Co-reporter:Tian-Yu Sun, Xiao Wang, Hao Geng, Yaoming Xie, Yun-Dong Wu, Xinhao Zhang and Henry F. Schaefer III
Chemical Communications 2016 vol. 52(Issue 31) pp:5371-5374
Publication Date(Web):26 Feb 2016
DOI:10.1039/C6CC00384B
Togni's reagents have become very popular trifluoromethylating reagents in organic synthesis. The existing form of Togni's reagent I is a hypervalent iodine compound which lies much higher in energy than its ether isomer. The high-energy hypervalent iodine form makes Togni's reagent I very effective and versatile. The energy differences between the two forms correlate with the trans influence of the substituents. The five-membered ring in the benziodoxole-based scaffold is an important reason for its existence in the higher-energy form. The relation to Buchwald's 2014 research is discussed.
Co-reporter:Megha Anand ; Raghavan B. Sunoj ; III
Journal of the American Chemical Society 2014 Volume 136(Issue 15) pp:5535-5538
Publication Date(Web):April 3, 2014
DOI:10.1021/ja412770h
The role of a widely employed additive (AgOAc) in a palladium acetate-catalyzed ortho-C–H bond activation reaction has been examined using the M06 density functional theory. A new hetero-bimetallic Pd-(μ-OAc)3-Ag is identified as the most likely active species. This finding could have far-reaching implications with respect to the notion of the active species in palladium catalysis in the presence of other metal salt additives.
Co-reporter:Yunxiang Lu, Hui Wang, Yaoming Xie, Honglai Liu, and Henry F. Schaefer
Inorganic Chemistry 2014 Volume 53(Issue 12) pp:6252-6256
Publication Date(Web):May 27, 2014
DOI:10.1021/ic500780h
Pioneering synthetic research by the groups of Grutzmacher and Goicoechea have made possible the preparation of 2-phosphaethynolates (PCO–). The obvious question arises: can progress be made toward AsCO–, SbCO–, and BiCO–? Here the properties of all five anion congeners ECO– (E = N, P, As, Sb, Bi) were systematically investigated using ab initio coupled-cluster methods with correlation-consistent basis sets cc-pVXZ (X = D, T, Q). These anions exhibit linear structures with significant natural bond orbital negative charge on both the E and O atoms. These species should react with electrophiles via attack at either center. On going from nitrogen to bismuth, with the atomic radius increasing, the bond between E and C becomes weaker, while the C–O bond tends to be slightly stronger. By the time one gets to BiCO–, the C–O bond distance is 1.181 Å, indicating a very strong double bond. Relative to the PCO– anion, which is reactive toward several unsaturated compounds, the As/Sb/BiCO– anions may undergo cycloaddition more readily with unsaturated substrates. The dissociation energy of the E–C bond, except for that of NCO–, is predicted to be much less than that of the C–O bond. These dissociation energies are 76 kcal/mol (P––CO), 58 kcal/mol (As––CO), 37 kcal/mol (Sb––CO), and 28 kcal/mol (Bi––CO). Even the BiCO– anion should be achievable in the laboratory. The vibrational frequencies for these anions are predicted, and our results should assist in the experimental characterization and exploration of the heavier congeners ECO–.
Co-reporter:Yanjun Hao, Jiande Gu, Yundong Guo, Meiling Zhang, Yaoming Xie and Henry F. Schaefer III
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 6) pp:2641-2646
Publication Date(Web):02 Jan 2014
DOI:10.1039/C3CP54031F
Using the CCSD(T) method with relativistic correlation consistent basis sets up to cc-pVQZ-PP, the entrance complex, transition state, and exit complex for the endothermic reaction I + H2O → HI + OH have been studied. The vibrational frequencies and the zero-point vibrational energies of the five stationary points for the title reaction are reported and compared with the limited available experimental results. Opposite to the valence isoelectronic F + H2O system, but similar to the Cl + H2O and Br + H2O reactions, the I + H2O reaction is endothermic, in this case by 46 kcal mol−1. After the zero-point vibrational energy and spin–orbit coupling corrections, the endothermic reaction energy is predicted to be 48 kcal mol−1, which agrees well with experimental values. For the reverse reaction HI + OH → I + H2O the transition state lies below the reactants, consistent with the experimental negative temperature dependence for the rate constants.
Co-reporter:Naziah B. Jaufeerally, Hassan H. Abdallah, Ponnadurai Ramasami and Henry F. Schaefer III
Dalton Transactions 2014 vol. 43(Issue 10) pp:4151-4162
Publication Date(Web):19 Dec 2013
DOI:10.1039/C3DT52294F
No stable germanetellone was described until Tbt(Dis)GeTe and Tbt(Tip)GeTe (Tbt = 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl, Dis = bis(trimethylsilyl)methyl and Tip = 2,4,6-triisopropylphenyl) were reported in 1997. Following these initial experiments, there has arisen considerable interest in GeTe systems. An obvious question is: why have the simple XYGeTe (X, Y = H, F, Cl, Br, I and CN) molecules not yet been isolated? In view of the present situation, theoretical information may be of great help for further advances in germanetellone chemistry. A systematic investigation of the XYGeTe molecules is carried out using the second order Møller−Plesset perturbation theory (MP2) and density functional theory (DFT). The structures and energetics, including ionization potentials (IPad and IPad(ZPVE)), four different forms of neutral–anion separations (EAad, EAad(ZPVE), VEA and VDE) and the singlet–triplet gaps, are reported. The electronegativity (χ) reactivity descriptor for the halogens (F, Cl, Br and I) and the natural charge separations of the GeTe moiety are used to assess the interrelated properties of germanetellone and its derivatives. The results are analyzed, discussed and compared with analogous studies of telluroformaldehyde, silanetellone and their derivatives. The thermodynamic viabilities of some of the novel germanetellones have also been evaluated in terms of the bond dissociation enthalpies of Tbt(Dis)GeTe and Tbt(Tip)GeTe. The simple mono-substituted germanetellones appear to be slightly more thermodynamically favored than Tbt(Dis)GeTe and Tbt(Tip)GeTe, since the bond dissociation enthalpies of these kinetically stabilized germanetellones are about 28 and 51 kcal mol−1 lower, respectively.
