Co-reporter:Kazuyuki Tokumaru
Chemical Science (2010-Present) 2017 vol. 8(Issue 4) pp:3187-3191
Publication Date(Web):2017/03/28
DOI:10.1039/C7SC00195A
The 1,3,4-oxadiazole is an aromatic heterocycle valued for its low-lipophilicity in drug development. Substituents at the 2- and/or 5-positions can modulate the heterocycle's electronic and hydrogen bond-accepting capability, while exploiting its use as a carbonyl bioisostere. A new approach to 1,3,4-oxadiazoles is described wherein α-bromo nitroalkanes are coupled to acyl hydrazides to deliver the 2,5-disubstituted oxadiazole directly, avoiding a 1,2-diacyl hydrazide intermediate. Access to new building blocks of oxadiazole-substituted secondary amines is improved by leveraging chiral α-bromo nitroalkane or amino acid hydrazide substrates. The non-dehydrative conditions for oxadiazole synthesis are particularly notable, in contrast to alternatives reliant on highly oxophilic reagents to effect cyclization of unsymmetrical 1,2-diacyl hydrazides. The mild conditions are punctuated by the straightforward removal of co-products by a standard aqueous wash.
Co-reporter:Kenneth E. Schwieter and Jeffrey N. Johnston
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14160-14169
Publication Date(Web):October 14, 2016
DOI:10.1021/jacs.6b08663
Peptide synthesis is a truly interdisciplinary tool, familiar to a broad group of scientists who do not otherwise overlap scientifically. For this reason, some may perceive even complex peptide synthesis to be a “solved problem”, while others might argue that immense opportunity remains untapped or simply inaccessible. At the extreme of complexity, what might a concise assessment of the state-of-the-art in peptide synthesis look like? As one of the most practiced forms of synthetic chemistry by chemists and non-chemists alike, what restrictions remain that constrain access to chemical space? Using popular terminology, what forms of peptide synthesis are appropriately termed “on-demand”? The purpose of this Perspective is to appraise synthetic access to complex peptides, particularly those containing unnatural α-amino amides. Several case studies in complex peptide synthesis are summarized here, each selected to characterize the challenges attendant to unnatural α-amino amide synthesis. As peptidic molecules find increasing value in therapeutic development, especially in clinical applications, their impact will ultimately be determined by efficient preparative methods.
Co-reporter:Brandon A. Vara and Jeffrey N. Johnston
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:13794-13797
Publication Date(Web):October 17, 2016
DOI:10.1021/jacs.6b07731
Co-reporter:Kenneth E. Schwieter and Jeffrey N. Johnston
Chemical Communications 2016 vol. 52(Issue 1) pp:152-155
Publication Date(Web):27 Oct 2015
DOI:10.1039/C5CC08415F
It has been over a half-century since Kornblum demonstrated the conversion of a primary nitroalkane to a carboxylic acid; addition of an amine results in carboxylic acid formation as well. We describe the formation of amides from terminal nitroalkanes in a two-step, one-pot reaction involving tandem halogenation/umpolung amide synthesis (UmAS).
Co-reporter:Sergey V. Tsukanov, Martin D. Johnson, Scott A. May, Morgan Rosemeyer, Michael A. Watkins, Stanley P. Kolis, Matthew H. Yates, and Jeffrey N. Johnston
Organic Process Research & Development 2016 Volume 20(Issue 2) pp:215-226
Publication Date(Web):February 1, 2016
DOI:10.1021/acs.oprd.5b00245
A stereoselective aza-Henry reaction between an arylnitromethane and Boc-protected aryl aldimine using a homogeneous Brønsted acid–base catalyst was translated from batch format to an automated intermittent-flow process. This work demonstrates the advantages of a novel intermittent-flow setup with product crystallization and slow reagent addition which is not amenable to the standard continuous equipment: plug flow tube reactor (PFR) or continuous stirred tank reactor (CSTR). A significant benefit of this strategy was the integration of an organocatalytic enantioselective reaction with straightforward product separation, including recycle of the catalyst, resulting in increased intensity of the process by maintaining high catalyst concentration in the reactor. A continuous campaign confirmed that these conditions could effectively provide high throughput of material using an automated system while maintaining high selectivity, thereby addressing nitroalkane safety and minimizing catalyst usage.
Co-reporter:Jeffrey N. Johnston;Suzanne M. Batiste
PNAS 2016 Volume 113 (Issue 52 ) pp:14893-14897
Publication Date(Web):2016-12-27
DOI:10.1073/pnas.1616462114
Macrocyclic small molecules are attractive tools in the development of sensors, new materials, and therapeutics. Within early-stage
drug discovery, they are increasingly sought for their potential to interact with broad surfaces of peptidic receptors rather
than within their narrow folds and pockets. Cyclization of linear small molecule precursors is a straightforward strategy
to constrain conformationally mobile motifs, but forging a macrocycle bond typically becomes more difficult at larger ring
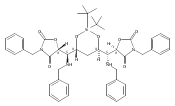
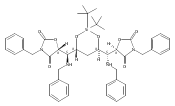
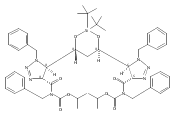
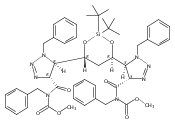
sizes. We report the development of a general approach to discrete collections of oligomeric macrocyclic depsipeptides using
an oligomerization/macrocyclization process governed by a series of Mitsunobu reactions of hydroxy acid monomers. Ring sizes
of 18, 24, 30, and 36 are formed in a single reaction from a didepsipeptide, whereas sizes of 24, 36, and 60 result from a
