Co-reporter:Hai-Fang Li, Yan-Xia Zhao, Zhen Yuan, Qing-Yu Liu, Zi-Yu Li, Xiao-Na Li, Chuan-Gang Ning, and Sheng-Gui He
The Journal of Physical Chemistry Letters 2017 Volume 8(Issue 3) pp:
Publication Date(Web):January 16, 2017
DOI:10.1021/acs.jpclett.6b02568
Methane activation by transition metals is of fundamental interest and practical importance, as this process is extensively involved in the natural gas conversion to fuels and value-added chemicals. While single-metal centers have been well recognized as active sites for methane activation, the active center composed of two or more metal atoms is rarely addressed and the detailed reaction mechanism remains unclear. Here, by using state-of-the-art time-of-flight mass spectrometry, cryogenic anion photoelectron imaging spectroscopy, and quantum-chemical calculations, the cooperation of the two Ta atoms in a dinuclear carbide cluster Ta2C4– for methane activation has been identified. The C–H bond activation takes place predominantly around one Ta atom in the initial stage of the reaction and the second Ta atom accepts the delivered H atom from the C–H bond cleavage. The well-resolved vibrational spectra of the cryogenically cooled anions agree well with theoretical simulations, allowing the clear characterization of the structure of Ta2C4– cluster. The reactivity comparison between Ta2C4– cluster and the carbon-less analogues (Ta2C3– and Ta2C2–) demonstrated that the cooperative effect of the two metal atoms can be well tuned by the carbon ligands in terms of methane activation and transformation.
Co-reporter:Lei Zhang;Xiang-Biao Zhang;Dan-Dan Zhang
RSC Advances (2011-Present) 2017 vol. 7(Issue 10) pp:5649-5659
Publication Date(Web):2017/01/16
DOI:10.1039/C6RA25501A
In spite of their widespread use as catalysts, 1,2,3,4,5-pentamethylcyclopentadienyl (Cp*) iridium complexes have been rarely employed in the synthesis of pyridine derivatives. Herein, we used density functional theory (DFT) calculations to predict the [Cp*Ir(OAc)]+-catalysed synthesis of 2,3-dihydropyridines, which are important starting materials for pharmaceuticals, from α,β-unsaturated oxime pivalates and alkenes. The corresponding Cp*Rh complex-catalysed processes were discussed in comparison. The simulated catalytic cycle consists of several elementary reactions, such as reversible acetate-assisted metalation–deprotonation, migratory insertion of the alkene, pivaloyl transfer, and reductive elimination. The migratory insertion of the alkene is identified as the rate-determining step, and the reductive elimination to furnish the product-ligated species makes the reaction irreversible (exergonic by about 48 kcal mol−1). The stabilities of the intermediates and the energy barrier of migratory insertion of the alkene can be affected by introducing substituent groups with different electronic characteristics into Cp* and the 2-position of α,β-unsaturated oxime pivalates, as well as by using polarised alkenes. The apparent activation energy of the reaction can be increased by increasing the electron-donating ability of the substituent group on Cp*, and by introducing electron-withdrawing groups into the terminus of alkenes. When a strong electron-donating group such as the amido group is introduced into the 2-position of α,β-unsaturated oxime pivalates, the apparent activation energy is greatly reduced so that the reaction can occur at room temperature. In contrast, changing phenyl into the highly electron-deficient p-CF3-phenyl makes the reaction more difficult. Diastereoselectivity of the reaction was examined using cyclohexylethylene as a substrate, and a method for enhancing diastereocontrol was suggested.
Co-reporter:Jiao-Jiao Chen, Zhen Yuan, Xiao-Na Li, Sheng-Gui He
International Journal of Mass Spectrometry 2017 Volume 422(Volume 422) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.ijms.2017.09.003
•A high-resolution VUV photoionization time-of-flight mass spectrometer has been newly designed.•The experimental apparatus is capable to study the generation, distribution, and the reactivity of nano-sized neutral manganese oxide clusters.•The mass resolution (m/Δm) of nano-sized neutral manganese oxide cluster Mn41O59 (∼1.3 nm) is about 7270.It is experimentally challenging to generate and detect nano-sized neutral metal oxide clusters with high-resolution mass spectrometry. Herein, a vacuum ultraviolet (VUV) photoionization time-of-flight (TOF) mass spectrometer based instrument has been home-made to study the generation, distribution, and reaction of nano-sized neutral metal oxide clusters. The VUV laser generation system has been designed to produce four 118 nm laser beams to ionize neutral metal oxide clusters in a supersonic beam with the “head-to-head” style. The TOF mass spectrometer is capable of analyzing nano-sized neutral metal oxide clusters with high mass resolution. Nano-sized neutral manganese oxide clusters MnmOn (m = 5–58; n = 8–82) have been generated and the mass resolution (m/Δm) for Mn41O59 (∼1.3 nm) is about 7270. The association products MnmOnC2H4 can be observed for the reaction of neutral manganese oxide clusters with C2H4. The rate constants for the reaction of (Mn2O3)N (N = 2–22) with C2H4 have been experimentally determined.A high-resolution VUV photoionization time-of-flight mass spectrometer has been home-made to study the generation, distribution, and reaction of nano-sized neutral metal oxide clusters. Nano-sized neutral manganese oxide clusters MnmOn have been studied and the mass resolution (m/Δm) for Mn41O59 (∼1.3 nm) is about 7270.Download high-res image (139KB)Download full-size image
Co-reporter:Mei-Qi Zhang, Yan-Xia Zhao, Qing-Yu Liu, Xiao-Na Li, and Sheng-Gui He
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:342-347
Publication Date(Web):December 12, 2016
DOI:10.1021/jacs.6b10839
Vanadium oxide cluster anions (V2O5)nVxOy– (n = 1–31; x = 0, 1; and x + y ≤ 5) with different oxygen deficiencies (Δ = 2y–1–5x = 0, ± 1, and ±2) have been prepared by laser ablation and reacted to abstract hydrogen atoms from alkane molecules (n-butane) in a fast flow reactor. When the cluster size n is less than 25, the Δ = 1 series [(V2O5)nO– clusters] that can contain atomic oxygen radical anions (O•–) generally have much higher reactivity than the other four cluster series (Δ = −2, −1, 0, and 2), indicating that each atom counts in the hydrogen-atom abstraction (HAA) reactivity. Unexpectedly, all of the five cluster series have similar HAA reactivity when the cluster size is greater than 25. The critical dimension of vanadia particles separating the cluster behavior (each atom counts) from the bulk behavior (each atom contributes a little part) is thus about 1.6 nm (∼V50O125). The strong electron–phonon coupling of the vanadia particles has been proposed to create the O•– radicals (V5+ = O2–+ heat → V4+–O•–) for the n > 25 clusters with Δ = −2, −1, 0, and 2. Such a mechanism is supported by a comparative study with the scandium system [(Sc2O3)nScxOy– (n = 1–29; x = 0, 1; and x + y ≤ 4)] for which the Δ = 1 series [(Sc2O3)nO– clusters] always have much higher HAA reactivity than the other cluster series.
Co-reporter:Ya-Ke Li, Zhen Yuan, Yan-Xia Zhao, Chongyang Zhao, Qing-Yu Liu, Hui Chen, and Sheng-Gui He
Journal of the American Chemical Society 2016 Volume 138(Issue 39) pp:12854-12860
Publication Date(Web):September 8, 2016
DOI:10.1021/jacs.6b05454
Laser ablation generated RhAl3O4+ heteronuclear metal oxide cluster cations have been mass-selected using a quadrupole mass filter and reacted with CH4 or CD4 in a linear ion trap reactor under thermal collision conditions. The reactions have been characterized by state-of-the-art mass spectrometry and quantum chemistry calculations. The RhAl3O4+ cluster can activate four C–H bonds of a methane molecule and convert methane to syngas, an important intermediate product in methane conversion to value-added chemicals. The Rh atom is the active site for activation of the C–H bonds of methane. The high electron-withdrawing capability of Rh atom is the driving force to promote the conversion of methane to syngas. The polarity of Rh oxidation state is changed from positive to negative after the reaction. This study has provided the first example of methane conversion to syngas by heteronuclear metal oxide clusters under thermal collision conditions. Furthermore, the molecular level origin has been revealed for the condensed-phase experimental observation that trace amounts of Rh can promote the participation of lattice oxygen of chemically very inert support (Al2O3) to oxidize methane to carbon monoxide.
Co-reporter:Zi-Yu Li; Hai-Fang Li; Yan-Xia Zhao
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9437-9443
Publication Date(Web):July 6, 2016
DOI:10.1021/jacs.6b03940
Gold in the +III oxidation state (AuIII) has been proposed as a promising species to mediate challenging chemical reactions. However, it is difficult to characterize the chemistry of individual AuIII species in condensed-phase systems mainly due to the interference from the AuI counterpart. Herein, by doping Au atoms into gas-phase vanadium oxide clusters, we demonstrate that the AuIII cation in the AuV2O6+ cluster is active for activation and transformation of methane, the most stable alkane molecule, into formaldehyde under mild conditions. In contrast, the AuV2O6+ cluster isomers with the AuI cation can only absorb CH4. The clusters were generated by laser ablation and mass selected to react with CH4, CD4, or CH2D2 in an ion trap reactor. The reactivity was characterized by mass spectrometry and quantum chemistry calculations. The structures of the reactant and product ions were identified by using collision-induced and 425 nm photo-induced dissociation techniques.
Co-reporter:Yan-Xia Zhao, Xiao-Na Li, Zhen Yuan, Qing-Yu Liu, Qiang Shi and Sheng-Gui He
Chemical Science 2016 vol. 7(Issue 7) pp:4730-4735
Publication Date(Web):29 Mar 2016
DOI:10.1039/C6SC00539J
The reactivity of closed-shell gas phase cluster anions AuTi3O7− and AuTi3O8− with methane under thermal collision conditions was studied by mass spectrometric experiments and quantum chemical calculations. Methane activation was observed with the formation of AuCH3 in both cases, while the formation of formaldehyde was also identified in the reaction system of AuTi3O8−. The cooperative effect of the separated Au+ and O2− ions on the clusters induces the cleavage of the first C–H bond of methane. Further activation of the second C–H bond by a peroxide ion O22− leads to the formation of formaldehyde. This study shows that closed-shell species on metal oxides can be reactive enough to facilitate thermal H–CH3 bond cleavage and the subsequent conversion.
Co-reporter:Yan-Xia Zhao, Qing-Yu Liu, Mei-Qi Zhang and Sheng-Gui He
Dalton Transactions 2016 vol. 45(Issue 28) pp:11471-11495
Publication Date(Web):13 Jun 2016
DOI:10.1039/C6DT01246A
The study of gas phase ion–molecule reactions by state-of-the-art mass spectrometric experiments in conjunction with quantum chemistry calculations offers an opportunity to clarify the elementary steps and mechanistic details of bond activation and conversion processes. In the past few decades, a considerable number of publications have been devoted to the ion–molecule reactions of metal clusters, the experimentally and theoretically tractable models for the active phase of condensed phase systems. The focus of this perspective concerns progress on activation and transformation of important inorganic and organic molecules by negatively charged metal clusters. The metal cluster anions cover bare metal clusters as well as ligated systems with oxygen, carbon, and nitrogen, among others. The following important issues have been summarized and discussed: (i) dependence of chemical reactivity and selectivity on cluster structures and sizes, metals and metal oxidation states, odd–even electron numbers, etc. and (ii) effects of doping, ligation, and pre-adsorption on the reactivity of metal clusters toward rather inert molecules.
Co-reporter:Jia-Bi Ma, Lin-Lin Xu, Jing-Heng Meng, Sheng-Gui He
International Journal of Mass Spectrometry 2016 Volume 401() pp:39-45
Publication Date(Web):25 April 2016
DOI:10.1016/j.ijms.2016.01.008
•A combination of mass spectrometry and quantum chemical calculations indicates that CeVO4+ clusters with closed-shell electronic structures are reactive toward C3H6.•The formation of CeVO3C3H4+/H2O and CeVO4C3H6+ are observed.•Compared with another closed-shell cluster V3O7+, possessing the similar mass with CeVO4+, the heteronuclear oxide cluster cations show higher reactivity toward propylene.Laser-ablation-generated cerium-vanadium oxide cluster cations CeVO4+ were mass-selected by applying a quadrupole mass filter, and reacted with propylene in a linear ion trap reactor at ambient conditions. Time-of-flight mass spectrometry and density functional theory calculations were used to characterize the cluster reactions. The CeVO4+ cluster has a closed-shell electronic structure, and is reactive toward propylene leading to the formation of CeVO3C3H4+/H2O and CeVO4C3H6+. The computed results indicate that this reaction involves the occurrence of spin conversions (singlet → triplet), and a new mechanism of dehydrogenation of alkenes, which is different from the reported ones, was proposed. Compared with another closed-shell cluster V3O7+, possessing the similar mass with CeVO4+, the heteronuclear oxide cluster cations show higher reactivity toward propylene. The computational results are in good agreement with the experimental observations. This work provides insights into nature of active sites and reaction mechanisms in related condensed-phase systems at a molecular level.
Co-reporter:Zhen Yuan, Qing-Yu Liu, Xiao-Na Li, Sheng-Gui He
International Journal of Mass Spectrometry 2016 Volume 407() pp:62-68
Publication Date(Web):20 August 2016
DOI:10.1016/j.ijms.2016.07.004
•The high-resolution tandem time-of-flight mass spectrometer has been home-made.•The experimental apparatus is capable to perform the photoreactivity study of nano-sized vanadium oxide clusters.•The maximum mass resolution is up to about m/Δm = 14175 for V62O155 – (∼1.8 nm).It is experimentally challenging to generate nano-sized metal oxide clusters with significant intensity as well as to resolve a single hydrogen atom by mass spectrometry (TOF-MS). Herein, the high-resolution tandem time-of-flight mass spectrometer is home-made and the experimental apparatus is capable to perform the reactivity study of nano-sized vanadium oxide clusters. The formation and distribution of nano-sized vanadium oxide cluster anions VxOy− (x up to 70) were analyzed and the photoreaction of alkene attached complex V40O101C2H4− (or V40O101C3H6−) was studied. Larger VxOy− cluster anions can be generated typically under higher O2 concentration and the intensity proportion (%) of vanadium oxide cluster with a certain oxygen-deficiency value Δ (Δ= 2y-5x-1 for VxOy−) in each Vx series is nearly invariant. In photoreaction of V40O101C2H4− (or V40O101C3H6−), the transfer of two oxygen atoms from V40O101− to C2H4 (or C3H6) and the generation of V40O99− dominants the channel. The mass resolution of the primary TOF-MS for V40O101− (∼1.5 nm) is up to 13002. The mass resolution of the secondary TOF-MS for photoreaction is calculated to be about 4530 to resolve parent (V40O101C2H4−) and daughter ions (V40O99−).Highlights The maximum mass resolution of our home-made tandem time-of-flight mass spectrometer is up to about m/Δm = 14175 for V62O155− (∼1.8 nm) and this resolution is high enough to resolve a hydrogen atom.
Co-reporter:Qing-Yu Liu
The Journal of Physical Chemistry C 2016 Volume 120(Issue 30) pp:17081-17086
Publication Date(Web):July 1, 2016
DOI:10.1021/acs.jpcc.6b05053
Photoinduced reactions of ethene (C2H4) bound to vanadium oxide nanoparticles (V2O5)nO– (n = 2–20) have been studied via a high-resolution tandem time-of-flight mass spectrometer coupled with a nanosecond ultraviolet laser system. Two major reaction channels, C2H4O2 elimination and photoelectron detachment, have been observed for all the examined cluster systems. As the cluster size increases, the efficiency of photoreaction increases. Kinetic analysis demonstrates that the C2H4O2 is generated by a single-photon absorption process (V2O5)nOC2H4– + hυ (first photon) → (V2O5)n-1V2O4– + C2H4O2, which is followed by the photoelectron detachment (V2O5)n-1V2O4– + hυ (second photon) → (V2O5)n-1V2O4 + e–. The fast dissociation to generate C2H4O2 within about 5 ns after absorption of a single photon demonstrates that the electron instead of phonon should be the energy carrier for photoreaction on the studied semiconducting nanoparticles as large as V40O101 (diameter around 1.5 nm).
