Co-reporter:Le Wang;Yong Ye;Vasiliki Lykourinou;Junliang Yang;Alexer Angerhofer;Yufen Zhao;Li-June Ming
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 42) pp:4899-4908
Publication Date(Web):2017/11/16
DOI:10.1002/ejic.201700811
Three ligands L1, L2, and L3 with 2, 4, and 6 1,4,7,10-tetraazacyclododecane (cyclen) moieties attached to a cyclotriphosphazene core, respectively, were synthesized, and oxidation activities of their CuII complexes were investigated. Aerobic oxidation of catechol by these complexes follows an intramolecular dinuclear pathway with significant cooperativity (i.e., θ ≈ 1.5 out of a maximum of 2 for two potential substrate binding sites) and kinetic constants (i.e., kcat = 17.5 × 10–3 s–1, Km = 2.8 mm, and quite remarkable catalytic specificity kcat/Km 12.5 m–1 s–1 per di-Cu center), while that by untethered CuII–cyclen follows a bimolecular dinuclear pathway without noticeable cooperativity (θ = 0.96) and fourfold lower kcat, despite their similar dinuclear mechanisms. The proximity of CuII centers is suggested by EPR spectra and relaxations, showing a broad spectral component particularly in Cu6L3. Thermodynamic parameters also indicate the significance of multi-CuII sites in the oxidative catalysis. Air is a more specific oxidation agent for the representative complex Cu2L1, showing 3.2-fold higher catalytic specificity kcat/Km than H2O2 toward a catechol substrate. The research provides further molecular basis for future design of O2/H2O2-specific oxidation of multi-domain Cu complexes.
Co-reporter:Wen-Zhu Bi;Chen Qu;Xiao-Lan Chen;Ling-Bo Qu;Zhi-Dong Liu;Kai Sun;Xu Li
European Journal of Organic Chemistry 2017 Volume 2017(Issue 34) pp:5125-5130
Publication Date(Web):2017/09/15
DOI:10.1002/ejoc.201701080
A practical and efficient method for the synthesis of 2-aroxy(alkoxy)quinolines has been developed by direct cross-dehydrogenative coupling reaction between quinoline N-oxides and readily available phenols and alcohols in the presence of H-phosphonate and CCl4 under mild conditions.
Co-reporter:Pengbo Zhang;Jianxi Ying;Guo Tang;Yufen Zhao
Organic Chemistry Frontiers 2017 vol. 4(Issue 10) pp:2054-2057
Publication Date(Web):2017/09/26
DOI:10.1039/C7QO00466D
The first phosphinodifluoroalkylation of alkynes is achieved and the simultaneous addition of both difluoromethylene and phosphinoyl groups across the alkynes with high regio- and stereoselectivity is of great significance. By using palladium(II) chloride as a catalyst and Xantphos as a ligand, the reaction of P(O)H compounds, alkynes, and ethyl difluoroiodoacetate proceeds with moderate to high yields, and provides an attractive approach for the construction of (E)-γ,γ-difluoroalkenylphosphine oxides.
Co-reporter:Yile Wu;Changkai Shan;Jianxi Ying;Jue Su;Jun Zhu;Liu Leo Liu;Yufen Zhao
Green Chemistry (1999-Present) 2017 vol. 19(Issue 17) pp:4169-4175
Publication Date(Web):2017/08/29
DOI:10.1039/C7GC01632H
Commercially available NaOH powder is shown to be an efficient transition-metal-free initiator for the catalytic hydroboration of aldehydes, ketones, alkynes and alkenes with HBpin and 9-BBN under mild conditions. Combined experimental and theoretical studies suggest that the catalytically active species is a boron hydride generated in situ from the reaction mixture.
Co-reporter:Peng Chen;Ying Sun;Yile Wu;Liu (Leo) Liu;Jun Zhu;Yufen Zhao
Organic Chemistry Frontiers 2017 vol. 4(Issue 8) pp:1482-1492
Publication Date(Web):2017/07/26
DOI:10.1039/C7QO00240H
Using density functional theory (DFT) calculations, the present study explores the mechanisms of two ruthenium(II)-catalyzed phosphoryl-directed ortho-selective C–H bond activation reactions. Depending on the nature of the phosphoryl groups, namely R2P(O) versus RP(O)OH, two different products could be selectively synthesized. For R2P(O), the overall catalytic cycle includes three basic steps: C–H bond activation, alkyne insertion, and protonation. The oxidation state of the Ru center does not change during this catalytic process. Alternatively, when RP(O)OH is used, the whole catalytic cycle involves four basic steps: C–H bond activation, alkyne insertion, reductive elimination, and catalyst recycling. This switchability is attributed to the hydroxy group of RP(O)OH, which facilitates the Ru(II)/Ru(0) catalytic cycle. Additionally, we found that most of the steps feature cationic intermediates and transition states. This is in line with experimental results showing that additives such as AgSbF6 and KPF6 are required for improved yields.
Co-reporter:Le Wang;Yong Ye;Vasiliki Lykourinou;Junliang Yang;Alexer Angerhofer;Yufen Zhao;Li-June Ming
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 42) pp:4885-4885
Publication Date(Web):2017/11/16
DOI:10.1002/ejic.201701238
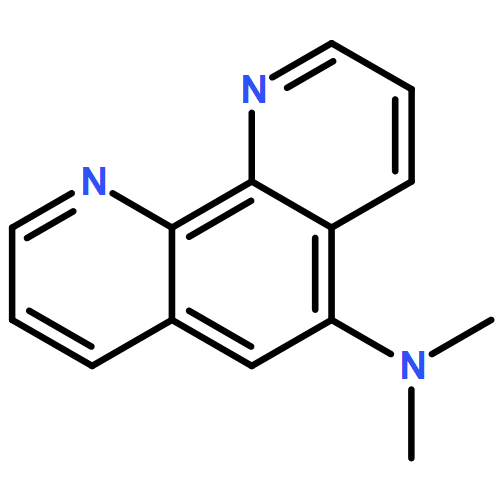
AbstractInvited for the cover of this issue are the groups of Y. Ye from Zhengzhou University, China, A. Angerhofer from the University of Florida, USA, Y. Zhao from Xiamen University, China, and L.-J. Ming from the University of South Florida, USA. The cover image shows the pathways for dinuclear catechol oxidation by CuII complexes of cyclotriphosphazene derivatives with up to six metal-binding sites.
Co-reporter:Le Wang;Yong Ye;Vasiliki Lykourinou;Junliang Yang;Alexer Angerhofer;Yufen Zhao;Li-June Ming
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 42) pp:4884-4884
Publication Date(Web):2017/11/16
DOI:10.1002/ejic.201701237
The Front Cover shows the pathways for catechol oxidation (foreground) by the copper complexes of cyclen-containing derivatives of cyclotriphosphazene (background) in aqueous media. Despite the mononuclear nature of the cyclen ligand, the reaction follows a dinuclear pathway determined by means of a “mechanistic Job plot” to reveal the stoichiometry of the transition state by monitoring the reaction rate. The dinuclear transition states imitate “The Creation” by Michelangelo to add some artistic flavor to the chemistry described herein. More information can be found in the Full Paper by Y. Ye, A. Angerhofer, Y. Zhao, L.-J. Ming et al. For more on the story behind the cover research, see the Cover Profile.
Co-reporter:Yile Wu, Liu Liu, Jue Su, Kaili Yan, Jun Zhu and Yufen Zhao
Chemical Communications 2016 vol. 52(Issue 8) pp:1582-1585
Publication Date(Web):13 Nov 2015
DOI:10.1039/C5CC09330A
Two newly discovered linear compounds tetraboronate and boroxine stabilized by digermylene are reported, which feature a B4O5 chain and a B3O3 ring, respectively. DFT calculations reveal that not only can digermylene stabilize the electron-deficient boron centers, but also increase the energies of the LUMOs of the boron moiety. Our results provide a hint for the development of boronate covalent organic frameworks.
Co-reporter:Han Ouyang, Chuan Fu, Songsen Fu, Zhe Ji, Ying Sun, Peiran Deng and Yufen Zhao
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 6) pp:1925-1929
Publication Date(Web):24 Dec 2015
DOI:10.1039/C5OB02603B
Protein phosphorylation is one of the most common and extensively studied post-translational modifications (PTMs). Compared to the O-phosphorylation on Ser, Thr and Tyr residues, our understanding of arginine phosphorylation is relatively limited, both in prokaryotes and eukaryotes, due to the intrinsic instability of phosphoarginine (pArg) and the lack of a feasible method to produce anti-pArg antibodies. We report the design and synthesis of a stable pArg analog, in which the labile N–P bond is replaced with a non-hydrolyzable C–P bond. Significantly, this analog was successfully used as a hapten to raise an immune response and the first mouse polyclonal antibody that specifically recognizes pArg-containing peptides and proteins was produced using analog-KLH conjugated as the immunogen. The generated antibody shows excellent specificity towards pArg-containing peptides and proteins, and could be used for a variety of biological detection methods. This provides us an invaluable tool to unravel the mystery of the biological function of pArg.
Co-reporter:Yile Wu, Liu Liu, Jue Su, Jun Zhu, Zhe Ji, and Yufen Zhao
Organometallics 2016 Volume 35(Issue 11) pp:1593-1596
Publication Date(Web):May 5, 2016
DOI:10.1021/acs.organomet.6b00187
The heavier cyclobutadiene analogue 2,4-digerma-1,3-diphosphacyclobutadiene ([L12Ge2P2], 4; L1 = CH{(CMe)(2,6-iPr2C6H3N)}2), featuring a planar Ge2P2 four-membered ring, has been synthesized via the elimination of carbon monoxide from the corresponding phosphaketenyl germylene [L1GePCO] (2) under UV irradiation.
Co-reporter:Liu (Leo) Liu, Peng Chen, Ying Sun, Yile Wu, Su Chen, Jun Zhu, and Yufen Zhao
The Journal of Organic Chemistry 2016 Volume 81(Issue 23) pp:11686-11696
Publication Date(Web):November 3, 2016
DOI:10.1021/acs.joc.6b02093
In textbooks, the low reactivity of amides is attributed to the strong resonance stability. However, Garg and co-workers recently reported the Ni-catalyzed activation of robust amide C–N bonds, leading to conversions of amides into esters, ketones, and other amides with high selectivity. Among them, the Ni-catalyzed Suzuki-Miyaura coupling (SMC) of N-benzyl-N-tert-butoxycarbonyl (N-Bn-N-Boc) amides with pinacolatoboronate (PhBpin) was performed in the presence of K3PO4 and water. Water significantly enhanced the reaction. With the aid of density functional theory (DFT) calculations, the present study explored the mechanism of the aforementioned SMC reaction as well as analyzed the weakening of amide C–N bond by N-functionalization. The most favorable pathway includes four basic steps: oxidative addition, protonation, transmetalation, and reductive elimination. Comparing the base- and water-free process, the transmetalation step with the help of K3PO4 and water is significantly more facile. Water efficiently protonates the basic N(Boc) (Bn) group to form a neutral HN(Boc) (Bn), which is easily removed. The transmetalation step is the rate-determining step with an energy barrier of 25.6 kcal/mol. Further, a DFT prediction was carried out to investigate the full catalytic cycle of a cyclic (amino) (aryl)carbene in the Ni-catalyzed SMC of amides, which provided clues for further design of catalysts.
Co-reporter:Xu Li, Xiao-Lan Chen, Qing Zhang, Ling-Bo Qu, Wen-Zhu Bi, Kai Sun, Jian-Yu Chen, Xin Chen and Yu-Fen Zhao
RSC Advances 2015 vol. 5(Issue 7) pp:5004-5009
Publication Date(Web):10 Dec 2014
DOI:10.1039/C4RA10617B
The readily available CuSO4-H-phosphonate catalytic system can catalyze the head-to-head dimerization of terminal alkynes to give the corresponding (E) conjugated enynes selectively in high yield. The present protocol provides an efficient and practical approach to various conjugated enynes with the advantages of cheap catalysts, operational simplicity and high stereo-and regioselectivity.
Co-reporter:Hang Yuan, Yile Wu, Wu Liu, Yan Liu, Xiang Gao, Jinming Lin, Yufen Zhao
Carbohydrate Research 2015 Volume 407() pp:5-9
Publication Date(Web):30 April 2015
DOI:10.1016/j.carres.2015.01.011
•MS technique was employed to dissect the intrinsic structure feature of sucrose.•Sucrose has the most labile glycosidic bond among sucrose isomers.•Sucrose has the most stable integral structure among sucrose isomers.•K+ is a better co-transporter for sucrose based on the MS results.•The above results are the new explanation for the nature selection of sucrose.Sucrose is the carbon skeletons and energy vector for plants, which is important for plants growth. Among thousands of disaccharides in Nature, why chose sucrose for plants? In this paper, we analyzed the intrinsic structural characteristics of four sucrose isomers with different glycosidic linkage by mass spectrometry (MS) technique. Our results show that sucrose has the most labile glycosidic bond compared with other three isomers, which is helpful for releasing glucose and fructose unit. Besides, sucrose has the most stable integral structure, which is hard to dehydrate and degrade into fragments through losing one or three even four-carbon units, just as its three isomers. In other words, sucrose is more easily holds an integral structure during the transport process, whenever it is necessary, and sucrose can be cleaved into glucose and fructose easily. Besides, we also investigate the internal relationship of sucrose with K+ by tandem mass spectrometry and viscosity measurement. The related results have shown that the K+ can stabilize sucrose to a greater extent than the Na+. Furthermore, under the same conditions, K+ ions reduce the viscosity of sucrose–water system much more than Na+. These results suggest that K+ is a better co-transporter for sucrose.Of course, the transport of sucrose in plants is a very complicated process, which is involved in many proteins. This paper directly accounts for the basic structure feature of sucrose, and the results discovered could provide the novel insight for the answer why Nature chose sucrose for plants.
Co-reporter:Feng Ni;Chuan Fu;Xiang Gao;Yan Liu;Pengxiang Xu;Liu Liu
Science China Chemistry 2015 Volume 58( Issue 3) pp:374-382
Publication Date(Web):2015 March
DOI:10.1007/s11426-015-5321-1
Post-translational modification of proteins by N-phosphorylation of the basic amino acid residues plays important roles in biological processes. The high-energy P-N bond might have contributed to the evolution of prebiotic chemistry. N-phosphoryl amino acids (PAAs) can serve as interesting small molecular models for the study of P-N bonds in prebiotic chemical evolution. PAAs are capable of simultaneously producing several important biomolecules such as polypeptides and oligonucleotides under mild reaction conditions. In this review, we describe the chemistry of PAAs, discusse their likely prebiotic origins and their reactivity and how they relate to biological P-N bond species. We also depict a possible prebiotic scenario mediated by PAAs in which PAAs may have acted as one of the essential forces driving prebiotic biomolecules to the first protocell.
