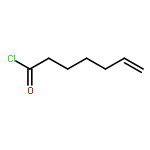
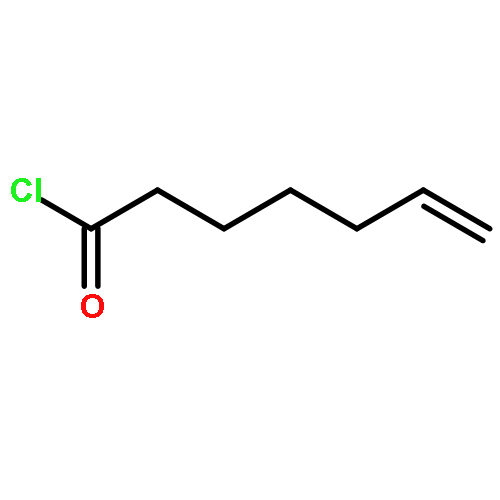
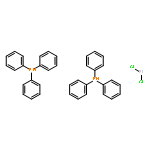
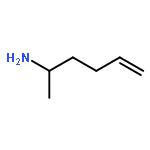
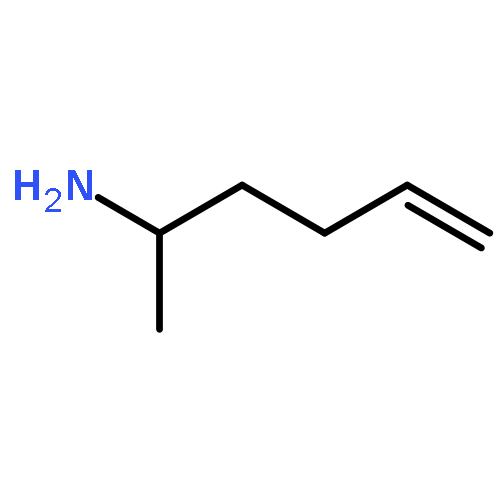
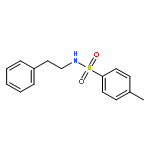
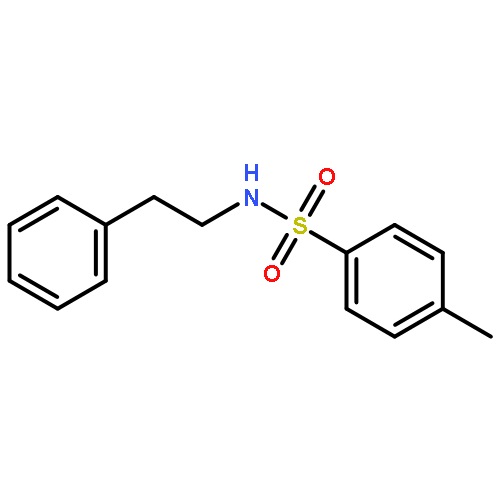
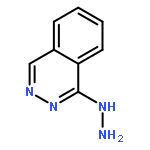
Co-reporter:Dr. Carolyn F. Rosewall;Dr. Erica L. Ingalls;Dr. Werner Kaminsky ;Dr. Forrest E. Michael
Angewandte Chemie 2015 Volume 127( Issue 15) pp:4640-4643
Publication Date(Web):
DOI:10.1002/ange.201412033
Abstract
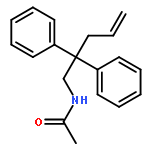
The formation of highly substituted carbon centers using catalysis has been a widely sought after goal, but complexes of highly substituted carbon atoms with transition metals are rare, and the factors that affect the relative stability of complexes with differentially substituted carbon atoms are poorly understood. In this study, a set of equilibrating alkyl–palladium complexes were subtly tuned to form either a primary or trisubstituted alkyl complex as the more thermodynamically favored state, depending on either the substrate or reaction conditions. An X-ray crystal structure of the trisubstituted alkyl–palladium complex is presented and compared with the corresponding primary alkyl complex. The mechanism for rearrangement and the factors that drive the change in stability are discussed.
Co-reporter:Dr. Carolyn F. Rosewall;Dr. Erica L. Ingalls;Dr. Werner Kaminsky ;Dr. Forrest E. Michael
Angewandte Chemie International Edition 2015 Volume 54( Issue 15) pp:4557-4560
Publication Date(Web):
DOI:10.1002/anie.201412033
Abstract
The formation of highly substituted carbon centers using catalysis has been a widely sought after goal, but complexes of highly substituted carbon atoms with transition metals are rare, and the factors that affect the relative stability of complexes with differentially substituted carbon atoms are poorly understood. In this study, a set of equilibrating alkyl–palladium complexes were subtly tuned to form either a primary or trisubstituted alkyl complex as the more thermodynamically favored state, depending on either the substrate or reaction conditions. An X-ray crystal structure of the trisubstituted alkyl–palladium complex is presented and compared with the corresponding primary alkyl complex. The mechanism for rearrangement and the factors that drive the change in stability are discussed.
Co-reporter:Megan L. Duda
Journal of the American Chemical Society 2013 Volume 135(Issue 49) pp:18347-18349
Publication Date(Web):November 25, 2013
DOI:10.1021/ja410686v
A mild palladium-catalyzed cross-coupling of unsubstituted and 2-alkyl-substituted aziridines with arylboronic acid nucleophiles is presented. The reaction is highly regioselective and compatible with diverse functionality. A catalytic amount of base, a sterically demanding triarylphosphine ligand, and a phenol additive are critical to the success of the reaction. Coupling of a deuterium-labeled substrate established that ring opening of the aziridine occurs with inversion of stereochemistry.
Co-reporter:Erica L. Ingalls ; Paul A. Sibbald ; Werner Kaminsky
Journal of the American Chemical Society 2013 Volume 135(Issue 24) pp:8854-8856
Publication Date(Web):June 4, 2013
DOI:10.1021/ja4043406
An enantioselective Pd-catalyzed vicinal diamination of unactivated alkenes using N-fluorobenzenesulfonimide as both an oxidant and a source of nitrogen is reported. The use of Ph-pybox and Ph-quinox ligands afforded differentially protected vicinal diamines in good yields with high enantioselectivities. Mechanistic experiments revealed that the high enantioselectivity arises from selective formation of only one of four possible diastereomeric aminopalladation products of the chiral Pd complex. The aminopalladation complex was characterized by X-ray crystallography.
Co-reporter:Alicia McGhee, Brian M. Cochran, Torrey A. Stenmark and Forrest E. Michael
Chemical Communications 2013 vol. 49(Issue 60) pp:6800-6802
Publication Date(Web):13 Jun 2013
DOI:10.1039/C3CC44117B
A palladium-catalyzed hydroamination reaction is the key step in a stereoselective synthesis of 2,5-disubstituted and 2,3,5-trisubsituted morpholines from carbamate-protected aziridines. Aziridines are selectively attacked at the more substituted position by unsaturated alcohol nucleophiles using Lewis acid catalysts. Palladium-catalyzed hydroamination of the resulting aminoalkenes gives morpholines as a single diastereomer in excellent yield.
Co-reporter:Justin M. Pierson;Erica L. Ingalls;Richard D. Vo ;Dr. Forrest E. Michael
Angewandte Chemie 2013 Volume 125( Issue 50) pp:13553-13555
Publication Date(Web):
DOI:10.1002/ange.201305766
Co-reporter:Justin M. Pierson;Erica L. Ingalls;Richard D. Vo ;Dr. Forrest E. Michael
Angewandte Chemie International Edition 2013 Volume 52( Issue 50) pp:13311-13313
Publication Date(Web):
