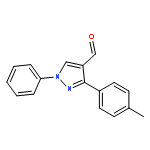
Co-reporter:Yong-Fang Yao, Zhong-Chang Wang, Song-Yu Wu, Qing-fang Li, ... Hai-Liang Zhu
Biochemical Pharmacology 2017 Volume 137(Volume 137) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bcp.2017.04.026
Microtubules are essential for the mitotic division of cells and have become an attractive target for anti-tumour drugs due to the increased incidence of cancer and significant mitosis rate of tumour cells. In this study, a total of six indole 1-position modified 1-indolyl acetate-5-nitroimidazole derivatives were designed, synthesized, and evaluated for their ability to inhibit tubulin polymerization caused by binding to the colchicine-binding site of tubulin. Among them, compound 3 displayed the best ability to inhibit tubulin polymerization; it also exhibited better anti-proliferative activities than colchicine against a panel of human cancer cells (with IC50 values ranging from 15 to 40 nM), especially HeLa cells (with IC50 values of 15 nM), based on the cellular cytotoxicity assay results. Moreover, cellular mechanism studies indicated that compound 3 could induce G2/M phase arrest and apoptosis of HeLa and MCF-7 cells, which were associated with alterations in the expression of cell cycle-checkpoint related proteins (Cyclin B1, Cdc2, and P21) and a reduction in the mitochondrial membrane potential as well as alterations in the levels of apoptosis-related proteins (PARP, Caspase 9, Bcl-2, and Bax) of these cells, respectively. Importantly, in vivo studies further revealed that compound 3 could dramatically suppress HeLa cell xenograft tumour growth compared with vehicle and CA-4 phosphate (CA-4P), and no signs of toxicity were observed in these mice. Collectively, these in vitro and in vivo results indicated that compound 3 might be a promising lead compound for further development as a potential anti-cancer drug.Download high-res image (182KB)Download full-size image
Co-reporter:Han-Yue Qiu, Jiang-Yan Fu, Min-Kai Yang, Hong-Wei Han, ... Yong-Hua Yang
Biochemical Pharmacology 2017 Volume 146(Volume 146) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.bcp.2017.10.009
The signal transducer and activator of transcription 3 is a constitutively activated oncogenic protein in various human tumors and represents a valid target for anticancer drug design. In this study, we have achieved a new type of STAT3 inhibitors based on structural modifications on shikonin scaffold, guided by computational modelling. By tests, PMMB-187 exhibited a more outstanding profile than shikonin on a small panel of human breast cancer cells, especially for the MDA-MB-231 cells. For the cellular mechanisms research, PMMB-187 was found to induce cell apoptosis in MDA-MB-231 cells, associated with the reduction of mitochondrial membrane potential, production of ROS and alteration of the levels of apoptosis-related proteins. Furthermore, PMMB-187 inhibited constitutive/inducible STAT3 activation, transcriptional activity, nuclear translocation and downstream target genes expression in STAT3-dependent breast cancer cells MDA-MB-231. Besides, no obvious inhibitory effect on activation of STAT1 and STAT5 was observed with PMMB-187 treatment. Most notably, the in vivo studies further revealed that PMMB-187 could dramatically suppress the MDA-MB-231 cells xenografted tumor growth. The in vitro and in vivo results collectively suggest that PMMB-187 may serve as a promising lead compound for the further development of potential therapeutic anti-neoplastic agents.Identification of new potent STAT3 inhibitors based on the interaction model of natural product shikonin (A), the lead compound (B), and its optimization (C).Download high-res image (244KB)Download full-size image
Co-reporter:Juan Sun, Shen-Zhen Ren, Xiao-Yuan Lu, Jing-Jing Li, Fa-Qian Shen, Chen Xu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 9(Issue 9) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bmc.2017.03.038
Focal adhesion kinase (FAK) is an important drug target that plays a fundamental role in mediating signal transduction system. We report herein the discovery of a novel class of 1,3,4-oxadiazole-2(3H)-thione derivatives containing piperazine skeleton with improved potency toward FAK. All of the 17 new synthesized compounds were assayed for the anticancer activities against four cancer cells, HepG2, Hela, SW116 and BGC823. Because of the combination of 1,4-benzodioxan, 1,3,4-oxadiazole and piperazine ring, most of them exhibited remarkable antitumor activities. Notably, compound 5m showed the most potent biological activities (IC50 = 5.78 μM for HepG2, and IC50 = 47.15 μM for SW1116), and its anti-FAK inhibitory activity (IC50 = 0.78 μM) was also the best. Computational docking studies also showed that compound 5m has interaction with FAK key residues in the active site.Download high-res image (64KB)Download full-size image
Co-reporter:Fa-Qian Shen, Zhong-Chang Wang, Song-Yu Wu, Shen-Zhen Ren, Ruo-Jun Man, Bao-Zhong Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.07.020
In our previous study, we designed a series of pyrazole derivatives as novel COX-2 inhibitors. In order to obtain novel dual inhibitors of COX-2 and 5-LOX, herein we designed and synthesized 20 compounds by hybridizing pyrazole with substituted coumarin who was reported to exhibit 5-LOX inhibition to select potent compounds using adequate biological trials sequentially including selective inhibition of COX-2 and 5-LOX, anti-proliferation in vitro, cells apoptosis and cell cycle. Among them, the most potent compound 11g (IC50 = 0.23 ± 0.16 μM for COX-2, IC50 = 0.87 ± 0.07 μM for 5-LOX, IC50 = 4.48 ± 0.57 μM against A549) showed preliminary superiority compared with the positive controls Celecoxib (IC50 = 0.41 ± 0.28 μM for COX-2, IC50 = 7.68 ± 0.55 μM against A549) and Zileuton (IC50 = 1.35 ± 0.24 μM for 5-LOX). Further investigation confirmed that 11g could induce human non-small cell lung cancer A549 cells apoptosis and arrest the cell cycle at G2 phase in a dose-dependent manner. Our study might contribute to COX-2, 5-LOX dual inhibitors thus exploit promising novel cancer prevention agents.As docking study exhibited, target compound 11g showed the lowest interaction energy of 64.38 kcal/mol and −66.62 kcal/mol after interacting with COX-2 (PDB code: 1CX2, a, b) and 5-LOX (PDB code: 3V99, c, d), respectively.Download high-res image (101KB)Download full-size image
Co-reporter:Wei Wei, Qi Liu, Zhen-Zhen Li, Wei-Kang Shi, Xing Fu, Jia Liu, Xuan Zhu, Xiao-Cong Wang, Ning Xu, Teng-Fei Li, Fu-Rui Jiang, Zhu-Ping Xiao, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2017 Volume 133(Volume 133) pp:
Publication Date(Web):16 June 2017
DOI:10.1016/j.ejmech.2017.03.074
•Adenosine containing 3-arylfuran-2(5H)-ones was firstly considered as TyrRS inhibitors.•The most active inhibitor shows excellent potency compared to the marketed antibiotic ciprofloxacin.•Adoption of the same pose as the enzyme co-crystallized ligand tyrosyl adenylate.•Being a lead for discovery of antibacterial agents.Tyrosyl-tRNA synthetase (TyrRS) is an aminoacyl-tRNA synthetase family protein that possesses an essential role in bacterial protein synthesis. The synthesis, structure-activity relationship, and evolution of a novel series of adenosine-containing 3-arylfuran-2(5H)-ones as TyrRS inhibitors are described. Advanced compound d3 from this series exhibited excellent affinity for TyrRS with IC50 of 0.61 ± 0.04 μM. Bacterial growth inhibition assays demonstrated that d3 showed submicromolar antibacterial potency against Escherichia coli and Pseudomonas aeruginosa, and compared to the marketed antibiotics ciprofloxacin.Download high-res image (216KB)Download full-size image
Co-reporter:Lin Lin, Xiangqiong Zeng, Yuning Shen, Hailiang Zhu, Yong Qian
Talanta 2017 Volume 165() pp:321-325
Publication Date(Web):1 April 2017
DOI:10.1016/j.talanta.2016.12.074
•TPP was a novel highly selective and sensitive fluorescent turn-on probe for thiophenols.•TPP was designed based on excited-state intramolecular proton transfer mechanism.•A kinetic study of TPP towards thiophenol displayed a fast response time (~150 s).•TPP could be used for thiophenols detection in aqueous media and real-water samples.An ultrasensitive fluorescent probe TPP was developed for the highly selective detection of thiophenols based on excited-state intramolecular proton transfer (ESIPT) mechanism, its synthesis through a simple straightforward combination of HBT fluorophore with 2,4-dinitrobenzene functional group. A kinetic study of TPP towards thiophenol displayed a fast response time (~150 s) and significant turn-on fluorescence enhancement (~60 fold). Selective and competitive experiments exhibited an excellent selectivity of TPP toward thiophenols over biothiols (Cys, GSH) and other aliphatic thiols or nucleophiles. Using this ultrasensitive probe (LOD, 1.05×10−8 M), we have successfully monitored and quantified highly toxic thiophenols in aqueous media and real-water samples.
Co-reporter:Zhu-Ping Xiao;Wei Wei;Qi Liu;Peng-Fei Wang;Xing Luo;Fang-Yuan Chen;Yang Cao;Hong-Xia Huang;Mi-Mi Liu
RSC Advances (2011-Present) 2017 vol. 7(Issue 11) pp:6193-6201
Publication Date(Web):2017/01/18
DOI:10.1039/C6RA28061G
Twenty C-7 modified flavonoids were designed and synthesized. Biological evaluation in vitro indicated that compounds generated by SYBYL-X with high scores also showed good inhibitory activities against TyrRS. Compounds containing the nargenin core exhibit better enzyme inhibitory activities than other flavonoid cores, with (S)-5-hydroxy-4′-hydroxy-7-(2-morpholino-2-oxoethoxy)-2,3-dihydroflavone (b1) being the most active (IC50 = 0.10 ± 0.01 μM) in all assayed compounds. All compounds were also assayed for antimicrobial activities against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa, and b1 also displayed excellent activity, showing 6-fold more potent than the marketed antibiotic ciprofloxacin. In comparison with Gram-positive organism, all these derivatives exhibited better activity against Gram-negative organism, and did not displayed significant differences between the two assayed Gram-negative strains (E. coli ATCC 8739 and P. aeruginosa ATCC 9027).
Co-reporter:Meng-Ru Yang, Ya-Juan Qin, Chen Chen, Ya-Liang Zhang, Bo-Yan Li, Tian-Bao Liu, Hai-Bin Gong, Bao-Zhong Wang and Hai-Liang Zhu
RSC Advances 2016 vol. 6(Issue 36) pp:30412-30424
Publication Date(Web):16 Mar 2016
DOI:10.1039/C5RA28141E
A series of novel compounds (6a–6v) containing 1-methylindol and 1-(4,5-dihydro-1H-pyrazol-1-yl)ethanone skeletons were designed, synthesized and biologically evaluated as potential tubulin polymerization inhibitors and anticancer agents. Among them, compound 6q showed the most potent tubulin polymerization inhibitory activity (IC50 = 1.98 μM) and in vitro growth inhibitory activity against A549, MCF-7 and HepG2 cell lines, with IC50 values of 0.15 μM, 0.17 μM, and 0.25 μM respectively, comparable to the positive control. Furthermore, compound 6q was a potent inducer of apoptosis in A549 cells and it had typical cellular effects for microtubule interacting agents, causing arrest of the cell cycle in the G2/M phase. Confocal microscopy assay and molecular docking results further demonstrated that 6q could bind tightly to the colchicine site of tubulin and act as an anti-tubulin agent. These studies, along with 3D-QSAR modeling provided an important basis for further optimization of compound 6q as a potential anticancer agent.
Co-reporter:Xiao-Yuan Lu, Zhong-Chang Wang, Ting Wei, Xiao-Qiang Yan, Peng-Fei Wang and Hai-Liang Zhu
RSC Advances 2016 vol. 6(Issue 27) pp:22917-22935
Publication Date(Web):09 Feb 2016
DOI:10.1039/C6RA02168A
Novel benzenesulfonamide-substituted 1,5-diarylpyrazoles containing phenylacetohydrazide derivatives have been designed, synthesized and evaluated for their biological activities as selective COX-2 inhibitors with anticancer potential. In vitro the bioassay results revealed that some of them displayed potent inhibitory activity in the enzymatic and cellular assays. Among them, compound 48 showed the most powerful and potent selective inhibitory activity (IC50 = 82.21 μM for COX-1 and IC50 = 0.37 μM for COX-2), comparable to the control positive compound Celecoxib (40.29 μM, 0.15 μM). Antiproliferative assay results indicated that compound 48 possess potent antiproliferative activity against A549 cells in vitro with an IC50 value of 0.78 μM. We then performed a PI staining assay and cell apoptosis analysis for compound 48 and found that it effectively causes A549 cell apoptosis. A docking simulation was further performed to position compound 48 into the COX-2 active site to determine the probable binding model. The 3D-QSAR models were built for reasonable design of selective COX-2 inhibitors in the future.
Co-reporter:Yu-Shun Yang, Bing Yang, Yan Zou, Guigen Li, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 13) pp:3052-3061
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmc.2016.05.012
•20 pyrazoline derivatives have been synthesized and all are novel except C1.•Their anti-cancer activities were evaluated for the first time.•Selectivity and cytotoxicity of representative compounds were tested.•Molecular docking and QSAR discussions indicated breaking the limit of previous pattern to get the most potent C6.A series of novel dioxin-containing triaryl pyrazoline derivatives C1–C20 have been synthesized. Their B-Raf inhibitory and anti-proliferation activities were evaluated. Compound C6 displayed the most potent biological activity against B-RafV600E and WM266.4 human melanoma cell line with corresponding IC50 value of 0.04 μM and GI50 value of 0.87 μM, being comparable with the positive controls and more potent than our previous best compounds. Moreover, C6 was selective for B-RafV600E from B-RafWT, C-Raf and EGFR and low toxic. The docking simulation suggested the potent bioactivity might be caused by breaking the limit of previous binding pattern. A new 3D QSAR model was built with the activity data and binding conformations to conduct visualized SAR discussion as well as to introduce new directions. Stretching the backbone to outer space or totally reversing the backbone are both potential orientations for future researches.A series of novel dioxin-containing triaryl pyrazoline derivatives C1–C20 have been synthesized with their B-Raf inhibitory activity and anti-proliferation activity tested. Selectivity and cytotoxicity of representative compounds have been tested. The docking simulation and 3D QSAR model indicated breaking the limit of previous binding pattern.
Co-reporter:Wei-Kang Shi, Rui-Cheng Deng, Peng-Fei Wang, Qin-Qin Yue, Qi Liu, Kun-Ling Ding, Mei-Hui Yang, Hong-Yu Zhang, Si-Hua Gong, Min Deng, Wen-Run Liu, Qiu-Ju Feng, Zhu-Ping Xiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 19) pp:4519-4527
Publication Date(Web):1 October 2016
DOI:10.1016/j.bmc.2016.07.052
•3-Arylpropionylhydroxamic acid derivatives showed a mixed inhibition mechanism.•Mixed urease inhibitors with enhanced inhibitory activities have been synthesized.•Urea showed substrate inhibition against H. pylori urease.Helicobacter pylori urease is involved in several physiologic responses such as stomach and duodenal ulcers, adenocarcinomas and stomach lymphomas. Thus, inhibition of urease is taken for a good chance to treat H. pylori-caused infections, we have therefore focused our efforts on seeking novel urease inhibitors. Here, a series of arylpropionylhydroxamic acids were synthesized and evaluated for urease inhibition. Out of these compounds, 3-(2-benzyloxy-5-chlorophenyl)-3-hydroxypropionylhydroxamic acid (d24) was the most active inhibitor with IC50 of 0.15 ± 0.05 μM, showing a mixed inhibition with both competitive and uncompetitive aspects. Non-linear fitting of kinetic data gives kinetics parameters of 0.13 and 0.12 μg·mL−1 for Ki and Ki′, respectively. The plasma protein binding assays suggested that d24 exhibited moderate binding to human and rabbit plasma proteins.
Co-reporter:Xiang-Xiang Tao, Yong-Tao Duan, Long-Wang Chen, Dan-Jie Tang, Meng-Ru Yang, Peng-Fei Wang, Chen Xu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:677-683
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.11.040
A series of novel pyrazole-nitroimidazole derivatives had been arranged and evaluated for their EGFR/HER-2 tyrosine kinase inhibitory activity as well as their antiproliferative properties on four kinds of cancer cell lines (MCF-7, Hela, HepG2, B16-F10). The bioassay results showed most of the designed compounds exhibited potential antiproliferation activity, with the IC50 values ranging from 0.13 μM to 128.06 μM in four tumor cell lines. Among them, compound 5c exhibited remarkable inhibitory activity against EGFR/HER-2 tyrosine kinase with IC50 value of 0.26 μM/0.51 μM, respectively, comparable to the positive control erlotinib (IC50 = 0.41 μM for HER-2 and IC50 = 0.20 μM for EGFR) and lapatinib (IC50 = 0.54 μM for HER-2 and IC50 = 0.28 μM for EGFR). Molecular modeling simulation studies were performed in order to predict the biological activity of the proposed compounds and activity relationship (SAR) of these pyrazole-nitroimidazole derivatives.
Co-reporter:Xiao-Yuan Lu, Zhong-Chang Wang, Shen-Zhen Ren, Fa-Qian Shen, Ruo-Jun Man, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3491-3498
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.06.037
Cyclooxygenase-2 is frequently overexpression in malignant tumors and the product PGE2 promotes cancer cell progression and metastasis. We designed novel series of coumarin sulfonamides derivatives to improve biological activities of COX-2 inhibition and anticancer. Among them, compound 7t showed most powerful selective inhibitory and antiproliferative activity (IC50 = 0.09 μM for COX-2, IC50 = 48.20 μM for COX-1, IC50 = 0.36 μM against HeLa cells), comparable to the control positive compound Celecoxib (0.31 μM, 43.37 μM, 7.79 μM). Cancer cell apoptosis assay were performed and results indicated that compound 7t effectively fuels HeLa cells apoptosis in a dose and time-dependent manner. Moreover, 7t could significantly suppress cancer cell adhesion, migration and invasion which were essential process of cancer metastasis. Docking simulations results was further indicated that compound 7t could bind well to the COX-2 active site and guided a reasonable design of selective COX-2 inhibitor with anticancer activities in future.Novel coumarin sulfonamides derivatives have been synthesized. Among them, compound 7t showed most powerful selective inhibitory and antiproliferative activity. 7t could effectively causing HeLa cells apoptosis in a dose and time-dependent manner and significantly suppress cancer cell adhesion, migration and invasion.
Co-reporter:Long-Wang Chen, Ze-Feng Wang, Bo Zhu, Ruo-Jun Man, Yan-Dong Liu, Yuan-Heng Zhang, Bao-Zhong Wang, Zhong-Chang Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 20) pp:4983-4991
Publication Date(Web):15 October 2016
DOI:10.1016/j.bmcl.2016.09.003
The increasingly acquired resistance to vemurafenib and side effects of known inhibitors motivate the search for new and more effective anti-melanoma drugs. In this Letter, virtual screening and scaffold growth were combined together to achieve new molecules as BRAFV600E inhibitors. Along with docking simulation, a primary screen in vitro was performed to filter the modifications for the lead compound, which was then substituted, synthesized and evaluated for their inhibitory activity against BRAFV600E and several melanoma cell lines. Out of the obtained compounds, derivative 3l was identified as a potent BRAFV600E inhibitor and exerted an anticancer effect through BRAFV600E inhibition. The following biological evaluation assays confirmed that 3l could induce cell apoptosis and marked DNA fragmentation. Furthermore, 3l could arrest the cell cycle at the G0/G1 phase in melanoma cells. The docking simulation displayed that 3l could tightly bind with the crystal structure of BRAFV600E at the active site. Overall, the biological profile of 3l suggests that this compound may be developed as a potential anticancer agent.Virtual screening and scaffold growth were combined together to achieve a series of novel compounds (3a–3n) bearing the benzo-α-pyrone scaffold. Among them, compound 3l showed most powerful antiproliferative activity (IC50 = 2.24 μM for A375 and IC50 = 1.35 μM for WM266-4) and enzyme inhibition activity (IC50 = 0.37 μM). 3l could effectively cause melanoma cells apoptosis in a dose-dependent manner, significantly induce DNA fragmentation and arrest cell cycle at the G0/G1 phase. Docking simulation was performed to explore the binding model of compound 3l with BRAFV600E.
Co-reporter:Yin Luo, Zhi-Jun Liu, Guo Chen, Jing Shi, Jing-Ran Li and Hai-Liang Zhu
MedChemComm 2016 vol. 7(Issue 2) pp:263-271
Publication Date(Web):19 Nov 2015
DOI:10.1039/C5MD00371G
Recent studies have proved that focal adhesion kinase (FAK) is a new potential therapeutic target in cancer therapy. In this study, a virtual screening was conducted to discover potential candidates for FAK inhibitors. Based on the results, a series of novel oxadiazole derivatives (5a–5q) bearing the benzotriazole group were designed and synthesized for FAK inhibitory evaluation. Among the compounds, 5h, which has an ortho methoxy group on the benzene ring, exhibited the most potent inhibitory activity for cancer cell growth with an IC50 value of 11 μM and 0.250 μM against Hela cells and FAK, respectively. Further, the apoptosis assay indicated that compound 5h induced the apoptosis of HeLa cells, and docking simulation showed that 5h could bind to the FAK protein catalytic region. Taking these together, 5h could be a lead for discovering novel FAK inhibitors.
Co-reporter:Ya-Liang Zhang;Ya-Juan Qin;Dan-Jie Tang;Meng-Ru Yang;Bo-Yan Li;Yan-Ting Wang;Hong-Yu Cai; Bao-Zhong Wang;Dr. Hai-Liang Zhu
ChemMedChem 2016 Volume 11( Issue 13) pp:1446-1458
Publication Date(Web):
DOI:10.1002/cmdc.201600137
Abstract
A series of 1-methyl-1H-indole–pyrazoline hybrids were designed, synthesized, and biologically evaluated as potential tubulin polymerization inhibitors. Among them, compound e19 [5-(5-bromo-1-methyl-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carboxamide] showed the most potent inhibitory effect on tubulin assembly (IC50=2.12 μm) and in vitro growth inhibitory activity against a panel of four human cancer cell lines (IC50 values of 0.21–0.31 μm). Further studies confirmed that compound e19 can induce HeLa cell apoptosis, cause cell-cycle arrest in G2/M phase, and disrupt the cellular microtubule network. These studies, along with molecular docking and 3D-QSAR modeling, provide an important basis for further optimization of compound e19 as a potential anticancer agent.
Co-reporter:Tianbao Liu, Jie Lin, Zhen Li, Lin Lin, Yuning Shen, Hailiang Zhu and Yong Qian
Analyst 2015 vol. 140(Issue 21) pp:7165-7169
Publication Date(Web):15 Sep 2015
DOI:10.1039/C5AN00119F
We have developed a novel colorimetric and ratiometric fluorescence probe for the selective and sensitive monitoring of hydrogen sulfide based on a dicyanoisophorone platform. An excellent linear relationship of fluorescence intensity ratio (I637/I558) (R2 = 0.9867) versus hydrogen sulfide concentration in the range of 1–12 μM was obtained. This probe exhibited a remarkable fluorescence response to hydrogen sulfide over other physiological thiols or biological species, which fluoresces in the red region with a large Stokes shift (172 nm). This probe was successfully utilized to monitor H2S under in vitro physiological conditions and for imaging H2S in living cells and living zebrafish in vivo.
Co-reporter:Shu-Fu Wang, Yong Yin, Ya-Liang Zhang, Shan-Wei Mi, Meng-Yue Zhao, Peng-Cheng Lv, Bao-Zhong Wang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2015 Volume 93() pp:291-299
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.018
•24 novel 5-phenyl-1H-pyrazol cinnamamide derivatives had been synthesized.•The compounds were evaluated for biological activities as tubulin inhibitors.•Compound 5j showed the most potent inhibitory activity.A series of novel 5-phenyl-1H-pyrazol derivatives (5a–5x) containing cinnamamide moiety were synthesized and their biological activities as potential tubulin polymerization inhibitors were evaluated. Among them, compound 5j exhibited the most potent inhibitory activity with an IC50 value of 1.02 μM for tubulin, which was superior to that of Colchicine (IC50 = 1.34 μM). Docking simulation was performed to insert compound 5j into the crystal structure of tubulin at colchicine binding site to determine the probable binding model. 3D-QSAR model was also built to provide more pharmacophore understanding that could be used to design new agents with more potent tubulin inhibitory activity.Compound 5j exhibited the most potent inhibitory activity with an IC50 value of 1.02 μM for tubulin, 0.35 μM for MCF-7, 0.62 μM for A549 and 0.57 μM for B16–F10.
Co-reporter:Ya-Juan Qin, Yu-jing Li, Ai-Qin Jiang, Meng-Ru Yang, Qi-Zhang Zhu, Hong Dong, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2015 Volume 94() pp:447-457
Publication Date(Web):13 April 2015
DOI:10.1016/j.ejmech.2015.02.058
•32 novel pyrazoline-containing derivatives has been designed and synthesized.•Crystal structure of compound 23 was determined.•Their biological activities were tested as potential tubulin assembling inhibitors.•The docking model were established and analyzed.•Compound 18 displayed the most potent antitumor activity.A series of novel pyrazoline-containing derivatives (15–47) has been designed, synthesized and evaluated for their biological activities. Among them, compound 18 displayed the most potent antiproliferative activity against A549, MCF-7 and HepG-2 cells line (IC50 = 0.07 μM, 0.05 μM, 0.03 μM, respectively) and the tubulin polymerization inhibitory activity (IC50 = 1.88 μM), being comparable to CA-4. Furthermore, we also tested that compound 18 was a potent inducer of apoptosis in HepG-2 cells and it had cellular effects typical for microtubule interacting agents, causing accumulation of cells in the G2/M phase of the cell cycle. These studies, along with molecular docking, provided a new molecular scaffold for the further development of antitumor agents that target tubulin.
Co-reporter:Zhu-Ping Xiao, Wei Wei, Peng-Fei Wang, Wei-Kang Shi, Na Zhu, Me-Qun Xie, Yu-Wen Sun, Ling-Xia Li, Yong-Xiang Xie, Liang-Song Zhu, Nian Tang, Hui Ouyang, Xian-Hui Li, Guang-Cheng Wang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2015 Volume 102() pp:631-638
Publication Date(Web):18 September 2015
DOI:10.1016/j.ejmech.2015.08.025
•N2-(Arylacetyl)glycinanilide as novel potent antibacterial agent.•Significant improvement of activity against Gram-negative bacterial strains.•The most potent compound showing 3-fold more potency than penicillin G.Tyrosyl-tRNA synthetase (TyrRS), an essential enzyme in bacterial protein biosynthesis, is an attractive therapeutic target for finding novel antibacterial agents, and a series of N2-(arylacetyl)glycinanilides has been herein synthesized and identified as TyrRS inhibitors. These efforts yielded several compounds, with IC50 in the low micromolar range against TyrRS from Staphylococcus aureus. Out of the obtained compounds, 3ap is the most active and exhibits excellent activity against both Gram-positive (S. aureus) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacterial strains. In comparison with the parent scaffold 3-arylfuran-2(5H)-one, N2-(arylacetyl)glycinanilide significantly improved the potency against Gram-negative bacterial strains, indicating that this scaffold offers a significant potential for developing new antibacterial drugs.
Co-reporter:Fang Wang, Xue Wang, Min-Xia Zhang, Yong-Hua Yang and Hai-Liang Zhu
RSC Advances 2015 vol. 5(Issue 91) pp:74425-74437
Publication Date(Web):25 Aug 2015
DOI:10.1039/C5RA13746B
A series of novel compounds (8a–21b) were designed and synthesized based on 1H-benzo[d]imidazole. Their biological activities were evaluated as potential tubulin polymerization inhibitors and anticancer agents. Among all the compounds, compound 18b showed the most potent in vitro growth inhibitory activity against A549, MCF-7, K562 cancer cell lines, with IC50 values of 0.12 μM, 0.15 μM, 0.21 μM, respectively. And, compound 18b also exhibited significant tubulin polymerization inhibitory activity (IC50 = 2.1 μM), which was comparable with the positive control. Furthermore, docking simulation and 3D-QSAR modeling of inhibitor analogues provide determination of the probable binding model and an important basis that compound 18b with potent inhibitory activity in tumor growth may be a potential anticancer agent.
Co-reporter:Syed Nasir Abbas Bukhari;Xin Zhang;Ibrahim Jantan;Muhammad Wahab Amjad;Vijay H. Mas
Chemical Biology & Drug Design 2015 Volume 85( Issue 6) pp:729-742
Publication Date(Web):
DOI:10.1111/cbdd.12457
A novel series of 1,3-diphenyl-2-propen-1-one (chalcone) derivatives was synthesized by a simple, eco-friendly, and efficient Claisen–Schmidt condensation reaction and used as precursors for the synthesis of new pyrazoline derivatives. All the synthesized compounds were screened for anti-inflammatory related activities such as inhibition of phospholipase A2 (PLA2), cyclooxygenases (COX-1 and COX-2), IL-6, and TNF-α. The results of the above studies show that the compounds synthesized are effective inhibitors of above pro-inflammatory enzymes and cytokines. Overall, the results of the studies reveal that the pyrazolines with chlorophenyl substitution (1b–6b) seem to be important for inhibition of enzymes and cytokines. Molecular docking experiments were performed to clarify the molecular aspects of the observed COX-inhibitory activities of the investigated compounds.
Co-reporter:Dong-Dong Li;Ya-Juan Qin;Xin Zhang;Yong Yin;Lin-Guo Zhao
Chemical Biology & Drug Design 2015 Volume 86( Issue 4) pp:731-745
Publication Date(Web):
DOI:10.1111/cbdd.12545
Interference with dynamic equilibrium of microtubule–tubulin has proven to be a useful tactics in the clinic. Based on investigation into the structure–activity relationship (SAR) studies of tubulin polymerization inhibitors obtained from several worldwide groups, we attempted to design 691 compounds covering several main heterocyclic scaffolds as novel colchicine-site inhibitors (CSIs). Evaluated by a series of combination of commonly used computer methods such as molecular docking, 3D-QSAR, and pharmacophore model, we can obtain the ultimate 16 target compounds derived from five important basic scaffolds in the field of medicinal chemistry. Among these compounds, compound A-132 with in silico moderate activity was synthesized, and subsequently validated for preliminary inhibition of tubulin polymerization by immunofluorescence assay. In additional, the work of synthesis and validation of biological activity for other 15 various structure compounds will be completed in our laboratory. This study not only developed a hierarchical strategy to screen novel tubulin inhibitors effectively, but also widened the spectrum of chemical structures of canonical CSIs.
Co-reporter:Yong Yin;Shao Sha;Yan-Ting Wang;Xun Wu;She-Feng Wang;Fang Qiao;Peng-Cheng Lv
Chemical Biology & Drug Design 2015 Volume 86( Issue 5) pp:1323-1329
Publication Date(Web):
DOI:10.1111/cbdd.12596
VEGFR2 has been proved to play a major role in the regulation of tumor angiogenesis. Twenty-one 4-alkoxyquinazoline-based derivatives have been designed and synthesized as vascular endothelial growth factor receptor 2 (VEGFR2) inhibitors, and their biological activities were evaluated. Among these compounds, compound 3h exhibited the most potent inhibitory activities against VEGFR2 tyrosine kinase and cell proliferation, with the IC50 values of 2.89 nm (for VEGFR2) and 0.25 μm (for MCF-7), which were comparable with the control compound. Docking simulation was performed to position compound 3h into the 4ASE active site, and the result showed that compound 3h could bind well at the 4ASE active site.
Co-reporter:Peng-Fei Wang;Han-Yue Qiu;Shahla Karim Baloch;Hai-Bin Gong;Zhong-Chang Wang
Chemical Biology & Drug Design 2015 Volume 86( Issue 6) pp:1405-1410
Publication Date(Web):
DOI:10.1111/cbdd.12604
In this study, we synthesized a series of dihydropyrazole sulfonamide derivatives containing 2-hydroxyphenyl moiety as antitumor agents to target the matrix metalloproteinase-2 (MMP-2). All of the synthesized compounds were examined by bioactivity assays, in which compound 4c turned out as a potential antagonist of MMP-2 along with potent anticancer activity against four tumor cell lines. Structure–activity relationship analysis was also performed to examine how structural changes impacted the bioactivity. Suggested to be caused by the induction of apoptosis, the antitumor mechanism of 4c was further confirmed by PI combining with annexin V-FITC staining assay using flow cytometry analysis. These new findings along with molecular docking observations suggested that compound 4c could be developed as a potential anticancer agent.
Co-reporter:Yu-Jia Ren, Zhong-Chang Wang, Xin Zhang, Han-Yue Qiu, Peng-Fei Wang, Hai-Bin Gong, Ai-Qin Jiang and Hai-Liang Zhu
RSC Advances 2015 vol. 5(Issue 28) pp:21445-21454
Publication Date(Web):21 Jan 2015
DOI:10.1039/C4RA10606G
A series of dihydropyridine containing thiazolinone derivatives (4a–4r) have been designed, synthesized and their biological activities evaluated as potential EGFR and HER-2 kinase inhibitors and in tumor cell antiproliferation. The synthesized compounds were analyzed by 1H-NMR and MS. In addition, compound 4m was scrutinized by X-ray structure analysis. Among them, compound 4r displayed the most potent inhibitory activity (IC50 = 0.099 μM for EGFR and IC50 = 3.26 μM for HER-2). Antiproliferative assay results indicated that compound 4r owned high antiproliferative activity against B16–F10, HeLa and MCF-7 in vitro, with IC50 values of 0.09 μM, 0.29 μM, and 0.56 μM, respectively. Docking simulations were further performed to position compound 4r into the EGFR active site to determine the probable binding mode. 3D-QSAR models were built for reasonable design of EGFR/HER-2 inhibitors in the present and the future.
Co-reporter:Fang Qiao, Yong Yin, Yu-Ning Shen, She-Feng Wang, Shao Sha, Xun Wu, Ai-Min Lu, Chen Xu, Wei-Ming Zhang and Hai-Liang Zhu
RSC Advances 2015 vol. 5(Issue 26) pp:19914-19923
Publication Date(Web):05 Feb 2015
DOI:10.1039/C4RA11780H
A series of 4-alkoxyquinazoline derivatives containing the 1,3,4-oxadiazole scaffold have been designed and synthesized, and their inhibitory activities were also tested against A549, MCF-7 and Hela. Of these compounds, 2-(3,4-dimethoxybenzyl)-5-((quinazolin-4-yloxy)methyl)-1,3,4-oxadiazole (compound 4j) showed the most potent inhibitory activity (IC50 = 0.23 μM for MCF-7, IC50 = 0.38 μM for A549 and IC50 = 0.32 μM for Hela) and the effect was better than the positive control drug Tivozanib (IC50 = 0.38 μM for MCF-7, IC50 = 0.62 μM for A549 and IC50 = 0.34 μM for Hela). A docking simulation was performed to position compound 4j into the VEGFR active site to determine the probable binding model. These results suggested that compound 4j with potent inhibitory activity in tumor growth inhibition may be a potential anticancer agent.
Co-reporter:Yu-Ning Shen, Lin Lin, Han-Yue Qiu, Wen-Yan Zou, Yong Qian and Hai-Liang Zhu
RSC Advances 2015 vol. 5(Issue 30) pp:23767-23777
Publication Date(Web):26 Feb 2015
DOI:10.1039/C4RA12108B
A series of novel 3,4-dimethoxylbenzenesulfonate derivatives containing a chalcone structure were synthesized and evaluated for their anti-proliferative activities against HepG2, HCT-116, MCF-7 and HeLa cell lines, as well as the effects of compound 10b on mitotic arrest and cell cycle of MCF-7 carcinoma cell line. Most importantly, the results of DAPI staining under co-focal microscope justified that compound 10b functioned even at relatively low concentration. The analogues showed a potent bio-activity towards tumor cells with IC50 values at nano-mole class, compared with those of positive control drug Colchicine, whose IC50 were 150.4 nM for MCF-7, 123.9 nM for HepG2, 125.4 nM for HCT-116 and 131.4 nM for HeLa cells. Also, a molecular docking modeling was utilized to reveal the binding mode of derivatives and microtubule. Among all the synthesized compounds, compound 10b stands out as IC50 values against all the selected cell lines were at average 80 nM (in which the values against MCF-7 and HepG2 were similar; about 79.2 nM). In this research, we gave strong evidence upon the optimized stratagem for ligands targeting the colchicine-binding site on microtubules, explaining the attribution that the analogues were designed upon the structure of chalcone and combretastatin A-4.
Co-reporter:Peng-Fei Wang, Han-Yue Qiu, Jun-Ting Ma, Xiao-Qiang Yan, Hai-Bin Gong, Zhong-Chang Wang and Hai-Liang Zhu
RSC Advances 2015 vol. 5(Issue 32) pp:24997-25005
Publication Date(Web):20 Feb 2015
DOI:10.1039/C4RA15201H
A series of dihydropyrazole derivatives containing morpholine was designed and synthesized as antimicrobial agents. All of the synthesized compounds were characterized by 1H-NMR and MS. Afterwards they were evaluated for in vitro antibacterial activity against four bacteria strains. Along with S. aureus TyrRS inhibition and cytotoxicity examination, some compounds proved to be of low toxicity and potent, especially against Gram-positive bacteria strains. Compound 4s exhibited the potential to be new a antibacterial drug with strong broad-spectrum antimicrobial activity and enzyme inhibitory activity. Docking simulation was performed to position compound 4s into the S. aureus TyrRS structure active site to investigate the probable binding mode. A 3D-QSAR model was also established to explain how structural alterations affect the activity and guide further study.
Co-reporter:Zhong-Cheng Song, Gao-Yuan Ma and Hai-Liang Zhu
RSC Advances 2015 vol. 5(Issue 32) pp:24824-24833
Publication Date(Web):20 Feb 2015
DOI:10.1039/C4RA15284K
A possible mechanism of dynamic kinetic resolution by the formation of N-tert-butoxycarbonyl-thiazolidine carboxylic acid was proposed and validated by a quantitative density functional theoretical calculation according to Curtin–Hammett principle. Such a mechanism of action of a nucleophilic substitution reaction through an intramolecular hydrogen bonding could be widely applied in the organic syntheses of particular enantiomer. Antibacterial activities showed that most of the N-tert-butoxycarbonyl-(2R)-arylthiazolidine-(4R)-carboxylic acid derivatives exhibited better antibacterial activities against the four bacterial strains than related (2RS)-arylthiazolidine-(4R)-carboxylic acid derivatives.
Co-reporter:Wei Wei, Wei-Kang Shi, Peng-Fei Wang, Xiao-Tong Zeng, Pan Li, Ji-Rong Zhang, Qian Li, Zhi-Ping Tang, Jia Peng, Lang-Zhou Wu, Mei-Qun Xie, Chan Liu, Xian-Hui Li, Ying-Chun Wang, Zhu-Ping Xiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 20) pp:6602-6611
Publication Date(Web):15 October 2015
DOI:10.1016/j.bmc.2015.09.018
Herein we describe the synthesis and evaluation of a series of adenosine analogs for in vitro antibacterial activity against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa. Out of these compounds, compound c6 has much stronger antibacterial potency against Pseudomonas aeruginosa than ciprofloxacin, and was determined to target tyrosyl-tRNA synthetase with IC50 of 0.8 ± 0.07 μM. Structure–activity relationship analysis suggested that introduction of a fluorine atom at the 3′-position of benzene ring of the phenylacetyl moiety significantly increased affinities to the enzyme. In comparison with isopropylidene analogs, 2′,3′-deprotected compounds displayed higher inhibitory activity. Molecular dockings provided an explanation for observations in biological assays.
Co-reporter:Xu-Dong Wang, Wei Wei, Peng-Fei Wang, Li-Cheng Yi, Wei-Kang Shi, Yong-Xiang Xie, Lang-Zhou Wu, Nian Tang, Liang-Song Zhu, Jia Peng, Chan Liu, Xian-Hui Li, Shi Tang, Zhu-Ping Xiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 15) pp:4860-4865
Publication Date(Web):1 August 2015
DOI:10.1016/j.bmc.2015.05.026
3-Arylfuran-2(5H)-one derivatives show good antibacterial activity and were determined as tyrosyl-tRNA synthetase (TyrRS) inhibitors. In a systematic medicinal chemistry exploration, we demonstrated chemical opportunities to treat infections caused by Helicobacter pylori. Twenty 3-arylfuran-2(5H)-ones were synthesized and evaluated for anti-H. pylori, antioxidant and anti-urease activities which are closely interconnected with H. pylori infection. The results displayed that some of the compounds show excellent antioxidant activity, and good anti-H. pylori and urease inhibitory activities. Out of these compounds, 3-(3-methylphenyl)furan-2(5H)-one (b9) showed the most potent antioxidant activity (IC50 = 8.2 μM) and good anti-H. pylori activity (MIC50 = 2.6 μg/mL), and it can be used as a good candidate for discovering novel anti-gastric ulcer agent.
