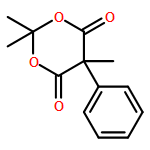
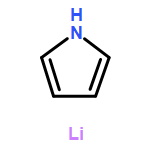
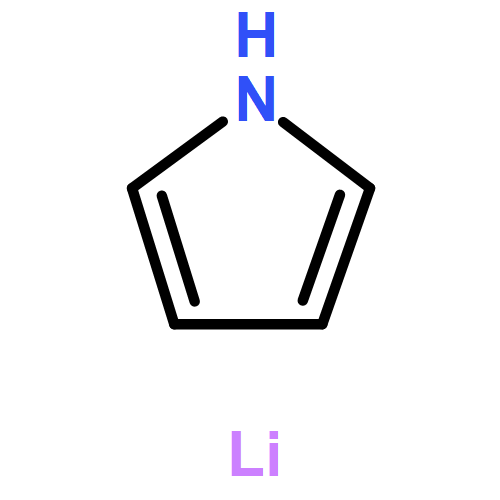
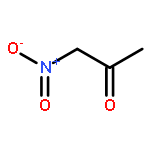
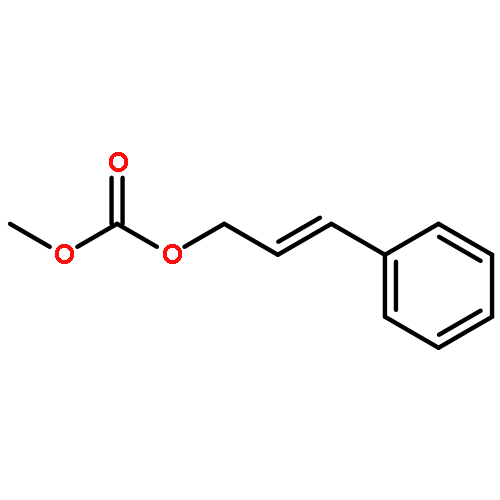
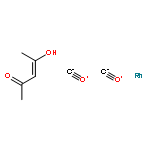
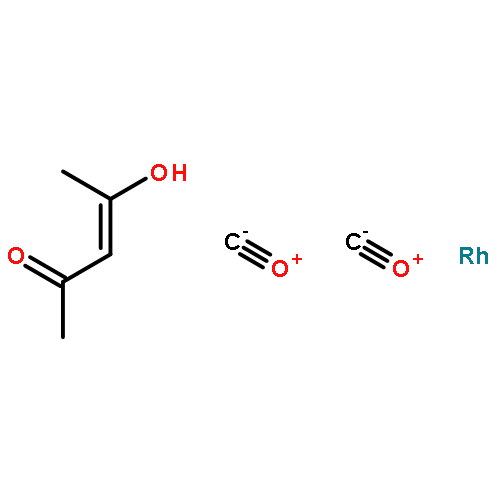
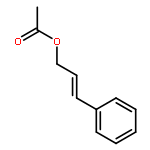
Co-reporter:Mary K. Smith and Jon A. Tunge
Organic Letters October 20, 2017 Volume 19(Issue 20) pp:5497-5497
Publication Date(Web):September 29, 2017
DOI:10.1021/acs.orglett.7b01751
A new strategy to access conjugated allenynes via a decarboxylative coupling of propargyl esters of propiolates has been developed. In this process, allenyl-palladium intermediates are coupled with acetylides that are generated in situ to form the conjugated allenynes. Finally, the coupling is demonstrated to be highly stereospecific, providing a route to enantioenriched allenes.
Co-reporter:Kinthada Ramakumar, Tapan Maji, James J. Partridge, and Jon A. Tunge
Organic Letters August 4, 2017 Volume 19(Issue 15) pp:
Publication Date(Web):July 24, 2017
DOI:10.1021/acs.orglett.7b01752
A new method is developed for the synthesis of spirooxindoles from amines and isatins via C–H functionalization. The reaction leverages the tert-amino effect to form an enolate–iminium intermediate via [1,5]-hydride shift followed by cyclization. Interestingly the hydride migrates to the N atom of a C═N, which is atypical for hydride additions to imines.
Co-reporter:Shehani N. Mendis and Jon A. Tunge
Chemical Communications 2016 vol. 52(Issue 49) pp:7695-7698
Publication Date(Web):23 May 2016
DOI:10.1039/C6CC03672D
We report the first example of a palladium-catalyzed decarboxylative dearomatization reaction that occurs via Pd-π-benzyl intermediates. In fact, the Pd-catalyzed decarboxylative cross-coupling reaction of benzyl enol carbonates can lead to either the dearomatized alicyclic ketones or α-monoarylated ketone products depending on the catalyst and ligand employed.
Co-reporter:Tapan Maji, Camina H. Mendis, Ward H. Thompson, Jon A. Tunge
Journal of Molecular Catalysis A: Chemical 2016 Volume 424() pp:145-152
Publication Date(Web):1 December 2016
DOI:10.1016/j.molcata.2016.08.021
•Study of one of the most selective hydroformylations of butadiene to adipaldehyde, which is industrially useful.•The first experimental observation of phosphine rhodium-catalyzed isomerizing hydroformylation to form adipaldehyde.•The selective formation of adipaldehyde from butadiene is due to standard hydroformylation and isomerizing hydroformylation.•DFT calculations show the intermediates, transition states and energetics of the key alkene isomerization.•Energy barriers for the formation of isomeric alkenes are similar.The (DIOP)rhodium-catalyzed hydroformylation of butadiene has been shown to give among the highest selectivities for formation of adipaldehyde, which is useful for the synthesis of nylon. Herein, isomerizing hydroformylation is shown to be a mechanism that is partially responsible for this selectivity and density functional theory studies are used to reveal the detailed pathway for the requisite alkene isomerization.
Co-reporter:Tapan Maji and Jon A. Tunge
Organic Letters 2015 Volume 17(Issue 19) pp:4766-4769
Publication Date(Web):September 16, 2015
DOI:10.1021/acs.orglett.5b02308
An interceptive decarboxylative allylation protocol has been developed utilizing pyrone as a C4 synthon. This palladium-catalyzed transformation difunctionalizes the pyrone moiety by in situ generation and activation of both the electrophile and nucleophile via a double decarboxylation pathway. Ultimately, allyl carbonates react smoothly with 2-carboxypyrone under mild reaction conditions to generate synthetically useful acyclic dienoic esters, forming carbon dioxide as the sole byproduct.
Co-reporter:Shehani N. Mendis and Jon A. Tunge
Organic Letters 2015 Volume 17(Issue 21) pp:5164-5167
Publication Date(Web):October 9, 2015
DOI:10.1021/acs.orglett.5b02410
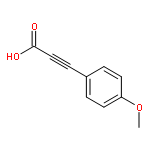
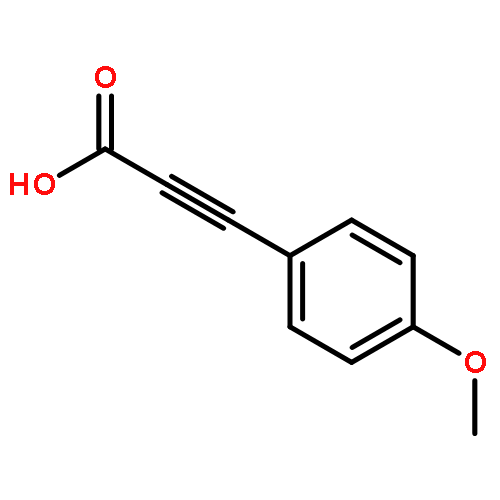
Enantioenriched benzyl esters of propiolic acids undergo highly stereospecific decarboxylative coupling to provide 1,1-diarylethynyl methanes. This sp–sp3 coupling does not require strongly basic conditions or preformed organometallics and produces CO2 as the sole byproduct. Ultimately, this method results in the successful transfer of stereochemical information from secondary benzyl alcohols to generate enantioenriched tertiary diarylmethanes.
Co-reporter:Simon B. Lang;Kathryn M. O'Nele;Dr. Justin T. Douglas ;Dr. Jon A. Tunge
Chemistry - A European Journal 2015 Volume 21( Issue 51) pp:18589-18593
Publication Date(Web):
DOI:10.1002/chem.201503644
Abstract
The room temperature radical decarboxylative allylation of N-protected α-amino acids and esters has been accomplished via a combination of palladium and photoredox catalysis to provide homoallylic amines. Mechanistic investigations revealed that the stability of the α-amino radical, which is formed by decarboxylation, dictates the predominant reaction pathway between competing mechanisms.
Co-reporter:Simon B. Lang ; Kathryn M. O’Nele
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13606-13609
Publication Date(Web):September 17, 2014
DOI:10.1021/ja508317j
A combination of photoredox and palladium catalysis has been employed to facilitate the room temperature decarboxylative allylation of recalcitrant α-amino and phenylacetic allyl esters. This operationally simple process produces CO2 as the only byproduct and provides direct access to allylated alkanes. After photochemical oxidation, the carboxylate undergoes radical decarboxylation to site-specifically generate radical intermediates which undergo allylation. A radical dual catalysis mechanism is proposed. Free phenylacetic acids were also allylated utilizing similar reactions conditions.
Co-reporter:Simon B. Lang, Theresa M. Locascio, and Jon A. Tunge
Organic Letters 2014 Volume 16(Issue 16) pp:4308-4311
Publication Date(Web):August 4, 2014
DOI:10.1021/ol502023d
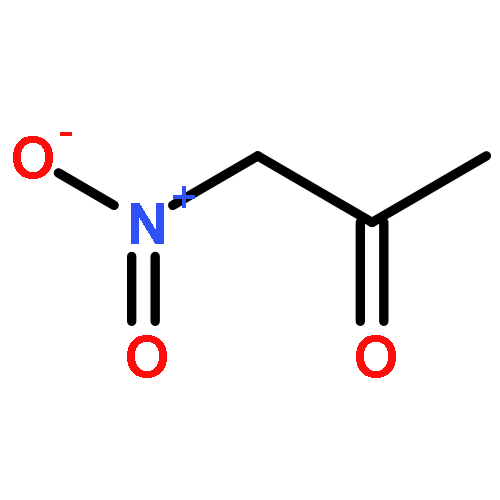
The direct coupling of allyl alcohols with nitroalkanes, nitriles, and aldehydes using catalytic Pd(PPh3)4 has been accomplished via activation of C–OH bonds with CO2. The in situ formation of carbonates from alcohols and CO2 facilitates oxidative addition to Pd to form reactive π-allylpalladium intermediates. In addition, the formation of a strong base activates nucleophiles toward the reaction with the π-allylpalladium electrophile. Overall, this atom economical reaction provides a new C–C bond without the use of an external base and generates water as the only byproduct.
Co-reporter:Tapan Maji, Kinthada Ramakumar and Jon A. Tunge
Chemical Communications 2014 vol. 50(Issue 90) pp:14045-14048
Publication Date(Web):24 Sep 2014
DOI:10.1039/C4CC07001A
A new strategy has been developed for the benzylation of nitriles directly from benzyl alcohols. In this process benzyl alcohols undergo retro-Claisen activation with cyanoacetic esters to generate an active electrophile and a carbanionic nucleophile. In the presence of Pd(0) these intermediates undergo catalytic coupling to generate a new C–C bond, resulting in the formation of phenyl propionitriles.
Co-reporter:Tapan Maji and Jon A. Tunge
Organic Letters 2014 Volume 16(Issue 19) pp:5072-5075