Co-reporter:Liu Liu, Jun Zhu and Yufen Zhao
Chemical Communications 2014 vol. 50(Issue 77) pp:11347-11349
Publication Date(Web):01 Aug 2014
DOI:10.1039/C4CC04610B
Density functional theory (DFT) calculations were carried out to investigate the [2+2], [3+2] and [4+2] cycloadditions of the phosphaethynolate anion (PCO−). The results reveal that the electronic properties of different unsaturated compounds play a crucial role in reactivity and regioselectivity.
Co-reporter:Xiaolan Chen;Xu Li;Zhibo Qu;Diian Ke;Lingbo Qu;Likun Duan;Wenpeng Mai;Jinwei Yuan;Jianyu Chen;Yufen Zhao
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 9) pp:1979-1985
Publication Date(Web):
DOI:10.1002/adsc.201301065
Co-reporter:Liu Liu, Yile Wu, Zeshu Wang, Jun Zhu, and Yufen Zhao
The Journal of Organic Chemistry 2014 Volume 79(Issue 15) pp:6816-6822
Publication Date(Web):July 1, 2014
DOI:10.1021/jo5007174
The reaction mechanism of copper-catalyzed phosphorylation of terminal alkynes under different conditions has been investigated experimentally and theoretically. The important role of dioxygen has been elucidated, including the formation of η1-superoxocopper(II), η2-superoxocopper(III), μ-η2:η2-peroxodicopper(II), and bis(μ-oxo)dicopper(III) complexes. More importantly, the proton transfer from the dialkyl phosphonate (in the form of phosphite) to the bridging oxygen atom entails the migration of the deprotonated phosphonate to the terminal alkyne, leading to the formation of a C–P bond with an activation barrier of only 1.8 kcal/mol. In addition, a particularly stable six-centered dicopper(I) species is formed with the migration of both of the Ph2P(O) groups from the copper atoms to the oxygen atoms of the bis(μ-oxo) bridge, explaining the experimental observation that secondary phosphine oxides can be oxidized to the phosphinic acids. Thus, the diphenylphosphine oxide was added to the reaction mixture dropwise to minimize the concentration during the reaction course. Gratifyingly, the coupling product was generated almost quantitatively when the reaction was completed.
Co-reporter:Chao Huang, Yulei Hao, Yufen Zhao, and Jun Zhu
Organometallics 2014 Volume 33(Issue 3) pp:817-822
Publication Date(Web):January 21, 2014
DOI:10.1021/om401188v
Metallaaromatics have attracted continuing interest of both theoretical and experimental chemists since the first metallabenzene was predicted by Hoffmann and isolated by Roper. In sharp contrast to metallabenzenes, metallaphosphabenzene (MPB) is much less developed and has not been synthesized so far. Thus, developing synthetic approaches is urgent. Here we present thorough density functional theory (DFT) calculations on the thermodynamics and kinetics of the rearrangement between MPBs and the corresponding η5-phosphacyclopentadiene (η5-PCp) complexes. The effects of metal centers, ligands, and substituents on the metallacycles were examined systematically. Our results reveal that the third-row metal osmium has the highest possibility to form MPB in comparison with the first-row metal iron and second-row metal ruthenium. Substituents were found to have a significant effect on the thermodynamics and kinetics of the rearrangement reactions, leading to an interconversion between osmaphosphabenzenes (OsPBs) and the corresponding η5-PCp complexes by simply tuning the substituents on the metallacycles. Thus, all of these findings should invite experimentalists to test these unconventional methods to realize the first MPB.
Co-reporter:Liu Liu, Yile Wu, Tao Wang, Xiang Gao, Jun Zhu, and Yufen Zhao
The Journal of Organic Chemistry 2014 Volume 79(Issue 11) pp:5074-5081
Publication Date(Web):May 9, 2014
DOI:10.1021/jo500616g
Density functional theory calculations (DFT) have been performed on Rh(III)-catalyzed phosphoryl-directed oxidative C–H activation/cyclization to investigate the detailed mechanism, including four basic steps: C–H activation, alkyne insertion, reductive elimination, and catalyst recycling, each of which consists of different steps. Interestingly, the Rh(III)–AgOAc catalyst system was found to be more favorable in the C–H activation step in comparison with the Rh(III)–Ag2CO3 system, whereas the Rh(I)–Ag2CO3 catalyst system was more efficient for catalyst recycling. Importantly, our calculations suggest that the alkyne insertion process is a reversible step. Reductive elimination is the rate-determining step with an activation energy of 25.0 kcal/mol. In addition, the origin of the reactivity and selectivity difference between diarylacetylenes and dialkylacetylenes or electron-rich and electron-deficient diarylacetylenes was probed by means of comparative DFT calculations. The calculation results show that the electronic effects of alkynes play a key role in the reactivity and selectivity, in line with the experimental observations that diarylacetylenes and electron-rich diarylacetylenes are more reactive than dialkylacetylenes and electron-deficient diarylacetylenes, respectively. Our findings should be useful for further developments of transition-metal-catalyzed C–H activation reactions.
Co-reporter:Liu Liu, Hang Yuan, Tingting Fu, Tao Wang, Xiang Gao, Zhiping Zeng, Jun Zhu, and Yufen Zhao
The Journal of Organic Chemistry 2014 Volume 79(Issue 1) pp:80-87
Publication Date(Web):December 5, 2013
DOI:10.1021/jo402307x
Density functional theory calculations have been carried out on Pd-catalyzed phosphoryl-directed ortho-olefination to probe the origin of the significant reactivity difference between methyl hydrogen benzylphosphonates and dimethyl benzylphosphonates. The overall catalytic cycle is found to include four basic steps: C–H bond activation, transmetalation, reductive elimination, and recycling of catalyst, each of which is constituted from different steps. Our calculations reveal that the hydroxy group of phosphoryl plays a crucial role almost in all steps, which can not only stabilize the intermediates and transition states by intramolecular hydrogen bonds but also act as a proton donor so that the η1-CH3COO– ligand could be protonated to form a neutral acetic acid for easy removal. These findings explain why only the methyl hydrogen benzylphosphonates and methyl hydrogen phenylphosphates were found to be suitable reaction partners. Our mechanistic findings are further supported by theoretical prediction of Pd-catalyzed ortho-olefination using methyl hydrogen phenylphosphonate, which is verified by experimental observations that the desired product was formed in a moderate yield.
Co-reporter:Chuan Fu, Xueyun Zheng, Yao Jiang, Yan Liu, Pengxiang Xu, Zhiping Zeng, Rihe Liu and Yufen Zhao
Chemical Communications 2013 vol. 49(Issue 27) pp:2795-2797
Publication Date(Web):18 Jan 2013
DOI:10.1039/C3CC38467E
We have synthesized γ-[18O4]-ATP and used it to develop a non-radioactive and multiplex method. Significantly, this novel approach can be applied to any kinases without using a purified enzyme or a fluorescent substrate. Using this approach, the effectiveness and specificity of inhibitors on several kinases could be readily determined.
Co-reporter:Wei-Zhu CHEN, Peng-Xiang XU, Rui-Zao YI, Yu-Fen ZHAO
Chinese Journal of Analytical Chemistry 2012 Volume 40(Issue 8) pp:1159-1163
Publication Date(Web):August 2012
DOI:10.1016/S1872-2040(11)60562-8
Co-reporter:Rongqiang Zhuang, Jian Xu, Zhenshi Cai, Guo Tang, Meijuan Fang, and Yufen Zhao
Organic Letters 2011 Volume 13(Issue 8) pp:2110-2113
Publication Date(Web):March 18, 2011
DOI:10.1021/ol200465z
A mild and efficient method was developed for the copper-catalyzed additions of H-phosphonate diesters to boronic acids under the copper catalyst system Cu2O/1,10-phenanthroline. To the best of our knowledge this finding is the first example of a copper-catalyzed synthesis of aryl phosphonates from arylboronic acids and H-phosphonate dialkyl esters.
Co-reporter:Zhiping Zeng, Ping Luo, Yao Jiang, Yan Liu, Guo Tang, Pengxiang Xu, Yufen Zhao and G. Michael Blackburn
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 20) pp:6973-6978
Publication Date(Web):22 Jul 2011
DOI:10.1039/C1OB06143G
A novel pyrrolidine-based chiral phosphoproline is an effective bifunctional organocatalyst for the asymmetric Michael addition of ketones to nitroolefins giving high levels of diastereo- and enantio-selectivities (up to > 99:1 dr and 96% ee). anti-SR Transition state has the lowest barrier which controls the stereoselectivity, in agreement with experimental results.
Co-reporter:Xiang Gao;Honggui Deng;Guo Tang;Yan Liu;Pengxiang Xu;Yufen Zhao
European Journal of Organic Chemistry 2011 Volume 2011( Issue 17) pp:3220-3228
Publication Date(Web):
DOI:10.1002/ejoc.201100234
Abstract
N-Phosphoryl amino acids (NPAAs) are a novel series of N-terminal-activated amino acids that act as the energy source and phosphoryl donor in intra- and intermolecular phosphoryl transfer to form “high-energy” species, such as acetyl phosphate and aminoacyl phosphates, and in the self-assembled synthesis of polypeptides under mild aqueous conditions. In this work, the chemical reactivity of N-mono(methoxyphosphoryl)glycine as a representative was investigated in detail by using a combination of the stable-isotope-labeling (15N) technique, 31P NMR, ESI-MS/MS and LC-MS. The phosphoryl group of NPAAs can be transferred intermolecularly to the carboxy group of another molecule through intramolecular cyclic pentacoordinate phosphoric–amino acid anhydride intermediates. In addition to C-terminal activation by phosphate anhydride, amino acids can also be self-activated by N-phosphorylation. This information not only provides some interesting clues for understanding the active role of the phosphoryl group in living systems, but also shows that the origin of life might be attributed to the chemical evolution of N-phosphoryl amino acids.
Co-reporter:Hu Chen, Zhongbin Huang, Xiaoming Hu, Guo Tang, Pengxiang Xu, Yufen Zhao, and Chien-Hong Cheng
The Journal of Organic Chemistry 2011 Volume 76(Issue 7) pp:2338-2344
Publication Date(Web):March 9, 2011
DOI:10.1021/jo2000034
The Suzuki−Miyaura cross-coupling of aryl phosphates using Ni(PCy3)2Cl2 as an inexpensive, bench-stable catalyst is described. Broad substrate scope and high efficiency are demonstrated by the syntheses of more than 40 biaryls and by constructing complex organic molecules. The poor reactivity of aryl phosphates relative to aryl halides is successfully employed to construct polyarenes by selective cross-coupling using Pd and Ni catalysts.
Co-reporter:Chao Li, Hua Tian, Shan Duan, Xuena Liu, Pengfei Xu, Renzhong Qiao, and Yufen Zhao
The Journal of Physical Chemistry B 2011 Volume 115(Issue 45) pp:13350-13354
Publication Date(Web):September 14, 2011
DOI:10.1021/jp206199b
The condensation of DNA is essential for biological processes such as DNA transcription and replication, and its study receives additional impetus from an interest in gene therapy. Although many efficacious condensing agents have been discovered and investigated, little is known about the conversation of condensation-release under suitable conditions. A novel class of DNA condensing agents based on small azaheterocyclic metal-binding molecules has been discovered and described. Both linear and plasmid DNA can be condensed to nanoparticles by the title compounds with 50 °C incubation, especially in the presence of divalent metal ions. Importantly, this condensation may be released to original forms with little or no damage to the DNA under incubation at physiological temperatures. These changes in DNA morphology over time have been analyzed by gel electrophoresis, circular dichroism (CD), and atomic force microscopy (AFM). The present work might help to develop strategies for the design and synthesis of controllable condensing agents, which may also be applied to control gene expression and delivery.
Co-reporter:Guochun Yang Dr.;Yunjie Xu Dr.;Jianbo Hou ;Hui Zhang ;Yufen Zhao Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 8) pp:2518-2527
Publication Date(Web):
DOI:10.1002/chem.200902501
Abstract
Vibrational circular dichroism (VCD) spectroscopic measurements and density functional theory (DFT) calculations have been used to obtain the absolute structural information about four sets of diastereomers of pentacoordinate spirophosphoranes derived separately from l- (or d-) valine and l- (or d-) leucine for the first time. Each compound contains three stereogenic centers: one at the phosphorus center and two at the amino acid ligands. Extensive conformational searches for the compounds have been carried out and their vibrational absorption (VA) and VCD spectra have been simulated at the B3LYP/6-311++G** level. Although both VA and VCD spectra are highly sensitive to the structural variation of the apical axis, that is, the OPO or NPO arrangement, the rotamers generated by the aliphatic amino side chains show little effect on both. The dominant experimental VCD features in the 1100–1500 cm−1 region were found to be controlled by the chirality at the phosphorus center, whereas those at the CO stretching region are determined by the chirality of the amino acid ligands. The good agreement between the experimental VA and VCD spectra in CDCl3 solution and the simulated ones allows us to assign the absolute configurations of these pentacoordinate phosphorus compounds with high confidence. This study shows that the VCD spectroscopy complemented with DFT calculations is a powerful and reliable method for determining the absolute configurations and dominating conformers of synthetic phosphorus coordination complexes in solution.
Co-reporter:Chao Li;Chao Du;Hua Tian;Chao Jiang;Min Du;Yan Liu; Ren-Zhong Qiao; Yan-Xing Jia; Yu-Fen Zhao