Co-reporter:Yong Yin, Yan-Qing Zhang, Biao Jin, Shao Sha, Xun Wu, Chetan B. Sangani, She-Feng Wang, Fang Qiao, Ai-Min Lu, Peng-Cheng Lv, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 6) pp:1231-1240
Publication Date(Web):15 March 2015
DOI:10.1016/j.bmc.2015.01.052
Twenty eight 6,7-dihydrobenzo[f]benzo[4,5]imidazo[1,2-d][1,4]oxazepine derivatives were synthesized and evaluated their biological activities as PI3K inhibitors. Biological evaluation against four human tumor cell lines revealed that most target compounds showed impressively better antiproliferative activities than that of LY294002. Among these compounds, compound 25 exhibited the most potent and selective activity for PI3Kα, with the IC50 value of 0.016 μM, an approximately 30-fold increase in comparison with LY294002, it also has an increased potency of approximately 11-fold for PI3Kβ. It indicated the potential of developing 6,7-dihydrobenzo[f]benzo[4,5]imidazo[1,2-d][1,4]oxazepine derivatives as the new PI3Kα selective inhibitors for tumor treatment.Twenty eight new 6,7-dihydrobenzo[f]benzo[4,5]imidazo[1,2-d][1,4]oxazepine derivatives have been synthesized and evaluated for biological activities as PI3Kα inhibitors. Compound 25 showed the most potent inhibitory activity.
Co-reporter:Meng-Yue Zhao, Yong Yin, Xiao-Wei Yu, Chetan B. Sangani, Shu-Fu Wang, Ai-Min Lu, Li-Fang Yang, Peng-Cheng Lv, Ming-Guo Jiang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 1) pp:46-54
Publication Date(Web):1 January 2015
DOI:10.1016/j.bmc.2014.11.029
Many reports implied that the BRAF serine/threonine kinase was mutated in various types of human tumors, which were related with cell growth, survival and differentiation. To provide new therapeutic opportunities, a series of novel 4,5-dihydro-1H-pyrazole derivatives (6a–10d) containing thiazole moiety as potential V600E mutant BRAF kinase (BRAFV600E) inhibitors were designed and synthesized. All compounds were evaluated in vitro for anticancer activities against WM266.4 human melanoma cell line and breast cancer MCF-7 cell line. Compound 10d displayed the most potential antiproliferative activity with an IC50 value of 0.12 μM against cell line WM266.4 and 0.16 μM against MCF-7 with positive control Sorafenib. Results of the inhibitory activity against BRAFV600E revealed that compound 10d was bearing the best bioactivity with IC50 of 0.05 μM as well. On the basis of the result of flow cytometry, with the dose of compound 10d increasing, more and more cancer cell gradually encountered apoptosis or died, which indicated the compound 10d could induce remarkable apoptosis of MCF-7 and WM266.4 cells in a dose dependent manner. Furthermore, docking simulation of inhibitor analogues and 3D-QSAR modeling provided potential binding model and further knowledge of pharmacophore.To provide new therapeutic opportunities, a series of novel 4,5-dihydro-1H-pyrazole derivatives (6a–10d) containing thiazole moiety as potential V600E mutant BRAF kinase (BRAFV600E) inhibitors were designed and synthesized. All compounds were evaluated in vitro for anticancer activities against WM266.4 and MCF-7 cell line. Compound 10d displayed the most potential antiproliferative activity.
Co-reporter:Shao Sha, Hong-Wei Han, Fei Gao, Tian-Bao Liu, Zhen Li, Chi Xu, Wei-Qing Zhong and Hai-Liang Zhu
MedChemComm 2015 vol. 6(Issue 12) pp:2233-2233
Publication Date(Web):06 Nov 2015
DOI:10.1039/C5MD90050F
Correction for ‘Discovery of fluoroquinolone derivatives as potent, selective inhibitors of PI3Kγ’ by Shao Sha et al., Med. Chem. Commun., 2015, DOI: 10.1039/c5md00364d.
Co-reporter:Shao Sha, Hong-Wei Han, Fei Gao, Tian-Bao Liu, Zhen Li, Chi Xu, Wei-Qing Zhong and Hai-Liang Zhu
MedChemComm 2015 vol. 6(Issue 11) pp:2029-2035
Publication Date(Web):29 Sep 2015
DOI:10.1039/C5MD00364D
Phosphoinositide 3-kinase (PI3K) is an attractive target to potentially treat a range of disease states as illustrated by more than 15 inhibitors now in clinical trials. We disclose herein the discovery of a new class of fluoroquinolone derivatives having improved potency toward PI3K. The activity of compound A3 as an inhibitor of PI3K was further investigated in human tumor cells. Notably, compound A3 with potent inhibitory activity was less toxic. By computational docking studies, it was predicted that, like CAL-101, a known small inhibitor of PI3K, compound A3 was tightly embedded into the ATP binding pocket with H bonds and π–cation interactions.
Co-reporter:Zhu-Ping Xiao, Wei-Kang Shi, Peng-Fei Wang, Wei Wei, Xiao-Tong Zeng, Ji-Rong Zhang, Na Zhu, Miao Peng, Bin Peng, Xiao-Yi Lin, Hui Ouyang, Xiao-Chun Peng, Guang-Cheng Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4508-4513
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.014
Co-reporter:Zhi-Hua Guo, Yong Yin, Cong Wang, Peng-Fei Wang, Xing-Tao Zhang, Zhong-Chang Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2015 23(18) pp: 6148-6156
Publication Date(Web):
DOI:10.1016/j.bmc.2015.07.075
Co-reporter:Yang Hu, Cui-Yun Li, Xiao-Ming Wang, Yong-Hua Yang, and Hai-Liang Zhu
Chemical Reviews 2014 Volume 114(Issue 10) pp:5572
Publication Date(Web):April 9, 2014
DOI:10.1021/cr400131u
Co-reporter:Yong Yin, Xun Wu, Hong-Wei Han, Shao Sha, She-Feng Wang, Fang Qiao, Ai-Min Lu, Peng-Cheng Lv and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 45) pp:9157-9165
Publication Date(Web):16 Sep 2014
DOI:10.1039/C4OB01589D
A series of novel 2-alkyl-chromeno[4,3-c]pyrazol-4(2H)-one derivatives were synthesized and evaluated for their biological activities as PI3K inhibitors. In vitro biological evaluation against four human tumor cell lines revealed that most target compounds showed impressively better antiproliferative activities than that of LY294002. Among these compounds, compound 4l exhibited the most potent and selective activity for PI3Kα, with the value of 0.014 μM, an approximately 30-fold increase in comparison with LY294002. Docking simulation was performed to position compound 4l into the PI3Kα active site and the result showed that compound 4l could bind well at the PI3Kα active site and it indicated that compound 4l could be a potential inhibitor of PI3Kα.
Co-reporter:Jia-Jia Liu, Juan Sun, Yun-Bin Fang, Yong-An Yang, Rui-Hua Jiao and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 6) pp:998-1008
Publication Date(Web):2013/11/08
DOI:10.1039/C3OB41953C
A series of novel 4,5-dihydropyrazole derivatives (4a–4t), containing the dinitrobenzotrifluoride moiety, as DNA gyrase inhibitors were designed and synthesized. Based on the preliminary results, compounds 4d, 4h and 4t with potent inhibitory activity in bacterial growth may be wonderful antibacterial agents; among them, compound 4t displayed the most potent activity with minimum inhibitory concentration (MIC) values of 3.125, 0.39, 0.39 and 0.39 μg mL−1 against Bacillus subtilis, Staphylococcus aureus, Pseudomonas aeruginosa, and Escherichia coli respectively, which was comparable with penicillin and kanamycin B with corresponding MIC values of 3.125, 3.125, 0.39, 0.39 μg mL−1 and 1.562, 1.562, 1.562, 1.562 μg mL−1, respectively. In particular, compound 4d showed the most potent anti-Gram-positive bacterial activity with a MIC value of 0.39 μg mL−1 against the tested Gram-positive bacterial strains and exhibited the most potent B. subtilis DNA gyrase and S. aureus DNA gyrase inhibitory activity with an IC50 of 0.125 μg mL−1. Docking simulation was performed to insert compound 4d into the S. aureus DNA gyrase active site to determine the probable binding conformation.
Co-reporter:Juan Sun, Peng-Cheng Lv, Feng-Jiao Guo, Xin-Yi Wang, Xiao- Han, Yang Zhang, Gui-Hua Sheng, Shao-Song Qian, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2014 Volume 81() pp:420-426
Publication Date(Web):23 June 2014
DOI:10.1016/j.ejmech.2014.05.026
•The synthesis and anticancer activities of these compounds have not been reported so far.•Our current findings are completely new.•Their biological activities are also evaluated for PLK1 inhibitory activity.A novel class of aromatic diacylhydrazine derivatives was designed as PLK1 inhibitors. All the 19 new synthesized compounds were assayed for antitumor activity against the respective cervical cancer cells. In which, nine compounds with better antitumor activities were further tested for their PLK1 inhibitory activity. Last, we have successfully found that compound 7k showed both the promising antitumor activity with IC50 of 0.17 μM against the cervical cancer cells, and also processed the most potent PLK1 inhibitory activity with IC50 of 0.03 μM. In addition, docking simulation also carried out in this study to give a potent prediction binding mode between the small molecule and PKL1 (PDB code: 1umw) protein.New diacylhydrazine derivatives have been designed and evaluated for their PLK1 inhibitory activity and anti-cervical cancer cell activity. The most active compound is 7k.
Co-reporter:Yong-Tao Duan, Ya-Li Sang, Jigar A. Makawana, Shashikant B. Teraiya, Yong-Fang Yao, Dan-Jie Tang, Xiang-Xiang Tao, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2014 Volume 85() pp:341-351
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.082
•18 novel 1-indolyl acetate-5-nitroimidazole 3a–3r analogs of combretastatin A-4 have been synthesized.•Their biological activities were evaluated as potential tubulin polymerization inhibitors.•Compound 3p displayed strong antitumor activity.A series of 18 novel 1-indolyl acetate-5-nitroimidazole 3a–3r were designed, synthesized, and evaluated for their in vitro biological activities as potential tubulin polymerization inhibitors. Among these compounds, 3p displayed strong antitumor activity with IC50 of 2.00, 1.05, 0.87 μM against A549, Hela and U251 respectively, and also showed the most potent PLK1 inhibitory activity with IC50 of 2.4 μM. Molecular docking studies within the colchicine binding site of tubulin were in good agreement with the tubulin polymerization inhibitory data and confirmed the importance of the configuration of the synthesized 1-indolyl acetate-5-nitroimidazolefor potential tubulin polymerization inhibitors.A series of 18 novel 1-indolyl acetate-5-nitroimidazole 3a–3r analogs of combretastatin A-4 were designed, synthesized, and evaluated for their in vitro biological activities as potential tubulin polymerization inhibitors.
Co-reporter:Jing-Ran Li, Dong-Dong Li, Rong-Rong Wang, Jian Sun, Jing-Jun Dong, Qian-Ru Du, Fei Fang, Wei-Ming Zhang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2014 Volume 75() pp:438-447
Publication Date(Web):21 March 2014
DOI:10.1016/j.ejmech.2013.11.020
•Thiazole derivatives have been synthesized and evaluated as potent FabH inhibitors.•Compound 5f has shown the best antibacterial activity.•Docking simulation and the QSAR study with DS 3.5 was conducted.Components of fatty acid biosynthetic pathway have been identified as attractive targets for the development of new antibacterial agents. Compounds of series A (4a–4g) and series B (5a–5g) were synthesized by the formation of an amine bond between aromatic acid and 4-phenylthiazol-2-amine or 4-(4-bromophenyl)thiazol-2-amine. These thiazole derivatives have evaluated as potent FabH inhibitors. Nineteen compounds (4b–4h, 4k, 4l, 5a–5h, 5k, 5l) are reported for the first time. Most of the synthesized compounds exhibited antibacterial activity in the MTT assay. The MIC value of these compounds ranged from 1.56 μg/mL to 100 μg/mL. Moreover, the tested compounds also showed FabH inhibition ability with IC50 value ranging from 5.8 μM to 48.1 μM. The IC50 values are near the MIC values. Compound 5f has exhibited the best antibacterial and Escherichia coli FabH inhibitory activity. Docking simulation and the QSAR study was conducted for learning about binding mode and the relationship between structure and activity.Compounds of novel thiazole derivatives containing amide skeleton were synthesized, isolated and evaluated for their antibacterial and FabH inhibitory activity. Docking simulation and the QSAR study was conducted.
Co-reporter:Chetan B. Sangani, Jigar A. Makawana, Xin Zhang, Shashikant B. Teraiya, Lin Lin, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2014 Volume 76() pp:549-557
Publication Date(Web):9 April 2014
DOI:10.1016/j.ejmech.2014.01.018
•A series of 4H-quinoline derivatives have been synthesized.•Synthesized derivatives have been screened for in vitro anticancer.•Synthesized derivatives have been screened for in vitro antibacterial.•Enzyme inhibitory activity against EGFR and FabH.A new series of pyrazole–quinoline–pyridine hybrids were designed based on molecular hybridization technique and synthesized by a base-catalyzed cyclocondensation reaction through one-pot multicomponent reaction. All compounds were tested for in vitro antibacterial and anticancer activities. Enzyme inhibitory activities of all compounds were carried out against FabH and EGFR. Of the compounds studied, majority of the compounds showed effective antibacterial as well as anticancer activity against used strains and cancer cell lines respectively. Compound 7k (IC50 = 0.51 ± 0.05 μM) against EGFR and 7b displayed the most potent inhibitory activity with IC50 of 3.1 μM against FabH as compared to other member of the series. In the molecular modeling study, compound 7k was bound in to the active pocket of EGFR with three hydrogen bond and one π–cation interaction with minimum binding energy ΔGb = −54.6913 kcal/mol, as well as compound 7b was bound in to the active site of FabH with hydrogen bond and π–sigma interactions with minimum binding energy ΔGb = −45.9125 kcal/mol.A new series of pyrazole–quinoline–pyridine hybrids were designed based on molecular hybridization technique and synthesized by a base-catalyzed cyclocondensation reaction through one-pot multicomponent reaction. All compounds were tested for in vitro antibacterial and anticancer activities. Enzyme inhibitory activities of all compounds were carried out against FabH and EGFR. Of the compounds studied, majority of the compounds showed effective antibacterial as well as anticancer activity against used strains and cancer cell lines respectively. Compound 7k (IC50 = 0.51 ± 0.05 μM) against EGFR and 7b displayed the most potent inhibitory activity with IC50 of 3.1 μM against FabH as compared to other member of the series.
Co-reporter:Yong-Tao Duan, Zhong-Chang Wang, Ya-Li Sang, Xiang-Xiang Tao, Shashikant B. Teraiya, Peng-Fei Wang, Qing Wen, Xiao-Jing Zhou, Liang Ding, Yong-Hua Yang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2014 Volume 76() pp:387-396
Publication Date(Web):9 April 2014
DOI:10.1016/j.ejmech.2014.02.004
•24 Nitroimidazole derivatives have been synthesized.•Their biological activities were evaluated as potential antibacterial and selective FabH inhibitors for the first time.•Compounds 35 and 37 showed the most potent FabH inhibition.A series of 2-Styryl-5-Nitroimidazole derivatives (25–48) have been synthesized and their biological activities were also evaluated against two Gram-negative bacterial strains: Escherichia coli and Pseudomonas aeruginosa and two Gram-positive bacterial strains: Bacillus subtilis and Bacillus thuringiensis as potential FabH inhibitors. All the compounds were structurally determined by 1H NMR, MS, and elemental analysis. E. coli β-ketoacyl-acyl carrier protein synthase III inhibitory assay and docking simulation indicated that compound 33 with IC50 of 9.0–36.4 μg/mL and compound 47 with IC50 of 6.3–34.3 μg/mL against bacterial strains were most potent inhibitors of E. coli FabH. And more, compounds 33 and 47 which possessed a broad-spectrum of antibacterial activities didn't exhibit any toxicity towards macrophage.A series of 2-ethanyl of 5-nitroimidazole derivatives have been synthesized and evaluated for their antibacterial activity as potential FabH inhibitors. Compounds 35 and 37 were the most active.
Co-reporter:Ya-Li Sang, Yong-Tao Duan, Han-Yue Qiu, Peng-Fei Wang, Jigar A. Makawana, Zhong-Chang Wang, Hai-Liang Zhu and Zhen-Xiang He
RSC Advances 2014 vol. 4(Issue 32) pp:16694-16704
Publication Date(Web):26 Mar 2014
DOI:10.1039/C4RA01444H
In the JAK/STAT pathway, sustainable activation of JAK with the capabilities of regulating cell growth and apoptosis can produce abnormal proliferation in tumor cells. A series of novel metronidazole derivatives containing the 1,4-benzodioxan moiety as potential inhibitors targeting JAK have been designed, synthesized and their biological activities were also evaluated. Among all synthesized compounds, compound 4t possessed the most potent antitumor activity against A549, Hela, HepG-2 and U251 in vitro, with IC50 values of 65, 21, 16 and 44 nM, respectively, which has been proved by the result of a flow cytometry (FCM) assay. Docking simulations demonstrated that compound 4t could bind tightly with the crystal structure of the JAK3 active site and act as a potential JAK3 inhibitor.
Co-reporter:Yong-Tao Duan, Yong-Fang Yao, Dan-Jie Tang, Nilesh j. Thumar, Shashikant B. Teraiya, Jigar A. Makawana, Ya-Li Sang, Zhong-Chang Wang, Xiang-Xiang Tao, Ai-Qin Jiang and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 39) pp:20382-20392
Publication Date(Web):24 Apr 2014
DOI:10.1039/C4RA01936A
A series of 1-isoquinoline-2-styryl-5-nitroimidazole derivatives has been synthesized and screened for telomerase inhibitory as well as antiproliferative properties on four variant cancer cell lines (U251, Hela, HepG-2 and A549). The bioassay results showed that most of the designed compounds exhibited moderate to potent in vitro inhibitory activity in the enzymatic and cellular assays, of which compounds 3u and 3v were revealed the most potent telomerase inhibitors, with IC50 values of 0.98 μM and 1.92 μM, respectively, compared to staurosporine. Docking simulation was performed to position compounds 3u and 3v into the telomerase active site to determine the probable binding pose. Twenty three compounds were scrutinized by the CoMFA and CoMSIA techniques of 3D Quantitative Structure–Activity Relationship (QSAR). These new findings along with molecular docking observations could provide important information that compounds 3u and 3v with potent inhibitory activity in tumor growth inhibition may be potential anticancer agents.
Co-reporter:Yin Luo, Yang Zhou, Jie Fu and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 45) pp:23904-23913
Publication Date(Web):07 May 2014
DOI:10.1039/C4RA02200A
Telomere and telomerase were closely related to the occurrence and development of some cancers. After the key active site of telomerase was identified, to enhance the ability of dihydropyrazole derivatives to inhibit telomerase, we designed a series of novel 4,5-dihydropyrazole derivatives containing heterocyclic oxygen moiety based on previous studies. The telomerase inhibition assay showed that compound 10a displayed the most potent inhibitory activity with an IC50 value of 0.6 μM for telomerase. The antiproliferative assay showed that 10a exhibited high activity against human gastric cancer cell SGC-7901 with an IC50 value of 10.95 ± 0.60 μM. Flow cytometric analysis and western blot results showed that 10a induced both apoptosis and autophagy. A docking simulation showed that 10a could bind well to the active site of telomerase and act as a telomerase inhibitor. The 3D-QSAR model was also built to provide a more pharmacological understanding that could be used to design new agents with more potent telomerase inhibitory activity.
Co-reporter:Yan-Ting Wang, Ya-Juan Qin, Ya-Liang Zhang, Yu-Jing Li, Bing Rao, Yan-Qing Zhang, Meng-Ru Yang, Ai-Qin Jiang, Jin-Liang Qi and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 61) pp:32263-32275
Publication Date(Web):02 Jul 2014
DOI:10.1039/C4RA03803G
Microtubule-targeted drugs are at present indispensable for various types of cancer therapy worldwide. A series of chalcone oxime derivatives were designed, synthesized and evaluated as potential tubulin polymerization inhibitors and for the cytotoxicity against anthropic cancer cell lines. These derivatives were completely demonstrated to have commendable inhibitory activity against tubulin polymerization by competing with the colchicine-binding site on tubulin, which was associated with G2/M phase cell cycle arrest as well as promising antiproliferative activity. Among the novel compounds, compound 13 was demonstrated to have the most potent inhibitory activity (tubulin IC50 = 1.6 μM). Antiproliferative assay results displayed that compound 13 had potent antiproliferative activity against A549, Hela and MCF-7 with GI50 values of 2.1, 3.5 and 3.6 μM, respectively, which were compared with the positive control colchicine and CA-4. Docking simulation showed that compound 13 could bind tightly to the colchicine domain of tubulin and act as a tubulin polymerization inhibitor. We also built a 3D-QSAR model to provide more information that could be applied to design new molecules with more potent tubulin inhibitory activity.
Co-reporter:Zhong-Chang Wang, Yong-Tao Duan, Han-Yue Qiu, Wan-Yun Huang, Peng-Fei Wang, Xiao-Qiang Yan, Shu-Feng Zhang and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 62) pp:33029-33038
Publication Date(Web):23 Jun 2014
DOI:10.1039/C4RA03819C
Metronidazole–sulfonamide derivatives 4a–4l, a new class of human carbonic anhydrase inhibitors (hCA), were designed, synthesized, isolated, and evaluated for their ability to inhibit the enzymatic activity of the physiologically dominant isozymes hCA II and the tumor-associated isozyme hCA IX (h = human). Many of these compounds inhibited CA II and IX in the range of 16–137 and 38–169 nM, respectively. Among all the compounds, the most potent inhibitor against hCA II and IX were compounds 4b (IC50 = 16 nM) and 4h (IC50 = 38 nM). Conversely compounds 4e and 4d displayed the most potent growth inhibitory activity against B16-F10 and MCF-7 cancer cell lines in vitro, respectively, with an IC50 value of 150 nM for B16-F10 and 6.5 nM for MCF-7. These metronidazole–sulfonamide derivatives may prove interesting lead candidates to target tumor-associated CA isozymes, wherein the CA domain is located extracellularly. All the new compounds were evaluated for cytotoxicity against human macrophages by MTT assay.
Co-reporter:Wei-Ming Zhang, Man Xing, Ting-Ting Zhao, Yu-Jia Ren, Xian-Hui Yang, Yu-Shun Yang, Peng-Cheng Lv and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 70) pp:37197-37207
Publication Date(Web):05 Aug 2014
DOI:10.1039/C4RA05257A
A series of pyrazole derivatives (1e–30e) has been designed and synthesized, and their biological activities were evaluated for EGFR and HER-2 inhibition and tumor cell antiproliferation. Among the compounds synthesized, compound 30e exhibited excellent enzyme inhibitory activity (IC50 = 0.21 ± 0.05 μM for EGFR and IC50 = 1.08 ± 0.15 μM for HER-2). Compound 30e also showed the most potent antiproliferative activity, which inhibited the growth of MCF-7 and B16-F10 cell lines with IC50 values of 0.30 ± 0.04 and 0.44 ± 0.05 μM, respectively. The molecular docking study was performed to analyze the probable binding models and the 3D-QSAR models were built for the rational design of EGFR/HER-2 inhibitors. Based on the results obtained, compound 30e with potent EGFR and HER-2 inhibitory activity may be a potential anticancer agent.
Co-reporter:Wan-Yun Huang, Ji Li, Shi-Lin Kong, Zhong-Chang Wang and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 66) pp:35193-35204
Publication Date(Web):01 Aug 2014
DOI:10.1039/C4RA05812G
Four complexes of the quinolone antibacterial agent enoxacin (HEn) and levofloxacin (HLevo) with Cu2+ and Co2+ have been synthesized and characterized by physicochemical and spectroscopic techniques. Complex 1 is a novel dinuclear structure, in which enoxacin exhibits a tridentate binding mode bound to two Cu(II) ions through the pyridone oxygen, the carboxylate oxygen and the piperazine nitrogen atom, so far as we know such a mode and structure has never been presented before in metal–quinolone complexes. In mononuclear complexes 2–4, enoxacin and levofloxacin act as a bidentate ligand bound to the metal through the ketone oxygen and a carboxylate oxygen atom. The antimicrobial activity of the complexes 1–4 against four bacterial species were tested and the results indicated that they exhibit enhanced or similar activity to the free ligands. Studies on the interaction of 1–4 with calf-thymus (CT) DNA through UV and circular dichroism (CD) spectroscopies indicated that they bind to CT DNA probably by the intercalative binding mode. Fluorescence competitive studies with ethidium bromide (EB) revealed that the complexes could compete with EB and displace it to bind to DNA using the intercalative binding site. In addition, the complexes 1–4 exhibited good binding propensity to human and bovine serum albumin proteins with relatively high binding constants.
Co-reporter:Han-Yue Qiu, Zhong-Chang Wang, Peng-Fei Wang, Xiao-Qiang Yan, Xiao-Ming Wang, Yong-Hua Yang and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 74) pp:39214-39225
Publication Date(Web):29 Aug 2014
DOI:10.1039/C4RA06438K
A novel series of MMPIs was designed, synthesized and purified using a scaffold modification strategy. The new compounds were also evaluated for biological activity against A549, MCF-7, HepG2 and Hela as potential inhibitors of MMP-2. The most potent inhibitor against MMP-2 was compound 19 (IC50 = 0.38 μM). Its antitumor effect is believed to be due to the induction of apoptosis, which is further confirmed by Annexin V-FITC/PI staining assay using flow cytometry analysis. Furthermore, all the compounds were evaluated for cytotoxicity against 293T. In addition, 3D-QSAR studies were conducted. The result showed that the benzosulfonamide benzenesulfonate MMPIs may prove interesting lead candidates to target MMP-2 associated tumor, where the MMP-2 domain is located extracellularly.
Co-reporter:Xin Zhang, Chetan B. Sangani, Li-Xin Jia, Pi-Xian Gong, Fang Wang, Jun-Fang Wang and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 97) pp:54217-54225
Publication Date(Web):24 Sep 2014
DOI:10.1039/C4RA08567A
Series of novel Schiff's base derivatives have been synthesized by combining 2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethyl 4-formylbenzoate 5, 6 with aromatic/heterocyclic amine 7a–r, 8, 9a–r in ethanol. All compounds were evaluated for antibacterial assay and inhibition against E. coli FabH. Among the compounds studied, most of the compounds showed effective antibacterial and potential inhibitory activity against E. coli FabH. Compound 10q showed most potent inhibitory activity (IC50 = 2.6883 μM) by binding tightly to the active site of the E. coli FabH receptor with minimum binding energy (ΔGb = −55.3117 kcal mol−1), in which molecular docking study indicated the binding mode was stabilized by one hydrogen bond and five π–π interactions.
Co-reporter:Ya-Juan Qin, Man Xing, Ya-Liang Zhang, Jigar A. Makawana, Ai-Qin Jiang and Hai-Liang Zhu
RSC Advances 2014 vol. 4(Issue 95) pp:52702-52711
Publication Date(Web):15 Oct 2014
DOI:10.1039/C4RA08708A
A series of (1,3-diphenyl-1H-pyrazol-4-yl) methyl benzoate derivatives (6a–10d) were designed, synthesized and evaluated as BRAFV600E inhibitors. Biological evaluation assays indicated that compound 10a showed the most potent inhibitory activity against A375, WM266.4 and BRAFV600E in vitro with IC50 values of 1.36 μM, 0.94 μM and 0.11 μM respectively compared with the positive compound vemurafenib. Furthermore, compound 10a showed highly selective BRAFV600E inhibitory activity in vitro. A docking simulation displayed that compound 10a could tightly bind the crystal structure of BRAFV600E at the active site. 3D-QSAR would provide a guideline to design and optimize more potent and positive BRAFV600E inhibitors based on the (1,3-diphenyl-1H-pyrazol-4-yl) methyl benzoate derivatives skeleton.
Co-reporter:She-Feng Wang, Yong Yin, Fang Qiao, Xun Wu, Shao Sha, Li Zhang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 8) pp:2409-2415
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmc.2014.03.004
Metronidazole has a broad-spectrum antibacterial activity. Hereby a series of novel metronidazole derivatives were designed and synthesized based on nitroimidazole scaffold in order to find some more potent antibacterial drugs. For these compounds which were reported for the first time, their antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus were tested. These compounds showed good antibacterial activities against Gram-positive strains. Compound 4m represented the most potent antibacterial activity against S. aureus ATCC 25923 with MIC of 0.003 μg/mL and it showed the most potent activity against S. aureus TyrRS with IC50 of 0.0024 μM. Molecular docking of 4m into S. aureus tyrosyl-tRNA synthetase active site were also performed to determine the probable binding mode.Metronidazole has a broad-spectrum antibacterial activity. Hereby a series of novel metronidazole derivatives were designed and synthesized based on nitroimidazole scaffold in order to find some more potent antibacterial drugs. For these compounds which were reported for the first time, their antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus were tested. These compounds showed good antibacterial activities against Gram-positive strains. Compound 4m represented the most potent antibacterial activity against S. aureus ATCC 25923 with MIC of 0.003 μg/mL and it showed the most potent activity against S. aureus TyrRS with IC50 of 0.0024 μM. Molecular docking of 4m into S. aureus tyrosyl-tRNA synthetase active site were also performed to determine the probable binding mode.
Co-reporter:Fei Zhang, Xiao-Liang Wang, Jing Shi, She-Feng Wang, Yong Yin, Yu-Shun Yang, Wei-Ming Zhang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:468-477
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.11.004
A series of new 1,3,4-oxadiazole derivatives (6a–6x) containing pyridine and acylhydrazone moieties were synthesized and developed as potential telomerase inhibitors. The bioassay tests demonstrated that compounds 6n, 6o, 6q, 6s and 6t exhibited significant broad-spectrum anticancer activity with IC50 range from 0.76 to 9.59 μM against the four cancer cell lines (HEPG2, MCF7, SW1116 and BGC823). Moreover, all the title compounds were assayed for telomerase inhibition using the TRAP-PCR-ELISA assay. Compound 6s showed the highest anticancer activity with IC50 of 0.76–1.54 μM against the tested cancer cell lines and exhibited the most potent telomerase inhibitory activity with IC50 of 1.18 ± 0.14 μM. The docking simulation was carried out to investigate a possible binding mode of compound 6s into the active site of telomerase (pdb. 3DU6) while the QSAR model was built to check the previous work as well as to introduce new directions.A series of new 1,3,4-oxadiazole derivatives (6a–6x) containing pyridine and acylhydrazone moieties were synthesized and developed as potential telomerase inhibitors. The bioassay tests demonstrated that compounds 6n, 6o, 6q, 6s and 6t exhibited significant broad-spectrum anticancer activity with IC50 range from 0.76 to 9.59 μM against the four cancer cell lines (HEPG2, MCF7, SW1116 and BGC823). Moreover, all the title compounds were assayed for telomerase inhibition using the TRAP-PCR-ELISA assay. Compound 6s showed the highest anticancer activity with IC50 of 0.76–1.54 μM against the tested cancer cell lines and exhibited the most potent telomerase inhibitory activity with IC50 of 1.18 ± 0.14 μM. The docking simulation was carried out to investigate a possible binding mode of compound 6s into the active site of telomerase (pdb. 3DU6) while the QSAR model was built to check the previous work as well as to introduce new directions.
Co-reporter:She-Feng Wang, Yong Yin, Xun Wu, Fang Qiao, Shao Sha, Peng-Cheng Lv, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:5727-5737
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.09.048
A series of 4-hydroxycoumarin derivatives were designed and synthesized in order to find some more potent antibacterial drugs. Their antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus were tested. These compounds showed good antibacterial activities against Gram-positive strains. Compound 4g represented the most potent antibacterial activity against Bacillus subtilis and S. aureus with MIC of 0.236, 0.355 μg/mL, respectively. What’s more, it showed the most potent activity against SaFabI with IC50 of 0.57 μM. Molecular docking of 4g into S. aureus Enoyl-ACP-reductase active site were performed to determine the probable binding mode, while the QSAR model was built to check the previous work as well as to introduce new directions.
Co-reporter:Ya-Juan Qin, Peng-Fei Wang, Jigar A. Makawana, Zhong-Chang Wang, Ze-Nan Wang, Yan-Gu, Ai-Qin Jiang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 22) pp:5279-5283
Publication Date(Web):15 November 2014
DOI:10.1016/j.bmcl.2014.09.054
A series of metronidazole–thiazole derivatives has been designed, synthesized and evaluated as potential antibacterial inhibitors. All the synthesized compounds were determined by elemental analysis, 1H NMR and MS. They were also tested for antibacterial activity against Escherichia coli, Bacillus thuringiensis, Bacillus subtilis and Pseudomonas aeruginosa as well as for the inhibition to FabH. The results showed that compound 5e exhibited the most potent inhibitory activity against E. coli FabH with IC50 of 4.9 μM. Molecular modeling simulation studies were performed in order to predict the biological activity of proposed compounds. Toxicity assay of compounds 5a, 5b, 5d, 5e, 5g and 5i showed that they were noncytotoxic against human macrophage. The results revealed that these compounds offered remarkable viability.A series of metronidazole–thiazole derivatives has been designed, synthesized and evaluated as potential antibacterial inhibitors. The results showed that compound 5e exhibited the most potent Escherichia coli FabH inhibitory activity with IC50 of 4.9 μM.
Co-reporter:Chetan B. Sangani, Jigar A. Makawana, Yong-Tao Duan, Yong Yin, Shashikant B. Teraiya, Nilesh J. Thumar, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 18) pp:4472-4476
Publication Date(Web):15 September 2014
DOI:10.1016/j.bmcl.2014.07.094
A new series of biquinoline–pyridine hybrids were designed and synthesized by a base-catalyzed cyclocondensation through one-pot multicomponent reaction. All compounds were tested for in vitro anticancer activities against two cancer cell lines A549 (adenocarcinomic human alveolar basal epithelial) and Hep G2 (liver cancer). Enzyme inhibitory activities of all compounds were carried out against EGFR and HER-2 kinase. Of the compounds studied, majority of the compounds showed effective anticancer activity against used cancer cell lines. Compound 9i (IC50 = 0.09 μM) against EGFR and (IC50 = 0.2 μM) against HER-2 kinase displayed the most potent inhibitory activity as compared to other member of the series. In the molecular modelling study, compound 9i was bound in to the active pocket of EGFR with four hydrogen bonds and two π–cation interactions having minimum binding energy ΔGb = −54.4 kcal/mol.A new series of biquinoline–pyridine hybrids were designed and synthesized by a base-catalyzed cyclocondensation through one-pot multicomponent reaction. All compounds were tested for in vitro anticancer activities against two cancer cell lines A549 (adenocarcinomic human alveolar basal epithelial) and Hep G2 (liver cancer). Enzyme inhibitory activities of all compounds were carried out against EGFR and HER-2 kinase. Of the compounds studied, majority of the compounds showed effective anticancer activity against used cancer cell lines. Compound 9i (IC50 = 0.09 μM) against EGFR and (IC50 = 0.2 μM) against HER-2 kinase displayed the most potent inhibitory activity as compared to other member of the series. In the molecular modelling study, compound 9i was bound in to the active pocket of EGFR with four hydrogen bonds and two π–cation interactions having minimum binding energy ΔGb = −54.4 kcal/mol.
Co-reporter:Ji-Wen Yuan, She-Feng Wang, Zhong-Liang Luo, Han-Yue Qiu, Peng-Fei Wang, Xin Zhang, Yong-An Yang, Yong Yin, Fei Zhang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 10) pp:2324-2328
Publication Date(Web):15 May 2014
DOI:10.1016/j.bmcl.2014.03.072
A series of compounds which contain pyrazole, thiazole and naphthalene ring (1a–7a, 1b–7b, 1c–7c, 1d–7d) were firstly synthesized and their anti-proliferative activity, EGFR inhibitory activity, cytotoxicity and inhibition to Hela cell migration were evaluated. Compound 2-(3-(3,4-dimethylphenyl)-5-(naphthalen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)thiazol-4(5H)-one (7d) displayed the most potent inhibitory activity (IC50 = 0.86 μM for Hela and IC50 = 0.12 μM for EGFR). Structure–activity relationship (SAR) analysis showed that the anti-proliferative activity was affected by A-ring-substituent (–OCH3 > –CH3 > –H > –Br > –Cl > –F). Docking simulation of compound 7d into EGFR active site showed that naphthalene ring of 7d with LYS721 formed two p–π bonds, which enhanced antitumor activity. Therefore, compound 7d may be developed as a potential antitumor agent.A series of compounds which contain pyrazole, thiazole and naphthalene ring (1a–7a, 1b–7b, 1c–7c, 1d–7d) were firstly synthesized and their anti-proliferative activity, EGFR inhibitory activity, cytotoxicity and inhibition to Hela cell migration were evaluated. Compound 2-(3-(3,4-dimethylphenyl)-5-(naphthalen-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)thiazol-4(5H)-one (7d) displayed the most potent inhibitory activity (IC50 = 0.86 μM for Hela and IC50 = 0.12 μM for EGFR). Structure–activity relationship (SAR) analysis showed that the anti-proliferative activity was affected by A-ring-substituent (–OCH3 > –CH3 > –H > –Br > –Cl > –F). Docking simulation of compound 7d into EGFR active site showed that naphthalene ring of 7d with LYS721 formed two p–π bonds, which enhanced antitumor activity. Therefore, compound 7d may be developed as a potential antitumor agent.
Co-reporter:Jigar A. Makawana, Chetan B. Sangani, Lin Lin, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 7) pp:1734-1736
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmcl.2014.02.041
New Schiff’s base derivatives 5a–j have been synthesized by reaction between 2-phenoxyquinoline-3-carbaldehydes 3a–j and 2-(2-methyl-5-nitro-1H-imidazol-1-yl)acetohydrazide 4 in presence of nickel(II) nitrate as a catalyst in ethanol under reflux in good yield (78–92%). All compounds were tested for anticancer and inhibition of EGFR. Of the compounds studied, majority of the compounds showed effective antiproliferation and inhibition of EGFR and HER-2 activities. Compound 5h showed most effective inhibition (IC50 = 0.12 ± 0.05 μM) by binding in to the active pocket of EGFR receptor with minimum binding energy (ΔGb = −58.3691 kcal/mol). The binding was stabilized by two hydrogen bonds, two π–cation and one π–sigma interactions. Compound 5d showed most effective inhibition (IC50 = 0.37 ± 0.04 μM).New Schiff’s base derivatives 5a–j have been synthesized by reaction between 2-phenoxyquinoline-3-carbaldehydes and 2-(2-methyl-5-nitro-1H-imidazol-1-yl)acetohydrazide in presence of nickel(II) nitrate as a catalyst in ethanol under reflux in good yield (78–92%). All compounds were tested for anticancer and inhibition of EGFR. Of the compounds studied, majority of the compounds showed effective antiproliferation and inhibition of EGFR activities. Compound 5h showed most effective inhibition (IC50 = 0.12 ± 0.05 μM) by binding in to the active pocket of EGFR receptor with minimum binding energy (ΔGb = −58.3691 kcal/mol). The binding was stabilized by two hydrogen bonds, two π–cation and one π–sigma interactions. Compound 5d showed most effective inhibition (IC50 = 0.37 ± 0.04 μM) against HER-2.
Co-reporter:Juan Sun, Jia-Jia Liu, Wei Zhou, Feng-Jiao Guo, Xin-Yi Wang, Hai-Liang Zhu
Journal of Molecular Structure 2014 Volumes 1056–1057() pp:104-109
Publication Date(Web):6 January 2014
DOI:10.1016/j.molstruc.2013.10.032
•A series of 4-hydroxy-3-(naphthalen-1-ylmethyl)thiophen-2(5H)-ones were first synthesized.•Compound 29 is the most potent agent against Staphylococcus aureus ATCC 25923 with MIC50 value of 0.21 μg/mL.•Their biological activities are also evaluated for tyrosyl-tRNA synthase inhibitory activity.A series of novel 4-hydroxy-3-(naphthalen-1-ylmethyl)thiophen-2(5H)-ones as tyrosyl-tRNA synthetase inhibitors were synthesized. Of these compounds, 4-(naphthalen-1-ylmethyl)-5-oxo-2,5-dihydrothiophen-3-yl-2-(4-hydroxyphenyl)acetate (29) was the most potent. The binding model and structure–activity relationship indicate that replacement of phenyl acetate in the side chain of 29 with a substituent containing more hydrophilic groups would be more suitable for further modification. Antibacterial assay revealed that the synthetic compounds are effective against growth of Gram-positive organisms, and 29 is the most potent agent against Staphylococcus aureus ATCC 25923 with MIC50 value of 0.21 μg/mL.