Publication Date(Web):September 19, 2014
DOI:10.1021/ol5024294
α-Cyano aldehydes undergo selective transition-metal-catalyzed monoallylation to provide α-allylated nitriles. The transformation leads to linear substitution products with palladium catalysts or branched allylated nitriles using an iridium catalyst. Facile TBD-catalyzed retro-Claisen cleavage is leveraged to attain selective monoallylation.
Co-reporter:Yamuna Ariyarathna and Jon A. Tunge
Chemical Communications 2014 vol. 50(Issue 90) pp:14049-14052
Publication Date(Web):25 Sep 2014
DOI:10.1039/C4CC07253G
The reaction of Meldrum's acid, pyrrolide, and allyl carbonates allows a multicomponent decarboxylative coupling to form allylated acyl pyrroles. This strategy is made possible by the in situ formation of β-oxo carboxylates from Meldrum's acid. Subsequent decarboxylative enolate formation and electrophilic allylation complete the reaction. Addition of benzylidene malononitriles as good Michael acceptors allow a 4-component interceptive decarboxylative allylation.
Co-reporter:Kinthada Ramakumar and Jon A. Tunge
Chemical Communications 2014 vol. 50(Issue 86) pp:13056-13058
Publication Date(Web):05 Sep 2014
DOI:10.1039/C4CC06369D
A redox amination strategy was developed for the synthesis of N-aryl-1-aminoindoles by N–N bond formation. Reaction of nitrosobenzenes with readily available indolines using Brønsted acid catalysis allows N–N bond formation under mild conditions. This method exploits the inherent reducing power of indoline to synthesize biologically relevant molecular architectures via redox amination. A one-pot synthesis of 1-aminoindoles starting from simple aniline and indolines is likewise described.
Co-reporter:Yamuna Ariyarathna and Jon A. Tunge
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 42) pp:8386-8389
Publication Date(Web):15 Sep 2014
DOI:10.1039/C4OB01752H
A variety of ester enolate equivalents are generated in situ and undergo α-allylation in high yields via palladium-catalyzed decarboxylative allylation. The transformations are complete within very short reaction times under ambient conditions. Synthesis of α-allylated acyl derivatives provides access to other carboxylic acid and alcohol derivatives via acyl group substitution or reduction.
Co-reporter:Alexander J. Grenning, Christie K. Van Allen, Tapan Maji, Simon B. Lang, and Jon A. Tunge
The Journal of Organic Chemistry 2013 Volume 78(Issue 14) pp:7281-7287
Publication Date(Web):June 5, 2013
DOI:10.1021/jo400793a
Herein we present the development of asymmetric deacylative allylation of ketone enolates. The reaction directly couples readily available ketone pronucleophiles with allylic alcohols using facile retro-Claisen cleavage to form reactive intermediates in situ. The simplicity and robustness of the reaction conditions is demonstrated by the preparation of >6 g of an allylated tetralone from commercially available materials. Furthermore, use of nonracemic PHOX ligands allows intermolecular formation of quaternary stereocenters directly from allylic alcohols.
Co-reporter:Antonio Recio, III, Jeffrey D. Heinzman and Jon A. Tunge
Chemical Communications 2012 vol. 48(Issue 1) pp:142-144
Publication Date(Web):07 Nov 2011
DOI:10.1039/C1CC16011G
Decarboxylative benzylation of nitriles is achieved via coupling of metallated nitriles with Pd-π-benzyl complexes that are generated in situ from cyanoacetic benzyl esters. In addition, decarboxylative couplings of α,α-disubstituted 2-methylfuranyl cyanoacetates can lead to either decarboxylative arylation or benzylation depending on the reaction conditions.
Co-reporter:Kalicharan Cattopadhyay, Antonio Recio III and Jon A. Tunge
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 34) pp:6826-6829
Publication Date(Web):12 Jul 2012
DOI:10.1039/C2OB25962A
We report the palladium-catalyzed, pyrrolidine-mediated α-benzylation of enamines generated from aldehydes and ketones. The method allows for direct coupling of medicinally relevant coumarin moieties with aldehydes and ketones in good yield under mild conditions. The reaction is believed to proceed via a Pd–π-benzyl complex generated from (coumarinyl)methyl acetates.
Co-reporter:Meghan Schmitt, Alexander J. Grenning, Jon A. Tunge
Tetrahedron Letters 2012 Volume 53(Issue 34) pp:4494-4497
Publication Date(Web):22 August 2012
DOI:10.1016/j.tetlet.2012.05.138
Using palladium-catalyzed decarboxylation, several cascade reactions of allyl and prenyl nitroalkanoates that lead to nitro-containing chemical building blocks are described. A nitronate Michael addition/Tsuji–Trost allylation cascade was developed, leading to functionally dense chemical building blocks. Likewise, a Tsuji–Trost/decarboxylative protonation sequence was developed for the synthesis of orthogonally functionalized 2° nitroalkanes. The latter method provides rapid access to the indolizidine core.
Co-reporter:Jimmie D. Weaver, Antonio Recio III, Alexander J. Grenning, and Jon A. Tunge
Chemical Reviews 2011 Volume 111(Issue 3) pp:1846
Publication Date(Web):January 14, 2011
DOI:10.1021/cr1002744
Co-reporter:Alexander J. Grenning
Journal of the American Chemical Society 2011 Volume 133(Issue 37) pp:14785-14794
Publication Date(Web):August 10, 2011
DOI:10.1021/ja205717f
A new method for allylic alkylation of a variety of relatively nonstabilized carbon nucleophiles is described herein. In this process of “deacylative allylation”, the coupling partners, an allylic alcohol and a ketone pronucleophile, undergo in situ retro-Claisen activation to generate an allylic acetate and a carbanion. In the presence of palladium, these reactive intermediates undergo catalytic coupling to form a new C–C bond. In comparison to unimolecular decarboxylative allylation, a commonly utilized method for allylation of carbon anions, deacylative allylation is an intermolecular process. Moreover, deacylative allylation allows the direct coupling of readily available allylic alcohols. Lastly, the full utility of deacylative allylation is demonstrated by the rapid construction of a variety 1,6-heptadienes via 3-component couplings.
Co-reporter:Ranjan Jana and Jon A. Tunge
The Journal of Organic Chemistry 2011 Volume 76(Issue 20) pp:8376-8385
Publication Date(Web):September 7, 2011
DOI:10.1021/jo201476h
A robust and practical polymer-supported, homogeneous, recyclable biphephos rhodium(I) catalyst has been developed for C–C bond formation reactions. Control of polymer molecular weight allowed tuning of the polymer solubility such that the polymer-supported catalyst is soluble in nonpolar solvents and insoluble in polar solvents. Using the supported rhodium catalysts, addition of aryl and vinylboronic acids to the electrophiles such as enones, aldehydes, N-sulfonyl aldimines, and alkynes occurs smoothly to provide products in high yields. Additions of terminal alkynes to enones and industrially relevant hydroformylation reactions have also been successfully carried out. Studies show that the leaching of Rh from the polymer support is low and catalyst recycle can be achieved by simple precipitation and filtration.
Co-reporter:Ranjan Jana;James J. Partridge ; Jon A. Tunge
Angewandte Chemie International Edition 2011 Volume 50( Issue 22) pp:5157-5161
Publication Date(Web):
DOI:10.1002/anie.201100765
Co-reporter:Alexer J. Grenning
