strong ionic interaction between Ag(I) and iodide leads to the efficient adsorption.
Co-reporter:Zhengjie Li
The Journal of Physical Chemistry C 2015 119(7) pp: 3674-3683
Publication Date(Web):February 2, 2015
DOI:10.1021/acs.jpcc.5b00019
The separation of H2S/CH4 mixture was computationally examined in the composites of ionic liquids (ILs) supported on metal–organic frameworks (MOFs) at room temperature. Cu-TDPAT was selected as supporter for four types of ILs combined from identical cation [BMIM]+ with different anions ([Cl]−, [Tf2N]−, [PF6]−, and [BF4]−). The results show that introducing ILs into Cu-TDPAT can greatly enhance the adsorption affinity toward H2S compared to the pristine MOF, and the strongest enhancement occurs in the composite containing the anion [Cl]− with the smallest size. The H2S/CH4 adsorption selectivities of each composite are significantly higher than those of the pristine Cu-TDPAT within the pressure range examined, and the selectivity generally shows an increasing trend with increasing the loading of the IL. By further taking the H2S working capacity into account, this work also reveals that the [BMIM][Cl]/Cu-TDPAT composite exhibits the best separation performance in both VSA and PSA processes. These findings may provide useful information for the design of new promising IL/MOF composites applied for H2S capture from natural gas.
Co-reporter:Jong Won Jun;Minman Tong;Beom K. Jung;Dr. Zubair Hasan; Chongli Zhong; Sung Hwa Jhung
Chemistry - A European Journal 2015 Volume 21( Issue 1) pp:347-354
Publication Date(Web):
DOI:10.1002/chem.201404658
Abstract
The adsorptive removal of organoarsenic compounds such as p-arsanilic acid (ASA) and roxarsone (ROX) from water using metal–organic frameworks (MOFs) has been investigated for the first time. A MOF, iron benzenetricarboxylate (also called MIL-100-Fe) exhibits a much higher adsorption capacity for ASA and ROX than activated carbon, zeolite (HY), goethite, and other MOFs. The adsorption of ASA and ROX over MIL-100-Fe is also much more rapid than that over activated carbon. Moreover, the used MIL-100-Fe can be recycled by simply washing with acidic ethanol. Therefore, it is determined that a MOF such as MIL-100-Fe can be used to remove organoarsenic compounds from contaminated water because of its high adsorption capacity, rapid adsorption, and ready regeneration. Moreover, only one of three analogous MIL-100 species (MIL-100-Fe, rather than MIL-100-Al or MIL-100-Cr) can effectively remove the organoarsenic compounds. This selective and high adsorption over MIL-100-Fe, different from other analogous MIL-100 species, can be explained (through calculations) by the facile desorption of water from MIL-100-Fe as well as the large (absolute value) replacement energy (difference between the adsorption energies of the organoarsenic compounds and water) exhibited by MIL-100-Fe. A plausible adsorption/desorption mechanism is proposed based on the surface charge of the MOFs, FTIR results, calculations, and the reactivation results with respect to the solvents used in the experiments.
Co-reporter:Minman Tong, Qingyuan Yang, Chongli Zhong
Microporous and Mesoporous Materials 2015 210() pp: 142-148
Publication Date(Web):
DOI:10.1016/j.micromeso.2015.02.034
Co-reporter:Dong Wu, Guillaume Maurin, Qingyuan Yang, Christian Serre, Hervé Jobic and Chongli Zhong
Journal of Materials Chemistry A 2014 vol. 2(Issue 6) pp:1657-1661
Publication Date(Web):28 Nov 2013
DOI:10.1039/C3TA13651E
A porous Zr-carboxylate based MOF functionalized with two free carboxylic groups on the terephthalate linkers was computationally explored for its membrane-based CO2 capture performances. This material in a pure or in a composite membrane was predicted to outperform Robeson's upper bound for two strategic gas mixtures (CO2/CH4 and CO2/N2).
Co-reporter:Minman Tong, Qingyuan Yang, Yuanlong Xiao and Chongli Zhong
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 29) pp:15189-15198
Publication Date(Web):30 May 2014
DOI:10.1039/C4CP02047B
With the aid of multi-scale computational methods, a diverse set of 46 covalent organic frameworks (COFs), covering the most typical COFs synthesized to date, were collected to study the structure–property relationship of COFs for CO2 capture. For this purpose, CO2 capture from postcombustion gas (CO2–N2 mixture) under industrial vacuum swing adsorption (VSA) conditions was considered as an example. This work shows that adsorption selectivity, CO2 working capacity and the sorbent selection parameter of COFs all exhibit strong correlation with the difference in the adsorbility of adsorbates (ΔAD), highlighting that realization of large ΔAD can be regarded as an important starting point for designing COFs with improved separation performance. Furthermore, it was revealed that the separation performance of 2D-layered COFs can be greatly enhanced by generating “splint effects”, which can be achieved through structural realignment to form slit-like pores with suitable size in the structures. Such “splint effects” in 2D-COFs can find their similar counterpart of “catenation effects” in 3D-COFs or MOFs. On the basis of these observations, a new design strategy was proposed to strengthen the separation performance of COFs. It could be expected that the information obtained in this work not only will enrich the knowledge of the structure–property relationship of COFs for separation, but also will largely facilitate their future applications to the fields related to energy and environmental science, such as natural gas purification, CO2, NOx and SOx capture, etc.
Co-reporter:Yuanlong Xiao, Tongtong Han, Gang Xiao, Yunpan Ying, Hongliang Huang, Qingyuan Yang, Dahuan Liu, and Chongli Zhong
Langmuir 2014 Volume 30(Issue 41) pp:12229-12235
Publication Date(Web):2017-2-22
DOI:10.1021/la5030795
Experimental measurements have been combined with molecular simulations to investigate the adsorption and separation of aniline/phenol mixtures from aqueous solutions by the aluminum terephthalate MIL-53. The results show that the framework flexibility of this material plays a crucial role in the adsorption process and thus can greatly enhance the separation of the aniline/phenol mixture from their solutions. Compared with the conventional adsorbents, MIL-53(Al) shows the best performance for such systems of interest, from the points of view of both the adsorption capacities and the selectivities for aniline. The findings obtained in this work may facilitate more investigations in connection with the application of flexible nanoporous materials for the separation of organic compounds from liquid-phase environments.
Co-reporter:Zubair Hasan ; Minman Tong ; Beom K. Jung ; Imteaz Ahmed ; Chongli Zhong ;Sung Hwa Jhung
The Journal of Physical Chemistry C 2014 Volume 118(Issue 36) pp:21049-21056
Publication Date(Web):August 21, 2014
DOI:10.1021/jp507074x
Pyridine (Py) was adsorbed over metal–organic frameworks (MOFs) (UiO-66 and UiO-66-NH2 with different incorporated amino group content) in both vapor and liquid phases to understand the interactions between the basic adsorbate Py and a basic or neutral adsorbent. Py was adsorbed more favorably over UiO-66-NH2 than over the pristine UiO-66. Not only the adsorbed amount of Py but also adsorption kinetics increased with increasing amino group content in UiO-66s, showing that amino groups in the MOFs benefit the adsorption of Py in both vapor and liquid phases. To understand the favorable interaction between Py and basic UiO-66, calculations were also carried out. The results, including calculations, suggested that hydrogen bonding is important for improving the adsorption of Py over UiO-66s. However, the expected repulsive interaction between basic Py and amino groups of UiO-66-NH2 was not observed. Therefore, it is vital that specific interaction mechanisms should be considered in order to understand selective adsorption processes. Moreover, the amount of adsorbed Py in the vapor phase increased with increasing adsorption temperature, suggesting that the window size of UiO-66 is very similar to the kinetic diameter or critical dimension of Py and that the window size probably is slightly increased with increasing temperature because of lattice vibrations.