Co-reporter:Hua-Lin Yang, Fei Fang, Chang-Po Zhao, Dong-Dong Li, Jing-Ran Li, Jian Sun, Qian-Ru Du and Hai-Liang Zhu
MedChemComm 2014 vol. 5(Issue 2) pp:219-225
Publication Date(Web):04 Dec 2013
DOI:10.1039/C3MD00301A
Due to the increasing evidence linking the PI3Kγ pathway to various disease states, PI3Kγ is becoming an important target for cancer treatment. Herein we designed and synthesized a novel series of N,4-diphenylpyrimidin-2-amine derivatives with low CDOCKER_INTERACTION_ENERGY and then evaluated their PI3Kγ in vitro inhibitory activities and in vitro antiproliferation assays against four human cancer cells. Among the compounds we synthesized, compound C8 (IC50 = 65 nM) demonstrated the most potent inhibitory activity against PI3Kγ kinase as well as at the cellular level, compared to the control drug TG100713 (IC50 = 127 nM). Moreover, molecular docking analysis was also performed to determine possible binding modes between PI3Kγ and the target compounds.
Co-reporter:Zhu-Ping Xiao, Xu-Dong Wang, Peng-Fei Wang, Yin Zhou, Jing-Wen Zhang, Lei Zhang, Jiao Zhou, Sha-Sha Zhou, Hui Ouyang, Xiao-Yi Lin, Manzira Mustapa, Asaimuguli Reyinbaike, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2014 80() pp: 92-100
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.04.037
Co-reporter:S. P. Xu;G. Xu;Y. Pei;H. L. Zhu
Russian Journal of Coordination Chemistry 2014 Volume 40( Issue 1) pp:63-67
Publication Date(Web):2014 January
DOI:10.1134/S1070328414010084
One mononuclear complex has been designed and synthesized by a β-dikertone ligand 4-chlorobenzoic acid 4-[3-(4-chlorophenyl)-3-hydroxyacryloyl]-3-hydroxyphenyl ester (L) with MnCl2 · 4H2O in microwave radiation assistance. The complex was characterized by X-ray crystallography, confirming that the central manganese(II) atom was coordinated by four oxygens from two L and two oxygens from two water. The complex was assayed for in vitro antibacterial (B. subtilis, S. aureus, S. faecalis, P. aeruginosa, E. coli, and E. cloacae) activities and showed better antimicrobial activity against Gram positive strains than Gram negative strains.
Co-reporter:Fei Zhang, Qing Wen, She-Feng Wang, Baloch Shahla Karim, Yu-Shun Yang, Jia-Jia Liu, Wei-Ming Zhang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2014 24(1) pp: 90-95
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.11.079
Co-reporter:Shu-Fu Wang, Yin-Ling Zhu, Ping-Ting Zhu, Jigar A. Makawana, Ya-Liang Zhang, Meng-Yue Zhao, Peng-Cheng Lv, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 22(21) pp: 6201-6208
Publication Date(Web):
DOI:10.1016/j.bmc.2014.08.029
Co-reporter:Xu-Dong Wang, Wei Wei, Peng-Fei Wang, Yun-Tao Tang, Rui-Cheng Deng, Biao Li, Sha-Sha Zhou, Jing-Wen Zhang, Lei Zhang, Zhu-Ping Xiao, Hui Ouyang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 22(14) pp: 3620-3628
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.018
Co-reporter:Yong-Tao Duan, Yong-Fang Yao, Wei Huang, Jigar A. Makawana, Shashikant B. Teraiya, Nilesh j. Thumar, Dan-Jie Tang, Xiang-Xiang Tao, Zhong-Chang Wang, Ai-Qin Jiang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 22(11) pp: 2947-2954
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.005
Co-reporter:Yong Yin, Fang Qiao, Lu-Yi Jiang, She-Feng Wang, Shao Sha, Xun Wu, Peng-Cheng Lv, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 22(15) pp: 4285-4292
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.029
Co-reporter:Yu-Jing Li, Ya-Juan Qin, Jigar A. Makawana, Yan-Ting Wang, Yan-Qing Zhang, Ya-Liang Zhang, Meng-Ru Yang, Ai-Qin Jiang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2014 22(15) pp: 4312-4322
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.017
Co-reporter:Yong Qian, Jie Lin, Lujing Han, Lin Lin, Hailiang Zhu
Biosensors and Bioelectronics 2014 Volume 58() pp:282-286
Publication Date(Web):15 August 2014
DOI:10.1016/j.bios.2014.02.059
•HFP was a novel selective, sensitive and colorimetric fluorescent probe for N2H4.•This probe shows a 117 nm red-shifted absorption spectra with addition of N2H4.•This probe can detect N2H4 in an aqueous solution by the fluorescence enhancement.•This probe can be used for imaging hydrazine in living cells.We report a novel colorimetric and red-emitting fluorescent probe for hydrazine detection based on resorufin platform. This OFF–ON fluorescent probe shows a large (117 nm) red-shifted absorption spectrum and the color changes from colorless to red upon addition of hydrazine in the aqueous solution, which can serve as a “naked-eye” probe for hydrazine. Moreover, this probe also shows a significant fluorescence increase (~16 folds) and excellent linear relationship at physiological pH. Utilizing this sensitive and selective probe, we have successfully detected hydrazine in living cells.
Co-reporter:Man Xing;Ting-Ting Zhao;Yu-Jia Ren;Na-Na Peng
Medicinal Chemistry Research 2014 Volume 23( Issue 7) pp:3274-3286
Publication Date(Web):2014 July
DOI:10.1007/s00044-014-0909-0
A series of pyrazolyl-acylhydrazone derivatives (1e–20e) have been designed and synthesized and their biologic activities were also evaluated for telomerase inhibition and tumor cell antiproliferation. Among all the compounds, 12e showed the most potent activity in vitro, which inhibited the growth of MCF-7 and B16-F10 cell lines with IC50 values of 0.57 ± 0.03 and 0.49 ± 0.07 μM, respectively. Compound 12e also exhibited significant telomerase inhibitory activity (IC50 = 1.9 ± 0.43 μM). The result of flow cytometry demonstrated that compound 12e induced cell apoptosis. Docking simulation was performed to insert compound 12e into the crystal structure of telomerase at ATP binding site to determine the probable binding model. Based on the preliminary results, compound 12e with potent inhibitory activity in tumor growth may be a potential anticancer agent.
Co-reporter:Youyi Xia, Xiang Lu, Hailiang Zhu
Composites Science and Technology 2013 Volume 77() pp:37-41
Publication Date(Web):22 March 2013
DOI:10.1016/j.compscitech.2013.01.008
Natural silk fibroin (SF) fibers were modified with methyl orange (MO) and, via an in situ oxidation technique, a composite with the core/shell coaxial-line structure was prepared. The morphology, formation and application as the substrate of cell proliferation of the as-prepared product, silk fibroin/polyaniline (core/shell) coaxial composite fiber, were investigated. By controlling the reaction time, the coating density of polyaniline on the composite fiber could be effectively adjusted. It is suggested that MO promotes the generation of polyaniline shell coated on the SF fibers. The composite fiber exhibited good biocompatibility, cell attachment and proliferation when L929 cell is selected as a model and cultured on this fibrous substrate.
Co-reporter:Jing-Ran Li, Dong-Dong Li, Fei Fang, Qian-Ru Du, Lin Lin, Jian Sun, Yong Qian and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 48) pp:8375-8386
Publication Date(Web):19 Sep 2013
DOI:10.1039/C3OB41161C
A series of 4,6-substituted-(diaphenylamino)quinazolines as c-Src inhibitors have been prepared and their biological activity has also been evaluated. All the compounds displayed potential antiproliferation activities, with IC50 values ranging from 3.42 μM to 118.81 μM in five human tumor cell lines. Particularly, compound 15 exhibited higher cytotoxicity against the tested five tumor cell lines compared to the other small molecules. Generally, most of these compounds showed selectivity between the A549 cells and the other four cells, according to their corresponding IC50 values. The results obtained from the in vitro enzyme assay indicated compound 15 has remarkable inhibitory activity against c-Src kinase with an IC50 value of 27.3 nM, which is comparable to the control compounds. Furthermore, molecular docking and QSAR study by means of DS 3.5 (Discovery Studio 3.5, Accelrys, Co. Ltd) explored the binding modes and the structure and activity relationship (SAR) of these derivatives.
Co-reporter:Zhu-Ping Xiao, Zhi-Yun Peng, Jing-Jun Dong, Juan He, Hui Ouyang, Yu-Ting Feng, Chun-Lei Lu, Wan-Qiang Lin, Jin-Xiang Wang, Yin-Ping Xiang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2013 Volume 63() pp:685-695
Publication Date(Web):May 2013
DOI:10.1016/j.ejmech.2013.03.016
In a continuing study for discovering urease inhibitors based on flavonoids, nineteen reductive derivatives of flavonoids were synthesized and evaluated against Helicobacter pylori urease. Analysis of structure–activity relationship disclosed that 4-deoxy analogues are more potent than other reductive products. Out of them, 4′,7,8-trihydroxyl-2-isoflavene (13) was found to be the most active with IC50 of 0.85 μM, being over 20-fold more potent than the commercial available urease inhibitor, acetohydroxamic acid (AHA). Kinetics study revealed that 13 is a competitive inhibitor of H. pylori urease with a Ki value of 0.641 μM, which is well matched with the results of molecular docking. Biological evaluation and mechanism study of 13 suggest that it is a good candidate for discovering novel anti-gastritis and anti-gastric ulcer agent.Graphical abstract4′,7,8-Trihydroxyl-2-isoflavene (13), derived from 4′,7,8-trihydroxylisoflavone is a potent Helicobacter pylori urease inhibitor with IC50 of 0.85 μM, and it is a pure competitive inhibitor with a Ki value of 0.641 μM.Highlights► Efficient methods were developed to access to 3-flavene and 2-isoflavene. ► 4-Deoxyflavonoids were determined as H. pylori urease inhibitors. ► The most active inhibitor showed IC50 over 20-fold lower than that of acetohydroxamic acid. ► Docking model and kinetic studies revealed a competitive inhibition mechanism.
Co-reporter:Yin Luo, Shuai Zhang, Zhi-Jun Liu, Wu Chen, Jie Fu, Qing-Fu Zeng, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2013 Volume 64() pp:54-61
Publication Date(Web):June 2013
DOI:10.1016/j.ejmech.2013.04.014
•Eighteen compounds are firstly reported and characterized fully.•Their antimicrobial activities are also evaluated.•Some of the synthesized compounds exhibited excellent activity.A series of novel 1,3,4-thiadiazole derivatives bearing 1,2,4-triazolo[1,5-a]pyrimidine moiety were synthesized by the method of splicing active substructures. Among these derivatives, compounds 12, 13, 15–22 and 24–31 were firstly reported. All the compounds were assayed for antimicrobial activities against five fungi strains and four bacteria strains. The preliminary results indicated that compounds 25 and 28–31 showed good antifungal activities against Physaclospora piricola and Rhizoctonia solani. Compound 26 exhibited good antifungal activities against Cercospora beticola and R. solani. Most of the compounds showed better antibacterial activities against Gram-negative bacteria strains than Gram-positive bacteria strains. Compounds 25 and 28 showed the best activities against Pseudomonas fluorescence while compounds 30–31 showed good activities against Escherichia coli.New 1,2,4-triazolo[1,5-a]pyrimidine derivatives have been synthesized and evaluated for their antimicrobial activity. Most of them exhibited good activities and some could be comparable to positive controls.
Co-reporter:Jian Sun, Xian-Hai Lv, Han-Yue Qiu, Yan-Ting Wang, Qian-Ru Du, Dong-Dong Li, Yong-Hua Yang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2013 Volume 68() pp:1-9
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.07.003
•Twenty novel pyrazole derivatives had been synthesized.•The compounds were evaluated for brand-range CDKs inhibitory activity.•Crystal structure of compound 7a was determined.It was discovered that a number of cyclin dependent kinase inhibitors containing the pyrazole core structure exhibited high inhibitory potency against broad-range CDKs and corresponding anti-proliferative activities. This information guided us to design and synthesize a series of 1,3-diphenyl-N-(phenylcarbamothioyl)-1H-pyrazole-4-carboxamide derivatives (5a–10d), and evaluate their biological activities as CDKs inhibitors. Among all the synthesized compounds, compound 10b inhibited CDK2 with an IC50 value of 25 nM, counteracting tumor cell proliferation of three cancer cell lines (H460, MCF-7, A549) in the micromolar range (from 0.75 μM to 4.21 μM), In addition, flow cytometry indicated that compound 10b could induce cycle G0/G1 phase arrest in A549 cells with a dose dependent. Taken together, compound 10b could be selected for further preclinical evaluation.Twenty new pyrazole derivatives have been designed and evaluated for their anticancer and CDKs inhibitory activity. Compound 10b demonstrated the most potent inhibitory activity.
Co-reporter:Zhu-Ping Xiao, Zhi-Yun Peng, Jing-Jun Dong, Rui-Cheng Deng, Xu-Dong Wang, Hui Ouyang, Pan Yang, Juan He, Yuan-Feng Wang, Man Zhu, Xiao-Chun Peng, Wan-Xi Peng, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2013 Volume 68() pp:212-221
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.07.047
•Novel hybrid urease inhibitors with hydroxamic acid were designed.•The first acid stable urease inhibitor with IC50 in nanomolar range was found.•A mixture of competitive and uncompetitive mechanism was proposed.•Two site binding model was proposed.Inhibition of urease results in Helicobacter pylori growth arrest in the stomach, promoting urease as promising targets for gastrointestinal ulcer therapy. Twenty hybrid derivatives of flavonoid scaffold and hydroxamic acid, β-hydroxy-β-phenylpropionylhydroxamic acids, were therefore synthesized and evaluated against H. pylori urease. Biological evaluation of these compounds showed improved urease inhibition exhibiting micromolar to mid-nanomolar IC50 values. Most importantly, 3-(3-chlorophenyl)-3-hydroxypropionyl-hydroxamic acid (6g) exhibited high potency with IC50 of 0.083 ± 0.004 μM and Ki of 0.014 ± 0.003 μM, indicating that 6g is an excellent candidate to develop novel antiulcer agent. A mixture of competitive and uncompetitive mechanism was putatively proposed to understand the inconsistency between the crystallographic and kinetic studies for the first time, which is supported by our molecular docking studies.β-Hydroxy-β-phenylpropionylhydroxamic acid inhibits urease with a mixture of competitive and uncompetitive mechanism.
Co-reporter:Jing-Jun Dong, Qing-Shan Li, Shu-Fu Wang, Cui-Yun Li, Xin Zhao, Han-Yue Qiu, Meng-Yue Zhao and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 37) pp:6328-6337
Publication Date(Web):05 Jul 2013
DOI:10.1039/C3OB40776D
The RAF–MEK–ERK cascade appears to be intimately involved in the regulation of cell cycle progression and apoptosis. The BRAFV600E mutant results in constitutive activation of the ERK pathway, which can lead to cellular growth dysregulation. A series of 5-phenyl-1H-pyrazol derivatives (3a–5e) have been designed and synthesized, and their biological activities were evaluated as potential BRAFV600E inhibitors. All the compounds were reported for the first time except 3e, and compound 1-(4-bromo-2-hydroxybenzyl)-3-phenyl-1-(5-phenyl-1H-pyrazol-3-yl)urea (5c) displayed the most potent inhibitory activity (BRAFV600E IC50 = 0.19 μM). Antiproliferative assay results indicated that compound 5c possessed high antiproliferative activity against cell lines WM266.4 and A375 in vitro, with IC50 values of 1.50 and 1.32 μM, respectively, which were comparable with the positive control vemurafenib. Docking simulations showed that compound 5c binds tightly to the BRAFV600E active site and acts as BRAFV600E inhibitor. A 3D-QSAR model was also built to provide more pharmacophore understanding towards designing new agents with more potent BRAFV600E inhibitory activity.
Co-reporter:Jian Sun, Dong-Dong Li, Jing-Ran Li, Fei Fang, Qian-Ru Du, Yong Qian and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 44) pp:7676-7686
Publication Date(Web):10 Sep 2013
DOI:10.1039/C3OB41136B
A series of novel 4-alkoxyquinazoline derivatives were prepared and synthesized and their biological activities were evaluated as potential inhibitors of vascular endothelial growth factor receptor 2 (VEGFR2). Of these compounds, compound 3j demonstrated the most potent inhibitory activities against VEGFR2 tyrosine kinase and cell proliferation, the IC50 values of this compound reaching up to 2.72 nM and 0.35 μM, respectively, compared with Tivozanib (3.40 nM and 0.38 μM). The obtained results, along with a 3D-QSAR study and molecular docking that was used for investigating the probable binding mode, could provide an important basis for further optimization of compound 3j as a potential tyrosine kinase inhibitor.
Co-reporter:Shuai Zhang, Yin Luo, Liang-Qiang He, Zhi-Jun Liu, Ai-Qin Jiang, Yong-Hua Yang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 13) pp:3723-3729
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmc.2013.04.043
1,3,4-Oxadiazole derivatives have drawn continuing interest over the years because of their varied biological activities. In order to search for novel anticancer agents, we designed and synthesized a series of new 1,3,4-oxadiazole derivatives containing benzotriazole moiety as potential focal adhesion kinase (FAK) inhibitors. All the synthesized compounds were firstly reported. Among the compounds, compound 4 shows the most potent inhibitory activity against MCF-7 and HT29 cell lines with IC50 values of 5.68 μg/ml and 10.21 μg/ml, respectively. Besides, all the compounds were assayed for FAK inhibitory activity using the TRAP–PCR–ELISA assay. The results showed compound 4 exhibited the most potent FAK inhibitory activity with IC50 values of 1.2 ± 0.3 μM. Docking simulation by positioning compound 4 into the FAK structure active site was performed to explore the possible binding mode. Apoptosis which was analyzed by flow cytometry, demonstrated that compound 4 induced apoptosis against MCF-7 cells. Therefore, compound 4 may be a potential anticancer agent against MCF-7 cancer cell.A series of 1,3,4-oxadiazole derivatives have been designed and synthesized, and their biological activities were also evaluated for FAK inhibitory activity. Compound 4 possessed the most potent enzyme inhibition activities (IC50 = 1.2 ± 0.3 μM for FAK) and anticancer activities (IC50 = 5.68 μg/ml for MCF-7 and IC50 = 10.21 μg/ml for HT29). Structure–activity relationship was also analysed to provide more pharmacophore understanding that could be used to design new agents with more potent FAK inhibitory activity. Docking simulation was performed to explore the binding model of compound 4 with FAK.
Co-reporter:Hai-Hong Wang, Ke-Ming Qiu, Hong-En Cui, Yu-Shun Yang, Yin-Luo, Man Xing, Xiao-Yang Qiu, Li-Fei Bai, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 2) pp:448-455
Publication Date(Web):15 January 2013
DOI:10.1016/j.bmc.2012.11.020
A series of novel thiazolyl-pyrazoline derivatives containing benzodioxole (C1–C20) have been designed and synthesized. Among of the synthesized compounds, 2-(5-(benzo[d][1,3]dioxol-5-yl)-3-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-(4-bromophenyl)thiazole (C6) displayed the most potent inhibitory activity for HER-2 (IC50 = 0.18 μM for HER-2). Antiproliferative assay results indicated that compound C6 owned high antiproliferative activity against MCF-7 and B16-F10 in vitro, with IC50 value of 0.09 and 0.12 μM, respectively, being comparable with the positive control Erlotinib. Docking simulation was further performed to determine the probable binding model. Based on the preliminary results, compound C6 with potent inhibitory activity in tumor growth would be a potential anticancer agent.A series of novel thiazolyl-pyrazoline derivatives containing benzodioxole (C1–C20) have been designed and synthesized. Among of the synthesized compounds, 2-(5-(benzo[d][1,3]dioxol-5-yl)-3-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-(4-bromophenyl)thiazole (C6) displayed the most potent inhibitory activity for HER-2 (IC50 = 0.18 μM for HER-2). Antiproliferative assay results indicated that compound C6 owned high antiproliferative activity against MCF-7 and B16-F10 in vitro, with IC50 value of 0.09 and 0.12 μM, respectively, being comparable with the positive control Erlotinib. Docking simulation was further performed to determine the probable binding model. Based on the preliminary results, compound C6 with potent inhibitory activity in tumor growth would be a potential anticancer agent.
Co-reporter:Xu-Dong Wang, Rui-Cheng Deng, Jing-Jun Dong, Zhi-Yun Peng, Xiao-Ming Gao, Shu-Ting Li, Wan-Qiang Lin, Chun-Lei Lu, Zhu-Ping Xiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 17) pp:4914-4922
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmc.2013.06.066
Thirty-eight 3-aryl-4-acyloxyethoxyfuran-2(5H)-ones were designed, prepared and tested for antibacterial activities. Some of them showed significant antibacterial activity against Gram-positive organism, Gram-negative organism and fungus. Out of these compounds, 4-(2-(3-chlorophenylformyloxy)ethoxy)-3-(4-chlorophenyl)furan-2(5H)-one (d40) showed the widest spectrum of activity with MIC50 of 2.0 μg/mL against Staphylococcus aureus, 4.3 μg/mL against Escherichia coli, 1.5 μg/mL against Pseudomonas aeruginosa and 1.2 μg/mL against Candida albicans. Our data disclosed that MIC50 values against whole cell bacteria are positive correlation with MIC50 values against tyrosyl-tRNA synthetase. Meanwhile, molecular docking of d40 into S. aureus tyrosyl-tRNA synthetase active site was also performed, and the inhibitor tightly fitting the active site might be an important reason why it has high antimicrobial activity.Thirty-eight 3-aryl-4-acyloxyethoxyfuran-2(5H)-ones were designed, prepared and tested for antibacterial activities. 4-(2-(3-Chlorophenylformyloxy)ethoxy)-3-(4-chlorophenyl)furan-2(5H)-one (d40) showed the widest spectrum of activity with MIC50 of 2.0 μg/mL against Staphylococcus aureus, 4.3 μg/mL against Escherichia coli, 1.5 μg/mL against Pseudomonas aeruginosa and 1.2 μg/mL against Candida albicans.
Co-reporter:Yao Li, Chang-Po Zhao, Hua-Ping Ma, Meng-Yue Zhao, Ya-Rong Xue, Xiao-Ming Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:3120-3126
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.03.023
FabH, β-ketoacyl-acyl carrier protein (ACP) synthase III, is critically important to the initiation of fatty acid biosynthesis and is highly conserved among Gram-positive and Gram-negative bacteria. A series of novel secnidazole derivatives (1–20) were synthesized and fully characterized by spectroscopic methods and elemental analysis. Among these compounds, 6, 8, 11, 13, 14, 16–20 were reported for the first time. These compounds were tested for antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. The compounds inhibitory assay and docking simulation indicated that compound 20 (E)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)-N′-(3,4,5-trimethylbenzylidene)acetohydrazide with MIC of 3.13–6.25 μg/mL against the tested bacterial strains was a potent inhibitor of Escherichia coli FabH.FabH, β-ketoacyl-acyl carrier protein (ACP) synthase III, is critically important to the initiation of fatty acid biosynthesis and is highly conserved among Gram-positive and Gram-negative bacteria. A series of novel secnidazole derivatives (1–20) were synthesized and fully characterized by spectroscopic methods and elemental analysis. Among these compounds, 6, 8, 11, 13, 14, 16–20 were reported for the first time. These compounds were tested for antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. The compounds inhibitory assay and docking simulation indicated that compound 20 (E)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)-N′-(3,4,5-trimethylbenzylidene)acetohydrazide with MIC of 3.13–6.25 μg/mL against the tested bacterial strains was a potent inhibitor of Escherichia coli FabH.
Co-reporter:Wen Yang, Yang Hu, Yu-Shun Yang, Fei Zhang, Yan-Bin Zhang, Xiao-Liang Wang, Jian-Feng Tang, Wei-Qing Zhong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 5) pp:1050-1063
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmc.2013.01.013
Two series of novel naphthalin-containing pyrazoline derivatives C1–C14 and D1–D14 have been synthesized and evaluated for their EGFR/HER-2 inhibitory and anti-proliferation activities. Compound D14 displayed the most potent activity against EGFR and A549 cell line (IC50 = 0.05 μM and GI50 = 0.11 μM), being comparable with the positive control Erlotinib (IC50 = 0.03 μM and GI50 = 0.03 μM) and more potent than our previous compounds C0–A (IC50 = 5.31 μM and GI50 = 33.47 μM) and C0–B (IC50 = 0.09 μM and GI50 = 0.34 μM). Meanwhile, compound C14 displayed the most potent activity against HER-2 and MCF-7 cell line (IC50 = 0.88 μM and GI50 = 0.35 μM), being a little less potent than Erlotinib (IC50 = 0.16 μM and GI50 = 0.08 μM) but far more potent than C0–A (IC50 = 6.58 μM and GI50 = 27.62 μM) and C0–B (IC50 = 2.77 μM and GI50 = 3.79 μM). The docking simulation was performed to analyze the probable binding models and the QSAR models were built for reasonable design of EGFR/HER-2 inhibitors at present and in future. The structural modification of introducing naphthalin moiety reinforced the combination of our compounds and the receptor, resulting in progress of bioactivity. Moreover, the replacement of thiourea skeleton by using benzene ring resulted in the slight diversity of the two series towards specific targets.
Co-reporter:Juan Sun, Ming-Hui Li, Shao-Song Qian, Feng-Jiao Guo, Xiao-Fang Dang, Xiao-Ming Wang, Ya-Rong Xue, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 10) pp:2876-2879
Publication Date(Web):15 May 2013
DOI:10.1016/j.bmcl.2013.03.068
A series of 1,3,4-oxadiazole derivatives containing 1,4-benzodioxan moiety (7a–7q) have been designed, synthesized and evaluated for their antitumor activity. Most of the synthesized compounds were proved to have potent antitumor activity and low toxicity. Among them, compound 7a showed the most potent biological activity against Human Umbilical Vein Endothelial cells, which was comparable to the positive control. The results of apoptosis and flow cytometry (FCM) demonstrated that compound 7a induce cell apoptosis by the inhibition of MetAP2 pathway. Molecular docking was performed to position compound 7a into MetAP2 binding site in order to explore the potential target.Compound 7a showed the most potent biological activity against Human Umbilical Vein Endothelial cells. The results of apoptosis and flow cytometry (FCM) demonstrated that compound 7a induce cell apoptosis by the inhibition of MetAP2 pathway.
Co-reporter:Yin Luo, Shuai Zhang, Ke-Ming Qiu, Zhi-Jun Liu, Yu-Shun Yang, Jie Fu, Wei-Qing Zhong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 4) pp:1091-1095
Publication Date(Web):15 February 2013
DOI:10.1016/j.bmcl.2012.12.010
A series of novel aryl-2H-pyrazole derivatives bearing 1,4-benzodioxan or 1,3-benzodioxole moiety were designed as potential telomerase inhibitors to enhance the ability of aryl-2H-pyrazole derivatives to inhibit telomerase, a target of anticancer. The telomerase inhibition tests showed that compound 16A displayed the most potent inhibitory activity with IC50 value of 0.9 μM for telomerase. The antiproliferative tests showed that compound 16A exhibited high activity against human gastric cancer cell SGC-7901 and human melanoma cell B16-F10 with IC50 values of 18.07 and 5.34 μM, respectively. Docking simulation showed that compound 16A could bind well with the telomerase active site and act as telomerase inhibitor. 3D-QSAR model was also built to provide more pharmacophore understanding that could be used to design new agents with more potent telomerase inhibitory activity.A series of aryl-2H-pyrazole derivatives have been designed and synthesized, and their biological activities were also evaluated for telomerase inhibitory activity. Compound 16A possessed the most potent enzyme inhibition activities (IC50 = 0.9 μM for telomerase) and anticancer activities (IC50 = 5.34 μM for B16-F10 and IC50 = 18.07 μM for SGC-7901). Docking simulation was performed to explore the binding model of compound 16A with telomerase. 3D-QSAR model was also built to provide more pharmacophore understanding that could be used to design new agents with more potent telomerase inhibitory activity.
Co-reporter:Jigar A. Makawana, Juan Sun, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 23) pp:6264-6268
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmcl.2013.09.086
New Schiff’s base derivatives 5a–5h have been synthesized by reaction between 1-(4-bromophenyl)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethanone 3 and various benzohydrazide 4a–4h in presence of nickel (II) nitrate as a catalyst in ethanol at room temperature in good yield (54–88%). All compounds were tested for antibacterial as well as anticancer and inhibition of EGFR. Of the compounds studied, compounds 5d, 5f and 5g in the case of antiproliferation and inhibition of EGFR as well as compounds 5b, 5c, 5e and 5h in the case of antibacterial activity were found to be most effective compounds in the series. Compound 5f shows effective inhibition (IC50 = 0.21 ± 0.02 μM) by binding in to the active pocket of EGFR receptor with minimum binding energy (ΔGb = −49.4869 kcal/mol).New Schiff’s base derivatives 5a–5h have been synthesized by reaction between 1-(4-bromophenyl)-2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethanone 3 and various benzohydrazide 4a–4h in presence of nickel (II) nitrate as a catalyst in ethanol at room temperature in good yield (54–88%). All compounds were tested for antibacterial as well as anticancer and inhibition of EGFR. Of the compounds studied, compounds 5d, 5f and 5g in the case of antiproliferation and inhibition of EGFR as well as compounds 5b, 5c, 5e and 5h in the case of antibacterial activity were found to be most effective compounds in the series. Compound 5f shows effective inhibition (IC50 = 0.21 ± 0.02 μM) by binding in to the active pocket of EGFR receptor with minimum binding energy (ΔGb = −49.4869 kcal/mol).
Co-reporter:Kui Cheng, Jia-Yu Xue, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4235-4238
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.05.006
Two series of thiazole derivatives containing amide skeleton were synthesized and developed as potent Escherichia coli β-ketoacyl-(acyl-carrier-protein) synthase III (ecKAS III) inhibitors. All the 24 new synthesized compounds were assayed for antibacterial activity against the respective Gram-negative and Gram-positive bacterial strains, including E. coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. In which, 10 compounds with broad-spectrum antibacterial activities were further tested for their ecKAS III inhibitory activity. Last, we have successfully found that compound 4e showed both the promising broad antibacterial activity with MIC of 1.56–6.25 μg/mL against the representative bacterial stains, and also processed the most potent ecKAS III inhibitory activity with IC50 of 5.3 μM. In addition, docking simulation also carried out in this study to give a potent prediction binding mode between the small molecule and ecKAS III (PDB code: 1hnj) protein.
Co-reporter:Juan Sun, Yong Yin, Gui-Hua Sheng, Zhi-Bo Yang, Hai-Liang Zhu
Journal of Molecular Structure 2013 Volume 1039() pp:214-218
Publication Date(Web):8 May 2013
DOI:10.1016/j.molstruc.2013.01.071
Two vanillin derivatives have been designed and synthesized and their biological activities were also evaluated for antimicrobial activity. Their chemical structures are characterized by single crystal X-ray diffraction studies, 1H NMR, MS, and elemental analysis. Structural stabilization of them followed by intramolecular as well as intermolecular H-bonds makes these molecules as perfect examples in molecular recognition with self-complementary donor and acceptor units within a single molecule. Docking simulations have been performed to position compounds into the FtsZ active site to determine their probable binding model. Compound 3a shows the most potent biological activity, which may be a promising antimicrobial leading compound for the further research.Highlights► The synthesis, crystal structure and antibacterial activities of these compounds have not been reported so far. ► Our current findings are completely new. ► Their biological activities are also evaluated for FtsZ inhibitory activity.
Co-reporter:Yong Qian, Bingya Yang, Yuning Shen, Qianru Du, Lin Lin, Jie Lin, Hailiang Zhu
Sensors and Actuators B: Chemical 2013 Volume 182() pp:498-503
Publication Date(Web):June 2013
DOI:10.1016/j.snb.2013.03.031
We report a sensitive and selective fluorescent probe SFP-BC for hydrogen sulfide detection based on BODIPY–coumarin platform. SFP-BC shows a significant emission increase in the fast response to sulfide at physiological pH. Utilizing this probe, we have successfully detected hydrogen sulfide in fresh mouse blood plasma and in live cells.
Co-reporter:Qian-Ru Du, Dong-Dong Li, Ya-Zhou Pi, Jing-Ran Li, Jian Sun, Fei Fang, Wei-Qing Zhong, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2013 21(8) pp: 2286-2297
Publication Date(Web):
DOI:10.1016/j.bmc.2013.02.008
Co-reporter:Yan-Bin Zhang;Wen Liu;Yu-Shun Yang;Xiao-Liang Wang
Medicinal Chemistry Research 2013 Volume 22( Issue 7) pp:3193-3203
Publication Date(Web):2013 July
DOI:10.1007/s00044-012-0306-5
There is an accumulating body of experimental evidences validating focal adhesion kinase (FAK) as a therapeutic target and offering opportunities for anti-tumor drug development. In present study, we sought to synthesize twenty-eight potential FAK inhibitors as anti-tumor agents based on 1,2,4-triazole skeleton. The bioassay assays demonstrated that compounds 3e and 6j showed the most potent activity, 3e inhibited the growth of HCT116 and HepG2 cell lines with IC50 values of 8.17 and 7.04 μM, while compound 6j showed the most potent biological activity against HCT116 cell line (IC50 = 1.99 μM). Besides, compound 6j also exhibited significant FAK inhibitory activity (IC50 = 2.41 μM). The results of flow cytometry and western-blot assay demonstrated that compounds 3e and 6j possessed good anti-proliferative activity. Docking simulations were performed to position compounds 3e and 6j into the active site of FAK to determine the probable binding model.
Co-reporter:Ru Yan;Peng-Gang Liu;Zhi-Ming Zhang;Xian-Ying Fang
Medicinal Chemistry Research 2013 Volume 22( Issue 12) pp:5707-5716
Publication Date(Web):2013 December
DOI:10.1007/s00044-013-0561-0
A series of Schiff bases derived from 4-methylsalicylic acid (4a–4s) were synthesized, 14 of which (4a–4h, 4j–4l, 4n, 4q, and 4s) were reported for the first time. All the synthesized compounds were evaluated for their immunosuppressive activities for the first time. Among them, compound 4o displayed the most potent biological activity against lymph node cells (IC50 = 1.02 μM for lymph node cells and IC50 = 2.17 μM for PI3Kγ). The results of apoptosis and western-blot assays demonstrated that the immunosuppressive activity of compound 4o might be mediated by the inhibition of PI3K/AKT signaling pathway. Docking simulation was performed to position compound 4o into the PI3Kγ structure active site to determine the probable binding model.
Co-reporter:Dr. Yang Zhou;Dr. Qian-Ru Du;Dr. Jian Sun;Dr. Jing-Ran Li;Dr. Fei Fang;Dr. Dong-Dong Li;Dr. Yong Qian;Dr. Hai-Bin Gong; Jing Zhao; Hai-Liang Zhu
ChemMedChem 2013 Volume 8( Issue 3) pp:433-441
Publication Date(Web):
DOI:10.1002/cmdc.201200587
Abstract
Fatty acid biosynthesis plays a vital role in bacterial survival and several key enzymes involved in this biosynthetic pathway have been identified as attractive targets for the development of new antibacterial agents. Of these promising targets, β-ketoacyl-acyl carrier protein (ACP) synthase III (FabH) is the most attractive target that could trigger the initiation of fatty acid biosynthesis and is highly conserved among Gram-positive and -negative bacteria. Designing small molecules with FabH inhibitory activity displays great significance for developing antibiotic agents, which should be highly selective, nontoxic and broad-spectrum. In this manuscript, a series of novel Schiff base compounds were designed and synthesized, and their biological activities were evaluated as potential inhibitors. Among these 21 new compounds, (E)-N-((3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)methylene)hexadecan-1-amine (10) showed the most potent antibacterial activity with a MIC value of 3.89–7.81 μM−1 against the tested bacterial strains and exhibited the most potent E. coli FabH inhibitory activity with an IC50 value of 1.6 μM. Docking simulation was performed to position compound 10 into the E. coli FabH active site to determine the probable binding conformation.
Co-reporter:Lei Shi, Lu Wang, Zhi Wang, Hai-Liang Zhu, Qiao Song
European Journal of Medicinal Chemistry 2012 Volume 47() pp:585-593
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.10.027
A series of new cinnamanilides (6–40) were synthesized and their immunosuppressive activity and cytotoxicity were evaluated. Most of the cinnamanilides showed good immunosuppressive activity. Among the synthesized compounds, (Z)-N-(4-bromophenyl)-2-methoxy-3-(4-methoxyphenyl)acrylamide (37) and (Z)-2-methoxy-3-(4-methoxyphenyl)-N-p-tolylacrylamide (38) exhibited potent immunosuppressive activity (IC50 = 1.77 ± 0.33 and 0.94 ± 0.13 μM) without significant cytotoxicity.A series of new cinnamanilides were synthesized and their immunosuppressive activities were evaluated. Compunds 37 and 38 exhibited most potent immunosuppressive activity (IC50 = 1.77 ± 0.33 and 0.94 ± 0.13 μM).Highlights► 35 novel cinnamanilides were synthesized for the first time. ► The immunosuppressive activity and cytotoxicity of these compounds were evaluated. ► Compounds 37 and 38 exhibited most potent immunosuppressive activity. ► These compounds did not exhibit significant cytotoxicity. ► This study may be helpful for the discovery of new immunosuppressant leads.
Co-reporter:Kai Liu, Xiang Lu, Hong-Jia Zhang, Juan Sun, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2012 Volume 47() pp:473-478
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.11.015
A series of 2-(benzylthio)-5-aryloxadiazole derivatives have been designed and synthesized, and their biological activities are also evaluated for EGFR inhibitory activity. Fourteen compounds among the twenty compounds are reported for the first time. Their chemical structures are characterized by 1H NMR, MS, and elemental analysis. Anti-proliferative and EGFR inhibition assay results have demonstrated that compound 3e shows the most potent biological activity (IC50 = 1.09 μM for MCF-7 and IC50 = 1.51 μM for EGFR). Docking simulation has been performed to position compound 3e into the EGFR active site to determine the probable binding model, with an estimated binding free energy value of −10.7 kcal/mol. Compound 3e with potent inhibitory activity in tumor growth inhibition may be a promising anti-tumor leading compound for the further research.A series of 2-benzylthio-5-aryloxadiazole derivatives have been designed and synthesized, and their biological activities were also evaluated for EGFR inhibitory activity. Compound 3e possessed the most potent biological activity (IC50 = 1.09 μM for MCF-7 and IC50 = 1.51 μM for EGFR). Docking simulation was performed to explore the binding model of compound 3e with EGFR kinase.Highlights► Fourteen compounds among the twenty compounds are reported for the first time. ► Their biological activities are also evaluated for EGFR inhibitory activity. ► Our current findings are completely new.
Co-reporter:Huan-Qiu Li, Dong-Dong Li, Xiang Lu, Yun-Yun Xu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 1) pp:317-323
Publication Date(Web):1 January 2012
DOI:10.1016/j.bmc.2011.10.085
A type of novel 4,6-substituted-(diaphenylamino)quinazolines, which designed based on the 4-(phenylamino)quinazoline moiety, have been discovered as potential EGFR inhibitors. These compounds displayed good antiproliferative activity and EGFR-TK inhibitory activity. Especially, 4-((4-(3-bromophenylamino)quinazolin-6-ylamino)methyl)phenol (5b), showed the most potent inhibitory activity (IC50 = 0.28 μM for Hep G2, IC50 = 0.59 μM for A16-F10 and IC50 = 0.87 μM for EGFR) and effectively induces apoptosis in a dose-dependent manner in the Hep G2 cell line. Molecular docking of 5b into EGFR TK active site was also performed. This inhibitor nicely fitting the active site might well explain its excellent inhibitory activity.A type of novel 4,6-substituted-(diaphenylamino)quinazolines, which designed based on the 4-(phenylamino)quinazoline moiety, have been discovered as potential EGFR inhibitors. These compounds displayed good antiproliferative activity and EGFR-TK inhibitory activity. Especially, 4-((4-(3-bromophenylamino)quinazolin-6-ylamino)methyl)phenol (5b), showed the most potent inhibitory activity (IC50 = 0.28 μM for Hep G2, IC50 = 0.59 μM for A16-F10 and IC50 = 0.87 μM for EGFR) and effectively induces apoptosis in a dose-dependent manner in the Hep G2 cell line. Molecular docking of 5b into EGFR TK active site was also performed. This inhibitor nicely fitting the active site might well explain its excellent inhibitory activity.