Co-reporter:Yundong Guo;Yanjun Hao;Hui Wang;Yaoming Xie III
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 29) pp:5015-5020
Publication Date(Web):
DOI:10.1002/ejic.201402510
Abstract
The geometries, proton affinities, and relative energies of protonated digermane (Ge2H7+), distannane (Sn2H7+), and diplumbane (Pb2H7+) have been investigated by density functional theory using the correlation-consistent cc-pVnZ-PP basis sets (n = D, T, Q). The results of the caluclations are consistent with the very limited experimental and theoretical results that are available. The lowest-lying structure of Ge2H7+ is predicted to have C2 symmetry with a bent 3-center-2-electron Ge–H–Ge bridge, analogous to Si2H7+. For Sn2H7+ and Pb2H7+, the lowest-lying structures are predicted to be different, with a D3d structure for Sn2H7+ and a C1 structure for Pb2H7+. The predicted proton affinities decrease in the order Pb2H6 (9.84 eV) > Sn2H6 (8.48 eV) > Ge2H6 (8.11 eV) > Si2H6 (7.72 eV) > C2H6 (6.18 eV).
Co-reporter:Yanjun Hao, Yaoming Xie and Henry F. Schaefer III
RSC Advances 2014 vol. 4(Issue 88) pp:47163-47168
Publication Date(Web):17 Sep 2014
DOI:10.1039/C4RA09829C
The SiOOH potential energy surface has become central to the understanding of recent experiments (Science 2013, 342, 463) by Chakraborty associated with nebular meteorite formation. The entrance complex, transition states, and exit complex for the title reaction SiO + OH→ SiO2 + H have been studied using the CCSD(T) method with correlation consistent basis sets as large as cc-pV(Q+d)Z. Reported here are characteristics of the reactants, products, six transition states, and four intermediate complexes for this reaction. These show four previously undiscovered stationary point geometries. The entrance complex OH⋯OSi is predicted to lie 28.6 kJ mol−1 below the separated reactants. The classical barriers cis-TS1 and trans-TS1 are predicted to lie 21.8 kJ mol−1 and 6.8 kJ mol−1, respectively, below the reactants. The exit complex HSiO2 is bound by 115.3 kJ mol−1 relative to the separated products. After zero-point vibrational energy corrections, the reaction energy is predicted to be −1.4 kJ mol−1. Vibrational frequencies of the stationary points are reported and compared with the limited available experimental results. The SiOOH potential surface is found to be very different from that for COOH, contrary to the analogy drawn by Chakraborty. Notwithstanding, the assumption of Chakraborty appears justified, because all the stationary points for the SiO + OH reaction have lower relative energies than known for the analogous carbon system.
Co-reporter:Chenyang Li, Jay Agarwal, and Henry F. Schaefer III
The Journal of Physical Chemistry B 2014 Volume 118(Issue 24) pp:6482-6490
Publication Date(Web):February 5, 2014
DOI:10.1021/jp412003s
The equilibrium geometries and vibrational frequencies of the extraordinary [ReH9]2– dianion (D3h symmetry) are investigated using Hartree–Fock (HF) theory, coupled cluster theory with single and double excitations (CCSD), and coupled cluster theory with single, double, and perturbative triple excitations [CCSD(T)]. The new generation of energy-consistent relativistic pseudopotentials and correlation consistent basis sets [cc-pVXZ-PP (Re) and cc-pVXZ (H) (X = D, T, Q)] are used. Anharmonicity was considered using second-order vibrational perturbation theory. The predicted geometries and vibrational frequencies generally agree with experimental findings. In order to stabilize the [ReH9]2– dianion, the M2ReH9 (M = Na, K) sandwich complexes (D3h symmetry) are studied at the CCSD(T)/VTZ (VTZ = cc-pVTZ-PP (Re) and cc-pVTZ (H, Na, K)) level of theory. Compared to the [ReH9]2– dianion, the predicted vibrational frequencies involving Re–H stretching modes are improved, indicating the importance of considering counterions in electronically dense systems. The natural bond orbital analysis shows that each H atom only bonds with the Re center, and the 5d orbitals of Re and 1s orbitals of H are major factors for the covalent Re–H bonding.
Co-reporter:Xiao Wang, Walter E. Turner II, Jay Agarwal, and Henry F. Schaefer III
The Journal of Physical Chemistry A 2014 Volume 118(Issue 35) pp:7560-7567
Publication Date(Web):April 2, 2014
DOI:10.1021/jp502282v
Ethylene is an exceptional example of a stable closed-shell singlet molecule with a low-lying triplet state of very different symmetry. Triplet C2H4 (the ã 3A1 state), which has a twisted D2d geometry, is studied herein with high-level theoretical methods, namely, CCSD(T) (coupled cluster theory with single, double, and perturbative triple excitations) and Dunning’s correlation-consistent quadruple-ζ basis set (cc-pVQZ). Geometric parameters, including equilibrium (re) and vibrationally corrected (rg) values, are reported for C2H4, C2D4, and 13C2H4. Harmonic and anharmonic vibrational frequencies are also predicted using second-order vibrational perturbation theory (VPT2). Challenges encountered for the wagging vibrational features are discussed.
Co-reporter:Kevin B. Moore III;Angela N. Migues; Henry F. Schaefer III; Robert A. Vergenz
Chemistry - A European Journal 2014 Volume 20( Issue 4) pp:990-998
Publication Date(Web):
DOI:10.1002/chem.201303231
Abstract
Carbon-donated hydrogen bonds (CDHBs) are weak forms of hydrogen bonding (0.5–1.0 kcal mol−1) that are difficult to detect, and thus their roles in the structure and functionality of chemical systems often go unrecognized. Utilizing a computational approach, the existence of a structurally significant CDHB in the medically relevant protein Streptococcus pneumoniae hyaluronate lyase (SpnHL) is affirmed. The structure of a tetrapeptide fragment model containing the CDHB was optimized with second-order perturbation theory. From this, a CDHB with bond distance and angle consistent with previously discovered CDHBs and comparable to neighboring traditional HBs in the fragment model was found. The CDHB competes with another donor T253 OH, whereby the two alternate in strength between protein conformations, imbuing αHelix 3 appreciable flexibility. The CDHB seems to exist in spite of torsional and steric strain on the donor methyl group. It is postulated that the CDHB could aid in either counteracting the macrodipole of αHelix 3 or protecting the A249 CO from destabilizing interactions with the adjacent solvent. Employing the energy gradients from the optimization, the torque generated by the fragment model was computed, which accurately predicts the direction of rotation of αHelix 3 observed from experiment. A strongly correlated motion between αHelix 3 and αHelices 2, 4, and 5 was noted, which the interactions of the fragment model help drive by generating a torque much larger than necessary to rotate just αHelix 3. Considering these results, we conclude that CDHBs should be considered as possible beneficial components of chemical and biological phenomena.