Co-reporter:Ting Zhang, Zi-Yu Li, Mei-Qi Zhang, and Sheng-Gui He
The Journal of Physical Chemistry A 2016 Volume 120(Issue 25) pp:4285-4293
Publication Date(Web):June 6, 2016
DOI:10.1021/acs.jpca.6b03836
To develop proper ionization methods for alkanes, the reactivity of bare or ligated transition metal ions toward alkanes has attracted increasing interests. In this study, the reactions of the gold cations with linear alkanes from ethane up to nonane (CnH2n+2, n = 2–9) under mild conditions have been characterized by mass spectrometry and density functional theory calculations. When reacting with Au+, small alkanes (n = 2–6) were confirmed to follow specific reaction channels of dehydrogenation for ethane and hydride transfer for others to generate product ions characteristic of the original alkanes, which indicates that Au+ can act as a reagent ion to ionize alkanes from ethane to n-hexane. Strong dependence of the chain length of alkanes was observed for the rate constants and reaction efficiencies. Extensive fragmentation took place for larger alkanes (n > 6). Theoretical results show that the fragmentation induced by the hydride transfer occurs after the release of AuH. Moreover, the fragmentation of n-heptane was successfully avoided when the reaction took place in a high-pressure reactor. This implies that Au+ is a potential reagent ion to ionize linear and even the branched alkanes.
Co-reporter:Dr. Xiao-Na Li;Dr. Zi-Yu Li;Hai-Fang Li;Dr. Sheng-Gui He
Chemistry - A European Journal 2016 Volume 22( Issue 26) pp:9024-9029
Publication Date(Web):
DOI:10.1002/chem.201600451
Abstract
Time-of-flight mass spectrometry experiments demonstrated that laser ablation generated and mass selected Au2TiO4− cluster anions can unexpectedly oxidize three CO molecules in an ion trap reactor. This is an improvement on the more commonly observed oxidation of at most two CO molecules by a doped cluster. Quantum chemistry calculations were performed to rationalize the reactions. The lowest energy isomer of Au2TiO4− contains a superoxide unit, the participation of which in CO oxidation can be promoted by the Au2 dimer. The Au2 dimer functions as a rather flexible electron reservoir in each CO oxidation step in terms of the release and storage of electrons to drive the dissociation of superoxide to peroxide and then to lattice oxygen atoms, which can be removed by reaction with CO molecules. This gas-phase study enriches our understanding toward the nature of reactive oxygen species involved in gold-catalyzed oxidation reactions.
Co-reporter:Zheng Meng; Ying Han; Li-Na Wang; Jun-Feng Xiang; Sheng-Gui He;Chuan-Feng Chen
Journal of the American Chemical Society 2015 Volume 137(Issue 30) pp:9739-9745
Publication Date(Web):July 17, 2015
DOI:10.1021/jacs.5b05758
The motions of biomolecular machines are usually multistep processes, and are involved in a series of conformational changes. In this paper, a novel triply interlocked [2](3)catenane composed of a tris(crown ether) host eTC and a circular ditopic guest with three dibenzyl ammonium (DBA) sites and three N-methyltriazolium (MTA) sites was reported. Due to the multivalency nature of the catenane, the acid–base triggered motion was performed by a stepwise manner. The coconformations of the four related stable states have been directly identified and quantified which confirmed the multistep process. In order to quantify the dynamics with environmental acidity changes, the values of the three levels of dissociation constant pKa have been determined. The special interlocked topology of the [2](3)catenane also endows the motion of each crown ether ring in the host with unexpected selectivity for the MTA sites. This study provides clues to comprehend the underlying motion mechanism of intricate biological molecular machines, and further design artificial molecular machine with excellent mechanochemistry properties.
Co-reporter:Jia-Bi Ma, Jing-Heng Meng and Sheng-Gui He
Dalton Transactions 2015 vol. 44(Issue 7) pp:3128-3135
Publication Date(Web):22 Dec 2014
DOI:10.1039/C4DT03398A
Cerium–vanadium oxide cluster cations CeVO5+ were generated by laser ablation, mass-selected using a quadrupole mass filter, thermalized through collisions with helium atoms, and then reacted with ethene molecules in a linear ion trap reactor. The cluster reactions have been characterized by time-of-flight mass spectrometry and density functional theory calculations. The CeVO5+ cluster has a closed-shell electronic structure and contains a peroxide (O22−) unit. The cluster bonded O22− species is reactive enough to oxidize a C2H4 molecule to generate C2H4O2 that can be an acetic acid molecule. Atomic oxygen radicals (O−˙), superoxide radicals (O2−˙), and peroxides are the three common reactive oxygen species. The reactivity of cluster bonded O−˙ and O2−˙ radicals has been widely studied while the O22− species were generally thought to be much less reactive or inert toward small molecules under thermal collision conditions. This work is among the first to report the reactivity of the peroxide unit on transition metal oxide clusters with hydrocarbon molecules, to the best of our knowledge.
Co-reporter:Ya-Ke Li, Jing-Heng Meng, Sheng-Gui He
International Journal of Mass Spectrometry 2015 Volumes 381–382() pp:10-16
Publication Date(Web):1 May 2015
DOI:10.1016/j.ijms.2015.02.001
•The generation of free H atoms during thermal reaction of simple alkane molecules has been identified.•The AuNbO3+ ion serves as the first hetero-nuclear oxide cluster species to activate hydrocarbons to produce the free H atoms.•The conversion of one AuO bond into one AuNb bond drives the H-atom generation.Single gold-atom doped niobium oxide cluster AuNbO3+ has been prepared and mass-selected to react with C2H6 in an ion trap reactor under thermal collision conditions. The reaction has been characterized by mass spectrometry with isotopic substitution (C2H6 → CH3CD3 and C2D6) and density functional theory calculations. In addition to hydrogen atom abstraction channels identified in a previous fast-flow reactor experiment, a rather unexpected channel of generating the free H atom was observed. The computational study revealed that the gold atom plays an important role in the reaction and the conversion of one AuO bond to one AuNb bond drives the generation of the free H atom. This study enriches the chemistry of gold atoms doped in atomic clusters and provides an ideal model reaction to generate the free H atom that can be a very important intermediate in hydrocarbon transformation.
Co-reporter:Jing-Heng Meng, Qing-Yu Liu, and Sheng-Gui He
The Journal of Physical Chemistry A 2015 Volume 119(Issue 46) pp:11265-11270
Publication Date(Web):October 28, 2015
DOI:10.1021/acs.jpca.5b05122
Laser ablation generated AuC3H– anions were skimmed into a time-of-flight mass spectrometer (TOF-MS) and selected with a mass gate. Photoelectron spectra of AuC3H– were recorded using the velocity map imaging technique at several photon energies. The experimental spectra, quantum chemistry calculations, and Franck–Condon simulations suggest that the AuC3H– cluster has four structure isomers, including one unexpected structure of [C═C═C–Au–H]−. When AuC3H– is compared with C3H2–, introduction of gold into the hydrocarbon system results in the much lower isomerization barriers.
Co-reporter:Hai-Fang Li; Zi-Yu Li; Qing-Yu Liu; Xiao-Na Li; Yan-Xia Zhao
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 12) pp:2287-2291
Publication Date(Web):June 3, 2015
DOI:10.1021/acs.jpclett.5b00937
Laser-ablation-generated and mass-selected iron-carbide cluster anions FeC6– were reacted with CH4 in a linear ion trap reactor under thermal collision conditions. The reactions were characterized by mass spectrometry and density functional theory calculations. Adsorption product of FeC6CH4– was observed in the experiments. The identified large kinetic isotope effect suggests that CH4 can be activated by FeC6– anions with a dissociative adsorption manner, which is further supported by the reaction mechanism calculations. The large dipole moment of FeC6– (19.21 D) can induce a polarization of CH4 and can facilitate the cleavage of C–H bond. This study reports the CH4 activation by transition-metal carbide anions, which provides insights into mechanistic understanding of iron–carbon centers that are important for condensed-phase catalysis.
Co-reporter:Li-Na Wang;Zi-Yu Li;Qing-Yu Liu;Jing-Heng Meng; Sheng-Gui He; Tong-Mei Ma
Angewandte Chemie International Edition 2015 Volume 54( Issue 40) pp:11720-11724
Publication Date(Web):
DOI:10.1002/anie.201505476
Abstract
Investigations on the reactivity of atomic clusters have led to the identification of the elementary steps involved in catalytic CO oxidation, a prototypical reaction in heterogeneous catalysis. The atomic oxygen species O.− and O2− bonded to early-transition-metal oxide clusters have been shown to oxidize CO. This study reports that when an Au2 dimer is incorporated within the cluster, the molecular oxygen species O22− bonded to vanadium can be activated to oxidize CO under thermal collision conditions. The gold dimer was doped into Au2VO4− cluster ions which then reacted with CO in an ion-trap reactor to produce Au2VO3− and then Au2VO2−. The dynamic nature of gold in terms of electron storage and release promotes CO oxidation and OO bond reduction. The oxidation of CO by atomic clusters in this study parallels similar behavior reported for the oxidation of CO by supported gold catalysts.
Co-reporter:Li-Na Wang;Zi-Yu Li;Qing-Yu Liu;Jing-Heng Meng; Sheng-Gui He; Tong-Mei Ma
Angewandte Chemie 2015 Volume 127( Issue 40) pp:11886-11890
Publication Date(Web):
DOI:10.1002/ange.201505476
Abstract
Investigations on the reactivity of atomic clusters have led to the identification of the elementary steps involved in catalytic CO oxidation, a prototypical reaction in heterogeneous catalysis. The atomic oxygen species O.− and O2− bonded to early-transition-metal oxide clusters have been shown to oxidize CO. This study reports that when an Au2 dimer is incorporated within the cluster, the molecular oxygen species O22− bonded to vanadium can be activated to oxidize CO under thermal collision conditions. The gold dimer was doped into Au2VO4− cluster ions which then reacted with CO in an ion-trap reactor to produce Au2VO3− and then Au2VO2−. The dynamic nature of gold in terms of electron storage and release promotes CO oxidation and OO bond reduction. The oxidation of CO by atomic clusters in this study parallels similar behavior reported for the oxidation of CO by supported gold catalysts.
Co-reporter:Xiao-Na Li
The Journal of Physical Chemistry C 2015 Volume 119(Issue 27) pp:15414-15420
Publication Date(Web):June 8, 2015
DOI:10.1021/acs.jpcc.5b04218
Single platinum-atom catalysts exhibit extraordinary activity in a large number of reactions. However, a consensus regarding the molecular origin of Pt catalysis is far from being reached. Here, benefiting from the study of atomic clusters, we propose the Electronegativity-Ladder (E-Ladder) effect to account for the origin of Pt catalysis. The concept was obtained from the study of single Pt-atom doped aluminum oxide clusters PtAl3O5–7–, which are catalytically active in CO oxidation by molecular O2. The undoped aluminum oxide clusters, however, cannot drive such a catalytic cycle. The reactions have been identified by mass spectrometry and density functional theory calculations. The key to drive the cycle lies in the unique structure of PtAl3O6–, in which the Pt atom that is not fully oxidized can coexist with the highly oxidative oxygen-centered radical (O–•). After the oxidation of one CO by PtAl3O7–, the resulting PtAl3O6– can also oxidize a second CO. The E-Ladder effect originates from the well-fitting electronegativity of the Pt atom (2.28) in between that of the Al atom (1.61) and the O atom (3.44), and this effect promotes the generation of an unpaired electron localized O–• radical, which results in the oxidative nature of PtAl3O6– toward CO. Thus, the large enthalpy in the catalytic reaction (2CO + O2 → 2CO2) can be distributed much more evenly into several elementary reactions in the Pt–Al–O system than in the pure Al–O system.
Co-reporter:Xiao-Na Li ; Zhen Yuan
Journal of the American Chemical Society 2014 Volume 136(Issue 9) pp:3617-3623
Publication Date(Web):February 14, 2014
DOI:10.1021/ja412608b
Laser ablation generated Aux(TiO2)yOz– (x = 0, 1; y = 2, 3; z = 1, 2) oxide cluster anions have been mass-selected using a quadrupole mass filter and reacted with CO in a hexapole collision cell. The reactions have been characterized by time-of-flight mass spectrometry and density functional theory calculations. Gold–titanium bimetallic oxide clusters Au(TiO2)yOz– are more reactive in CO oxidation than pure titanium oxide clusters (TiO2)yOz–. The computational studies identify the dual roles that the gold atom plays in CO oxidation: functioning as a CO trapper and electron acceptor. Both factors are important for the high reactivity of Au(TiO2)yOz– clusters. To the best of our knowledge, this is the first example of CO oxidation by gold-containing heteronuclear oxide clusters, which provides molecular-level insights into the roles of gold in CO oxidation over oxide supports.
Co-reporter:Zi-Yu Li ; Zhen Yuan ; Xiao-Na Li ; Yan-Xia Zhao
Journal of the American Chemical Society 2014 Volume 136(Issue 40) pp:14307-14313
Publication Date(Web):September 12, 2014
DOI:10.1021/ja508547z
The single gold atom doped aluminum oxide clusters AuAl3O3+, AuAl3O4+, and AuAl3O5+ have been prepared and mass-selected to react with CO, O2, and mixtures of CO and O2 in an ion trap reactor under thermal collision conditions. The reactions have been characterized by mass spectrometry with isotopic substitution (16O2 → 18O2) and density functional theory calculations. The AuAl3O5+ cluster can oxidize two CO molecules consecutively to form AuAl3O4+ and then AuAl3O3+, the latter of which can react with one O2 molecule to regenerate AuAl3O5+. The AuAl316O3+ ions interact with a mixture of C16O and 18O2 to produce the fully substituted 18O species AuAl318O3–5+, which firmly identifies a catalytic cycle for CO oxidation by O2. The oxidation catalysis is driven by electron cycling primarily through making and breaking a gold–aluminum chemical bond. To the best of our knowledge, this is the first identification of catalytic CO oxidation by O2 mediated with gas-phase cluster catalysts with single-noble-metal atoms, which serves as an important step to understand single-atom catalysis at strictly a molecular level.
Co-reporter:Dr. Yan-Xia Zhao;Zi-Yu Li;Zhen Yuan;Dr. Xiao-Na Li ;Dr. Sheng-Gui He
Angewandte Chemie 2014 Volume 126( Issue 36) pp:9636-9640
Publication Date(Web):
DOI:10.1002/ange.201403953
Abstract
Identification and mechanistic study of thermal methane conversion mediated by gas-phase species is important for finding potentially useful routes for direct methane transformation under mild conditions. Negatively charged oxide species are usually inert with methane. This work reports an unexpected result that the bi-metallic oxide cluster anions PtAl2O4− can transform methane into a stable organic compound, formaldehyde, with high selectivity. The clusters are prepared by laser ablation and reacted with CH4 in an ion trap reactor. The reaction is characterized by mass spectrometry and density functional theory calculations. It is found that platinum rather than oxygen activates CH4 at the beginning of the reaction. The Al2O4− moiety serves as the support of Pt atom and plays important roles in the late stage of the reaction. A new mechanism for selective methane conversion is provided and new insights into the surface chemistry of single Pt atoms may be obtained from this study.