Co-reporter:Yang Hu, Xiang Lu, Ke Chen, Ru Yan, Qing-Shan Li, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 2) pp:903-909
Publication Date(Web):15 January 2012
DOI:10.1016/j.bmc.2011.11.057
A total of 20 novel 1,3,4-oxadiazoline analogs (6a–6t) of combretastatin A-4 with naphthalene ring were designed, synthesized, and evaluated for biological activities as potential tubulin polymerization inhibitors. Among these compounds, 6n showed the most potent antiproliferative activities against multiple cancer cell lines and retained the microtubule disrupting effects. Docking simulation was performed to insert compound 6n into the crystal structure of tubulin to determine the probable binding model. These results indicated oxadiazoline compounds bearing the naphthyl moiety are promising tubulin inhibitors.A new series of novel 1,3,4-oxadiazoline analogs (6a–6t) of combretastatin A-4 with naphthalene ring were designed, synthesized, and evaluated for biological activities as potential tubulin polymerization inhibitors. Among these compounds, 6n showed the most potent antiproliferative activities against multiple cancer cell lines and retained the microtubule disrupting effects. Docking simulation was performed to insert compound 6n into the crystal structure of tubulin to determine the probable binding model.
Co-reporter:Ru Yan, Zhi-Ming Zhang, Xian-Ying Fang, Yang Hu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 4) pp:1373-1379
Publication Date(Web):15 February 2012
DOI:10.1016/j.bmc.2012.01.023
A series of novel 1,3,4-oxadiazole derivatives (5a–5s) have been designed, synthesized and evaluated for their immunosuppressive activity. Most of these synthesized compounds were proved to have potent immunosuppressive activity and low toxicity. Among them, compounds (5m–5r) showed the most potent biological activity against lymph node cells. The results of flow cytometry (FCM) and western blotting demonstrated that compound 5q induce cell apoptosis by the inhibition of PI3 K/AKT pathway. Molecular docking was performed to position compound 5q into PI3Kγ binding site in order to explore the potential target.A series of novel 1,3,4-oxadiazole derivatives (5a–5s) have been designed, synthesized and evaluated for their immunosuppressive activity. Most of these synthesized compounds were proved to have potent immunosuppressive activity and low toxicity. Among them, compounds (5m–5r) showed the most potent biological activity against lymph node cells. The results of flow cytometry (FCM) and western blotting demonstrated that compound 5q induce cell apoptosis by the inhibition of PI3 K/AKT pathway. Molecular docking was performed to position compound 5q into PI3 K binding site in order to explore the potential target.
Co-reporter:Cui-Yun Li, Qing-Shan Li, Li Yan, Xiao-Guang Sun, Ran Wei, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 12) pp:3746-3755
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmc.2012.04.047
A series of novel 4,5-dihydropyrazole derivatives containing niacinamide moiety as potential V600E mutant BRAF kinase (BRAFV600E) inhibitors were designed and synthesized. Results of the bioassays against BRAFV600E and WM266.4 human melanoma cell line showed several compounds to be endowed potent activities with IC50 and GI50 value in low micromolar range, among which compound 27e, (5-(4-Chlorophenyl)-3-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)6-methylpyridin-3-yl methanone (IC50 = 0.20 μM, GI50 = 0.89 μM) was bearing the best bioactivity comparable with the positive control Sorafenib. Docking simulation was performed to determine the probable binding model and 3D-QSAR model was built to provide more pharmacophore understanding that could use to design new agents with more potent BRAFV600E inhibitory activity.A new series of novel 4,5-dihydropyrazole derivatives containing niacinamide moiety were designed, synthesized and evaluated for biological activities as potential V600E mutant BRAF kinase (BRAFV600E) inhibitors. Among these compounds, 27e ((5-(4-Chlorophenyl)-3-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)6-methylpyridin-3-yl methanone) showed the most potent agent against BRAF and WM266.4 human melanoma cell line with IC50 value of 0.20 μM and GI50 value of 0.89 μM. Docking simulation was performed to insert compound 27e into the crystal structure of BRAF to determine the probable binding model. QSAR model was also built to provide a reliable tool for rational design of novel BRAF inhibitors.
Co-reporter:Hui Zhang, Xiang Lu, Li-Rong Zhang, Jia-Jia Liu, Xian-Hui Yang, Xiao-Ming Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 4) pp:1411-1416
Publication Date(Web):15 February 2012
DOI:10.1016/j.bmc.2012.01.004
A series of novel N-phenylsulfonylnicotinamide derivatives (1–24) have been synthesized and evaluated as potential EGFR tyrosine kinase (TK) inhibitors. Among all the compounds, compound 10 (5-bromo-N-(4-chlorophenylsulfonyl)nicotinamide) showed the most potent growth inhibitory activity against EGFR TK and antiproliferative activity of MCF-7 cancer cell line in vitro, with IC50 value of 0.09 and 0.07 μM. Docking simulation was performed to insert compound 10 into the EGFR TK active site to determine the probable binding model. Based on the preliminary results, compound 10 with potent inhibitory activity to tumor growth may be a potential anticancer agent.A series of novel N-phenylsulfonylnicotinamide derivatives (1–24) have been synthesized and evaluated as potential EGFR tyrosine kinase (TK) inhibitors. Among all the compounds, compound 10 (5-bromo-N-(4-chlorophenylsulfonyl)nicotinamide) showed the most potent growth inhibitory activity against EGFR TK and antiproliferative activity of MCF-7 cancer cell line in vitro, with IC50 value of 0.09 and 0.07 μM. Docking simulation was performed to insert compound 10 into the EGFR TK active site to determine the probable binding model.
Co-reporter:Ting-Ting Zhao, Xiang Lu, Xian-Hui Yang, Li-Ming Wang, Xi Li, Zhong-Chang Wang, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 10) pp:3233-3241
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmc.2012.03.057
A series of 2,6-dinitro-4-(trifluoromethyl)phenoxysalicylaldoxime derivatives (1h–20h) have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and tubulin polymerization inhibitors. Among all the compounds, 2h showed the most potent activity in vitro, which inhibited the growth of MCF-7, Hep-G2 and A549 cell lines with IC50 values of 0.70 ± 0.05, 0.68 ± 0.02 and 0.86 ± 0.05 μM, respectively. Compound 2h also exhibited significant tubulin polymerization inhibitory activity (IC50 = 3.06 ± 0.05 μM). The result of flow cytometry (FCM) demonstrated that compound 2h induced cell apoptosis. Docking simulation was performed to insert compound 2h into the crystal structure of tubulin at colchicine binding site to determine the probable binding model. Based on the preliminary results, compound 2h with potent inhibitory activity in tumor growth may be a potential anticancer agent.A series of 2,6-dinitro-4-(trifluoromethyl)phenoxysalicylaldoxime derivatives (1h–20h) have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and tubulin polymerization inhibitors. Among all the compounds, 2h showed the most potent activity in vitro, which inhibited the growth of MCF-7, Hep-G2 and A549 cell lines with IC50 values of 0.70 ± 0.05, 0.68 ± 0.02 and 0.86 ± 0.05 μM, respectively. Compound 2h also exhibited significant tubulin polymerization inhibitory activity (IC50 = 3.06 ± 0.05 μM). The result of flow cytometry (FCM) demonstrated that compound 2h induced cell apoptosis. Docking simulation was performed to insert compound 2h into the crystal structure of tubulin at colchicine binding site to determine the probable binding model. Based on the preliminary results, compound 2h with potent inhibitory activity in tumor growth may be a potential anticancer agent.
Co-reporter:Xian-Hui Yang, Qing Wen, Ting-Ting Zhao, Jian Sun, Xi Li, Man Xing, Xiang Lu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 3) pp:1181-1187
Publication Date(Web):1 February 2012
DOI:10.1016/j.bmc.2011.12.057
A series of cinnamic acyl 1,3,4-thiadiazole amide derivatives (6a–10e) have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and tubulin polymerization inhibitors. Among all the compounds, 10e showed the most potent activity in vitro, which inhibited the growth of MCF-7 and A549 cell lines with IC50 values of 0.28 and 0.52 μg/mL, respectively. Compound 10e also exhibited significant tubulin polymerization inhibitory activity (IC50 = 1.16 μg/mL). Docking simulation was performed to insert compound 10e into the crystal structure of tubulin at colchicine binding site to determine the probable binding model. Based on the preliminary results, compound 10e with potent inhibitory activity in tumor growth may be a potential anticancer agent.A series of cinnamic acyl 1,3,4-thiadiazole amide derivatives have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and tubulin polymerization inhibitors. Compound 10e possessed the most potent tubulin polymerization inhibitory activity (IC50 = 1.16 μg/mL) and anticancer activities (IC50 = 0.28 μg/mL for MCF-7 and IC50 = 0.52 μg/mL for A549). Docking simulation was performed to insert compound 10e into the crystal structure of tubulin to determine the probable binding model.
Co-reporter:Zhu-Ping Xiao, Xu-Dong Wang, Zhi-Yun Peng, Shen Huang, Pan Yang, Qing-Shan Li, Li-Hu Zhou, Xiao-Jun Hu, Li-Jun Wu, Yin Zhou, and Hai-Liang Zhu
Journal of Agricultural and Food Chemistry 2012 Volume 60(Issue 42) pp:10572-10577
Publication Date(Web):October 1, 2012
DOI:10.1021/jf303393n
It was disclosed in our group for the first time that the flavonoids in Lonicera japonica Thunb. are related to its therapy for gastric ulcer. Based on this finding, 20 flavonoids were selected for Helicobacter pylori urease inhibitory activity evaluation, and quercetin showed excellent potency with IC50 of 11.2 ± 0.9 μM. Structure–activity relationship analysis revealed that removal of the 5-, 3-, or 3′-OH in quercetin led to a sharp decrease in activity. Thus, 3- and 5-OH as well as 3′,4′-dihydroxyl groups are believed to be the key structural characteristics for active compounds, which was supported by the molecular docking study. Meanwhile, the results obtained from molecular docking and enzymatic kinetics research strongly suggested that quercetin is a noncompetitive urease inhibitor, indicating that quercetin may be able to tolerate extensive structural modification irrespective of the shape of the active site cavity and could be used as a lead candidate for the development of novel urease inhibitors.
Co-reporter:Jia-Jia Liu, Hui Zhang, Juan Sun, Zhong-Chang Wang, Yu-Shun Yang, Dong-Dong Li, Fei Zhang, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 20) pp:6089-6096
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmc.2012.08.020
A series of novel 4,5-dihydropyrazole derivatives (3a–3t) containing hydroxyphenyl moiety as potential V600E mutant BRAF kinase (BRAFV600E) inhibitors were designed and synthesized. Docking simulation was performed to insert compounds 3d (1-(5-(5-chloro-2-hydroxyphenyl)-3-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone) and 3m (1-(3-(4-chlorophenyl)-5-(3,5-dibromo-2-hydroxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone) into the crystal structure of BRAFV600E to determine the probable binding model, respectively. Based on the preliminary results, compound 3d and 3m with potent inhibitory activity in tumor growth may be a potential anticancer agent. Results of the bioassays against BRAFV600E, MCF-7 human breast cancer cell line and WM266.4 human melanoma cell line all showed several compounds had potent activities IC50 value in low micromolar range, among them, compound 3d and compound 3m showed strong potent anticancer activity, which were proved by that 3d: IC50 = 1.31 μM for MCF-7 and IC50 = 0.45 μM for WM266.5, IC50 = 0.22 μM for BRAFV600E, 3m: IC50 = 0.97 μM for MCF-7 and IC50 = 0.72 μM for WM266.5, IC50 = 0.46 μM for BRAFV600E, which were comparable with the positive control Erlotinib.A series of novel 4,5-dihydropyrazole derivatives (3a–3t) containing hydroxyphenyl moiety as potential V600E mutant BRAF kinase (BRAFV600E) inhibitors were designed and synthesized. Docking simulation was performed to insert compounds 3d and 3m into the crystal structure of BRAFV600E to determine the probable binding model, respectively. Based on the preliminary results, compound 3d and 3m with potent inhibitory activity in tumor growth may be a potential anticancer agent. Results of the bioassays against BRAFV600E, MCF-7 human breast cancer cell line and WM266.4 human melanoma cell line all showed several compounds had potent activities IC50 value in low micromolar range, among them, compound 3d and compound 3m showed strong potent anticancer activity, which were proved by that 3d: IC50 = 1.31 μM for MCF-7 and IC50 = 0.45 μM for WM266.5, IC50 = 0.22 μM for BRAFV600E, 3m: IC50 = 0.97 μM for MCF-7 and IC50 = 0.72 μM for WM266.5, IC50 = 0.46 μM for BRAFV600E, which were comparable with the positive control Erlotinib.
Co-reporter:Xi Li, Jia-Lin Liu, Xian-Hui Yang, Xiang Lu, Ting-Ting Zhao, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 14) pp:4430-4436
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmc.2012.05.031
In present study, a series of 3-(1,3-diphenyl-1H-pyrazol-4-yl)-N-phenylacrylamide derivatives (5a–8d) were designed, synthesized, and evaluated for HDAC inhibition and tumor cell antiproliferation. All of these compounds are reported for the first time, the chemical structures of these compounds were confirmed by means of 1H NMR, ESI-MS and elemental analyzes. Among the compounds, compound 8c showed the most potent biological activity against HCT116 cancer cell line (IC50 of 0.42 ± 0.02 μM for HDAC-1 and IC50 = 0.62 ± 0.02 for HCT116). Docking simulation was performed to position compound 8c into the HDAC active site to determine the probable binding model. The results of antiproliferative assay and western-blot demonstrated that compound 8c with potent inhibitory activity in tumor growth inhibition may be a potential anticancer agent against HCT116 cancer cell.In present study, a series of 3-(1,3-diphenyl-1H-pyrazol-4-yl)-N-phenylacrylamid derivatives (5a–8d) were designed, synthesized, and evaluated for HDAC inhibition and tumor cell antiproliferation. All of these compounds are reported for the first time, the chemical structures of these compounds were confirmed by means of 1H NMR, ESI-MS and elemental analyzes. Among the compounds, compound 8c showed the most potent biological activity against HCT116 cancer cell line (IC50 of 0.42 ± 0.02 μM for HDAC-1 and IC50 = 0.62 ± 0.02 for HCT116). The results of Docking simulation and Western-blot demonstrated that compound 8c with potent inhibitory activity in tumor growth inhibition may be a potential anticancer agent against HCT116 cancer cell.
Co-reporter:Hui Zhang, Jia-Jia Liu, Jian Sun, Xian-Hui Yang, Ting-Ting Zhao, Xiang Lu, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 10) pp:3212-3218
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmc.2012.03.055
A series of novel chalcone derivatives have been designed and synthesized, and their biological activities were also evaluated as potential inhibitors of tubulin. These compounds were assayed for growth-inhibitory activity against MCF-7 and A549 cell lines in vitro. Compound 3d showed the most potent antiproliferative activity against MCF-7 and A549 cell lines with IC50 values of 0.03 and 0.95 μg/mL and exhibited the most potent tubulin inhibitory activity with IC50 of 1.42 μg/mL. Docking simulation was performed to insert compound 3d into the crystal structure of tubulin at colchicines binding site to determine the probable binding model. Based on the preliminary results, compound 3d with potent inhibitory activity in tumor growth may be a potential anticancer agent.A series of novel chalcone derivatives have been designed and synthesized, and their biological activities were also evaluated as potential inhibitors of tubulin. These compounds were assayed for growth-inhibitory activity against MCF-7 and A549 cell lines in vitro. Compound 3d showed the most potent antiproliferative activity against MCF-7 and A549 cell lines with IC50 values of 0.03 and 0.95 μg/mL and exhibited the most potent tubulin inhibitory activity with IC50 of 1.42 μg/mL. Docking simulation was performed to insert compound 3d into the crystal structure of tubulin at colchicines binding site to determine the probable binding model. Based on the preliminary results, compound 3d with potent inhibitory activity in tumor growth may be a potential anticancer agent.
Co-reporter:Jian-Feng Tang, Xian-Hai Lv, Xiao-Liang Wang, Jian Sun, Yan-Bin Zhang, Yu-Shun Yang, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 14) pp:4226-4236
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmc.2012.05.055
In present study, a series of novel 1,3,4-oxadiazole derivatives have been designed, synthesized and purified. All of these compounds are reported for the first time, the chemical structures of these compounds were confirmed by means of 1H NMR, ESI-MS and elemental analyses. Besides, we evaluated their immunosuppressive activity. Most of these synthesized compounds were proved to have potent immunosuppressive activity and low toxicity. Among them, the bioassay results demonstrated that compounds 5c, 5n, 5p, 5o, 6f and 6g exhibited immunosuppressive activities with IC50 concentration range from 1.25 μM to 7.60 μM against the T cells, and the IC50 of positive control (csa) is 2.12 μM. Moreover, all the title compounds were assayed for PI3K/AKT signaling pathway inhibition using the ELISA assay. We examined the compounds with potent inhibitory activities against IL-1, IL-6 and IL-10 released in ConA-simulated mouse lymph node cells. The results showed compounds 5o and 6f displayed the most potential biological activity against T cells (IC50 = 1.25 μM and 4.75 μM for T cells). The preliminary mechanism of compound 5o inhibition effects was also detected by flow cytometry (FCM). The results of apoptosis and ELISA assay demonstrated that the immunosuppressive activity of compounds 5o and 6f against T cells may be mediated by the inhibition of PI3Kγ/AKT signaling pathway. Molecular docking was performed to position compounds 5o and 6f into PI3Kγ binding site in order to indicate the potential target.A series of 1,3,4-oxadiazole derivatives (5a–5q, 6a–6q) have been first synthesized for their potential immunosuppressive activity. Among them, compound 5o and 6f displayed the most potential biological activity against T cells (IC50 = 1.25 μM and 4.75 μM for T cells) and the IC50 of positive control (csa) is 2.12 μM. The preliminary mechanism of compound 5o and 6f inhibition effects was also detected by flow cytometry (FCM) and the compounds exerted immunosuppressive activity via inducing the apoptosis of activated lymph node cells in a dose dependent manner. Docking simulation was performed to position compound 5o and 6f into the PI3Kγ structure active site to determine the probable binding model.
Co-reporter:Ke-Ming Qiu, Hai-Hong Wang, Li-Ming Wang, Yin Luo, Xian-Hui Yang, Xiao-Ming Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 6) pp:2010-2018
Publication Date(Web):15 March 2012
DOI:10.1016/j.bmc.2012.01.051
A series of pyrazolyl-thiazolinone derivatives (E1–E36) have been designed and synthesized and their biological activities were also evaluated as potential EGFR and HER-2 kinase inhibitors. Thirty-four of the 36 compounds were reported for the first time. Among them, compound 2-(5-(4-bromophenyl)-3-p-tolyl-4,5-dihydro-1H-pyrazol-1-yl)thiazol-4(5H)-one (E28) displayed the most potent inhibitory activity (IC50 = 0.24 μM for EGFR and IC50 = 1.07 μM for HER-2). Antiproliferative assay results indicated that compound E28 owned high antiproliferative activity against MCF-7, B16-F10 and HCT-116 in vitro, with IC50 value of 0.30, 0.54, and 0.70 μM, respectively. Docking simulation was further performed to position compound E28 into the EGFR active site to determine the probable binding model. Based on the preliminary results, compound E28 with potent inhibitory activity in tumor growth would be a potential anticancer agent.A series of pyrazolyl-thiazolinone derivatives (E1–E36) have been designed and synthesized and their biological activities were also evaluated as potential EGFR and HER-2 kinase inhibitors. Thirty-four of the 36 compounds were reported for the first time. Among them, compound 2-(5-(4-bromophenyl)-3-p-tolyl-4,5-dihydro-1H-pyrazol-1-yl)thiazol-4(5H)-one (E28) displayed the most potent inhibitory activity (IC50 = 0.24 μM for EGFR and IC50 = 1.07 μM for HER-2). Antiproliferative assay results indicated that compound E28 owned high antiproliferative activity against MCF-7, B16-F10 and HCT-116 in vitro, with IC50 value of 0.30, 0.54, and 0.70 μM, respectively. Docking simulation was further performed to position compound E28 into the EGFR active site to determine the probable binding model. Based on the preliminary results, compound E28 with potent inhibitory activity in tumor growth would be a potential anticancer agent.
Co-reporter:Ke-Ming Qiu, Ru Yan, Man Xing, Hai-Hong Wang, Hong-En Cui, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 22) pp:6648-6654
Publication Date(Web):15 November 2012
DOI:10.1016/j.bmc.2012.09.021
Co-reporter:Zhi-Ming Zhang, Xue-Wei Zhang, Zong-Zheng Zhao, Ru Yan, Rui Xu, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 10) pp:3359-3367
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmc.2012.03.064
A series of 1,3,4-oxadiazole derivatives derived from 4-methoxysalicylic acid or 4-methylsalicylic acid (6a–6z) have been first synthesized for their potential immunosuppressive activity. Among them, compound 6z displayed the most potent biological activity against lymph node cells (inhibition = 38.76% for lymph node cells and IC50 = 0.31 μM for PI3Kγ). The preliminary mechanism of compound 6z inhibition effects was also detected by flow cytometry (FCM) and the compound exerted immunosuppressive activity via inducing the apoptosis of activated lymph node cells in a dose dependent manner. Docking simulation was performed to position compound 6z into the PI3Kγ structure active site to determine the probable binding model.A series of 1,3,4-oxadiazole derivatives derived from 4-methoxysalicylic acid or 4-methylsalicylic acid (6a–6z) have been first synthesized for their potential immunosuppressive activity. Among them, compound 6z displayed the most potent biological activity against lymph node cells (inhibition = 38.76% for lymph node cells and IC50 = 0.31 μM for PI3Kγ). The preliminary mechanism of compound 6z inhibition effects was also detected by flow cytometry (FCM) and the compound exerted immunosuppressive activity via inducing the apoptosis of activated lymph node cells in a dose dependent manner. Docking simulation was performed to position compound 6z into the PI3Kγ structure active site to determine the probable binding model.
Co-reporter:Xian-Hui Yang, Lu Xiang, Xi Li, Ting-Ting Zhao, Hui Zhang, Wen-Ping Zhou, Xiao-Ming Wang, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 9) pp:2789-2795
Publication Date(Web):1 May 2012
DOI:10.1016/j.bmc.2012.03.040
A series of 1,3,4-thiadiazol-2-amide derivatives (5a–5y) have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and FAK inhibitors. Among all the compounds, 5h showed the most potent activity in vitro, which inhibited the growth of MCF-7 and B16-F10 cell lines with IC50 values of 0.45 and 0.31 μM, respectively. Compound 5h also exhibited significant FAK inhibitory activity (IC50 = 5.32 μM). Docking simulation was performed to position compound 5h into the FAK structure active site to determine the probable binding model. The results of antiproliferative and Western-blot assay demonstrated that compound 5h possessed good antiproliferative activity. Therefore, compound 5h with potent FAK inhibitory activity may be a potential anticancer agent.A series of 1,3,4-thiadiazol-2-amide derivatives have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and FAK inhibitors. Compound 5h possessed the most potent FAK inhibitory activity (IC50 = 5.32 μM) and anticancer activities (IC50 = 0.45 μM for MCF-7 and IC50 = 0.31 μM for B16-F10). Docking simulation was performed to insert compound 5h into the crystal structure of FAK to determine the probable binding model.
Co-reporter:Yao Li, Yin Luo, Yang Hu, Di-Di Zhu, Shuai Zhang, Zhi-Jun Liu, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 14) pp:4316-4322
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmc.2012.05.050
Nitroimidazoles and their derivatives have drawn continuing interest over the years because of their varied biological activities, recently found application in drug development for antimicrobial chemotherapeutics and antiangiogenic hypoxic cell radiosensitizers. In order to search for novel antibacterial agents, we designed and synthesized a series of secnidazole analogs based on oxadiazole scaffold (4–21). Among these compounds, 4 and 7–21 were reported for the first time. These compounds were tested for antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. This new nitroimidazole derivatives class demonstrated strong antibacterial activities. Escherichia coli β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitory assay and docking simulation indicated that the compounds 2-(2-methoxyphenyl)-5-((2-methyl-5-nitro-1H-imidazol-1-yl)methyl)-1,3,4-oxadiazole (11) with MIC of 1.56–3.13 μg/mL against the tested bacterial strains and 2-((2-methyl-5-nitro-1H-imidazol-1-yl)methyl)-5-(2-methylbenzyl)-1,3,4-oxadiazole (12) with MIC of 1.56–6.25 μg/mL were most potent inhibitors of Escherichia coli FabH.Nitroimidazoles and their derivatives have drawn continuing interest over the years because of their varied biological activities, recently found application in drug development for antimicrobial chemotherapeutics and antiangiogenic hypoxic cell radiosensitizers. In order to search for novel antibacterial agents, we designed and synthesized a series of secnidazole analogs based on oxadiazole scaffold (4–21). Among these compounds, 4 and 7–21 were reported for the first time. These compounds were tested for antibacterial activities against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. This new nitroimidazole derivatives class demonstrated strong antibacterial activities. Escherichia coli β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitory assay and docking simulation indicated that the compounds 2-(2-methoxyphenyl)-5-((2-methyl-5-nitro-1H-imidazol-1-yl)methyl)-1,3,4-oxadiazole (11) with MIC of 1.56–3.13 μg/mL against the tested bacterial strains and 2-((2-methyl-5-nitro-1H-imidazol-1-yl)methyl)-5-(2-methylbenzyl)-1,3,4-oxadiazole (12) with MIC of 1.56–6.25 μg/mL were most potent inhibitors of Escherichia coli FabH.
Co-reporter:Yan-Bin Zhang, Xiao-Liang Wang, Wen Liu, Yu-Shun Yang, Jian-Feng Tang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 21) pp:6356-6365
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmc.2012.08.059
Three series of novel heterocyclic azoles derivatives containing pyrazine (5a–5k, 8a–8k and 11a–11k) have been designed, synthesized, structurally determined, and their biological activities were evaluated as potential telomerase inhibitors. Among the oxadiazole derivatives, compound 5c showed the most potent biological activity against SW1116 cancer cell line (IC50 = 2.46 μM against SW1116 and IC50 = 3.55 μM for telomerase). Compound 8h performed the best in the thiadiazole derivatives (IC50 = 0.78 μM against HEPG2 and IC50 = 1.24 μM for telomerase), which was comparable to the positive control. While compound 11f showed the most potent biological activity (IC50 = 4.12 μM against SW1116 and IC50 = 15.03 μM for telomerase) among the triazole derivatives. Docking simulation by positioning compounds 5c, 8h and 11f into the telomerase structure active site was performed to explore the possible binding model. The results of apoptosis demonstrated that compound 8h possessed good antitumor activity against HEPG2 cancer cell line. Therefore, compound 8h with potent inhibitory activity in tumor growth inhibition may be a potential antitumor agent against HEPG2 cancer cell. Therefore, the introduction of oxadiazole, thiadiazole and triazole structures reinforced the combination of our compounds and the receptor, resulting in progress of bioactivity.Three series of novel heterocyclic azoles derivatives containing pyrazine (5a–5k, 8a–8k and 11a–11k) have been designed, synthesized, structurally determined, and their biological activities were evaluated as potential telomerase inhibitors. Among the oxadiazole derivatives, compound 5c showed the most potent biological activity against SW1116 cancer cell line (IC50 = 2.46 μM against SW1116 and IC50 = 3.55 μM for telomerase). Compound 8h performed the best in the thiadiazole derivatives (IC50 = 0.78 μM against HEPG2 and IC50 = 1.24 μM for telomerase), which was comparable to the positive control. While compound 11f showed the most potent biological activity (IC50 = 4.12 μM against SW1116 and IC50 = 15.03 μM for telomerase) among the triazole derivatives. Docking simulation by positioning compounds 5c, 8h and 11f into the telomerase structure active site was performed to explore the possible binding model. The results of apoptosis demonstrated that compound 8h possessed good antitumor activity against HEPG2 cancer cell line. Therefore, compound 8h with potent inhibitory activity in tumor growth inhibition may be a potential antitumor agent against HEPG2 cancer cell. Therefore, the introduction of oxadiazole, thiadiazole and triazole structures reinforced the combination of our compounds and the receptor, resulting in progress of bioactivity.
Co-reporter:Li-Rong Zhang, Zhi-Jun Liu, Hui Zhang, Jian Sun, Yin Luo, Ting-Ting Zhao, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 11) pp:3615-3621
Publication Date(Web):1 June 2012
DOI:10.1016/j.bmc.2012.03.061
In present study, a series of new 2-(1,3,4-oxadiazol-2-ylthio)-1-phenylethanone derivatives (6a–6x) as potential focal adhesion kinase (FAK) inhibitors were synthesized. The bioassay assays demonstrated that compound 6i showed the most potent activity, which inhibited the growth of MCF-7 and A431 cell lines with IC50 values of 140 ± 10 nM and 10 ± 1 nM, respectively. Compound 6i also exhibited significant FAK inhibitory activity (IC50 = 20 ± 1 nM). Docking simulation was performed to position compound 6i into the active site of FAK to determine the probable binding model.A series of new 2-(1,3,4-oxadiazol-2-ylthio)-1-phenylethanone derivatives (6a–6x) as potential focal adhesion kinase (FAK) inhibitors were synthesized. The bioassay tests demonstrated that compound 6i showed the most potent activity, which inhibited the growth of MCF-7 and A431 cell lines with IC50 values of 0.14 and 0.01 μM, respectively. Compound 6i also exhibited significant FAK inhibitory activity (IC50 = 0.02 μM). Docking simulation was performed to position compound 6i into the active site of FAK to determine the probable binding model.
Co-reporter:Xian-Feng Huang, Xiang Lu, Yong Zhang, Guo-Qiang Song, Qi-Long He, Qing-Shan Li, Xian-Hui Yang, Yao Wei, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 16) pp:4895-4900
Publication Date(Web):15 August 2012
DOI:10.1016/j.bmc.2012.06.056
A series of N-((1,3-diphenyl-1H-pyrazol-4-yl)methyl)aniline derivatives (5a–8d) have been designed and synthesized, and their biological activities were also evaluated as potential antitumor and cyclin dependent kinase 2 (CDK2) inhibitors. Among all the compounds, compound 5a displayed the most potent CDK2/cyclin E inhibitory activity in vitro, with an IC50 of 0.98 ± 0.06 μM. Antitumor assays indicated that compound 5a owned high antiproliferative activity against MCF-7 and B16-F10 cancer cell lines with IC50 values of 1.88 ± 0.11 and 2.12 ± 0.15 μM, respectively. Docking simulation was performed to insert compound 5a into the crystal structure of CDK2 at active site to determine the probable binding model. Based on the preliminary results, compound 5a with potent inhibitory activity in tumor growth may be a potential anticancer agent.A series of novel N-((1,3-diphenyl-1H-pyrazol-4-yl)methyl)aniline derivatives (5a–8d) have been designed and synthesized and evaluated as potential antitumor and cyclin dependent kinase 2 (CDK2) inhibitors. Among all the compounds, compound 5a displayed the most potent CDK2/cyclin E inhibitory activity in vitro, with an IC50 of 0.98 ± 0.06 μM. Compound 5a also owned high antiproliferative activity against MCF-7 and B16-F10 cancer cell lines with IC50 values of 1.88 ± 0.11 and 2.12 ± 0.15 μM, respectively.
Co-reporter:Yu-Shun Yang, Qing-Shan Li, Shuai Sun, Yan-Bin Zhang, Xiao-Liang Wang, Fei Zhang, Jian-Feng Tang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 20) pp:6048-6058
Publication Date(Web):15 October 2012
DOI:10.1016/j.bmc.2012.08.043
Two series of novel 2,3-dihydrobenzo[b][1,4]dioxin-containing 4,5-dihydro-1H-pyrazole derivatives C1–C15 and D1–D15 have been synthesized and evaluated for their B-Raf inhibitory and anti-proliferation activities. Compound C14 ((3-(4-bromophenyl)-5-(2-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methanone) showed the most potent biological activity against B-RafV600E (IC50 = 0.11 μM) and WM266.4 human melanoma cell line (GI50 = 0.58 μM), being comparable with the positive control Erlotinib and more potent than our previous best compound, while D10 ((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)(5-(3-fluorophenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone) performed the best in the D series (IC50 = 1.70 μM; GI50 = 1.45 μM). The docking simulation was performed to analyze the probable binding models and poses and the QSAR model was built for reasonable design of B-Raf inhibitors in future. The introduction of 2,3-dihydrobenzo[b][1,4]dioxin structure reinforced the combination of our compounds and the receptor, resulting in progress of bioactivity.Two series of novel 2,3-dihydrobenzo[b][1,4]dioxin-containing 4,5-dihydro-1H-pyrazole derivatives C1–C15 and D1–D15 have been synthesized and evaluated for their B-Raf inhibitory and anti-proliferation activities. Compound C14 ((3-(4-bromophenyl)-5-(2-fluorophenyl)-4,5-dihydro-1H-pyrazol-1-yl)(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methanone) showed the most potent biological activity against B-RafV600E (IC50 = 0.11 μM) and WM266.4 human melanoma cell line (GI50 = 0.58 μM), being comparable with the positive control Erlotinib and more potent than our previous best compound, while D10 ((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)(5-(3-fluorophenyl)-3-phenyl-4,5-dihydro-1H-pyrazol-1-yl)methanone) performed the best in the D series (IC50 = 1.70 μM; GI50 = 1.45 μM). The docking simulation was performed to analyze the probable binding models and poses and the QSAR model was built for reasonable design of B-Raf inhibitors in future. The introduction of 2,3-dihydrobenzo[b][1,4]dioxin structure reinforced the combination of our compounds and the receptor, resulting in progress of bioactivity.
Co-reporter:Qing-Shan Li, Xian-Hai Lv, Yan-Bin Zhang, Jing-Jun Dong, Wen-Ping Zhou, Yang Yang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 21) pp:6596-6601
Publication Date(Web):1 November 2012
DOI:10.1016/j.bmcl.2012.09.004
There is an accumulating body of experimental evidences validating oncogenic BRAFV600E as a therapeutic target and offering opportunities for anti-melanoma drug development. Encouraged by the positive results of pyrazole derivatives as BRAFV600E inhibitors, we sought to design diverse novel potential BRAFV600E inhibitors as antitumor agents based on pyrazole skeleton. In silico and in vitro screening of our designed pyrazole derivatives has identified Hit 1 as BRAFV600E inhibitor. Based on its structure and through further structure modification, compound 25, which exhibited the most potent inhibitory activity with an IC50 value of 0.16 μM for BRAFV600E and GI50 value of 0.24 μM for mutant BRAF-dependent melanoma cells, was obtained. The 3D-QSAR models and the molecular docking simulation were introduced to analyze the structure–activity relationship.Based on in silico screening results, a series of novel 3,5-diarylpyrazoline salicylamide derivatives were designed and synthesized. Compound 25 exhibited the most potent inhibitory activity with an IC50 value of 0.16 μM for BRAFV600E and GI50 value of 0.24 μM for mutant BRAF-dependent WM266.4 melanoma cells.
Co-reporter:Xi Li, Xiang Lu, Man Xing, Xian-Hui Yang, Ting-Ting Zhao, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 11) pp:3589-3593
Publication Date(Web):1 June 2012
DOI:10.1016/j.bmcl.2012.04.066
A series of N,1,3-triphenyl-1H-pyrazole-4-carboxamide derivatives have been designed, synthesized and evaluated for their potential antiproliferation activity and Aurora-A kinase inhibitory activity. Among all the compounds, compound 10e possessed the most potent biological activity against HCT116 and MCF-7 cell lines with IC50 values of 0.39 ± 0.06 μM and 0.46 ± 0.04 μM, respectively, which were comparable to the positive control. Compound 10e also exhibited significant Aurora-A kinase inhibitory activity (IC50 = 0.16 ± 0.03 μM). Docking simulation was performed to position compound 10e into the active site of Aurora-A kinase, in order to get the probable binding model for further study. The results of Western-blot assay demonstrated that compound 10e possessed good Aurora-A kinase inhibitory activity against HCT116. Based on the preliminary results, it is deduced that compound 10e with potent Aurora-A kinase inhibitory activity may be a potential anticancer agent.A series of N,1,3-triphenyl-1H-pyrazole-4-carboxamide derivatives have been designed, synthesized and evaluated for their potential antiproliferation activity against Aurora-A kinase. Among all the compounds, compound 10e possessed the most potent biological activity against HCT116 with IC50 values of 0.39 ± 0.06 μM, which was comparable to the positive control. The results of Docking simulation and Western-blot assay futher demonstrated that compound 10e possessed good Aurora-A kinase inhibitory activity against HCT116. Based on the preliminary results, it is deduced that compound 10e with potent Aurora-A kinase inhibitory activity may be a potential anticancer agent.
Co-reporter:Dong-Dong Li, Fei Fang, Jing-Ran Li, Qian-Ru Du, Jian Sun, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 18) pp:5870-5875
Publication Date(Web):15 September 2012
DOI:10.1016/j.bmcl.2012.07.079
It had been reported that some dioxygenated rings fusing with the quinazoline scaffold could lead to new EGFR inhibitors. Based on this, several kinds of oxygenated alkane quinazoline derivatives were synthetized and evaluated as EGFR inhibitors. Their antiproliferative activities were tested against four cancer cell lines: A431, MCF-7, A549, and B16-F10. Most derivatives could counteract EGF-induced EGFR phosphorylation, and their potency was comparable to the reference compound Erlotinib. The size of the fused dioxygenated ring was crucial for the biological activity and the heptatomic ring derivative 19 showed potent in vitro inhibitory activity in the enzymatic assay as well as in the cellular assay.It had been reported that some dioxygenated rings fusing with the quinazoline scaffold could lead to new EGFR inhibitors. Based on this, several kinds of oxygenated alkane quinazoline derivatives were synthetized and evaluated as EGFR inhibitors. Their antiproliferative activities were tested against four cancer cell lines: A431, MCF-7, A549, and B16-F10. Most derivatives could counteract EGF-induced EGFR phosphorylation, and their potency was comparable to the reference compound Erlotinib. The size of the fused dioxygenated ring was crucial for the biological activity and the heptatomic ring derivative 19 showed potent in vitro inhibitory activity in the enzymatic assay as well as in the cellular assay.
Co-reporter:Yu-Shun Yang, Fei Zhang, Chao Gao, Yan-Bin Zhang, Xiao-Liang Wang, Jian-Feng Tang, Jian Sun, Hai-Bin Gong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4619-4624
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.091