Co-reporter:Yi Zeng, Hao Feng, Yaoming Xie, and Henry F. Schaefer III
The Journal of Organic Chemistry 2014 Volume 79(Issue 7) pp:2926-2933
Publication Date(Web):March 7, 2014
DOI:10.1021/jo402841h
Yamamoto and co-workers synthesized two cyclic aromatic carbenes with remote amino groups. Here we theoretically studied related compounds to explore tuning effects on the singlet–triplet splitting by variations of functional groups. For the Yamamoto compound, the lowest singlet state lies 15.7 kcal/mol below the lowest triplet. The singlet–triplet separation is reduced by ∼7 kcal/mol when the dimethylamino groups are replaced by H. In one set of carbenes, when X = O, we substitute S, Se, Te, SO, SeO, and TeO for X; the resulting ΔE(S–T) predictions are 9.9, 7.3, 3.9, 4.3, 2.3, and −0.1 kcal/mol, respectively. A different set of X fragments yields triplet electronic ground states with ΔE(S–T) values of −8.6 (X = BH), −6.8 (X = AlH), −7.2 (X = GaH), −7.5 (X = InH), and −7.0 kcal/mol (X = TlH). We also predicted ΔE(S–T) with N(CH3)2 replaced by PH2, AsH2, SbH2, BiH2, BH2, CH3, OH, and F. Of all the molecules considered, that with N(CH3)2 replaced with BH2 and X = BH most favors the triplet state, lying 13.7 kcal/mol below the singlet. Finally, we have relocated the N(CH3)2 and NH2 groups from the (3, 6) positions to the (4, 5), (2, 7), and (1, 8) terminal ring positions, with very interesting results.
Co-reporter:Valia Nikolova, Sonia Ilieva, Boris Galabov, and Henry F. Schaefer III
The Journal of Organic Chemistry 2014 Volume 79(Issue 15) pp:6823-6831
Publication Date(Web):July 8, 2014
DOI:10.1021/jo500732m
IR spectroscopic experiments and theoretical DFT computations reveal the effects of aromatic substituents on π-hydrogen bonding between monosubstituted phenol derivatives and benzene. Simultaneous formation of two π-hydrogen bonds (red-shifting O–H···π and blue-shifting ortho-C–H···π) contribute to the stability of these complexes. The interaction of the acidic phenol O–H proton-donating group with the benzene π-system dominates the complex formation. The experimental shifts of O–H stretching frequencies for the different phenol complexes vary in the range 45–74 cm–1. Strong effects on hydrogen-bonding energies and frequency shifts of electron-withdrawing aromatic substituents and very weak influence of electron-donating groups have been established. Experimental quantities and theoretical parameters are employed in rationalizing the properties of these complexes. The acidities of the proton-donating phenols describe quantitatively the hydrogen-bonding process. The results obtained provide clear evidence that, when the structural variations are in the proton-donating species, the substituent effects on π-hydrogen bonding follow classic mechanisms, comprising both resonance and direct through-space influences. The performance of three alternative DFT functionals (B3LYP, B97-D, and PBE0 combined with the 6-311++G(2df,2p) basis set) in predicting the O–H frequency shifts upon complexation is examined. For comparison, O–H frequency shifts for several complexes were also determined at MP2/6-31++G(d,p).
Diatomic Silylynes, Germylynes, Stannylynes, and Plumbylynes: Structures, Dipole Moments, Dissociation Energies, and Quartet-Doublet Gaps of EH and EX (E = Si, Ge, Sn, Pb; X = F, Cl, Br, I)
Co-reporter:Huidong Li, Hao Feng, Weiguo Sun, Yaoming Xie, and Henry F. Schaefer
Inorganic Chemistry 2013 Volume 52(Issue 12) pp:6849-6859
Publication Date(Web):June 3, 2013
DOI:10.1021/ic3025099
Systematic theoretical studies of the carbyne and halocarbyne analogues E-H and E-X (E = Si, Ge, Sn, Pb; X = F, Cl, Br, I) were carried out with ab initio coupled-cluster methods using very large basis sets. The 2Π state is the ground electronic state for all these compounds. The quartet-doublet energy separations, equilibrium distances, and dissociation energies for these species are predicted. The quartet-doublet splittings fall in the order EF > ECl > EBr > EI > EH for a given metal E; and PbX > GeX > SnX > SiX for the same halogen atom X. The dipole moments span a large range, from 0.08 debye (GeH) to 3.58 debye (PbCl). The dissociation energies range from 1.84 eV (PbH) to 6.15 eV (SiF).
Co-reporter:Beulah S. Narendrapurapu, Nancy A. Richardson, Andreas V. Copan, Marissa L. Estep, Zheyue Yang, and Henry F. Schaefer III
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 7) pp:2930-2938
Publication Date(Web):May 20, 2013
DOI:10.1021/ct4002398
Density functional theory (DFT) is a widely used method for predicting equilibrium geometries of organometallic compounds involving transition metals, with a wide choice of functional and basis set combinations. A study of the role of basis set size in predicting the structural parameters can be insightful with respect to the effectiveness of using small basis sets to optimize larger molecular systems. For many organometallic systems, the metal–metal and metal–carbon distances are the most important structural features. In this study, we compare the equilibrium metal–ligand and metal–metal distances of six transition metal carbonyl compounds predicted by the Hood-Pitzer double-ζ polarization (DZP) basis set, against those predicted employing the standard correlation consistent cc-pVXZ (X = D,T,Q) basis sets, for 35 different DFT methods. The effects of systematically increasing the basis set size on the structural parameters are carefully investigated. The Mn–Mn bond distance in Mn2(CO)10 shows a greater dependence on basis set size compared to the other M–M bonds. However, the DZP predictions for re(Mn–Mn) are closer to experiment than those obtained with the much larger cc-pVQZ basis set. Our results show that, in general, DZP basis sets predict structural parameters with an accuracy comparable to the triple and quadruple-ζ basis sets. This finding is very significant, because the quadruple-ζ basis set for Mn2(CO)10 includes 1308 basis functions, while the equally effective double-ζ set (DZP) includes only 366 basis functions. Overall, the DZP M06-L method predicts structures that are very consistent with experiment.