Angewandte Chemie 2011 Volume 123( Issue 7) pp:1726-1729
Publication Date(Web):
DOI:10.1002/ange.201006273
Co-reporter:Ranjan Jana;James J. Partridge ; Jon A. Tunge
Angewandte Chemie 2011 Volume 123( Issue 22) pp:5263-5267
Publication Date(Web):
DOI:10.1002/ange.201100765
Co-reporter:Alexer J. Grenning
Angewandte Chemie International Edition 2011 Volume 50( Issue 7) pp:1688-1691
Publication Date(Web):
DOI:10.1002/anie.201006273
Co-reporter:Robert R. P. Torregrosa ; Yamuna Ariyarathna ; Kalicharan Chattopadhyay
Journal of the American Chemical Society 2010 Volume 132(Issue 27) pp:9280-9282
Publication Date(Web):June 21, 2010
DOI:10.1021/ja1035557
Benzyl esters of propiolic and β-keto acids undergo catalytic decarboxylative coupling when treated with appropriate palladium catalysts. Such decarboxylative couplings allow the benzylation of alkynes without the use of strong bases and/or organometallics. This allows the synthesis of sensitive benzylic alkynes that are prone to undergo isomerizations under basic conditions. Additionally, decarboxylation facilitates the site-specific benzylation of diketones and ketoesters under mild, base-free conditions. Ultimately, the methodology described expands our ability to cross-couple medicinally relevant heterocycles.
Co-reporter:Jimmie D. Weaver ; Being J. Ka ; David K. Morris ; Ward Thompson
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12179-12181
Publication Date(Web):August 17, 2010
DOI:10.1021/ja104196x
Allyl sulfonylacetic esters undergo highly stereospecific, palladium-catalyzed decarboxylative allylation. The reaction allows the stereospecific formation of tertiary homoallylic sulfones in high yield. In contrast to related reactions that proceed at −100 °C and require highly basic preformed organometallics, the decarboxylative coupling described herein occurs under mild nonbasic conditions and requires no stoichiometric additives. Allylation of the intermediate α-sulfonyl anion is more rapid than racemization, leading to a highly enantiospecific process. Density functional theory calculations indicate that the barrier for racemization is 9.9 kcal/mol, so the barrier for allylation must be <9.9 kcal/mol.
Co-reporter:Kalicharan Chattopadhyay, Ranjan Jana, Victor W. Day, Justin T. Douglas and Jon A. Tunge
Organic Letters 2010 Volume 12(Issue 13) pp:3042-3045
Publication Date(Web):June 10, 2010
DOI:10.1021/ol101042x
A stereochemical test has been used to probe the mechanism of decarboxylative allylation. This probe suggests that the mechanism of DcA reactions can change based on the substitution pattern at the α-carbon of the nucleophile; however, reaction via stabilized malonate nucleophiles is the lower energy pathway. Lastly, this mechanistic proposal has predictive power and can be used to explain chemoselectivities in decarboxylative reactions that were previously confounding.
Co-reporter:Alexander J. Grenning and Jon A. Tunge
Organic Letters 2010 Volume 12(Issue 4) pp:740-742
Publication Date(Web):January 20, 2010
DOI:10.1021/ol902828p
Allyl nitroacetates undergo decarboxylative allylation to provide tertiary nitroalkanes in high yield. Moreover, the transformations are complete within several minutes under ambient conditions. High yields result because O-allylation of the intermediate nitronates, which is typically problematic, is reversible under conditions of the decarboxylative allylation process. Lastly, the preparation of substrate allyl nitroacetates by tandem Knoevenagel/Diels−Alder sequences allows the facile synthesis of relatively complex substrates that undergo diastereoselective decarboxylative allylation.
Co-reporter:Nirmal K. Pahadi ; Miranda Paley ; Ranjan Jana ; Shelli R. Waetzig
Journal of the American Chemical Society 2009 Volume 131(Issue 46) pp:16626-16627
Publication Date(Web):November 3, 2009
DOI:10.1021/ja907357g
A wide variety of aldehydes, ketones, and lactols undergo redox amination when allowed to react with 3-pyrroline in the presence of a mild Brønsted acid catalyst. This reaction utilizes the inherent reducing power of 3-pyrroline to perform the equivalent of a reductive amination to form alkyl pyrroles. In doing so, the reaction avoids stoichiometric reducing agents that are typically associated with reductive aminations. Moreover, the redox amination protocol allows access to alkyl pyrroles that cannot be made via standard reductive amination.
Co-reporter:Rushi Trivedi and Jon A. Tunge
Organic Letters 2009 Volume 11(Issue 24) pp:5650-5652
Publication Date(Web):November 18, 2009
DOI:10.1021/ol902291z
An anionic iron complex catalyzes the decarboxylative allylation of phenols to form allylic ethers in high yield. The allylation is regioselective rather than regiospecific. This suggests that the allylation proceeds through π-allyl iron intermediates in contrast to related allylations of carbon nucleophiles that have been proposed to proceed via σ-allyl complexes. Ultimately, iron catalysts have the potential to replace more expensive palladium catalysts that are typically utilized for decarboxylative couplings.
Co-reporter:Ranjan Jana, Rushi Trivedi and Jon A. Tunge
Organic Letters 2009 Volume 11(Issue 15) pp:3434-3436
Publication Date(Web):July 9, 2009
DOI:10.1021/ol901288r
Allyl esters of 3-carboxylcoumarins undergo facile decarboxylative coupling at just 25−50 °C. This represents the first extension of decarboxylative C−C bond-forming reactions to the coupling of aromatics with sp3-hybridized electrophiles. Finally, the same concept can be applied to the sp2−sp3 couplings of pyrones and flavones. Thus, a variety of biologically important heteroaromatics can be readily functionalized without the need for strong bases or stoichiometric organometallics that are typically required for more standard cross-coupling reactions.
Co-reporter:Antonio Recio III and Jon A. Tunge
Organic Letters 2009 Volume 11(Issue 24) pp:5630-5633
Publication Date(Web):November 18, 2009
DOI:10.1021/ol902065p
Palladium-catalyzed decarboxylative α-allylation of nitriles readily occurs with use of Pd2(dba)3 and rac-BINAP. This catalyst mixture also allows the highly regiospecific α-allylation of nitriles in the presence of much more acidic α-protons. Thus, the reported method provides access to compounds that are not readily available via base-mediated allylation chemistries. Lastly, mechanistic investigations indicate that there is a competition between C- and N-allylation of an intermediate nitrile-stabilized anion and that N-allylation is followed by a rapid [3,3]-sigmatropic rearrangement.
Co-reporter:Shaofeng Duan, Ranjan Jana and Jon A. Tunge
The Journal of Organic Chemistry 2009 Volume 74(Issue 12) pp:4612-4614
Publication Date(Web):May 21, 2009
DOI:10.1021/jo900367g
Herein we report that simple Lewis acids catalyze the hydroarylation of benzylidene malonates with phenols. Ultimately, 3,4-disubstituted dihydrocoumarins are obtained via a hydroarylation−lactonization sequence. Moreover, the dihydrocoumarins are formed with a high degree of diastereoselectivity favoring the trans stereoisomer.
Co-reporter:Chao Wang, Nirmal Pahadi, Jon A. Tunge
Tetrahedron 2009 65(26) pp: 5102-5109
Publication Date(Web):
DOI:10.1016/j.tet.2009.04.071
Co-reporter:Shelli R. Waetzig and Jon A. Tunge
Chemical Communications 2008 (Issue 28) pp:3311-3313
Publication Date(Web):10 Jun 2008