A series of sulfur-containing heterocyclic pyrazoline derivatives (C1–C18; D1–D9) have been synthesized and purified (all are new except one) to be screened for FabH inhibitory activity. Compound C14 showed the most potent biological activity against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Bacillus subtilis (MIC values: 1.56–3.13 μg/mL), being comparable with the positive control, while D6 performed the best in the thiazolidinone series (MIC values: 3.13–6.25 μg/mL). They also demonstrated strong broad-spectrum antimicrobial activity. Compounds C14 and D6 exhibited the most potent E. coli FabH inhibitory activity with IC50 of 4.6 and 8.4 μM, respectively, comparable with the positive control DDCP (IC50 = 2.8 μM). Docking simulation was performed to position compound C14 and D6 into the E. coli FabH structure active site to determine the probable binding model. The structurally modification of previous compounds and the attempt in innovative target have brought a positive progress.A series of sulfur-containing heterocyclic pyrazoline derivatives (C1–C18; D1–D9) have been synthesized and purified (all are new except one) to screen for FabH inhibitory activity. Compound C14 showed the most potent biological activity against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Bacillus subtilis (MIC values: 1.56–3.13 μg/mL), being comparable with the positive control, while D6 performed the best in the thiazolidinone series (MIC values: 3.13–6.25 μg/mL). They also demonstrated strong broad-spectrum antimicrobial activity. Compounds C14 and D6 exhibited the most potent E. coli FabH inhibitory activity with IC50 of 4.6 and 8.4 μM, respectively, comparable with the positive control DDCP (IC50 = 2.8 μM). Docking simulation was performed to position compound C14 and D6 into the E. coli FabH structure active site to determine the probable binding model. The structurally modification of previous compounds and the attempt in innovative target have brought a positive progress.
Co-reporter:Yin Luo, Ran Song, Yao Li, Shuai Zhang, Zhi-Jun Liu, Jie Fu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 9) pp:3039-3043
Publication Date(Web):1 May 2012
DOI:10.1016/j.bmcl.2012.03.080
A series of deoxybenzoin oximes were recently reported as potent immunosuppressive agents by our group. In order to continue the original research for potential immunosuppressive agents with high efficacy and low toxicity, we synthesized a series of new chalcone oximes and evaluated them for their cytotoxicities and immunosuppressive activities. Among the synthesized compounds, chalcone oximes 25 and 27 exhibited lower cytotoxicities and higher inhibitory activities on anti-CD3/anti-CD28 co-stimulated lymph node cells than other compounds. Specially, compound 27 displayed 200-fold lower cytotoxicity (CC50 = 2174.39 μM) than cyclosporin A (CC50 = 10.10 μM) and showed SI value (SI = 176.69) close to cyclosporin A (SI = 154.13). Besides, the preliminary mechanism of inhibition effect of compounds 25 and 27 was also detected by flow cytometry, and the compounds exerted immunosuppressive activities via inducing the apoptosis of activated lymph node cells in a dose dependent manner. Also, the deep mechanism of apoptosis was detected by Western blot analysis.A series of new chalcone oximes were synthesized and evaluated for their cytotoxicities and immunosuppressive activities. Chalcone oximes 25 and 27 exhibited lower cytotoxicities and higher inhibitory activities on anti-CD3/anti-CD28 co-stimulated lymph node cell than other compounds. Compound 27 displayed 200-fold lower cytotoxicity and potent inhibition activity (SI = 176.69) close to cyclosporin A (SI = 154.13). The preliminary mechanism of inhibition effect of compounds 25 and 27 was also detected by flow cytometry, and the compounds exerted immunosuppressive activities via inducing the apoptosis of activated lymph node cells in a dose dependent manner. The deep mechanism of apoptosis was detected by Western blot analysis.
Co-reporter:Zhu-Ping Xiao;Li-Cheng Yi;Tian-Fang Yi
Journal of Chemical Crystallography 2012 Volume 42( Issue 4) pp:323-329
Publication Date(Web):2012 April
DOI:10.1007/s10870-011-0246-9
Crystalline hydrate of the title compound (5), C19H26N2O5·2(H2O), was structurally characterized by single crystal X-ray diffraction. It crystallizes in monoclinic system space group P 21/c with a = 7.3987(7) Å, b = 17.8691(16) Å, c = 17.0022(13) Å, β = 112.944(3)°, V = 2070.0(3) Å3, Z = 4, R1 = 0.0592, wR2 = 0.1016, and T = 298(2) K. The X-ray structure determination revealed that the center furanone ring is nearly coplanar with 3,4-dimethoxybenzene ring, making a dihedral angle of 0.860(69)°. Two kinds of centrosymmetric tetramers characterized by graph-set motifs of R78(36) and R46(32) are formed through O–H···O, O–H···N and C–H···O hydrogen bonding interactions, which generate a sheet of edge-fused rings parallel to the (011) plane. These sheets are further linked into a three dimensional network by C–H···π interactions. Nine 3-(3,4-dimethoxyphenyl)furan-2(5H)-ones were synthesized and fully characterized by elemental analysis, MS and 1H NMR. All of them were evaluated for antimicrobial activities against three Gram-positive organisms and a Gram-negative organism, and compound 5 was the most active against Staphylococcus aureus ATCC 25923.
Co-reporter:Shao-Song Qian;Yue-Hu Chen;Hong-You Cui
Journal of Chemical Crystallography 2012 Volume 42( Issue 6) pp:611-614
Publication Date(Web):2012 June
DOI:10.1007/s10870-012-0290-0
Two crystalline forms of 1,4-dimethyl-2-(4-(methyl-sulfonyl) styryl)benzene (1) and 1,3-diisopropyl-2-(4-(methyl-sulfonyl)styryl) benzene (2) were obtained and their structures were determined by X-ray diffraction technique. Both compound 1 and 2 crystallized in a triclinic space group P-1 with cell parameters a = 5.375(2) Å, b = 18.402(7) Å, c = 23.109(9) Å, α = 95.838(4), β = 94.579(4)º, γ = 91.407(4)º, V = 2265.3(15) Å3, Dc = 1.264 g/cm3, Z = 6, and a = 9.332(7) Å, b = 11.028(8) Å, c = 11.574(9) Å, α = 111.795(6)º, β = 94.162(8)º, γ = 92.177(7)º, V = 1100.3(14) Å3, Dc = 1.161 g/cm3, Z = 2, respectively. Both of them display a trans-configuration with respect to the C=N double bond. Their crystal packings are stabilized by weak C–H···O hydrogen bonds and van der Waals interactions.
Co-reporter:Dr. Yin Luo;Dr. Li-Rong Zhang;Dr. Yang Hu;Dr. Shuai Zhang;Dr. Jie Fu; Xiao-Ming Wang; Hai-Liang Zhu
ChemMedChem 2012 Volume 7( Issue 9) pp:1587-1593
Publication Date(Web):
DOI:10.1002/cmdc.201200225
Abstract
Forty-three oxime derivatives were synthesized by allowing O-benzylhydroxylamines to react with primary benzaldehydes or salicylaldehydes; these products were gauged as potential inhibitors of β-ketoacyl-(acyl-carrier-protein) synthase III (FabH). Among the 43 compounds, 38 are reported herein for the first time. These compounds were assayed for antimicrobial activities against Escherichia coli, Pseudomonas aeruginosa, Pseudomonas fluorescens, Bacillus subtilis, Staphylococcus aureus, and Enterococcus faecalis. Compounds with prominent antibacterial activities were tested for their E. coli FabH inhibitory activities. 3-((2,4-Dichlorobenzyloxyimino)methyl)benzaldehyde O-2,4-dichlorobenzyl oxime (44) showed the best antibacterial activity, with minimum inhibitory concentrations of 3.13–6.25 μg mL−1 against the tested bacterial strains, exhibiting the best E. coli FabH inhibitory activity, with an IC50 value of 1.7 mM. Docking simulations were performed to position compound 44 into the E. coli FabH active site in order to determine the most probable binding conformation.
Co-reporter:Qing-Shan Li, Cui-Yun Li, Xiang Lu, Hui Zhang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2012 50() pp: 288-295
Publication Date(Web):
DOI:10.1016/j.ejmech.2012.02.007
Co-reporter:S. P. Xu;J. F. Tang;J. T. Liu;B. F. Ruan
Russian Journal of Coordination Chemistry 2012 Volume 38( Issue 6) pp:426-429
Publication Date(Web):2012 June
DOI:10.1134/S1070328412050107
2-(p-Tolyliminomethyl)phenolcopper(II) (I) was synthesized and its structure was determined by X-ray diffraction. It crystallizes in the monoclinic system, space group P21/c, with the cell parameters a = 12.0266(15), b = 27.714(2), c = 11.0043(11) Å, β = 107.7510(10)°, V = 3493.1(6) Å3, and Z = 6. The Cu atom is surrounded by two O atoms and two N atoms from two 2-(p-tolyliminomethyl)phenol molecules to form a tetrahedral coordination environment. The complex is linked into a column by weak intermolecular interactions.
Co-reporter:Youyi Xia and Hailiang Zhu
Soft Matter 2011 vol. 7(Issue 19) pp:9388-9393
Publication Date(Web):25 Aug 2011
DOI:10.1039/C1SM05890H
A novel composite hydrogel embedded with polyaniline (PANI) nanofibers has been fabricated via rapid polymerization of aniline in a poly (acrylic acid) (PAA) hydrogel. The PAA/PANI nanofiber composite hydrogel displays a compress fracture stress as high as 1.7 MPa and a pH-responsive hysteresis due to the formation of polyaniline (PANI) nanofibers within the hydrogel. The PANI nanofiber content and the conductivity of the composite hydrogel can be facilely modulated by adjusting the concentration of the aniline monomers during the preparation. The as-prepared composite hydrogel would find many applications in various fields, and the present findings are also of crucial significance due to the combination of a common polymer hydrogel and a conductive polymer nanostructure with unique morphology.
Co-reporter:Peng-Cheng Lv, Tian-Tian Cai, Yong Qian, Juan Sun, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 1) pp:393-398
Publication Date(Web):January 2011
DOI:10.1016/j.ejmech.2010.10.034
A series of novel chrysin derivatives was firstly synthesized and evaluated on their immunosuppressive activity in the search for potential immunosuppressive agents. Among them, compounds 5c displayed the most potent immunosuppressive inhibitory activity with IC50 of 0.78 μM, which was comparable to that of cyclosporin A (IC50 = 0.06 μM). The preliminary mechanism of compound 5c inhibition effects was also detected by flow cytometry (FCM), and the compound exerted immunosuppressive activity via inducing the apoptosis of activated lymph node cells in a dose dependent manner. Furthermore, the estimated LD50 (in mg/kg) in vivo of compound 5c is 738.2, which indicated that compound 5c was low toxic.Compound 5c exhibited lower cytotoxicity and higher inhibition activity on CD3/CD28 co-stimulated T cell proliferation than other compounds.Research highlights► A series of novel chrysin derivatives were firstly synthesized and evaluated for their immunosuppressive activity on CD3/CD28 co-stimulated T cell proliferation. ► Compound 5c exhibited lower cytotoxicity and higher inhibition activity on CD3/CD28 co-stimulated T cell proliferation than other compounds. ► The preliminary mechanism study indicated that compound 5c exerted immunosuppressive activity via inducing the apoptosis of activated lymph node cells in a dose dependent manner.
Co-reporter:Zhu-Ping Xiao, Tao-Wu Ma, Mei-Lin Liao, Yu-Ting Feng, Xiao-Chun Peng, Jia-Liang Li, Zhi-Ping Li, Ying Wu, Qun Luo, Yang Deng, Xiao Liang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:4904-4914
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.07.047
Thirty-five 3-aryl-4-arylaminofuran-2(5H)-one derivatives were designed, prepared and tested for their inhibitory activity against tyrosyl-tRNA synthetase. Out of these compounds, 3-(3-bromophenyl)-4-(3,5-dichlorophenylamino)furan-2(5H)-one (35) was the most active with IC50 of 0.09 ± 0.02 μM. The structure–activity relationship revealed that introduction of chlorine atoms at both meta positions of aniline moiety significantly increased the enzyme inhibitory activity. The results of antibacterial assay revealed that the tested compounds showed good activity against Gram-positive bacteria, with 35 being the most potent with MIC50 of 0.06 μg/mL against Staphylococcus aureus ATCC 25923. Molecular docking of 35 into S. aureus tyrosyl-tRNA synthetase active site was also performed. The inhibitor snugly fitting the active site may well explain its excellent inhibitory activity.3-(3-Bromophenyl)-4-(3,5-dichlorophenylamino)furan-2(5H)-one (35) is a potent tyrosyl-tRNA synthetase inhibitor with IC50 of 0.09 ± 0.02 μM, and showed excellent antibacterial activity against Staphyloccocus aureus with MIC50 value of 0.06 μg/mL.Highlights► Thirty-five furan-2(5H)-ones were determined as tyrosyl-tRNA synthetase inhibitors. ► The most potent inhibitor showed MIC50 18-fold lower than that of kanamycin. ► Docking model of the most potent inhibitor could well explain its excellent activity.
Co-reporter:Ban-Feng Ruan, Xiang Lu, Jian-Feng Tang, Yao Wei, Xiao-Liang Wang, Yan-Bin Zhang, Li-Sheng Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 8) pp:2688-2695
Publication Date(Web):15 April 2011
DOI:10.1016/j.bmc.2011.03.001
Twenty-three resveratrol derivatives possessing chalcone moiety were synthesized and characterized, and their biological activities were also evaluated as potential antiproliferation and tubulin polymerization inhibitors. Compound C19 exhibited the most potent activity in vitro, which inhibited the growth of HepG2, B16-F10, and A549 cell lines with IC50 values of 0.2, 0.1, and 1.4 μg/mL, respectively. Compound C19 also exhibited significant tubulin polymerization inhibitory activity (IC50 = 2.6 μg/mL). Docking simulation was performed to position compound C19 into the tubulin–colchicine binding site to determine the probable binding mode.Twenty-three resveratrol derivatives possessing chalcone moiety were synthesized and characterized, and their biological activities were also evaluated as potential antiproliferation and antitubulin polymerization inhibitors. Docking simulation was performed to position compound C19 into the colchicine binding site to determine the probable binding mode.
Co-reporter:Zhu-Ping Xiao, Hui Ouyang, Xu-Dong Wang, Peng-Cheng Lv, Ze-Jun Huang, She-Rong Yu, Tian-Fang Yi, Ye-Ling Yang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 13) pp:3884-3891
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmc.2011.05.042
A series of novel 4-alkoxy-3-arylfuran-2(5H)-ones as tyrosyl-tRNA synthetase inhibitors were synthesized. Of these compounds, 3-(4-hydroxyphenyl)-4-(2-morpholinoethoxy)furan-2(5H)-one (27) was the most potent. The binding model and structure–activity relationship indicate that replacement of morpholine-ring in the side chain of 27 with a substituent containing more hydrophilic groups would be more suitable for further modification. Antibacterial assay revealed that the synthetic compounds are effective against growth of Gram-positive organisms, and 27 is the most potent agent against Staphylococcus aureus ATCC 25923 with MIC50 value of 0.23 μg/mL.A series of novel 4-alkoxy-3-arylfuran-2(5H)-ones were synthesized and evaluated for their inhibitory activity against tyrosyl-tRNA synthetase from Staphylococcus aureus. 3-(4-Hydroxyphenyl)-4-(2-morpholinoethoxy)furan-2(5H)-one (27) is the most active with IC50 = 0.10 ± 0.03 μM and is the most potent antibacterial agent against S. aureus ATCC 25923 with MIC50 value of 0.23 μg/mL.
Co-reporter:Juan Sun, Ning Cao, Xiao-Min Zhang, Yu-Shun Yang, Yan-Bin Zhang, Xiao-Ming Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 16) pp:4895-4902
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmc.2011.06.061
A series of oxadiazole derivatives containing 1,4-benzodioxan (4a–4s) have been first synthesized for their potential immunosuppressive activity. Among the compounds, compound 4i showed the most potent biological activity against RAW264.7 cells (inhibition = 37.66 ± 2.34% for NO overproduction and IC50 = 0.05 μM for iNOS). Docking simulation was performed to position compound 4i into the iNOS structure active site to determine the probable binding model. RT-PCR experiment results demonstrated that some of these compounds possessed good immunosuppressive activity against iNOS, especially for compound 4i. Therefore, compound 4i with potent inhibitory activity may be a potential agent.A series of oxadiazole derivatives containing 1,4-benzodioxan (4a–4s) have been designed, synthesized, structurally determined, and their biological activities were also evaluated as potential immunosuppressive agents. All the synthesized compounds were first synthesized. Among the compounds, compound 4i showed the most potent biological activity against RAW264.7 cells (inhibition = 37.66 ± 2.34% for NO overproduction and IC50 = 0.05 μM for iNOS). Docking simulation was performed to position compound 4i into the iNOS structure active site to determine the probable binding model. RT-PCR experiment results demonstrated that some of these compounds possessed good immunosuppressive activity against iNOS, especially for compound 4i. Therefore, compound 4i with potent inhibitory activity may be a potential agent.
Co-reporter:Dong-Dong Li, Peng-Cheng Lv, Hui Zhang, Hong-Jia Zhang, Ya-Ping Hou, Kai Liu, Yong-Hao Ye, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 16) pp:5012-5022
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmc.2011.06.044
A novel type of cinnamic acid quinazoline amide derivatives (20–42), which designed the combination between quinazoline as the backbone and various substituted cinnamic acid as the side chain, have been synthesized and their biological activities were evaluated within cytotoxicity assay firstly and then potent EGFR inhibitory activity. Compound 42 demonstrated the most potent inhibitory activity (IC50 = 0.94 μM for EGFR), which could be optimized as a potential EGFR inhibitor in the further study. Docking simulation was performed to position compound 42 into the EGFR active site to determine the probable binding model. Analysis of the binding conformation of 42 in active site displayed compound 42 was stabilized by hydrogen bonding interactions with Lys822, which was different from other derivatives. In the further study, Compounds 43 and 44 had been synthesized and their biological activities were also evaluated, which were the same as that we expected. Compound 43 has demonstrated significant EGFR (IC50 = 0.12 μM) and tumor growth inhibitory activity as a potential anticancer agent.A novel type of cinnamic acid quinazoline amide derivatives (20–42), which designed the combination between quinazoline as the backbone and various substituted cinnamic acid as the side chain, have been synthesized and their biological activities were evaluated within cytotoxicity assay firstly and then potent EGFR inhibitory activity. Compound 42 demonstrated the most potent inhibitory activity (IC50 = 0.94 μM for EGFR), which could be optimized as a potential EGFR inhibitor in the further study. Docking simulation was performed to position compound 42 into the EGFR active site to determine the probable binding model. Analysis of the binding conformation of 42 in active site displayed compound 42 was stabilized by hydrogen bonding interactions with Lys822, which was different from other derivatives. In the further study, compounds 43 and 44 had been synthesized and their biological activities were also evaluated, which were the same as that we expected. Compound 43 has demonstrated significant EGFR (IC50 = 0.12 μM) and tumor growth inhibitory activity as a potential anticancer agent.
Co-reporter:Ya-Ping Hou, Juan Sun, Zhong-Hua Pang, Peng-Cheng Lv, Dong-Dong Li, Li Yan, Hong-Jia Zhang, Emily Xi Zheng, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 20) pp:5948-5954
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmc.2011.08.063
A series of 1,2,4-triazole derivatives containing 1,4-benzodioxan (5a–5q) have been designed, synthesized, structurally determined, and their biological activities were evaluated as potential MetAP2 inhibitors. All the synthesized compounds were first reported. Among the compounds, compound 5k showed the most potent biological activity against HEPG2 cancer cell line (IC50 = 0.81 μM for HEPG2 and IC50 = 0.93 μM for MetAP2), which was comparable to the positive control. Docking simulation by positioning compound 5k into the MetAP2 structure active site was performed to explore the possible binding model. The results of apoptosis and Western-blot assay demonstrated that compound 5k possessed good antitumor activity against HEPG2 cancer cell line. Therefore, compound 5k with potent inhibitory activity in tumor growth inhibition may be a potential antitumor agent against HEPG2 cancer cell.A series of 1,2,4-triazole derivatives containing 1,4-benzodioxan (5a–5q) have been designed, synthesized, structurally determined, and their biological activities were evaluated as potential MetAP2 inhibitors. All the synthesized compounds were first reported. Among the compounds, compound 5k showed the most potent biological activity against HEPG2 cancer cell line (IC50 = 0.81 μM for HEPG2 and IC50 = 0.93 μM for MetAP2), which was comparable to the positive control. Docking simulation by positioning compound 5k into the MetAP2 structure active site was performed to explore the possible binding model. The results of apoptosis and Western-blot assay demonstrated that compound 5k possessed good antitumor activity against HEPG2 cancer cell line. Therefore, compound 5k with potent inhibitory activity in tumor growth inhibition may be a potential antitumor agent against HEPG2 cancer cell.
Co-reporter:Xiao-Min Zhang, Min Qiu, Juan Sun, Yan-Bin Zhang, Yu-Shun Yang, Xiao-Liang Wang, Jian-Feng Tang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 21) pp:6518-6524
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmc.2011.08.013
In present study, a series of new 1,3,4-oxadiazole derivatives containing 1,4-benzodioxan moiety (6a–6s) as potential telomerase inhibitors were synthesized. The bioassay tests demonstrated that compounds 6k, 6l, 6m, 6n and 6s exhibited broad-spectrum antitumor activity with IC50 concentration range from 7.21 μM to 25.87 μM against the four cancer cell lines, HEPG2, HELA, SW1116 and BGC823. Moreover, all the title compounds were assayed for telomerase inhibition using the TRAP-PCR-ELISA assay. The results showed compound 6k possessed the most potent telomerase activity (IC50 = 1.27 ± 0.05 μM). Docking simulation was performed to position compound 6k into the active site of telomerase (3DU6) to determine the probable binding model.A series of new 1,3,4-oxadiazole derivatives containing 1,4-benzodioxan moiety (6a–6s) as potential telomerase inhibitors have been designed, synthesized, structurally determined, and their biological activities were also evaluated as potential anticancer agents. The bioassay tests demonstrated that compounds 6k, 6l, 6m, 6n and 6s exhibited broad-spectrum antitumor activity with IC50 concentration range from 7.21 μM to 25.87 μM against the four cancer cell lines HEPG2, HELA, SW1116 and BGC823. All the title compounds were assayed for telomerase inhibition using the TRAP-PCR-ELISA assay. The results showed compound 6k possessed the most potent telomerase activity (IC50 = 1.27 ± 0.05 μM). Docking simulation was performed to position compound 6k into the active site of telomerase (3DU6) to determine the probable binding model.
Co-reporter:Hong-Jia Zhang, Xuan Qin, Kai Liu, Di-Di Zhu, Xiao-Ming Wang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5708-5715
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.06.077
A series of novel Schiff base derivatives have been designed and synthesized, and their biological activities were also evaluated as potential inhibitors of FabH. These compounds were assayed for antibacterial activity against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. Compounds with potent antibacterial activities were tested for their E. coli FabH inhibitory activity. Compound 3v showed the most potent antibacterial activity with MIC of 1.56–6.25 μg/mL against the tested bacterial strains and exhibited the most potent E. coli FabH inhibitory activity with IC50 of 4.3 μM. Docking simulation was performed to position compound 3v into the E. coli FabH active site to determine the probable binding conformation.A series of novel Schiff base derivatives have been designed and synthesized, and their biological activities were also evaluated as potential inhibitors of FabH. These compounds were assayed for antibacterial activity against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis, and Staphylococcus aureus. Compounds with potent antibacterial activities were tested for their E. coli FabH inhibitory activity. Compound 3v showed the most potent antibacterial activity with MIC of 1.56–6.25 μg/mL against the tested bacterial strains and exhibited the most potent E. coli FabH inhibitory activity with IC50 of 4.3 μM. Docking simulation was performed to position compound 3v into the E. coli FabH active site to determine the probable binding conformation.
Co-reporter:Xiang Lu, Hui Zhang, Xi Li, Guo Chen, Qing-Shan Li, Yin Luo, Ban-Feng Ruan, Xian-Wei Chen, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 22) pp:6827-6832
Publication Date(Web):15 November 2011
DOI:10.1016/j.bmc.2011.09.034
A series of pyridine acyl sulfonamide derivatives (1–24) have been designed and synthesized and their biological activities were also evaluated as potential cyclooxygenase-2 (COX-2) inhibitors. Among all the compounds, compound 23 displayed the most potent COX-2 inhibitory activity with an IC50 of 0.8 μM. Antitumor and anti-inflammatory assays indicated that compound 23 owned high antiproliferative activity against B16-F10, HepG2 and MCF-7 cancer cell lines as well as COX-2-derived prostaglandin E2 (PGE2) inhibitory activity of murine macrophage RAW 264.7 cell line with IC50 values of 2.8, 1.2, 1.8 and 0.15 μM, respectively. Docking simulation was performed to position compound 23 into the COX-2 active site to determine the probable binding model.
Co-reporter:Zi-Lin Li, Qing-Shan Li, Hong-Jia Zhang, Yang Hu, Di-Di Zhu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 15) pp:4413-4420
Publication Date(Web):1 August 2011
DOI:10.1016/j.bmc.2011.06.049
FabH, β-ketoacyl-acyl carrier protein (ACP) synthase III, is a particularly attractive target, since it is central to the initiation of fatty acid biosynthesis and is highly conserved among Gram-positive and Gram-negative bacteria. A series of o-hydroxybenzylamines 1–16 and its corresponding new urea derivatives 17–32 were synthesized and fully characterized by spectroscopic methods and elemental analysis. This new urea derivatives class demonstrates strong antibacterial activity. Escherichia coli FabH inhibitory assay and docking simulation indicated that the compounds 1-(5-bromo-2-hydroxybenzyl)-1-(4-fluorophenyl)-3-phenylurea (18) and 1-(5-bromo-2-hydroxybenzyl)-1-(4-chlorophenyl)-3-phenylurea (20) were potent inhibitors of E. coli FabH.FabH, β-ketoacyl-acyl carrier protein (ACP) synthase III, is a particularly attractive target, since it is central to the initiation of fatty acid biosynthesis and is highly conserved among Gram-positive and Gram-negative bacteria. A series of o-hydroxybenzylamines 1–16 and its corresponding new urea derivatives 17–32 were synthesized and fully characterized by spectroscopic methods and elemental analysis. This new urea derivatives class demonstrates strong antibacterial activity. Escherichia coli FabH inhibitory assay and docking simulation indicated that the compounds 1-(5-bromo-2-hydroxybenzyl)-1-(4-fluorophenyl)-3-phenylurea (18) and 1-(5-bromo-2-hydroxybenzyl)-1-(4-chlorophenyl)-3-phenylurea (20) were potent inhibitors of E. coli FabH.
Co-reporter:Yin Luo, Yao Li, Ke-Ming Qiu, Xiang Lu, Jie Fu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 20) pp:6069-6076
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmc.2011.08.038
A series of novel metronidazole derivatives were recently reported as potent anticancer agents targeting EGFR and HER-2 by our group [Qian, Y.; Zhang, H. J.; Zhang, H.; Xu, C.; Zhao, J.; Zhu, H. L. Bioorg. Med. Chem.2010, 18, 4991]. Based on the previous results, we designed and synthesized a new series of metronidazole acid acyl sulfonamide derivatives and a new series of phenylacetyl benzenesulfonamide derivatives and their anticancer activities were evaluated as potential EGFR and HER-2 kinase inhibitors. Among all the compounds, compound 12 displayed the most potent inhibitory activity EGFR and HER-2 (IC50 = 0.39 μM for EGFR and IC50 = 1.53 μM for HER-2) and it also showed the most potent growth inhibitory activity against A549 and B16-F10 cancer cell line in vitro, with an IC50 value of 1.26 μg/mL for A549 and 0.35 μg/mL for B16-F10. Docking simulation was further performed to position compound 12 into the EGFR active site to determine the probable binding model.Two series of benzenesulfonamide derivatives have been designed and synthesized, and their biological activities were also evaluated for EGFR inhibitory activity. Compound 12 possessed the most potent enzyme inhibition activities (IC50 = 0.39 μM for EGFR and IC50 = 1.53 μM for HER-2) and anticancer activities (IC50 = 1.26 μg/mL for A549 and IC50 = 0.35 μg/mL for B16-F10). Docking simulation was performed to explore the binding model of compound 12 with EGFR.
Co-reporter:Hong-Jia Zhang, Di-Di Zhu, Zi-Lin Li, Juan Sun, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 15) pp:4513-4519
Publication Date(Web):1 August 2011
DOI:10.1016/j.bmc.2011.06.021
A series of novel cinnamic acid secnidazole ester derivatives have been designed and synthesized, and their biological activities were also evaluated as potential inhibitors of FabH. These compounds were assayed for antibacterial activity against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. Compounds with potent antibacterial activities were tested for their E. coli FabH inhibitory activity. Compound 3n showed the most potent antibacterial activity with MIC of 1.56–6.25 μg/mL against the tested bacterial strains and exhibited the most potent E. coli FabH inhibitory activity with IC50 of 2.5 μM. Docking simulation was performed to position compound 3n into the E. coli FabH active site to determine the probable binding conformation.A series of novel cinnamic acid secnidazole ester derivatives have been designed and synthesized, and their biological activities were also evaluated as potential inhibitors of FabH. These compounds were assayed for antibacterial activity against Escherichia coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus. Compounds with potent antibacterial activities were tested for their E. coli FabH inhibitory activity. Compound 3n showed the most potent antibacterial activity with MIC of 1.56–6.25 μg/mL against the tested bacterial strains and exhibited the most potent E. coli FabH inhibitory activity with IC50 of 2.5 μM. Docking simulation was performed to position compound 3n into the E. coli FabH active site to determine the probable binding conformation.
Co-reporter:Zhu-Ping Xiao, Xing-Bing He, Zhi-Yun Peng, Tao-Ju Xiong, Juan Peng, Li-Hua Chen, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 5) pp:1571-1579
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmc.2011.01.051
Thirty-one 3-aryl-4-alkylaminofuran-2(5H)-ones were designed, prepared and tested for their antibacterial activity. Some of them showed significant antibacterial activity against Gram-positive organisms, especially against Staphylococcus aureus ATCC 25923, but all were inactive against Gram-negative organisms. Out of these compounds, 3-(4-bromophenyl)-4-(2-(4-nitrophenyl)hydrazinyl)furan-2(5H)-one (4a11) showed the most potent antibacterial activity against S. aureus ATCC 25923 with MIC50 of 0.42 μg/mL. The enzyme assay revealed that the possible antibacterial mechanism of the synthetic compounds might be due to their inhibitory activity against tyrosyl-tRNA synthetase. Molecular dockings of 4a11 into S. aureus tyrosyl-tRNA synthetase active site were also performed. This inhibitor snugly fitting the active site might well explain its excellent inhibitory activity. Meanwhile, this modeling disclosed that a more suitable optimization strategy might be to modify the benzene ring at 3-position of furanone with hydrophilic groups.Thirty-one 3-aryl-4-alkylaminofuran-2(5H)-one derivatives were prepared and tested for their antimicrobial activities. 3-(4-Bromophenyl)-4-(2-(4-nitrophenyl)hydrazinyl)furan-2(5H)-one (4a11) showed the most potent antibacterial activity with MIC50 of 0.42 μg/mL against Staphylococcus aureus ATCC 25923 and was the most potent inhibitor against tyrosyl-tRNA synthetase from S. aureus with IC50 of 0.12 ± 0.04 μM.
Co-reporter:Lei Shi, Zi-Lin Li, Ying Yang, Zhen-Wei Zhu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 1) pp:121-124
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmcl.2010.11.062
A series of N-phenylnicotinamides (1–40) were designed and evaluated in vitro for their COX inhibitory activities. Most of the synthesized compounds were proved to be potent and selective inhibitors of COX-1. Compound 28 showed the most potent COX-1 inhibitory activity (COX-1 IC50 = 0.68 ± 0.07 μM) and good selectivity (COX-2 IC50 >100 μM). This compound may be useful as a lead compound for superior COX-1 inhibitors. On the basis of the biological results, structure–activity relationships for the COX-1-inhibitory activities of the synthesized N-phenylnicotinamides were discussed concisely.A series of N-phenylnicotinamides (1–40) were designed and evaluated in vitro for their COX inhibitory activities. Most of the synthesized compounds were proved to be potent and selective inhibitors of COX-1. Compound 28 showed the most potent COX-1 inhibitory activity (COX-1 IC50 = 0.68 ± 0.07 μM) and good selectivity (COX-2 IC50 >100 μM). This compound may be useful as a lead compound for superior COX-1 inhibitors. On the basis of the biological results, structure–activity relationships for the COX-1-inhibitory activities of the synthesized N-phenylnicotinamides were discussed concisely.
Co-reporter:Juan Sun, Yu-Shun Yang, Wei Li, Yan-Bin Zhang, Xiao-Liang Wang, Jian-Feng Tang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 20) pp:6116-6121
Publication Date(Web):15 October 2011
DOI:10.1016/j.bmcl.2011.08.039
A series of 1,3,4-thiadiazole derivatives containing 1,4-benzodioxan (2a–2s) have been synthesized to screen for FAK inhibitory activity. Compound 2p showed the most potent biological activity against HEPG2 cancer cell line (EC50 = 10.28 μg/mL for HEPG2 and EC50 = 10.79 μM for FAK), which was comparable to the positive control. Docking simulation was performed to position compound 2p into the FAK structure active site to determine the probable binding model. The results of antiproliferative and Western-blot assay demonstrated that compound 2p possessed good antiproliferative activity against HEPG2 cancer cell line. Therefore, compound 2p with potent FAK inhibitory activity may be a potential anticancer agent against HEPG2 cancer cell.Compound 2p displayed the most potent FAK inhibitory activity with EC50 of 10.79 μM, which was comparable to the positive control staurosporine. Molecular docking study indicated that compound 2p was nicely bound to the FAK with two hydrogen bonds, and Gly/Ile were very important residues in FAK.Compound 2p also showed significant antiproliferative activity against HEPG2 with EC50 of 10.28 μM. The results of apoptosis and Western-blot assay demonstrated that compound 2p would be a potential anticancer agent against HEPG2 cancer cell.
Co-reporter:Peng-Cheng Lv, Dong-Dong Li, Qing-Shan Li, Xiang Lu, Zhu-Ping Xiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 18) pp:5374-5377
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmcl.2011.07.010
Fourty-two thiazolyl-pyrazoline derivatives were synthesized to screen for their EGFR kinase inhibitory activity. Compound 4-(4-chlorophenyl)-2-(3-(3,4-dimethylphenyl)-5-p-tolyl-4,5-dihydro-1H-pyrazol-1-yl)thiazole (11) displayed the most potent EGFR TK inhibitory activity with IC50 of 0.06 μM, which was comparable to the positive control. Molecular docking results indicated that compound 11 was nicely bound to the EGFR kinase. Compound 11 also showed significant antiproliferative activity against MCF-7 with IC50 of 0.07 μM, which would be a potential anticancer agent.Compound 11 displayed the most potent EGFR TK inhibitory activity with IC50 of 0.06 μM, which was comparable to the positive control. Molecular docking results indicated that compound 11 was nicely bound to the EGFR kinase with three hydrogen bonds. Compound 11 also showed significant antiproliferative activity against MCF-7 with IC50 of 0.07 μM, which would be a potential anticancer agent.
Co-reporter:Xin-Hua Liu, Ban-Feng Ruan, Jing-Xin Liu, Bao-An Song, Ling-Hong Jing, Jun Li, Yang Yang, Hai-Liang Zhu, Xing-Bao Qi
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 10) pp:2916-2920
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmcl.2011.03.066
A series of novel N-phenylacetyl (sulfonyl) 4,5-dihydropyrazole derivatives as potential telomerase inhibitors were synthesized. The bioassay tests show that compound 4a exhibited high activity against human gastric cancer cell SGC-7901, liver cancer Hep-G2 and human prostate PC-3 cell lines with IC50 values of 21.23 ± 0.99, 29.43 ± 0.32 and 30.89 ± 1.07 μM, respectively. All title compounds were assayed for telomerase inhibition by a modified TRAP assay, the results show that compound 4a can inhibit telomerase with IC50 value of 4.0 ± 0.32 μM. Docking simulation was performed to position compound 4a into the telomerase (3DU6) active site to determine the probable binding model.Novel 5-(2-hydroxyphenyl)-3-methyl-4,5-dihydropyrazol-1-yl-anone as potential telomerase inhibitors were synthesized. The bioassay tests show that compound 4a exhibited high activity against human gastric cancer cell SGC-7901, Hep-G2 and PC-3 cell lines. All title compounds were assayed for telomerase inhibition, the results show that compound 4a can inhibit telomerase. Docking simulation was performed to position compound 4a into the telomerase (3DU6) active site to determine the probable binding model.
Co-reporter:Suo-Ping Xu;Xiao-Liang Wang;Jian-Feng Tang
Journal of Chemical Crystallography 2011 Volume 41( Issue 1) pp:82-84
Publication Date(Web):2011 January
DOI:10.1007/s10870-010-9920-6
One new complex, 2-hydroxy-3-iodo-benzaldehyde-copper (II) has been designed and microwave synthesized. The structure was determined by UV, IR and single X-ray crystallography study. The title complex C14H8CuI2O4 crystallizes in the orthorhombic space group Pna21 with the cell parameters a = 12.7897(12) Å, b = 6.1132(8) Å, c = 19.5114(18) Å, V = 1525.5(3) Å3 and Z = 4. The central copper (II) is four-coordinated by four oxygen atoms from two 3-iodosalicylaldehyde. The complex is linked into rhombic crystals by weak intermolecular interactions.
Co-reporter:Zhu-Ping Xiao;Zhi-Yun Peng;Zhu-Xiang Liu
Journal of Chemical Crystallography 2011 Volume 41( Issue 5) pp:649-653
Publication Date(Web):2011 May
DOI:10.1007/s10870-010-9946-9
The title compound, C17H13NO6, was synthesized and structurally characterized by elemental analysis, MS, 1H NMR and single crystal X-ray diffraction. It crystallizes in monoclinic system space group C 2/c with a = 27.981(6) Å, b = 12.996(3) Å, c = 8.0900(16) Å, β = 91.06(3)°, V = 2941.4(10) Å3, Z = 8, R1 = 0.0675, wR2 = 0.1626, and T = 298(2) K. The X-ray structure determination revealed that the center furanone ring is nearly coplanar with p-methoxybenzene ring and forms a dihedral angle of 87.2(1)° with the nitrobenzene ring. O–H···O Intermolecular hydrogen bonds link pairs of molecules into centrosymmetric dimers, making a graph set motif of R22(10). The dimers are further assembled into a chain of edge-fused R44(34) rings running along the [001] direction. The final three-dimensional supramolecular architecture is stabilized by weak π–π interactions.
Co-reporter:Jie Fu;Sheng Sheng;Teng Wen;Zhi-Ming Zhang;Qing Wang;Qiu-Xiang Hu
Ecotoxicology 2011 Volume 20( Issue 5) pp:940-950
Publication Date(Web):2011 July
DOI:10.1007/s10646-011-0622-4
The Jialu River, an important branch of the Huaihe River in China, was seriously polluted because of rapid economic growth and urbanization. In order to evaluate the potential for serious environmental consequences as a result of anthropogenic contamination, the distribution of polycyclic aromatic hydrocarbons (PAHs) has been investigated in surface sediment samples collected in connection with field surveys of 19 sites along the Jialu River. The total concentration of the 16 USEPA priority PAHs ranged from 466.0 to 2605.6 ng/g dry weight with a mean concentration of 1363.2 ng/g. Sediment samples with the highest PAH concentrations were from the upper reaches of the river, where Zhengzhou City is located; the PAH levels in the middle and lower reaches were relatively low. According to the observed molecular indices, PAHs originated largely from the high-temperature pyrolytic process. According to the numerical effect-based sediment quality guidelines (SQGs) of the United States, the levels of PAHs in the Jialu River should not exert adverse biological effects. The total benzo[a]pyrene toxicity equivalent (TEQ) values calculated for samples varied from 50.4 to 312.8 ng/g dry weight with an average of 167.4 ng/g. The relationships between PAHs and environmental factors, including chemical properties of sediments, water quality, aquatic organisms, hydrological conditions, and anthropogenic activities, are also discussed. PAHs exerted a potential negative impact on the benthos. Settlement percentage, population density and industrial GDP per capita had a significant influence on the distribution of PAHs.