Co-reporter:Qiong Luo, Katherine R. Randall and Henry F. Schaefer
RSC Advances 2013 vol. 3(Issue 18) pp:6572-6585
Publication Date(Web):20 Feb 2013
DOI:10.1039/C3RA40538A
Polyfluorocyclohexanes present an interesting challenge to our current understanding of fundamental organic chemistry. In part to improve molecular mechanics methods and facilitate drug design, a systematic survey of cyclohexanes with up to six fluorine substituents has been carried out, using theoretical methods. The preferred conformers are determined by delocalization effects, such as hyperconjugation, and do not necessarily follow the common assumption that substituents prefer the equatorial position. Thus, accurate ab intio results, which can capture electronic effects, are required. The lowest energy conformations of fluorocyclohexane, difluorocyclohexanes (six structural isomers), trifluorocyclohexanes (nine structural isomers), tetrafluorocyclohexanes (seventeen structural isomers), pentafluorocyclohexane (ten structural isomers), and hexafluorocyclohexanes (seven structural isomers) have been determined; relative energies, geometries, dipole moments, and population distributions are reported. We present a model for predicting the relative energies of polyfluorocyclohexane conformers based on the number of 1,2; 1,3; and 1,4 interactions present. The model is based on the energies from the difluorocyclohexanes; the correlation coefficient (R2) between computed relative energies and model relative energies is 0.967.
Co-reporter:J. Wayne Mullinax;David S. Hollman ; Henry F. Schaefer III
Chemistry - A European Journal 2013 Volume 19( Issue 23) pp:7487-7495
Publication Date(Web):
DOI:10.1002/chem.201203481
Abstract
Germanium has been a central feature in the renaissance of main-group inorganic chemistry. Herein, we present the stationary-point geometries of tetragermacyclobutadiene and its related isomers on the singlet potential energy surface at the CCSD(T)/cc-pVTZ level of theory. Three of these 12 structures are reported for the first time and one of them is predicted to lie only 0.4 kcal mol−1 above the previously reported global minimum. Focal-point analyses has provided electronic energies at the CCSD(T) level of theory, which are extrapolated to the complete basis-set limit and demonstrate the convergence behavior of the electronic energies with improving levels of theory and increasing basis-set size. The lowest-energy structure is the bicyclic structure, which lies 35 kcal mol−1 below the “all-Ge” cyclobutadiene structure. The reaction energies for the association of known Ge hydrides (e.g., digermene) to form Ge4H4 indicate that Ge4H4 could be observed experimentally. We investigate the bonding patterns by examining the frontier molecular orbitals. Our results demonstrate that: 1) the cyclic isomers of (GeH)4 distort to maximize the mixing of the p orbitals that are involved in the π system of tetragermacyclobutadiene and 2) the lowest-energy isomers exhibit unusual bonding arrangements (e.g., bridging H bonds) that maximize the nonbonding electron density at the Ge centers.
Co-reporter:Qunchao Fan, Huidong Li, Hao Feng, Weiguo Sun, Tongxiang Lu, Andrew C. Simmonett, Yaoming Xie, and Henry F. Schaefer III
The Journal of Physical Chemistry A 2013 Volume 117(Issue 39) pp:10027-10033
Publication Date(Web):April 15, 2013
DOI:10.1021/jp400541a
The existing potential energy surfaces for the Li + HF system have been challenged by the experiments of Loesch, Stienkemeier, and co-workers. Here a very accurate potential energy surface has been obtained with rather rigorous theoretical methods. Methods up to full CCSDT have been pursued with basis sets as large as core correlated quintuple ζ. Reported here are the reactants, products, two transition states, and three intermediate complexes for this reaction. These reveal one previously undiscovered equilibrium geometry. The stationary point relative energies are very sensitive to level of theory. The reaction has a classical endothermicity of 2.6 kcal mol–1. The complex Li···HF in the entrance valley lies 6.1 kcal/mol below the reactants. The expected transition state Li···H···F is bent with an angle of 72.2° and lies 4.5 kcal/mol above the reactants. The latter predicted classical barrier should be no more than one kcal/mol above the exact barrier. Not one but two product complexes lie 1.6 and 2.2 kcal/mol above reactants, respectively. Between the two product complexes, a second transition state, very broad, is found. The vibrational frequencies and zero-point vibrational energies (ZPVE) of all stationary points are reported, and significantly affect the relative energies.
Co-reporter:Jun Guan, Katherine R. Randall, Henry F. Schaefer III, and Huidong Li
The Journal of Physical Chemistry A 2013 Volume 117(Issue 10) pp:2152-2159
Publication Date(Web):February 13, 2013
DOI:10.1021/jp311681u
The ground triplet state and lowest singlet state of formylmethylene have been proposed as important intermediates in the Wolff rearrangement of α-diazo ketones into ketenes. The ground triplet state of formylmethylene has been examined experimentally, but the lowest singlet state has yet to be observed. We predict equilibrium geometries, energies, bonding, dipole moments, and harmonic vibrational frequencies for these two lowest states of formylmethylene at the cc-pVQZ CCSD(T) level of theory. The singlet–triplet energy difference [ΔE(S-T)] is quite sensitive to the level of theory. The highly accurate cc-pVQZ CCSD(T) level of theory yields the most reliable result of only 2.0 kcal mol–1. An estimate based on the experimentally characterized CH2 molecule yields ΔE(S-T) = 1.27 kcal mol–1. In addition, accurate quartic force fields have been determined at the cc-pVTZ CCSD(T) level of theory. Fundamental vibrational frequencies, anharmonic constants, and vibration–rotation coupling constants were determined using vibrational second-order perturbation theory (VPT2). Our results should aid in experimental detection and characterization of the lowest singlet state of formylmethylene, which is highly desirable for better understanding the mechanism of the Wolff rearrangement.
Co-reporter:Naziah B. Jaufeerally, Hassan H. Abdallah, Ponnadurai Ramasami, and Henry F. Schaefer III
The Journal of Physical Chemistry A 2013 Volume 117(Issue 27) pp:5567-5577
Publication Date(Web):June 5, 2013
DOI:10.1021/jp403341z
The unavailability of monomeric heavy ketone analogues has been ascribed to the evanescence of the very reactive A═E double bond (A and E are the heavier group 14 and group 16 elements, respectively). Can the isolation of any of the monomeric telluro-ketones be assisted by an energetic favorability on its potential energy surface (PES)? In this light, the reaction pathways for the isomerization and decomposition reactions of H2A═Te and HFA═Te (A = C, Si, and Ge) molecules on their singlet state PES have been studied using second-order Møller–Plesset perturbation theory (MP2). The barrier heights reported suggest that telluroformaldehyde, silanetellone, and germatellone are kinetically more resistant to unimolecular reactions than the corresponding lighter chalcogen analogues. However, upon replacing a hydrogen atom by fluorine, the barrier heights of most of the isomerization and decomposition reactions are lowered. Among the unimolecular reactions studied for the H2A═Te and HFA═Te (A = C, Si, and Ge) molecules, the decomposition of cis-FGeTeH into HF and GeTe is found to be the most facile reaction, with a barrier height of only 4.6 kcal/mol. We also predict the ground state telluro-ketones to be viable molecules, as they have no imaginary vibrational frequencies and their lowest vibrational frequencies are always >100 cm–1. In view of the scarcity of information on the chemistry of the mentioned telluro-ketones, the molecular parameters of various isomers and decomposition products have been reported, and should be useful for future experimental investigations.