Co-reporter:Qingyuan Yang, Dahuan Liu, Chongli Zhong, and Jian-Rong Li
Chemical Reviews 2013 Volume 113(Issue 10) pp:8261
Publication Date(Web):July 5, 2013
DOI:10.1021/cr400005f
Co-reporter:Chunjuan Zhang, Yuanlong Xiao, Dahuan Liu, Qingyuan Yang and Chongli Zhong
Chemical Communications 2013 vol. 49(Issue 6) pp:600-602
Publication Date(Web):22 Nov 2012
DOI:10.1039/C2CC37621K
A ZIF-9-67 hybrid membrane on α-Al2O3 support was prepared using mixed-linker synthesis. The gas permeation and selectivity data demonstrate that this membrane may have potential applications for efficient CO2 capture from several industrial mixtures.
Co-reporter:Wenjuan Zhang, Hongliang Huang, Dahuan Liu, Qingyuan Yang, Yuanlong Xiao, Qintian Ma, Chongli Zhong
Microporous and Mesoporous Materials 2013 Volume 171() pp:118-124
Publication Date(Web):1 May 2013
DOI:10.1016/j.micromeso.2013.01.003
A new porous Zr-based MOF was synthesized by the solvothermal method using 2,6-naphthalenedicarboxylic acid (NDC) as the organic linker, and its luminescent performance and stability were investigated systematically. The material synthesized exhibits quite high chemical stability in addition to exceptional high thermal stability, better than the existing luminescent MOFs. The experiments combining with computations indicate that the electron transfer from inorganic moieties to organic moieties contributes to the luminescent behavior of the Zr-NDC MOF besides the NDC ligand. As far as we know, this is the first study of investigating the luminescent behaviors of such Zr-based MOFs. Moreover, the cycling measurements demonstrate an interesting prospect for the long-term reusability of this material. The results obtained in this work may provide useful information for the design of physicochemically stable MOFs with permanent porosity in sensing applications in the future.Graphical abstractHighlights► A new porous zirconium-based luminescent MOF with high physicochemical stability has been synthesized. ► The nitrogen adsorption data indicates that the specific pore volume is 0.67 cm3 g−1and the surface area is 1720 m2 g−1. ► The emission intensity is enhanced compared to that of the free ligand NDC. ► The luminescent behavior in different solvent solutions suggests the potential ability for sensing applications. ► The reusability is excellent.
Co-reporter:Pascal G. Yot, Qintian Ma, Julien Haines, Qingyuan Yang, Aziz Ghoufi, Thomas Devic, Christian Serre, Vladimir Dmitriev, Gérard Férey, Chongli Zhong and Guillaume Maurin
Chemical Science 2012 vol. 3(Issue 4) pp:1100-1104
Publication Date(Web):20 Dec 2011
DOI:10.1039/C2SC00745B
A joint experimental–modelling study has demonstrated a large flexibility of the MIL-47(VIV) upon mechanical pressure which strongly deviates from its rigid behaviour in presence of guest molecules. A structural transition suspected by mercury intrusion and further confirmed by X-ray powder diffraction and molecular dynamics simulations, leads to a closed MIL-47(VIV) form never observed so far corresponding to a cell contraction of up to 43%. The microscopic key features that govern this transition are then elucidated from complementary Raman experiments.
Co-reporter:Qintian Ma, Qingyuan Yang, Aziz Ghoufi, Gérard Férey, Chongli Zhong and Guillaume Maurin
Dalton Transactions 2012 vol. 41(Issue 14) pp:3915-3919
Publication Date(Web):23 Dec 2011
DOI:10.1039/C2DT12002J
Molecular dynamics simulations evidenced a structural transition of the flexible MIL-53(Cr) under a relatively moderate applied pressure ∼50 MPa. The incorporation of CO2 within its porosity significantly shifts the onset of such a transformation at lower pressure while it decreases the bulk modulus of this solid.
Co-reporter:Wenjuan Zhang, Hongliang Huang, Chongli Zhong and Dahuan Liu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 7) pp:2317-2325
Publication Date(Web):08 Dec 2011
DOI:10.1039/C2CP23839J
In this work, the cooperative effect of temperature and linker functionality on CO2 capture in metal–organic frameworks (MOFs) was investigated using experimental measurements in combination with molecular simulations. To do this, four MOFs with identical topology but different functional groups on the linkers and three important CO2-containing industrial gas mixtures were adopted. The interplay between linker functionality and temperature was analyzed in terms of CO2 storage capacity, adsorption selectivity, working capacity of CO2 in temperature swing adsorption (TSA) processes, as well as sorbent selection parameter (Sssp). The results show that the effect of linker functionality on CO2 capture performance in the MOFs is strongly interconnected with temperature: up to moderate pressures, the lower the temperature, the larger the effect of the functional groups. Furthermore, the modification of a MOF by introducing more complex functional groups can not only improve the affinity of framework for CO2, but also reduce the free volume, and thus may contribute negatively to CO2 capture capability when the packing effect is obvious. Therefore, when we design a new MOF for a certain CO2 capture process operated at a certain temperature, the MOF should be designed to have maximized affinity for CO2 but with a negligible or small effect caused by the reduction of free volume at that temperature and the corresponding operating pressure.
Co-reporter:Hongliang Huang, Wenjuan Zhang, Dahuan Liu, and Chongli Zhong
Industrial & Engineering Chemistry Research 2012 Volume 51(Issue 30) pp:10031-10038
Publication Date(Web):April 13, 2012
DOI:10.1021/ie202699r
In this work, molecular simulations were performed to investigate the effect of trace amount of water on CO2 capture in natural gas upgrading process in a diverse collection of 25 metal–organic frameworks (MOFs). The results show that the interaction between water molecules and MOFs plays a crucial role: at the condition of weak interaction, water molecules move freely in the materials and show a negligible effect on the adsorption selectivity of CO2/CH4; while when the interaction is strong enough that water molecules are adsorbed to the preferential adsorption sites in MOFs, the effect can be significant, depending on the strength of water adsorption. In this case, the electrostatic interaction produced by the MOF framework is the dominant factor. This work provides a better understanding of the different behaviors of water effect on CO2 capture observed previously that may guide the future application of MOFs in industrial separations.