Co-reporter:Yin Luo, Ke-Ming Qiu, Xiang Lu, Kai Liu, Jie Fu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2011 19(16) pp: 4730-4738
Publication Date(Web):
DOI:10.1016/j.bmc.2011.06.088
Co-reporter:Jie Fu, Yan-Hua Ding, Luo Li, Sheng Sheng, Teng Wen, Lu-Ji Yu, Wu Chen, Shu-Qing An and Hai-Liang Zhu
Environmental Science: Nano 2011 vol. 13(Issue 3) pp:597-604
Publication Date(Web):12 Jan 2011
DOI:10.1039/C0EM00604A
The distribution, source, ecological risk and ecotoxicity of polycyclic aromatic hydrocarbons (PAHs) of sediments from 7 sampling sites, named as Xinyang (XY), Huainan (HN), Bengbu (BB), Xuyi (XuY), Fuyang (FY), Mengcheng (MC) and Zhengzhou (ZZ), in the Huaihe River basin, China, have been investigated. The total concentrations of 16 USEPA priority PAHs ranged from 62.9 to 2232.4 ng g−1 dry weight (d.w.) with a mean concentration of 1056.8 ng g−1d.w. Through the assessment of ecological risk, we found that the levels of PAHs in the Huaihe River should not exert adverse biological effects. The total benzo[a]pyrene toxicity equivalent (TEQ) values calculated for samples varied from 0.01 to 194.1 ng g−1d.w., with an average of 65.9 ng g−1. The toxicity data were accordant with the chemical analysis results in this study. HN, BB and ZZ showed the greatest pollution extent both in the chemical analysis and the study of ecotoxicological effects.
Co-reporter:Jie Fu, Kui Cheng, Zhi-ming Zhang, Rui-qin Fang, Hai-liang Zhu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 6) pp:2638-2643
Publication Date(Web):June 2010
DOI:10.1016/j.ejmech.2010.01.066
A series of caffeic acid amides 1–23 were synthesized and nine of which (13–17, 19–21 and 23) were reported for the first time. The chemical structures of these compounds were confirmed by means of 1H NMR, ESI MS and elemental analyses. Compound 15 was determined by single-crystal X-ray diffraction analysis. All of the compounds were assayed for antibacterial (Bacillus subtilis, Escherichia coli, Pseudomonas fluorescens and Staphylococcus aureus) and antifungal (Aspergillus niger, Candida albicans and Trichophyton rubrum) activities by MTT method. Compounds 10–12, 15, 18 and 21 showed considerable antibacterial activities against B. subtilis with MICs of 7.95, 6.25, 3.89, 1.18, 3.12 and 15.5 μg/mL, respectively. Structure–activity relationship analysis disclosed that caffeic acid anilides with electron-donating groups at p-position of benzene ring have better inhibitory activities.A series of caffeic acid amides were synthesized and (E)-3-(3,4-Dihydroxyphenyle)-N-p-tolylacrylamide (15) showed considerable antibacterial activity against Bacillus subtilis with MIC of 1.18 ug/mL.
Co-reporter:Yong-Miao Shen, Peng-Cheng Lv, Wu Chen, Peng-Gang Liu, Ming-Zhu Zhang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 7) pp:3184-3190
Publication Date(Web):July 2010
DOI:10.1016/j.ejmech.2010.02.056
Indolizine and annulated indolizine derivatives incorporating a cyclopropylcarbonyl group were synthesized in a one pot procedure by the tanden reactions of [3 + 2] cycloaddition of the corresponding N-ylide with electron deficient alkene. Seventeen indolizine derivatives were reported for the first time. All the compounds were examined for their antiproliferative activity against the human hepatocellular liver carcinoma (Hep-G2) cell line by MTT method. Among the compounds tested, 5a, 5d, 5g and 5j showed the most favorable activities with IC50 values of 0.39, 0.48, 0.29 and 0.20 μg/mL. Especially, compound 5j displayed potent antiproliferative activities with IC50 value of 0.20 μg/mL, and showed significant EGFR kinase inhibitory activity with IC50 value of 0.085 μM. Docking simulations of 5j were carried out to illustrate the binding mode of the molecular into the EGFR active site.Indolizine and annulated indolizine derivatives incorporating a cyclopropylcarbonyl group were synthesized by the tanden reactions of [3 + 2] cycloaddition of the corresponding N-ylide with electron deficient alkene.
Co-reporter:Qing-Zhong Zheng, Kui Cheng, Xiao-Min Zhang, Kai Liu, Qing-Cai Jiao, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 7) pp:3207-3212
Publication Date(Web):July 2010
DOI:10.1016/j.ejmech.2010.03.027
A series of N-alkyl substituted urea derivatives were synthesized and evaluated for their in vitro antibacterial and antifungal activities. The N-alkyl substituted urea derivatives bearing morpholine moiety (3a–3k) showed better activities than those bearing diethylamine moiety (2a–2f). Compounds having fluoro substituent at ortho (3c) and para (3b) positions of the phenyl ring exhibited potent antimicrobial activities against Gram-positive and Gram-negative bacteria as well as fungi.
Co-reporter:Zhu-Ping Xiao, Tao-Wu Ma, Wei-Chang Fu, Xiao-Chun Peng, Ai-Hua Zhang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 11) pp:5064-5070
Publication Date(Web):November 2010
DOI:10.1016/j.ejmech.2010.08.015
Some pyrogallol and catechol derivatives were synthesized, and their urease inhibitory activity was evaluated by using acetohydroxamic acid (AHA), a well known Helicobacter pylori urease inhibitor, as positive control. The assay results indicate that many compounds have showed potential inhibitory activity against H. pylori urease. 4-(4-Hydroxyphenethyl)phen-1,2-diol (2a) was found to be the most potent urease inhibitor with IC50s of 1.5 ± 0.2 μM for extracted fraction and 4.2 ± 0.3 μM for intact cell, at least 10 times and 20 times lower than those of AHA (IC50 of 17.2 ± 0.9 μM, 100.6 ± 13 μM), respectively. This finding indicate that 2a would be a potential urease inhibitor deserves further research. Molecular dockings of 2a into H. pylori urease active site were performed for understanding the good activity observed.A series of polyphenols were synthesized and evaluated for inhibitory activity against Helicobacter pylori urease. 4-(4-Hydroxyphenethyl)phen-1,2-diol (2a) was the most potent inhibitors with IC50 = 1.5±μM.
Co-reporter:Lei Shi, Ying Yang, Zi-Lin Li, Zhen-Wei Zhu, Chang-Hong Liu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 4) pp:1659-1664
Publication Date(Web):15 February 2010
DOI:10.1016/j.bmc.2009.12.065
A series of N-(2-morpholinoethyl)nicotinamide (1–13) and N-(3-morpholinopropyl)nicotinamide derivatives (14–26) have been designed, synthesized and evaluated in vitro for their monoamine oxidase (MAO) A and B inhibitory activity and selectivity. Most of these synthesized compounds proved to be potent, and selective inhibitors of MAO-A rather than of MAO-B. 5-Chloro-6-hydroxy-N-(2-morpholinoethyl)nicotinamide (13) displayed the highest MAO-A inhibitory potency (IC50 = 0.045 μM) and a good selectivity. 2-Bromo-N-(2-morpholinoethyl)nicotinamide (3) was the most potent MAO-B inhibitor with the IC50 value of 0.32 μM, but it was not selective. Molecular dockings of compound 13 were performed in order to give structural insights regarding the MAO-A selectivity.A series of nicotinamide derivatives (1–26) have been designed, synthesized and evaluated in vitro for their monoamine oxidase inhibitory activity and selectivity. Most of these synthesized compounds proved to be potent, and selective inhibitors of MAO-A rather than of MAO-B. 5-Chloro-6-hydroxy-N-(2-morpholinoethyl)nicotinamide (13) displayed the highest MAO-A inhibitory potency (IC50 = 0.045 μM) and a good selectivity. 2-Bromo-N-(2-morpholinoethyl)nicotinamide (3) was the most potent MAO-B inhibitor with the IC50 value of 0.32 μM, but it was not selective. Molecular dockings of compound 13 were performed in order to give structural insights regarding the MAO-A selectivity.
Co-reporter:Jie Fu, Ying Yang, Xue-Wei Zhang, Wen-Jun Mao, Zhi-Ming Zhang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 24) pp:8457-8462
Publication Date(Web):15 December 2010
DOI:10.1016/j.bmc.2010.10.049
Twenty-one benzotriazoles (3–16 and 18–24) were synthesized and half of them (5, 8–16, 20, and 21) were reported for the first time. Their antiproliferative activities against three human cancer cells were assayed. It revealed that 1H-benzo[d][1,2,3]triazol-1-yl 3,4,5-trimethoxybenzoate (9) showed considerable activity against three human cancer cell lines with the half maximal inhibitory concentration (IC50) values of 1.2–2.4 nM, which were close to the value of the positive control, doxorubicin. Further investigation indicated compound 9 was a potential histone deacetylase inhibitor (IC50 = 9.4 μM) and its binding mode was simulated using docking method.
Co-reporter:Yong Qian, Hong-Jia Zhang, Peng-Cheng Lv, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 23) pp:8218-8225
Publication Date(Web):1 December 2010
DOI:10.1016/j.bmc.2010.10.008
A series of novel chalcone guanidine derivatives (4a–4q) have been designed and synthesized, and their biological activity were also evaluated as potential antiproliferative and antitubulin polymerization inhibitors. Compound 4q showed the most potent biological activity (IC50 = 0.09 ± 0.01 μM for MCF-7 and IC50 = 8.4 ± 0.6 μM for tubulin), which is comparable to the positive controls. Docking simulation was performed to position compound 4q into the colchicine binding site to determine the probable binding model, which suggested probable inhibition mechanism.Compound 4q showed the most potent biological activity (IC50 = 0.09 ± 0.01 μM for MCF-7 and IC50 = 8.4 ± 0.6 μM for tubulin), which is comparable to the positive controls. Docking simulation was performed to position compound 4q into the colchicine binding site to determine the probable binding model, which suggested probable inhibition mechanism. Antitubulin polymerization, antiproliferative assay and docking simulation results showed the compound 4q was a potential anticancer agent.
Co-reporter:Peng-Cheng Lv, Kai-Rui Wang, Qing-Shan Li, Jin Chen, Juan Sun, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 3) pp:1117-1123
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmc.2009.12.048
A series of long-chain derivatives of chrysin (compounds 3–22) were synthesized to evaluate for their antiproliferative activities against the human liver cancer cell line HT-29 and EGFR inhibitory activity. Among the compounds tested, compounds hexadecyl 2-(5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yloxy)acetate (10) and N-hexadecyl 2-(5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yloxy)acetamide (20) displayed potent EGFR inhibitory activity with IC50 values of 0.048 μM and 0.035 μM), comparable to the positive control erlotinib. Docking simulation of compounds 10 and 20 was carried out to illustrate the binding mode of the molecular into the EGFR active site, and the result suggested that compound 10 and 20 can bind the EGFR kinase well. Thus, compounds 10 and 20 with potent EGFR inhibitory activity would be potential anticancer agents.A series of long-chain derivatives of chrysin (compounds 3–22) were synthesized to evaluate for their antiproliferative activities against the human liver cancer cell line HT-29 and EGFR inhibitory activity. Compounds 10 and 20 displayed potent EGFR inhibitory activity with IC50 values of 0.048 μM and 0.035 μM, comparable to the positive control erlotinib.
Co-reporter:Yong Qian, Gao-Yuan Ma, Ying Yang, Kui Cheng, Qing-Zhong Zheng, Wen-Jun Mao, Lei Shi, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 12) pp:4310-4316
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmc.2010.04.091
A series of novel dithiocarbamate compounds with the chalcone scaffold have been designed and synthesized, and their biological activities were also evaluated as potential antiproliferation and antitubulin polymerization inhibitors. Compound 2n showed the most potent biological activity in vitro, which inhibited the growth of MCF-7 cells with IC50 of 0.04 ± 0.01 μM and the polymerization of tubulin with IC50 of 6.8 ± 0.6 μM. To understand the tubulin–inhibitor interaction and the selectivity of the most active compound towards tubulin, molecular modeling studies were performed to dock compound 2n into the colchicine binding site, which suggested probable inhibition mechanism.Compound 2n showed the most potent biological activity in vitro, which inhibited the growth of MCF-7 human breast carcinoma cells with IC50 of 0.04 ± 0.01 μM and the polymerization of tubulin with IC50 of 6.8 ± 0.6 μM. To understand the tubulin–inhibitor interaction and the selectivity of the most active compound towards tubulin, molecular modeling studies were performed to dock compound 2n into colchicine binding site and this study provided probable inhibition mechanism. The result indicated that compound 2n was a potent inhibitor of tubulin polymerization.
Co-reporter:Peng-Cheng Lv, Huan-Qiu Li, Juan Sun, Yang Zhou, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 13) pp:4606-4614
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmc.2010.05.034
Two series of pyrazole derivatives designing for potential EGFR kinase inhibitors have been discovered. Some of them exhibited significant EGFR inhibitory activity. Compound 3-(3,4-dimethylphenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (C5) displayed the most potent EGFR inhibitory activity with IC50 of 0.07 μM, which was comparable to the positive control erlotinib. Docking simulation was performed to position compound C5 into the EGFR active site to determine the probable binding model. Antiproliferative assay results indicating that some of the pyrazole derivatives own high antiproliferative activity against MCF-7. Compound C5 showed significant antiproliferative activity against MCF-7 with IC50 of 0.08 μM. Therefore, compound C5 with potent inhibitory activity in tumor growth inhibition would be a potential anticancer agent.Compound C5 exhibited the most potent EGFR inhibitory activity with IC50 of 0.07 μM, which was comparable to the positive control erlotinib. Docking simulation was performed to position compound C5 into the EGFR active site to determine the probable binding model. Besides, compound C5 showed significant antiproliferative activity against MCF-7 with IC50 of 0.08 μM, which would be a potential anticancer agent.
Co-reporter:Qing-Zhong Zheng, Fei Zhang, Kui Cheng, Ying Yang, Yu Chen, Yong Qian, Hong-Juan Zhang, Huan-Qiu Li, Chang-Fang Zhou, Shu-Qing An, Qing-Cai Jiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 2) pp:880-886
Publication Date(Web):15 January 2010
DOI:10.1016/j.bmc.2009.11.037
A series of amide-coupled benzoic nitrogen mustard derivatives as potential EGFR and HER-2 kinase inhibitors were synthesized and reported for the first time. Some of them exhibited significant EGFR and HER-2 inhibitory activity. Of all the studied compounds, compounds 5b and 5t exhibited the most potent inhibitory activity, which was comparable to the positive control erlotinib. Docking simulation was performed to position compounds 5b and 5t into the EGFR active site to determine the probable binding model. Antiproliferative assay results indicated that some of the benzoic nitrogen mustard derivatives possessed high antiproliferative activity against MCF-7. In particular, compounds 5b and 5t with potent inhibitory activity in tumor growth inhibition may function as potential antitumor agents.Of all the studied compounds, compounds 5b and 5t exhibited the most potent EGFR and HER-2 inhibitory activity. Docking simulation was performed to position compounds 5b and 5t into the EGFR active site to determine the probable binding model. Antiproliferative assay results indicated that some of the benzoic nitrogen mustard derivatives possessed high antiproliferative activity against MCF-7. In particular, compounds 5b and 5t with potent inhibitory activity in tumor growth inhibition may function as potential antitumor agents.
Co-reporter:Huan-Qiu Li, Tao Yan, Ying Yang, Lei Shi, Chang-Fang Zhou, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 1) pp:305-313
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmc.2009.10.054
Two series of novel N-benzyl-N-(X-2-hydroxybenzyl)-N′-phenylureas and thioureas (1a–18a; 1b–18b) as potential EGFR and HER-2 kinase inhibitors have been discovered. These compounds displayed good EGFR and HER-2 inhibitory activity and the SARs are also been studied. Especially compound 7b demonstrated significant EGFR and HER-2 inhibitory activity (IC50 = 0.08 μM for EGFR and IC50 = 0.35 μM for HER-2). Docking simulation was performed to position compound 7b into the EGFR active site to determine the probable binding conformation and antiproliferative assay results indicating that these series of urea and thioureas own high antiproliferative activity against MCF-7. Above all, thiourea 7b would be a potential anticancer agent deserves further research.
Co-reporter:Peng-Cheng Lv, Chang-Fang Zhou, Jin Chen, Peng-Gang Liu, Kai-Rui Wang, Wen-Jun Mao, Huan-Qiu Li, Ying Yang, Jing Xiong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 1) pp:314-319
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmc.2009.10.051
Two series of thiazolidinone derivatives designing for potential EGFR and HER-2 kinase inhibitors have been discovered. Some of them exhibited significant EGFR and HER-2 inhibitory activity. Compound 2-(2-(5-bromo-2-hydroxybenzylidene)hydrazinyl)thiazol-4(5H)-one (12) displayed the most potent inhibitory activity (IC50 = 0.09 μM for EGFR and IC50 = 0.42 μM for HER-2), comparable to the positive control erlotinib. Docking simulation was performed to position compound 12 into the EGFR active site to determine the probable binding model. Antiproliferative assay results indicating that some of the thiazolidinone derivatives own high antiproliferative activity against MCF-7. Compound 12 with potent inhibitory activity in tumor growth inhibition would be a potential anticancer agent.Compound 12 displayed the most potent inhibitory activity (IC50 = 0.09 μM for EGFR and IC50 = 0.42 μM for HER-2), comparable to the positive control erlotinib. Docking simulation was performed to position compound 12 into the EGFR active site to determine the probable binding model and antiproliferative assay results indicating that some of the thiazolidinone derivatives own high antiproliferative activity against MCF-7. Compound 12 with potent inhibitory activity in tumor growth inhibition would be a potential anticancer agent.
Co-reporter:Qing-Zhong Zheng, Xiao-Min Zhang, Ying Xu, Kui Cheng, Qing-Cai Jiao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 22) pp:7836-7841
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmc.2010.09.051
A series of new 2-chloropyridine derivatives possessing 1,3,4-oxadiazole moiety were synthesized. Antiproliferative assay results indicated that compounds 6o and 6u exhibited the most potent activity against gastric cancer cell SGC-7901, which was more potent than the positive control. Especially, compound 6o exhibited significant telomerase inhibitory activity (IC50 = 2.3 ± 0.07 μM), which was comparable to the positive control ethidium bromide. Docking simulation was performed to position compound 6o into the active site of telomerase (3DU6) to determine the probable binding model.Antiproliferative assay results indicated that compounds 6o and 6u exhibited the most potent activity against gastric cancer cell SGC-7901, which was more potent than the positive control. Especially, compound 6o exhibited significant telomerase inhibitory activity (IC50 = 2.3 ± 0.07 μM), which was comparable to the positive control ethidium bromide. Docking simulation was performed to position compound 6o into the active site of telomerase (3DU6) to determine the probable binding model.
Co-reporter:Yong Qian, Hong-Jia Zhang, Hao Zhang, Chen Xu, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 14) pp:4991-4996
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmc.2010.06.003
A series of novel cinnamic acid metronidazole ester derivatives have been designed and synthesized, and their biological activities were also evaluated as potential EGFR and HER-2 kinase inhibitors. Compound 3h showed the most potent biological activity (IC50 = 0.62 μM for EGFR and IC50 = 2.15 μM for HER-2). Docking simulation was performed to position compound 3h into the EGFR active site to determine the probable binding model. Antiproliferative assay results demonstrated that some of these compounds possessed good antiproliferative activity against MCF-7. Compound 3h with potent inhibitory activity in tumor growth inhibition may be a potential anticancer agent.Compound 3h showed the most potent biological activity (IC50 = 0.62 μM for EGFR and IC50 = 2.15 μM for HER-2). Docking simulation was performed to position compound 3h into the EGFR active site to determine the probable binding model. Antiproliferative assay results demonstrated that some of these compounds possessed good antiproliferative activity against MCF-7. Compound 3h with potent inhibitory activity in tumor growth inhibition may be a potential anticancer agent.
Co-reporter:Kui Cheng, Qing-Zhong Zheng, Jin Hou, Yang Zhou, Chang-Hong Liu, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 7) pp:2447-2455
Publication Date(Web):1 April 2010
DOI:10.1016/j.bmc.2010.02.052
We described here the design, synthesis, molecular modeling, and biological evaluation of a series of peptide and Schiff bases (PSB) small molecules, inhibitors of Escherichia coli β-Ketoacyl-acyl carrier protein synthase III (ecKAS III). The initial lead compound was reported by us previously, we continued to carry out structure–activity relationship studies and optimize the lead structure to potent inhibitors in this research. The results demonstrated that both N-(2-(3,5-dichloro-2-hydroxybenzylideneamino)propyl)-2-hydroxybenzamide (1f) and 2-hydroxy-N-(2-(2-hydroxy-5-iodobenzylideneamino)propyl)-4-methylbenzamide (3e) posses good ecKAS III inhibitory activity and well binding affinities by bonding Gly152/Gly209 of ecKAS III and fit into the mouth of the substrate tunnel, and can be as potential antibiotics agent, displaying minimal inhibitory concentration values in the range 0.20–3.13 μg/mL and 0.39–3.13 μg/mL against various bacteria.We described here the design, synthesis, molecular modeling, and biological evaluation of a series of peptide and Schiff bases (PSB) small molecules, inhibitors of Escherichia coli β-Ketoacyl-acyl carrier protein synthase III (ecKAS III). The initial lead compound was reported by us previously, we continued to carry out structure–activity relationship studies and optimize the lead structure to potent inhibitors in this research. The results demonstrated that both N-(2-(3,5-dichloro-2-hydroxybenzylideneamino)propyl)-2-hydroxybenzamide (1f) and 2-hydroxy-N-(2-(2-hydroxy-5-iodobenzylideneamino)propyl)-4-methylbenzamide (3e) were posses good ecKAS III inhibitory activity and well binding affinities by bonding Gly152/Gly209 of ecKAS III and fit into the mouth of the substrate tunnel, and can be as potential antibiotics agent, displaying minimal inhibitory concentration values in the range 0.20–3.13 μg/mL and 0.39–3.13 μg/mL against various bacteria.
Co-reporter:Suo-Ping Xu;Peng-Cheng Lv;Lei Shi
Archiv der Pharmazie 2010 Volume 343( Issue 5) pp:282-290
Publication Date(Web):
DOI:10.1002/ardp.200900129
Abstract
A series of 3,5-diiodo-salicylalidene Schiff bases (compounds 1–35) has been synthesized and tested for antimicrobial activity. The compounds were assayed for antibacterial activities by the MTT method. Some of the compounds inhibit the growth of a broad range of bacteria including the species of Bacillus subtilis, Staphylococcus aureus, Streptococcus faecalis, Pseudomonas aeruginosa, Escherichia coli, and Enterobacter cloacae. Among them, compounds 2-[(4-chloro-phenylimino)methyl]-4,6-diiodo-phenol 11 and 2,4-diiodo-6-[(2-morpholin-4-yl-ethylimino)methyl]phenol 19 showed the most potent antibacterial activity with MIC of 3.1, 12.9, 3.3, 6.5, 12.9, 3.3 and 3.2, 12.8, 3.2, 12.8, 12.8, 3.2 μM against B. subtilis, S. aureus, S. faecalis, P. aeruginosa, E. Coli, and E. cloacae, respectively.
Co-reporter:Huan-Qiu Li, Yin Luo, Peng-Cheng Lv, Lei Shi, Chang-Hong Liu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 6) pp:2025-2028
Publication Date(Web):15 March 2010
DOI:10.1016/j.bmcl.2010.01.032
β-Ketoacyl-acyl carrier protein synthase III (FabH) catalyzes the initial step of fatty acid biosynthesis via a type II fatty acid synthase in most bacteria. The important role of this essential enzyme combined with its unique structural features and ubiquitous occurrence in bacteria has made it an attractive new target for the development of new FabH inhibitors. The synthesis and biological evaluation halide-deoxybenzoins derivatives are described in this Letter. Potent FabH inhibitory and selective anti-Gram-negative bacteria activities were observed in deoxybenzoin derivatives. Furthermore, compound 19 was able to reduce the ECE-induced IL-8 production in gastric mucosal cells significantly. Based on the biological data and molecular docking, compound 19 is a potential FabH inhibitor and anti-inflammatory agent deserving further research.A novel series of deoxybenzoin derivatives were designed and synthesized. Based on the biological data obtained in this study, it can be concluded that compound 19 would be a potential Escherichia coli FabH inhibitor and promising anti-inflammatory agent. The binding model of compound 19 and E. coli FabH was also been researched.
Co-reporter:Peng-Cheng Lv, Juan Sun, Yin Luo, Ying Yang, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 15) pp:4657-4660
Publication Date(Web):1 August 2010
DOI:10.1016/j.bmcl.2010.05.105
Fatty acid biosynthesis is essential for bacterial survival. FabH, β-ketoacyl-acyl carrier protein (ACP) synthase III, is a particularly attractive target, since it is central to the initiation of fatty acid biosynthesis and is highly conserved among Gram-positive and -negative bacteria. Fifty-six 1-acetyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazole derivatives were synthesized and developed as potent inhibitors of FabH. This inhibitor class demonstrates strong antibacterial activity. Escherichia coli FabH inhibitory assay and docking simulation indicated that the compounds 1-(5-(4-fluorophenyl)-3-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone (12) and 1-(5-(4-chlorophenyl)-3-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone (13) were potent inhibitors of E. coli FabH.Fifty-six 1-acetyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazole derivatives were synthesized and developed as potent inhibitors of FabH. Escherichia coli FabH inhibitory assay was undertaken and the results suggested that compounds 12 and 13 were potent E. coli FabH inhibitors.
Co-reporter:Xin-Hua Liu, Hui-Feng Liu, Jin Chen, Yang Yang, Bao-An Song, Lin-Shan Bai, Jing-Xin Liu, Hai-Liang Zhu, Xing-Bao Qi
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 19) pp:5705-5708
Publication Date(Web):1 October 2010
DOI:10.1016/j.bmcl.2010.08.017
A series of novel coumarin derivatives containing 4,5-dihydropyrazole moiety as potential telomerase inhibitors were synthesized. The bioassay tests show that compound 3d exhibited potentially high activity against human gastric cancer cell SGC-7901 with IC50 value of 2.69 ± 0.60 μg/mL. All title compounds were assayed for telomerase inhibition by a modified TRAP assay, the results show that compounds 3d and 3f can strongly inhibit telomerase with IC50 values of 2.0 ± 0.07 and 1.8 ± 0.35 μM, respectively. Docking simulation was performed to position compound 3d into the telomerase (3DU6) active site to determine the probable binding model.Novel coumarin derivatives containing 4,5-dihydropyrazole moiety as potential telomerase inhibitors were synthesized. The bioassay tests showed that compound 3d exhibited potentially high activity against human gastric cancer cell SGC-7901 with the IC50 value was 2.69 ± 0.60 μg/mL.
Co-reporter:Xin-Hua Liu, Hui-Feng Liu, Xu Shen, Bao-An Song, Pinaki S. Bhadury, Hai-Liang Zhu, Jin-Xing Liu, Xing-Bao Qi
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 14) pp:4163-4167
Publication Date(Web):15 July 2010
DOI:10.1016/j.bmcl.2010.05.080
A series of novel 2-chloro-pyridine derivatives containing flavone, chrome or dihydropyrazole moieties as potential telomerase inhibitors were synthesized. The bioassay tests showed that compounds 6e and 6f exhibited some effect against gastric cancer cell SGC-7901 with IC50 values of 22.28 ± 6.26 and 18.45 ± 2.79 μg/mL, respectively. All title compounds were assayed for telomerase inhibition by a modified TRAP assay, the results showed that compound 6e can strongly inhibit telomerase with IC50 value of 0.8 ± 0.07 μM. Docking simulation was performed to position compound 6e into the active site of telomerase (3DU6) to determine the probable binding model.The bioassay tests showed that compounds 6e and 6f exhibited certain effective against gastric SGC-7901 cell with the IC50 values were 22.28 ± 6.26 and 18.45 ± 2.79 μg/mL, respectively. Compound 6e can strongly inhibit telomerase with IC50 value of 0.8 ± 0.07 μg/mL. The docking simulation result shows that some 2-chloro-pyridine containing flavone (6e) can combine well with the telomerase active site may use as potential telomerase inhibitors.
Co-reporter:Ying Yang, Lei Shi, Yang Zhou, Huan-Qiu Li, Zhen-Wei Zhu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 22) pp:6653-6656
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmcl.2010.09.014
Vascular endothelial growth factor receptor-2 (VEGFR-2) plays a crucial role in the process of cancer angiogenesis. A series of quinoline amide derivatives were prepared and found to be good inhibitors of VEGFR-2. The inhibitory activities were investigated against VEGFR-2 kinase and human umbilical vein endothelial cells (HUVEC) in vitro. Compound 6 (5-chloro-2-hydroxy-N-(quinolin-8-yl)benzamide) exhibited the most potent inhibitory activity (IC50 = 3.8 and 5.5 nM for VEGFR-2 kinase and HUVEC, respectively). Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of VEGFR-2, which demonstrates that compound 6 is a potential agent for cancer therapy deserving further researching.Two series of quinoline amide derivatives were prepared and found to be good inhibitors of vascular endothelial growth factor receptor-2 (VEGFR-2). The inhibitory activities were investigated against VEGFR-2 kinase and human umbilical vein endothelial cells (HUVEC) in vitro. Compound 6 (5-chloro-2-hydroxy-N-(quinolin-8-yl)benzamide) exhibited the most potent inhibitory activity (IC50 = 3.8 and 5.5 nM for VEGFR-2 kinase and HUVEC, respectively). Docking simulation was performed to position compound 6 into the VEGFR-2 ATP-binding site to determine the probable binding model. The results supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of VEGFR-2.
Co-reporter:Jun Wan;Xia Yan;Cuiping Ma;Sai Bi
Medicinal Chemistry Research 2010 Volume 19( Issue 8) pp:970-983
Publication Date(Web):2010 November
DOI:10.1007/s00044-009-9243-3
Ten novel benzotriazole compounds were synthesized. Their chemical structures were confirmed by 1H NMR, IR, and elemental analyses, coupled with three selected single-crystal structures (compounds A2, B3, and B5). Their antimycotic and antitumor activities were also investigated. The title compounds showed some antitumor activities, especially in the case of A3 and A4, which showed the most potent activity of propagation inhibition in liver and galactophore cancer cells.
Co-reporter:Huan-Qiu Li Dr.;Yin Luo Dr.;Ran Song Dr.;Zi-Lin Li;Tao Yan Dr.
ChemMedChem 2010 Volume 5( Issue 7) pp:1117-1122
Publication Date(Web):
DOI:10.1002/cmdc.201000107
Abstract
In the search for potential immunosuppressive agents with high efficacy and low toxicity, a series of new deoxybenzoins were synthesized and evaluated for their cytotoxicity and immunosuppressive activity. Among the synthesized compounds, four deoxybenzoin oximes (compounds 31, 32, 37, and 38) exhibited lower cytotoxicity and higher inhibitory activity toward anti-CD3/anti-CD28 co-stimulated T-cell proliferation than other compounds. More significantly, compound 31 is >100-fold less cytotoxic than cyclosporine A (CsA) and is also more potent (31: SI>684.64, CsA: SI=235.44). The preliminary inhibition mechanism of compound 31 was also identified by flow cytometry, and this compound exerts immunosuppressive activity by inducing apoptosis in activated lymph node cells in a dose-dependent manner. In addition, the mechanism of apoptosis was detected by western blot analysis.
Co-reporter:Yin Luo Dr.;Huan-Qiu Li Dr.;Yang Zhou Dr.;Zi-Lin Li;Tao Yan Dr.
ChemMedChem 2010 Volume 5( Issue 7) pp:1110-1116
Publication Date(Web):
DOI:10.1002/cmdc.201000126
Abstract
A series of new metronidazole–deoxybenzoin derivatives were synthesized and evaluated for their antimicrobial activity against Helicobacter pylori. Highly selective anti-H. pylori activity was also observed in synthesized compounds. Compound 34 exhibited the most potent activity, similar to the positive control amoxicillin. Furthermore, compounds 17 and 34 were able to significantly decrease H. pylori water extract (HPE)-induced production of interleukin-8 (IL-8) in gastric mucosal cells, which did not show any effect on the cell viability.
Co-reporter:Jin Hou;Ying Yang
Journal of Chemical Crystallography 2010 Volume 40( Issue 8) pp:661-663
Publication Date(Web):2010 August
DOI:10.1007/s10870-010-9714-x
A Schiff base complex, Bis{2-[3-(diethylamino)propyliminiomethyl]-5-methoxyphenolate}copper(II) nitrate, has been synthesized and structurally characterized by elemental analyses and X-ray diffraction. In the complex, the Cu(II) atom is four-coordinated by two O and two N atoms from the two Schiff base ligands, 2-[3-(diethylamino)propyliminiomethyl]-5-methoxyphenolate, forming a distorted square-planar coordination.
Co-reporter:Lei Shi
Journal of Chemical Crystallography 2010 Volume 40( Issue 4) pp:384-386
Publication Date(Web):2010 April
DOI:10.1007/s10870-010-9715-9
A new Schiff base complex, Bis {2-[(cyclohexylimino)methyl]-4,6-dihydroselenophenol}copper(II), has been synthesized and structurally characterized by elemental analyses and X-ray diffraction. The title complex C26H32N2O2·Se4Cu crystallizes in the monoclinic space group P21/c with the cell parameters a = 15.308(3) Å, b = 12.857(2) Å, c = 14.161(3) Å, β = 93.23(3)°, V = 2782.8(10) Å3 and Z = 4. The central Copper(II) atom is four-coordinated by two O and two N atoms from the two Schiff base ligands, 2-[(cyclohexylimino)methyl]-4,6-dihydroselenophenol, forming a distorted square-planar coordination.
Co-reporter:Jun Wan;Peng-Cheng Lv;Na-Na Tian
Journal of Chemical Sciences 2010 Volume 122( Issue 4) pp:597-606
Publication Date(Web):2010 July
DOI:10.1007/s12039-010-0094-8
A series of benzotriazole derivatives (compounds 1–27) were synthesized, and 24 (compounds 1–5, 9–27) of which were first reported. Their chemical structures were confirmed by means of 1H NMR, IR and elemental analyses, coupled with one selected single crystal structure (compound 1). All the compounds were assayed for antibacterial activities against three Gram positive bacterial strains (Bacillus subtilis, Staphylococcus aureus and Streptococcus faecalis) and three Gram negative bacterial strains (Escherichia coli, Pseudomonas aeruginosa and Enterobacter cloacae) by MTT method. Among the compounds tested, most of them exhibited potent antibacterial activity against the six bacterial strains. Most importantly, compound 3-benzotriazol-1-yl-1-(4-bromo-phenyl)-2-[1,2,4]triazol-1-ylpropan-1-one (19) showed the most favourable antibacterial activity against B. subtilis, S. aureus, S. faecalis, P. aeruginosa, E. coli and E. cloacae with MIC of 1.56 µg/mL, 1.56 µg/mL, 1.56 µg/mL, 3.12 µg/mL, 6.25 µg/mL and 6.25 µg/mL, respectively.
Co-reporter:Peng-Cheng Lv, Huan-Qiu Li, Jia-Yu Xue, Lei Shi, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 2) pp:908-914
Publication Date(Web):February 2009
DOI:10.1016/j.ejmech.2008.01.013
A series of luteolin derivatives 2–20 were prepared, 3–20 of which were first reported. The chemical structures of these compounds were confirmed by means of 1H NMR, ESI-MS and elemental analyses. The compounds were assayed for antibacterial (Bacillus subtilis, Staphylococcus aureus, Pseudomonas fluorescens and Escherichia coli) activities by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl trtrazolium bromide) method. Among the compounds tested, most of them displayed significant activity against the tested strains, and 2-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-5-hydroxy-7-(2-(3-morpholinopropylamino)ethoxy)-4H-chromen-4-one (17) showed the most favorable antibacterial activity in vitro with MICs of 1.562, 3.125, 3.125, and 6.25 μg/mL against B. subtilis, S. aureus, P. fluorescens and E. coli, respectively. Structure–activity relationships (SAR) were also discussed based on the obtained experimental data.A series of luteolin derivatives containing a 2-carbon spacer at C-7 position and potential pharmacophore 1,4-benzodioxin were prepared and evaluated for antibacterial activity against four bacteria.
Co-reporter:Huan-Qiu Li, Tao-Tao Zhu, Tao Yan, Yin Luo, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 2) pp:453-459
Publication Date(Web):February 2009
DOI:10.1016/j.ejmech.2008.04.011
Twenty-four new 1,3-disubstituted urea derivatives (compounds 1–24) were synthesized and reported for the first time. The antiproliferative activities of these compounds were evaluated against a panel of one human liver cell line (L02) and two human tumor cell lines (KB and K562) by applying the MTT colorimetric assay. The series of 1,3-disubstituted urea derivatives show good antiproliferative activity against human cancer cell lines (KB and K562) and no antiproliferative activity against liver cell line (L02). The potent in vitro antiproliferative activity of these derivatives and their selectivity for L02 are quite important points for an anticancer drug candidate with fewer side effects. Structure–activity relationships were also discussed based on the obtained experimental data. The hydroxyl groups on the phenyl ring reduced the antiproliferative activities of 1,3-disubstituted urea derivatives. The OH groups could be responsible for a reduction in the permeability of the cell membrane. Generally, an aromatic ring on N-3 seems to be in favor of enhancing the inhibitory activity, compounds introduced a nitro group substituent at C-3 position on the aromatic ring approved to generally decrease activity.
Co-reporter:Peng-Cheng Lv, Zhu-Ping Xiao, Rui-Qin Fang, Huan-Qiu Li, Hai-Liang Zhu, Chang-Hong Liu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 4) pp:1779-1787
Publication Date(Web):April 2009
DOI:10.1016/j.ejmech.2008.04.019
Twenty-six depsides were synthesized to screen for their antibacterial activity. All of them were reported for the first time. Their chemical structures were clearly determined by 1H NMR, 13C NMR, ESI mass spectra and elemental analyses, coupled with one selected single-crystal structure. All the compounds were assayed for antibacterial activities against three Gram-positive bacterial strains (Bacillus subtilis ATCC 6633, Staphylococcus aureus ATCC 6538 and Streptococcus faecalis ATCC 9790) and three Gram-negative bacterial strains (Escherichia coli ATCC 35218, Pseudomonas aeruginosa ATCC 13525 and Enterobacter cloacae ATCC 13047) by MTT method. Compound 2-(2-methoxy-2-oxoethyl)phenyl 3-nitrobenzoate (C10) and 2-(2-ethoxy-2-oxoethyl)phenyl 3-nitrobenzoate (C23) showed powerful antibacterial activities against B. subtilis with MIC of 0.78 μg/mL while compound 2-(2-methoxy-2-oxoethyl)phenyl 2-(3,4-diethoxyphenyl)acetate (C8) and 2-(2-ethoxy-2-oxoethyl)phenyl 2-(3,4-diethoxyphenyl)acetate (C21) exhibited significant antibacterial activities against E. coli with MIC of 1.562 μg/mL, which were superior to the positive controls penicillin G and kanamycin B, respectively. On the basis of the biological results, structure–activity relationships were discussed.Twenty-six depsides were synthesized to screen for their antibacterial activity. Compounds C10 and C23 showed potent antibacterial activities against Bacillus subtilis with MIC of 0.78 μg/mL while compounds C8 and C21 exhibited significant antibacterial activities against Escherichia coli with MIC of 1.562 μg/mL, which were superior to the positive controls penicillin and kanamycin, respectively.
Co-reporter:Huan-Qiu Li, Zhu-Ping Xiao, Yin-Luo, Tao Yan, Peng-Cheng Lv, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 5) pp:2246-2251
Publication Date(Web):May 2009
DOI:10.1016/j.ejmech.2008.06.001