DOI:10.1002/anie.201305766
Co-reporter:Jessica M. Hoover ; Antonio DiPasquale ; James M. Mayer
Journal of the American Chemical Society 2010 Volume 132(Issue 14) pp:5043-5053
Publication Date(Web):March 24, 2010
DOI:10.1021/ja906563z
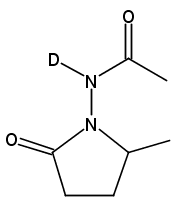
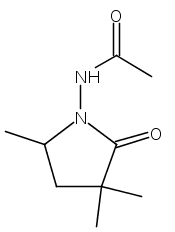
Dicationic (bpy)Pt(II) complexes were found to catalyze the intramolecular hydrohydrazination of alkenes. Reaction optimization revealed Pt(bpy)Cl2 (10 mol %) and AgOTf (20 mol %) in DMF-d7 to be an effective catalyst system for the conversion of substituted hydrazides to five- and six-membered N-amino lactams (N-amino = N-acetamido at 120 °C, N-phthalimido at 80 °C, −OTf = trifluoromethanesulfonate). Of the four possible regioisomeric products, only the product of 5-exo cyclization at the proximal nitrogen is formed, without reaction at the distal nitrogen or 6-endo cyclization. The resting states were found to be a 2:1 Pt-amidate complex (25, for N-acetamido) of the deprotonated hydrazide and a Pt-alkyl complex of the cyclized pyrrolidinone (20 for N-phthalimido). Both complexes are catalytically competent. Catalysis using 25 as the precatalyst shows no rate dependence on added acid (HOTf) or base (2,6-lutidine). The available mechanistic data are all consistent with a mechanism involving N−H activation of the hydrazide, followed by insertion of the alkene into the Pt−N bond, and finally protonation of the resulting cyclized alkyl complex by hydrazide to release the hydrohydrazination product and regenerate the active Pt-amidate catalyst.
Co-reporter:Helena M. Lovick
Journal of the American Chemical Society 2010 Volume 132(Issue 4) pp:1249-1251
Publication Date(Web):January 8, 2010
DOI:10.1021/ja906648w
We report a highly regioselective metal-free oxidative cyclization of sulfonamides onto tethered, unactivated alkenes using hypervalent iodine and Brønsted acids. Under these conditions the acid counterion is incorporated into the cyclized products providing an overall aminotrifluoroacetoxylation of the alkene. An unusual preference for endo ring closure is exhibited in contrast to existing exo selective methods. Multiple ring sizes can be formed to access functionalized pyrrolidines, piperidines, and azepanes with a general preference for endo cyclization. A variety of substrate substitution patterns were tolerated to provide nitrogen-containing heterocycles with high regioselectivities and good to excellent diastereoselectivities.
Co-reporter:Dmitry V. Liskin, Paul A. Sibbald, Carolyn F. Rosewall, and Forrest E. Michael
The Journal of Organic Chemistry 2010 Volume 75(Issue 18) pp:6294-6296
Publication Date(Web):August 26, 2010
DOI:10.1021/jo101171g
A Pd-catalyzed alkoxyamination of protected aminoalkenes promoted by N-fluorobenzenesulfonimide is described. This mild transformation allows the direct formation of ethers from carbon−carbon double bonds. An unusual switch from exo to endo selectivity in polar solvents was discovered, allowing the selective formation of either regioisomer by careful choice of reaction conditions.
Co-reporter:Paul A. Sibbald ; Carolyn F. Rosewall ; Rodney D. Swartz
Journal of the American Chemical Society 2009 Volume 131(Issue 43) pp:15945-15951
Publication Date(Web):October 13, 2009
DOI:10.1021/ja906915w
The mechanism of the Pd-catalyzed diamination and carboamination of alkenes promoted by N-fluorobenzenesulfonimide (NFBS) was investigated. Stereochemical labeling experiments established that the diamination reaction proceeds via overall syn addition of the two nitrogen groups, whereas carboamination is the result of an anti addition of arene and nitrogen to the alkene. The intermediate Pd-alkyl complex arising from aminopalladation was observed, and an X-ray crystal structure of its 2,2′-bipyridine (bipy) complex was obtained, revealing strong chelation of the amide protecting group to palladium. Aminopalladation was shown to be an anti-selective process in both the presence and the absence of added ligands, proceeding via external attack of the nitrogen on a Pd-coordinated alkene. The intermediate Pd-alkyl complex was converted to diamination product upon exposure to NFBS with inversion of configuration via oxidative addition followed by dissociation of the benzenesulfonimide anion and SN2 displacement of the Pd−C bond. Conversely, arylation of the Pd-alkyl complex proceeds via retention of stereochemistry, consistent with C−H activation of the arene at the Pd(IV) center. A small intermolecular isotope effect (kH/kD = 1.1) and a large intramolecular isotope effect (kH/kD = 4) were measured for this process, indicating that C−H activation occurs via a poorly selective product-determining coordination of the arene followed by a highly selective C−H activation. Competition between arenes reveals an unusual reactivity order of toluene > benzene > bromobenzene > anisole.