DOI:10.1039/B806949B
This communication details the Pd-catalyzed decarboxylation of selenocarbonates; use of a chiral nonracemic catalyst affords enantioenriched allyl selenides which undergo stereospecific [2,3]-sigmatropic rearrangements to form enantioenriched allylic amines and chlorides.
Co-reporter:Kelin Li and Jon A. Tunge
ACS Combinatorial Science 2008 Volume 10(Issue 2) pp:170
Publication Date(Web):January 31, 2008
DOI:10.1021/cc700150q
Phenols provide a useful template for diversification via sequential hydroarylation reactions. Specifically, a protocol has been developed that begins with the hydroarylation of cinnamic acids by 3,5-dimethoxyphenol to produce dihydrocoumarins. This activated ester undergoes facile ring-opening with amines to form a C−N bond and regenerate a phenol. The resulting phenol can be further functionalized via a second hydroarylation reaction. Thus, in 3–4 steps, a phenol is coupled with a cinnamic acid, an amine, and a cinnamic or propiolic acid.
Co-reporter:Shelli R. Waetzig;Dinesh K. Rayabarapu;Jimmie D. Weaver
Angewandte Chemie 2006 Volume 118(Issue 30) pp:
Publication Date(Web):3 JUL 2006
DOI:10.1002/ange.200600721
Freie Wahl: Die palladiumkatalysierte decarboxylierende Kupplung von Estern mit zwei Allylgruppen führt zur kinetischen Allylierung in α-Position zu elektronenziehenden Gruppen. Die Tandem-Allylierung/Cope-Umlagerung eröffnet den Zugang zu γ-Kupplungsprodukten (siehe Schema). Somit kann man das gewünschte Regioisomer einfach über die Reaktionstemperatur auswählen.
Co-reporter:Shelli R. Waetzig;Dinesh K. Rayabarapu;Jimmie D. Weaver
Angewandte Chemie International Edition 2006 Volume 45(Issue 30) pp:
Publication Date(Web):3 JUL 2006
DOI:10.1002/anie.200600721
Have it both ways: The palladium-catalyzed decarboxylative coupling of esters with two allyl groups results in kinetic allylation at a position α to electron-withdrawing groups. Tandem allylation/Cope rearrangement provides access to γ-coupling products (see scheme). Thus, the desired regioisomer is obtained simply by controlling the temperature of the reaction mixture.
Co-reporter:Erin C. Burger and Jon A. Tunge
Chemical Communications 2005 (Issue 22) pp:2835-2837
Publication Date(Web):21 Apr 2005
DOI:10.1039/B503568F
The ruthenium-catalyzed stereospecific decarboxylative allylation of ketone enolates provides access to γ,δ-unsaturated ketones with good yields and enantioenrichments.
Co-reporter:Chao Wang and Jon Tunge
Chemical Communications 2004 (Issue 23) pp:2694-2695
Publication Date(Web):07 Oct 2004
DOI:10.1039/B410576A
Through a 2e- oxidation-reduction cycle, phenylselenides catalytically activate N-chlorosuccinimide toward electrophilic reactions with ketones, resulting in α-haloketones.
Co-reporter:Kinthada Ramakumar and Jon A. Tunge
Chemical Communications 2014 - vol. 50(Issue 86) pp:NaN13058-13058
Publication Date(Web):2014/09/05
DOI:10.1039/C4CC06369D
A redox amination strategy was developed for the synthesis of N-aryl-1-aminoindoles by N–N bond formation. Reaction of nitrosobenzenes with readily available indolines using Brønsted acid catalysis allows N–N bond formation under mild conditions. This method exploits the inherent reducing power of indoline to synthesize biologically relevant molecular architectures via redox amination. A one-pot synthesis of 1-aminoindoles starting from simple aniline and indolines is likewise described.
Co-reporter:Kalicharan Cattopadhyay, Antonio Recio III and Jon A. Tunge
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 34) pp:NaN6829-6829
Publication Date(Web):2012/07/12
DOI:10.1039/C2OB25962A
We report the palladium-catalyzed, pyrrolidine-mediated α-benzylation of enamines generated from aldehydes and ketones. The method allows for direct coupling of medicinally relevant coumarin moieties with aldehydes and ketones in good yield under mild conditions. The reaction is believed to proceed via a Pd–π-benzyl complex generated from (coumarinyl)methyl acetates.
Co-reporter:Yamuna Ariyarathna and Jon A. Tunge
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 42) pp:NaN8389-8389
Publication Date(Web):2014/09/15
DOI:10.1039/C4OB01752H
A variety of ester enolate equivalents are generated in situ and undergo α-allylation in high yields via palladium-catalyzed decarboxylative allylation. The transformations are complete within very short reaction times under ambient conditions. Synthesis of α-allylated acyl derivatives provides access to other carboxylic acid and alcohol derivatives via acyl group substitution or reduction.
Co-reporter:Shelli R. Waetzig and Jon A. Tunge
Chemical Communications 2008(Issue 28) pp:NaN3313-3313
Publication Date(Web):2008/06/10
DOI:10.1039/B806949B
This communication details the Pd-catalyzed decarboxylation of selenocarbonates; use of a chiral nonracemic catalyst affords enantioenriched allyl selenides which undergo stereospecific [2,3]-sigmatropic rearrangements to form enantioenriched allylic amines and chlorides.
Co-reporter:Antonio Recio, III, Jeffrey D. Heinzman and Jon A. Tunge
Chemical Communications 2012 - vol. 48(Issue 1) pp:NaN144-144
Publication Date(Web):2011/11/07
DOI:10.1039/C1CC16011G
Decarboxylative benzylation of nitriles is achieved via coupling of metallated nitriles with Pd-π-benzyl complexes that are generated in situ from cyanoacetic benzyl esters. In addition, decarboxylative couplings of α,α-disubstituted 2-methylfuranyl cyanoacetates can lead to either decarboxylative arylation or benzylation depending on the reaction conditions.
Co-reporter:Yamuna Ariyarathna and Jon A. Tunge
Chemical Communications 2014 - vol. 50(Issue 90) pp:NaN14052-14052
Publication Date(Web):2014/09/25
DOI:10.1039/C4CC07253G
The reaction of Meldrum's acid, pyrrolide, and allyl carbonates allows a multicomponent decarboxylative coupling to form allylated acyl pyrroles. This strategy is made possible by the in situ formation of β-oxo carboxylates from Meldrum's acid. Subsequent decarboxylative enolate formation and electrophilic allylation complete the reaction. Addition of benzylidene malononitriles as good Michael acceptors allow a 4-component interceptive decarboxylative allylation.
Co-reporter:Tapan Maji, Kinthada Ramakumar and Jon A. Tunge
Chemical Communications 2014 - vol. 50(Issue 90) pp:NaN14048-14048
Publication Date(Web):2014/09/24
DOI:10.1039/C4CC07001A
A new strategy has been developed for the benzylation of nitriles directly from benzyl alcohols. In this process benzyl alcohols undergo retro-Claisen activation with cyanoacetic esters to generate an active electrophile and a carbanionic nucleophile. In the presence of Pd(0) these intermediates undergo catalytic coupling to generate a new C–C bond, resulting in the formation of phenyl propionitriles.
Co-reporter:Shehani N. Mendis and Jon A. Tunge
Chemical Communications 2016 - vol. 52(Issue 49) pp:NaN7698-7698
Publication Date(Web):2016/05/23
DOI:10.1039/C6CC03672D
We report the first example of a palladium-catalyzed decarboxylative dearomatization reaction that occurs via Pd-π-benzyl intermediates. In fact, the Pd-catalyzed decarboxylative cross-coupling reaction of benzyl enol carbonates can lead to either the dearomatized alicyclic ketones or α-monoarylated ketone products depending on the catalyst and ligand employed.
.jpg)






































