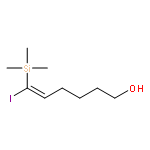
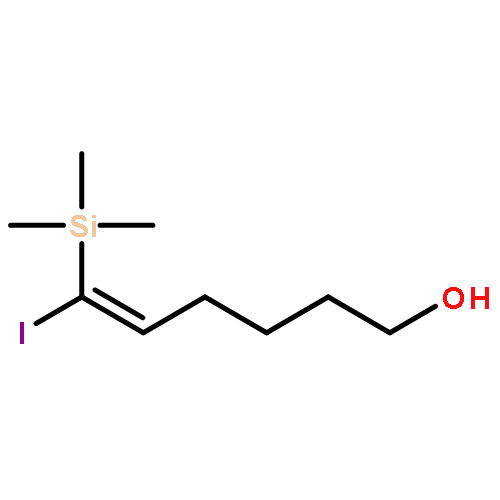
Co-reporter:Ming Xiang, Yiwei Wu, Jason P. Burke, and Jason J. Chruma
The Journal of Organic Chemistry 2016 Volume 81(Issue 18) pp:8508-8519
Publication Date(Web):August 26, 2016
DOI:10.1021/acs.joc.6b01677
An unconstrained exocyclic stereogenic center and a removable trimethylsilyl group are combined to induce high π-facial selectivity and near-exclusive exo-selectivity in the intramolecular Diels–Alder cycloaddition of dodeca-3,9,11-trien-5-ones. This strategy provides direct access to polysubstituted trans-1-decalones related to the symbioimines in good yield and acceptable diastereoselectivity.
Co-reporter:Shaojian Tang, Jong Yeun Park, Andrew A. Yeagley, Michal Sabat, and Jason J. Chruma
Organic Letters 2015 Volume 17(Issue 9) pp:2042-2045
Publication Date(Web):April 17, 2015
DOI:10.1021/acs.orglett.5b00107
Condensation between the tetrabutylammonium salt of 2,2-diphenylglycine and aldehydes results in a decarboxylative Erlenmeyer reaction, affording 1,2-diaryl-2-iminoalcohols as a mixture of diastereomers in good yields. The diastereomeric ratio shifts over time, with the anti diastereomer and the syn oxazolidine tautomer serving as the kinetic and thermodynamic products, respectively. Addition of Lewis acids can catalyze the rates of reaction and product equilibration. The results highlight the stereochemical promiscuity of 1,2-diaryl-2-iminoalcohols in the presence of Lewis acids and Brønsted bases.
Co-reporter:Xiaoyan Qian, Pengfei Ji, Chang He, Jean-Olivier Zirimwabagabo, Michelle M. Archibald, Andrew A. Yeagley, and Jason J. Chruma
Organic Letters 2014 Volume 16(Issue 19) pp:5228-5231
Publication Date(Web):September 22, 2014
DOI:10.1021/ol502693r
A palladium-catalyzed asymmetric decarboxylative allylic alkylation of allyl 2,2-diphenylglycinate imines using (S,S)-f-binaphane as a chiral supporting ligand has been developed. This transformation allows for decarboxylative generation and enantioselective allylation of nonenolate α-imino (2-azaallyl anions) to afford α-aryl homoallylic imines.
Co-reporter:Dr. Zhe Li;Yuan-Ye Jiang;Dr. Andrew A. Yeagley;James P. Bour; Lei Liu; Jason J. Chruma; Yao Fu
Chemistry - A European Journal 2012 Volume 18( Issue 45) pp:14527-14538
Publication Date(Web):
DOI:10.1002/chem.201201425
Abstract
The Pd-catalyzed decarboxylative allylation of α-(diphenylmethylene)imino esters (1) or allyl diphenylglycinate imines (2) is an efficient method to construct new C(sp3)C(sp3) bonds. The detailed mechanism of this reaction was studied by theoretical calculations [ONIOM(B3LYP/LANL2DZ+p:PM6)] combined with experimental observations. The overall catalytic cycle was found to consist of three steps: oxidative addition, decarboxylation, and reductive allylation. The oxidative addition of 1 to [(dba)Pd(PPh3)2] (dba=dibenzylideneacetone) produces an allylpalladium cation and a carboxylate anion with a low activation barrier of +9.1 kcal mol−1. The following rate-determining decarboxylation proceeds via a solvent-exposed α-imino carboxylate anion rather than an O-ligated allylpalladium carboxylate with an activation barrier of +22.7 kcal mol−1. The 2-azaallyl anion generated by this decarboxylation attacks the face of the allyl ligand opposite to the Pd center in an outer-sphere process to produce major product 3, with a lower activation barrier than that of the minor product 4. A positive linear Hammett correlation [ρ=1.10 for the PPh3 ligand] with the observed regioselectivity (3 versus 4) supports an outer-sphere pathway for the allylation step. When Pd combined with the bis(diphenylphosphino)butane (dppb) ligand is employed as a catalyst, the decarboxylation still proceeds via the free carboxylate anion without direct assistance of the cationic Pd center. Consistent with experimental observations, electron-withdrawing substituents on 2 were calculated to have lower activation barriers for decarboxylation and, thus, accelerate the overall reaction rates.
.jpg)




![Silane,[[(2R)-4,4-dibromo-2-methyl-3-butenyl]oxy](1,1-dimethylethyl)diphenyl-](http://img.cochemist.com/ccimg/194700/194610-23-6.png)
![Silane,[[(2R)-4,4-dibromo-2-methyl-3-butenyl]oxy](1,1-dimethylethyl)diphenyl-](http://img.cochemist.com/ccimg/194700/194610-23-6_b.png)