Co-reporter:Lei Zhao;Qingyuan Yang;Qintian Ma
Journal of Molecular Modeling 2011 Volume 17( Issue 2) pp:227-234
Publication Date(Web):2011 February
DOI:10.1007/s00894-010-0720-x
A new force field that can describe the flexibility of Cu-BTC metal-organic framework (MOF) was developed in this work. Part of the parameters were obtained using density functional theory calculations, and the others were taken from other force fields. The new force field could reproduce well the experimental crystal structure, negative thermal expansion, vibrational properties as well as adsorption behavior in Cu-BTC. In addition, the bulk modulus of Cu-BTC was predicted using the new force field. We believe the new force field is useful in understanding the structure-property relationships for MOFs, and the approach can be extended to other MOFs.
Co-reporter:Qing Xu, Dahuan Liu, Qingyuan Yang, Chongli Zhong and Jianguo Mi
Journal of Materials Chemistry A 2010 vol. 20(Issue 4) pp:706-714
Publication Date(Web):03 Dec 2009
DOI:10.1039/B912407A
In this work three Li-modified metal–organic frameworks (MOFs) were constructed from MOF-5, by substituting the H atoms with O–Li groups in the organic linkers. A multiscale approach combining grand canonical Monte Carlo (GCMC) simulation and density functional theory (DFT) calculation was adopted to investigate the separation of CO2/CH4 mixtures in these new Li-modified MOFs, as well as in a previously proposed Li-doped MOF-5 for hydrogen storage and the original MOF-5. The results show that the selectivity of CO2 from CH4/CO2 mixtures in Li-modified MOFs is greatly improved, due to the enhancement of electrostatic potential in the materials by the presence of the metals. One of the new Li-modified MOFs, chem-4Li, shows a higher CO2 selectivity than any other known MOFs. Therefore, this work provides a route to improve the separation performance of MOFs for gas mixtures with components that have large differences in dipole and/or quadrupole moments. In addition, the mechanisms for selectivity enhancement in the Li-modified MOFs were elucidated at the molecular level, and we found that the location of doped metals can change the adsorption sites for CO2, and in turn may change the active sites in MOFs when used as catalysts.
Co-reporter:Dahuan Liu and Chongli Zhong
Journal of Materials Chemistry A 2010 vol. 20(Issue 46) pp:10308-10318
Publication Date(Web):07 Sep 2010
DOI:10.1039/C0JM01045F
Metal–organic frameworks (MOFs) are a new family of nanoporous materials that combine the advantages of both inorganic and organic materials with great variety in functionality, pore size and topology. Gas separation is one of the fields that the first practical application of MOFs may be applied to; however, the study of MOFs as adsorbents in gas separation is still in its early stage, and their separation characteristics are not quite clear. Here, we summarize the recent advances on gas separation in MOFs using computer modeling, and show how computer modeling can help to understand the separation characteristics of MOFs. In addition, several strategies are proposed to improve the separation efficiency of MOFs, which are expected to be useful for designing new MOFs with improved separation performance for targeted properties.
Co-reporter:Yunhua Liu, Dahuan Liu, Qingyuan Yang, Chongli Zhong and Jianguo Mi
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 6) pp:2902-2906
Publication Date(Web):February 15, 2010
DOI:10.1021/ie901488f
In this work, grand canonical Monte Carlo (GCMC) simulations were performed to evaluate the separation performance of covalent organic frameworks (COFs) compared with that of metal−organic frameworks (MOFs) for CH4/CO2/H2 mixtures. The simulation results show that the adsorption selectivities of COFs and MOFs are similar. The electrostatic contribution of framework charges in COFs should be taken into account, although it is smaller than that in MOFs. In addition, the present work shows that the ideal adsorbed solution theory (IAST) is applicable to most COFs.
Co-reporter:Qintian Ma, Qingyuan Yang, Chongli Zhong, Jianguo Mi and Dahuan Liu
Langmuir 2010 Volume 26(Issue 7) pp:5160-5166
Publication Date(Web):December 2, 2009
DOI:10.1021/la903643f
Capillary phase transitions of CH4 confined in a series of metal−organic frameworks (MOFs) were investigated in this work using gauge cell Monte Carlo simulations. The results show that capillary phase transitions can occur in MOFs, and the effects of temperature, pore size, and adsorption energy are very significant. Furthermore, this work shows the confinement can induce a shift in critical point for fluids confined in MOFs, leading to a decrease in critical temperature and an increase in critical density. The critical point shift is more obvious for MOFs with small pore size and large adsorption energy.
Co-reporter:Dahuan Liu and Chongli Zhong
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 1) pp:97-101
Publication Date(Web):November 6, 2009
DOI:10.1021/jz900055k
The properties of Lewis acid sites in two MOFs, Cu−BTC and Cu−MIPT, were studied by using density functional theory (DFT). The strengths of these sites were investigated through exploring the geometry parameters, the natural bond orbital (NBO) charge, and the vibrational frequency as well as the adsorption energy of the probe CO molecule. The results show that both MOFs have Lewis acid sites, and the strength of the Lewis acid sites in Cu−BTC is stronger than that in Cu−MIPT. In addition, proposals for enhancing the acid strength of MOFs are discussed; increasing the number of atoms with high electronegativity in organic linkers and modification of the metal cation by the one with more empty d orbits are suggested as the two possible ways. Keywords (keywords): catalysis; density functional theory; Lewis acid site; metal−organic frameworks; modification;
Co-reporter:Chengcheng Zheng and Chongli Zhong
The Journal of Physical Chemistry C 2010 Volume 114(Issue 21) pp:9945-9951
Publication Date(Web):May 12, 2010
DOI:10.1021/jp102409a
In our previous work, an approach named connectivity-based atom contribution method (CBAC) was developed for estimating framework charges in metal−organic frameworks (MOFs). In this work, we extend the approach to covalent organic frameworks (COFs), another emerging family of nanoporous materials. The results show that those framework atoms with the same bonding connectivity (same atom type) in COFs have identical charges as that in MOFs; this further validates the suitability of the CBAC method and makes it possible to apply to other nanoporous materials. The CBAC charge databank previously established was extended to include 11 new atom types that are commonly involved in COFs, and excellent agreement was obtained between molecular simulations based on CBAC charges and those on quantum mechanically calculated charges.
Co-reporter:Dong Wu, Qing Xu, Dahuan Liu, and Chongli Zhong
The Journal of Physical Chemistry C 2010 Volume 114(Issue 39) pp:16611-16617
Publication Date(Web):September 13, 2010
DOI:10.1021/jp105899t
In this work, a computational study is performed on CO2 capture from various practical systems in a Li-modified metal−organic framework (MOF), chem-4Li MOF. The results demonstrate that this material shows exceptional CO2 capture capability, due to the enhancement of the electrostatic potential in it by the presence of lithium. This study shows that not only the strength and gradient but also the distribution of the electrostatic potential can be controlled by metal doping, leading to more obvious heterogeneity in electrostatic potential in the material, resulting in the occurrence of molecular-level segregation for some systems. The present work observes for the first time that molecular-level segregation can occur in MOFs with simple cubic pores of different sizes and reveals that if the preferential adsorption sites of the less selective component can be shifted to its less preferential adsorption sites, the material may exhibit an exceptionally high selectivity, and such a shift can be achieved by various methods, for which metal doping is an effective way.