![L-METHIONINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-, 2-PROPENYL ESTER](http://img.cochemist.com/ccimg/887700/887646-46-0.png)
![L-METHIONINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-, 2-PROPENYL ESTER](http://img.cochemist.com/ccimg/887700/887646-46-0_b.png)


![Benzeneacetic acid, 4-[bis(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/833500/833484-70-1.png)
![Benzeneacetic acid, 4-[bis(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/833500/833484-70-1_b.png)





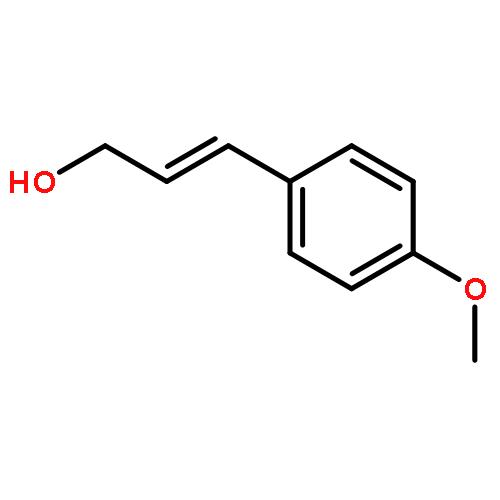
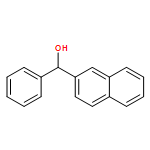
![2-Naphthalenemethanol, α-[4-(methylthio)phenyl]-](/data/chemimg/1973400/774235-32-4.png)
![2-Naphthalenemethanol, α-[4-(methylthio)phenyl]-](/data/chemimg/1973400/774235-32-4_b.png)