Co-reporter:Dr. Yan-Xia Zhao;Zi-Yu Li;Zhen Yuan;Dr. Xiao-Na Li ;Dr. Sheng-Gui He
Angewandte Chemie International Edition 2014 Volume 53( Issue 36) pp:9482-9486
Publication Date(Web):
DOI:10.1002/anie.201403953
Abstract
Identification and mechanistic study of thermal methane conversion mediated by gas-phase species is important for finding potentially useful routes for direct methane transformation under mild conditions. Negatively charged oxide species are usually inert with methane. This work reports an unexpected result that the bi-metallic oxide cluster anions PtAl2O4− can transform methane into a stable organic compound, formaldehyde, with high selectivity. The clusters are prepared by laser ablation and reacted with CH4 in an ion trap reactor. The reaction is characterized by mass spectrometry and density functional theory calculations. It is found that platinum rather than oxygen activates CH4 at the beginning of the reaction. The Al2O4− moiety serves as the support of Pt atom and plays important roles in the late stage of the reaction. A new mechanism for selective methane conversion is provided and new insights into the surface chemistry of single Pt atoms may be obtained from this study.
Co-reporter:Mei-Ye Jia, Bo Xu, Ke Deng, Sheng-Gui He, and Mao-Fa Ge
The Journal of Physical Chemistry A 2014 Volume 118(Issue 37) pp:8106-8114
Publication Date(Web):January 3, 2014
DOI:10.1021/jp411961q
Vanadium oxide cluster anions Vm16On– and Vm18On– were prepared by laser ablation and reacted with hydrogen sulfide (H2S) in a fast flow reactor under thermal collision conditions. A time-of-flight mass spectrometer was used to detect the cluster distributions before and after the interactions with H2S. The experiments suggest that the oxygen-for-sulfur (O/S) exchange reaction to release water was evidenced in the reactor for most of the cluster anions: VmOn– + H2S → VmOn–1S– + H2O. For reactions of clusters VO3– and VO4– with H2S, consecutive O/S exchange reactions led to the generation of sulfur containing vanadium oxide cluster anions VO3–kSk– (k = 1–3) and VO4–kSk– (k = 1–4). Density functional theory calculations were performed for the reactions of VO3–4– with H2S, and the results indicate that the O/S exchange reactions are both thermodynamically and kinetically favorable, which supports the experimental observations. The reactions of VmOn+ cluster cations with H2S have been reported previously (Jia, M.-Y.; Xu, B.; Ding, X.-L.; Zhao, Y.-X.; He, S.-G.; Ge, M.-F. J. Phys. Chem. C 2012, 116, 9043), and this study of cluster anions provides further new insights into the transformations of H2S over vanadium oxides at the molecular level.
Co-reporter:Dr. Jia-Bi Ma;Zhen Yuan;Jing-Heng Meng;Qing-Yu Liu; Dr. Sheng-Gui He
ChemPhysChem 2014 Volume 15( Issue 18) pp:4117-4125
Publication Date(Web):
DOI:10.1002/cphc.201402347
Abstract
The reactivity of metal oxide clusters toward hydrocarbon molecules can be changed, tuned, or controlled by doping. Cerium-doped vanadium cluster cations CeV2O7+ are generated by laser ablation, mass-selected by a quadrupole mass filter, and then reacted with C2H4 in a linear ion trap reactor. The reaction is characterized by a reflectron time-of-flight mass spectrometer. Three types of reaction channels are observed: 1) single oxygen-atom transfer , 2) double oxygen-atom transfer , and 3) CC bond cleavage. This study provides the first bimetallic oxide cluster ion, CeV2O7+, which gives rise to CC bond cleavage of ethene. Neither CexOy± nor VxOy± alone possess the necessary topological and electronic properties to bring about such a reaction.
Co-reporter:Zhen Yuan ; Zi-Yu Li ; Zhen-Xun Zhou ; Qing-Yu Liu ; Yan-Xia Zhao
The Journal of Physical Chemistry C 2014 Volume 118(Issue 27) pp:14967-14976
Publication Date(Web):June 18, 2014
DOI:10.1021/jp5040344
Vanadium oxide cluster ions are prepared by laser ablation and the (V2O5)nO– (n = 1–3) clusters (V2O6–, V4O11–, and V6O16–) are selected by a quadrupole mass filter and interacted with ethylene (C2H4) and propylene (C3H6) in an ion trap reactor. Molecular association (MA) is observed for all of the reactions and oxygen atom transfer (OAT) is also observed for the V4O11– and V6O16– cluster systems. The branching ratio of the OAT versus the MA channel increases as the size of (V2O5)nO– increases and the OAT becomes the major channel in V6O16– + C2H4 and V6O16– + C3H6. Density functional theory computations are carried out for the reactions of V2O6– and V4O11– with C2H4 and the results are consistent with the experimental observations. Anionic metal oxide clusters are usually much less reactive than their cationic and neutral counterparts and this work is among the first to identify the OAT reactivity for cluster anions with hydrocarbon molecules under thermal collision conditions, to the best of our knowledge.
Co-reporter:Bo Xu ; Jing-Heng Meng
The Journal of Physical Chemistry C 2014 Volume 118(Issue 32) pp:18488-18495
Publication Date(Web):July 21, 2014
DOI:10.1021/jp5009187
VO2(V2O5)nCH3OH+ (n = 1–3) complexes are generated from the interactions of pregenerated VO2(V2O5)n+ clusters with CH3OH in a fast flow reactor. The photoreactions of these three adsorption complexes at 355 nm are characterized by a high-resolution tandem time-of-flight mass spectrometer. The major photoproducts of V3O7CH3OH+ are CH2O and V3O7H2+, whereas those of V5O12CH3OH+ and V7O17CH3OH+ are CH3OH and the corresponding VxOy+ (x, y = 5,12 and 7,17). Collision-induced dissociation and density functional theory/RRKM calculations suggest that CH3OH is dissociatively adsorbed on V3O7+, whereas it is nondissociatively adsorbed on V5O12+ and V7O17+. This study suggests that the thermal dissociation of CH3OH to CH3O is the prerequisite for its photo-oxidation into CH2O, which may shed light on the photochemistry of methanol over transition metal oxide surfaces.
Co-reporter:Jing-Heng Meng and Sheng-Gui He
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 21) pp:3890-3894
Publication Date(Web):October 22, 2014
DOI:10.1021/jz502057n
Laser-ablation-generated AuCeO2+ and CeO2+ oxide clusters were mass-selected using a quadrupole mass filter and reacted with H2 in an ion trap reactor at ambient conditions. The reactions were characterized by mass spectrometry and density functional theory calculations. The gold–cerium bimetallic oxide cluster AuCeO2+ is more reactive in H2 activation than the pure cerium oxide cluster CeO2+. The gold atom is the active adsorption site and facilitates the heterolytic cleavage of H2 in collaboration with the separated O2– ion of the CeO2 support. To the best of our knowledge, this is the first example of thermal H2 activation by a closed-shell atomic cluster, which provides molecular-level insights into the single gold atom catalysis over metal oxide supports.Keywords: atomic clusters; density functional calculations; dihydrogen activation; gold; mass spectrometry;
Co-reporter:Zhen Yuan, Xiao-Na Li, and Sheng-Gui He
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 9) pp:1585-1590
Publication Date(Web):April 15, 2014
DOI:10.1021/jz500509j
Time-of-flight mass spectrometry experiment shows that upon the interactions with carbon monoxide, the mass-selected AuFeO3– oxide cluster anions can evaporate neutral gold atoms in a hexapole collision cell and oxidize CO into CO2 in an ion trap reactor. The computational studies identify that the gold atom is loosely attached in the AuFeO3– cluster, and the different reaction channels can be attributed to different cluster velocities. The structure of the AuFeO3– cluster is very flexible, and the approach of CO induces significant geometrical and electronic structure changes of AuFeO3–, which facilitates the exposure of the positively charged gold atom to trap and oxidize CO. The CO oxidation by the AuFeO3– cluster follows the Au-assisted Mars–van Krevelen mechanism, in which the direct participation of the surface lattice oxygen (O2–) is proposed.Keywords: atomic clusters; CO oxidation; density functional calculations; gold; mass spectrometry;
Co-reporter:Mei-Ye Jia, Zhixun Luo, Sheng-Gui He, and Mao-Fa Ge
The Journal of Physical Chemistry A 2014 Volume 118(Issue 37) pp:8163-8169
Publication Date(Web):February 20, 2014
DOI:10.1021/jp500837g
We present here a study of gas-phase reactivity of cobalt sulfide cluster anions ComSn– with molecular oxygen. Nascent ComSn– clusters were prepared via a laser ablation source and reacted with oxygen in a fast flow reactor under thermal collision conditions. We chose 18O2 in place of 16O2 to avoid mass degeneration with sulfur, and a time-of-flight (TOF) mass spectrometer was used to detect the cluster distributions in the absence and presence of the reactant. It was found that oxygen–sulfur exchange occurs in the reactions for those with specific compositions (CoS)n– and (CoS)nS– (n = 2–5) according to a consistent pathway, “ComSn– + 18O2 → ComSn–118O– + S18O”. Typically, for “Co2S2– + 18O2” we have calculated the reaction coordinates by employing the density functional theory (DFT), where both the oxygen–sulfur exchange and SO molecule release are thermodynamically and kinetically favorable. It is noteworthy that the reaction with molecular oxygen (triplet ground state) needs to overcome a spin excitation as well as a large O–O activation energy. This study sheds light on the activation of molecular oxygen by cobalt sulfides on one hand and also provides insight into the regeneration mechanism of cobalt oxides from the counterpart sulfides in the presence of oxygen gas on the other hand.
Co-reporter:Li-Hua Tian ;Jing-Heng Meng ;Dr. Xiao-Nan Wu;Dr. Yan-Xia Zhao;Dr. Xun-Lei Ding;Dr. Sheng-Gui He;Dr. Tong-Mei Ma
Chemistry - A European Journal 2014 Volume 20( Issue 4) pp:1167-1175
Publication Date(Web):
DOI:10.1002/chem.201302705
Abstract
The activation of CH bonds in alkanes is currently a hot research topic in chemistry. The atomic oxygen radical anion (O−.) is an important species in CH activation. The mechanistic details of CH activation by O−. radicals can be well understood by studying the reactions between O−. containing transition metal oxide clusters and alkanes. Here the reactivity of scandium oxide cluster anions toward n-butane was studied by using a high-resolution time-of-flight mass spectrometer coupled with a fast flow reactor. Hydrogen atom abstraction (HAA) from n-butane by (Sc2O3)NO− (N=1–18) clusters was observed. The reactivity of (Sc2O3)NO− (N=1–18) clusters is significantly sizedependent and the highest reactivity was observed for N=4 (Sc8O13−) and 12 (Sc24O37−). Larger (Sc2O3)NO− clusters generally have higher reactivity than the smaller ones. Density functional theory calculations were performed to interpret the reactivity of (Sc2O3)NO− (N=1–5) clusters, which were found to contain the O−. radicals as the active sites. The local charge environment around the O−. radicals was demonstrated to control the experimentally observed size-dependent reactivity. This work is among the first to report HAA reactivity of cluster anions with dimensions up to nanosize toward alkane molecules. The anionic O−. containing scandium oxide clusters are found to be more reactive than the corresponding cationic ones in the CH bond activation.
Co-reporter:Zi-Yu Li;Zhen Yuan;Dr. Yan-Xia Zhao;Dr. Sheng-Gui He
Chemistry - A European Journal 2014 Volume 20( Issue 14) pp:4163-4169
Publication Date(Web):
DOI:10.1002/chem.201304042
Abstract
Metal carbide species have been proposed as a new type of chemical entity to activate methane in both gas-phase and condensed-phase studies. Herein, methane activation by the diatomic cation MoC+ is presented. MoC+ ions have been prepared and mass-selected by a quadrupole mass filter and then allowed to interact with methane in a hexapole reaction cell. The reactant and product ions have been detected by a reflectron time-of-flight mass spectrometer. Bare metal Mo+ and MoC2H2+ ions have been observed as products, suggesting the occurrence of ethylene elimination and dehydrogenation reactions. The branching ratio of the C2H4 elimination channel is much larger than that of the dehydrogenation channel. Density functional theory calculations have been performed to explore in detail the mechanism of the reaction of MoC+ with CH4. The computed results indicate that the ethylene elimination process involves the occurrence of spin conversions in the CC coupling (doubletquartet) and hydrogen atom transfer (quartetsextet) steps. The carbon atom in MoC+ plays a key role in methane activation because it becomes sp3 hybridized in the initial stages of the ethylene elimination reaction, which leads to much lower energy barriers and more stable intermediates. This study provides insights into the CH bond activation and CC coupling involved in methane transformation over molybdenum carbide-based catalysts.
Co-reporter:Jing-Heng Meng;Xiao-Jiao Deng;Zi-Yu Li;Dr. Sheng-Gui He;Dr. Wei-Jun Zheng
Chemistry - A European Journal 2014 Volume 20( Issue 19) pp:5580-5583
Publication Date(Web):
DOI:10.1002/chem.201400218
Abstract
The first example of a metal oxide cluster anion, La6O10− that can activate methane under ambient conditions is reported. This reaction is facilitated by the oxygen-centered radical (O−⋅) and follows the hydrogen atom transfer mechanism. The La6O10− has a high vertical electron detachment energy (VDE=4.06 eV) and a high symmetry (C4v).
Co-reporter:Jia-Bi Ma ; Bo Xu ; Jing-Heng Meng ; Xiao-Nan Wu ; Xun-Lei Ding ; Xiao-Na Li
Journal of the American Chemical Society 2013 Volume 135(Issue 8) pp:2991-2998
Publication Date(Web):January 31, 2013
DOI:10.1021/ja311695t
Titanium and zirconium oxide cluster anions with dimensions up to nanosize are prepared by laser ablation and reacted with carbon monoxide in a fast low reactor. The cluster reactions are characterized by time-of-flight mass spectrometry and density functional theory calculations. The oxygen atom transfers from (TiO2)nO– (n = 3–25) to CO and formations of (TiO2)n– are observed, whereas the reactions of (ZrO2)nO– (n = 3–25) with CO generate the CO addition products (ZrO2)nOCO–, which lose CO2 upon the collisions (studied for n = 3–9) with a crossed helium beam. The computational study indicates that the (MO2)nO– (M = Ti, Zr; n = 3–8) clusters are atomic radical anion (O–) bonded systems, and the energetics for CO oxidation by the O– radicals to form CO2 is strongly dependent on the metals as well as the cluster size for the titanium system. Atomic oxygen radical anions are important reactive intermediates, while it is difficult to capture and characterize them for condensed phase systems. The reactivity pattern of the O–-bonded (TiO2)nO– and (ZrO2)nO– correlates very well with different behaviors of titania and zirconia supports in the low-temperature catalytic CO oxidation.