Co-reporter:Carolyn F. Rosewall ; Paul A. Sibbald ; Dmitry V. Liskin
Journal of the American Chemical Society 2009 Volume 131(Issue 27) pp:9488-9489
Publication Date(Web):June 22, 2009
DOI:10.1021/ja9031659
This report describes a unique Pd-catalyzed oxidative carboamination of protected aminoalkenes in which inexpensive unactivated nucleophilic arenes are incorporated to give carboamination products in good yields. A variety of protected amide and carbamate groups are tolerated, and various five-, six-, and seven-membered rings are formed in good yields. Under these conditions, halobenzenes are activated at the C−H bond rather than the C−X bond, and very high regioselectivity for the para substitution product is observed in all cases. We propose that this carboamination takes place via electrophilic aromatic substitution of a Pd(IV) alkyl intermediate.
Co-reporter:Helena M. Lovick, Forrest E. Michael
Tetrahedron Letters 2009 50(9) pp: 1016-1019
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.12.058
Co-reporter:Jessica M. Hoover, John Freudenthal, Forrest E. Michael and James M. Mayer
Organometallics 2008 Volume 27(Issue 10) pp:2238-2245
Publication Date(Web):April 16, 2008
DOI:10.1021/om701192s
Three complexes, IrCl(PEt3)3, Rh(NNN)Cl (NNN = 2,6-(CyN═CH)2C5H3N), and (NN)PtMe2 [NN = 1,10-phenanthroline (phen) or 2,2′-bipyridine (bpy)], have been investigated for N−N oxidative addition reactivity with di- and tetrasubstituted hydrazines. The reaction of IrCl(PEt3)3with 1,2-diphenylhydrazine (PhNHNHPh) forms the cyclometalated azobenzene complex (Et3P)2Cl(H)Ir(C6H4N═NPh) (1) and the 2:1 complex [(Et3P)2Cl(H)Ir]2(μ-C6H4N═NC6H4) (2). The Rh(NNN)Cl pincer complex catalyzes the disproportionation of PhNHNHPh to azobenzene and aniline with no change in the Rh complex. The reaction of (bpy)PtMe2 with the hydrazine AcNHNHC(O)CH2CH2CH═CH2 (3) yields the platinum metallacycle (bpy)Pt(η2-AcN-NC(O)(CH2)2CHCH2 (4). Although IrCl(PEt3)3, Rh(NNN)Cl, and (NN)PtMe2 all undergo facile oxidative addition of other X−Y bonds, such reactivity is not observed for hydrazines; in the Ir and Pt systems there is a clear preference for cleavage of the stronger N−H bond over the N−N bond. The tetrasubstituted hydrazines tetraphenylhydrazine, 1,2-diphenyl-1,2-dimethylhydrazine, bisuccinimide, biphthalimide, and N-dimethylamino phthalimide do not react with IrCl(PEt3)3, Rh(NNN)Cl, and (NN)PtMe2. This lack of reactivity appears to be due to kinetic rather than thermochemical factors.
Co-reporter:Erica L. Ingalls, G. Andrew Holtzen, Werner Kaminsky, Forrest E. Michael
Journal of Organometallic Chemistry (15 March 2017) Volume 832() pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.jorganchem.2017.01.008
•New chiral bidentate N-heterocyclic carbene palladium complexes have been synthesized.•Ligand synthesis is concise and high yielding from easily available aminoalcohols.•The 6-membered chelate ring adopts a boat conformation.A new class of chiral Pd(II) complexes bearing a rigid, bidentate C,N-ligand are synthesized and characterized. The ligands are obtained in good yield via a simple five-step synthetic route beginning with cheap and commercially available amino alcohols. The structure of one of these new complexes is determined by X-ray crystallography. This data shows the expected large trans effect difference between the N and C donors (0.07 Å PdCl bond length difference) as well as a boat conformation of the 6-membered chelate ring.A new class of chiral bidentate N-heterocyclic carbene ligands are easily made in high yield from amino alcohols. The X-ray crystal structure reveals a rigid chiral environment and a boat conformation of the chelate ring.
Co-reporter:Alicia McGhee, Brian M. Cochran, Torrey A. Stenmark and Forrest E. Michael
Chemical Communications 2013 - vol. 49(Issue 60) pp:NaN6802-6802
Publication Date(Web):2013/06/13
DOI:10.1039/C3CC44117B
A palladium-catalyzed hydroamination reaction is the key step in a stereoselective synthesis of 2,5-disubstituted and 2,3,5-trisubsituted morpholines from carbamate-protected aziridines. Aziridines are selectively attacked at the more substituted position by unsaturated alcohol nucleophiles using Lewis acid catalysts. Palladium-catalyzed hydroamination of the resulting aminoalkenes gives morpholines as a single diastereomer in excellent yield.
.jpg)





























































































































































































































































































































































































































































































































