tetradepsipeptide. The ring-size selectivity inherent to the approach can be modulated by salt additives that enhance the
formation of specific ring sizes. Use of chemical synthesis to prepare the monomers suggests broad access to functionally
and stereochemically diverse collections of natural product-like oligodepsipeptide macrocycles. Two cyclodepsipeptide natural
products were prepared along with numerous unnatural oligomeric congeners to provide rapid access to discrete collections
of complex macrocyclic small molecules from medium (18) to large (60) ring sizes.
Co-reporter:Brandon A. Vara; Thomas J. Struble; Weiwei Wang; Mark C. Dobish
Journal of the American Chemical Society 2015 Volume 137(Issue 23) pp:7302-7305
Publication Date(Web):June 3, 2015
DOI:10.1021/jacs.5b04425
Carbon dioxide exhibits many of the qualities of an ideal reagent: it is nontoxic, plentiful, and inexpensive. Unlike other gaseous reagents, however, it has found limited use in enantioselective synthesis. Moreover, unprecedented is a tool that merges one of the simplest biological approaches to catalysis—Brønsted acid/base activation—with this abundant reagent. We describe a metal-free small molecule catalyst that achieves the three component reaction between a homoallylic alcohol, carbon dioxide, and an electrophilic source of iodine. Cyclic carbonates are formed enantioselectively.
Co-reporter:Kenneth E. Schwieter and Jeffrey N. Johnston
ACS Catalysis 2015 Volume 5(Issue 11) pp:6559
Publication Date(Web):October 5, 2015
DOI:10.1021/acscatal.5b01901
This report details the enantioselective synthesis of β-amino-α-bromo nitroalkanes with β-alkyl substituents, using homogeneous catalysis to prepare either antipode. Use of a bifunctional Brønsted base/acid catalyst allows equal access to either enantiomer of the products, enabling the use of Umpolung Amide Synthesis (UmAS) to prepare the corresponding L- or D-α-amino amide bearing alkyl side chains—overall, in only four steps from aldehyde. The approach also addresses an underlying incompatibility between bromonitromethane and solid hydroxide bases.Keywords: enantioselective catalysis; homogeneous catalysis; organocatalysis; peptides; umpolung amide synthesis
Co-reporter:Kenneth E. Schwieter and Jeffrey N. Johnston
Chemical Science 2015 vol. 6(Issue 4) pp:2590-2595
Publication Date(Web):27 Feb 2015
DOI:10.1039/C5SC00064E
Peptides consisting of D-amino amides are highly represented among both biologically active natural products and non-natural small molecules used in therapeutic development. Chemical synthesis of D-amino amides most often involves approaches based on enzymatic resolution or fractional recrystallization of their diastereomeric amino acid salt precursors, techniques that produce an equal amount of the L-amino acid. Enantioselective synthesis, however, promises selective and general access to a specific α-amino amide, and may enable efficient peptide synthesis regardless of the availability of the corresponding α-amino acid. This report describes the use of a cinchona alkaloid-catalyzed aza-Henry reaction using bromonitromethane, and the integration of its product with umpolung amide synthesis. The result is a straightforward 3-step protocol beginning from aliphatic aldehydes that provides homologated peptides bearing an aliphatic side chain at the resulting D-α-amino amide.
Co-reporter:Michael W. Danneman, Ki Bum Hong, and Jeffrey N. Johnston
Organic Letters 2015 Volume 17(Issue 15) pp:3806-3809
Publication Date(Web):July 17, 2015
DOI:10.1021/acs.orglett.5b01783
An operationally straightforward and metal-free inter-/intramolecular oxidative diamination of vinyl aminopyridines is a common gateway to access all four azaindoline heterocycle families. 3-Amino azaindolines are formed by the reaction of ortho-vinyl N-tosyl anilines with electron-rich amines using phenyliododiaceate (PIDA) and an iodide additive.
Co-reporter:Michael W. Danneman, Ki Bum Hong, and Jeffrey N. Johnston
Organic Letters 2015 Volume 17(Issue 10) pp:2558-2561
Publication Date(Web):May 5, 2015
DOI:10.1021/acs.orglett.5b01177
Doubly intermolecular alkene diamination is achieved with electron-rich, terminal alkenes through the use of a hypervalent iodine (PhI(OAc)2) reagent, iodide, and electron-rich amines. Mono- and disubstituted amines combine with electron-rich alkenes, particularly o-hydroxystyrenes, to achieve the greatest level of generality. This operationally straightforward protocol, unreliant on conventional metal-based activation, is compatible with a broad range of functional groups.
Co-reporter:Daniel J. Sprague, Benjamin M. Nugent, Ryan A. Yoder, Brandon A. Vara, and Jeffrey N. Johnston
Organic Letters 2015 Volume 17(Issue 4) pp:880-883
Publication Date(Web):January 31, 2015
DOI:10.1021/ol503626w
Chiral diamine-derived hydrogen-bond donors were evaluated for their ability to effect stereocontrol in an intramolecular hetero-Diels–Alder (HDA) reaction hypothesized in the biosynthesis of brevianamides A and B. Collectively, these results provide proof of principle that small-molecule hydrogen-bond catalysis, if even based on a hypothetical biosynthesis construct, holds significant potential within enantioselective natural product synthesis.
Co-reporter:Yasunori Toda ; Maren Pink
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14734-14737
Publication Date(Web):October 14, 2014
DOI:10.1021/ja5088584
The first highly diastereo- and enantioselective additions of a halogen and phosphoramidic acid to unactivated alkenes have been developed, catalyzed by a chiral Brønsted acid. A unique feature of these additions is the opportunity for stereocontrol at two noncontiguous chiral centers, carbon and phosphorus, leading to cyclic P-chiral phosphoramidates. In addition to their inherent value, the phosphoramidates are precursors to enantioenriched epoxy allylamines.