Co-reporter:Zhen Yuan, Yan-Xia Zhao, Xiao-Na Li, Sheng-Gui He
International Journal of Mass Spectrometry 2013 Volumes 354–355() pp:105-112
Publication Date(Web):15 November 2013
DOI:10.1016/j.ijms.2013.06.004
•Reactions of V4O10+ ions with H2, CO, CH4, C2H2, C2H4, and C2H6 were studied by QH-TOF-MS.•The homolytic HH splitting rather than the water formation was identified.•Double oxygen atom transfer reaction with C2H2 was identified.•The increased velocity of the cluster ions favors the oxygen atom transfer reaction with C2H6.The reactivity of mass-selected V4O10+ cluster ions toward hydrocarbon molecules including CH4, C2H4, and C2H6 was explored in the references case by case. Herein, further systematic studies on the reactions of V4O10+ with simple inorganic and organic molecules (H2, CO, CH4, C2H2, C2H4, and C2H6) are presented. The vanadium oxide cluster ions are prepared by laser ablation and the V4O10+ clusters are selected by a quadrupole mass filter and interacted with the simple molecules in a hexapole reaction cell. The reactant and product ions are detected by a reflectron time-of-flight mass spectrometer. Hydrogen and oxygen atom transfer reactions are observed. Density functional theory calculations are carried out for the reaction mechanism of V4O10+ + H2. The oxygen atom transfer (OAT) channel V4O10+ + H2 → V4O9+ + H2O is much more exothermic than the hydrogen atom transfer (HAT) channel V4O10+ + H2 → V4O10H+ + H whereas the former is less favorable than the later in terms of the reaction kinetics. The computational result is in good agreement with the experiment that the HAT (H2 splitting) rather than the OAT (water formation) is observed for V4O10+ + H2.Figure optionsDownload full-size imageDownload high-quality image (180 K)Download as PowerPoint slide
Co-reporter:Bo Xu, Yan-Xia Zhao, Xun-Lei Ding, Sheng-Gui He
International Journal of Mass Spectrometry 2013 Volume 334() pp:1-7
Publication Date(Web):15 January 2013
DOI:10.1016/j.ijms.2012.09.004
Scandium and lanthanum oxide cluster anions Sc2O4− and La2O4− are prepared by laser ablation and reacted with CO in a fast flow reactor. A time of flight mass spectrometer is used to detect the cluster distribution before and after the reactions. In the reaction of Sc2O4− with CO, Sc2O3− anion is observed as a product. In sharp contrast, no product anion is observed for La2O4− + CO although the CO reactant gas causes a fast depletion of La2O4−. The experiment thus provides evidence of electron auto-detachment in the reaction of La2O4− with CO. Density functional theory calculations are performed to study the mechanisms for the above two reactions and the results well interpret the experimental observations. This study is among the first to report electron auto-detachment in the oxidation of CO by metal oxide cluster anions.Graphical abstractHighlights► Oxygen atom transfer reaction is observed in the reaction of Sc2O4− with CO. ► Electron auto-detachment rather than the oxygen atom transfer occurs in the reaction of La2O4− with CO. ► Oxidation of CO by atomic oxygen radical anions provides enough energy to evaporate electrons from the studied reaction systems. ► Formation of high-symmetry intermediate or product anions in which all metal atoms have high-coordination number is important for electron auto-detachment.
Co-reporter:Jing-Heng Meng, Yan-Xia Zhao, and Sheng-Gui He
The Journal of Physical Chemistry C 2013 Volume 117(Issue 34) pp:17548-17556
Publication Date(Web):July 25, 2013
DOI:10.1021/jp4039286
Lanthanum oxide cluster cations are prepared by laser ablation and reacted with alkane molecules (n-butane and methane) in a fast flow reactor under thermal collision conditions. A reflectron time-of-flight mass spectrometer is used to detect the cluster distributions before and after the reactions. Hydrogen atom abstraction (HAA) from n-butane by (La2O3)N+ (N = 1–8, N ≠ 6) is observed, while the HAA from methane is only observed for (La2O3)5+. The experimentally determined rate constants for HAA vary significantly with the cluster sizes. Density functional theory (DFT) calculations are performed to study the structures and reactivity of (La2O3)N+ (N = 1–6) clusters. The DFT results suggest that the experimentally observed C–H bond activation by (La2O3)N+ is facilitated by oxygen-centered radicals. The position of oxygen-centered radicals binding onto the clusters can heavily influence the reactivity in C–H bond activation. This gas-phase study improves our understanding about the chemistry of oxygen-centered radicals.
Co-reporter:Mei-Ye Jia, Xun-Lei Ding, Sheng-Gui He, and Mao-Fa Ge
The Journal of Physical Chemistry A 2013 Volume 117(Issue 35) pp:8377-8387
Publication Date(Web):August 12, 2013
DOI:10.1021/jp4044623
Transition metal oxide cluster anions Mm18On– (M = Fe, Co, Ni, Cu, and Zn) were prepared by laser ablation and reacted with H2S in a fast flow reactor under thermal collision conditions. A time-of-flight mass spectrometer was used to detect the cluster distributions before and after the interactions with H2S. The experiments reveal a suite of oxygen/sulfur (O/S) exchange and oxygen/sulfydryl (O/SH) exchange reactions. The O/S exchange reaction to release water was evidenced for all of the MO2– cluster anions: MO2– + H2S → MOS– + H2O, whereas the O/SH exchange reaction to derive MOSH– and OH species was only observed for reactions of NiO2–, CuO2–, and ZnO2–. Density functional theory calculations were performed for reaction mechanisms of MO2– + H2S (M = Fe, Co, Ni, Cu, and Zn). The computational results are generally in good agreement with the experimental results. This gas-phase study provides an insight into the metal dependent reactivity in the removal of H2S over metal oxides.
Co-reporter:Bo Xu, Yan-Xia Zhao, Xun-Lei Ding, Qing-Yu Liu, and Sheng-Gui He
The Journal of Physical Chemistry A 2013 Volume 117(Issue 14) pp:2961-2970
Publication Date(Web):March 18, 2013
DOI:10.1021/jp401169p
V5O12CH4+ and V5O13CH4+ clusters are generated from interactions of pregenerated V5O12+ and V5O13+ with CH4 in a fast flow reactor, respectively. The two adsorption complexes are then characterized by collision-induced dissociation (CID) and infrared photodissociation (IRPD) methods. The CID studies indicate that CH4 is molecularly adsorbed on V5O12+ and V5O13+. Each of the IRPD spectra of V5O12CH4+ and V5O13CH4+ has a broad red band located around 2770 cm–1 and a narrow blue band located around 2990 cm–1. The red and blue bands have large and small red shifts with respect to the symmetric and antisymmetric C–H stretch vibrations of free CH4, respectively. Density functional theory calculations are carried out for the structures and vibrational frequencies of V5O12+ and V5O12CH4+. The computed results suggest that the anharmonicity including Fermi resonance should be taken into account to interpret the observed IRPD spectrum. In V5O12CH4+, the CH4 unit adsorbs on the 3-fold coordinated V5+ site with an η2 configuration. The stretch of the two C–H bonds close to the V5+ ion is associated with the red band and the stretch of the other two C–H bonds is associated with the blue band. This study may shed light on the nature of methane adsorption onto vanadium pentoxide surfaces.
Co-reporter:Dr. Xiao-Nan Wu;Dr. Xiao-Na Li;Dr. Xun-Lei Ding ;Dr. Sheng-Gui He
Angewandte Chemie International Edition 2013 Volume 52( Issue 9) pp:2444-2448
Publication Date(Web):
DOI:10.1002/anie.201207016
Co-reporter:Jia-Bi Ma;Dr. Zhe-Chen Wang;Dr. Maria Schlangen;Dr. Sheng-Gui He;Dr. Helmut Schwarz
Angewandte Chemie International Edition 2013 Volume 52( Issue 4) pp:1226-1230
Publication Date(Web):
DOI:10.1002/anie.201208559
Co-reporter:Dr. Xiao-Nan Wu;Dr. Xiao-Na Li;Dr. Xun-Lei Ding ;Dr. Sheng-Gui He
Angewandte Chemie 2013 Volume 125( Issue 9) pp:2504-2508
Publication Date(Web):
DOI:10.1002/ange.201207016
Co-reporter:Xun-Lei Ding, Xiao-Nan Wu, Yan-Xia Zhao, and Sheng-Gui He
Accounts of Chemical Research 2012 Volume 45(Issue 3) pp:382
Publication Date(Web):October 21, 2011
DOI:10.1021/ar2001364
Saturated hydrocarbons, or alkanes, are major constituents of natural gas and oil. Directly transforming alkanes into more complex organic compounds is a value-adding process, but the task is very difficult to achieve, especially at low temperature. Alkanes can react at high temperature, but these reactions (with oxygen, for example) are difficult to control and usually proceed to carbon dioxide and water, the thermodynamically stable byproducts. Consequently, a great deal of research effort has been focused on generating and studying chemical entities that are able to react with alkanes or efficiently activate C–H bonds at lower temperatures, preferably room temperature.To identify low-temperature methods of C–H bond activation, researchers have investigated free radicals, that is, species with open-shell electronic structures. Oxygen-centered radicals are typical of the open-shell species that naturally occur in atmospheric, chemical, and biological systems. In this Account, we survey atomic clusters that contain oxygen-centered radicals (O–•), with an emphasis on radical generation and reaction with alkanes near room temperature. Atomic clusters are an intermediate state of matter, situated between isolated atoms and condensed-phase materials. Atomic clusters containing the O–• moiety have generated promising results for low-temperature C–H bond activation.After a brief introduction to the experimental methods and the compositions of atomic clusters that contain O–• radicals, we focus on two important factors that can dramatically influence C–H bond activation. The first factor is spin. The O–•-containing clusters have unpaired spin density distributions over the oxygen atoms. We show that the nature of the unpaired spin density distribution, such as localization and delocalization within the clusters, heavily influences the reactivity of O–• radicals in C–H bond activation.The second factor is charge. The O–•-containing clusters can be negatively charged, positively charged, or neutral overall. We discuss how the charge state may influence C–H bond activation. Moreover, for a given charge state, such as the cationic state, it can be demonstrated that local charge distribution around the O–• centers can also significantly change the reactivity in C–H bond activation. Through judicious synthetic choices, spin and charge can be readily controllable physical quantities in atomic clusters. The adjustment of these two properties can impact C–H bond activation, thus constituting an important consideration in the rational design of catalysts for practical alkane transformations.
Co-reporter:Xiao-Na Li, Bo Xu, Xun-Lei Ding and Sheng-Gui He
Dalton Transactions 2012 vol. 41(Issue 18) pp:5562-5570
Publication Date(Web):14 Mar 2012
DOI:10.1039/C2DT12174C
Vanadium oxide cluster anions (VxOy−, x = 2–3; y = 3–7) are produced by laser ablation and reacted with water in a fast flow reactor. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. Reaction channels of molecular hydrogen elimination (for V2,3O3−), water association (for V2O5− and V3O6,7−) and the coexistence of both channels (for V2O4− and V3O4,5−) are observed. V2O6− and V3O8− are nearly inert toward water. Density functional theory (DFT) calculations are performed to study the reaction mechanism of V2O3− in different spin states with water and the results support the experimental observation. The reaction mechanism of V2O3+ with water is also studied, which is in agreement with the experimental report in previous literature [Eur. J. Inorg. Chem., 2008, 4961] that molecular hydrogen elimination is a minor reaction channel for V2O3+ + H2O. The influence of cluster charge states and oxidation states of vanadium atoms on the cluster reactivity are presented based on the experimental and theoretical studies.
Co-reporter:Mei-Ye Jia, Bo Xu, Xun-Lei Ding, Yan-Xia Zhao, Sheng-Gui He, and Mao-Fa Ge
The Journal of Physical Chemistry C 2012 Volume 116(Issue 16) pp:9043-9048
Publication Date(Web):April 5, 2012
DOI:10.1021/jp3005458
Vanadium oxide cluster cations (Vm16On+ and Vm18On+) are prepared by laser ablation and reacted with hydrogen sulfide (H2S) in a fast flow reactor under thermal collision conditions. A time-of-flight mass spectrometer is used to detect the cluster distributions before and after the interactions with H2S. The experiments suggest that, in addition to H2S adsorption to form association products VmOnH2S+, three types of reactions are evidenced in the reactor: (1) VmOn+ + H2S → VmOnH+ + SH, (2) VmOn+ + H2S → VmOnH2+ + S, and (3) VmOn+ + H2S → VmOn–1S+ + H2O. Density functional theory calculations are performed for reaction of VO2+ with H2S, and the results indicate that the above three types of reaction channels are both thermodynamically and kinetically favorable, which supports the experimental observations. This gas-phase cluster study may provide insights into selective oxidation of H2S to elemental sulfur over vanadium-based oxide catalysts.
Co-reporter:Jia-Bi Ma;Dr. Zhe-Chen Wang;Dr. Maria Schlangen;Dr. Sheng-Gui He;Dr. Helmut Schwarz
Angewandte Chemie International Edition 2012 Volume 51( Issue 24) pp:5991-5994
Publication Date(Web):
DOI:10.1002/anie.201201698
Co-reporter:Li-Hua Tian;Dr. Yan-Xia Zhao;Xiao-Nan Wu;Dr. Xun-Lei Ding; Dr. Sheng-Gui He;Dr. Tong-Mei Ma
ChemPhysChem 2012 Volume 13( Issue 5) pp:1282-1288
Publication Date(Web):
DOI:10.1002/cphc.201100973
Abstract
Oxygen-rich scandium cluster anions ScO3–5− are prepared by laser ablation and allowed to react with n-butane in a fast-flow reactor. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. The ScO3− and ScO4− clusters can react with n-butane to produce ScO3H−, ScO3H2−, and ScO4H−, while the more oxygen-rich cluster ScO5− is inert. The experiment suggests that unreactive cluster isomers of ScO3− and ScO4− are also present in the cluster source. Density functional theory and ab initio methods are used to calculate the structures and reaction mechanisms of the clusters. The theoretical results indicate that the unreactive and reactive cluster isomers of ScO3,4− contain peroxides (O22−) and oxygen-centered radicals (O.−), respectively. The mechanisms and energetics for conversion of unreactive O22− to reactive O.− species are also theoretically studied.
Co-reporter:Jia-Bi Ma, Yan-Xia Zhao, Sheng-Gui He, and Xun-Lei Ding
The Journal of Physical Chemistry A 2012 Volume 116(Issue 9) pp:2049-2054
Publication Date(Web):February 8, 2012
DOI:10.1021/jp300279u
Vanadium oxide cluster cations VxOy+ (x = 2–6) are prepared by laser ablation and are reacted with D2O in a fast flow reactor under room temperature conditions. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. Observation of the products (V2O5)1–3D+ indicates the deuterium atom abstraction reaction (V2O5)1–3+ + D2O → (V2O5)1–3D+ + OD. In addition, significant association products (V2O5)1–3D2O+ are also observed in the experiments. Density functional theory calculations are performed to study the reaction mechanisms of V4O10+ with H2O. The calculated results are in agreement with the experimental observations and indicate that H2O is dissociatively rather than molecularly adsorbed in V4O10H2O+ complex.
Co-reporter:Xiao-Nan Wu, Bo Xu, Jing-Heng Meng, Sheng-Gui He
International Journal of Mass Spectrometry 2012 310() pp: 57-64
Publication Date(Web):
DOI:10.1016/j.ijms.2011.11.011
Co-reporter:Dr. Xiao-Na Li;Dr. Xiao-Nan Wu;Dr. Xun-Lei Ding;Bo Xu; Dr. Sheng-Gui He
Chemistry - A European Journal 2012 Volume 18( Issue 35) pp:10998-11006
Publication Date(Web):
DOI:10.1002/chem.201201467
Abstract
Vanadium–silver bimetallic oxide cluster ions (VxAgyOz+; x=1–4, y=1–4, z=3–11) are produced by laser ablation and reacted with ethane in a fast-flow reactor. A reflectron time of flight (Re-TOF) mass spectrometer is used to detect the cluster distribution before and after the reactions. Hydrogen atom abstraction (HAA) reactions are identified over VAgO3+, V2Ag2O6+, V2Ag4O7+, V3AgO8+, V3Ag3O9+, and V4Ag2O11+ ions, in which the oxygen-centered radicals terminally bonded on V atoms are active sites for the facile HAA reactions. DFT calculations are performed to study the structures, bonding, and reactivity. The reaction mechanisms of V2Ag2O6++C2H6 are also given. The doped Ag atoms with a valence state of +1 are highly dispersed at the periphery of the VxAgyOz+ cluster ions. The reactivity can be well-tuned gradually by controlling the number of Ag atoms. The steric protection due to the peripherally bonded Ag atoms greatly enhances the selectivity of the V–Ag bimetallic oxide clusters with respect to the corresponding pure vanadium oxide systems.
