Co-reporter:Yuqin Cai, Shuang Ding, Nicholas E. Geacintov, and Suse Broyde
Chemical Research in Toxicology 2011 Volume 24(Issue 4) pp:522
Publication Date(Web):February 28, 2011
DOI:10.1021/tx1004002
Among the polycyclic aromatic hydrocarbon class of chemical carcinogens, dibenzo[a,l]pyrene (DB[a,l]P) is the most potent tumorigen that has been identified to date. Structurally, it is bulky with six aromatic rings, and it contains the nonplanar fjord-region. The conformational properties of DB[a,l]P-derived DNA adducts responsible for its extraordinary carcinogenicity are hence of great interest. We have carried out molecular modeling and MD simulations for the 14R (+)- and 14S (−)-trans-anti-DB[a,l]P-N6-dA adducts derived from the reactions of the DB[a,l]P diol epoxides with adenine in double-stranded DNA. The structures are based on the classically intercalated NMR solution structures of the analogous fjord-region benzo[c]phenanthrene-derived-N6-dA adducts. One objective was to gain insight on the impact of the more bulky DB[a,l]P ring system on the structural characteristics of the intercalative adduct conformations. A further objective was to elucidate the effect of the flexible twist associated with the sterically hindered aromatic ring in the fjord-region on the intercalated conformations, for comparison with the intercalated but planar bay-region benzo[a]pyrene-derived-N6-dA adducts. For the DB[a,l]P-N6-dA adducts, our results show that the 14R (+)-adduct is more favorably intercalated on the 5′-side of the modified adenine than the stereoisomeric 14S (−)-adduct, intercalated on its 3′-side. The 14R (+)-adduct manifests better van der Waals stacking interactions with flanking base pairs, less perturbed Watson−Crick hydrogen bonding, less local groove enlargement, less unwinding, and a lower solvent exposure than the 14S (−)-adduct. These structural findings are consistent with observed thermodynamic melting data, UV absorption properties, and fluorescence quenching studies. By contrast, the NMR solution structures for the analogous but less bulky B[c]Ph-derived adducts reveal no such stereoisomeric effect, while the planar bay-region benzo[a]pyrene-derived-N6-dA adducts do. Differences in nucleotide excision repair susceptibilities of the fjord and bay region adducts stem from distinctions in their intercalative conformations, produced by the intrinsic topological variations in their polycyclic aromatic ring systems.
Co-reporter:Han Zheng, Yuqin Cai, Shuang Ding, Yijin Tang, Konstantin Kropachev, Yanzi Zhou, Lihua Wang, Shenglong Wang, Nicholas E. Geacintov, Yingkai Zhang, and Suse Broyde
Chemical Research in Toxicology 2010 Volume 23(Issue 12) pp:1868
Publication Date(Web):November 23, 2010
DOI:10.1021/tx1003613
Lesion-induced thermodynamic destabilization is believed to facilitate β-hairpin intrusion by the human XPC/hHR23B nucleotide excision repair (NER) recognition factor, accompanied by partner-base flipping, as suggested by the crystal structure of the yeast orthologue (Min, J. H. and Pavletich, N. P. (2007) Nature449, 570−575). To investigate this proposed mechanism, we employed the umbrella sampling method to compute partner base flipping free energies for the repair susceptible 14R (+)-trans-anti-DB[a,l]P-N2-dG modified duplex 11-mer, derived from the fjord region polycyclic aromatic hydrocarbon dibenzo[a,l]pyrene, and for the undamaged duplex. Our flipping free energy profiles show that the adduct has a lower flipping barrier by ∼7.7 kcal/mol, consistent with its thermally destabilizing impact on the damaged DNA duplex and its susceptibility to NER.
Co-reporter:Jian Liang ; Jing Zhang ; Lei Zhu ; Alexander Duarandin ; Victor G. Young; ; Jr.; Nicholas Geacintov ;James W. Canary
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11196-11208
Publication Date(Web):October 30, 2009
DOI:10.1021/ic901662z
Metal complexes of tris((6-phenyl-2-pyridyl)methyl)amine (2) have hydrophobic cavities that potentially accommodate small molecules. However, the utility of this attractive motif has been hampered by the poor solubility of such complexes in many common solvents. In this study, two tripodal ligands (3, tris-[6-(3,4,5-trimethoxy-phenyl)-pyridin-2-ylmethyl]-amine, and 4, tris((6-(3,4,5-tris(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)phenyl)pyridin-2-yl)methyl)amine) derived from 2 were prepared with enhanced solubility in organic and aqueous solvents. The X-ray crystallographic analyses of selected ligands and complexes revealed that the hydrophobic cavities inside the zinc complexes were retained after derivatization. Fluorescence, nuclear magnetic resonance (NMR), and potentiometric titration studies, which were enabled by the improved solubility, were performed to investigate the binding properties of the soluble ligands (3 and 4) with metal ions such as Zn2+ and Cu2+. When saturating quantities of Zn2+ ions are added to ligand 3 in acetonitrile, the fluorescence emission maximum exhibits a pronounced red shift of ∼80 nm (from 376 to 457 nm) and is enhanced by a factor of >100 when measured at 520 nm. The fluorescence properties of the Zn2+ ion-coordinated ligands in the Zn(3) complex are consistent with a charge-transfer character in the excited state, with possible contributions from a planarization of the pyridyl-trimethoxyphenyl groups in the excited state, and from excitonic interactions.
.jpg)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-](/data/chemimg/124700/426265-32-9.png)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-](/data/chemimg/124700/426265-32-9_b.png)