Co-reporter:Qing Xu and Chongli Zhong
The Journal of Physical Chemistry C 2010 Volume 114(Issue 11) pp:5035-5042
Publication Date(Web):March 2, 2010
DOI:10.1021/jp910522h
Although the number of metal−organic frameworks (MOFs) is nearly infinite, the atom types included are quite limited. On the basis of this thought, this work provides a strategy for estimating framework charges in MOFs. We developed a so-called connectivity-based atom contribution method (CBAC), in which it is assumed that the atoms with same bonding connectivity have identical charges in different MOFs. The results for 43 MOFs including a training set of 30 MOFs and a test set of 13 MOFs show that the CBAC charges give nearly identical results to those from quantum mechanical calculations for adsorption isotherms of CO2, CO, and N2 in them. Since the method will readily include new atom types, it is applicable to any MOF as long as its structure is known. The strategy, which is applicable to other porous materials, paves a way for large-scale computational screening of MOFs for specific applications as well as contributes to a better understanding of the structure−property relationships for MOFs and eventually contributes to the development and application of MOFs.
Co-reporter:Chengcheng Zheng, Dahuan Liu, Qingyuan Yang, Chongli Zhong and Jianguo Mi
Industrial & Engineering Chemistry Research 2009 Volume 48(Issue 23) pp:10479
Publication Date(Web):October 8, 2009
DOI:10.1021/ie901000x
This work involved a computational study to investigate the influences of framework charges on CO2 uptake in metal−organic frameworks (MOFs), in which a total of 20 MOFs with different topologies, pore sizes, and chemical characteristics were examined. The results showed that, at atmospheric pressure, the contribution of the framework charges is generally large, and a linear relationship with pore size was found, showing that, when the pore size is larger than 3.3 nm, the contribution becomes smaller than 10%. On the other hand, the framework charge contribution was found to decrease rapidly with increasing pressure and to become less than 10% at pressures higher than 2.0 MPa. This work shows that the framework charge contribution in MOFs cannot be ignored in computational screening of MOF materials for CO2 capture under low-pressure conditions, whereas at moderate operating pressures, the contribution can be ignored in large-scale prescreening such as in the natural gas upgrading process.
Co-reporter:Chunyu XUE
Chinese Journal of Chemistry 2009 Volume 27( Issue 3) pp:472-478
Publication Date(Web):
DOI:10.1002/cjoc.200990077
Abstract
The modified MM3 force field for describing flexible IRMOF-1 was extended to include other IRMOFs, and a molecular dynamics simulation study was performed on hexane diffusion in IRMOF-1 and IRMOF-16. The self-diffusion coefficients and diffusion pathways of hexane, as well as the mobility of the frameworks were investigated, as a function of both temperature and loading. The results revealed that the diffusion pathway of hexane was largely influenced by loading, and the flexibility of IRMOF-16 was much larger than that of IRMOF-1. The microscopic information obtained is useful for understanding the diffusion mechanism of chain molecules in dynamic MOF.
Co-reporter:Jing Xin, Dahuan Liu and Chongli Zhong
The Journal of Physical Chemistry B 2009 Volume 113(Issue 28) pp:9364-9372
Publication Date(Web):June 18, 2009
DOI:10.1021/jp902300g
Control of the overall morphology and inner structure of multicompartment micelles from binary blends of triblock copolymers in solution was studied by dissipative dynamics simulations. The effects of the block sequence, block ratio, block length, and chain architecture on the morphology and structure of mixed multicompartment micelles were investigated systematically. This work shows that by changing the block composition or chain architecture of one copolymer in the binary blends, the mixed degree, relative distance, and degree of participation of core-forming blocks from two triblock copolymers can be tuned, and diverse morphologies of mixed micelles with alterable domain arrangements and overall size can be obtained. This work shows that blending of copolymers is an effective way to control the morphology and inner structure of multicompartment micelles, providing useful information on the preparation of mixed micelles with unique properties for practical applications.
Co-reporter:Qingyuan Yang and Chongli Zhong
Langmuir 2009 Volume 25(Issue 4) pp:2302-2308
Publication Date(Web):January 20, 2009
DOI:10.1021/la8035902
In this work, grand canonical Monte Carlo simulations were performed to investigate the adsorption behaviors of three important gases (CO2, CH4 and H2) in two two-dimensional (2D) covalent organic frameworks (COFs) with different pore sizes. The simulation results show that stepped behavior is common in gas adsorption in 2D COFs, and multilayer formation is likely to be the underlying mechanism. For CO2 adsorption in 2D COFs, stepped phenomena easily occur, and the electrostatic interactions between CO2−CO2 molecules play a dominant role, while, within the temperature range studied, no stepped behaviors were found in isotherms for H2 adsorption in 2D COFs because of the too weak interactions in the systems. In addition, this work demonstrates that the stepped behaviors are highly affected by temperature, pore size, and the interaction strengths between adsorbates as well as those between adsorbates and adsorbents.
Co-reporter:Dahuan Liu, Chengcheng Zheng, Qingyuan Yang and Chongli Zhong
The Journal of Physical Chemistry C 2009 Volume 113(Issue 12) pp:5004-5009
Publication Date(Web):2017-2-22
DOI:10.1021/jp809373r
In this work, atomic partial charges in the framework atoms of two typical zeolitic imidazolate frameworks (ZIFs), ZIF-68 and ZIF-69, were calculated using density functional theory, and a suitable force field for describing CO2 adsorption in ZIFs was identified. On the basis of this, a combined grand canonical Monte Carlo (GCMC) and molecular dynamics (MD) simulation study was performed to investigate the adsorption and diffusion behaviors of CO2 in ZIFs. The results show that the small pores formed by the nIM linkers in ZIF-68 and ZIF-69 are the preferential adsorption sites for CO2 molecules, with the corners formed by the phenyl rings in the large pores being less preferential adsorption sites. This work demonstrates that the chlorine atoms in cbIM linkers in ZIF-69 lead to enhanced binding energy but reduced diffusivity for CO2, the electrostatic interactions produced by the frameworks are important that cannot be ignored, and, down to 180 K, no steps are found in isotherms. In addition, this work demonstrates that the diffusion of CO2 in ZIFs is likely to be much slower than that in other MOFs.
Co-reporter:Lei Zhao and Chongli Zhong
The Journal of Physical Chemistry C 2009 Volume 113(Issue 39) pp:16860-16862
Publication Date(Web):September 9, 2009
DOI:10.1021/jp906806k
This work performs a computational study on the thermal expansion behavior of covalent organic frameworks (COFs). The results demonstrate that COFs show negative thermal expansion (NTE), and the origin of the NTE behavior is the motion of the aromatic carbon rings with temperature, providing a better understanding of this new family of materials.
Co-reporter:Bei Liu, Qingyuan Yang, Chunyu Xue, Chongli Zhong and Berend Smit
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 22) pp:3244-3249
Publication Date(Web):08 Apr 2008
DOI:10.1039/B801494A
In this work a combined molecular dynamics simulation and dynamically corrected transition-state theory (dcTST) study was performed to investigate the effect of interpenetration (catenation) on hydrogen diffusion in metal–organic frameworks (MOFs) as well as their relationships. The results on 10 isoreticular MOFs (IRMOFs) with and without interpenetration show that catenation can reduce hydrogen diffusivity by a factor of 2 to 3 at room temperature, and for the interpenetrated IRMOFs with multi-pores of different sizes, free volume can serve as a measure for hydrogen diffusivity: the bigger the free volume, the larger the hydrogen diffusivity. In addition, the present work shows that dcTST can directly reveal the influence of the MOF structure on hydrogen diffusivity, which is a powerful tool for providing a better understanding of the relationship between gas diffusivity and MOF structure.