Co-reporter:Mei-Ye Jia, Bo Xu, Xun-Lei Ding, Sheng-Gui He, and Mao-Fa Ge
The Journal of Physical Chemistry C 2012 Volume 116(Issue 45) pp:24184-24192
Publication Date(Web):October 22, 2012
DOI:10.1021/jp309004s
Manganese oxide cluster anions (Mnm16On¯ and Mnm18On¯) were prepared by laser ablation and reacted with hydrogen sulfide (H2S) in a fast flow reactor under thermal collision conditions. A time-of-flight mass spectrometer was used to detect the cluster distributions before and after interaction with H2S. The experiments suggest that an oxygen-for-sulfur (O/S) exchange reaction to release water occurred in the reactor for most of the manganese oxide cluster anions: MnmOn¯ + H2S → MnmOn–1S¯ + H2O. The O/S exchange reactivity of MnmOn¯ was generally found to decrease when the ratio (n/m) of oxygen to manganese atoms in the cluster increased. Density functional theory (DFT) calculations were performed for reaction mechanisms of MnO2¯ + H2S, MnO3¯ + H2S, and Mn2O4¯ + H2S. The computational results were in good agreement with the experimental observations. This gas-phase cluster study provides molecular-level insights into the adsorptive removal of H2S by bulk manganese oxides.
Co-reporter:Yan-Xia Zhao, Xiao-Nan Wu, Jia-Bi Ma, Sheng-Gui He and Xun-Lei Ding
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 6) pp:1925-1938
Publication Date(Web):05 Jan 2011
DOI:10.1039/C0CP01171A
We introduce chemical structures and reactivity of oxygen-centred radicals (O−˙) over transition metal oxide (TMO) clusters based on mass spectrometric and density functional theory studies. Two main issues will be discussed: (1) the compositions of TMO clusters that have the bonding characteristics of (or contain) the O−˙ radicals; and (2) the dependences (cluster structures, sizes, charge states, metal types, etc.) of the chemical reactivity and selectivity for the O−˙ radicals over TMO clusters. One of the goals of cluster chemistry is to understand the elementary reactions involved with complex heterogeneous catalysis. The study of the O−˙ containing TMO clusters permits rather detailed descriptions for how mono-nuclear oxygen-centred radicals may exist and react with small molecules over TMO based catalysts.
Co-reporter:Xiao-Nan Wu, Jia-Bi Ma, Bo Xu, Yan-Xia Zhao, Xun-Lei Ding, and Sheng-Gui He
The Journal of Physical Chemistry A 2011 Volume 115(Issue 21) pp:5238-5246
Publication Date(Web):May 10, 2011
DOI:10.1021/jp200984r
Zirconium oxide cluster cations and anions are produced by laser ablation and reacted with CO in a fast flow reactor. The CO adsorption products ZrxOyCO+ are observed for most of the generated cationic clusters (ZrxOy+ = Zr2O5,6+, Zr3O7,8+, Zr4O9,10+...) while only specific anionic systems (ZrxOy– = Zr3O7–, Zr4O9–...) absorb CO to produce ZrxOyCO–. To study how the CO molecule is adsorbed on the clusters, the ZrxOyCO± products are mass-selected by a time-of-flight mass spectrometer (TOF-MS) and collided with a crossed helium beam. The fragment ions from collision-induced dissociation (CID) are detected by a secondary TOF-MS. Loss of CO and CO2 is observed upon the collision of the helium beam with ZrxOyCO+ and ZrxO2x+1CO–, respectively. Density functional theory calculations indicate that oxidative and nonoxidative adsorption of CO takes place over Zr3O7– and Zr3O7+, respectively. This is consistent with the CID experiments.
Co-reporter:Bo Xu, Yan-Xia Zhao, Xiao-Na Li, Xun-Lei Ding, and Sheng-Gui He
The Journal of Physical Chemistry A 2011 Volume 115(Issue 37) pp:10245-10250
Publication Date(Web):August 11, 2011
DOI:10.1021/jp203990w
Lanthanum oxide cluster anions are prepared by laser ablation and reacted with n-C4H10 in a fast flow reactor. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. (La2O3)m=1–3OH– and La3O7H– are observed as products, which suggests the occurrence of hydrogen atom abstraction reactions: (La2O3)m=1–3O– + n-C4H10 → (La2O3)m=1–3OH– + C4H9 and La3O7– + n-C4H10 → La3O7H– + C4H9. Density functional theory (DFT) calculations are performed to study the structures and bonding properties of La2O4–, La3O7–, and La4O7– clusters. The calculated results show that each of La2O4– and La4O7– contains one oxygen-centered radical (O–•) which is responsible for the high reactivity toward n-C4H10. La3O7– contains one oxygen-centered radical (O–•) and one superoxide unit (O2–•), and the O–• is responsible for its high reactivity toward n-C4H10. The O–• and O2–• can be considered to be generated by the adsorption of an O2 molecule onto the singlet La3O5– with electron transfer from a terminally bonded oxygen ion (O2–) to the O2. This may help us understand the mechanism of the formation of O–• and O2–• radicals in lanthanum oxide systems. The reaction mechanisms of La2O4– + n-C4H10 and La3O7– + n-C4H10 are also studied by the DFT calculations, and the calculated results are in good agreement with the experimental observations.
Co-reporter:Dr. Xun-Lei Ding;Xiao-Nan Wu;Dr. Yan-Xia Zhao;Jia-Bi Ma; Sheng-Gui He
ChemPhysChem 2011 Volume 12( Issue 11) pp:2110-2117
Publication Date(Web):
DOI:10.1002/cphc.201100216
Abstract
Cerium oxide cluster cations (CemOn+, m=2–16; n=2m, 2m±1 and 2m±2) are prepared by laser ablation and reacted with acetylene (C2H2) in a fast-flow reactor. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. Reactions of stoichiometric CemO2m+ (m=2–6) with C2H2 produce CemO2m−2+ clusters, which indicates a “double-oxygen-atom transfer” reaction CemO2m++C2H2CemO2m−2++(CHO)2 (ethanedial). A single-oxygen-atom transfer reaction channel is also identified as CemO2m++C2H2CemO2m−1++C2H2O (at least for m=2 and 3). Density functional theory calculations are performed to study reaction mechanisms of Ce2O4++C2H2, and the calculated results confirm that both the single- and double-oxygen-atom transfer channels are thermodynamically and kinetically favourable.
Co-reporter:Dr. Xun-Lei Ding;Xiao-Nan Wu;Dr. Yan-Xia Zhao;Jia-Bi Ma; Sheng-Gui He
ChemPhysChem 2011 Volume 12( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/cphc.201190055
Co-reporter:Xiao-Nan Wu ; Xun-Lei Ding ; Shu-Ming Bai ; Bo Xu ; Sheng-Gui He ;Qiang Shi
The Journal of Physical Chemistry C 2011 Volume 115(Issue 27) pp:13329-13337
Publication Date(Web):June 9, 2011
DOI:10.1021/jp202077s
Reactions of cerium oxide cluster anions with carbon monoxide are investigated by time-of-flight mass spectrometry and density functional theory computations aided with molecular dynamics simulations. Interesting size-dependent reactivity of the CenO2n+1– cluster series with n = 1–21 is observed: (1) the small n = 1–3 clusters have no or very low reactivity toward CO, (2) the large n = 4–21 clusters can oxidize CO to produce CO2, and (3) the n = 4 (Ce4O9–), 6 (Ce6O13–), 7 (Ce7O15–), and 12 (Ce12O25–) clusters have relatively higher reactivity than their neighboring systems Ce3O7–, Ce5O11–, Ce8O17–, etc. Theoretical study indicates that the CenO2n+1– clusters contain oxygen-centered radicals (O–•) and the nature of the spin density distributions within the clusters controls the experimentally observed size-dependent reactivity. The experiment and theory in this study suggest that the metal oxide clusters as large as Ce21O43– can contain the reactive O–• centers, at which the size may be large enough to mimic related active sites in condensed phase catalysts. Oxidation of CO by O2 at low temperature is of widespread importance and reactive oxygen species including O–• are usually involved. The nature of the O–• radicals is demonstrated to be able to further address the goodness of nanocrystalline CeO2 in the low-termperautre CO oxidation.
Co-reporter:Zi-Yu Li;Dr. Yan-Xia Zhao;Xiao-Nan Wu;Dr. Xun-Lei Ding; Sheng-Gui He
Chemistry - A European Journal 2011 Volume 17( Issue 42) pp:11728-11733
Publication Date(Web):
DOI:10.1002/chem.201102055
Co-reporter:Yan-Xia Zhao, Xiao-Nan Wu, Zhe-Chen Wang, Sheng-Gui He and Xun-Lei Ding
Chemical Communications 2010 vol. 46(Issue 10) pp:1736-1738
Publication Date(Web):05 Feb 2010
DOI:10.1039/B924603G
Stoichiometric early transition metal oxide cations (TiO2)1–5+, (ZrO2)1–4+, (HfO2)1–2+, (V2O5)1–5+, (Nb2O5)1–3+, (Ta2O5)1–2+, (MoO3)1–2+, (WO3)1–3+, and Re2O7+ are able to activate the C–H bond of methane under near room temperature conditions.
Co-reporter:Jia-Bi Ma, Xiao-Nan Wu, Xian-Xia Zhao, Xun-Lei Ding and Sheng-Gui He
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 38) pp:12223-12228
Publication Date(Web):16 Aug 2010
DOI:10.1039/C0CP00360C
A series of vanadium and phosphorus heteronuclear oxide cluster cations (VxPyOz+) are prepared by laser ablation and the reactions of V3PO10˙+ and V4O10˙+ with methane in a fast flow reactor under the same conditions are studied. A time of flight mass spectrometer is used to detect the cluster distribution before and after reactions. In addition to previously identified reaction of V4O10˙+ + CH4 → V4O10H+ + CH3˙, the observation of hydrogen atom pickup cluster V3PO10H+ suggests the reaction: V3PO10˙+ + CH4 → V3PO10H+ + CH3˙. The rate of the reaction of V4O10˙+ with CH4 is approximately 2.5 times faster than that of V3PO10˙+ with CH4. Density functional theory (DFT) calculations predict that structure of V3PO10˙+ is topologically similar to that of V4O10˙+, as well as that of P4O10˙+, which is very similar to V4O10˙+ in terms of methane activation in previous studies. The facile methane activation by the homo- and hetero-nuclear oxide clusters can all be attributed to the presence of an oxygen-centered radical (O˙) in these clusters. Further theoretical study indicates that the O˙ radical (or spin density of the cluster) can transfer within the high symmetry V4O10˙+ and P4O10˙+ clusters quite easily, and CH4 molecule further enhances the rate of intra-cluster spin density transfer. In contrast, the intra-cluster spin density transfer within low symmetry V3PO10˙+ is thermodynamically forbidden. The experimentally observed reactivity difference is consistent with the theoretical consideration of the intra-cluster spin density transfer.
Co-reporter:Xiao-Nan Wu, Yan-Xia Zhao, Wei Xue, Zhe-Chen Wang, Sheng-Gui He and Xun-Lei Ding
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 16) pp:3984-3997
Publication Date(Web):20 Mar 2010
DOI:10.1039/B925294K
Cerium oxide cluster cations (CemOn+, m = 2–16; n = 2m, 2m ± 1 and 2m ± 2) are prepared by laser ablation and reacted with carbon monoxide (CO) and small hydrocarbon molecules (CH4, C2H4, and C2H6) in a fast flow reactor. A time of flight mass spectrometer is used to detect the cluster distribution before and after the reactions. The observation of oxygen reduction and hydrogen pickup of CemO2m+ clusters strongly suggests the following reactions: (1) CemO2m+ + C2H4 → CemO2m−1+ + C2H4O (m = 2–6); (2) CemO2m+ + CO → CemO2m−1+ + CO2 (m = 4–6); and (3) CemO2m+ + CH4/C2H6 → CemO2mH+ + CH3/C2H5 (m = 2–4). Density functional theory (DFT) calculations are performed to study reaction mechanisms of Ce2O4+ + X (X = CO, CH4, C2H4, and C2H6). The calculated results are in good agreement with the experimental observations. The structural and bonding properties of CemO2m+ (m = 2–5) clusters are also investigated by the DFT calculations. The unpaired electron in each of the clusters is mainly distributed over one Ce atom (4f and 5p orbitals) and two O atoms (2p orbital) in a CeO2 moiety, which can be considered as the active site in the cluster. To further understand the nature of the active sites in CemO2m+ clusters, the fast flow reaction experiments are also carried out on zirconium oxide clusters ZrmOn+, because both Zr ([Kr]4d25s2) and Ce ([Xe]4f15d16s2) have the same number of valence electrons while the latter has one more f and one less d electrons. In addition to the oxygen transfer reactions such as ZrmO2m+ + C2H4 → ZrmO2m−1+ + C2H4O (m = 1–4) reported in the literature, hydrogen abstraction reactions ZrmO2m+ + CH4/C2H6 → ZrmO2mH+ + CH3/C2H5 are also identified. The rate constants of CO oxidation as well as hydrogen abstraction by CemO2m+ and ZrmO2m+ are very different. The reactivity and selectivity of CemO2m+versus ZrmO2m+ can be well rationalized based on the DFT calculations. The oxygen transfer and hydrogen abstraction reactions studied in this work are of widespread importance. The nature of the active sites of CemO2m+ clusters is unique and may be considered in the use and design of cerium oxide based catalysts.
Co-reporter:Yan-Xia Zhao;Xun-Lei Ding;Yan-Ping Ma;Zhe-Chen Wang
Theoretical Chemistry Accounts 2010 Volume 127( Issue 5-6) pp:449-465
Publication Date(Web):2010 November
DOI:10.1007/s00214-010-0732-8
Density functional theory (DFT) calculations are applied to study the structure and bonding properties of groups 3–7 transition metal oxide clusters Mx=1–3Oyq and Scx=4–6Oyq with 2y − nx + q = 1, in which n is the number of metal valence electrons and q is the charge number. These clusters include MO2, M2O3+, M2O4−, and M3O5 (M = Sc, Y, La); MO2+, MO3−, M2O4+, M2O5−, M3O6+, and M3O7− (M = Ti, Zr, Hf), and so on. The obtained lowest energy structures of most of these clusters are with character of oxygen-centered radical (O·). That is, the clusters contain oxygen atom(s) with the unpaired electron being localized on the 2p orbital(s). Chromium and manganese oxide clusters (except CrO4−) do not contain O· with the adopted DFT methods. The binding energies of the radical oxygen with the clusters are also calculated. The DFT results are supported by available experimental investigations and predict that a lot of other transition metal oxide clusters including those with mixed-metals (such as TiVO5 and CrVO6) may have high oxidative reactivity that has not been experimentally identified. The chemical structures of radical oxygen over V2O5/SiO2 and MoO3/SiO2 catalysts are suggested and the balance between high reactivity and low concentration of the radical oxygen in condensed phase catalysis is discussed.