Twenty amines and oximes from deoxybenzoins were prepared and evaluated for their inhibitory activity against Helicobacter pylori urease. Among these compounds, high inhibitory activities were observed in amines and oximes, especially amines 1b (IC50 = 0.011 mM) and 6b (IC50 = 0.047 mM) exhibited good in vitro activities, and were comparable to acetohydroxamic acid (AHA). The hydroxyl groups on deoxybenzoin skeleton may be responsible for the inhibitory activity and coordinate with the nickel (active site) on enzyme. A direct interaction may exist between the OH group of hydroxylamines or NH group of amines and His α323 of H. pylori urease, which is on the flap of the enzyme active site.
Co-reporter:Zhong-Cheng Song, Gao-Yuan Ma, Peng-Cheng Lv, Huan-Qiu Li, Zhu-Ping Xiao, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 10) pp:3903-3908
Publication Date(Web):October 2009
DOI:10.1016/j.ejmech.2009.04.014
Nine 2-arylthiazolidine-4-carboxylic acid derivatives and nine 3-tert-butoxycarbonyl-2-arylthiazolidine-4-carboxylic acid derivatives were synthesized to screen for their antibacterial activities. Compounds 5, 14–18 were first reported. Their chemical structures were clearly determined by 1H NMR, 13C NMR, ESI mass spectra and elemental analyses, coupled with one selected single-crystal structure. All the compounds were assayed for antibacterial activities against two Gram-positive bacterial strains (Bacillus subtilis ATCC 6633 and Staphylococcus aureus ATCC 6538) and two Gram-negative bacterial strains (Escherichia coli ATCC 35218 and Pseudomonas aeruginosa ATCC 13525) by MTT method. Most of the 3-tert-butoxycarbonyl-2-arylthiazolidine-4-carboxylic acid derivatives exhibited better antibacterial activities against the four bacterial strains than relative 2-arylthiazolidine-4-carboxylic acid derivatives. Compound (2RS,4R)-3-(tert-butoxycarbonyl)-2-(5-fluoro-2-hydroxyphenyl)thiazolidine-4-carboxylic acid (14) showed powerful antibacterial activities against P. aeruginosa with IC50 value of 0.195 μg/mL, which was superior to the positive controls Penicillin G and Kanamycin B, respectively. On the basis of the biological results, structure–activity relationships were discussed.The newly synthesized (2RS,4R)-3-(tert-butoxycarbonyl)-2-(5-fluoro-2-hydroxyphenyl) thiazolidine-4-carboxylic acid (14) exhibited higher selective antibacterial activities against pathogenic bacterium Pseudomonas aeruginosa with IC50 value of 0.195 μg/mL than positive controls Kanamycin G and Penicillin B did respectively. The crystal structure of (2R,4R)-3-(tert-butoxycarbonyl)-2-(4-methoxyphenyl)thiazolidine-4-carboxylic acid was also first reported.
Co-reporter:Xin-Hua Liu, Peng-Cheng Lv, Jia-Yu Xue, Bao-An Song, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 10) pp:3930-3935
Publication Date(Web):October 2009
DOI:10.1016/j.ejmech.2009.04.019
Microwave irradiation promotes the rapid O,N-acylation–cyclodehydration cascade reaction of oximes and acid chloride. Twenty novel 2,4,5-trisubstituted oxazole derivatives containing heterocycle moiety were synthesized and evaluated for their antiproliferative activity. The twenty compounds are all first reported and their structures were established by elemental analysis, 1H NMR and 13C NMR spectra. The bioassay tests showed that compounds 2-(2-(2-fluorophenyl)-4-(2,3,4-trimethoxyphenyl)oxazol-5-ylthio)benzo[d]thiazole (6af), 2-(2-(pyridin-3-yl)-4-(2,3,4-trimethoxyphenyl)oxazol-5-ylthio)pyrimidine (6bg) and 2-(2-(2-fluorophenyl)-4-(2,3,4-trimethoxyphenyl)oxazol-5-ylthio)-5-methyl-1,3,4-thiadiazole (6cf) displayed good antiproliferative activity in vitro, which were comparable to the positive control (5-fluorouracil).
Co-reporter:Huan-Qiu Li, Lei Shi, Qing-Shan Li, Peng-Gang Liu, Yin Luo, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 17) pp:6264-6269
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmc.2009.07.046
As a naturally wide distributed flavone, chrysin exhibits numerous biological activities including anticancer, anti-inflammatory, and antimicrobials activities. β-Ketoacyl-acyl carrier protein synthase III (FabH) catalyzes the initial step of fatty acid biosynthesis via a type II fatty acid synthase in most bacteria. The important role of this essential enzyme combined with its unique structural features and ubiquitous occurrence in bacteria has made it an attractive new target for the development of antibacterial agents. We first used a structure-based approach to develop 18 novel chrysin analogues that target FabH for the development of new antibiotics. Structure-based design methods were used for the expansion of the chrysin derivatives including molecular docking and SAR research. Based on the results, 5-hydroxy-2-phenyl-7-(2-(piperazin-1-yl)ethoxy)-4H-chromen-4-one (3g) showed the most potent antibacterial activity with MIC of 1.56–6.25 μg/mL against the test bacterial stains, and docking simulation was performed to position compound 3g into the Escherichiacoli FabH active site to determine the probable binding conformation. The biological assays indicated that compound 3g is a potent inhibitor of E.coli FabH as antibiotics.
Co-reporter:Wen-Jun Mao, Peng-Cheng Lv, Lei Shi, Huan-Qiu Li, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 21) pp:7531-7536
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmc.2009.09.018
Fourteen metronidazole derivatives (compounds 3a–f and 4b–h) have been synthesized by coupling of metronidazole and salicylic acid derivatives. All of them are reported for the first time. Their chemical structures are characterized by 1H NMR, MS, and elemental analysis. The inhibitory activities against Helicobacter pylori urease have been investigated in vitro and many compounds have showed promising potential inhibitory activities of H. pylori urease. The effect of compounds 4b (IC50 = 26 μM) and 4g (IC50 = 12 μM) was comparable with that of acetohydroxamic acid, a well known H. pylori urease inhibitor used as a positive control. The experimental values of IC50 showed that inhibitor was potent urease inhibitor. A docking analysis using the autodock 4.0 program could explain the inhibitory activities of compound 4g against H. pylori urease.Fourteen metronidazole derivatives (compounds 3a–f and 4b–h) have been synthesized by coupling of metronidazole and salicylic acid derivatives. All of them are reported for the first time. Their chemical structures are characterized by 1H NMR, MS, and elemental analysis. The inhibitory activities against Helicobacter pylori urease have been investigated in vitro and many compounds have showed promising potential inhibitory activities of H. pylori urease. The effect of compounds 4b (IC50 = 26 μM) and 4g (IC50 = 12 μM) was comparable with that of acetohydroxamic acid, a well known H. pylori urease inhibitor used as a positive control. The experimental values of IC50 showed that inhibitor was potent urease inhibitor. A docking analysis using the autodock 4.0 program could explain the inhibitory activities of compound 4g against H. pylori urease.
Co-reporter:Kui Cheng, Qing-Zhong Zheng, Yong Qian, Lei Shi, Jing Zhao, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 23) pp:7861-7871
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmc.2009.10.037
A series of peptide and Schiff bases (PSB) were synthesized by reacting salicylic acid, primary diamines with salicylaldehyde or its derivatives, and 40 of which were newly reported. The inhibitory activities against Escherichia coli β-ketoacyl-acyl carrier protein synthase III (ecKAS III) were investigated in vitro and molecular docking simulation also surveyed. Top 10 PSB compounds which posses both good inhibitory activity and well binding affinities were picked out, and their antibacterial activities against Gram-negative and Gram-positive bacterial strains were tested, expecting to exploit potent antibacterial agent with broad-spectrum antibiotics activity. The results demonstrate compound N-(3-(5-bromo-2-hydroxybenzylideneamino)propyl)-2-hydroxybenzamide (2d) can be as a potential antibiotics agent, displaying minimal inhibitory concentration values in the range of 0.39–3.13 μg/mL against various bacteria.The inhibitory activities of 44 PSB against Escherichia coli β-ketoacyl-acyl carrier protein synthase III (ecKAS III) were investigated in vitro and molecular docking simulation also surveyed. The results demonstrate compound N-(3-(5-bromo-2-hydroxybenzylideneamino)propyl)-2-hydroxybenzamide (2d), bonding two active site of ecKAS III and fit into the mouth of the substrate tunnel, can be as a potential antibiotics agent, displaying minimal inhibitory concentration values in the range 0.39–3.13 μg/mL against various bacteria.
Co-reporter:Xinhua Liu;Linshan Bai;Chunxiu Pan;Baoan Song;Hailiang Zhu
Chinese Journal of Chemistry 2009 Volume 27( Issue 10) pp:1957-1961
Publication Date(Web):
DOI:10.1002/cjoc.200990329
Abstract
Eleven novel 5-methyl-2-[(un)substituted phenyl]-4-{4,5-dihydro-3-[(un)substituted phenyl]-5-(1,2,3,4-tetrahydroisoquinoline-2-yl)pyrazol-1-yl}-oxazole derivatives were synthesized and characterized by elemental analysis, ESI-MS, 1H NMR and 13C NMR. All of the compounds have been screened for their antiproliferative activities against PC-3 cell (human prostate cancer) and A431 cell (human epidermoid carcinoma cancer) lines in vitro. The results revealed that compounds 4g, 4j and 4k exhibited the strong inhibitory activities against the PC-3 cell lines (with IC50 values of 2.8±0.11, 3.1±0.10 and 3.0±0.06 μg/mL, respectively).
Co-reporter:Peng-Cheng Lv, Kai-Rui Wang, Ying Yang, Wen-Jun Mao, Jin Chen, Jing Xiong, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6750-6754
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.09.111
Fatty acid biosynthesis is essential for bacterial survival. Components of this biosynthetic pathway have been identified as attractive targets for the development of new antibacterial agents. FabH, β-ketoacyl-acyl carrier protein (ACP) synthase III, is a particularly attractive target, since it is central to the initiation of fatty acid biosynthesis and is highly conserved among Gram positive and negative bacteria. Three series of Schiff bases containing thiazole template were synthesized and developed as potent inhibitors of FabH. This inhibitor class demonstrates strong antibacterial activity. Escherichia coli FabH inhibitory assay and docking simulation indicated that the compounds 11 and 18 were potent inhibitors of E. coli FabH.Docking results, along with the data of Escherichia coli FabH inhibitory activity assay indicated that compounds 11 and 18 would be potential inhibitors of E. coli FabH with potent antibacterial activity.
Co-reporter:Kui Cheng, Qing-Zhong Zheng, Hai-Liang Zhu
Inorganic Chemistry Communications 2009 Volume 12(Issue 11) pp:1116-1119
Publication Date(Web):November 2009
DOI:10.1016/j.inoche.2009.09.001
[CoIII(L1)2] (NO3) 1, Co3II(L2)2(C5H5N)6·2(ClO4)n2 (where L1 = (3-carboxysalicylidene)-(2-hydroxyaminoethylamine), L2 = (3-carboxysalicylidene)-phenylmethylamine) were synthesized and characterized by elemental analysis, single crystal X-ray diffraction and tested for inhibitive enzymatic activity on Jack Bean Urease(jbU). 1 is a mononuclear complex and the center cobalt atom is chelated by donors of N4O2 possessing a well defined octahedral configuration, and 2 contains two environmentally different cobalt centers, by the μ2 carboxyl groups bridging the repeat units extending into a one-dimension configuration. Both of the mononuclear neutral molecules 1 and polymeric (1D chain) 2 are future connected via huge inter- and intra-molecular O-H⋯O and C–H⋯O bonds exhibiting a network structure. The enzymatic activity study indicated that the two complexes are showed potent inhibitions against jbU, with IC50 20.31 ± 0.53 μM of 1 and 22.24 ± 0.67 μM of 2, which are about 2 times more than 42.12 ± 0.08 μM of acetohydroxamic acid as positive reference.As an expansion to the previous characterization of enzyme inhibitors from silver and metal Schiff base complexes, here describe the Cobalt Schiff base complexes [CoIII(L1)2] (NO3) 1 and Co3II(L2)2(C5H5N)6·2(ClO4)n2 with ligands derived from 3-formylsalicylic acid as potent inhibitors on Jack Bean Urease (jbU).
Co-reporter:Peng-Cheng Lv Dr.;Kai-Rui Wang;Wen-Jun Mao;Jing Xiong;Huan-Qiu Li Dr.;Ying Yang;Lei Shi Dr. Dr.
ChemMedChem 2009 Volume 4( Issue 9) pp:1421-1424
Publication Date(Web):
DOI:10.1002/cmdc.200900167
Co-reporter:Suo-Ping Xu;Peng-Cheng Lv;Rui-Qin Fang;Lei Shi
Journal of Chemical Crystallography 2009 Volume 39( Issue 12) pp:
Publication Date(Web):2009 December
DOI:10.1007/s10870-009-9612-2
One new complex, 2,4-Diiodo-6-[(2-morpholin-4-yl-ethylimino)- methyl]- phenolato-zinc(II) has been designed and synthesized. The structure was determined by UV, IR and single X-ray crystallography study. The title complex C26H30O4N4I4Zn crystallizes in the triclinic space group P − 1 with the cell parameters a = 9.938(2) Å, b = 11.937(2) Å, c = 14.527(3) Å, α = 87.14(3)°, β = 79.03(3)°, γ = 76.20(3)°, V = 1565.1(5) Å3 and Z = 2. The central zinc(II) is four coordinate and bonds to two nitrogen atoms and two oxygen atoms from two 3,5-diiodosalicylaldehyde-2- morpholinoethylamine Schiff bases. The complex is linked into a column by weak intermolecular interactions.
Co-reporter:Peng-Cheng Lv;Kai-Rui Wang;Wen-Jun Mao
Journal of Chemical Crystallography 2009 Volume 39( Issue 12) pp:
Publication Date(Web):2009 December
DOI:10.1007/s10870-009-9622-0
A depside derivative, 2-(2-methoxy-2-oxoethyl)phenyl 2-(3,4-dimethoxyphenyl)acetate (3), was synthesized through a facile approach in high yields. Its structure was confirmed by 1H NMR, 13C NMR, ESI mass spectra, elemental analyses and X-ray single crystal diffraction study. Crystal structure analysis revealed that compound 3 crystallized in the monoclinic, space group P21/c with the following unit cell parameters: a = 10.173(2) Å, b = 10.459(2) Å, c = 16.516(3) Å, α = 90°, β = 102.30(3)°, γ = 90°, V = 1717.0(6) Å3, Z = 4.
Co-reporter:Yong Qian;Peng-Cheng Lv;Lei Shi;Rui-Qin Fang
Journal of Chemical Sciences 2009 Volume 121( Issue 4) pp:463-470
Publication Date(Web):2009 July
DOI:10.1007/s12039-009-0055-2
Antiepileptic drug lamotrigine and its thirteen ammonium salt complexes (4a–4m) were synthesized and characterized by IR, elemental analysis, 1H-NMR, and MS spectral methods. Many of the ammonium salts (4a–4m) were first reported. Furthermore, the crystal structure of compound 3 was determined by single crystal X-ray diffraction analysis. All these complexes were tested in vitro for their antibacterial activity (Bacillus subtilis, Staphylococcus aureus, Enterococus faecalis, Escherichia coli, Pseudomonas aeruginosa and Enterobacter cloacae). The results indicated that most of the complexes showed good antibacterial activity against Gram-positive (B. subtilis, S. aureus and S. faecalis), but showed mild, even inactive against Gram-negative bacterial strains.
Co-reporter:Zhong-Lu You, Da-Hua Shi, Chen Xu, Qiang Zhang, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 4) pp:862-871
Publication Date(Web):April 2008
DOI:10.1016/j.ejmech.2007.06.015
Twenty transition metal complexes with Schiff bases were evaluated for their inhibitory activities on xanthine oxidase (XO), of which 11 were newly synthesized and characterized by X-ray single crystal diffraction. It was found that 9 of the 20 complexes showed potent inhibitory activities against XO near to the standard inhibitor allopurinol. The cadmium(II) complex (8) had the most potent inhibitory activity with the IC50 value of 2.16 μM. Relationships between the structures and the activities showed that the ligands and the metal ions influenced the inhibitory activities. The XO inhibition of the Schiff base metal complexes most probably resulted from their direct interactions with the enzymes “in the whole complex form”. These results demonstrated that the Schiff base transition metal complexes could be potential selective XO inhibitors.Twenty transition metal complexes with Schiff bases were evaluated for their inhibitory activities on xanthine oxidase (XO), of which 11 were newly synthesized and characterized by X-ray single crystal diffraction. It was found that 9 of the 20 complexes showed the potent inhibitory activities against XO near to the standard inhibitor allopurinol. Cadmium complex [Cd(C12H16N2)(μ-NCS)2] (8)++6 possessed the most potent inhibitory activity with the IC50value of 2.16 μM.
Co-reporter:Zhu-Ping Xiao, Rui-Qin Fang, Huan-Qiu Li, Jia-Yu Xue, Yi Zheng, Hai-Liang Zhu
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 9) pp:1828-1836
Publication Date(Web):September 2008
DOI:10.1016/j.ejmech.2007.11.026
Twenty-six enamines were synthesized to screen for the antimicrobial activity. Out of the compounds, 22 were reported for the first time. Their chemical structures including E/Z-configurations were clearly determined by 1H NMR, ESI mass spectra and elemental analyses, coupled with three selected single-crystal structures. In general, these synthetic compounds were shown to be more effective to inhibit growth of bacteria than fungi. The most active compound, (E)-ethyl 3-(4-hydroxyphenylamino)-2-(4-chlorophenyl)acrylate (1b), showed considerable antibacterial activities against Staphylococcus aureus ATCC 6538 with MIC of 0.5 μg/mL and against Pseudomonas fluorescens ATCC 13525 with MIC of 1.5 μg/mL, which was superior to the positive controls penicillin and kanamycin, respectively. Structure–activity relationship analysis revealed: as for A-ring, the compounds substituted at 3,5-positions were more active than 2,4-position-substituted derivatives, and halo-substituted analogs at 2-position had essentially same activities as the 4-position-substituted derivatives. Increase of steric hindrance around the nitrogen atom led to an inactive compound.Figure optionsThe most active compound, (E)-ethyl 3-(4-hydroxphenylamino)-2-(4-chlorophenyl)acrylate (1b), showed considerable antibacterial activities against Staphylococcus aureus ATCC 6538 and against Pseudomonas fluorescens ATCC 13525, which was superior to the positive controls penicillin and kanamycin, respectively.
Co-reporter:Xian-Feng Huang;Huan-Qiu Li;Lei Shi;Jia-Yu Xue;Ban-Feng Ruan
Chemistry & Biodiversity 2008 Volume 5( Issue 4) pp:636-642
Publication Date(Web):
DOI:10.1002/cbdv.200890059
Abstract
Thirteen resveratrol (=5-[(E)-2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol) analogues with a CHO group have been prepared by partial synthesis from resveratrol. The synthesized compounds have been evaluated for their cytotoxic activity against a human nasopharyngeal epidermoid tumor cell line KB, as well as for their xanthine oxidase inhibitory activity. Compounds 2, 3, and 6a showed the most significant cytotoxic activities against the cell line KB, and compound 2 also exhibited strong xanthine oxidase inhibitory activity.
Co-reporter:Huan-Qiu Li;Zhu-Ping Xiao;Rui-Qin Fang
Journal of Chemical Crystallography 2008 Volume 38( Issue 6) pp:461-466
Publication Date(Web):2008 June
DOI:10.1007/s10870-008-9343-9
Metronidazole (MET-OH), widely used as an antibacterial agent, is found to have some side effects on human bodies. Due to these disadvantages, people have been looking for its modification compounds for substituents. In this article, four MET-OH derivatives were designed, prepared, and structurally characterized by single crystal X-ray diffraction. These compounds are MET-OTs (1), MET-Br (2), MET-Cl (3), and MET-I (4). X-ray structure analyses revealed that, 1 crystallized in the monoclinic system with space group P21/c, with a = 16.1178, b = 7.5473, c = 13.4161 Å, V = 1520.3 Å3, β = 111.3210o and Z = 4. 2 crystallized in the monoclinic system with space group P21/c, with a = 12.079, b = 11.089, c = 6.380 Å, V = 847.1 Å3, β = 97.57o and Z = 4. 3 crystallized in the monoclinic system with space group P21/c, with a = 12.098, b = 11.007, c = 6.295 Å, V = 830.3 Å3, β = 97.886o and Z = 4. 4 crystallized in the triclinic system with space group P1, with a = 6.192, b = 7.740, c = 10.001 Å, V = 457.9 Å3, α = 89.073, β = 86.903, γ = 73.097o and Z = 2. In this article, metronidazole-derived compounds were prepared and structurally characterized by single crystal X-ray diffraction
Co-reporter:Zhu-Ping Xiao Dr.;Huan-Qiu Li Dr.;Lei Shi Dr.;Peng-Cheng Lv Dr.;Zhong-Cheng Song Dr.
ChemMedChem 2008 Volume 3( Issue 7) pp:1077-1082
Publication Date(Web):
DOI:10.1002/cmdc.200800057
Abstract
The antiproliferative activities of 36 3-aryl-1H-quinolin-4-ones were determined against two cancer cell lines (Hep G2 and KB) in vitro. The results indicate that most of these compounds show good cytotoxic activity against human cancer cell lines, but no cytotoxicity against a human normal cell line (L02). The positive control compounds genistein and 5-fluorouracil show no selectivity at inhibiting the growth of the above three cell lines. Generally, compounds that bear a halogen atom at the 8 position and a methoxy group at the 3′ position exhibited remarkable cytotoxicity toward human cancer cell lines. Electron-withdrawing substituents at the 6 position decrease the antiproliferative activity significantly. We also put forward a pharmacophore model for 3-aryl-4-quinolinones binding with epidermal growth factor receptor protein tyrosine kinases (EGFR PTK). Out of the 36 synthetic compounds, 34 are reported for the first time.
Co-reporter:Zhu-Ping Xiao;Peng-cheng Lv;Suo-Ping Xu;Tao-Tao Zhu
ChemMedChem 2008 Volume 3( Issue 10) pp:1516-1519
Publication Date(Web):
DOI:10.1002/cmdc.200800160
Co-reporter:Zhu-Ping Xiao, Da-Hua Shi, Huan-Qiu Li, Li-Na Zhang, Chen Xu, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 11) pp:3703-3710
Publication Date(Web):1 June 2007
DOI:10.1016/j.bmc.2007.03.045
Twenty polyphenols were synthesized and evaluated for their effect on Helicobacter pylori urease. Among these compounds, 4-(p-hydroxyphenethyl)pyrogallol (15) (IC50 = 0.03 mM) and 7,8,4′-trihydroxyisoflavone (19) (IC50 = 0.14 mM) showed potent inhibitory activities, and inhibited Helicobacter pylori urease in a time-dependent manner. The structure–activity relationship of these polyphenols revealed: the two ortho hydroxyl groups were essential for inhibitory activity of polyphenol. When the C-ring of isoflavone was broken, the inhibitory activity markedly decreased. As for deoxybenzoin, the carboxyl group was clearly detrimental.A series of polyphenols were synthesized and evaluated for inhibitory activity against Helicobacter pylori urease. Compounds 15 and 17 were the potent inhibitors with IC50 = 0.03 and 0.14 mM, respectively.
Co-reporter:Zhu-Ping Xiao, Jia-Yu Xue, Shu-Hua Tan, Huan-Qiu Li, Hai-Liang Zhu
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 12) pp:4212-4219
Publication Date(Web):15 June 2007
DOI:10.1016/j.bmc.2007.03.060
Twenty-four enamines were synthesized and reported for the first time. Their chemical structures were confirmed by means of 1H NMR, ESI mass spectra, and elemental analyses, and four of them were determined by single crystal X-ray diffraction analysis. All of the compounds were assayed for antibacterial (Bacillus subtilis ATCC 6633, Escherichia coli ATCC 35218, Pseudomonas fluorescens ATCC 13525, and Staphylococcus aureus ATCC 6538) and antifungal (Aspergillus niger ATCC 16404, Candida albicans ATCC 10231, and Trichophyton rubrum ATCC 10218) activities by MTT method. Compounds (E)-ethyl 3-(4-hydroxyphenylamino)-2-(4-methoxyphenyl)acrylate (9b), (E)-ethyl 3-(3,5-difluorophenylamino)-2-(4-chlorophenyl)acrylate (11b), (E)-ethyl 3-(3,5-dichlorophenylamino)-2-(4-chlorophenyl)acrylate (12b), and (E)-ethyl 3-(4-methylphenylamino)-2-(4-chlorophenyl)acrylate (15b) showed considerable antibacterial activities against S. aureus ATCC 6538 with MICs of 3.8, 1.9, 1.1, and 0.9 μg/mL, respectively. Structure–activity relationship (SAR) analysis disclosed, generally, an E-isomer exhibited higher antibacterial activity than the corresponding Z-isomer. An electron-withdrawing group on A-ring led to some decrease in activity, while on B-ring, a similar substitution provided higher activity.Figure optionsThe tested enamines were shown more effective to inhibiting growth of bacteria than fungi. An E-isomer exhibited higher antibacterial activity than the corresponding Z-isomer and four compounds (9b, 11b, 12b and 15b) showed considerable antibacterial activities against Staphylococcus aureus.
Co-reporter:Yu-Guang Li, Da-Hua Shi, Hai-Liang Zhu, Hong Yan, Seik Weng Ng
Inorganica Chimica Acta 2007 Volume 360(Issue 9) pp:2881-2889
Publication Date(Web):10 June 2007
DOI:10.1016/j.ica.2007.02.019
Six new transition metal complexes (M = Cu(II), Ni(II) and Mn(III)) of tridentate (H2L1, HL2) and/or bidentate (HL3, HL4) Schiff-base ligands, obtained from the condensation of salicylaldehyde with glycine, N-(2-aminoethyl)morpholine, 4-(2-aminoethyl)phenylic acid and 4-(2-aminoethyl)benzsulfamide, respectively, were synthesized and structurally determined by single-crystal X-ray analysis. Complexes 1–6 were evaluated for their effect on the jack bean urease and xanthine oxidase (XO). Copper(II) complexes 1–3 (IC50 = 0.43–2.25 μM) showed potent inhibitory activity against jack bean urease, comparable with acetohydroxamicacid (IC50 = 42.12 μM), which is a positive reference. And these copper(II) complexes (IC50 = 10.26–15.82 μM) also exhibited strong ability to inhibit activity of XO, comparable to allopurinol (IC50 = 10.37 μM), which was used as a positive reference. Nickel(II) and manganese(III) complexes 4–6 showed weak inhibitory activity to jack bean urease (IC50 = 4.36–8.25 μM) and no ability to inhibit XO (IC50 > 100 μM).Six new transition metal complexes (M = Cu(II), Ni(II) and Mn(III)) of Schiff base were synthesized and structurally determined by single-crystal X-ray analysis. And these complexes were evaluated for their effect on the jack bean urease and xanthine oxidase (XO). Copper(II) complexes 1–3 not only showed potent inhibitory activity against jack bean urease (IC50 = 0.43–2.25 μM), these complexes also exhibited strong ability to inhibit activity of XO (IC50 = 10.26–15.82 μM). However, nickel(II) and manganese(III) complexes 4–6 showed weak inhibitory activity to jack bean urease (IC50 = 4.36–8.25 μM) and no ability to inhibit XO (IC50 > 100 μM). We have demonstrated for the first time that Schiff-base complexes show urease and XO inhibitory activities. The mechanisms of the inhibitory activity require to be further investigated.
Co-reporter:Li-Na Zhang;Zhu-Ping Xiao;Hui Ding;Hui-Ming Ge;Chen Xu;Ren-Xiang Tan
Chemistry & Biodiversity 2007 Volume 4(Issue 2) pp:248-255
Publication Date(Web):20 FEB 2007
DOI:10.1002/cbdv.200790030
Two series of genistein (=5,7-dihydroxy-3-(4-hydroxyphenyl)-4H-1-benzopyran-4-one) derivatives with heterocycles were prepared, in which genistein and heterocyclic moieties were separated by C2 and C3 spacers. Among the 24 compounds we prepared, 22, i.e., 3a–3k and 4a–4k, were reported for the first time, while the preparation of 2a and 2b was reported in our recent paper. The cytotoxic activities of these compounds were evaluated against human chronic myeloid leukemia cells (K562) and a human nasopharyngeal epidermoid tumor cell line (KB). Compounds 4a, 4d, 4e, 4h, and 4i showed remarkable anticancer activities in vitro that are comparable with 5-fluorouracil, an canonical anticancer drug. Structure–effect relationships were also discussed based on the experimental data obtained.
Co-reporter:Ping Cao;Hui Ding;Xian-Feng Huang;Hui-Ming Ge;Ban-Feng Ruan;Huan-Qiu Li
Chemistry & Biodiversity 2007 Volume 4(Issue 5) pp:881-886
Publication Date(Web):18 MAY 2007
DOI:10.1002/cbdv.200790075
A series of substituted urea derivatives, compounds 1–14, were synthesized and evaluated for their cytotoxic activities against the human-leukemia K562 cell line. Two structurally simple compounds, 7 and 12, both incorporating a morpholine ring, were found to be highly active, with IC50 values of ca. 0.25 μM.
Co-reporter:Huan-Qiu Li Dr.;Chen Xu Dr.;Hong-Sen Li Dr.;Zhu-Ping Xiao Dr.;Lei Shi Dr.
ChemMedChem 2007 Volume 2(Issue 9) pp:
Publication Date(Web):12 JUL 2007
DOI:10.1002/cmdc.200700097
Three series of metronidazole–flavonoid derivatives were generated and evaluated for antimicrobial activity against H. pylori. Among these compounds, high anti-H. pylori activities were observed in isoflavones derivatives 4–7, 19, and 20 but exhibited no inhibitory activity against other sorts of bacteria and fungi, for example, Streptococcus pneumoniae, Bacillus subtilis, Escherichia coli, Pseudomonas fluorescence, and Aspergillus niger. Genistein derivative 6 with the potent activity (MIC=0.39 μg mL−1) was >50-fold more than metronidazole, and comparable to the positive control amoxicillin. Additionally, compound 6 can significantly attenuate the increase in interleukin-8 (IL-8) levels in the AGS cells stimulated by H. pylori water extract (HPE) at concentrations of 15, 30, and 60 μmol L−1, which did not show any effects on the cell viability.
Co-reporter:Xian-Feng Huang;Lei Shi;Huan-Qiu Li
Journal of Chemical Crystallography 2007 Volume 37( Issue 11) pp:739-742
Publication Date(Web):2007 November
DOI:10.1007/s10870-007-9242-5
The title compound, 3, 6-dihydroxy-2-[2-(4-hydroxy-phenyl)-vinyl]- benzene-1,3-dicarbaldehyde was synthesized by Vilslmeier reaction from resveratrol (trans-3,4′,5-trihydroxystilbene). Its structure was determined by X-ray single crystal diffraction. The crystal belongs to monoclinic, space group P21/n with crystallographic parameters: a = 7.2950(15) Å, b = 14.781(3) Å, c = 12.202(2) Å, β = 96.57(3)°, μ = 0.108 mm−1, V = 1307.1(5) Å, Z = 4, Dx = 1.445 g/cm3, F(000) = 592, T = 293(2) K, 2.17°≤ θ ≤ 26.00°. The X-ray results demonstrated that the Vilslmeier reaction of resveratrol with DMF, POCl3 and CH3CN yielded 4,6-dhydroxy-2-[2-(4-hydroxy-phenyl)-vinyl]-benzene-1,3-dicarbaldehyde.
Co-reporter:Hui-Ming Ge;Huan-Qiu Li;Yun-Xi Chen;Lei Shi;Hui Ding;Ren-Xiang Tan;Chen Xu
Chemistry & Biodiversity 2006 Volume 3(Issue 4) pp:463-472
Publication Date(Web):19 APR 2006
DOI:10.1002/cbdv.200690049
Thirty genistein (=5,7-dihydroxy-3-(4-hydroxyphenyl)-4H-1-benzopyran-4-one; GEN) derivatives were synthesized from genistein through a facile approach in high yields. Compounds 9, 11, 12, 23–30 were reported for the first time, while 13–22 have already been reported in our recent paper. The cytotoxic activities of these compounds were evaluated against a human nasopharyngeal epidermoid tumor cell line KB. Compounds 7–9, 12, 14, 16–19, 21, 24, 27, 29 showed remarkable antitumor activities in vitro, which was comparable with 5-fluorouracil, an anticancer drug. On the basis of the obtained experimental data, structure–effect relationships were discussed.
Co-reporter:Xian-Feng Huang;Hui Ding;Ban-Feng Ruan;Hui-Ming Ge;Ren-Xiang Tan;Chen Xu
Chemistry & Biodiversity 2006 Volume 3(Issue 9) pp:975-981
Publication Date(Web):22 SEP 2006
DOI:10.1002/cbdv.200690106
A total of 17 resveratrol (=(E)-5-[2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol) derivatives were synthesized from resveratrol (RES) through a facile approach. Among them, 13 compounds, 2 and 6–17, were reported for the first time, while 1 and 3–5 had already been reported several years ago. The cytotoxic activities of these compounds were evaluated against human nasopharyngeal epidermoid tumor cell line KB, and compounds 1 and 8–11 showed strong anticancer activities in vitro, comparable with that of 5-fluorouracil, an anticancer drug. On the basis of the experimental data obtained, structure–activity relationships are discussed.
Co-reporter:Li Shen;Yong-Hao Ye;Xiao-Ting Wang Dr.;Chen Xu Dr.;Yong-Cun Song Dr.;Hai Li;Ren-Xiang Tan Dr.
Chemistry - A European Journal 2006 Volume 12(Issue 16) pp:
Publication Date(Web):23 MAR 2006
DOI:10.1002/chem.200501423
Aspernigerin (1), a novel cytotoxic alkaloid consisting of an unprecedented structural framework has been isolated from the extract of a culture of Aspergillus niger IFB-E003, an endophyte in Cyndon dactylon. Its structure was elucidated on the basis of comprehensive NMR spectral analysis and confirmed by single-crystal X-ray analysis. Aspernigerin (1) has been shown to be cytotoxic to the tumor cell lines nasopharynyeal epidermoid KB, cervical carcinoma Hela, and colorectal carcinoma SW1116 with corresponding IC50 values of 22, 46, and 35 μM, respectively. A feasible total synthetic route for aspernigerin (1) has been established for further pharmacological research.
Co-reporter:Li Shen;Rui H. Jiao;Yong H. Ye;Xiao T. Wang;Chen Xu Dr.;Yong C. Song Dr.;Hai L. Zhu ;Ren X. Tan
Chemistry - A European Journal 2006 Volume 12(Issue 21) pp:
Publication Date(Web):4 MAY 2006
DOI:10.1002/chem.200600084
Three new cytotoxic 10,13-cyclotrichothecane-derived macrolides, myrothecines A–C (1–3), were characterized from the extracts of two Myrothecium roridum strains, IFB-E009 and IFB-E012, isolated as endophytic fungi found on the traditional Chinese medicinal plants Trachelospermum jasminoides and Artemisia annua, respectively. The absolute configuration of myrothecines A–C was elucidated by a combination of spectral techniques (UV, IR, MS, circular dichroism (CD), 1H and 13C NMR, DEPT, 1H–1H COSY, NOESY, HMQC, and HMBC spectrascopic analyses), Mosher's ester analysis, and single-crystal X-ray diffraction. The absolute configuration of the reported bioactive analogue, mytoxin B was established by correlating its spectral data with that of known absolute configurational structures. Furthermore, the significance in endophytism (or symbiosis) and biocatabolism, highlighted by production of those macrolides by the endophytic strains, is discussed in brief.
Co-reporter:Peng-Fei Wang, Han-Yue Qiu, Ze-Feng Wang, Yong-Jiao Zhang, Zhong-Chang Wang, Dong-Dong Li, Hai-Liang Zhu
Biochemical Pharmacology (15 May 2017) Volume 132() pp:63-76
Publication Date(Web):15 May 2017
DOI:10.1016/j.bcp.2017.02.022
Co-reporter:Jie Fu, Yan-Hua Ding, Luo Li, Sheng Sheng, Teng Wen, Lu-Ji Yu, Wu Chen, Shu-Qing An and Hai-Liang Zhu
Environmental Science: Nano 2011 - vol. 13(Issue 3) pp:NaN604-604
Publication Date(Web):2011/01/12
DOI:10.1039/C0EM00604A
The distribution, source, ecological risk and ecotoxicity of polycyclic aromatic hydrocarbons (PAHs) of sediments from 7 sampling sites, named as Xinyang (XY), Huainan (HN), Bengbu (BB), Xuyi (XuY), Fuyang (FY), Mengcheng (MC) and Zhengzhou (ZZ), in the Huaihe River basin, China, have been investigated. The total concentrations of 16 USEPA priority PAHs ranged from 62.9 to 2232.4 ng g−1 dry weight (d.w.) with a mean concentration of 1056.8 ng g−1d.w. Through the assessment of ecological risk, we found that the levels of PAHs in the Huaihe River should not exert adverse biological effects. The total benzo[a]pyrene toxicity equivalent (TEQ) values calculated for samples varied from 0.01 to 194.1 ng g−1d.w., with an average of 65.9 ng g−1. The toxicity data were accordant with the chemical analysis results in this study. HN, BB and ZZ showed the greatest pollution extent both in the chemical analysis and the study of ecotoxicological effects.
Co-reporter:Jian Sun, Dong-Dong Li, Jing-Ran Li, Fei Fang, Qian-Ru Du, Yong Qian and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 44) pp:NaN7686-7686
Publication Date(Web):2013/09/10
DOI:10.1039/C3OB41136B
A series of novel 4-alkoxyquinazoline derivatives were prepared and synthesized and their biological activities were evaluated as potential inhibitors of vascular endothelial growth factor receptor 2 (VEGFR2). Of these compounds, compound 3j demonstrated the most potent inhibitory activities against VEGFR2 tyrosine kinase and cell proliferation, the IC50 values of this compound reaching up to 2.72 nM and 0.35 μM, respectively, compared with Tivozanib (3.40 nM and 0.38 μM). The obtained results, along with a 3D-QSAR study and molecular docking that was used for investigating the probable binding mode, could provide an important basis for further optimization of compound 3j as a potential tyrosine kinase inhibitor.
Co-reporter:Jing-Jun Dong, Qing-Shan Li, Shu-Fu Wang, Cui-Yun Li, Xin Zhao, Han-Yue Qiu, Meng-Yue Zhao and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 37) pp:NaN6337-6337
Publication Date(Web):2013/07/05
DOI:10.1039/C3OB40776D
The RAF–MEK–ERK cascade appears to be intimately involved in the regulation of cell cycle progression and apoptosis. The BRAFV600E mutant results in constitutive activation of the ERK pathway, which can lead to cellular growth dysregulation. A series of 5-phenyl-1H-pyrazol derivatives (3a–5e) have been designed and synthesized, and their biological activities were evaluated as potential BRAFV600E inhibitors. All the compounds were reported for the first time except 3e, and compound 1-(4-bromo-2-hydroxybenzyl)-3-phenyl-1-(5-phenyl-1H-pyrazol-3-yl)urea (5c) displayed the most potent inhibitory activity (BRAFV600E IC50 = 0.19 μM). Antiproliferative assay results indicated that compound 5c possessed high antiproliferative activity against cell lines WM266.4 and A375 in vitro, with IC50 values of 1.50 and 1.32 μM, respectively, which were comparable with the positive control vemurafenib. Docking simulations showed that compound 5c binds tightly to the BRAFV600E active site and acts as BRAFV600E inhibitor. A 3D-QSAR model was also built to provide more pharmacophore understanding towards designing new agents with more potent BRAFV600E inhibitory activity.
Co-reporter:Yong Yin, Xun Wu, Hong-Wei Han, Shao Sha, She-Feng Wang, Fang Qiao, Ai-Min Lu, Peng-Cheng Lv and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 45) pp:NaN9165-9165
Publication Date(Web):2014/09/16
DOI:10.1039/C4OB01589D
A series of novel 2-alkyl-chromeno[4,3-c]pyrazol-4(2H)-one derivatives were synthesized and evaluated for their biological activities as PI3K inhibitors. In vitro biological evaluation against four human tumor cell lines revealed that most target compounds showed impressively better antiproliferative activities than that of LY294002. Among these compounds, compound 4l exhibited the most potent and selective activity for PI3Kα, with the value of 0.014 μM, an approximately 30-fold increase in comparison with LY294002. Docking simulation was performed to position compound 4l into the PI3Kα active site and the result showed that compound 4l could bind well at the PI3Kα active site and it indicated that compound 4l could be a potential inhibitor of PI3Kα.
Co-reporter:Jing-Ran Li, Dong-Dong Li, Fei Fang, Qian-Ru Du, Lin Lin, Jian Sun, Yong Qian and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 48) pp:NaN8386-8386
Publication Date(Web):2013/09/19
DOI:10.1039/C3OB41161C
A series of 4,6-substituted-(diaphenylamino)quinazolines as c-Src inhibitors have been prepared and their biological activity has also been evaluated. All the compounds displayed potential antiproliferation activities, with IC50 values ranging from 3.42 μM to 118.81 μM in five human tumor cell lines. Particularly, compound 15 exhibited higher cytotoxicity against the tested five tumor cell lines compared to the other small molecules. Generally, most of these compounds showed selectivity between the A549 cells and the other four cells, according to their corresponding IC50 values. The results obtained from the in vitro enzyme assay indicated compound 15 has remarkable inhibitory activity against c-Src kinase with an IC50 value of 27.3 nM, which is comparable to the control compounds. Furthermore, molecular docking and QSAR study by means of DS 3.5 (Discovery Studio 3.5, Accelrys, Co. Ltd) explored the binding modes and the structure and activity relationship (SAR) of these derivatives.
Co-reporter:Jia-Jia Liu, Juan Sun, Yun-Bin Fang, Yong-An Yang, Rui-Hua Jiao and Hai-Liang Zhu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 6) pp:NaN1008-1008
Publication Date(Web):2013/11/08
DOI:10.1039/C3OB41953C
A series of novel 4,5-dihydropyrazole derivatives (4a–4t), containing the dinitrobenzotrifluoride moiety, as DNA gyrase inhibitors were designed and synthesized. Based on the preliminary results, compounds 4d, 4h and 4t with potent inhibitory activity in bacterial growth may be wonderful antibacterial agents; among them, compound 4t displayed the most potent activity with minimum inhibitory concentration (MIC) values of 3.125, 0.39, 0.39 and 0.39 μg mL−1 against Bacillus subtilis, Staphylococcus aureus, Pseudomonas aeruginosa, and Escherichia coli respectively, which was comparable with penicillin and kanamycin B with corresponding MIC values of 3.125, 3.125, 0.39, 0.39 μg mL−1 and 1.562, 1.562, 1.562, 1.562 μg mL−1, respectively. In particular, compound 4d showed the most potent anti-Gram-positive bacterial activity with a MIC value of 0.39 μg mL−1 against the tested Gram-positive bacterial strains and exhibited the most potent B. subtilis DNA gyrase and S. aureus DNA gyrase inhibitory activity with an IC50 of 0.125 μg mL−1. Docking simulation was performed to insert compound 4d into the S. aureus DNA gyrase active site to determine the probable binding conformation.