Co-reporter:Stefan Vogt-Geisse, Alexander Yu. Sokolov, Shane R. McNew, Yukio Yamaguchi, and Henry F. Schaefer III
The Journal of Physical Chemistry A 2013 Volume 117(Issue 28) pp:5765-5774
Publication Date(Web):June 17, 2013
DOI:10.1021/jp402395v
In this study a systematic theoretical investigation of Ge2CH2 is carried out. The singlet potential energy surface (PES) was explored using state-of-the-art theoretical methods including self-consistent field (SCF), coupled cluster theory incorporating single and double excitation (CCSD), perturbative triple [CCSD(T)] and full triples [CCSDT] with perturbative quadruple (Q), together with a variety of correlation-consistent polarized valence basis sets cc-pVXZ (where X = D, T, and Q). A total of eleven stationary points have been located on the Ge2CH2 singlet ground state PES. Among them, seven structures are minima (1S–7S), two are transition states (TS1 and TS2), and two are second-order saddle points (SSP1 and SSP2). The global minimum is predicted to be an exotic hydrogen-bridged structure 1S. The energy ordering of the seven minima (in kcal mol–1) obtained from focal point analysis using the extrapolation to complete basis set (CBS) limit with zero point vibrational energy (ZPVE), core correlation, diagonal Born–Oppenheimer (DBOC) and relativistic correction is 1S [0.0] < 2S [17.2] < 3S [18.3] < 4S [31.7] < 5S [39.9] < 6S [58.1] < 7S [82.1].
Co-reporter:Yudong Qiu, Alexander Yu. Sokolov, Yukio Yamaguchi, and Henry F. Schaefer III
The Journal of Physical Chemistry A 2013 Volume 117(Issue 38) pp:9266-9273
Publication Date(Web):August 23, 2013
DOI:10.1021/jp406579w
The simplest metal carbene, BeCH2, is experimentally unknown. Its isomer, HBeCH, lies higher in energy, but has been detected by the infrared matrix isolation [J. Am. Chem. Soc. 1998, 120, 6097]. In the present study the ground and low-lying excited states of the BeCH2 and HBeCH isomers were investigated using state-of-the-art ab initio methods, including coupled-cluster theory with up to full quadruple excitations (CCSDTQ), and complete active space self-consistent field (CASSCF) with multireference configuration interaction with single and double excitations (MRCISD). The relative energies were obtained using the focal point analysis combined with large correlation-consistent cc-pCVXZ basis sets (X = D, T, Q, 5) and were extrapolated to the complete basis set (CBS) limit. The 3B1 state of BeCH2 (C2v symmetry) is the global minimum on the ground triplet potential energy surface (PES). The 3Σ– state of the linear isomer HBeCH is located 4.9 kcal mol–1 above the global minimum, at the CCSDTQ/CBS level of theory. The BeCH2 and HBeCH isomers are connected through the 3A″ transition state lying 46.1 kcal mol–1 above the global minimum. The higher-lying energy HBeCH structure has much larger Be–C bond dissociation energy (126.6 kcal mol–1, cf. BDE(BeCH2) = 62.1 kcal mol–1). The lowest excited state of BeCH2 is the open-shell 1B1 state, with a relative energy of only 4.9 kcal mol–1 above the global minimum, followed by 1A1 state (16.8 kcal mol–1) at the MRCISD/cc-pCVQZ level of theory. For the HBeCH isomer the lowest-energy excited states are 1Δ and 1Σ+, lying about 30 kcal mol–1 above the global minimum. For the ground state of BeCH2 the fundamental vibrational frequencies computed using second-order vibrational perturbation theory (VPT2) at the CCSD(T)/cc-pCVQZ level are reported. We hope that our highly accurate theoretical results will assist in the experimental identification of BeCH2.
Co-reporter:Jiande Gu, Jerzy Leszczynski, and Henry F. Schaefer III
Chemical Reviews 2012 Volume 112(Issue 11) pp:5603
Publication Date(Web):June 13, 2012
DOI:10.1021/cr3000219
Co-reporter:Ashwini Bundhun, Hassan H. Abdallah, Ponnadurai Ramasami, Peter P. Gaspar, and Henry F. Schaefer III
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12152-12164
Publication Date(Web):November 7, 2012
DOI:10.1021/ic301225w
A systematic investigation is carried out using the B3LYP, BLYP, and BHLYP functionals and MP2 level of theory to characterize the low-lying electronic singlet and triplet GeC2N2 isomers. The basis sets used are of double-ζ plus polarization quality with additional s- and p-type diffuse functions, DZP++. Three bent isomers Ge(CN)2, CNGeCN, and Ge(NC)2 are located on the singlet and triplet potential energy surfaces. In visualizing the reaction pathways for the singlet isomerization of the bent isomers, two three-membered [Ge, C, N] cyclic systems, with exocyclic −C–C≡N and −C–N≡C bonding, appear on the energy surface. Four types of electron affinities reported are: the adiabatic electron affinity, the zero-point vibrationally corrected electron affinity, the vertical electron affinity, and the vertical detachment energy of the anion. The ionization energies and singlet–triplet gaps for all isomers are also reported. The energetic ordering (kcal mol–1) (B3LYP) with zero-point vibrational energy corrections for the singlet ground state isomers follows: Ge(CN)2 (global minimum) < CNGeCN (2.3) < Ge(NC)2 (3.3) < Cyc_exo_CCN (15.3) < Cyc_exo_CNC (30.6). All the bent and cyclic isomers are found to be below the dissociation limit to Ge (3P) + C2N2 (1Σg). The rate constants for all interconversions are evaluated using transition state theory.