Co-reporter:Bei Liu ; Qingyuan Yang ; Chunyu Xue ; Chongli Zhong ; Biaohua Chen ;Berend Smit
The Journal of Physical Chemistry C 2008 Volume 112(Issue 26) pp:9854-9860
Publication Date(Web):June 10, 2008
DOI:10.1021/jp802343n
In this work a systematic molecular simulation study was performed to study the effect of interpenetration on gas mixture separation in metal−organic frameworks (MOFs). To do this, three pairs of isoreticular MOFs (IRMOFs) with and without interpenetration were adopted to compare their adsorption separation selectivity for CH4/H2 mixtures at room temperature. The results show that methane selectivity is greatly enhanced in the interpenetrated IRMOFs compared with their noninterpenetrated counterparts, due to the formation of additional small pores and adsorption sites by the interpenetration of frameworks. Furthermore, this work shows methane selectivity behavior is more complex in the former and selectivity differs largely in the different areas of the pores, attributed to the existence of various small pores of different sizes. In addition, the present work shows the ideal adsorbed solution theory is likely to be applicable to interpenetrated MOFs with complex structures.
Co-reporter:Hongfeng Qi, Dahuan Liu and Chongli Zhong
The Journal of Physical Chemistry B 2008 Volume 112(Issue 51) pp:16409-16414
Publication Date(Web):November 26, 2008
DOI:10.1021/jp806664f
Density functional theory was applied to investigate the cooperative aggregation of comblike copolymer and linear homopolymer blends in selective solvents, where the linear homopolymers are set to be identical to the hydrophobic side chains of the comblike copolymers. The effects of the composition and the length of the linear homopolymers, as well as solvent quality, on the cooperative aggregation were studied systematically. The results show that large aggregates of macrophase separation can always be formed with the addition of linear homopolymers, where the homopolymers accumulate at the center and the comblike copolymers as the corona with the hydrophobic blocks distribute at the core/corona interface. This work demonstrates that the structure and size of micelles can be tuned by the controlled addition of appropriate linear homopolymers, leading to a better understanding of the controlled synthesis of micelles with target structure and properties.
Co-reporter:Dahuan Liu
Macromolecular Rapid Communications 2007 Volume 28(Issue 3) pp:292-297
Publication Date(Web):30 JAN 2007
DOI:10.1002/marc.200600696
In this work, the formation of two-compartment micelles from symmetric pentablock copolymers in selective solvents was studied using the dissipative particle dynamics simulation technique, and the effects of block lengths and solvent quality were investigated. The simulations revealed several new morphologies and their formation mechanisms were elucidated at the molecular level, providing useful information that may contribute to the future rational design and synthesis of novel multicompartment micelles with tailored structures.
Co-reporter:Chongli Zhong;Dahuan Liu
Macromolecular Theory and Simulations 2007 Volume 16(Issue 2) pp:
Publication Date(Web):26 FEB 2007
DOI:10.1002/mats.200790002
Cover: The picture on the cover shows the structures of copolymer chains and the morphologies of multicompartment micelles formed from them in a selective solvent, illustrating that various morphologies can be formed by changing chain architecture, composition, and/or solvent quality. Further details can be found in the article by C. Zhong* and D. Liu on page 141.
Co-reporter:Chongli Zhong;Dahuan Liu
Macromolecular Theory and Simulations 2007 Volume 16(Issue 2) pp:141-157
Publication Date(Web):26 FEB 2007
DOI:10.1002/mats.200600074
Multicompartment micelles are a new class of nanomaterials that may find wide applications in the fields of drug delivery, nanotechnology and catalysis. Due to their structural complexity, as well as the wide parameter space to explore, experimental investigations are a difficult task, to which molecular simulation may contribute greatly. In this paper, the application of the dissipative particle dynamics simulation technique to the understanding of multicompartment micelles is introduced, illustrating that DPD is a powerful tool for identifying new morphologies by varying block length, block ratio and solvent quality in a systematic way. The formation process of multicompartment micelles, as well as shear effects and the self-assembly of nanoparticle mixtures in multicompartment micelles, can also be studied well by DPD simulation. The present work shows that DPD, as well as other simulation techniques and theories, can complement experiments greatly, not only in exploring properties in a wider parameter space, but also by giving a preview of phenomena prior to experiments. DPD, as a mesoscopic dynamic simulation technique, is particularly useful for understanding the dynamic processes of multicompartment micelles at a microscopic level.
Co-reporter:Jun XIA;Chong-Li ZHONG
Chinese Journal of Chemistry 2007 Volume 25(Issue 11) pp:1732-1738
Publication Date(Web):13 NOV 2007
DOI:10.1002/cjoc.200790320
Dissipative particle dynamics simulations were performed on the morphology and structure of multicompartment micelles formed from π-shaped ABC block copolymers in water. The influences of chain architectures were studied in a systematic way, and a rich variety of morphologies were observed, such as spherical, wormlike, X-shaped, Y-shaped, ribbon-like, layered rod-like, layered disk-like, as well as network morphologies. The simulations show that the distance between the two grafts plays an important role in control of the morphology. Since π-shaped ABC block copolymers can be reduced to linear ABC and star ABC block copolymers, they are good model copolymers for studying the self-assembly of complex block copolymers into micelles. The knowledge obtained in this work as well as the new morphologies identified provide useful information for future rational design and synthesis of novel multicompartment micelles.
Co-reporter:Dahuan Liu
Macromolecular Rapid Communications 2006 Volume 27(Issue 6) pp:458-462
Publication Date(Web):6 MAR 2006
DOI:10.1002/marc.200500827
Summary: Dissipative particle dynamics simulations are performed on the distribution of binary nanoparticle mixtures in lamellar diblock copolymers. The results show that the self-assembly of nanoparticle mixtures in polymer matrix is a cooperative assembly that is affected by various factors, providing molecular-level information for the rational design of new polymer nanocomposites with tailored properties.
Co-reporter:Chongli Zhong;Jun Xia
Macromolecular Rapid Communications 2006 Volume 27(Issue 14) pp:1110-1114
Publication Date(Web):17 JUL 2006
DOI:10.1002/marc.200600187
Summary: Dissipative particle dynamics simulation was performed to study the formation of multicompartment micelles from ABC star triblock copolymers in water. The study revealed some new morphologies that had not been observed before and also provided a direct visualization of the evolution of wormlike multicompartment micelles that follows the fusion process. Thus, this work provides molecular understanding of multicompartment micelles which will be useful for the future rational synthesis of novel micelles.