Co-reporter:Jia-Bi Ma, Xiao-Nan Wu, Yan-Xia Zhao, Xun-Lei Ding, and Sheng-Gui He
The Journal of Physical Chemistry A 2010 Volume 114(Issue 37) pp:10024-10027
Publication Date(Web):August 26, 2010
DOI:10.1021/jp106857y
Zirconium oxide cluster anions ZrxOy− are prepared by laser ablation and are reacted with n-butane in a fast flow reactor under near room temperature conditions. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. Observation of a hydrogen atom pickup product Zr2O8H− indicates a C−H activation reaction: Zr2O8− + n-C4H10 → Zr2O8H− + C4H9. Density functional theory calculations predict that the oxygen-very-rich cluster Zr2O8− contains one mononuclear oxygen-centered radical (O−•), which leads to a high C−H activation reactivity, in agreement with the experiments. This study provides one example for how the highly oxidative O−• radical may be generated by adsorption of O2 onto unreactive metal oxide clusters.
Co-reporter:Yan-Ping Ma Dr.;Xun-Lei Ding Dr.;Yan-Xia Zhao Dr.
ChemPhysChem 2010 Volume 11( Issue 8) pp:1718-1725
Publication Date(Web):
DOI:10.1002/cphc.200900903
Abstract
Density functional theory (DFT) calculations are used to investigate the reaction mechanism of V3O8+C2H4. The reaction of V3O8 with C2H4 produces V3O7CH2+HCHO or V3O7+CH2OCH2 overall barrierlessly at room temperature, whereas formation of hydrogen-transfer products V3O7+CH3CHO is subject to a tiny overall free energy barrier (0.03 eV), although the formation of the last-named pair of products is thermodynamically more favorable than that of the first two. These DFT results are in agreement with recent experimental observations. The (Ob)2V(OtOt). (b=bridging, t=terminal) moiety containing the oxygen radical in V3O8 is the active site in the reaction with C2H4. Similarities and differences between the reactivities of (Ob)2V(OtOt). in V3O8 and the small VO3 cluster [(Ot)2VOt.] are discussed. Moreover, the effect of the support on the reactivity of the (Ob)2V(OtOt). active site is evaluated by investigating the reactivity of the cluster VX2O8, which is obtained by replacing the V atoms in the (Ob)3VOt support moieties of V3O8 with X atoms (X=P, As, Sb, Nb, Ta, Si, and Ti). Support X atoms with different electronegativities influence the oxidative reactivity of the (Ob)2V(OtOt). active site through changing the net charge of the active site. These theoretical predictions of the mechanism of V3O8+C2H4 and the effect of the support on the active site may be helpful for understanding the reactivity and selectivity of reactive O. species over condensed-phase catalysts.
Co-reporter:Yan-Xia Zhao, Xiao-Nan Wu, Jia-Bi Ma, Sheng-Gui He and Xun-Lei Ding
The Journal of Physical Chemistry C 2010 Volume 114(Issue 28) pp:12271-12279
Publication Date(Web):June 29, 2010
DOI:10.1021/jp1045244
Vanadium and silicon heteronuclear oxide cluster anions VxSiyOz− (x + y ≥ 2, z ≥ 4) are prepared by laser ablation and reacted with n-butane (C4H10) in a fast flow reactor. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. The observation of hydrogen-containing products (V2O5)m(SiO2)nOH− (m = 1, n = 1−4; m = 2, n = 1) strongly suggests the following reactions: (V2O5)m(SiO2)nO− + C4H10 → (V2O5)m(SiO2)nOH− + C4H9. Although V2O6− is produced in the cluster source, no V2O6H− product is produced under the same experimental condition. It indicates that specific heteronuclear oxide clusters V2O5(SiO2)1−4O− and (V2O5)2SiO2O− are more reactive than the homonuclear oxide cluster V2O6− (or V2O5O−). Density functional theory (DFT) calculations are performed to study reaction mechanisms of V2O5SiO2O− (or V2SiO8−) + C4H10. The calculated results are in good agreement with the experimental observations. The structural and bonding properties of (V2O5)m(SiO2)nO− (m = 1, n = 1−4; m = 2, n = 1) are also investigated by the DFT calculations. The unpaired electron in each of the clusters is mainly distributed over one or two O atoms (2p orbitals) bonded with Si rather than V atom(s). Furthermore, the experimentally observed higher reactivity of the V−Si heteronuclear oxide cluster (V2O5)m(SiO2)nO− over the homonuclear V2O6− in the reaction with C4H10 is interpreted based on the theoretical results.
Co-reporter:Xun-Lei Ding, Wei Xue, Yan-Ping Ma, Yan-Xia Zhao, Xiao-Nan Wu and Sheng-Gui He
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:3161-3169
Publication Date(Web):February 2, 2010
DOI:10.1021/jp9112415
The mechanisms of the selective oxidation of methanol to formaldehyde facilitated by monomeric vanadium oxide species on silica support are studied theoretically. The active sites are modeled by clusters with “umbrella” structures: O═V(OH)2−O−Si(OH)3 (hydrated VO4) and O═V(O−O)−O−Si(OH)3 (dehydrated VO4). The possible reaction pathways are studied by density functional theory, and the reaction kinetics is studied using statistical mechanics and absolute rate theory. The reaction mechanisms and catalytic cycles are proposed: the hydrated VO4 species reacts with methanol to produce formaldehyde and the dehydrated VO4 species; the dehydrated VO4 species has high reactivity toward methanol to recycle the hydrated VO4. Hydrogen transfer is the rate-limiting step. The umbrella models of the active sites are found to be capable of giving a reliable description of the mechanisms of the methanol oxidation reaction, which supports the possibility of the existence of VO4 species with umbrella structure on the catalyst surface.
Co-reporter:Dr. Xun-Lei Ding;Yan-Xia Zhao;Xiao-Nan Wu;Zhe-Chen Wang;Jia-Bi Ma; Sheng-Gui He
Chemistry - A European Journal 2010 Volume 16( Issue 37) pp:
Publication Date(Web):
DOI:10.1002/chem.201090184
Co-reporter:Dr. Xun-Lei Ding;Yan-Xia Zhao;Xiao-Nan Wu;Zhe-Chen Wang;Jia-Bi Ma; Sheng-Gui He
Chemistry - A European Journal 2010 Volume 16( Issue 37) pp:11463-11470
Publication Date(Web):
DOI:10.1002/chem.201001297
Abstract
Vanadium–silicon heteronuclear oxide cluster cations were prepared by laser ablation of a V/Si mixed sample in an O2 background. Reactions of the heteronuclear oxide cations with methane in a fast-flow reactor were studied with a time-of-flight (TOF) mass spectrometer to detect the cluster distribution before and after the reactions. Hydrogen abstraction reactions were identified over stoichiometric cluster cations [(V2O5)n(SiO2)m]+ (n=1, m=1–4; n=2, m=1), and the estimated first-order rate constants for the reactions were close to that of the homonuclear oxide cluster V4O10+ with methane. Density functional calculations were performed to study the structural, bonding, electronic, and reactivity properties of these stoichiometric oxide clusters. Terminal-oxygen-centered radicals (Ot.) were found in all of the stable isomers. These Ot. radicals are active sites of the clusters in reaction with CH4. The Ot. radicals in [V2O5(SiO2)1–4]+ clusters are bonded with Si rather than V atoms. All the hydrogen abstraction reactions are favorable both thermodynamically and kinetically. This work reveals the unique properties of metal/nonmetal heteronuclear oxide clusters, and may provide new insights into CH4 activation on silica-supported vanadium oxide catalysts.
Co-reporter:Yan-Ping Ma, Sheng-Gui He, Xun-Lei Ding, Zhe-Chen Wang, Wei Xue and Qiang Shi
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 14) pp:2543-2552
Publication Date(Web):13 Feb 2009
DOI:10.1039/B815010A
The intermolecular interactions between two meso-tetraphenylporphyrin diacid H4TPPCl2 monomers are investigated by density functional theory with the PBE1PBE functional and 6-31G* basis set. Structures of five stable isomers of (H4TPPCl2)2 are determined. It is found that the interaction (IMHB-1) of Cl with orthoH atoms in two phenyl groups of H4TPPCl2 is unique in that it is the strongest interaction between two H4TPPCl2 monomers. Natural bond orbital analysis is carried out to explain the subtle differences of hydrogen bondings in these isomers. To understand the interactions of water molecule with H4TPPCl2 and (H4TPPCl2)2, structures of H4TPPCl2·H2O and nine isomers of (H4TPPCl2)2·H2O are also determined. The binding energy of H4TPPCl2·H2O is 36.47 kJ mol−1, less than that of the most stable structure of (H4TPPCl2)2 dimers, 41.59 kJ mol−1. The dimers containing the special IMHB-1 interactions may be the elementary building blocks for the aggregation of H4TPPCl2, which is supported by the crystal structure of H4TPPCl2·H2O·2CH3CN. Thus, study of the interactions between two or a small number of gas molecules can provide important information for understanding the main interactions and stable structures in related condensed-phase systems.
Co-reporter:Shi Yin, Wei Xue, Xun-Lei Ding, Wei-Gang Wang, Sheng-Gui He, Mao-Fa Ge
International Journal of Mass Spectrometry 2009 Volume 281(1–2) pp:72-78
Publication Date(Web):15 March 2009
DOI:10.1016/j.ijms.2008.12.014
A time of flight mass spectrometer coupled with a laser ablation/supersonic expansion cluster source is used to study the formation and distribution of cationic iron and cobalt oxide clusters. Although the distributions of iron oxide clusters (FemOnq, q = 0, ±1) have been extensively reported in literature, new and very interesting distribution of FemOn+ clusters is observed in this study. Under saturated O2 growth conditions, the smallest (leading) cluster in m = 2k + 1 (k = 2−14) cluster series is with stoichiometry of Fe2kO3kFeO+, which is perfect (iron atoms are perfectly oxidized) in terms of average oxidation states of iron (Fe3+) and oxygen (O2−) atoms. For m = 2k (k = 2–15) cluster series, the leading cluster is either Fe2kO3k+ (the least over-oxidized) or Fe2kO3k−1+ (the least under-oxidized). Density functional theory (DFT) calculations indicate that these leading clusters are with unexpected structures although their appearance in the mass spectra is predictable. These clusters may serve as good models for predicting or interpreting novel properties of Fe2O3 nano-materials. The distribution of the cobalt oxide clusters (ComOn+) under saturated O2 growth conditions is complex and very different from that of FemOn+. A very interesting result for cobalt species is that two clusters Co11O13+ and Co12O13+ are missing in the cluster distribution although their oxygen-neighbor clusters Co11O12,14+ and Co12O12,14+ are generated. This suggests relatively high stability for Co11O12+ and Co12O12+ clusters. The DFT calculations predict that Co12O12 cluster are with tower or cage structure rather than the compact NaCl-like arrangement that is found for bulk CoO.
Co-reporter:Wei Xue, Shi Yin, Xun-Lei Ding, Sheng-Gui He and Mao-Fa Ge
The Journal of Physical Chemistry A 2009 Volume 113(Issue 18) pp:5302-5309
Publication Date(Web):April 7, 2009
DOI:10.1021/jp810426s
Reactions of small cationic iron oxide clusters (Fe2O4−6+) with N2 are investigated by experiments and first principle calculations. The cationic iron oxide clusters are generated by reaction of laser ablated iron plasma with O2 in a supersonic expansion, and are reacted with N2 in a fast flow reactor at near room temperature conditions. Cluster cations are detected by a time-of-flight mass spectrometer. The substitution reaction Fe2On+ + N2 → Fe2On-2N2+ + O2 is observed for n = 5 but not for n = 4 and 6. Density functional theory calculations predict that the low-lying energy structures of Fe2O4−6+ are with side-on (η1-O2) or end-on (η2-O2) bonded molecular oxygen unit(s). The calculations further predict that the substitution of η1-O2 and η2-O2 in Fe2O4,6+ clusters by N2 is exothermic and subject to negative and positive overall reaction barriers, respectively, at room temperature. We thus propose that the ground state structures of Fe2O4+ and Fe2O6+ contain η2-O2. In contrast, both the experiment and theory favor a η1-O2 in the ground state structure of Fe2O5+.
Co-reporter:ZheChen Wang;XunLei Ding;YanPing Ma;Hai Cao;XiaoNan Wu
Science Bulletin 2009 Volume 54( Issue 16) pp:2814-2821
Publication Date(Web):2009 August
DOI:10.1007/s11434-009-0276-2
Density functional theory (DFT) study of reaction between vanadium trioxide cluster cation (VO3+) and ethylene (C2H4) to yield VO2+ + CH3CHO (acetaldehyde) and VO2CH2+ + HCHO (formaldehyde) is carried out. Structures of all reactants, products, intermediates, and transition state in the reaction have been optimized and characterized. The results show unexpected barriers in the reaction due to the existence of a η2-O2 moiety in the ground state structure of VO3+. The initial reaction steps combining ethylene adsorption, C=C activation and O-O cleavage are proposed as rate limiting processes. Comparison of reactions of VO3+ + C2H4 with VO3 + C2H4 and VO2+ + C2H4 in the previous studies is made in detail. The results of this work may shed light on the understanding of C=C bond cleavage in related heterogeneous catalysis.
Co-reporter:Sheng-Gui He, Dennis J. Clouthier
Computer Physics Communications 2008 Volume 178(Issue 9) pp:676-684
Publication Date(Web):1 May 2008
DOI:10.1016/j.cpc.2007.11.017
In this work we present Windows and Fortran programs which can be used for the Renner–Teller analysis of the vibronic levels of Π2 states of linear, triatomic molecules. The programs can do least squares fitting of term values relative to the lowest energy level within a single Π2 state, of combination differences between vibronic levels within a single Π2 state or of transitions between the levels of two different Π2 states. The algorithm allows for the inclusion of Renner–Teller, spin–orbit, vibrational anharmonicity, Fermi resonance and Sears resonance terms in the Hamiltonian matrices. The Windows program RT3, written in Visual Basic 6.0 with a Fortran DLL engine for the numerically intensive computations, facilitates the construction and editing of input files and comparisons of input and output. For very large calculations, the Fortran code may be run as a stand-alone program on mainframe or other computers, but the Windows version offers significant advantages for common usage.Program summaryTitle of program: RT3Catalogue identifier: AEAL_v1_0Program summary URL:http://cpc.cs.qub.ac.uk/summaries/AEAL_v1_0.htmlProgram obtainable from: CPC Program Library, Queen's University of Belfast, N. IrelandLicensing provisions: Standard CPC licence, http://cpc.cs.qub.ac.uk/licence/licence.htmlNo. of lines in distributed program, including test data, etc.: 37 477No. of bytes in distributed program, including test data, etc.: 3 665 414Distribution format: tar.gzProgramming language: Visual Basic 6.0, Fortran 90Operating systems: Windows 98, Windows XP, Windows Vista, UNIXClassification: 16.2Nature of problem: The vibronic levels of triatomic molecules in Π2 states are complicated by spin–orbit and vibrational-orbital angular momentum coupling (Renner–Teller) effects that make them difficult to assign and analyze. The RT3 program, available in Windows and Fortran versions, can be used to calculate the vibronic energy levels of such systems and fit experimental data to a Hamiltonian that includes Renner–Teller, spin–orbit, vibrational anharmonicity, Fermi resonance and Sears resonance effects.Solution method: The RT3 program solves for the Renner–Teller levels of a Π2 state by diagonalizing large truncated Hamiltonian matrices whose elements are generated in a harmonic oscillator basis set. Given an initial input set of constants, the resulting energy levels can be used to least squares fit or predict vibronic term values (relative to the lowest energy level), combination differences between vibronic levels within a electronic state, and transitions between the vibronic levels of two different Π2 states.Running time: 6.0 S to run the Transitions.ir3 example file on an Intel Pentium 4 2 GHz CPU with the Windows XP operating system.