Co-reporter:Tongxiang Lu, Qiang Hao, Jeremiah J. Wilke, Yukio Yamaguchi, De-Cai Fang, Henry F. Schaefer III
Journal of Molecular Structure 2012 Volume 1009() pp:103-110
Publication Date(Web):15 February 2012
DOI:10.1016/j.molstruc.2011.10.032
Three equilibrium structures and two associated isomerization transition states for the SiCH2–HSiCH–CSiH2 system have been theoretically investigated using highly correlated ab initio methods combined with the correlation-consistent polarized core-valence basis sets. Vibrational second-order perturbation theory (VPT2) has been employed to obtain the zero-point vibration corrected rotational constants, centrifugal distortion constants, and fundamental vibrational frequencies for each equilibrium structure. The comparison of the theoretically predicted rotational constants and fundamental frequencies for silylidene (SiCH2) with existing experimental observations shows excellent agreement. The CH2 (CD2) rocking modes (ν6) are the most anharmonic among the six vibrational modes, consistent with limited experimental observations. The five stationary point structures and their relative energies on the ground state potential energy surface (PES) are accurately determined using the focal point extrapolation technique. Silylidene (SiCH2) has been confirmed to be the global minimum. Silaacetylene (HSiCH) with a trans-bent structure is located 34.8 ± 0.3 kcal mol−1 (with zero-point vibrational energy, scalar relativistic effects, and DBOC corrections) above the global minimum. The barrier height for the critical reverse isomerization process [HSiCH → SiCH2] is predicted to be 4.1 ± 0.3 kcal mol−1. The third isomer silavinylidene (CSiH2) is predicted to lie 84.6 ± 0.3 kcal mol−1 above the global minimum, with the barrier height for the reverse isomerization process [CSiH2 → HSiCH] being only 2.6 ± 0.3 kcal mol−1. The present research should assist the future experimental characterization of silylidene (SiCH2) isomers.Highlights► The potential energy surface for SiCH2 system has been accurately established. ► The B0 rotational constants for SiCH2 and its isotopologues have been evaluated. ► The fundamental vibrational frequencies for SiCH2 system have been predicted. ► The theoretical predictions of frequencies are in accord with experimental results.
Co-reporter:Dr. Qianyi Cheng;Dr. Jie Gu;Katherine R. Compaan;Dr. Henry F. Schaefer III
Chemistry - A European Journal 2012 Volume 18( Issue 16) pp:4877-4886
Publication Date(Web):
DOI:10.1002/chem.201102415
Abstract
Several possible mechanisms underlying isoguanine formation when OH radical attacks the C2 position of adenine (A) are investigated theoretically for the first time. Two steps are involved in this process. In the first step, one of two low-lying A⋅⋅⋅OH reactant complexes is formed, leading to C2H2 bond cleavage. Between the two reactant complexes there is a small isomerization barrier, which lies well below separated adenine plus OH radical. The complex dissociates to free molecular hydrogen and an isoguanine tautomer (isoG 1 or isoG 2). The local and activation barriers for the two pathways are very similar. This evidence suggests that the two pathways are competitive. After dehydrogenation, there are two possible routes for the second step of the reaction. One is direct hydrogen transfer, via enol–keto tautomerization, which has high local barriers for both tautomers and is not favored. The other option is indirect hydrogen transfer involving microsolvation by one water molecule. The water lowers the reaction barrier by over 20 kcal mol−1, indicating that water-mediated hydrogen transfer is much more favorable. Both A+OH.isoG+H. reactions are exothermic and spontaneous. Among four isoguanine tautomers, isoG 1 has the lowest energy. Our findings explain why only the N1H and O2H tautomers of isolated isoguanine and isoguanosine have been observed experimentally.
Co-reporter:Peter N. Ascik;René Rugango;Dr. Andrew C. Simmonett;Katherine R. Compaan ; Henry F. Schaefer III
ChemPhysChem 2012 Volume 13( Issue 5) pp:1255-1260
Publication Date(Web):
DOI:10.1002/cphc.201101008
Abstract
Recent high-resolution spectroscopic studies by Merritt, Bondybey, and Heaven (Science2009, 324, 1548) have heightened the anticipation that small beryllium clusters will soon be observed in the laboratory. Beryllium clusters are important discrete models for the theoretical study of metals. The trigonal bipyramidal Be5 molecule is studied using high-level coupled cluster methods. We obtain the optimized geometry, atomization and dissociation energies, and vibrational frequencies. The c∼CCSDT(Q) method is employed to compute the atomization and dissociation energies. In this approach, complete basis set (CBS) extrapolations at the CCSD(T) level of theory are combined with an additive correction for the effect of iterative triple and perturbative quadruple excitations. Harmonic vibrational frequencies are obtained using analytic gradients computed at the CCSD(T) level of theory. We report an atomization energy of 129.6 kcal mol−1 at the trigonal bipyramid global minimum geometry. The Be5Be4+Be dissociation energy is predicted to be 39.5 kcal mol−1. The analogous dissociation energies for the smaller beryllium clusters are 64.0 kcal mol−1 (Be4Be3+Be), 24.2 kcal mol−1 (Be3Be2+Be), and 2.7 kcal mol−1 (Be2Be+Be). The trigonal bipyramidal Be5 structure has an equatorial–equatorial bond length of 2.000 Å and an axial–equatorial distance of 2.060 Å. Harmonic frequencies of 730, 611, 456, 583, 488, and 338 cm−1 are obtained at the CCSD(T)/cc-pCVQZ level of theory. Quadruple excitations are found to make noticeable contributions to the energetics of the pentamer, which exhibits a significant level of static correlation.
Co-reporter:Ashutosh Gupta, Heather M. Jaeger, Katherine R. Compaan, and Henry F. Schaefer III
The Journal of Physical Chemistry B 2012 Volume 116(Issue 19) pp:5579-5587
Publication Date(Web):April 24, 2012
DOI:10.1021/jp211608b
The guanine–cytosine (GC) radical anion and its interaction with a single water molecule is studied using ab initio and density functional methods. Z-averaged second-order perturbation theory (ZAPT2) was applied to GC radical anion for the first time. Predicted spin densities show that the radical character is localized on cytosine. The Watson–Crick monohydrated GC anion is compared to neutral GC·H2O, as well as to the proton-transferred analogue on the basis of structural and energetic properties. In all three systems, local minima are identified that correspond to water positioned in the major and minor grooves of macromolecular DNA. On the anionic surface, two novel structures have water positioned above or below the GC plane. On the neutral and anionic surfaces, the global minimum can be described as water interacting with the minor groove. These structures are predicted to have hydration energies of 9.7 and 11.8 kcal mol–1, respectively. Upon interbase proton-transfer (PT), the anionic global minimum has water positioned in the major groove, and the hydration energy increases to 13.4 kcal mol–1. PT GC·H2O•– has distonic character; the radical character resides on cytosine, while the negative charge is localized on guanine. The effects of proton transfer are further investigated through the computed adiabatic electron affinities (AEA) of GC and monohydrated GC, and the vertical detachment energies (VDE) of the corresponding anions. Monohydration increases the AEAs and VDEs by only 0.1 eV, while proton-transfer increases the VDEs substantially (0.8 eV). The molecular charge distribution of monohydrated guanine–cytosine radical anion depends heavily on interbase proton transfer.