Chemistry - A European Journal 2010 Volume 16( Issue 43) pp:12935-12940
Publication Date(Web):
DOI:10.1002/chem.201000552
Abstract
A new family of artificial transcription factor (ATF)-based conjugates have been designed and synthesized as potent chemical nucleases. Polyamides as the important and efficient ATFs were used to modify and activate several anchor compounds. The results demonstrate that the resulting conjugates remarkably promote the rate accelerations and non-random double-strand DNA cleavage activity. Interestingly, the cleavage activity of both the hydrolytic and oxidative agents was promoted efficiently through the modification of the ATFs.
Co-reporter:Gang Wang, Ruwei Shen, Qing Xu, Midori Goto, Yufen Zhao and Li-Biao Han
The Journal of Organic Chemistry 2010 Volume 75(Issue 11) pp:3890-3892
Publication Date(Web):May 5, 2010
DOI:10.1021/jo100473s
The reaction of H-phosphinates and secondary phosphine oxides with amines and alcohols proceeds highly stereospecifically to give the corresponding coupling products with inversion of configuration at the phosphorus center under the Atherton−Todd reaction conditions. This finding leads to the establishment of a general and efficient method for the synthesis of a variety of optically active organophosphorus acid derivatives from the easily available chiral H-phosphinates and secondary phosphine oxides.
Co-reporter:Feng Ni, Shuting Sun, Chao Huang and Yufen Zhao
Green Chemistry 2009 vol. 11(Issue 4) pp:569-573
Publication Date(Web):17 Feb 2009
DOI:10.1039/B817013D
Inspired by a reactivity study between sodium trimetaphosphate (P3m) and amino acids in prebiotic chemistry, a one-step reaction with efficient purification procedure in aqueous media has been developed for the synthesis of N-phosphono-amino acids (NPAA). P3m was used to phosphorylate amino acids to NPAA with yields of 60∼91%. The by-products, inorganic polyphosphates, were recycled to regenerate the phosphorylation reagent P3m.
Co-reporter:Jian-Bo Hou, Hui Zhang, Jian-Nan Guo, Yan Liu, Peng-Xiang Xu, Yu-Fen Zhao and G. Michael Blackburn
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 15) pp:3020-3023
Publication Date(Web):25 Jun 2009
DOI:10.1039/B909786D
Two pairs of enantiomers of stable chiral pentacoordinate spirophosphoranes with two bonds from the amino and two bonds from the carboxyl groups of amino acids have been synthesized and analysed. The results show that differences in chirality at phosphorus are linked to distinct differences in physical properties.
Co-reporter:Feng Ni;Xiang Gao;Zeng-Xiang Zhao;Chao Huang
European Journal of Organic Chemistry 2009 Volume 2009( Issue 18) pp:3026-3035
Publication Date(Web):
DOI:10.1002/ejoc.200900227
Abstract
Cyclic acylphosphoramidates (2-hydroxy-2-oxy-1,3,2-oxazaphospholidine-5-ones, CAPAs) are phosphate-activated amino acids possessing both a carboxyl–phosphoryl anhydride and a phosphoramidate bond in a ring. They structurally resemble NCAs (amino acid N-carboxyanhydrides,Leuchs' anhydrides), which have been extensively investigated. By contrast, the chemistry of CAPAs has remained almost unexplored since it was proposed in a prebiotic reaction of inorganic polyphosphates (PolyPs) with amino acids. In the present work, the bielectrophilicity of α-CAPAs (Gly-CAPA, Ala-CAPA) was identified by isotopic analysis (18O, 15N) and further proved by trapping α-CAPA with nucleophiles such as water, amino acids, phosphate and methanol in alkaline media, which yielded N-phosphoamino acids and peptide phosphoanhydride and phosphate ester derivatives. By comparison with the reactivity of NCAs, the bielectrophilicity of CAPAs indicates that CAPAs can provide rich chemistry.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Y. Liu;J. -B. Hou;X. -X. Liu;F. -M. Miao;Y. -F. Zhao
Journal of Structural Chemistry 2009 Volume 50( Issue 5) pp:835-840
Publication Date(Web):2009 October
DOI:10.1007/s10947-009-0125-9
L-Seryl-L-histidine dipeptide is of scientific interest mainly because of its various biological activities. In this paper, the lowest energy structure of the dipeptide was determined by molecular modeling, and further validated by quantum chemistry calculations and 1H NMR spectroscopy.
Co-reporter:Jian-Bo Hou, Guo Tang, Jian-Nan Guo, Yan Liu, Hui Zhang, Yu-Fen Zhao
Tetrahedron: Asymmetry 2009 Volume 20(Issue 11) pp:1301-1307
Publication Date(Web):19 June 2009
DOI:10.1016/j.tetasy.2009.05.009
Two sets of diastereomers of pentacoordinate spirophosphoranes separately derived from l-valine (or d-valine) and l-leucine (or d-leucine) were synthesized, isolated, and structurally characterized both in the solid state (X-ray crystallography and solid-state CD spectroscopy) and in the solution (1H NMR). The Λ and Δ absolute configurations of a pair of enantiomers 3a and 4a with distorted trigonal bipyramids (TBPs) geometry are directly determined by X-ray diffraction analysis, respectively. The chiral-at-phosphorus features of the related diastereomers were correlated with their solid-state CD and 1H NMR spectra.(3S,5Λ,8S)-3,8-Di(propan-2-yl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC10H19N2O4P[α]D20=-60.3 (c 1.0, DMSO)Absolute configuration: (3S,5Λ,8S)(3S,5Δ,8S)-3,8-Di(propan-2-yl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC10H19N2O4P[α]D20=+22.6 (c 1.0, DMSO)Absolute configuration: (3S,5Δ,8S)(3R,5Δ,8R)-3,8-Di(propan-2-yl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC10H19N2O4P[α]D20=+60.1 (c 1.0, DMSO)Absolute configuration: (3R,5Δ,8R)(3R,5Λ,8R)-3,8-Di(propan-2-yl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC10H19N2O4P[α]D20=-22.6 (c 1.0, DMSO)Absolute configuration: (3R,5Λ,8R)(3S,5Λ,8S)-3,8-Bis(2-methylpropyl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC12H23N2O4P[α]D20=-55.7 (c 1.0, acetone)Absolute configuration: (3S,5Λ,8S)(3S,5Δ,8S)-3,8-Bis(2-methylpropyl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC12H23N2O4P[α]D20=+31.7 (c 1.0, acetone)Absolute configuration: (3S,5Δ,8S)(3R,5Δ,8R)-3,8-Bis(2-methylpropyl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC12H23N2O4P[α]D20=+55.4 (c 1.0, acetone)Absolute configuration: (3R,5Δ,8R)(3R,5Λ,8R)-3,8-Bis(2-methylpropyl)-1,6-dioxa-4,9-diaza-5λ5-phosphaspiro[4.4]-nonane-2,7-dioneC12H23N2O4P[α]D20=-31.9 (c 1.0, acetone)Absolute configuration: (3R,5Λ,8R)
Co-reporter:Lei Ding, Fang Wang, Leiqi Chen, Hui Zhang, YuFen Zhao
Tetrahedron: Asymmetry 2008 Volume 19(Issue 23) pp:2653-2658
Publication Date(Web):1 December 2008
DOI:10.1016/j.tetasy.2008.12.009
Unsymmetric Schiff base complexes have attracted more attention in recent years due to their diverse effects in catalytic reactions. Due to their high dissymmetry, unsymmetric metallosalen complexes are harder to prepare than symmetric ones. This means that X-ray crystallographic structural determination is sometimes unavailable, so their absolute configurations are determined by circular dichroism (CD) spectroscopy instead. Herein, some quadridentate unsymmetric metallosalen nickel(II) complexes were synthesized and their structures were characterized by CD spectra. An empirical rule for assignment of the absolute configurations of tetra-coordinated pseudo-planar Ni(II) complexes was put forward. Furthermore, a fingerprint was found to judge whether the metallosalen complexes are symmetric or unsymmetric.
Co-reporter:Qingle Zeng;Heqing Wang;Tongjian Wang;Yimin Cai;Wen Weng;Yufen Zhao
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 15) pp:
Publication Date(Web):2 DEC 2005
DOI:10.1002/adsc.200505259
Simple, inexpensive, preformed vanadium-Schiff base complexes were facilely prepared and used in enantioselective sulfoxidation. Both the amount of aqueous H2O2 and reaction time greatly influenced the ee values and yields of chiral sulfoxides. High enantioselectivities (up to 99% ee) and reasonable yields (>40%) for various chiral sulfoxides were achieved by combining enantioselective sulfoxidation and appropriate concomitant kinetic resolution.
Co-reporter:Qingle Zeng, Heqing Wang, Wen Weng, Wenshi Lin, Yuxing Gao, Xiantong Huang and Yufen Zhao
New Journal of Chemistry 2005 vol. 29(Issue 9) pp:1125-1127
Publication Date(Web):21 Jul 2005
DOI:10.1039/B505237H
The effects of substituents of the Schiff base ligands on oxovanadium-catalyzed enantioselective sulfoxidation were first systematically studied, and a rational mechanism of enantioselective sulfoxidation based on our experimental data and the reported data is proposed.
Co-reporter:Peng Chen, Ying Sun, Yile Wu, Liu (Leo) Liu, Jun Zhu and Yufen Zhao
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 8) pp:NaN1492-1492
Publication Date(Web):2017/06/01
DOI:10.1039/C7QO00240H
Using density functional theory (DFT) calculations, the present study explores the mechanisms of two ruthenium(II)-catalyzed phosphoryl-directed ortho-selective C–H bond activation reactions. Depending on the nature of the phosphoryl groups, namely R2P(O) versus RP(O)OH, two different products could be selectively synthesized. For R2P(O), the overall catalytic cycle includes three basic steps: C–H bond activation, alkyne insertion, and protonation. The oxidation state of the Ru center does not change during this catalytic process. Alternatively, when RP(O)OH is used, the whole catalytic cycle involves four basic steps: C–H bond activation, alkyne insertion, reductive elimination, and catalyst recycling. This switchability is attributed to the hydroxy group of RP(O)OH, which facilitates the Ru(II)/Ru(0) catalytic cycle. Additionally, we found that most of the steps feature cationic intermediates and transition states. This is in line with experimental results showing that additives such as AgSbF6 and KPF6 are required for improved yields.
Co-reporter:Chuan Fu, Xueyun Zheng, Yao Jiang, Yan Liu, Pengxiang Xu, Zhiping Zeng, Rihe Liu and Yufen Zhao
Chemical Communications 2013 - vol. 49(Issue 27) pp:NaN2797-2797
Publication Date(Web):2013/01/18
DOI:10.1039/C3CC38467E
We have synthesized γ-[18O4]-ATP and used it to develop a non-radioactive and multiplex method. Significantly, this novel approach can be applied to any kinases without using a purified enzyme or a fluorescent substrate. Using this approach, the effectiveness and specificity of inhibitors on several kinases could be readily determined.
Co-reporter:Jian-Bo Hou, Hui Zhang, Jian-Nan Guo, Yan Liu, Peng-Xiang Xu, Yu-Fen Zhao and G. Michael Blackburn
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 15) pp:NaN3023-3023
Publication Date(Web):2009/06/25
DOI:10.1039/B909786D
Two pairs of enantiomers of stable chiral pentacoordinate spirophosphoranes with two bonds from the amino and two bonds from the carboxyl groups of amino acids have been synthesized and analysed. The results show that differences in chirality at phosphorus are linked to distinct differences in physical properties.
Co-reporter:Zhiping Zeng, Ping Luo, Yao Jiang, Yan Liu, Guo Tang, Pengxiang Xu, Yufen Zhao and G. Michael Blackburn
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 20) pp:NaN6978-6978
Publication Date(Web):2011/07/22
DOI:10.1039/C1OB06143G
A novel pyrrolidine-based chiral phosphoproline is an effective bifunctional organocatalyst for the asymmetric Michael addition of ketones to nitroolefins giving high levels of diastereo- and enantio-selectivities (up to > 99:1 dr and 96% ee). anti-SR Transition state has the lowest barrier which controls the stereoselectivity, in agreement with experimental results.
Co-reporter:Han Ouyang, Chuan Fu, Songsen Fu, Zhe Ji, Ying Sun, Peiran Deng and Yufen Zhao
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 6) pp:NaN1929-1929
Publication Date(Web):2015/12/24
DOI:10.1039/C5OB02603B
Protein phosphorylation is one of the most common and extensively studied post-translational modifications (PTMs). Compared to the O-phosphorylation on Ser, Thr and Tyr residues, our understanding of arginine phosphorylation is relatively limited, both in prokaryotes and eukaryotes, due to the intrinsic instability of phosphoarginine (pArg) and the lack of a feasible method to produce anti-pArg antibodies. We report the design and synthesis of a stable pArg analog, in which the labile N–P bond is replaced with a non-hydrolyzable C–P bond. Significantly, this analog was successfully used as a hapten to raise an immune response and the first mouse polyclonal antibody that specifically recognizes pArg-containing peptides and proteins was produced using analog-KLH conjugated as the immunogen. The generated antibody shows excellent specificity towards pArg-containing peptides and proteins, and could be used for a variety of biological detection methods. This provides us an invaluable tool to unravel the mystery of the biological function of pArg.
Co-reporter:Liu Liu, Jun Zhu and Yufen Zhao
Chemical Communications 2014 - vol. 50(Issue 77) pp:NaN11349-11349
Publication Date(Web):2014/08/01
DOI:10.1039/C4CC04610B
Density functional theory (DFT) calculations were carried out to investigate the [2+2], [3+2] and [4+2] cycloadditions of the phosphaethynolate anion (PCO−). The results reveal that the electronic properties of different unsaturated compounds play a crucial role in reactivity and regioselectivity.
Co-reporter:Yile Wu, Liu Liu, Jue Su, Kaili Yan, Jun Zhu and Yufen Zhao
Chemical Communications 2016 - vol. 52(Issue 8) pp:NaN1585-1585
Publication Date(Web):2015/11/13
DOI:10.1039/C5CC09330A
Two newly discovered linear compounds tetraboronate and boroxine stabilized by digermylene are reported, which feature a B4O5 chain and a B3O3 ring, respectively. DFT calculations reveal that not only can digermylene stabilize the electron-deficient boron centers, but also increase the energies of the LUMOs of the boron moiety. Our results provide a hint for the development of boronate covalent organic frameworks.