Co-reporter:Dawn M. Makley and Jeffrey N. Johnston
Organic Letters 2014 Volume 16(Issue 11) pp:3146-3149
Publication Date(Web):May 14, 2014
DOI:10.1021/ol501297a
We report that N-(trimethylsilyl)imines serve in the Bis(AMidine)-catalyzed addition of bromonitromethane with a high degree of enantioselection. This allows for the production of a range of protected α-bromo nitroalkane donors (including Fmoc) for use in Umpolung Amide Synthesis (UmAS). Hence, peptide homologation with nonnatural aryl glycine amino acids is achieved in three steps from aromatic aldehydes, which are plentiful and inexpensive. Epimerization during the homologation step is circumvented by avoiding an α-amino acid intermediate.
Co-reporter:Kenneth E. Schwieter, Bo Shen, Jessica P. Shackleford, Matthew W. Leighty, and Jeffrey N. Johnston
Organic Letters 2014 Volume 16(Issue 18) pp:4714-4717
Publication Date(Web):September 8, 2014
DOI:10.1021/ol502089v
Umpolung Amide Synthesis (UmAS) provides direct access to amides from an α-bromo nitroalkane and an amine. Based on its mechanistic bifurcation after convergent C–N bond formation, depending on the absence or presence of oxygen, UmAS using substoichiometric N-iodosuccinimide (NIS) under aerobic conditions has been developed. In combination with the enantioselective preparation of α-bromo nitroalkane donors, this protocol realizes the goal of enantioselective α-amino amide and peptide synthesis based solely on catalytic methods.
Co-reporter:Ki Bum Hong and Jeffrey N. Johnston
Organic Letters 2014 Volume 16(Issue 14) pp:3804-3807
Publication Date(Web):July 1, 2014
DOI:10.1021/ol501693j
A combined inter-/intramolecular oxidative diamination of terminal alkenes is described that uses a hypervalent iodine oxidant and a nucleophilic amine to produce 3-aminoindolines at room temperature. This operationally straightforward and metal-free protocol is compatible with a broad range of functional groups. A mechanism involving the conversion of the amine to an electrophilic nitrogen source is advanced and used to identify a protocol effective with substoichiometric amounts of iodide and commercially available phenyl iodobenzene diacetate (PIDA) as the stoichiometric oxidant.
Co-reporter:Brandon A. Vara, Anand Mayasundari, John C. Tellis, Michael W. Danneman, Vanessa Arredondo, Tyler A. Davis, Jaeki Min, Kristin Finch, R. Kiplin Guy, and Jeffrey N. Johnston
The Journal of Organic Chemistry 2014 Volume 79(Issue 15) pp:6913-6938
Publication Date(Web):July 14, 2014
DOI:10.1021/jo501003r
The finding by scientists at Hoffmann-La Roche that cis-imidazolines could disrupt the protein–protein interaction between p53 and MDM2, thereby inducing apoptosis in cancer cells, raised considerable interest in this scaffold over the past decade. Initial routes to these small molecules (i.e., Nutlin-3) provided only the racemic form, with enantiomers being enriched by chromatographic separation using high-pressure liquid chromatography (HPLC) and a chiral stationary phase. Reported here is the first application of an enantioselective aza-Henry approach to nonsymmetric cis-stilbene diamines and cis-imidazolines. Two novel mono(amidine) organocatalysts (MAM) were discovered to provide high levels of enantioselection (>95% ee) across a broad range of substrate combinations. Furthermore, the versatility of the aza-Henry strategy for preparing nonsymmetric cis-imidazolines is illustrated by a comparison of the roles of aryl nitromethane and aryl aldimine in the key step, which revealed unique substrate electronic effects providing direction for aza-Henry substrate–catalyst matching. This method was used to prepare highly substituted cis-4,5-diaryl imidazolines that project unique aromatic rings, and these were evaluated for MDM2-p53 inhibition in a fluorescence polarization assay. The diversification of access to cis-stilbene diamine-derived imidazolines provided by this platform should streamline their further development as chemical tools for disrupting protein–protein interactions.
Co-reporter:Tyler A. Davis, Anna E. Vilgelm, Ann Richmond, and Jeffrey N. Johnston
The Journal of Organic Chemistry 2013 Volume 78(Issue 21) pp:10605-10616
Publication Date(Web):October 15, 2013
DOI:10.1021/jo401321a
Chiral nonracemic cis-4,5-bis(aryl)imidazolines have emerged as a powerful platform for the development of cancer chemotherapeutics, stimulated by the Hoffmann-La Roche discovery that Nutlin-3 can restore apoptosis in cells with wild-type p53. The lack of efficient methods for the enantioselective synthesis of cis-imidazolines, however, has limited their more general use. Our disclosure of the first enantioselective synthesis of (−)-Nutlin-3 provided a basis to prepare larger amounts of this tool used widely in cancer biology. Key to the decagram-scale synthesis described here was the discovery of a novel bis(amidine) organocatalyst that provides high enantioselectivity at warmer reaction temperature (−20 °C) and low catalyst loadings. Further refinements to the procedure led to the synthesis of (−)-Nutlin-3 in a 17 g batch and elimination of all but three chromatographic purifications.
Co-reporter:Julie A. Pigza, Jeong-Seok Han, Aroop Chandra, Daniel Mutnick, Maren Pink, and Jeffrey N. Johnston
The Journal of Organic Chemistry 2013 Volume 78(Issue 3) pp:822-843
Publication Date(Web):December 28, 2012
DOI:10.1021/jo302333s