Co-reporter:Qiang Hao, Tongxiang Lu, Jeremiah J. Wilke, Andrew C. Simmonett, Yukio Yamaguchi, De-Cai Fang, and Henry F. Schaefer
The Journal of Physical Chemistry A 2012 Volume 116(Issue 18) pp:4578-4589
Publication Date(Web):April 10, 2012
DOI:10.1021/jp211880r
Theoretical investigations of three equilibrium structures and two associated isomerization reactions of the GeCH2 - HGeCH - H2GeC system have been systematically carried out. This research employed ab initio self-consistent-field (SCF), coupled cluster (CC) with single and double excitations (CCSD), and CCSD with perturbative triple excitations [CCSD(T)] wave functions and a wide variety of correlation-consistent polarized valence cc-pVXZ and cc-pVXZ-DK (where X = D, T, Q) basis sets. For each structure, the total energy, geometry, dipole moment, harmonic vibrational frequencies, and infrared intensities are predicted. Complete active space SCF (CASSCF) wave functions are used to analyze the effects of correlation on physical properties and energetics. For each of the equilibrium structures, vibrational second-order perturbation theory (VPT2) has been utilized to obtain the zero-point vibration corrected rotational constants, centrifugal distortion constants, and fundamental vibrational frequencies. The predicted rotational constants and anharmonic vibrational frequencies for 1-germavinylidene are in good agreement with available experimental observations. Extensive focal point analyses, including CCSDT and CCSDT(Q) energies and basis sets up to quintuple zeta, are used to obtain complete basis set (CBS) limit energies. At all levels of theory employed in this study, the global minimum of the GeCH2 potential energy surface (PES) is confirmed to be 1-germavinylidene (GeCH2, 1). The second isomer, germyne (HGeCH, 2) is predicted to lie 40.4(41.1) ± 0.3 kcal mol–1 above the global minimum, while the third isomer, 2-germavinylidene (H2GeC, 3) is located 92.3(92.7) ± 0.3 kcal mol–1 above the global minimum; the values in parentheses indicate core–valence and zero-point vibration energy (ZPVE) corrected energy differences. The barriers for the forward (1→2) and reverse (2→1) isomerization reactions between isomers 1 and 2 are 48.3(47.7) ± 0.3 kcal mol–1 and 7.9(6.6) ± 0.3 kcal mol–1, respectively. On the other hand, the barriers of the forward (2→3) and reverse (3→2) isomerization reactions between isomers 2 and 3 are predicted to be 55.2(53.2) ± 0.3 kcal mol–1 and 3.3(1.6) ± 0.3 kcal mol–1, respectively.
Co-reporter:Tian-Yu Sun, Xiao Wang, Hao Geng, Yaoming Xie, Yun-Dong Wu, Xinhao Zhang and Henry F. Schaefer III
Chemical Communications 2016 - vol. 52(Issue 31) pp:NaN5374-5374
Publication Date(Web):2016/02/26
DOI:10.1039/C6CC00384B
Togni's reagents have become very popular trifluoromethylating reagents in organic synthesis. The existing form of Togni's reagent I is a hypervalent iodine compound which lies much higher in energy than its ether isomer. The high-energy hypervalent iodine form makes Togni's reagent I very effective and versatile. The energy differences between the two forms correlate with the trans influence of the substituents. The five-membered ring in the benziodoxole-based scaffold is an important reason for its existence in the higher-energy form. The relation to Buchwald's 2014 research is discussed.
Co-reporter:Yanjun Hao, Jiande Gu, Yundong Guo, Meiling Zhang, Yaoming Xie and Henry F. Schaefer III
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 6) pp:NaN2646-2646
Publication Date(Web):2014/01/02
DOI:10.1039/C3CP54031F
Using the CCSD(T) method with relativistic correlation consistent basis sets up to cc-pVQZ-PP, the entrance complex, transition state, and exit complex for the endothermic reaction I + H2O → HI + OH have been studied. The vibrational frequencies and the zero-point vibrational energies of the five stationary points for the title reaction are reported and compared with the limited available experimental results. Opposite to the valence isoelectronic F + H2O system, but similar to the Cl + H2O and Br + H2O reactions, the I + H2O reaction is endothermic, in this case by 46 kcal mol−1. After the zero-point vibrational energy and spin–orbit coupling corrections, the endothermic reaction energy is predicted to be 48 kcal mol−1, which agrees well with experimental values. For the reverse reaction HI + OH → I + H2O the transition state lies below the reactants, consistent with the experimental negative temperature dependence for the rate constants.
Co-reporter:Naziah B. Jaufeerally, Hassan H. Abdallah, Ponnadurai Ramasami and Henry F. Schaefer III
Dalton Transactions 2014 - vol. 43(Issue 10) pp:NaN4162-4162
Publication Date(Web):2013/12/19
DOI:10.1039/C3DT52294F
No stable germanetellone was described until Tbt(Dis)GeTe and Tbt(Tip)GeTe (Tbt = 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl, Dis = bis(trimethylsilyl)methyl and Tip = 2,4,6-triisopropylphenyl) were reported in 1997. Following these initial experiments, there has arisen considerable interest in GeTe systems. An obvious question is: why have the simple XYGeTe (X, Y = H, F, Cl, Br, I and CN) molecules not yet been isolated? In view of the present situation, theoretical information may be of great help for further advances in germanetellone chemistry. A systematic investigation of the XYGeTe molecules is carried out using the second order Møller−Plesset perturbation theory (MP2) and density functional theory (DFT). The structures and energetics, including ionization potentials (IPad and IPad(ZPVE)), four different forms of neutral–anion separations (EAad, EAad(ZPVE), VEA and VDE) and the singlet–triplet gaps, are reported. The electronegativity (χ) reactivity descriptor for the halogens (F, Cl, Br and I) and the natural charge separations of the GeTe moiety are used to assess the interrelated properties of germanetellone and its derivatives. The results are analyzed, discussed and compared with analogous studies of telluroformaldehyde, silanetellone and their derivatives. The thermodynamic viabilities of some of the novel germanetellones have also been evaluated in terms of the bond dissociation enthalpies of Tbt(Dis)GeTe and Tbt(Tip)GeTe. The simple mono-substituted germanetellones appear to be slightly more thermodynamically favored than Tbt(Dis)GeTe and Tbt(Tip)GeTe, since the bond dissociation enthalpies of these kinetically stabilized germanetellones are about 28 and 51 kcal mol−1 lower, respectively.