![2-Decenyl, 10-carboxy-1-[(1Z)-1-heptenyl]-, (2Z)-](http://img.cochemist.com/ccimg/851600/851536-93-1.png)
![2-Decenyl, 10-carboxy-1-[(1Z)-1-heptenyl]-, (2Z)-](http://img.cochemist.com/ccimg/851600/851536-93-1_b.png)




![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-](/data/chemimg/124700/334830-01-2.png)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-](/data/chemimg/124700/334830-01-2_b.png)








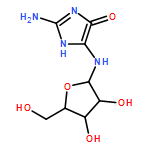
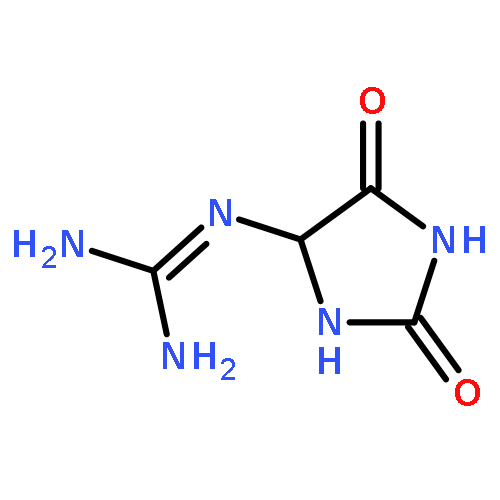
![Guanidine, [3-(2-deoxy-β-D-erythro-pentofuranosyl)-2,5-dioxo-4-imidazolidinyl]-](/data/chemimg/119300/265986-31-0.png)
![Guanidine, [3-(2-deoxy-β-D-erythro-pentofuranosyl)-2,5-dioxo-4-imidazolidinyl]-](/data/chemimg/119300/265986-31-0_b.png)




![(11R,12S,12aS,13aR)-11,12,12a,13a-tetrahydronaphtho[4',3',2',1':1,12]tetrapheno[10,11-b]oxirene-11,12-diol](http://img.cochemist.com/ccimg/154000/153926-04-6.png)
![(11R,12S,12aS,13aR)-11,12,12a,13a-tetrahydronaphtho[4',3',2',1':1,12]tetrapheno[10,11-b]oxirene-11,12-diol](http://img.cochemist.com/ccimg/154000/153926-04-6_b.png)






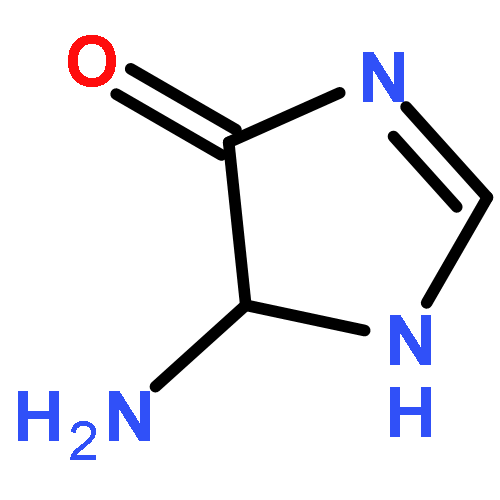
![4H-Imidazol-4-one, 2-amino-5-[(2-deoxy-β-D-erythro-pentofuranosyl)amino]-](/data/chemimg/124700/142559-87-3.png)
![4H-Imidazol-4-one, 2-amino-5-[(2-deoxy-β-D-erythro-pentofuranosyl)amino]-](/data/chemimg/124700/142559-87-3_b.png)