Serratezomine A is a member of the structurally diverse class of compounds known as the Lycopodium alkaloids. The key supporting studies and successful total synthesis of serratezomine A are described in this account. Significant features of the synthesis include the first application of free radical mediated vinyl amination and Hwu’s oxidative allylation in a total synthesis and an intramolecular lactonization via a transannular SNi reaction. Minimal use of protecting groups and the highly diastereoselective formation of a hindered, quaternary stereocenter using an umpolung allylation are also highlights from a strategy perspective. Observation of quaternary carbon epimerization via a retro-Mannich/Mannich sequence highlights the additional challenge presented by the axial alcohol at C8 in serratezomine A.
Co-reporter:Mark C. Dobish
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6068-6071
Publication Date(Web):March 30, 2012
DOI:10.1021/ja301858r
Highly enantioselective halolactonizations have been developed that employ a chiral proton catalyst–N-iodosuccinimide (NIS) reagent system in which the Brønsted acid is used at catalyst loadings as low as 1 mol %. An approach that modulates the achiral counterion (equimolar to the neutral chiral ligand–proton complex present at low catalyst loadings) to optimize the enantioselection is documented for the first time in this transformation. In this way, unsaturated carboxylic acids are converted to γ-lactones in high yields (up to 98% ee) using commercially available NIS.
Co-reporter:Matthew W. Leighty ; Bo Shen
Journal of the American Chemical Society 2012 Volume 134(Issue 37) pp:15233-15236
Publication Date(Web):September 11, 2012
DOI:10.1021/ja306225u
α-Oxy amides are prepared through enantioselective synthesis using a sequence beginning with a Henry addition of bromonitromethane to aldehydes and finishing with Umpolung Amide Synthesis (UmAS). Key to high enantioselection is the finding that ortho-iodo benzoic acid salts of the chiral copper(II) bis(oxazoline) catalyst deliver both diastereomers of the Henry adduct with high enantiomeric excess, homochiral at the oxygen-bearing carbon. Overall, this approach to α-oxy amides provides an innovative complement to alternatives that focus almost entirely on the enantioselective synthesis of α-oxy carboxylic acids.
Co-reporter:Tyler A. Davis, Michael W. Danneman and Jeffrey N. Johnston
Chemical Communications 2012 vol. 48(Issue 45) pp:5578-5580
Publication Date(Web):30 Apr 2012
DOI:10.1039/C2CC32225K
The first enantioselective synthesis of a potent GlyT1 inhibitor is described. A 3-nitroazetidine donor is used in an enantioselective aza-Henry reaction catalyzed by a bis(amidine)-triflic acid salt organocatalyst, delivering the key intermediate with 92% ee. This adduct is reductively denitrated and converted to the target through a short sequence, thereby allowing assignment of the absolute configuration of the more potent enantiomer.
Co-reporter:Mark C. Dobish, Fernando Villalta, Michael R. Waterman, Galina I. Lepesheva, and Jeffrey N. Johnston
Organic Letters 2012 Volume 14(Issue 24) pp:6322-6325
Publication Date(Web):December 7, 2012
DOI:10.1021/ol303092v
VNI is a potent inhibitor of CYP51 and was recently shown to achieve a parasitological cure of mice infected with T. cruzi in both acute and chronic stages of infection. T. cruzi is the causative parasite of Chagas disease, a neglected tropical disease. The first enantioselective chemical synthesis of VNI (at a materials cost of less than $0.10/mg) is described. Furthermore, the key enantioselective step is performed at the 10 g scale.
Co-reporter:Jessica P. Shackleford;Bo Shen
PNAS 2012 Volume 109 (Issue 1 ) pp:
Publication Date(Web):2012-01-03
DOI:10.1073/pnas.1113553108
The mechanism of umpolung amide synthesis was probed by interrogating potential sources for the oxygen of the product amide
carbonyl that emanates from the α-bromo nitroalkane substrate. Using a series of 18O-labeled substrates and reagents, evidence is gathered to advance two pathways from the putative tetrahedral intermediate.
Under anaerobic conditions, a nitro-nitrite isomerization delivers the amide oxygen from nitro oxygen. The same homolytic
nitro-carbon fragmentation can be diverted by capture of the carbon radical intermediate with oxygen gas (O2) to deliver the amide oxygen from O2. This understanding was used to develop a straightforward protocol for the preparation of 18O-labeled amides in peptides by simply performing the umpolung amide synthesis reaction under an atmosphere of .
Co-reporter:Tyler A. Davis and Jeffrey N. Johnston
Chemical Science 2011 vol. 2(Issue 6) pp:1076-1079
Publication Date(Web):25 Mar 2011
DOI:10.1039/C1SC00061F
The first highly diastereo- and enantioselective additions of aryl nitromethane pronucleophiles to aryl aldimines are described. Identification of an electron rich chiral Bis(AMidine) catalyst for this aza-Henry variant was key to this development, leading ultimately to differentially protected cis-stilbene diamines in two steps. This method then became the lynchpin for an enantioselective synthesis of (−)-Nutlin-3 (Hoffmann-La Roche), a potent cis-imidazoline small molecule inhibitor of p53-MDM2 used extensively as a probe of cell biology and currently in drug development.
Co-reporter:Jayasree M. Srinivasan, Priya A. Mathew, Amie L. Williams, John C. Huffman and Jeffrey N. Johnston
Chemical Communications 2011 vol. 47(Issue 13) pp:3975-3977
Publication Date(Web):24 Feb 2011
DOI:10.1039/C0CC05734G
A concise synthesis of a highly functionalized intermediate lacking only C10 of the mitomycin backbone is described. The key to this development is the Brønsted acid-catalyzed aza-Darzens reaction used to forge the cis-aziridine. Additionally an oxidative ketalization fortuitously occurs during the quinone–enamine coupling step, leading to an orthogonally protected hydroquinone.