.jpg)


![3-AZABICYCLO[3.2.1]OCTA-2,6-DIENE](http://img.cochemist.com/ccimg/848700/848615-17-8.png)
![3-AZABICYCLO[3.2.1]OCTA-2,6-DIENE](http://img.cochemist.com/ccimg/848700/848615-17-8_b.png)
![2-AZABICYCLO[3.2.1]OCTA-3,6-DIENE](http://img.cochemist.com/ccimg/848700/848615-15-6.png)
![2-AZABICYCLO[3.2.1]OCTA-3,6-DIENE](http://img.cochemist.com/ccimg/848700/848615-15-6_b.png)
![Tricyclo[3.3.1.13,7]decane, 1,1'-(1,1,2,2-tetramethyl-1,2-ethanediyl)bis-](http://img.cochemist.com/ccimg/99700/99663-24-8.png)
![Tricyclo[3.3.1.13,7]decane, 1,1'-(1,1,2,2-tetramethyl-1,2-ethanediyl)bis-](http://img.cochemist.com/ccimg/99700/99663-24-8_b.png)



![4-METHYL-1-AZABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/45700/45651-41-0.png)
![4-METHYL-1-AZABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/45700/45651-41-0_b.png)

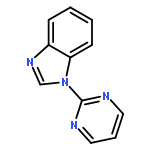
![2-(Pyrimidin-2-yl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/18200/18107-02-3.png)
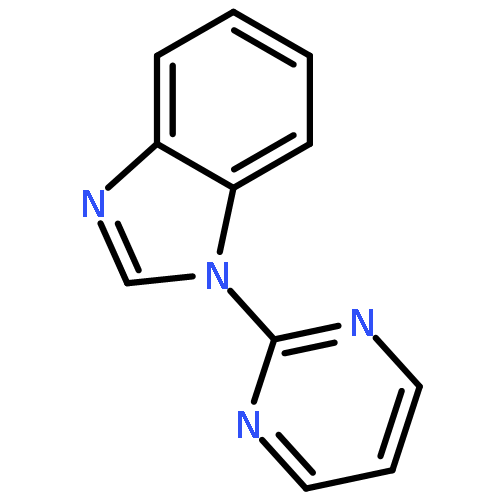
![2-(Pyrimidin-2-yl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/18200/18107-02-3_b.png)



![undecacyclo[9.9.0.0~2,9~.0~3,7~.0~4,20~.0~5,18~.0~6,16~.0~8,15~.0~10,14~.0~12,19~.0~13,17~]icosane (non-preferred name)](/data/chemimg/51900/4493-23-6.png)
![undecacyclo[9.9.0.0~2,9~.0~3,7~.0~4,20~.0~5,18~.0~6,16~.0~8,15~.0~10,14~.0~12,19~.0~13,17~]icosane (non-preferred name)](/data/chemimg/51900/4493-23-6_b.png)







![Indeno[1,2-a]indene](http://img.cochemist.com/ccimg/300/206-71-3.png)
![Indeno[1,2-a]indene](http://img.cochemist.com/ccimg/300/206-71-3_b.png)



![Acetamide,N-[2-[(triphenylmethyl)thio]ethyl]-2-[[2-[(triphenylmethyl)thio]ethyl]amino]-](http://img.cochemist.com/ccimg/152900/152863-83-7.png)
![Acetamide,N-[2-[(triphenylmethyl)thio]ethyl]-2-[[2-[(triphenylmethyl)thio]ethyl]amino]-](http://img.cochemist.com/ccimg/152900/152863-83-7_b.png)
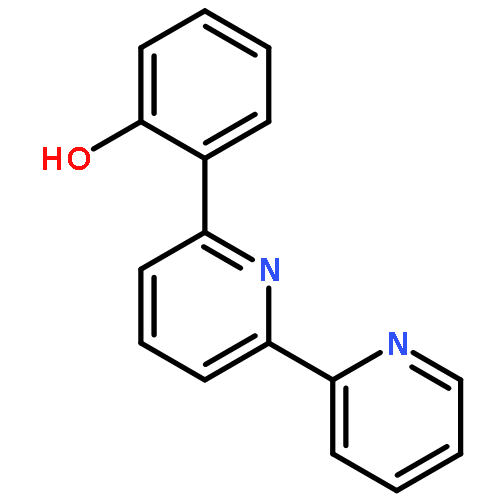










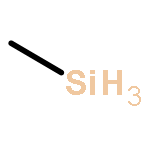
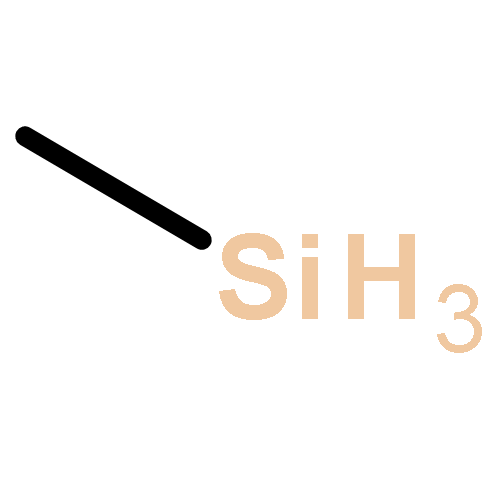
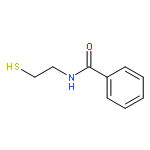




![(7R,8S,8aR,9aS)-7,8,8a,9a-tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/63400/63357-09-5.png)
![(7R,8S,8aR,9aS)-7,8,8a,9a-tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/63400/63357-09-5_b.png)










![Bicyclo[2.2.1]hept-2-ylium](/data/chemimg/1161800/24321-81-1.png)
![Bicyclo[2.2.1]hept-2-ylium](/data/chemimg/1161800/24321-81-1_b.png)





















![Ethanamine,2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/1100/1095-85-8.png)
![Ethanamine,2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/1100/1095-85-8_b.png)