Co-reporter:Chongli Zhong;Yuanyuan Cui;Jun Xia
Macromolecular Rapid Communications 2006 Volume 27(Issue 17) pp:1437-1441
Publication Date(Web):24 AUG 2006
DOI:10.1002/marc.200600326
Summary: Dissipative particle dynamics simulations were performed to study the effect of shear on the rheological behavior of multicompartment micellar solutions, demonstrating that both shear thickening and thinning can occur, and the macroscopic behavior was elucidated at a molecular level. In addition, a novel shear-induced morphology of “sphere-on-rod” was observed. This work provides useful information towards a complete understanding of the properties and morphologies of multicompartment micelles that is useful for future rational synthesis of novel micelles.
Co-reporter:Jun Xia
Macromolecular Rapid Communications 2006 Volume 27(Issue 19) pp:1654-1659
Publication Date(Web):27 SEP 2006
DOI:10.1002/marc.200600411
Summary: Dissipative particle dynamics simulations are performed on the distributions of two agents in a core-shell-corona multicompartment micelle. The simulated results show that when the agents are weakly hydrophobic, their distributions in the multicompartment micelle are largely affected by the interactions between the agents and the blocks; while for strongly hydrophobic agents, the self-assembly of solubilized species in the micelle is also affected largely by the interactions between the species. This work confirms that a multicompartment micelle can store two agents within separate nanoscopic compartments simultaneously, and shows that the distributions of the agents can be tailored easily by changing the interactions presented. This provides molecular-level information that is useful for the future rational design of new micellar systems with tailored properties.
Co-reporter:Jun Xia
Macromolecular Rapid Communications 2006 Volume 27(Issue 19) pp:
Publication Date(Web):13 OCT 2006
DOI:10.1002/marc.200690038
Co-reporter:Qingyuan Yang Dr. Dr.
ChemPhysChem 2006 Volume 7(Issue 7) pp:1417-1421
Publication Date(Web):30 JUN 2006
DOI:10.1002/cphc.200600191
Ordered microdomains with different electrostatic field strengths exist in certain metal-organic frameworks (MOFs; see picture), and the electrostatic interactions in MOFs can enhance the separation of some gas mixtures, demonstrating that MOFs have great potential for adsorption separations.
Co-reporter:Chongli Zhong;Dahuan Liu
Macromolecular Rapid Communications 2005 Volume 26(Issue 24) pp:1960-1964
Publication Date(Web):5 DEC 2005
DOI:10.1002/marc.200500505
Summary: Dissipative particle dynamic simulations were performed on the microphase separation, end-to-end distance, and shear viscosity of linear–dendritic diblock copolymers under steady shear flow. The results show that their microstructure and properties depend on both the shear rate and the degree of branching, and they can incorporate the characteristics of both linear and dendritic polymers to lead to new materials with unique properties.
Co-reporter:Xinping Bu, Chongli Zhong, A.F Jalbout
Chemical Physics Letters 2004 Volume 387(4–6) pp:410-414
Publication Date(Web):1 April 2004
DOI:10.1016/j.cplett.2004.02.062
The MHen+(M=Be, Mg) complexes with n=1–4 were investigated by ab initio calculations at the levels of HF, MP2 and MP2(full)/6-311+G(3df, 3pd). The complexes were found to be stable, and the calculated results show that the C3V geometry is stable for the MHe3+ complexes, and the C2V geometry is stable for the MHe4+ complexes.
Co-reporter:Jianguo Mi, Chongli Zhong, Yi-Gui Li, Jian Chen
Chemical Physics 2004 Volume 305(1–3) pp:37-45
Publication Date(Web):25 October 2004
DOI:10.1016/j.chemphys.2004.06.031
Co-reporter:Chongli Zhong;Qinghua Hu
Journal of Pharmaceutical Sciences 2003 Volume 92(Issue 11) pp:2284-2294
Publication Date(Web):25 AUG 2003
DOI:10.1002/jps.10499
A correlation for estimation of the aqueous solubility of organic compounds that is based on a training set of 120 chemicals is proposed. The new model proposed is predictive and requires only molecular connectivity indices in the calculations. The calculated results of the new model are comparable to those from the existing general solubility equation (GSE) and the Klopman–Zhu models. The new model was also applied to a testing set of 80 compounds, and the predictions show that the new model is reliable with good predictive accuracy. Because the new model does not require any experimental physicochemical properties in the calculation, it is simple and easy to apply. This work shows again that molecular connectivity indices are useful structural descriptors in quantitative structure–property (QSPR) studies in pharmaceutical research. © 2003 Wiley-Liss, Inc. and the American Pharmacists Association J Pharm Sci 92:2284–2294, 2003
Co-reporter:Qingyuan YANG, Qing XU, Bei LIU, Chongli ZHONG, Smit Berend
Chinese Journal of Chemical Engineering (October 2009) Volume 17(Issue 5) pp:781-790
Publication Date(Web):1 October 2009
DOI:10.1016/S1004-9541(08)60277-3
In this work grand canonical Monte Carlo simulations were performed to study gas separation in three pairs of isoreticular metal-organic frameworks (IRMOFs) with and without catenation at room temperature. Mixture composed of CO2 and H2 was selected as the model system to separate. The results show that CO2 selectivity in catenated MOFs with multi-porous frameworks is much higher than their non-catenated counterparts. The simulations also show that the electrostatic interactions are very important for the selectivity, and the contributions of different electrostatic interactions are different, depending on pore size, pressure and mixture composition. In fact, changing the electrostatic interactions can even qualitatively change the adsorption behavior. A general conclusion is that the electrostatic interactions between adsorbate molecules and the framework atoms play a dominant role at low pressures, and these interactions in catenated MOFs have much more pronounced effects than those in their non-catenated counterparts, while the electrostatic interactions between adsorbate molecules become evident with increasing pressure, and eventually dominant.
Co-reporter:Qing Xu, Dahuan Liu, Qingyuan Yang, Chongli Zhong and Jianguo Mi
Journal of Materials Chemistry A 2010 - vol. 20(Issue 4) pp:NaN714-714
Publication Date(Web):2009/12/03
DOI:10.1039/B912407A
In this work three Li-modified metal–organic frameworks (MOFs) were constructed from MOF-5, by substituting the H atoms with O–Li groups in the organic linkers. A multiscale approach combining grand canonical Monte Carlo (GCMC) simulation and density functional theory (DFT) calculation was adopted to investigate the separation of CO2/CH4 mixtures in these new Li-modified MOFs, as well as in a previously proposed Li-doped MOF-5 for hydrogen storage and the original MOF-5. The results show that the selectivity of CO2 from CH4/CO2 mixtures in Li-modified MOFs is greatly improved, due to the enhancement of electrostatic potential in the materials by the presence of the metals. One of the new Li-modified MOFs, chem-4Li, shows a higher CO2 selectivity than any other known MOFs. Therefore, this work provides a route to improve the separation performance of MOFs for gas mixtures with components that have large differences in dipole and/or quadrupole moments. In addition, the mechanisms for selectivity enhancement in the Li-modified MOFs were elucidated at the molecular level, and we found that the location of doped metals can change the adsorption sites for CO2, and in turn may change the active sites in MOFs when used as catalysts.