![Phenol, 2,6-bis(1,1-dimethylethyl)-4-[(diphenylphosphinyl)methyl]-](http://img.cochemist.com/ccimg/15400/15367-77-8.png)
![Phenol, 2,6-bis(1,1-dimethylethyl)-4-[(diphenylphosphinyl)methyl]-](http://img.cochemist.com/ccimg/15400/15367-77-8_b.png)


![Benzenamine, N-[3-(4-methoxyphenyl)-2-propynyl]-](http://img.cochemist.com/ccimg/848800/848738-23-8.png)
![Benzenamine, N-[3-(4-methoxyphenyl)-2-propynyl]-](http://img.cochemist.com/ccimg/848800/848738-23-8_b.png)
![6H-Dibenz[c,e][1,2]oxaphosphorin, 6-phenyl-, 6-oxide](http://img.cochemist.com/ccimg/36300/36240-32-1.png)
![6H-Dibenz[c,e][1,2]oxaphosphorin, 6-phenyl-, 6-oxide](http://img.cochemist.com/ccimg/36300/36240-32-1_b.png)


![Benzenamine, N-[3-(2-thienyl)-2-propyn-1-yl]-](http://img.cochemist.com/ccimg/918900/918866-66-7.png)
![Benzenamine, N-[3-(2-thienyl)-2-propyn-1-yl]-](http://img.cochemist.com/ccimg/918900/918866-66-7_b.png)