![Ethanone, 1-[2-(2-propen-1-yl)-1-pyrrolidinyl]-](http://img.cochemist.com/ccimg/918700/918631-41-1.png)
![Ethanone, 1-[2-(2-propen-1-yl)-1-pyrrolidinyl]-](http://img.cochemist.com/ccimg/918700/918631-41-1_b.png)



















































![2-Cyclohexen-1-one, 2-[hydroxy(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/203200/203111-43-7.png)
![2-Cyclohexen-1-one, 2-[hydroxy(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/203200/203111-43-7_b.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/194000/193904-48-2.png)
![Aziridine, 1-[(4-nitrophenyl)sulfonyl]-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/194000/193904-48-2_b.png)
![2,6-Bis((3aS,8aR)-8,8a-dihydro-3aH-indeno[1,2-d]oxazol-2-yl)pyridine](/data/chemimg/119100/185346-09-2.png)
![2,6-Bis((3aS,8aR)-8,8a-dihydro-3aH-indeno[1,2-d]oxazol-2-yl)pyridine](/data/chemimg/119100/185346-09-2_b.png)






![Benzenesulfonamide, N-[2-(2-cyclohexen-1-yl)ethyl]-4-methyl-](http://img.cochemist.com/ccimg/172700/172606-16-5.png)
![Benzenesulfonamide, N-[2-(2-cyclohexen-1-yl)ethyl]-4-methyl-](http://img.cochemist.com/ccimg/172700/172606-16-5_b.png)















