![Chryseno[3,4-b]oxirene-1,2-diol,1,2,2a,3a-tetrahydro-5-methyl-, (1S,2R,2aR,3aS)-](http://img.cochemist.com/ccimg/112000/111901-45-2.png)
![Chryseno[3,4-b]oxirene-1,2-diol,1,2,2a,3a-tetrahydro-5-methyl-, (1S,2R,2aR,3aS)-](http://img.cochemist.com/ccimg/112000/111901-45-2_b.png)
![Benzo[5,6]phenanthro[3,4-b]oxirene-2,3-diol,1a,2,3,11d-tetrahydro- (9CI)](http://img.cochemist.com/ccimg/111100/111001-48-0.png)
![Benzo[5,6]phenanthro[3,4-b]oxirene-2,3-diol,1a,2,3,11d-tetrahydro- (9CI)](http://img.cochemist.com/ccimg/111100/111001-48-0_b.png)
![1-Methyl-6-phenyl-1H-imidazo[4,5-b]pyridin-2-amine](http://img.cochemist.com/ccimg/105700/105650-23-5.png)
![1-Methyl-6-phenyl-1H-imidazo[4,5-b]pyridin-2-amine](http://img.cochemist.com/ccimg/105700/105650-23-5_b.png)


![Dibenzo[def,p]chrysene-11,12-diol,11,12-dihydro-](http://img.cochemist.com/ccimg/88200/88191-01-9.png)
![Dibenzo[def,p]chrysene-11,12-diol,11,12-dihydro-](http://img.cochemist.com/ccimg/88200/88191-01-9_b.png)

![2'-deoxy-N-[(7R,8S,9S,10R)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]guanosine](http://img.cochemist.com/ccimg/79200/79172-11-5.png)
![2'-deoxy-N-[(7R,8S,9S,10R)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]guanosine](http://img.cochemist.com/ccimg/79200/79172-11-5_b.png)

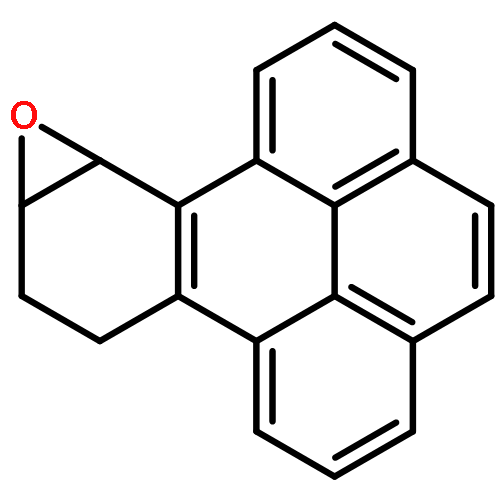
![(7R,8S,8aR,9aS)-7,8,8a,9a-tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/63400/63357-09-5.png)
![(7R,8S,8aR,9aS)-7,8,8a,9a-tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/63400/63357-09-5_b.png)
![(7S,8R,8aS,9aR)-7,8,8a,9a-tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/63400/63323-29-5.png)
![(7S,8R,8aS,9aR)-7,8,8a,9a-tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/63400/63323-29-5_b.png)
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8.png)
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8_b.png)
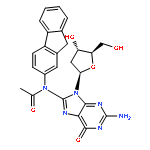
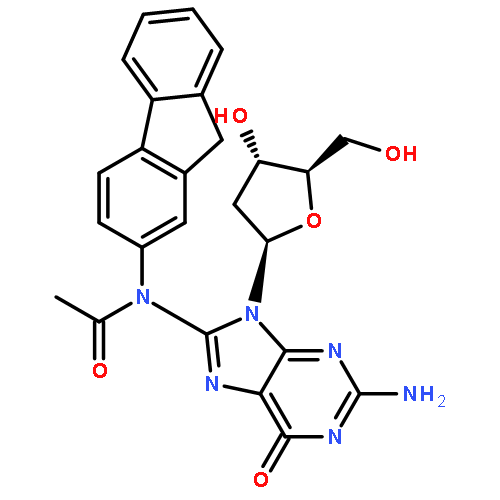
![N-[2-(acetylamino)-9H-fluoren-3-yl]-2'-deoxyguanosine](http://img.cochemist.com/ccimg/61400/61370-84-1.png)
![N-[2-(acetylamino)-9H-fluoren-3-yl]-2'-deoxyguanosine](http://img.cochemist.com/ccimg/61400/61370-84-1_b.png)