![Benzenesulfonamide, 4-methyl-N-[3-(phenylethynyl)-2-pyridinyl]-](http://img.cochemist.com/ccimg/665000/664981-97-9.png)
![Benzenesulfonamide, 4-methyl-N-[3-(phenylethynyl)-2-pyridinyl]-](http://img.cochemist.com/ccimg/665000/664981-97-9_b.png)

![Phosphonic acid, [2-hydroxy-2-(2-thienyl)ethyl]-, bis(1-methylethyl) ester](http://img.cochemist.com/ccimg/719300/719277-52-8.png)
![Phosphonic acid, [2-hydroxy-2-(2-thienyl)ethyl]-, bis(1-methylethyl) ester](http://img.cochemist.com/ccimg/719300/719277-52-8_b.png)
![Formamide, N-(4'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/926700/926644-15-7.png)
![Formamide, N-(4'-methoxy[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/926700/926644-15-7_b.png)
![Benzoic acid, 4-[(3-phenyl-2-propynyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/848800/848738-27-2.png)
![Benzoic acid, 4-[(3-phenyl-2-propynyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/848800/848738-27-2_b.png)
![Ethanone, 1-[4-[3-(phenylamino)-1-propynyl]phenyl]-](http://img.cochemist.com/ccimg/848800/848738-25-0.png)
![Ethanone, 1-[4-[3-(phenylamino)-1-propynyl]phenyl]-](http://img.cochemist.com/ccimg/848800/848738-25-0_b.png)
![Phosphonic acid, [(1E)-2-phenylethenyl]-, bis(phenylmethyl) ester](http://img.cochemist.com/ccimg/848500/848420-33-7.png)
![Phosphonic acid, [(1E)-2-phenylethenyl]-, bis(phenylmethyl) ester](http://img.cochemist.com/ccimg/848500/848420-33-7_b.png)
![Phosphinic acid, phenyl[(1E)-2-phenylethenyl]-, ethyl ester](http://img.cochemist.com/ccimg/83000/82943-05-3.png)
![Phosphinic acid, phenyl[(1E)-2-phenylethenyl]-, ethyl ester](http://img.cochemist.com/ccimg/83000/82943-05-3_b.png)
![Benzenesulfonamide, N-[2-(1-hexynyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/665000/664981-87-7.png)
![Benzenesulfonamide, N-[2-(1-hexynyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/665000/664981-87-7_b.png)





![Formamide, N-(2'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/744300/744262-41-7.png)
![Formamide, N-(2'-methyl[1,1'-biphenyl]-2-yl)-](http://img.cochemist.com/ccimg/744300/744262-41-7_b.png)





![Phosphonic acid, [(1Z)-2-phenylethenyl]-, diethyl ester](http://img.cochemist.com/ccimg/25400/25362-01-0.png)
![Phosphonic acid, [(1Z)-2-phenylethenyl]-, diethyl ester](http://img.cochemist.com/ccimg/25400/25362-01-0_b.png)











![PHOSPHONIC ACID, [1,1'-BIPHENYL]-4-YL-, DIETHYL ESTER](http://img.cochemist.com/ccimg/78000/77918-44-6.png)
![PHOSPHONIC ACID, [1,1'-BIPHENYL]-4-YL-, DIETHYL ESTER](http://img.cochemist.com/ccimg/78000/77918-44-6_b.png)



![Phosphine oxide, [(1E)-2-(4-methoxyphenyl)ethenyl]diphenyl-](http://img.cochemist.com/ccimg/59700/59675-60-4.png)
![Phosphine oxide, [(1E)-2-(4-methoxyphenyl)ethenyl]diphenyl-](http://img.cochemist.com/ccimg/59700/59675-60-4_b.png)
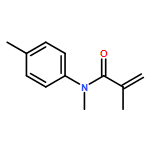
![Benzoic acid, 4-[methyl(2-methyl-1-oxo-2-propenyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/294700/294642-82-3.png)
![Benzoic acid, 4-[methyl(2-methyl-1-oxo-2-propenyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/294700/294642-82-3_b.png)
![Phosphonic acid, [(4-methylphenyl)ethynyl]-, diethyl ester](http://img.cochemist.com/ccimg/176200/176100-86-0.png)
![Phosphonic acid, [(4-methylphenyl)ethynyl]-, diethyl ester](http://img.cochemist.com/ccimg/176200/176100-86-0_b.png)
![[1,1'-Biphenyl]-4-amine, 3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/500/449-42-3.png)
![[1,1'-Biphenyl]-4-amine, 3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/500/449-42-3_b.png)





![Phosphonic acid, [2-(4-bromophenyl)-2-oxoethyl]-, diethyl ester](http://img.cochemist.com/ccimg/18300/18276-83-0.png)
![Phosphonic acid, [2-(4-bromophenyl)-2-oxoethyl]-, diethyl ester](http://img.cochemist.com/ccimg/18300/18276-83-0_b.png)






![[1,1'-Biphenyl]-4-amine, N,N-diphenyl-](http://img.cochemist.com/ccimg/4500/4432-94-4.png)
![[1,1'-Biphenyl]-4-amine, N,N-diphenyl-](http://img.cochemist.com/ccimg/4500/4432-94-4_b.png)



![3-Buten-1-ol, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/173600/173546-40-2.png)
![3-Buten-1-ol, 4-[4-(trifluoromethyl)phenyl]-, (3E)-](http://img.cochemist.com/ccimg/173600/173546-40-2_b.png)









![Acetonitrile, [(2,2,6,6-tetramethyl-1-piperidinyl)oxy]-](http://img.cochemist.com/ccimg/102300/102261-91-6.png)
![Acetonitrile, [(2,2,6,6-tetramethyl-1-piperidinyl)oxy]-](http://img.cochemist.com/ccimg/102300/102261-91-6_b.png)









![BENZENAMINE, N-[3-(4-FLUOROPHENYL)-2-PROPYNYL]-](http://img.cochemist.com/ccimg/848800/848738-24-9.png)
![BENZENAMINE, N-[3-(4-FLUOROPHENYL)-2-PROPYNYL]-](http://img.cochemist.com/ccimg/848800/848738-24-9_b.png)

![[1,1'-BIPHENYL]-2-AMINE, 4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/400/363-08-6.png)
![[1,1'-BIPHENYL]-2-AMINE, 4-(TRIFLUOROMETHYL)-](http://img.cochemist.com/ccimg/400/363-08-6_b.png)






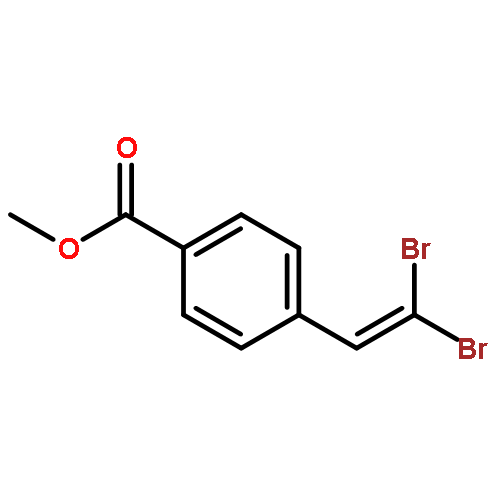
![Benzene, [(1E)-4,4-dibromo-1,3-butadienyl]-](http://img.cochemist.com/ccimg/77300/77295-76-2.png)
![Benzene, [(1E)-4,4-dibromo-1,3-butadienyl]-](http://img.cochemist.com/ccimg/77300/77295-76-2_b.png)
![[1,1'-Biphenyl]-4-amine, 3-chloro-](http://img.cochemist.com/ccimg/7300/7285-66-7.png)
![[1,1'-Biphenyl]-4-amine, 3-chloro-](http://img.cochemist.com/ccimg/7300/7285-66-7_b.png)
![Phosphine oxide, [1,1'-biphenyl]-4-yldiphenyl-](http://img.cochemist.com/ccimg/2000/1942-83-2.png)
![Phosphine oxide, [1,1'-biphenyl]-4-yldiphenyl-](http://img.cochemist.com/ccimg/2000/1942-83-2_b.png)
![Phosphonic acid, [2-(4-methoxyphenyl)-2-oxoethyl]-, diethyl ester](http://img.cochemist.com/ccimg/18300/18276-85-2.png)
![Phosphonic acid, [2-(4-methoxyphenyl)-2-oxoethyl]-, diethyl ester](http://img.cochemist.com/ccimg/18300/18276-85-2_b.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)