Co-reporter:Dr. Jeffrey N. Johnston
Angewandte Chemie International Edition 2011 Volume 50( Issue 13) pp:2890-2891
Publication Date(Web):
DOI:10.1002/anie.201006951
Co-reporter:Aroop Chra ;Dr. Jeffrey N. Johnston
Angewandte Chemie International Edition 2011 Volume 50( Issue 33) pp:7641-7644
Publication Date(Web):
DOI:10.1002/anie.201100957
Co-reporter:Dr. Jeffrey N. Johnston
Angewandte Chemie 2011 Volume 123( Issue 13) pp:2942-2943
Publication Date(Web):
DOI:10.1002/ange.201006951
Co-reporter:Aroop Chra ;Dr. Jeffrey N. Johnston
Angewandte Chemie 2011 Volume 123( Issue 33) pp:7783-7786
Publication Date(Web):
DOI:10.1002/ange.201100957
Co-reporter:Tyler A. Davis ; Jeremy C. Wilt
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:2880-2882
Publication Date(Web):February 12, 2010
DOI:10.1021/ja908814h
The reactivity of a series of symmetrical chiral Brønsted acids (polar ionic hydrogen-bond donors) follows the counterintuitive trend wherein the more Brønsted basic member is a more effective catalyst for the aza-Henry (nitro-Mannich) reaction. This new design element leads to a substantially more reactive catalyst for the aza-Henry reaction, one that can promote the addition of a secondary nitroalkane. Additionally, when an achiral Brønsted acid (TfOH) is used in slight excess of the neutral, chiral bisamidine ligand, diastereoselection can be optimized to levels generally greater than 15:1 while the enantioselection remains unchanged at generally >90% ee.
Co-reporter:Mark C. Dobish and Jeffrey N. Johnston
Organic Letters 2010 Volume 12(Issue 24) pp:5744-5747
Publication Date(Web):November 19, 2010
DOI:10.1021/ol1025712
A Brønsted base-catalyzed reaction of nitroalkanes with alkyl electrophiles provides indole heterocycles substituted at C3 bearing a sec-alkyl group with good enantioselectivity (up to 90% ee). Denitration by hydrogenolysis provides a product with equally high ee. An indolenine intermediate is implicated in the addition step, and surprisingly, water cosolvent was found to have a beneficial effect in this step, leading to a one-pot protocol for elimination/enantioselective addition using PBAM, a bis(amidine) chiral nonracemic base.
Co-reporter:JeffreyN. Johnston ;Hubert Muchalski ;TimothyL. Troyer Dr.
Angewandte Chemie 2010 Volume 122( Issue 13) pp:2340-2349
Publication Date(Web):
DOI:10.1002/ange.200904828
Abstract
In den letzten Jahren wurden neue Aktivierungsmöglichkeiten für Diazoalkane entdeckt und bereits umfangreich angewendet. Mit chiralen und achiralen Brønsted-Säuren als Katalysatoren gelangen Kohlenstoff-Kohlenstoff- und Kohlenstoff-Heteroatom-Verknüpfungen mit einer zunehmenden Zahl von Diazoalkan-Derivaten. Auf diesem Weg lassen sich sehr einfach in struktureller und stereochemischer Hinsicht komplizierte Verbindungen erhalten. Außerdem wird auch die konkurrierende Protonierung der Diazoverbindung umgangen – eine Reaktion, die seit langem zur effizienten Veresterung von Carbonsäuren genutzt wird. Achirale Brønsted-Säuren als Katalysatoren erreichen hohe Diastereoselektivitäten, und chirale Brønsted-Säuren führen zu den gewünschten Produkten mit sowohl hoher Diastereo- als auch Enantioselektivität. Seit einiger Zeit gibt es auch Systeme zur Kohlenstoff-Kohlenstoff-Verknüpfung, die eine Brønsted-Säure mit einer Lewis-Säure oder einer Übergangsmetallverbindung kombinieren.
Co-reporter:JeffreyN. Johnston ;Hubert Muchalski ;TimothyL. Troyer Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 13) pp:2290-2298
Publication Date(Web):
DOI:10.1002/anie.200904828
Abstract
A new means to activate diazoalkanes has been discovered and applied broadly over the past few years. Brønsted acids, both achiral and chiral, have been used to promote the formation of carbon–carbon and carbon–heteroatom bonds with a growing number of diazoalkane derivatives. Aside from their straightforward ability to build structural and stereochemical complexity in innovative new ways, these transformations are remarkable owing to their ability to skirt competitive diazo protonation—a reaction that has long been used to prepare esters efficiently and cleanly from carboxylic acids. In cases where achiral Brønsted acids are used, high diastereoselection can be achieved. Meanwhile, chiral Brønsted acids can deliver products with both high diastereo- and enantioselectivity. More recently, systems have emerged that combine Brønsted acids and either Lewis acids or transition metals to promote carbon–carbon bond formation from diazoalkanes.
Co-reporter:Timothy L. Troyer, Hubert Muchalski and Jeffrey N. Johnston
Chemical Communications 2009 (Issue 41) pp:6195-6197
Publication Date(Web):14 Sep 2009
DOI:10.1039/B913785H
A novel α-diazo imide reagent and its activation by strong Brønsted acid is shown to produce the product of a syn-glycolate Mannich transform with high diastereoselection.
Co-reporter:Jeremy C. Wilt, Maren Pink and Jeffrey N. Johnston
Chemical Communications 2008 (Issue 35) pp:4177-4179
Publication Date(Web):06 Aug 2008
DOI:10.1039/B808393B
Highly diastereo- and enantioselective additions of α-nitrophosphonates to imines catalyzed by a chiral Brønsted acid are described.
Co-reporter:Kenneth E. Schwieter and Jeffrey N. Johnston
Chemical Communications 2016 - vol. 52(Issue 1) pp:NaN155-155
Publication Date(Web):2015/10/27
DOI:10.1039/C5CC08415F
It has been over a half-century since Kornblum demonstrated the conversion of a primary nitroalkane to a carboxylic acid; addition of an amine results in carboxylic acid formation as well. We describe the formation of amides from terminal nitroalkanes in a two-step, one-pot reaction involving tandem halogenation/umpolung amide synthesis (UmAS).