![2,6-Bis[(4S)-4-tert-butyl-2-oxazolin-2yl]pyridine](/data/chemimg/119100/118949-63-6.png)
![2,6-Bis[(4S)-4-tert-butyl-2-oxazolin-2yl]pyridine](/data/chemimg/119100/118949-63-6_b.png)


























![Benzenesulfonamide, 4-methyl-N-[[1-(2-propenyl)cyclohexyl]methyl]-](http://img.cochemist.com/ccimg/81100/81097-22-5.png)
![Benzenesulfonamide, 4-methyl-N-[[1-(2-propenyl)cyclohexyl]methyl]-](http://img.cochemist.com/ccimg/81100/81097-22-5_b.png)
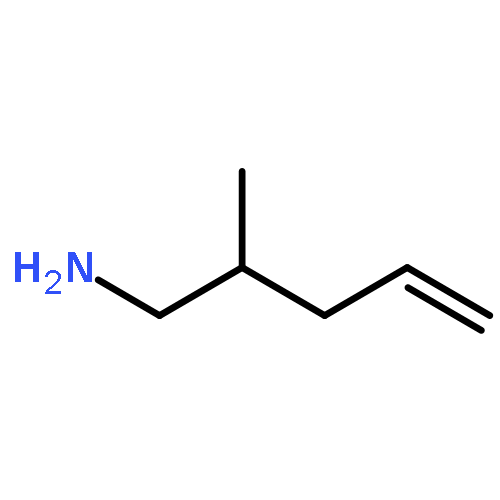
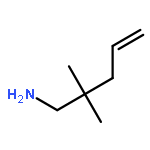
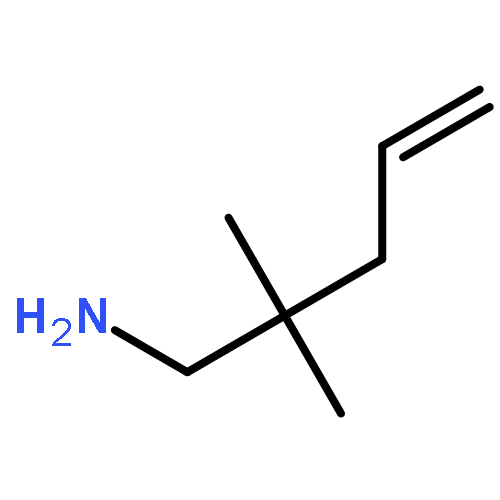














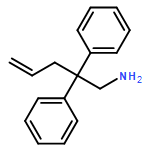







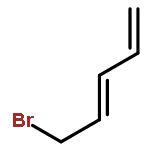

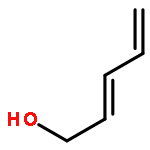
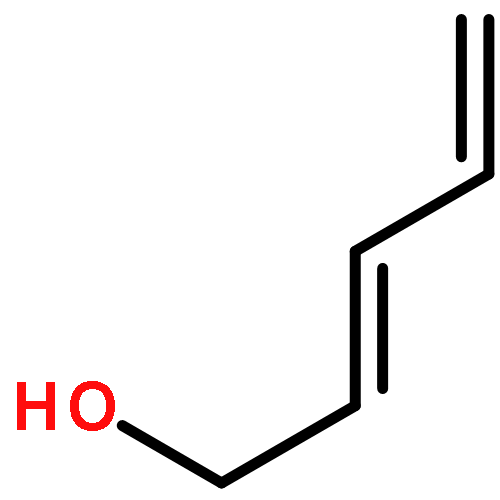












![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/25900/25856-77-3.png)
![Aziridine, 2-methyl-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/25900/25856-77-3_b.png)