![[1,1'-Biphenyl]-4-amine, 3-fluoro-](http://img.cochemist.com/ccimg/76400/76302-56-2.png)
![[1,1'-Biphenyl]-4-amine, 3-fluoro-](http://img.cochemist.com/ccimg/76400/76302-56-2_b.png)
![Phosphonic acid, P-[phenyl(phenylamino)methyl]-, diethyl ester](/data/chemimg/1951600/7236-88-6.png)
![Phosphonic acid, P-[phenyl(phenylamino)methyl]-, diethyl ester](/data/chemimg/1951600/7236-88-6_b.png)


![Phosphonic acid, [(1E)-2-phenylethenyl]-, bis(1-methylethyl) ester](http://img.cochemist.com/ccimg/78500/78463-00-0.png)
![Phosphonic acid, [(1E)-2-phenylethenyl]-, bis(1-methylethyl) ester](http://img.cochemist.com/ccimg/78500/78463-00-0_b.png)






![[1,1'-BIPHENYL]-2-CARBOXYLIC ACID, 5-AMINO-, METHYL ESTER](http://img.cochemist.com/ccimg/52000/51990-95-5.png)
![[1,1'-BIPHENYL]-2-CARBOXYLIC ACID, 5-AMINO-, METHYL ESTER](http://img.cochemist.com/ccimg/52000/51990-95-5_b.png)
![1-[bromo(ethoxy)phosphoryl]oxyethane](http://img.cochemist.com/ccimg/51800/51761-27-4.png)
![1-[bromo(ethoxy)phosphoryl]oxyethane](http://img.cochemist.com/ccimg/51800/51761-27-4_b.png)
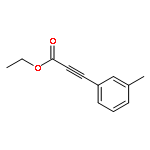
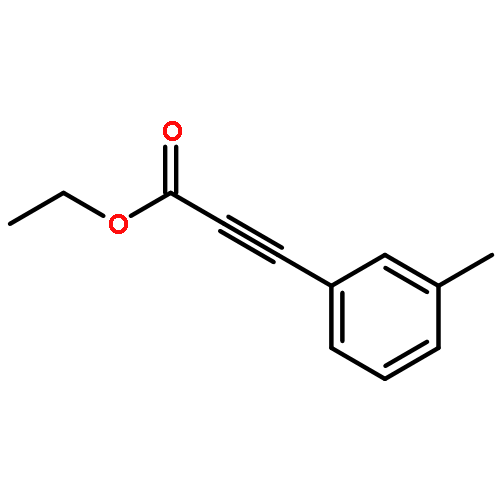


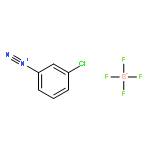
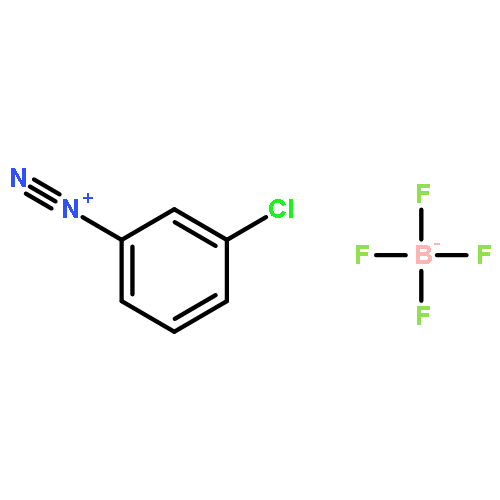
![2-Propynoic acid, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/3800/3792-88-9.png)
![2-Propynoic acid, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/3800/3792-88-9_b.png)
















![2-Propynoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/220100/220006-53-1.png)
![2-Propynoic acid, 3-[4-(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/220100/220006-53-1_b.png)








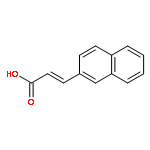
![2-Propynoic acid, 3-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/32400/32340-38-8.png)
![2-Propynoic acid, 3-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/32400/32340-38-8_b.png)







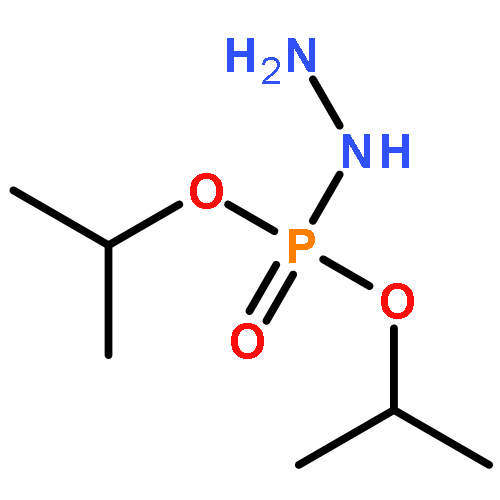



![2-Propenoic acid, 3-[4-(dimethylamino)phenyl]-, (2E)-](/data/chemimg/1851700/5676-65-3.png)
![2-Propenoic acid, 3-[4-(dimethylamino)phenyl]-, (2E)-](/data/chemimg/1851700/5676-65-3_b.png)
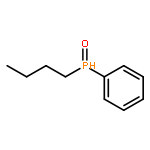
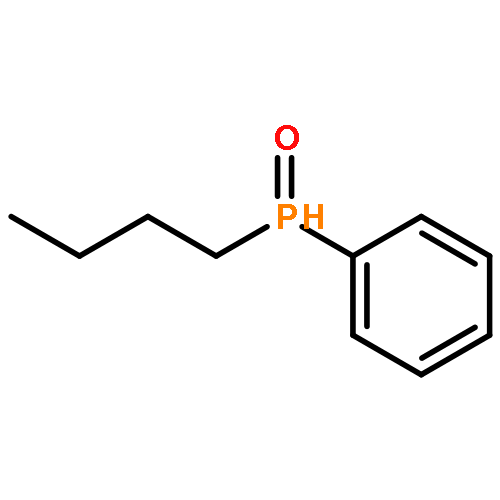

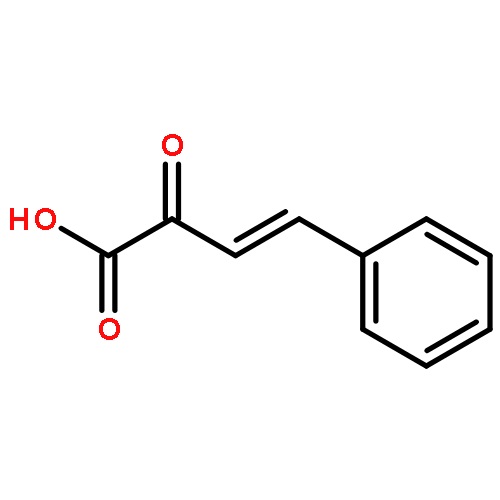
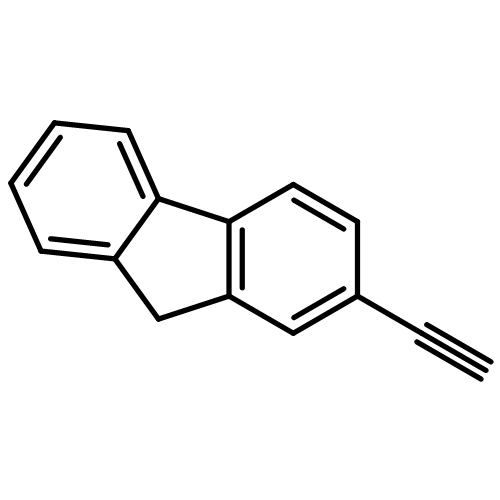
![Phosphonic acid, [2-(4-chlorophenyl)-2-oxoethyl]-, diethyl ester](http://img.cochemist.com/ccimg/18300/18276-82-9.png)
![Phosphonic acid, [2-(4-chlorophenyl)-2-oxoethyl]-, diethyl ester](http://img.cochemist.com/ccimg/18300/18276-82-9_b.png)
![Phosphonic acid, P-[2-(4-methylphenyl)-2-oxoethyl]-, diethyl ester](/data/chemimg/1767000/18276-81-8.png)
![Phosphonic acid, P-[2-(4-methylphenyl)-2-oxoethyl]-, diethyl ester](/data/chemimg/1767000/18276-81-8_b.png)
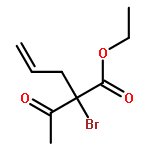

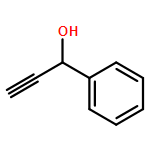
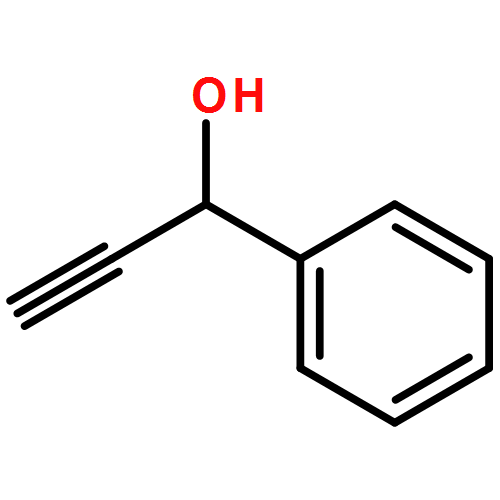
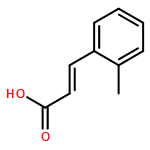
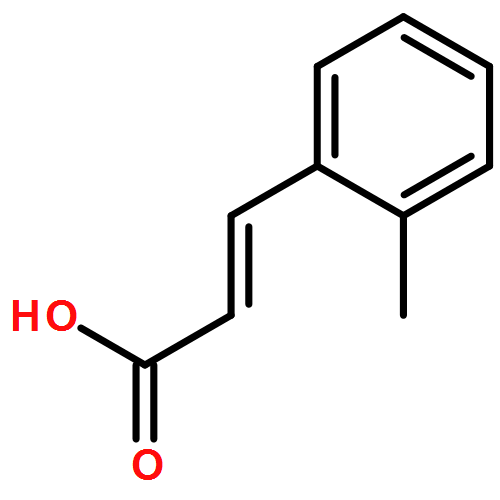
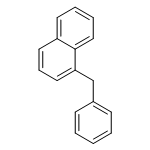
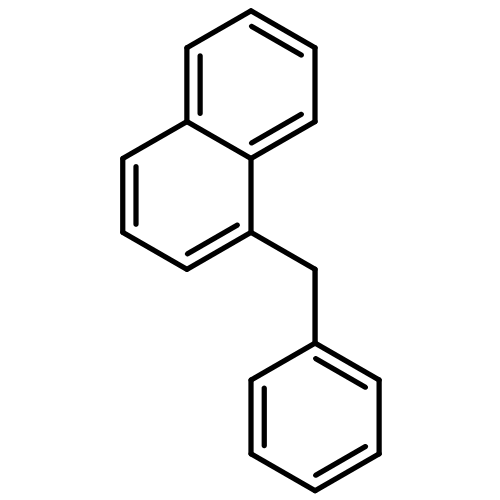