Co-reporter:Jayasree M. Srinivasan, Priya A. Mathew, Amie L. Williams, John C. Huffman and Jeffrey N. Johnston
Chemical Communications 2011 - vol. 47(Issue 13) pp:NaN3977-3977
Publication Date(Web):2011/02/24
DOI:10.1039/C0CC05734G
A concise synthesis of a highly functionalized intermediate lacking only C10 of the mitomycin backbone is described. The key to this development is the Brønsted acid-catalyzed aza-Darzens reaction used to forge the cis-aziridine. Additionally an oxidative ketalization fortuitously occurs during the quinone–enamine coupling step, leading to an orthogonally protected hydroquinone.
Co-reporter:Jeremy C. Wilt, Maren Pink and Jeffrey N. Johnston
Chemical Communications 2008(Issue 35) pp:NaN4179-4179
Publication Date(Web):2008/08/06
DOI:10.1039/B808393B
Highly diastereo- and enantioselective additions of α-nitrophosphonates to imines catalyzed by a chiral Brønsted acid are described.
Co-reporter:Timothy L. Troyer, Hubert Muchalski and Jeffrey N. Johnston
Chemical Communications 2009(Issue 41) pp:NaN6197-6197
Publication Date(Web):2009/09/14
DOI:10.1039/B913785H
A novel α-diazo imide reagent and its activation by strong Brønsted acid is shown to produce the product of a syn-glycolate Mannich transform with high diastereoselection.
Co-reporter:Tyler A. Davis and Jeffrey N. Johnston
Chemical Science (2010-Present) 2011 - vol. 2(Issue 6) pp:NaN1079-1079
Publication Date(Web):2011/03/25
DOI:10.1039/C1SC00061F
The first highly diastereo- and enantioselective additions of aryl nitromethane pronucleophiles to aryl aldimines are described. Identification of an electron rich chiral Bis(AMidine) catalyst for this aza-Henry variant was key to this development, leading ultimately to differentially protected cis-stilbene diamines in two steps. This method then became the lynchpin for an enantioselective synthesis of (−)-Nutlin-3 (Hoffmann-La Roche), a potent cis-imidazoline small molecule inhibitor of p53-MDM2 used extensively as a probe of cell biology and currently in drug development.
Co-reporter:Kenneth E. Schwieter and Jeffrey N. Johnston
Chemical Science (2010-Present) 2015 - vol. 6(Issue 4) pp:NaN2595-2595
Publication Date(Web):2015/02/27
DOI:10.1039/C5SC00064E
Peptides consisting of D-amino amides are highly represented among both biologically active natural products and non-natural small molecules used in therapeutic development. Chemical synthesis of D-amino amides most often involves approaches based on enzymatic resolution or fractional recrystallization of their diastereomeric amino acid salt precursors, techniques that produce an equal amount of the L-amino acid. Enantioselective synthesis, however, promises selective and general access to a specific α-amino amide, and may enable efficient peptide synthesis regardless of the availability of the corresponding α-amino acid. This report describes the use of a cinchona alkaloid-catalyzed aza-Henry reaction using bromonitromethane, and the integration of its product with umpolung amide synthesis. The result is a straightforward 3-step protocol beginning from aliphatic aldehydes that provides homologated peptides bearing an aliphatic side chain at the resulting D-α-amino amide.
Co-reporter:Tyler A. Davis, Michael W. Danneman and Jeffrey N. Johnston
Chemical Communications 2012 - vol. 48(Issue 45) pp:NaN5580-5580
Publication Date(Web):2012/04/30
DOI:10.1039/C2CC32225K
The first enantioselective synthesis of a potent GlyT1 inhibitor is described. A 3-nitroazetidine donor is used in an enantioselective aza-Henry reaction catalyzed by a bis(amidine)-triflic acid salt organocatalyst, delivering the key intermediate with 92% ee. This adduct is reductively denitrated and converted to the target through a short sequence, thereby allowing assignment of the absolute configuration of the more potent enantiomer.
Co-reporter:Kazuyuki Tokumaru and Jeffrey N. Johnston
Chemical Science (2010-Present) 2017 - vol. 8(Issue 4) pp:NaN3191-3191
Publication Date(Web):2017/02/23
DOI:10.1039/C7SC00195A
The 1,3,4-oxadiazole is an aromatic heterocycle valued for its low-lipophilicity in drug development. Substituents at the 2- and/or 5-positions can modulate the heterocycle's electronic and hydrogen bond-accepting capability, while exploiting its use as a carbonyl bioisostere. A new approach to 1,3,4-oxadiazoles is described wherein α-bromo nitroalkanes are coupled to acyl hydrazides to deliver the 2,5-disubstituted oxadiazole directly, avoiding a 1,2-diacyl hydrazide intermediate. Access to new building blocks of oxadiazole-substituted secondary amines is improved by leveraging chiral α-bromo nitroalkane or amino acid hydrazide substrates. The non-dehydrative conditions for oxadiazole synthesis are particularly notable, in contrast to alternatives reliant on highly oxophilic reagents to effect cyclization of unsymmetrical 1,2-diacyl hydrazides. The mild conditions are punctuated by the straightforward removal of co-products by a standard aqueous wash.
.jpg)
![4-((9-Chloro-7-(2-fluoro-6-methoxyphenyl)-5H-benzo[c]pyrimido[4,5-e]azepin-2-yl)amino)-2-methoxybenzoic acid](http://img.cochemist.com/ccimg/1028500/1028486-01-2.png)
![4-((9-Chloro-7-(2-fluoro-6-methoxyphenyl)-5H-benzo[c]pyrimido[4,5-e]azepin-2-yl)amino)-2-methoxybenzoic acid](http://img.cochemist.com/ccimg/1028500/1028486-01-2_b.png)
![BENZENE, [(1S)-1-METHYL-2-NITROETHYL]-](http://img.cochemist.com/ccimg/746700/746647-15-4.png)
![BENZENE, [(1S)-1-METHYL-2-NITROETHYL]-](http://img.cochemist.com/ccimg/746700/746647-15-4_b.png)