![1H-Benzotriazole, 1-[[5-(4-nitrophenyl)-1,3,4-oxadiazol-2-yl]methyl]-](http://img.cochemist.com/ccimg/881900/881880-04-2.png)
![1H-Benzotriazole, 1-[[5-(4-nitrophenyl)-1,3,4-oxadiazol-2-yl]methyl]-](http://img.cochemist.com/ccimg/881900/881880-04-2_b.png)










![[1]Benzopyrano[4,3-c]pyrazol-4(2H)-one,2-methyl-](http://img.cochemist.com/ccimg/803000/802911-49-5.png)
![[1]Benzopyrano[4,3-c]pyrazol-4(2H)-one,2-methyl-](http://img.cochemist.com/ccimg/803000/802911-49-5_b.png)






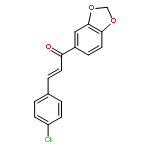









![1H-Benzotriazole, 1-[[5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl]methyl]-](http://img.cochemist.com/ccimg/307000/306990-71-6.png)
![1H-Benzotriazole, 1-[[5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl]methyl]-](http://img.cochemist.com/ccimg/307000/306990-71-6_b.png)
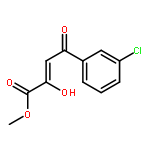













![1H-Pyrazole-3-carboxylic acid,1-[4-(aminosulfonyl)phenyl]-5-(4-methoxyphenyl)-, methyl ester](http://img.cochemist.com/ccimg/170600/170571-24-1.png)
![1H-Pyrazole-3-carboxylic acid,1-[4-(aminosulfonyl)phenyl]-5-(4-methoxyphenyl)-, methyl ester](http://img.cochemist.com/ccimg/170600/170571-24-1_b.png)
![1H-Pyrazole-3-carboxylic acid,1-[4-(aminosulfonyl)phenyl]-5-(4-fluorophenyl)-, methyl ester](http://img.cochemist.com/ccimg/170600/170570-80-6.png)
![1H-Pyrazole-3-carboxylic acid,1-[4-(aminosulfonyl)phenyl]-5-(4-fluorophenyl)-, methyl ester](http://img.cochemist.com/ccimg/170600/170570-80-6_b.png)
![Benzenesulfonamide,4-[5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-](http://img.cochemist.com/ccimg/170600/170569-93-4.png)
![Benzenesulfonamide,4-[5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-](http://img.cochemist.com/ccimg/170600/170569-93-4_b.png)






















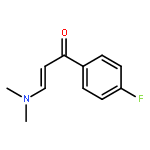









![TETRAZOLO[1,5-A]QUINOLINE-4-CARBALDEHYDE](http://img.cochemist.com/ccimg/121500/121497-03-8.png)
![TETRAZOLO[1,5-A]QUINOLINE-4-CARBALDEHYDE](http://img.cochemist.com/ccimg/121500/121497-03-8_b.png)
![2-Propen-1-one, 3-[4-(4-morpholinyl)phenyl]-1-phenyl-](http://img.cochemist.com/ccimg/120300/120217-00-7.png)
![2-Propen-1-one, 3-[4-(4-morpholinyl)phenyl]-1-phenyl-](http://img.cochemist.com/ccimg/120300/120217-00-7_b.png)
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6.png)
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6_b.png)
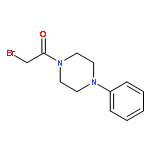





![Benzoic acid, 4-[3-(4-methoxyphenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-53-1.png)
![Benzoic acid, 4-[3-(4-methoxyphenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-53-1_b.png)
![Benzoic acid, 4-[3-(4-methylphenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-52-0.png)
![Benzoic acid, 4-[3-(4-methylphenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-52-0_b.png)
![Benzoic acid, 4-[3-(4-nitrophenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-51-9.png)
![Benzoic acid, 4-[3-(4-nitrophenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-51-9_b.png)
![Benzoic acid, 4-[3-(4-chlorophenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-50-8.png)
![Benzoic acid, 4-[3-(4-chlorophenyl)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/106400/106315-50-8_b.png)
![Phenol, 2-[5-(trifluoromethyl)-1H-benzimidazol-2-yl]-](http://img.cochemist.com/ccimg/104700/104619-92-3.png)
![Phenol, 2-[5-(trifluoromethyl)-1H-benzimidazol-2-yl]-](http://img.cochemist.com/ccimg/104700/104619-92-3_b.png)




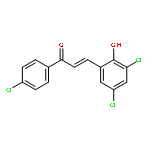

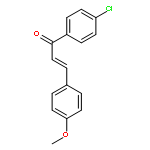
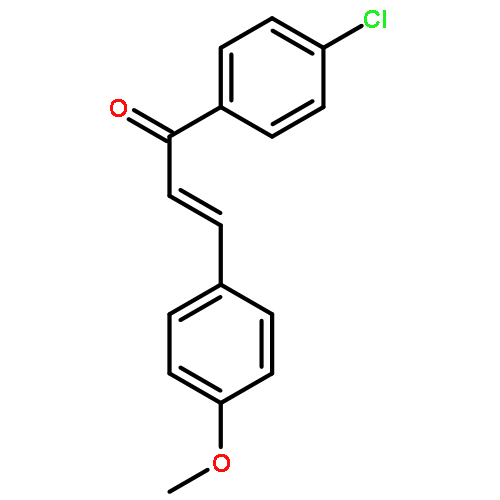
![5-[(2R)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/82900/82855-09-2.png)
![5-[(2R)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/82900/82855-09-2_b.png)
![1H-Imidazole-1-ethanol, 2-[2-(4-bromophenyl)ethenyl]-5-nitro-](http://img.cochemist.com/ccimg/82700/82603-57-4.png)
![1H-Imidazole-1-ethanol, 2-[2-(4-bromophenyl)ethenyl]-5-nitro-](http://img.cochemist.com/ccimg/82700/82603-57-4_b.png)
![1H-IMIDAZOLE-1-ETHANOL, 2-[2-(3-CHLOROPHENYL)ETHENYL]-5-NITRO-](http://img.cochemist.com/ccimg/82700/82603-56-3.png)
![1H-IMIDAZOLE-1-ETHANOL, 2-[2-(3-CHLOROPHENYL)ETHENYL]-5-NITRO-](http://img.cochemist.com/ccimg/82700/82603-56-3_b.png)
![1H-Imidazole-1-ethanol, 5-nitro-2-[2-(3-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/82700/82603-54-1.png)
![1H-Imidazole-1-ethanol, 5-nitro-2-[2-(3-nitrophenyl)ethenyl]-](http://img.cochemist.com/ccimg/82700/82603-54-1_b.png)
![1H-IMIDAZOLE-1-ETHANOL, 2-[2-(4-FLUOROPHENYL)ETHENYL]-5-NITRO-](http://img.cochemist.com/ccimg/82700/82603-42-7.png)
![1H-IMIDAZOLE-1-ETHANOL, 2-[2-(4-FLUOROPHENYL)ETHENYL]-5-NITRO-](http://img.cochemist.com/ccimg/82700/82603-42-7_b.png)
![(2R)-5-hydroxy-2,4-dimethyl-2-[(1E)-2-methylbuta-1,3-dienyl]thiophen-3-one](http://img.cochemist.com/ccimg/82100/82079-32-1.png)
![(2R)-5-hydroxy-2,4-dimethyl-2-[(1E)-2-methylbuta-1,3-dienyl]thiophen-3-one](http://img.cochemist.com/ccimg/82100/82079-32-1_b.png)






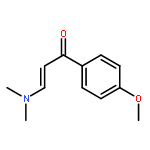


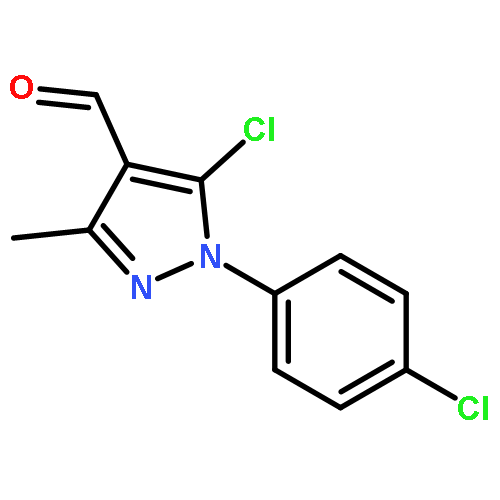
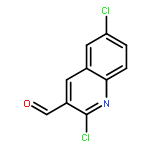




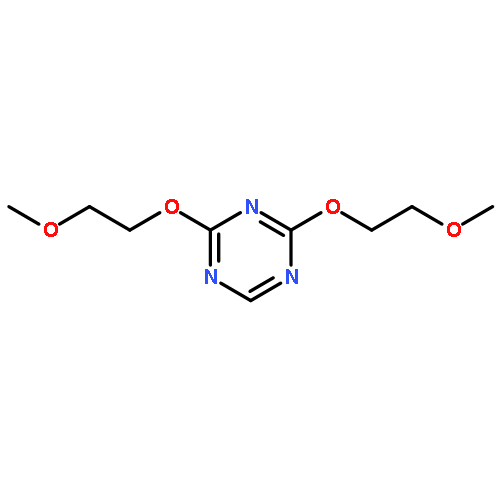
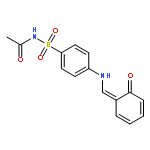





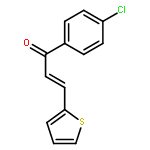



![Ethyl 2-(1H-benzo[d][1,2,3]triazol-1-yl)acetate](http://img.cochemist.com/ccimg/69300/69218-46-8.png)
![Ethyl 2-(1H-benzo[d][1,2,3]triazol-1-yl)acetate](http://img.cochemist.com/ccimg/69300/69218-46-8_b.png)







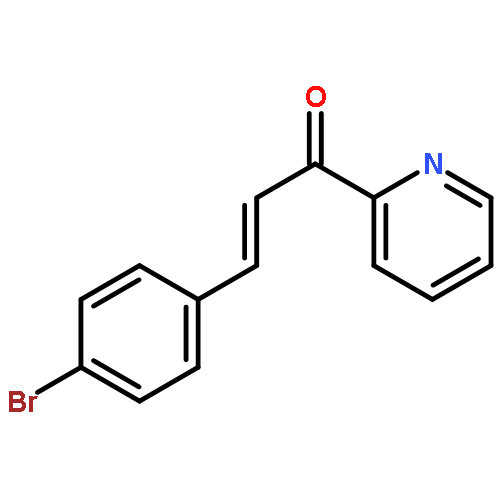
![Benzenesulfonamide,4-[[(2-hydroxyphenyl)methylene]amino]-N-(4-methyl-2-pyrimidinyl)-](http://img.cochemist.com/ccimg/59600/59526-77-1.png)
![Benzenesulfonamide,4-[[(2-hydroxyphenyl)methylene]amino]-N-(4-methyl-2-pyrimidinyl)-](http://img.cochemist.com/ccimg/59600/59526-77-1_b.png)


![Spiro[benzo[1,2-b:5,4-c']dipyran-2(3H),2'(3'H)-naphtho[2,3-b]furan]-7-carboxylicacid, 4,5',8',9-tetrahydro-4,4',9',10-tetrahydroxy-7'-methoxy-5',8',9-trioxo-,methyl ester, (2R,4S)-rel-](http://img.cochemist.com/ccimg/54000/53969-01-0.png)
![Spiro[benzo[1,2-b:5,4-c']dipyran-2(3H),2'(3'H)-naphtho[2,3-b]furan]-7-carboxylicacid, 4,5',8',9-tetrahydro-4,4',9',10-tetrahydroxy-7'-methoxy-5',8',9-trioxo-,methyl ester, (2R,4S)-rel-](http://img.cochemist.com/ccimg/54000/53969-01-0_b.png)






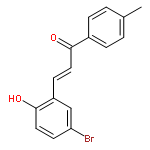
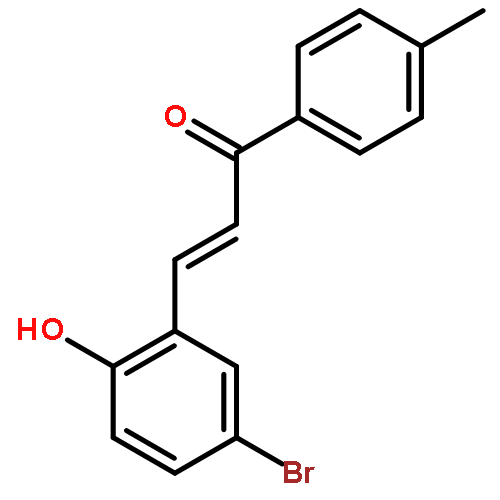




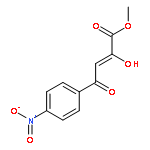
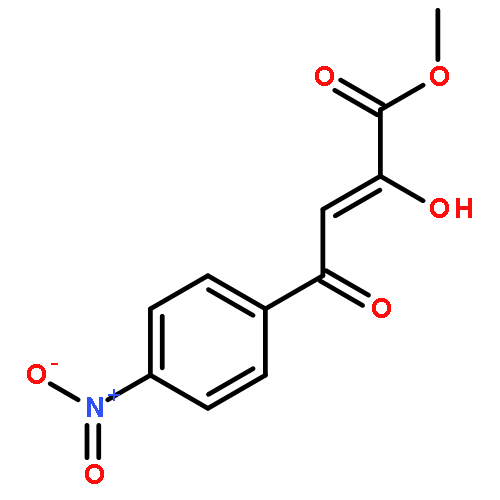






![2,3-DIHYDROBENZO[1,4]DIOXINE-5-CARBONYL CHLORIDE](http://img.cochemist.com/ccimg/38900/38871-41-9.png)
![2,3-DIHYDROBENZO[1,4]DIOXINE-5-CARBONYL CHLORIDE](http://img.cochemist.com/ccimg/38900/38871-41-9_b.png)
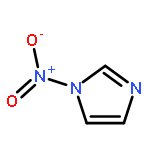
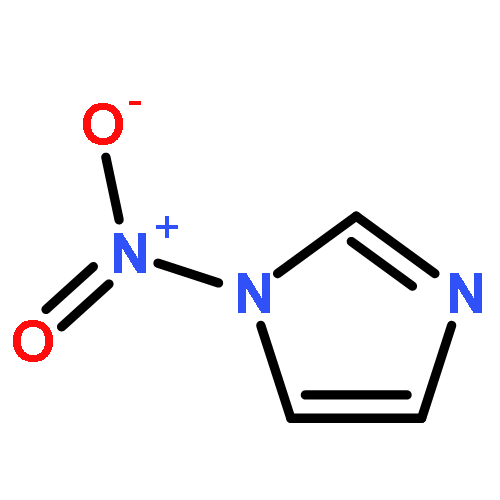







![1H-Imidazole-1-ethanol, 2-[2-(4-methoxyphenyl)ethenyl]-5-nitro-](http://img.cochemist.com/ccimg/30600/30579-51-2.png)
![1H-Imidazole-1-ethanol, 2-[2-(4-methoxyphenyl)ethenyl]-5-nitro-](http://img.cochemist.com/ccimg/30600/30579-51-2_b.png)
![2-{2-[2-(3-methoxyphenyl)ethenyl]-5-nitro-1H-imidazol-1-yl}ethanol](http://img.cochemist.com/ccimg/30600/30579-48-7.png)
![2-{2-[2-(3-methoxyphenyl)ethenyl]-5-nitro-1H-imidazol-1-yl}ethanol](http://img.cochemist.com/ccimg/30600/30579-48-7_b.png)
![1H-Imidazole-1-ethanol, 2-[2-(2-chlorophenyl)ethenyl]-5-nitro-](http://img.cochemist.com/ccimg/30600/30579-26-1.png)
![1H-Imidazole-1-ethanol, 2-[2-(2-chlorophenyl)ethenyl]-5-nitro-](http://img.cochemist.com/ccimg/30600/30579-26-1_b.png)



























![8,11-Ethanofuro[2,3-e]naphtho[2,3-c:6,7-c']dipyran-2,6,13(9H)-trione,3,3a,5,8,11,13b-hexahydro-7,8,12,15-tetrahydroxy-5,9-dimethyl-,(3aS,5S,8S,9R,11R,13bS,15R)-](http://img.cochemist.com/ccimg/19900/19879-06-2.png)
![8,11-Ethanofuro[2,3-e]naphtho[2,3-c:6,7-c']dipyran-2,6,13(9H)-trione,3,3a,5,8,11,13b-hexahydro-7,8,12,15-tetrahydroxy-5,9-dimethyl-,(3aS,5S,8S,9R,11R,13bS,15R)-](http://img.cochemist.com/ccimg/19900/19879-06-2_b.png)






![2-Oxiranecarboxamide,3-[(4E,7E)-1-oxo-4,7-nonadien-1-yl]-, (2R,3S)-](http://img.cochemist.com/ccimg/17400/17397-89-6.png)
![2-Oxiranecarboxamide,3-[(4E,7E)-1-oxo-4,7-nonadien-1-yl]-, (2R,3S)-](http://img.cochemist.com/ccimg/17400/17397-89-6_b.png)


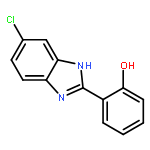
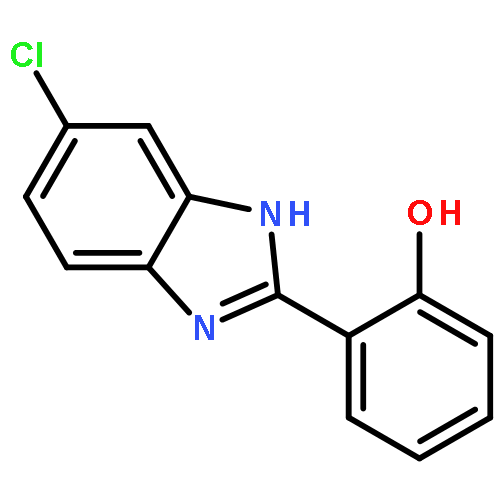








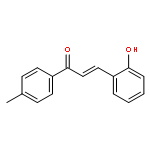

![2,3-Dihydrobenzo[b][1,4]dioxine-6-carbonyl chloride](http://img.cochemist.com/ccimg/6800/6761-70-2.png)
![2,3-Dihydrobenzo[b][1,4]dioxine-6-carbonyl chloride](http://img.cochemist.com/ccimg/6800/6761-70-2_b.png)














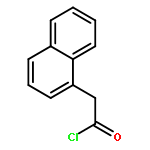



![2-(1H-Benzo[d]imidazol-2-yl)phenol](http://img.cochemist.com/ccimg/3000/2963-66-8.png)
![2-(1H-Benzo[d]imidazol-2-yl)phenol](http://img.cochemist.com/ccimg/3000/2963-66-8_b.png)
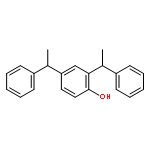





![4-{[(6-oxocyclohexa-2,4-dien-1-ylidene)methyl]amino}benzenesulfonamide](http://img.cochemist.com/ccimg/2100/2067-06-3.png)
![4-{[(6-oxocyclohexa-2,4-dien-1-ylidene)methyl]amino}benzenesulfonamide](http://img.cochemist.com/ccimg/2100/2067-06-3_b.png)
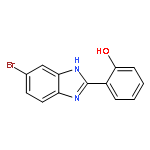





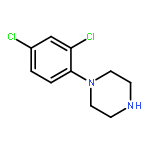
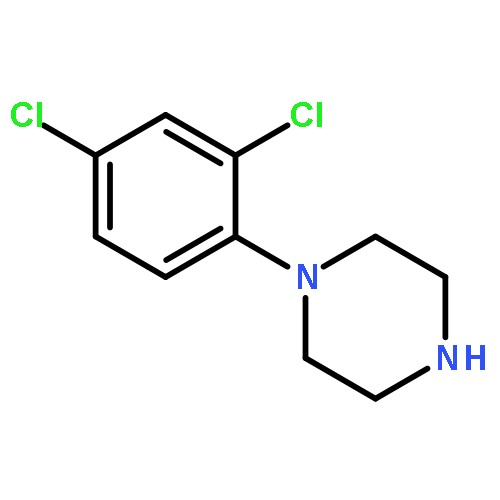
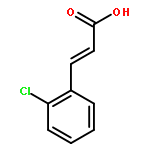
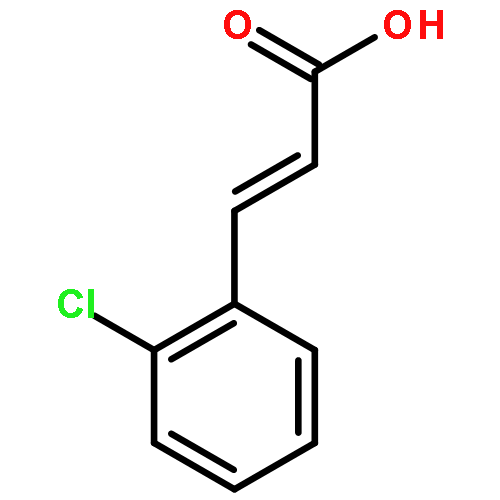
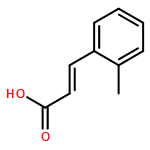
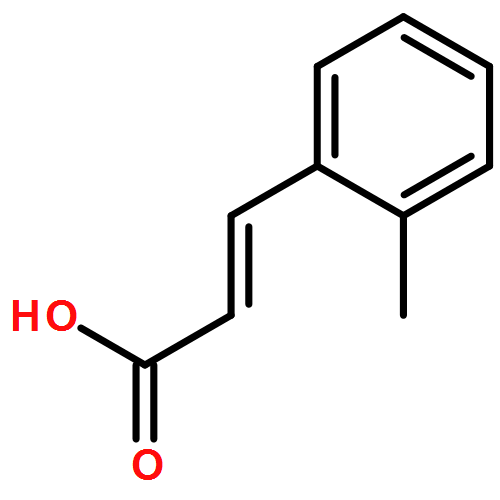
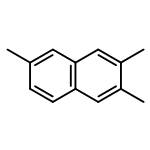
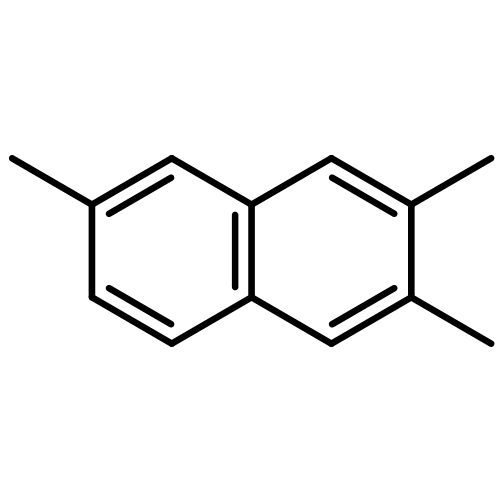


![L-Phenylalanine,N-[[(3R)-5-chloro-3,4-dihydro-8-hydroxy-3-methyl-1-oxo-1H-2-benzopyran-7-yl]carbonyl]-](http://img.cochemist.com/ccimg/400/303-47-9.png)
![L-Phenylalanine,N-[[(3R)-5-chloro-3,4-dihydro-8-hydroxy-3-methyl-1-oxo-1H-2-benzopyran-7-yl]carbonyl]-](http://img.cochemist.com/ccimg/400/303-47-9_b.png)




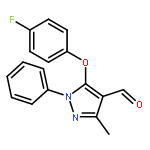

![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1_b.png)










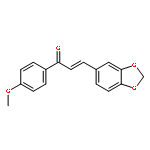



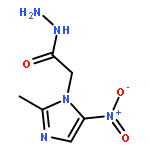


























![N-[1-(4-methylphenyl)ethylideneamino]aniline](http://img.cochemist.com/ccimg/54800/54779-81-6.png)
![N-[1-(4-methylphenyl)ethylideneamino]aniline](http://img.cochemist.com/ccimg/54800/54779-81-6_b.png)












![ETHYL [(2,4-DIMETHYLPHENYL)SULFONYL]ACETATE](http://img.cochemist.com/ccimg/36700/36663-00-0.png)
![ETHYL [(2,4-DIMETHYLPHENYL)SULFONYL]ACETATE](http://img.cochemist.com/ccimg/36700/36663-00-0_b.png)