![1,2,7,8-TETRACHLORODIBENZO[B,D]FUR](http://img.cochemist.com/ccimg/800/767-87-3.png)
![1,2,7,8-TETRACHLORODIBENZO[B,D]FUR](http://img.cochemist.com/ccimg/800/767-87-3_b.png)




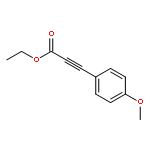
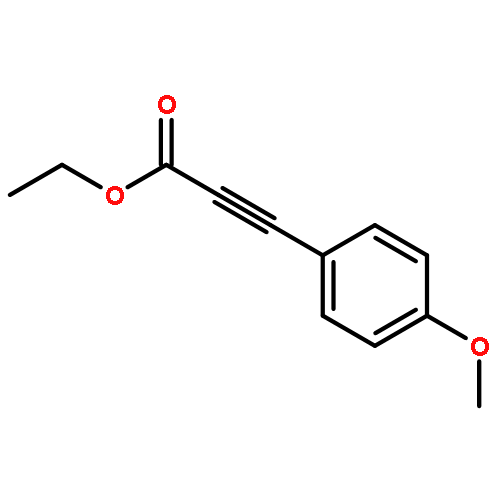
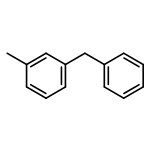
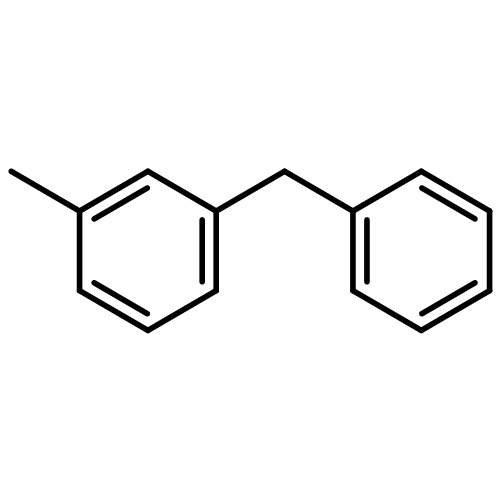
![Phosphine oxide, diphenyl[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/62800/62754-67-0.png)
![Phosphine oxide, diphenyl[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/62800/62754-67-0_b.png)

![Phosphine oxide, diphenyl[(1Z)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/78100/78045-10-0.png)
![Phosphine oxide, diphenyl[(1Z)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/78100/78045-10-0_b.png)
![[1,1'-Biphenyl]-2-amine, 3'-methyl-](/data/chemimg/670700/73818-73-2.png)
![[1,1'-Biphenyl]-2-amine, 3'-methyl-](/data/chemimg/670700/73818-73-2_b.png)
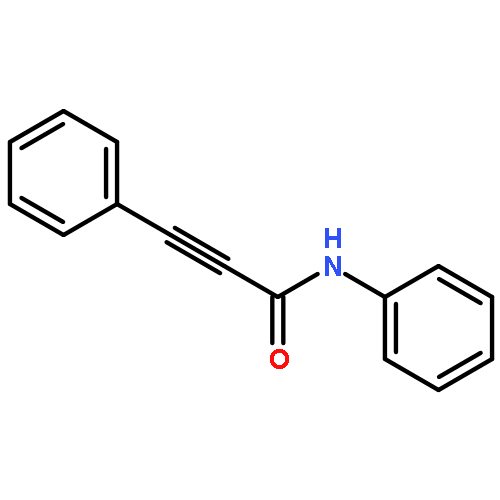


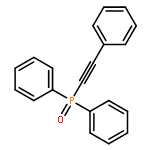
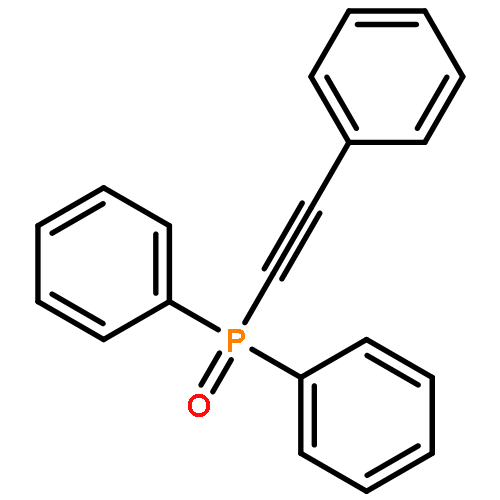
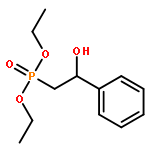
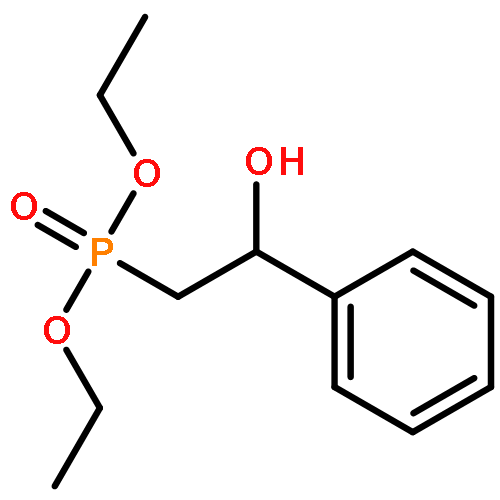



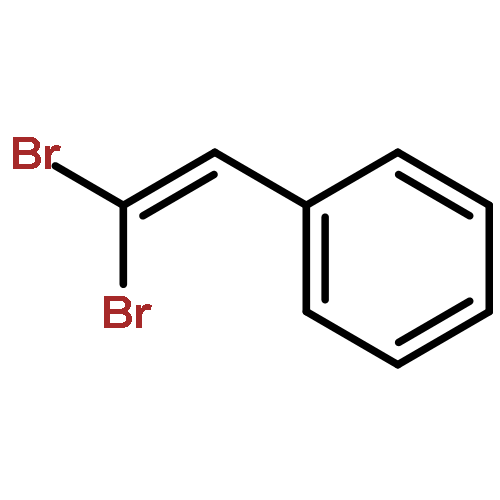
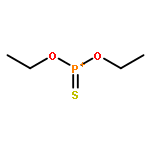



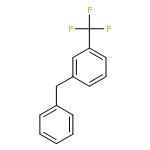

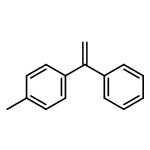
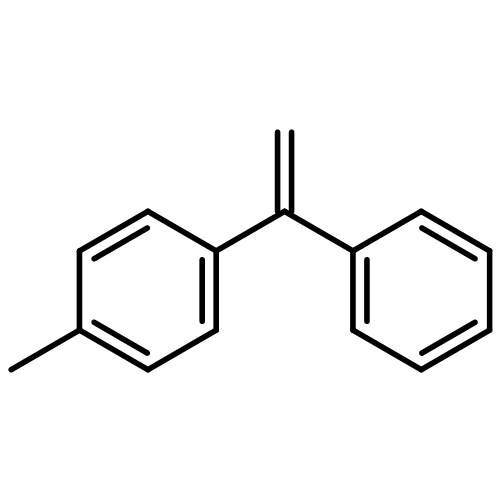


![Phosphonic acid, [(1E)-2-(4-methoxyphenyl)ethenyl]-, bis(1-methylethyl)ester](http://img.cochemist.com/ccimg/168100/168025-52-3.png)
![Phosphonic acid, [(1E)-2-(4-methoxyphenyl)ethenyl]-, bis(1-methylethyl)ester](http://img.cochemist.com/ccimg/168100/168025-52-3_b.png)











![Acetamide, N-[2-(phenylethynyl)phenyl]-](http://img.cochemist.com/ccimg/26400/26385-33-1.png)
![Acetamide, N-[2-(phenylethynyl)phenyl]-](http://img.cochemist.com/ccimg/26400/26385-33-1_b.png)




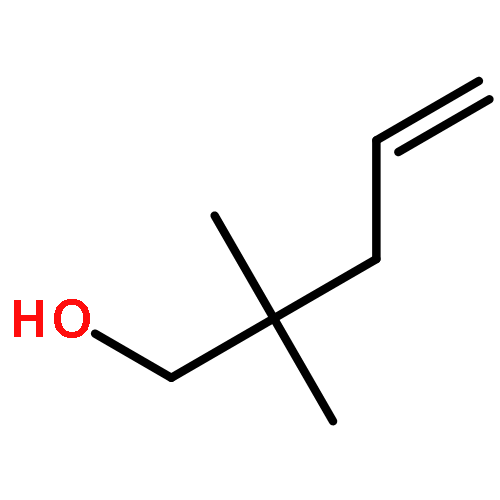
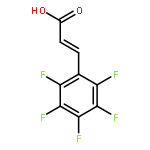
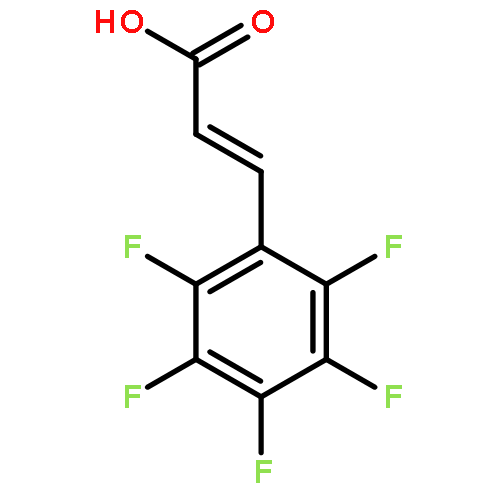