![Thiourea, N'-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexyl-](/data/chemimg/108000/454203-57-7.png)
![Thiourea, N'-[3,5-bis(trifluoromethyl)phenyl]-N-cyclohexyl-](/data/chemimg/108000/454203-57-7_b.png)
















![Cyclo[(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl-(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl-(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl-(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl]](http://img.cochemist.com/ccimg/64800/64763-82-2.png)
![Cyclo[(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl-(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl-(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl-(2R)-2-hydroxy-3-methylbutanoyl-N-methyl-L-leucyl]](http://img.cochemist.com/ccimg/64800/64763-82-2_b.png)
![Spiro[5H,6H-5a,9a-(iminomethano)-1H-cyclopent[f]indolizine-7(8H),2'-[2H]indole]-3',5,10(1'H)-trione, 2,3,8a,9-tetrahydro-8,8-dimethyl-,](/data/chemimg/1106200/23402-09-7.png)
![Spiro[5H,6H-5a,9a-(iminomethano)-1H-cyclopent[f]indolizine-7(8H),2'-[2H]indole]-3',5,10(1'H)-trione, 2,3,8a,9-tetrahydro-8,8-dimethyl-,](/data/chemimg/1106200/23402-09-7_b.png)









![N-[(1S)-1-phenylethyl]benzamide](http://img.cochemist.com/ccimg/4200/4108-58-1.png)
![N-[(1S)-1-phenylethyl]benzamide](http://img.cochemist.com/ccimg/4200/4108-58-1_b.png)