![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-6,7-dimethyl-](http://img.cochemist.com/ccimg/485900/485821-94-1.png)
![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-6,7-dimethyl-](http://img.cochemist.com/ccimg/485900/485821-94-1_b.png)
![2H-1-BENZOPYRAN-2-ONE, 4-[(ACETYLOXY)METHYL]-5,7-DIMETHYL-](http://img.cochemist.com/ccimg/485900/485821-93-0.png)
![2H-1-BENZOPYRAN-2-ONE, 4-[(ACETYLOXY)METHYL]-5,7-DIMETHYL-](http://img.cochemist.com/ccimg/485900/485821-93-0_b.png)
![3H-Naphtho[2,1-b]pyran-3-one, 1-(1-piperidinylmethyl)-](http://img.cochemist.com/ccimg/401600/401523-10-2.png)
![3H-Naphtho[2,1-b]pyran-3-one, 1-(1-piperidinylmethyl)-](http://img.cochemist.com/ccimg/401600/401523-10-2_b.png)




![Benzenesulfonamide, N-[(2E)-1,3-diphenyl-2-propenyl]-4-methyl-](http://img.cochemist.com/ccimg/292700/292639-13-5.png)
![Benzenesulfonamide, N-[(2E)-1,3-diphenyl-2-propenyl]-4-methyl-](http://img.cochemist.com/ccimg/292700/292639-13-5_b.png)
![Bicyclo[2.2.1]heptan-2-ol,1,7,7-trimethyl-3-(4-morpholinyl)-, (1R,2S,3R,4S)-](http://img.cochemist.com/ccimg/287200/287105-48-0.png)
![Bicyclo[2.2.1]heptan-2-ol,1,7,7-trimethyl-3-(4-morpholinyl)-, (1R,2S,3R,4S)-](http://img.cochemist.com/ccimg/287200/287105-48-0_b.png)