![11-Methylbenz[a]anthracene](http://img.cochemist.com/ccimg/6200/6111-78-0.png)
![11-Methylbenz[a]anthracene](http://img.cochemist.com/ccimg/6200/6111-78-0_b.png)
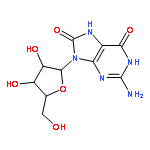
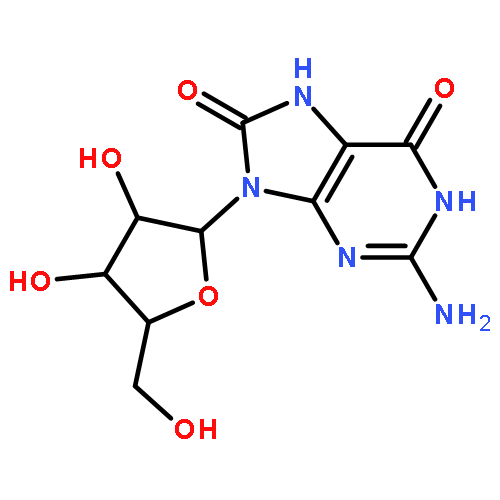
![N-[(4S)-2,5-dioxo-4-imidazolidinyl]-Urea](http://img.cochemist.com/ccimg/3900/3844-67-5.png)
![N-[(4S)-2,5-dioxo-4-imidazolidinyl]-Urea](http://img.cochemist.com/ccimg/3900/3844-67-5_b.png)




![6-({[5-(DIMETHYLAMINO)-1-NAPHTHYL]SULFONYL}AMINO)HEXANOIC ACID](http://img.cochemist.com/ccimg/3400/3353-33-1.png)
![6-({[5-(DIMETHYLAMINO)-1-NAPHTHYL]SULFONYL}AMINO)HEXANOIC ACID](http://img.cochemist.com/ccimg/3400/3353-33-1_b.png)


![Benz[a]anthracene, 12-methyl-](http://img.cochemist.com/ccimg/2500/2422-79-9.png)
![Benz[a]anthracene, 12-methyl-](http://img.cochemist.com/ccimg/2500/2422-79-9_b.png)
![Benz[a]anthracene, 8-methyl-](http://img.cochemist.com/ccimg/2400/2381-31-9.png)
![Benz[a]anthracene, 8-methyl-](http://img.cochemist.com/ccimg/2400/2381-31-9_b.png)





![Acetic acid,2-[(aminocarbonyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/600/585-05-7.png)
![Acetic acid,2-[(aminocarbonyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/600/585-05-7_b.png)






![Benzo[rst]pentaphene](http://img.cochemist.com/ccimg/200/189-55-9.png)
![Benzo[rst]pentaphene](http://img.cochemist.com/ccimg/200/189-55-9_b.png)