![Carbamic acid, [2-methyl-1-[(4-methylphenyl)sulfonyl]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/271700/271600-32-9.png)
![Carbamic acid, [2-methyl-1-[(4-methylphenyl)sulfonyl]propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/271700/271600-32-9_b.png)


































































































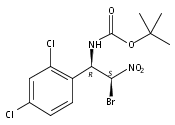





























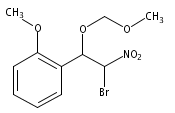













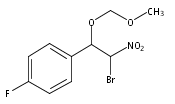







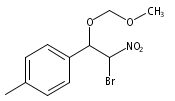
























































































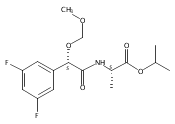




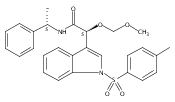



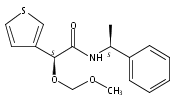




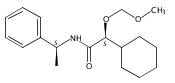





























































































































































































































































![3,4-dihydro-2,3,3-trimethyl-Cyclopent[b]indol-1(2H)-one](http://img.cochemist.com/ccimg/1335100/1335030-82-4.png)
![3,4-dihydro-2,3,3-trimethyl-Cyclopent[b]indol-1(2H)-one](http://img.cochemist.com/ccimg/1335100/1335030-82-4_b.png)



















































































































































































































































































![1H-Indole, 3-[(4-methoxyphenyl)[(4-methylphenyl)sulfonyl]methyl]-2-methyl-](/data/chemimg/197900/1099592-74-1.png)
![1H-Indole, 3-[(4-methoxyphenyl)[(4-methylphenyl)sulfonyl]methyl]-2-methyl-](/data/chemimg/197900/1099592-74-1_b.png)
![1H-Indole, 3-[(4-bromophenyl)[(4-methylphenyl)sulfonyl]methyl]-2-methyl-](/data/chemimg/197900/1099592-73-0.png)
![1H-Indole, 3-[(4-bromophenyl)[(4-methylphenyl)sulfonyl]methyl]-2-methyl-](/data/chemimg/197900/1099592-73-0_b.png)





















![1H-Indole, 2-methyl-3-[[(4-methylphenyl)sulfonyl]phenylmethyl]-](/data/chemimg/119600/911303-07-6.png)
![1H-Indole, 2-methyl-3-[[(4-methylphenyl)sulfonyl]phenylmethyl]-](/data/chemimg/119600/911303-07-6_b.png)
![1H-Indole, 3-[[(4-methylphenyl)sulfonyl]phenylmethyl]-](/data/chemimg/119600/911303-02-1.png)
![1H-Indole, 3-[[(4-methylphenyl)sulfonyl]phenylmethyl]-](/data/chemimg/119600/911303-02-1_b.png)




![1H-Indole, 2-methyl-3-[(1S)-2-nitro-1-phenylethyl]-](/data/chemimg/119500/871731-06-5.png)
![1H-Indole, 2-methyl-3-[(1S)-2-nitro-1-phenylethyl]-](/data/chemimg/119500/871731-06-5_b.png)















![Carbamic acid,N-[(4-methylphenyl)methylene]-, 1,1-dimethylethyl ester, [N(E)]-](http://img.cochemist.com/ccimg/743500/743430-45-7.png)
![Carbamic acid,N-[(4-methylphenyl)methylene]-, 1,1-dimethylethyl ester, [N(E)]-](http://img.cochemist.com/ccimg/743500/743430-45-7_b.png)































![(7S)-7-Amino-5,7-dihydro-5-methyl-6H-dibenz[b,d]azepin-6-one](http://img.cochemist.com/ccimg/365300/365242-16-6.png)
![(7S)-7-Amino-5,7-dihydro-5-methyl-6H-dibenz[b,d]azepin-6-one](http://img.cochemist.com/ccimg/365300/365242-16-6_b.png)








![L-Tryptophan,1-[(1,1-dimethylethoxy)carbonyl]-, 1,1-dimethylethyl ester, monohydrochloride(9CI)](http://img.cochemist.com/ccimg/219000/218938-67-1.png)
![L-Tryptophan,1-[(1,1-dimethylethoxy)carbonyl]-, 1,1-dimethylethyl ester, monohydrochloride(9CI)](http://img.cochemist.com/ccimg/219000/218938-67-1_b.png)

















![Acetic acid, [[(1R)-1-phenylethyl]imino]-, methyl ester](http://img.cochemist.com/ccimg/153900/153831-92-6.png)
![Acetic acid, [[(1R)-1-phenylethyl]imino]-, methyl ester](http://img.cochemist.com/ccimg/153900/153831-92-6_b.png)




































![[BROMO(NITRO)METHYL]BENZENE](/data/chemimg/62600/42157-97-1.png)
![[BROMO(NITRO)METHYL]BENZENE](/data/chemimg/62600/42157-97-1_b.png)
























![Methyl 2-[(2-phenylacetyl)amino]acetate](http://img.cochemist.com/ccimg/5300/5259-87-0.png)
![Methyl 2-[(2-phenylacetyl)amino]acetate](http://img.cochemist.com/ccimg/5300/5259-87-0_b.png)