Co-reporter:Shi Yin;YanPing Ma;Lin Du;ShengGui He;MaoFa Ge
Science Bulletin 2008 Volume 53( Issue 24) pp:3829-3838
Publication Date(Web):2008 December
DOI:10.1007/s11434-008-0502-3
The time of flight mass spectrometer coupled with a laser ablation/supersonic expansion cluster source and a fast flow reactor was adopted to study the reactivity of cationic vanadium oxide clusters (VmOn+) toward acetylene (C2H2) molecules under gas phase (P, ∼ 1.14 kPa), under near room temperature (T, ∼ 350 K) conditions. Association products, VmOnC2H2+ (m,n = 2,4; 2,6; 3,7–8; 4,9–11; 5,12–13; 6,13–16, and 7,17), are observed. The oxidation of C2H2 by (V2O5)n+ (n = 1–3) is experimentally identified. The reactivity of (V2O5)n+ decreases as n increases. Density functional theory (DFT) calculations were carried out to interpret the reaction mechanisms. The DFT results indicate that a terminal oxygen atom from V2O5+ can transfer overall barrierlessly to C2H2 at room temperature, which is in agreement with the experimental observation. Other experimental results such as the observation of V2O6C2H2+ and nonobservation of V2O7,8C2H2+ in the experiments are also well interpreted based on the DFT calculations. The reactivity of vanadium oxide clusters toward acetylene and other hydrocarbons may be considered in identifying molecular level mechanisms for related heterogeneous catalysis.
Co-reporter:Zhe-Chen Wang, Wei Xue, Yan-Ping Ma, Xun-Lei Ding, Sheng-Gui He, Feng Dong, Scott Heinbuch, Jorge J. Rocca and Elliot R. Bernstein
The Journal of Physical Chemistry A 2008 Volume 112(Issue 26) pp:5984-5993
Publication Date(Web):June 6, 2008
DOI:10.1021/jp7115774
Density functional theory (DFT) calculations are carried out to investigate partial oxidation of propylene over neutral VO3 clusters. C═C bond cleavage products CH3CHO + VO2CH2 and HCHO + VO2CHCH3 can be formed overall barrierlessly from the reaction of propylene with VO3 at room temperature. Formation of hydrogen transfer products H2O + VO2C3H4, CH2═CHCHO + VO2H2, CH3CH2CHO + VO2, and (CH3)2CO + VO2 is subject to tiny (0.01 eV) or small (0.06 eV, 0.19 eV) overall free energy barriers, although their formation is thermodynamically more favorable than the formation of C═C bond cleavage products. These DFT results are in agreement with recent experimental observations. VO3 regeneration processes at room temperature are also investigated through reaction of O2 with the C═C bond cleavage products VO2CH2 and VO2CHCH3. The following barrierless reaction channels are identified: VO2CH2 + O2 → VO3 + CH2O; VO2CH2 + O2 → VO3C + H2O, VO3C + O2 → VO3 + CO2; VO2CHCH3 + O2 → VO3 + CH3CHO; and VO2CHCH3 + O2 → VO3C + CH3OH, VO3C + O2 → VO3 + CO2. The kinetically most favorable reaction products are CH3CHO, H2O, and CO2 in the gas phase model catalytic cycles. The results parallel similar behavior in the selective oxidation of propylene over condensed phase V2O5/SiO2 catalysts.
Co-reporter:Jia-Bi Ma, Xiao-Nan Wu, Xian-Xia Zhao, Xun-Lei Ding and Sheng-Gui He
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 38) pp:NaN12228-12228
Publication Date(Web):2010/08/16
DOI:10.1039/C0CP00360C
A series of vanadium and phosphorus heteronuclear oxide cluster cations (VxPyOz+) are prepared by laser ablation and the reactions of V3PO10˙+ and V4O10˙+ with methane in a fast flow reactor under the same conditions are studied. A time of flight mass spectrometer is used to detect the cluster distribution before and after reactions. In addition to previously identified reaction of V4O10˙+ + CH4 → V4O10H+ + CH3˙, the observation of hydrogen atom pickup cluster V3PO10H+ suggests the reaction: V3PO10˙+ + CH4 → V3PO10H+ + CH3˙. The rate of the reaction of V4O10˙+ with CH4 is approximately 2.5 times faster than that of V3PO10˙+ with CH4. Density functional theory (DFT) calculations predict that structure of V3PO10˙+ is topologically similar to that of V4O10˙+, as well as that of P4O10˙+, which is very similar to V4O10˙+ in terms of methane activation in previous studies. The facile methane activation by the homo- and hetero-nuclear oxide clusters can all be attributed to the presence of an oxygen-centered radical (O˙) in these clusters. Further theoretical study indicates that the O˙ radical (or spin density of the cluster) can transfer within the high symmetry V4O10˙+ and P4O10˙+ clusters quite easily, and CH4 molecule further enhances the rate of intra-cluster spin density transfer. In contrast, the intra-cluster spin density transfer within low symmetry V3PO10˙+ is thermodynamically forbidden. The experimentally observed reactivity difference is consistent with the theoretical consideration of the intra-cluster spin density transfer.
Co-reporter:Xiao-Nan Wu, Yan-Xia Zhao, Wei Xue, Zhe-Chen Wang, Sheng-Gui He and Xun-Lei Ding
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 16) pp:NaN3997-3997
Publication Date(Web):2010/03/20
DOI:10.1039/B925294K
Cerium oxide cluster cations (CemOn+, m = 2–16; n = 2m, 2m ± 1 and 2m ± 2) are prepared by laser ablation and reacted with carbon monoxide (CO) and small hydrocarbon molecules (CH4, C2H4, and C2H6) in a fast flow reactor. A time of flight mass spectrometer is used to detect the cluster distribution before and after the reactions. The observation of oxygen reduction and hydrogen pickup of CemO2m+ clusters strongly suggests the following reactions: (1) CemO2m+ + C2H4 → CemO2m−1+ + C2H4O (m = 2–6); (2) CemO2m+ + CO → CemO2m−1+ + CO2 (m = 4–6); and (3) CemO2m+ + CH4/C2H6 → CemO2mH+ + CH3/C2H5 (m = 2–4). Density functional theory (DFT) calculations are performed to study reaction mechanisms of Ce2O4+ + X (X = CO, CH4, C2H4, and C2H6). The calculated results are in good agreement with the experimental observations. The structural and bonding properties of CemO2m+ (m = 2–5) clusters are also investigated by the DFT calculations. The unpaired electron in each of the clusters is mainly distributed over one Ce atom (4f and 5p orbitals) and two O atoms (2p orbital) in a CeO2 moiety, which can be considered as the active site in the cluster. To further understand the nature of the active sites in CemO2m+ clusters, the fast flow reaction experiments are also carried out on zirconium oxide clusters ZrmOn+, because both Zr ([Kr]4d25s2) and Ce ([Xe]4f15d16s2) have the same number of valence electrons while the latter has one more f and one less d electrons. In addition to the oxygen transfer reactions such as ZrmO2m+ + C2H4 → ZrmO2m−1+ + C2H4O (m = 1–4) reported in the literature, hydrogen abstraction reactions ZrmO2m+ + CH4/C2H6 → ZrmO2mH+ + CH3/C2H5 are also identified. The rate constants of CO oxidation as well as hydrogen abstraction by CemO2m+ and ZrmO2m+ are very different. The reactivity and selectivity of CemO2m+versus ZrmO2m+ can be well rationalized based on the DFT calculations. The oxygen transfer and hydrogen abstraction reactions studied in this work are of widespread importance. The nature of the active sites of CemO2m+ clusters is unique and may be considered in the use and design of cerium oxide based catalysts.
Co-reporter:Yan-Xia Zhao, Xiao-Nan Wu, Zhe-Chen Wang, Sheng-Gui He and Xun-Lei Ding
Chemical Communications 2010 - vol. 46(Issue 10) pp:NaN1738-1738
Publication Date(Web):2010/02/05
DOI:10.1039/B924603G
Stoichiometric early transition metal oxide cations (TiO2)1–5+, (ZrO2)1–4+, (HfO2)1–2+, (V2O5)1–5+, (Nb2O5)1–3+, (Ta2O5)1–2+, (MoO3)1–2+, (WO3)1–3+, and Re2O7+ are able to activate the C–H bond of methane under near room temperature conditions.
Co-reporter:Yan-Xia Zhao, Xiao-Na Li, Zhen Yuan, Qing-Yu Liu, Qiang Shi and Sheng-Gui He
Chemical Science (2010-Present) 2016 - vol. 7(Issue 7) pp:NaN4735-4735
Publication Date(Web):2016/03/29
DOI:10.1039/C6SC00539J
The reactivity of closed-shell gas phase cluster anions AuTi3O7− and AuTi3O8− with methane under thermal collision conditions was studied by mass spectrometric experiments and quantum chemical calculations. Methane activation was observed with the formation of AuCH3 in both cases, while the formation of formaldehyde was also identified in the reaction system of AuTi3O8−. The cooperative effect of the separated Au+ and O2− ions on the clusters induces the cleavage of the first C–H bond of methane. Further activation of the second C–H bond by a peroxide ion O22− leads to the formation of formaldehyde. This study shows that closed-shell species on metal oxides can be reactive enough to facilitate thermal H–CH3 bond cleavage and the subsequent conversion.
Co-reporter:Yan-Xia Zhao, Qing-Yu Liu, Mei-Qi Zhang and Sheng-Gui He
Dalton Transactions 2016 - vol. 45(Issue 28) pp:NaN11495-11495
Publication Date(Web):2016/06/13
DOI:10.1039/C6DT01246A
The study of gas phase ion–molecule reactions by state-of-the-art mass spectrometric experiments in conjunction with quantum chemistry calculations offers an opportunity to clarify the elementary steps and mechanistic details of bond activation and conversion processes. In the past few decades, a considerable number of publications have been devoted to the ion–molecule reactions of metal clusters, the experimentally and theoretically tractable models for the active phase of condensed phase systems. The focus of this perspective concerns progress on activation and transformation of important inorganic and organic molecules by negatively charged metal clusters. The metal cluster anions cover bare metal clusters as well as ligated systems with oxygen, carbon, and nitrogen, among others. The following important issues have been summarized and discussed: (i) dependence of chemical reactivity and selectivity on cluster structures and sizes, metals and metal oxidation states, odd–even electron numbers, etc. and (ii) effects of doping, ligation, and pre-adsorption on the reactivity of metal clusters toward rather inert molecules.
Co-reporter:Jia-Bi Ma, Jing-Heng Meng and Sheng-Gui He
Dalton Transactions 2015 - vol. 44(Issue 7) pp:NaN3135-3135
Publication Date(Web):2014/12/22
DOI:10.1039/C4DT03398A
Cerium–vanadium oxide cluster cations CeVO5+ were generated by laser ablation, mass-selected using a quadrupole mass filter, thermalized through collisions with helium atoms, and then reacted with ethene molecules in a linear ion trap reactor. The cluster reactions have been characterized by time-of-flight mass spectrometry and density functional theory calculations. The CeVO5+ cluster has a closed-shell electronic structure and contains a peroxide (O22−) unit. The cluster bonded O22− species is reactive enough to oxidize a C2H4 molecule to generate C2H4O2 that can be an acetic acid molecule. Atomic oxygen radicals (O−˙), superoxide radicals (O2−˙), and peroxides are the three common reactive oxygen species. The reactivity of cluster bonded O−˙ and O2−˙ radicals has been widely studied while the O22− species were generally thought to be much less reactive or inert toward small molecules under thermal collision conditions. This work is among the first to report the reactivity of the peroxide unit on transition metal oxide clusters with hydrocarbon molecules, to the best of our knowledge.
Co-reporter:Yan-Xia Zhao, Xiao-Nan Wu, Jia-Bi Ma, Sheng-Gui He and Xun-Lei Ding
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 6) pp:NaN1938-1938
Publication Date(Web):2011/01/05
DOI:10.1039/C0CP01171A
We introduce chemical structures and reactivity of oxygen-centred radicals (O−˙) over transition metal oxide (TMO) clusters based on mass spectrometric and density functional theory studies. Two main issues will be discussed: (1) the compositions of TMO clusters that have the bonding characteristics of (or contain) the O−˙ radicals; and (2) the dependences (cluster structures, sizes, charge states, metal types, etc.) of the chemical reactivity and selectivity for the O−˙ radicals over TMO clusters. One of the goals of cluster chemistry is to understand the elementary reactions involved with complex heterogeneous catalysis. The study of the O−˙ containing TMO clusters permits rather detailed descriptions for how mono-nuclear oxygen-centred radicals may exist and react with small molecules over TMO based catalysts.
Co-reporter:Xiao-Na Li, Bo Xu, Xun-Lei Ding and Sheng-Gui He
Dalton Transactions 2012 - vol. 41(Issue 18) pp:NaN5570-5570
Publication Date(Web):2012/03/14
DOI:10.1039/C2DT12174C
Vanadium oxide cluster anions (VxOy−, x = 2–3; y = 3–7) are produced by laser ablation and reacted with water in a fast flow reactor. A time-of-flight mass spectrometer is used to detect the cluster distribution before and after the reactions. Reaction channels of molecular hydrogen elimination (for V2,3O3−), water association (for V2O5− and V3O6,7−) and the coexistence of both channels (for V2O4− and V3O4,5−) are observed. V2O6− and V3O8− are nearly inert toward water. Density functional theory (DFT) calculations are performed to study the reaction mechanism of V2O3− in different spin states with water and the results support the experimental observation. The reaction mechanism of V2O3+ with water is also studied, which is in agreement with the experimental report in previous literature [Eur. J. Inorg. Chem., 2008, 4961] that molecular hydrogen elimination is a minor reaction channel for V2O3+ + H2O. The influence of cluster charge states and oxidation states of vanadium atoms on the cluster reactivity are presented based on the experimental and theoretical studies.
Co-reporter:Yan-Ping Ma, Sheng-Gui He, Xun-Lei Ding, Zhe-Chen Wang, Wei Xue and Qiang Shi
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 14) pp:NaN2552-2552
Publication Date(Web):2009/02/13
DOI:10.1039/B815010A
The intermolecular interactions between two meso-tetraphenylporphyrin diacid H4TPPCl2 monomers are investigated by density functional theory with the PBE1PBE functional and 6-31G* basis set. Structures of five stable isomers of (H4TPPCl2)2 are determined. It is found that the interaction (IMHB-1) of Cl with orthoH atoms in two phenyl groups of H4TPPCl2 is unique in that it is the strongest interaction between two H4TPPCl2 monomers. Natural bond orbital analysis is carried out to explain the subtle differences of hydrogen bondings in these isomers. To understand the interactions of water molecule with H4TPPCl2 and (H4TPPCl2)2, structures of H4TPPCl2·H2O and nine isomers of (H4TPPCl2)2·H2O are also determined. The binding energy of H4TPPCl2·H2O is 36.47 kJ mol−1, less than that of the most stable structure of (H4TPPCl2)2 dimers, 41.59 kJ mol−1. The dimers containing the special IMHB-1 interactions may be the elementary building blocks for the aggregation of H4TPPCl2, which is supported by the crystal structure of H4TPPCl2·H2O·2CH3CN. Thus, study of the interactions between two or a small number of gas molecules can provide important information for understanding the main interactions and stable structures in related condensed-phase systems.