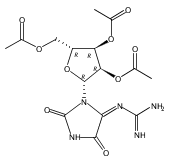

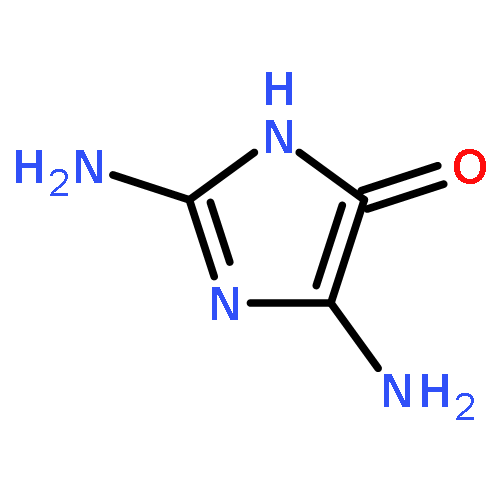

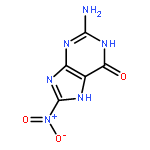
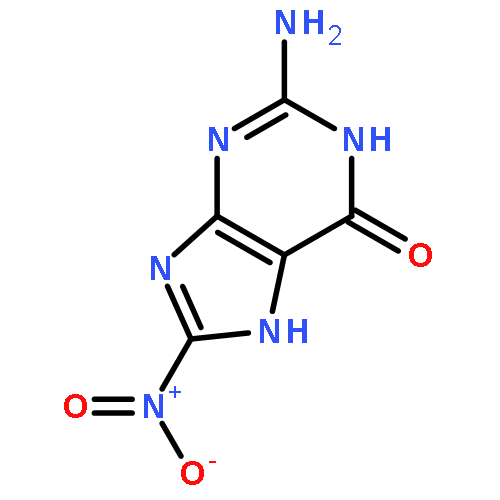
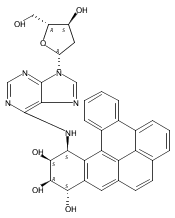
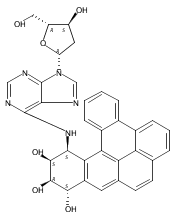
![7,10-Epoxy-6H-azepino[1,2-e]purine-6,8-diol,4-amino-7,8,9,10-tetrahydro-, (6S,7S,8S,10R)-](http://img.cochemist.com/ccimg/117200/117182-88-4.png)
![7,10-Epoxy-6H-azepino[1,2-e]purine-6,8-diol,4-amino-7,8,9,10-tetrahydro-, (6S,7S,8S,10R)-](http://img.cochemist.com/ccimg/117200/117182-88-4_b.png)
![Chryseno[3,4-b]oxirene-1,2-diol,1,2,2a,3a-tetrahydro-5-methyl-, (1R,2S,2aS,3aR)-](http://img.cochemist.com/ccimg/112000/111901-44-1.png)
![Chryseno[3,4-b]oxirene-1,2-diol,1,2,2a,3a-tetrahydro-5-methyl-, (1R,2S,2aS,3aR)-](http://img.cochemist.com/ccimg/112000/111901-44-1_b.png)
![Chryseno[3,4-b]oxirene-1,2-diol,1,2,2a,3a-tetrahydro-4-methyl-, (1S,2R,2aR,3aS)-](http://img.cochemist.com/ccimg/112000/111901-40-7.png)
![Chryseno[3,4-b]oxirene-1,2-diol,1,2,2a,3a-tetrahydro-4-methyl-, (1S,2R,2aR,3aS)-](http://img.cochemist.com/ccimg/112000/111901-40-7_b.png)
![7,10-Epoxy-4H-azepino[1,2-e]purin-4-one, 2-amino-1,6,7,8,9,10-hexahydro-6,8-dihydroxy-, (6S,7S,8S,10R)-](http://img.cochemist.com/ccimg/106000/105929-27-9.png)
![7,10-Epoxy-4H-azepino[1,2-e]purin-4-one, 2-amino-1,6,7,8,9,10-hexahydro-6,8-dihydroxy-, (6S,7S,8S,10R)-](http://img.cochemist.com/ccimg/106000/105929-27-9_b.png)
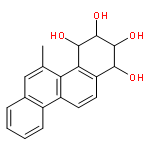



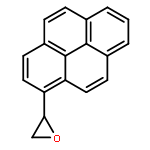
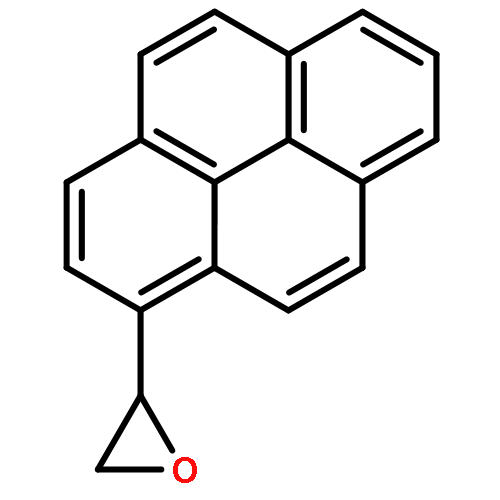
![Guanosine,N-[(7S,8R,9S,10R)-7,8,9,10-tetrahydro-7,8,9-trihydroxybenzo[a]pyren-10-yl]-](http://img.cochemist.com/ccimg/60700/60653-69-2.png)
![Guanosine,N-[(7S,8R,9S,10R)-7,8,9,10-tetrahydro-7,8,9-trihydroxybenzo[a]pyren-10-yl]-](http://img.cochemist.com/ccimg/60700/60653-69-2_b.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6_b.png)