![Benzeneacetic acid, α-cyano-α-[(2E)-3-phenyl-2-propen-1-yl]-, ethyl ester](/data/chemimg/1790400/214982-17-9.png)
![Benzeneacetic acid, α-cyano-α-[(2E)-3-phenyl-2-propen-1-yl]-, ethyl ester](/data/chemimg/1790400/214982-17-9_b.png)








![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/180700/180635-56-7.png)
![2-Propen-1-ol, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/180700/180635-56-7_b.png)






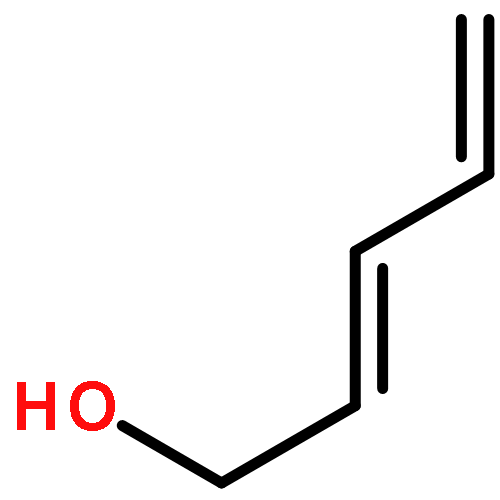
![Benzene, 1-methoxy-4-[(3-methyl-3-butenyl)oxy]-](http://img.cochemist.com/ccimg/169400/169310-73-0.png)
![Benzene, 1-methoxy-4-[(3-methyl-3-butenyl)oxy]-](http://img.cochemist.com/ccimg/169400/169310-73-0_b.png)


![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/160800/160788-63-6.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/160800/160788-63-6_b.png)




![Benzene, [(1-methyl-2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/153000/152959-29-0.png)
![Benzene, [(1-methyl-2-phenylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/153000/152959-29-0_b.png)














![2-Naphthalenemethanol, α-[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/1071400/124419-75-6.png)
![2-Naphthalenemethanol, α-[4-(1,1-dimethylethyl)phenyl]-](/data/chemimg/1071400/124419-75-6_b.png)
![Cyclohexanone, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/123000/122902-06-1.png)
![Cyclohexanone, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/123000/122902-06-1_b.png)
![Dibenzo[d,f][1,3,2]dioxaphosphepin,6,6'-[[3,3'-bis(1,1-dimethylethyl)-5,5'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis(oxy)]bis-](http://img.cochemist.com/ccimg/121700/121627-17-6.png)
![Dibenzo[d,f][1,3,2]dioxaphosphepin,6,6'-[[3,3'-bis(1,1-dimethylethyl)-5,5'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis(oxy)]bis-](http://img.cochemist.com/ccimg/121700/121627-17-6_b.png)




![L-Alanine, N-[(1,1-dimethylethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/113800/113707-00-9.png)
![L-Alanine, N-[(1,1-dimethylethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/113800/113707-00-9_b.png)


![3H-Naphtho[2,1-b]pyran-3-one, 1-[(acetyloxy)methyl]-](http://img.cochemist.com/ccimg/106300/106236-97-9.png)
![3H-Naphtho[2,1-b]pyran-3-one, 1-[(acetyloxy)methyl]-](http://img.cochemist.com/ccimg/106300/106236-97-9_b.png)













![Glycine, N-[(phenylmethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/97000/96921-56-1.png)
![Glycine, N-[(phenylmethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/97000/96921-56-1_b.png)


![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-](http://img.cochemist.com/ccimg/88900/88861-50-1.png)
![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-](http://img.cochemist.com/ccimg/88900/88861-50-1_b.png)

![Benzene, 1-[(1E)-1,2-diphenylethenyl]-4-methyl-](http://img.cochemist.com/ccimg/84300/84224-86-2.png)
![Benzene, 1-[(1E)-1,2-diphenylethenyl]-4-methyl-](http://img.cochemist.com/ccimg/84300/84224-86-2_b.png)
![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-7-methyl-](http://img.cochemist.com/ccimg/84200/84105-43-1.png)
![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-7-methyl-](http://img.cochemist.com/ccimg/84200/84105-43-1_b.png)











![[(1-methyl-1H-pyrrol-2-yl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/79700/79691-33-1.png)
![[(1-methyl-1H-pyrrol-2-yl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/79700/79691-33-1_b.png)
![Dibenzo[d,f][1,3,2]dioxaphosphepin, 6-phenoxy-](http://img.cochemist.com/ccimg/78600/78560-02-8.png)
![Dibenzo[d,f][1,3,2]dioxaphosphepin, 6-phenoxy-](http://img.cochemist.com/ccimg/78600/78560-02-8_b.png)