![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,5'-[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[2,9-bis(2-ethylhexyl)-13-(2-methoxyethoxy)-](/data/chemimg/139600/1614219-32-7.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,5'-[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[2,9-bis(2-ethylhexyl)-13-(2-methoxyethoxy)-](/data/chemimg/139600/1614219-32-7_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,5'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[2,9-bis(2-ethylhexyl)-13-(2-methoxyethoxy)-](/data/chemimg/139600/1614219-31-6.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,5'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[2,9-bis(2-ethylhexyl)-13-(2-methoxyethoxy)-](/data/chemimg/139600/1614219-31-6_b.png)
![Glycine, N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[2-(6-fluoro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-, ethyl ester](/data/chemimg/139500/1412448-74-8.png)
![Glycine, N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[2-(6-fluoro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-, ethyl ester](/data/chemimg/139500/1412448-74-8_b.png)
![Glycine, N-[2-(6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1412448-71-5.png)
![Glycine, N-[2-(6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1412448-71-5_b.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1412448-68-0.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1412448-68-0_b.png)
![Glycine, N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[2-(6-fluoro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-](/data/chemimg/139500/1401247-34-4.png)
![Glycine, N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[2-(6-fluoro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-](/data/chemimg/139500/1401247-34-4_b.png)




![Benzo[g]quinazoline-1,3(2H,4H)-diacetic acid, 2,4-dioxo-](/data/chemimg/139500/1401247-39-9.png)
![Benzo[g]quinazoline-1,3(2H,4H)-diacetic acid, 2,4-dioxo-](/data/chemimg/139500/1401247-39-9_b.png)
![Benzo[g]quinazoline-3(2H)-acetic acid, 1,4-dihydro-2,4-dioxo-](/data/chemimg/139500/1401247-38-8.png)
![Benzo[g]quinazoline-3(2H)-acetic acid, 1,4-dihydro-2,4-dioxo-](/data/chemimg/139500/1401247-38-8_b.png)
![Glycine, N-[2-(6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1401247-33-3.png)
![Glycine, N-[2-(6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1401247-33-3_b.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1401247-32-2.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1401247-32-2_b.png)
![Benzo[g]quinazoline-2,4(1H,3H)-dione, potassium salt (1:2)](http://img.cochemist.com/data/images/1401247-31-1.png)
![Benzo[g]quinazoline-2,4(1H,3H)-dione, potassium salt (1:2)](http://img.cochemist.com/data/images/1401247-31-1_b.png)
![Benzo[g]quinazoline-2,4(1H,3H)-dione, sodium salt (1:2)](http://img.cochemist.com/data/images/1401247-30-0.png)
![Benzo[g]quinazoline-2,4(1H,3H)-dione, sodium salt (1:2)](http://img.cochemist.com/data/images/1401247-30-0_b.png)
![Benzo[g]quinazoline-2,4(1H,3H)-dione, lithium salt (1:2)](http://img.cochemist.com/data/images/1401247-29-7.png)
![Benzo[g]quinazoline-2,4(1H,3H)-dione, lithium salt (1:2)](http://img.cochemist.com/data/images/1401247-29-7_b.png)


















![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-[(4-methylphenyl)methoxy]-](/data/chemimg/139500/1356240-97-5.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-[(4-methylphenyl)methoxy]-](/data/chemimg/139500/1356240-97-5_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(phenylmethoxy)-](/data/chemimg/139500/1356240-93-1.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(phenylmethoxy)-](/data/chemimg/139500/1356240-93-1_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-[(4-bromophenyl)methoxy]-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-88-4.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-[(4-bromophenyl)methoxy]-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-88-4_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-(1,1-dimethylethoxy)-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-83-9.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-(1,1-dimethylethoxy)-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-83-9_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(octadecyloxy)-](/data/chemimg/139500/1356240-79-3.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(octadecyloxy)-](/data/chemimg/139500/1356240-79-3_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-(decyloxy)-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-75-9.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-(decyloxy)-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-75-9_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(pentyloxy)-](/data/chemimg/139500/1356240-70-4.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(pentyloxy)-](/data/chemimg/139500/1356240-70-4_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-butoxy-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-65-7.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-12-butoxy-2,9-bis(2-ethylhexyl)-](/data/chemimg/139500/1356240-65-7_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-methoxy-](/data/chemimg/139500/1356240-60-2.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-methoxy-](/data/chemimg/139500/1356240-60-2_b.png)
![Carbamic acid, N-[2-[[12-bromo-2,9-bis(2-ethylhexyl)-1,2,3,8,9,10-hexahydro-1,3,8,10-tetraoxoanthra[2,1,9-def:6,5,10-d'e'f']diisoquinolin-5-yl]oxy]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/139500/1356240-49-7.png)
![Carbamic acid, N-[2-[[12-bromo-2,9-bis(2-ethylhexyl)-1,2,3,8,9,10-hexahydro-1,3,8,10-tetraoxoanthra[2,1,9-def:6,5,10-d'e'f']diisoquinolin-5-yl]oxy]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/139500/1356240-49-7_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-dicyclohexyl-12-(2-methoxyethoxy)-](/data/chemimg/139500/1356240-34-0.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-dicyclohexyl-12-(2-methoxyethoxy)-](/data/chemimg/139500/1356240-34-0_b.png)
![Stannane, 1,1'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-trimethyl-](/data/chemimg/139400/1352642-37-5.png)
![Stannane, 1,1'-[4,8-bis[5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-trimethyl-](/data/chemimg/139400/1352642-37-5_b.png)
![Propanedinitrile, 2-[2,7-bis(5-bromo-4-decyl-2-thienyl)-9H-fluoren-9-ylidene]-](/data/chemimg/139500/1324007-39-7.png)
![Propanedinitrile, 2-[2,7-bis(5-bromo-4-decyl-2-thienyl)-9H-fluoren-9-ylidene]-](/data/chemimg/139500/1324007-39-7_b.png)
![Benzonitrile, 4,4'-[[2,9-bis(2-ethylhexyl)-1,2,3,8,9,10-hexahydro-1,3,8,10-tetraoxoanthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-5,12-diyl]bis(oxy)]bis-](/data/chemimg/139500/1313710-48-3.png)
![Benzonitrile, 4,4'-[[2,9-bis(2-ethylhexyl)-1,2,3,8,9,10-hexahydro-1,3,8,10-tetraoxoanthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-5,12-diyl]bis(oxy)]bis-](/data/chemimg/139500/1313710-48-3_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-5,12-bis(4-pyridinyloxy)-, hydrochloride (1:2)](http://img.cochemist.com/data/images/1313710-47-2.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-5,12-bis(4-pyridinyloxy)-, hydrochloride (1:2)](http://img.cochemist.com/data/images/1313710-47-2_b.png)





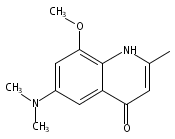
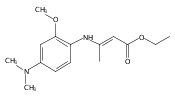

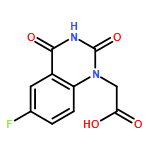
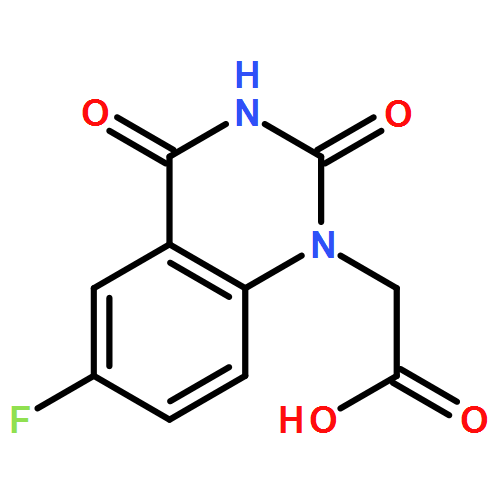
![Glycine, N-[2-(3,4-dihydro-6,7-dimethoxy-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-37-2.png)
![Glycine, N-[2-(3,4-dihydro-6,7-dimethoxy-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-37-2_b.png)
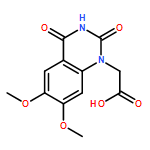
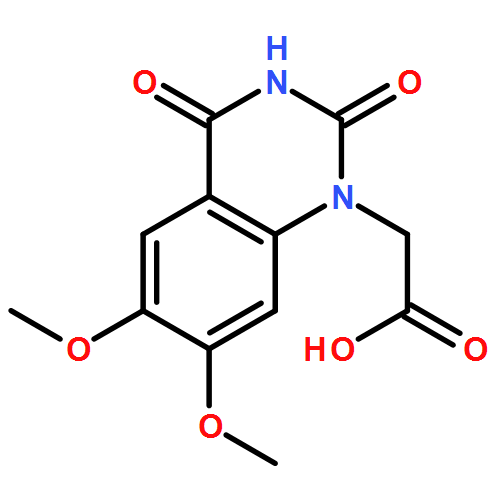
![Glycine, N-[2-(3,4-dihydro-2,4-dioxobenzo[g]quinazolin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-32-7.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxobenzo[g]quinazolin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-32-7_b.png)
![Benzo[g]quinazoline-1(2H)-acetic acid, 3,4-dihydro-2,4-dioxo-](/data/chemimg/139500/1258302-31-6.png)
![Benzo[g]quinazoline-1(2H)-acetic acid, 3,4-dihydro-2,4-dioxo-](/data/chemimg/139500/1258302-31-6_b.png)
![Glycine, N-[2-(3,4-dihydro-6-methyl-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-29-2.png)
![Glycine, N-[2-(3,4-dihydro-6-methyl-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-29-2_b.png)
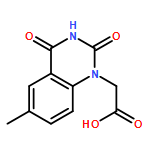
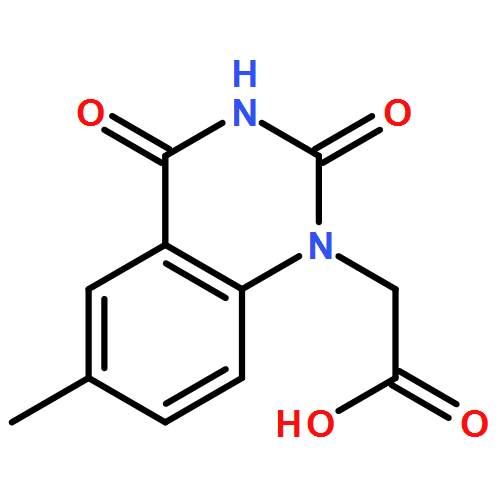
![Glycine, N-[2-(3,4-dihydro-2,4-dioxopyrido[2,3-d]pyrimidin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-24-7.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxopyrido[2,3-d]pyrimidin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-, ethyl ester](/data/chemimg/139500/1258302-24-7_b.png)
![Glycine, N-[2-(3,4-dihydro-6,7-dimethoxy-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-20-3.png)
![Glycine, N-[2-(3,4-dihydro-6,7-dimethoxy-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-20-3_b.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxobenzo[g]quinazolin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-18-9.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxobenzo[g]quinazolin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-18-9_b.png)
![Glycine, N-[2-(3,4-dihydro-6-methyl-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-17-8.png)
![Glycine, N-[2-(3,4-dihydro-6-methyl-2,4-dioxo-1(2H)-quinazolinyl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-17-8_b.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxopyrido[2,3-d]pyrimidin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-15-6.png)
![Glycine, N-[2-(3,4-dihydro-2,4-dioxopyrido[2,3-d]pyrimidin-1(2H)-yl)acetyl]-N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-](/data/chemimg/139500/1258302-15-6_b.png)
![Pyrido[2,3-d]pyrimidine-1(2H)-acetic acid, 3,4-dihydro-2,4-dioxo-](/data/chemimg/139500/1258302-13-4.png)
![Pyrido[2,3-d]pyrimidine-1(2H)-acetic acid, 3,4-dihydro-2,4-dioxo-](/data/chemimg/139500/1258302-13-4_b.png)
![Stannane, 1,1'-(3,4'-didodecyl[2,2'-bithiophene]-5,5'-diyl)bis[1,1,1-trimethyl-](/data/chemimg/139500/1239898-47-5.png)
![Stannane, 1,1'-(3,4'-didodecyl[2,2'-bithiophene]-5,5'-diyl)bis[1,1,1-trimethyl-](/data/chemimg/139500/1239898-47-5_b.png)
![4H-Dithieno[3,2-b:2',3'-d]pyrrole, 4-(2-ethylhexyl)-2,6-bis(trimethylstannyl)-](/data/chemimg/139500/1234499-56-9.png)
![4H-Dithieno[3,2-b:2',3'-d]pyrrole, 4-(2-ethylhexyl)-2,6-bis(trimethylstannyl)-](/data/chemimg/139500/1234499-56-9_b.png)
![Stannane, 1,1'-[4,8-bis(dodecyloxy)benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-trimethyl-](/data/chemimg/139400/1044795-08-5.png)
![Stannane, 1,1'-[4,8-bis(dodecyloxy)benzo[1,2-b:4,5-b']dithiophene-2,6-diyl]bis[1,1,1-trimethyl-](/data/chemimg/139400/1044795-08-5_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,12-dibromo-2,9-bis(2-ethylhexyl)-](/data/chemimg/102800/851786-15-7.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,12-dibromo-2,9-bis(2-ethylhexyl)-](/data/chemimg/102800/851786-15-7_b.png)


![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,6,12,13-tetrakis[4-(1,1-dimethylethyl)phenoxy]-2,9-bis(2-ethylhexyl)- (9CI)](/data/chemimg/102700/318293-22-0.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5,6,12,13-tetrakis[4-(1,1-dimethylethyl)phenoxy]-2,9-bis(2-ethylhexyl)- (9CI)](/data/chemimg/102700/318293-22-0_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dicyclohexyl-6,13-bis[4-(1,1-dimethylethyl)phenoxy]-](/data/chemimg/102800/192126-72-0.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dicyclohexyl-6,13-bis[4-(1,1-dimethylethyl)phenoxy]-](/data/chemimg/102800/192126-72-0_b.png)




![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-](/data/chemimg/3730400/82531-03-1.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-](/data/chemimg/3730400/82531-03-1_b.png)
![Iron, chloro[5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/27600/36995-20-7.png)
![Iron, chloro[5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-5-12)-](/data/chemimg/27600/36995-20-7_b.png)
![Acetamide,N-[4-(dimethylamino)-2-methoxyphenyl]-](http://img.cochemist.com/ccimg/7500/7475-00-5.png)
![Acetamide,N-[4-(dimethylamino)-2-methoxyphenyl]-](http://img.cochemist.com/ccimg/7500/7475-00-5_b.png)



![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(2-methoxyethoxy)-](/data/chemimg/139400/1356240-55-5.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 5-bromo-2,9-bis(2-ethylhexyl)-12-(2-methoxyethoxy)-](/data/chemimg/139400/1356240-55-5_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dicyclohexyl-5-(2-methoxyethoxy)-12-(4-pyridinyloxy)-](/data/chemimg/139400/1356240-42-0.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dicyclohexyl-5-(2-methoxyethoxy)-12-(4-pyridinyloxy)-](/data/chemimg/139400/1356240-42-0_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dicyclohexyl-5,12-bis(4-pyridinyloxy)-](/data/chemimg/139400/1356240-37-3.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dicyclohexyl-5,12-bis(4-pyridinyloxy)-](/data/chemimg/139400/1356240-37-3_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-5,12-bis(2-methoxyethoxy)-](/data/chemimg/139400/1313710-49-4.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-5,12-bis(2-methoxyethoxy)-](/data/chemimg/139400/1313710-49-4_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 6,13-bis[4-(1,1-dimethylethyl)phenoxy]-2,9-bis(2-ethylhexyl)-](/data/chemimg/102700/952684-26-3.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 6,13-bis[4-(1,1-dimethylethyl)phenoxy]-2,9-bis(2-ethylhexyl)-](/data/chemimg/102700/952684-26-3_b.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-5,12-bis(4-pyridinyloxy)-](/data/chemimg/139400/1239352-59-0.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2-ethylhexyl)-5,12-bis(4-pyridinyloxy)-](/data/chemimg/139400/1239352-59-0_b.png)