Co-reporter:Chunjuan Zhang, Yuanlong Xiao, Dahuan Liu, Qingyuan Yang and Chongli Zhong
Chemical Communications 2013 - vol. 49(Issue 6) pp:NaN602-602
Publication Date(Web):2012/11/22
DOI:10.1039/C2CC37621K
A ZIF-9-67 hybrid membrane on α-Al2O3 support was prepared using mixed-linker synthesis. The gas permeation and selectivity data demonstrate that this membrane may have potential applications for efficient CO2 capture from several industrial mixtures.
Co-reporter:Minman Tong, Qingyuan Yang, Qintian Ma, Dahuan Liu and Chongli Zhong
Journal of Materials Chemistry A 2016 - vol. 4(Issue 1) pp:NaN131-131
Publication Date(Web):2015/11/12
DOI:10.1039/C5TA06707C
Ultrathin films have the intrinsic feature to show high flux, which may become ideal membranes if they are fabricated to also have high selectivity. 2D covalent organic frameworks (COFs) are a class of crystalline materials with well-defined layered porous structures. Utilizing this feature, 2D-COF nanosheets can be stacked into few-layered ultrathin membranes, and thus the newly formed interlayer flow passages can be regulated to tune their separation properties, making them potential candidates for high-performance membranes. In this work, a series of few-layered 2D-COF membranes were constructed to explore their capability for gas separation as well as the underlying gas transport mechanisms by taking CO2/N2 separation as an example. The results showed that various few-layered 2D-COF membranes can be fabricated to show very different separation performances, from nonselective to highly selective, and even to the molecular sieving level. Furthermore, it was revealed that the energetic microenvironment around the narrow interlayer passages plays a crucial role, and introduction of interacting surfaces to generate van der Waals potential sites near these passages by tuning stacking modes can achieve few-layered membranes with both high CO2 flux and high CO2/N2 selectivity, leading to their separation performance far above the Robeson's upper bound. The mechanisms revealed and the design strategies proposed in this work may provide useful guidance for preparing ultrathin membranes with outstanding performance for gas separation.
Co-reporter:Dahuan Liu and Chongli Zhong
Journal of Materials Chemistry A 2010 - vol. 20(Issue 46) pp:NaN10318-10318
Publication Date(Web):2010/09/07
DOI:10.1039/C0JM01045F
Metal–organic frameworks (MOFs) are a new family of nanoporous materials that combine the advantages of both inorganic and organic materials with great variety in functionality, pore size and topology. Gas separation is one of the fields that the first practical application of MOFs may be applied to; however, the study of MOFs as adsorbents in gas separation is still in its early stage, and their separation characteristics are not quite clear. Here, we summarize the recent advances on gas separation in MOFs using computer modeling, and show how computer modeling can help to understand the separation characteristics of MOFs. In addition, several strategies are proposed to improve the separation efficiency of MOFs, which are expected to be useful for designing new MOFs with improved separation performance for targeted properties.
Co-reporter:Jing Ma, Yunpan Ying, Qingyuan Yang, Yujie Ban, Hongliang Huang, Xiangyu Guo, Yuanlong Xiao, Dahuan Liu, Yanshuo Li, Weishen Yang and Chongli Zhong
Chemical Communications 2015 - vol. 51(Issue 20) pp:NaN4251-4251
Publication Date(Web):2015/01/27
DOI:10.1039/C5CC00384A
To enhance dispersion and adhesion, functionalized porous metal–organic polyhedrons were incorporated into polysulfone as a filler to obtain mixed-matrix membranes, which exhibit largely improved gas permeability and separation factor simultaneously for CO2–CH4 separation.
Co-reporter:Yuyao Huang, Yuanlong Xiao, Hongliang Huang, Ziping Liu, Dahuan Liu, Qingyuan Yang and Chongli Zhong
Chemical Communications 2015 - vol. 51(Issue 97) pp:NaN17284-17284
Publication Date(Web):2015/10/06
DOI:10.1039/C5CC05061H
A ZIF-9 membrane covered by ionic liquid (ILs) functionalized carbon nanotubes (CNTs) was grown using heat treatment of the layer-by-layer deposition method. This hybrid membrane exhibits a high selectivity for H2/CO2 due to the cooperative effect of ZIFs, CNTs and ILs.
Co-reporter:Qintian Ma, Qingyuan Yang, Aziz Ghoufi, Gérard Férey, Chongli Zhong and Guillaume Maurin
Dalton Transactions 2012 - vol. 41(Issue 14) pp:NaN3919-3919
Publication Date(Web):2011/12/23
DOI:10.1039/C2DT12002J
Molecular dynamics simulations evidenced a structural transition of the flexible MIL-53(Cr) under a relatively moderate applied pressure ∼50 MPa. The incorporation of CO2 within its porosity significantly shifts the onset of such a transformation at lower pressure while it decreases the bulk modulus of this solid.
Co-reporter:Bei Liu, Qingyuan Yang, Chunyu Xue, Chongli Zhong and Berend Smit
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 22) pp:NaN3249-3249
Publication Date(Web):2008/04/08
DOI:10.1039/B801494A
In this work a combined molecular dynamics simulation and dynamically corrected transition-state theory (dcTST) study was performed to investigate the effect of interpenetration (catenation) on hydrogen diffusion in metal–organic frameworks (MOFs) as well as their relationships. The results on 10 isoreticular MOFs (IRMOFs) with and without interpenetration show that catenation can reduce hydrogen diffusivity by a factor of 2 to 3 at room temperature, and for the interpenetrated IRMOFs with multi-pores of different sizes, free volume can serve as a measure for hydrogen diffusivity: the bigger the free volume, the larger the hydrogen diffusivity. In addition, the present work shows that dcTST can directly reveal the influence of the MOF structure on hydrogen diffusivity, which is a powerful tool for providing a better understanding of the relationship between gas diffusivity and MOF structure.
Co-reporter:Jing Ma, Yunpan Ying, Xiangyu Guo, Hongliang Huang, Dahuan Liu and Chongli Zhong
Journal of Materials Chemistry A 2016 - vol. 4(Issue 19) pp:NaN7288-7288
Publication Date(Web):2016/04/11
DOI:10.1039/C6TA02611G
Mixed-matrix membranes (MMMs) have exhibited advantages in membrane-based gas separation in recent years, however, there is still intensive demand for the development of a proper method to design effective fillers to further enhance the gas separation performance of MMMs. In this work, a nanoporous material to selectively facilitate CO2 transport was proposed through the loading of a task-specific ionic liquid (TSIL) into a metal–organic framework (MOF). [C3NH2bim][Tf2N] and NH2-MIL-101(Cr) were selected as a demonstrative TSIL and MOF, respectively. The amine-containing TSIL worked as a selective CO2 transport carrier, which can be beneficial for the improvement of CO2 permeability and CO2/N2 selectivity. Simultaneously, NH2-MIL-101(Cr) is an appropriate porous host material that can control the good dispersion of TSIL and can effectively expose more active adsorption sites of the TSIL. Meanwhile, the amine-containing porous MOF is helpful for rapid CO2 transport and further increases the CO2 permeability. We further incorporated the porous composite into PIM-1 to fabricate MMMs with different loadings. The prepared TSIL@NH2-MIL-101(Cr)/PIM-1 membrane exhibits largely improved gas permeability and selectivity for CO2/N2 separation, with CO2 permeation values of 2979 Barrer and a CO2/N2 separation selectivity of 37 at 5 wt% loading. Compared with NH2-MIL-101(Cr)/PIM-1 and PIM-1 membranes, the CO2/N2 separation selectivity was increased by 116% and 119%, respectively, at the same loading.