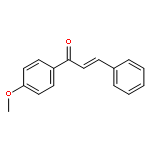
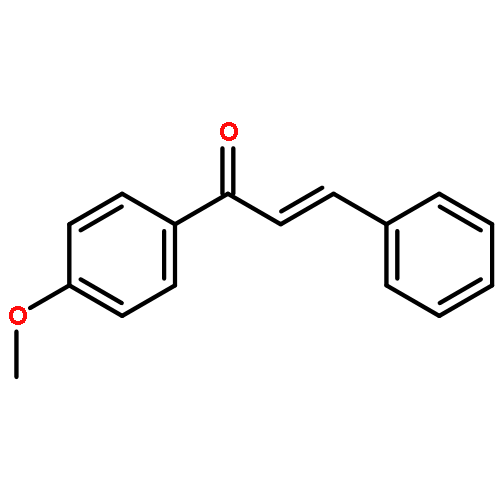
![Benzene, 1-[(1E)-1,2-diphenylethenyl]-4-methoxy-](http://img.cochemist.com/ccimg/72500/72474-43-2.png)
![Benzene, 1-[(1E)-1,2-diphenylethenyl]-4-methoxy-](http://img.cochemist.com/ccimg/72500/72474-43-2_b.png)

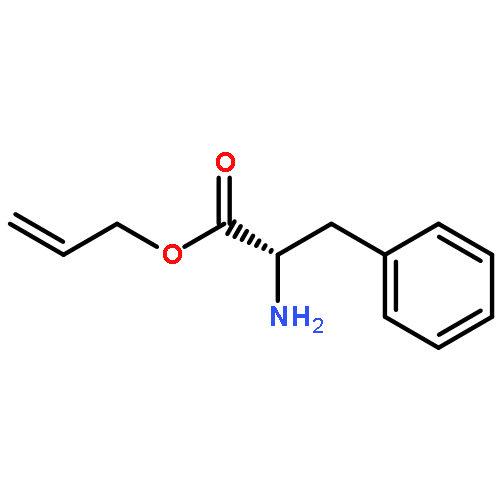
![L-Phenylalanine, N-[(phenylmethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/64300/64286-85-7.png)
![L-Phenylalanine, N-[(phenylmethoxy)carbonyl]-, 2-propenyl ester](http://img.cochemist.com/ccimg/64300/64286-85-7_b.png)




![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/61000/60951-30-6.png)
![2H-1-Benzopyran-2-one, 4-[(acetyloxy)methyl]-7-methoxy-](http://img.cochemist.com/ccimg/61000/60951-30-6_b.png)















![2-[[2-(TRIFLUOROMETHYL)PHENYL]METHYLIDENE]PROPANEDINITRILE](http://img.cochemist.com/ccimg/37000/36937-89-0.png)
![2-[[2-(TRIFLUOROMETHYL)PHENYL]METHYLIDENE]PROPANEDINITRILE](http://img.cochemist.com/ccimg/37000/36937-89-0_b.png)
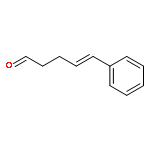
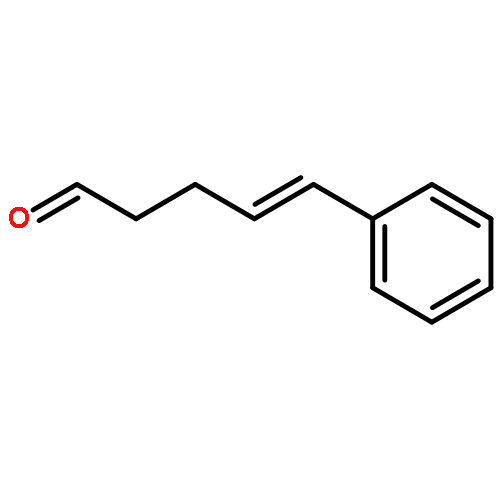
![Bicyclo[3.1.1]hept-2-ene-2-methanol,6,6-dimethyl-, 2-acetate](http://img.cochemist.com/ccimg/35700/35670-93-0.png)
![Bicyclo[3.1.1]hept-2-ene-2-methanol,6,6-dimethyl-, 2-acetate](http://img.cochemist.com/ccimg/35700/35670-93-0_b.png)
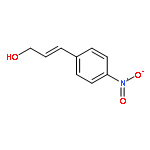
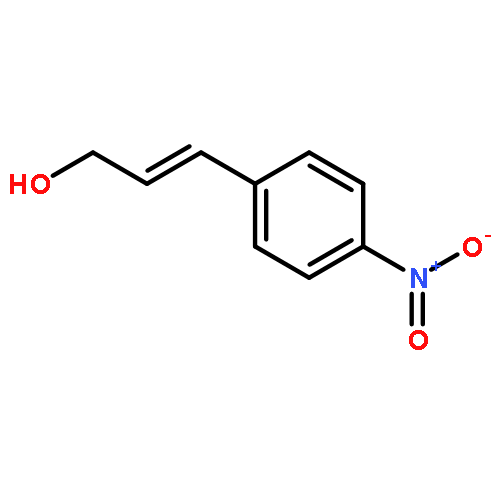
















![2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]acetic Acid](http://img.cochemist.com/ccimg/25700/25616-33-5.png)
![2-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-3-phenylpropanoyl]amino]acetic Acid](http://img.cochemist.com/ccimg/25700/25616-33-5_b.png)













![6-chlorodibenzo[d,f][1,3,2]dioxaphosphepine](http://img.cochemist.com/ccimg/16700/16611-68-0.png)
![6-chlorodibenzo[d,f][1,3,2]dioxaphosphepine](http://img.cochemist.com/ccimg/16700/16611-68-0_b.png)














![2-[(2,4-dioxo-3-phenyl-1,3-thiazolidin-5-yl)(methyl)amino]benzoic acid](http://img.cochemist.com/ccimg/5900/5840-58-4.png)
![2-[(2,4-dioxo-3-phenyl-1,3-thiazolidin-5-yl)(methyl)amino]benzoic acid](http://img.cochemist.com/ccimg/5900/5840-58-4_b.png)