![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-, (5S)-](http://img.cochemist.com/ccimg/852400/852314-86-4.png)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-, (5S)-](http://img.cochemist.com/ccimg/852400/852314-86-4_b.png)
![1,3,6,8-TETRAAZASPIRO[4.4]NON-6-ENE-2,4,9-TRIONE, 7-AMINO-, (5R)-](http://img.cochemist.com/ccimg/852400/852314-85-3.png)
![1,3,6,8-TETRAAZASPIRO[4.4]NON-6-ENE-2,4,9-TRIONE, 7-AMINO-, (5R)-](http://img.cochemist.com/ccimg/852400/852314-85-3_b.png)
![2-AMINO-1,3,6,8-TETRAZASPIRO[4.4]NON-1-ENE-4,7,9-TRIONE](http://img.cochemist.com/ccimg/481000/480996-13-2.png)
![2-AMINO-1,3,6,8-TETRAZASPIRO[4.4]NON-1-ENE-4,7,9-TRIONE](http://img.cochemist.com/ccimg/481000/480996-13-2_b.png)

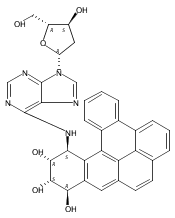


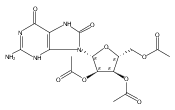

![(7S,8R,8aR)-10-Methyl-7,8,8a,9a-tetrahydrochryseno[3,4-b]oxirene- 7,8-diol](http://img.cochemist.com/ccimg/81900/81851-68-5.png)
![(7S,8R,8aR)-10-Methyl-7,8,8a,9a-tetrahydrochryseno[3,4-b]oxirene- 7,8-diol](http://img.cochemist.com/ccimg/81900/81851-68-5_b.png)
![Guanosine,N-(7,8,9,10-tetrahydro-7,8,9-trihydroxybenzo[a]pyren-10-yl)-, (7R,8S,9R,10S)-](http://img.cochemist.com/ccimg/60900/60872-70-0.png)
![Guanosine,N-(7,8,9,10-tetrahydro-7,8,9-trihydroxybenzo[a]pyren-10-yl)-, (7R,8S,9R,10S)-](http://img.cochemist.com/ccimg/60900/60872-70-0_b.png)


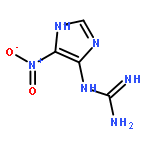
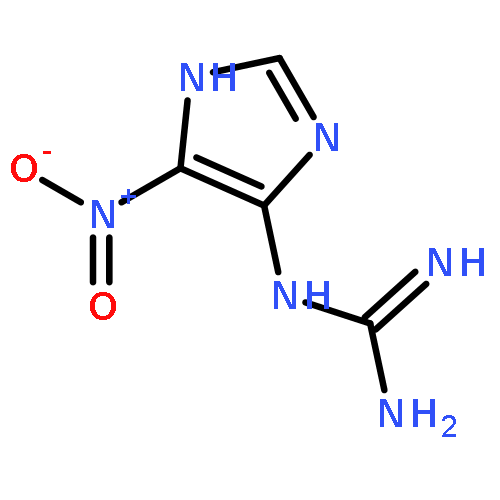
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-, (5S)-](/data/chemimg/121800/365568-86-1.png)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-, (5S)-](/data/chemimg/121800/365568-86-1_b.png)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-, (5R)-](/data/chemimg/121800/321742-81-8.png)
![1,3,6,8-Tetraazaspiro[4.4]non-6-ene-2,4,9-trione, 7-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-, (5R)-](/data/chemimg/121800/321742-81-8_b.png)
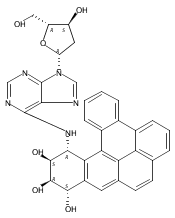
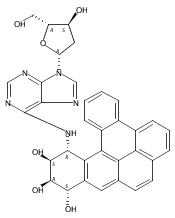
![Adenosine,2'-deoxy-N-[(1R,2S,3R,4S)-1,2,3,4-tetrahydro-2,3,4-trihydroxybenzo[c]phenanthren-1-yl]-(9CI)](http://img.cochemist.com/ccimg/107100/107033-10-3.png)
![Adenosine,2'-deoxy-N-[(1R,2S,3R,4S)-1,2,3,4-tetrahydro-2,3,4-trihydroxybenzo[c]phenanthren-1-yl]-(9CI)](http://img.cochemist.com/ccimg/107100/107033-10-3_b.png)