Co-reporter:Wenjuan Zhang, Hongliang Huang, Chongli Zhong and Dahuan Liu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 7) pp:NaN2325-2325
Publication Date(Web):2011/12/08
DOI:10.1039/C2CP23839J
In this work, the cooperative effect of temperature and linker functionality on CO2 capture in metal–organic frameworks (MOFs) was investigated using experimental measurements in combination with molecular simulations. To do this, four MOFs with identical topology but different functional groups on the linkers and three important CO2-containing industrial gas mixtures were adopted. The interplay between linker functionality and temperature was analyzed in terms of CO2 storage capacity, adsorption selectivity, working capacity of CO2 in temperature swing adsorption (TSA) processes, as well as sorbent selection parameter (Sssp). The results show that the effect of linker functionality on CO2 capture performance in the MOFs is strongly interconnected with temperature: up to moderate pressures, the lower the temperature, the larger the effect of the functional groups. Furthermore, the modification of a MOF by introducing more complex functional groups can not only improve the affinity of framework for CO2, but also reduce the free volume, and thus may contribute negatively to CO2 capture capability when the packing effect is obvious. Therefore, when we design a new MOF for a certain CO2 capture process operated at a certain temperature, the MOF should be designed to have maximized affinity for CO2 but with a negligible or small effect caused by the reduction of free volume at that temperature and the corresponding operating pressure.
Co-reporter:Pascal G. Yot, Qintian Ma, Julien Haines, Qingyuan Yang, Aziz Ghoufi, Thomas Devic, Christian Serre, Vladimir Dmitriev, Gérard Férey, Chongli Zhong and Guillaume Maurin
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1104-1104
Publication Date(Web):2011/12/20
DOI:10.1039/C2SC00745B
A joint experimental–modelling study has demonstrated a large flexibility of the MIL-47(VIV) upon mechanical pressure which strongly deviates from its rigid behaviour in presence of guest molecules. A structural transition suspected by mercury intrusion and further confirmed by X-ray powder diffraction and molecular dynamics simulations, leads to a closed MIL-47(VIV) form never observed so far corresponding to a cell contraction of up to 43%. The microscopic key features that govern this transition are then elucidated from complementary Raman experiments.
Co-reporter:Dong Wu, Guillaume Maurin, Qingyuan Yang, Christian Serre, Hervé Jobic and Chongli Zhong
Journal of Materials Chemistry A 2014 - vol. 2(Issue 6) pp:NaN1661-1661
Publication Date(Web):2013/11/28
DOI:10.1039/C3TA13651E
A porous Zr-carboxylate based MOF functionalized with two free carboxylic groups on the terephthalate linkers was computationally explored for its membrane-based CO2 capture performances. This material in a pure or in a composite membrane was predicted to outperform Robeson's upper bound for two strategic gas mixtures (CO2/CH4 and CO2/N2).
Co-reporter:Minman Tong, Qingyuan Yang, Yuanlong Xiao and Chongli Zhong
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 29) pp:NaN15198-15198
Publication Date(Web):2014/05/30
DOI:10.1039/C4CP02047B
With the aid of multi-scale computational methods, a diverse set of 46 covalent organic frameworks (COFs), covering the most typical COFs synthesized to date, were collected to study the structure–property relationship of COFs for CO2 capture. For this purpose, CO2 capture from postcombustion gas (CO2–N2 mixture) under industrial vacuum swing adsorption (VSA) conditions was considered as an example. This work shows that adsorption selectivity, CO2 working capacity and the sorbent selection parameter of COFs all exhibit strong correlation with the difference in the adsorbility of adsorbates (ΔAD), highlighting that realization of large ΔAD can be regarded as an important starting point for designing COFs with improved separation performance. Furthermore, it was revealed that the separation performance of 2D-layered COFs can be greatly enhanced by generating “splint effects”, which can be achieved through structural realignment to form slit-like pores with suitable size in the structures. Such “splint effects” in 2D-COFs can find their similar counterpart of “catenation effects” in 3D-COFs or MOFs. On the basis of these observations, a new design strategy was proposed to strengthen the separation performance of COFs. It could be expected that the information obtained in this work not only will enrich the knowledge of the structure–property relationship of COFs for separation, but also will largely facilitate their future applications to the fields related to energy and environmental science, such as natural gas purification, CO2, NOx and SOx capture, etc.

![Silicic acid (H4SiO4), tetraethyl ester, polymer with α-hydro-ω-hydroxypoly[oxy(dimethylsilylene)]](http://img.cochemist.com/ccimg/155900/155827-81-9.png)
![Silicic acid (H4SiO4), tetraethyl ester, polymer with α-hydro-ω-hydroxypoly[oxy(dimethylsilylene)]](http://img.cochemist.com/ccimg/155900/155827-81-9_b.png)
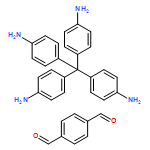
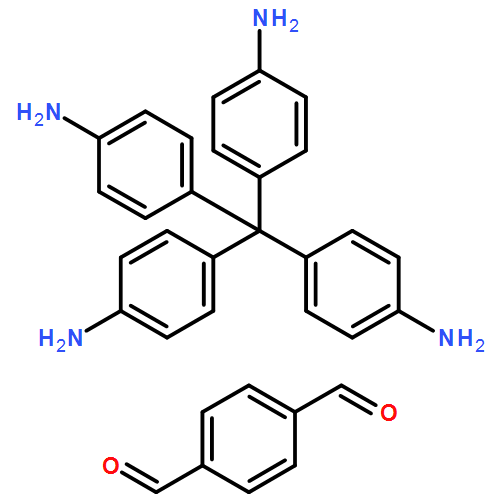
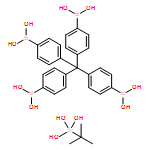
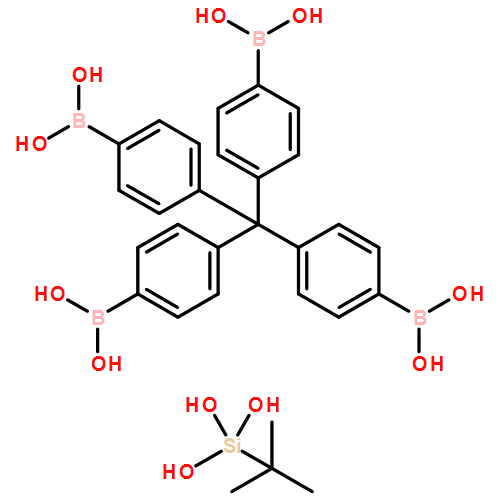









![Copper, tetrakis[m-(acetato-kO:kO')]di-, (Cu-Cu)](http://img.cochemist.com/ccimg/23700/23686-23-9.png)
![Copper, tetrakis[m-(acetato-kO:kO')]di-, (Cu-Cu)](http://img.cochemist.com/ccimg/23700/23686-23-9_b.png)