![(1aR,2R,3S,11dS)-1a,2,3,11d-tetrahydrobenzo[5,6]phenanthro[3,4-b]oxirene-2,3-diol](http://img.cochemist.com/ccimg/82600/82510-57-4.png)
![(1aR,2R,3S,11dS)-1a,2,3,11d-tetrahydrobenzo[5,6]phenanthro[3,4-b]oxirene-2,3-diol](http://img.cochemist.com/ccimg/82600/82510-57-4_b.png)
![Benzo[10,11]chryseno[3,4-b]oxirene-7,8-diol,7,8,8a,9a-tetrahydro-, (7R,8S,8aR,9aS)-rel-](http://img.cochemist.com/ccimg/59000/58917-91-2.png)
![Benzo[10,11]chryseno[3,4-b]oxirene-7,8-diol,7,8,8a,9a-tetrahydro-, (7R,8S,8aR,9aS)-rel-](http://img.cochemist.com/ccimg/59000/58917-91-2_b.png)



![Benzo[a]pyrene-7,8,9,10-tetrol,7,8,9,10-tetrahydro-](http://img.cochemist.com/ccimg/60000/59957-91-4.png)
![Benzo[a]pyrene-7,8,9,10-tetrol,7,8,9,10-tetrahydro-](http://img.cochemist.com/ccimg/60000/59957-91-4_b.png)





![2'-deoxy-N-[(7R,8S,9R,10S)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]adenosine](http://img.cochemist.com/ccimg/72800/72777-36-7.png)
![2'-deoxy-N-[(7R,8S,9R,10S)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]adenosine](http://img.cochemist.com/ccimg/72800/72777-36-7_b.png)


![Benzo[a]pyrene-7,8,9,10-tetrol,7,8,9,10-tetrahydro-, (7R,8S,9R,10R)-rel-](http://img.cochemist.com/ccimg/61500/61490-68-4.png)
![Benzo[a]pyrene-7,8,9,10-tetrol,7,8,9,10-tetrahydro-, (7R,8S,9R,10R)-rel-](http://img.cochemist.com/ccimg/61500/61490-68-4_b.png)

![Dibenzo[def,p]chrysene](/data/chemimg/1053400/191-30-0.png)
![Dibenzo[def,p]chrysene](/data/chemimg/1053400/191-30-0_b.png)

![7,8,8a,9a-Tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/55100/55097-80-8.png)
![7,8,8a,9a-Tetrahydrobenzo[1,12]tetrapheno[10,11-b]oxirene-7,8-diol](http://img.cochemist.com/ccimg/55100/55097-80-8_b.png)
![2'-deoxy-N-[(7S,8R,9S,10R)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]guanosine](http://img.cochemist.com/ccimg/85100/85026-87-5.png)
![2'-deoxy-N-[(7S,8R,9S,10R)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]guanosine](http://img.cochemist.com/ccimg/85100/85026-87-5_b.png)

![Benzo[10,11]chryseno[3,4-b]oxirene-7,8-diol,7,8,8a,9a-tetrahydro-, (7R,8S,8aS,9aR)-rel-](http://img.cochemist.com/ccimg/59000/58917-67-2.png)
![Benzo[10,11]chryseno[3,4-b]oxirene-7,8-diol,7,8,8a,9a-tetrahydro-, (7R,8S,8aS,9aR)-rel-](http://img.cochemist.com/ccimg/59000/58917-67-2_b.png)
![2'-deoxy-N-[(7R,8S,9R,10S)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]guanosine](http://img.cochemist.com/ccimg/65500/65437-20-9.png)
![2'-deoxy-N-[(7R,8S,9R,10S)-7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[pqr]tetraphen-10-yl]guanosine](http://img.cochemist.com/ccimg/65500/65437-20-9_b.png)