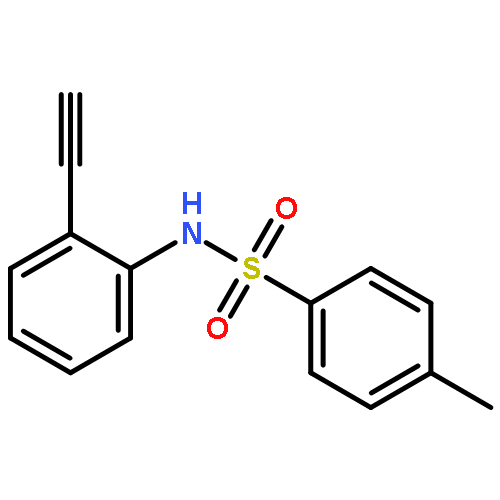







![Phosphonic acid, [phenyl[(phenylmethyl)amino]methyl]-, diethyl ester](http://img.cochemist.com/ccimg/63100/63000-01-1.png)
![Phosphonic acid, [phenyl[(phenylmethyl)amino]methyl]-, diethyl ester](http://img.cochemist.com/ccimg/63100/63000-01-1_b.png)









![Phosphoric acid, diethyl [3-(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/128800/128732-38-7.png)
![Phosphoric acid, diethyl [3-(trifluoromethyl)phenyl]methyl ester](http://img.cochemist.com/ccimg/128800/128732-38-7_b.png)




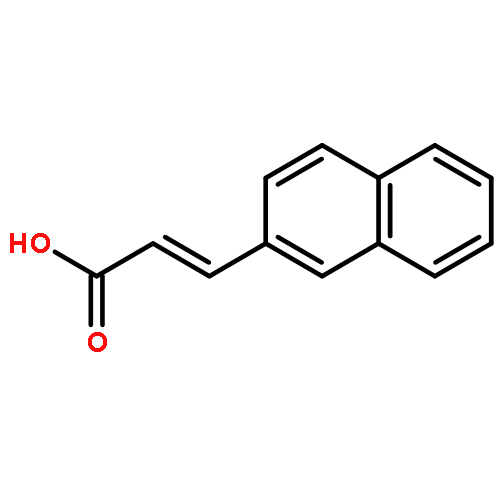
![2-Propenoic acid,3-[1,1'-biphenyl]-4-yl-, (2E)-](http://img.cochemist.com/ccimg/88300/88241-65-0.png)
![2-Propenoic acid,3-[1,1'-biphenyl]-4-yl-, (2E)-](http://img.cochemist.com/ccimg/88300/88241-65-0_b.png)








![[PHENYL(PHENYLSULFANYL)PHOSPHORYL]BENZENE](http://img.cochemist.com/ccimg/5600/5510-78-1.png)
![[PHENYL(PHENYLSULFANYL)PHOSPHORYL]BENZENE](http://img.cochemist.com/ccimg/5600/5510-78-1_b.png)



![Phosphonic acid, [(1E)-2-phenylethenyl]-, diethyl ester](http://img.cochemist.com/ccimg/20500/20408-33-7.png)
![Phosphonic acid, [(1E)-2-phenylethenyl]-, diethyl ester](http://img.cochemist.com/ccimg/20500/20408-33-7_b.png)
![4'-Fluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/5800/5731-10-2.png)
![4'-Fluoro-[1,1'-biphenyl]-4-carboxylic acid](http://img.cochemist.com/ccimg/5800/5731-10-2_b.png)












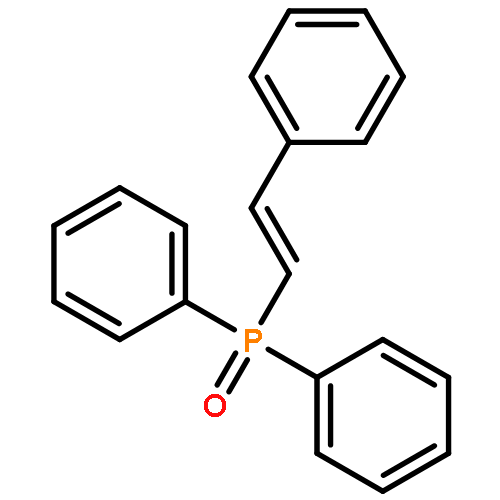
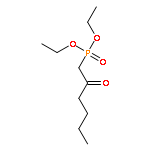
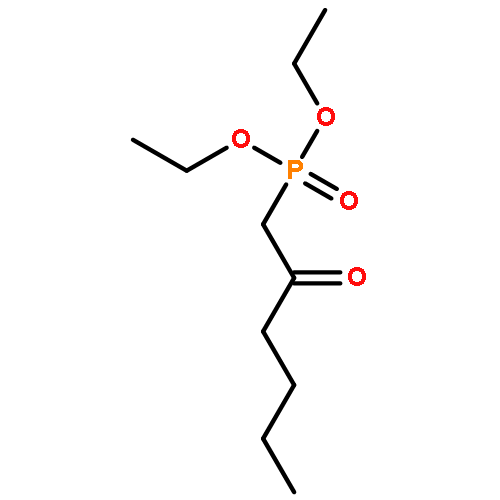



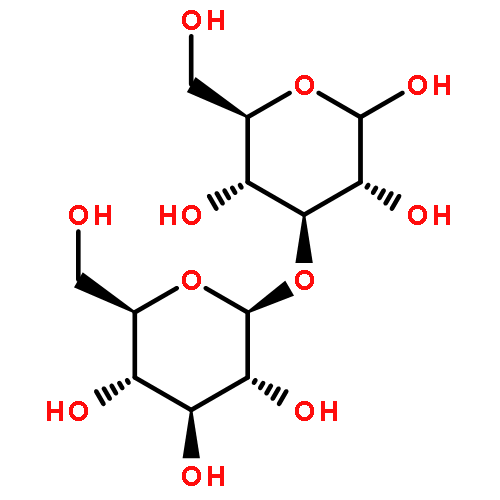



![1H-Pyrrole-2-carboxamide,N-[5-[[(3-amino-3-iminopropyl)amino]carbonyl]-1-methyl-1H-pyrrol-3-yl]-4-[[[4-(formylamino)-1-methyl-1H-pyrrol-2-yl]carbonyl]amino]-1-methyl-](http://img.cochemist.com/ccimg/700/636-47-5.png)
![1H-Pyrrole-2-carboxamide,N-[5-[[(3-amino-3-iminopropyl)amino]carbonyl]-1-methyl-1H-pyrrol-3-yl]-4-[[[4-(formylamino)-1-methyl-1H-pyrrol-2-yl]carbonyl]amino]-1-methyl-](http://img.cochemist.com/ccimg/700/636-47-5_b.png)







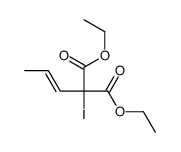


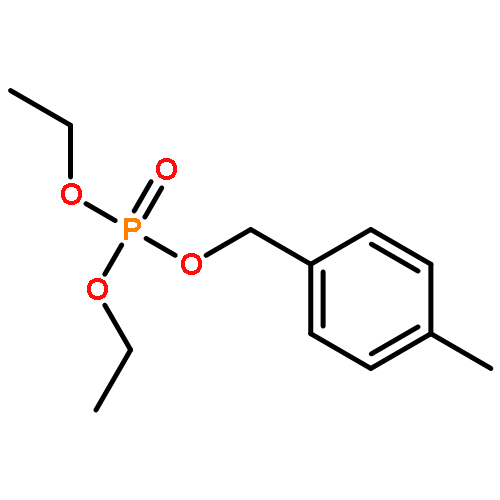
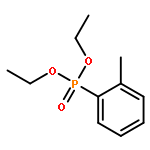
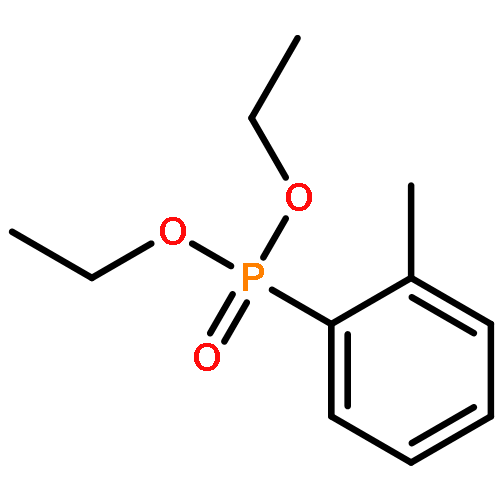


![Phosphine oxide, diphenyl[(1E)-2-[4-(trifluoromethyl)phenyl]ethenyl]-](/data/chemimg/202600/1429762-73-1.png)
![Phosphine oxide, diphenyl[(1E)-2-[4-(trifluoromethyl)phenyl]ethenyl]-](/data/chemimg/202600/1429762-73-1_b.png)




![Phosphine oxide, diphenyl[(1E)-2-(2-thienyl)ethenyl]-](/data/chemimg/120700/362514-10-1.png)
![Phosphine oxide, diphenyl[(1E)-2-(2-thienyl)ethenyl]-](/data/chemimg/120700/362514-10-1_b.png)



















![6,7-diethoxy-1-[4-methoxy-3-(1-methylethyl)phenyl]-1,2,3,4-tetrahydroisoquinoline](http://img.cochemist.com/ccimg/5100/5068-23-5.png)
![6,7-diethoxy-1-[4-methoxy-3-(1-methylethyl)phenyl]-1,2,3,4-tetrahydroisoquinoline](http://img.cochemist.com/ccimg/5100/5068-23-5_b.png)



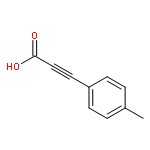
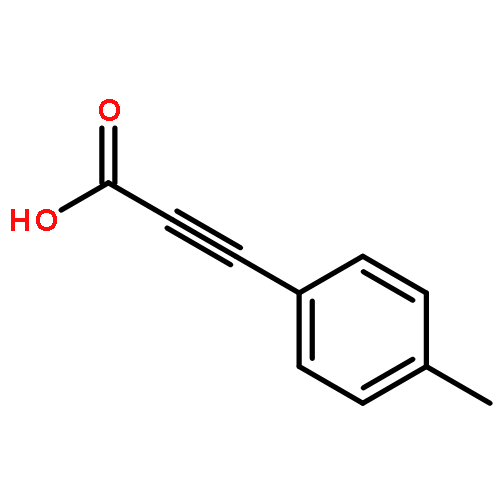


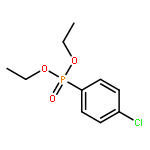
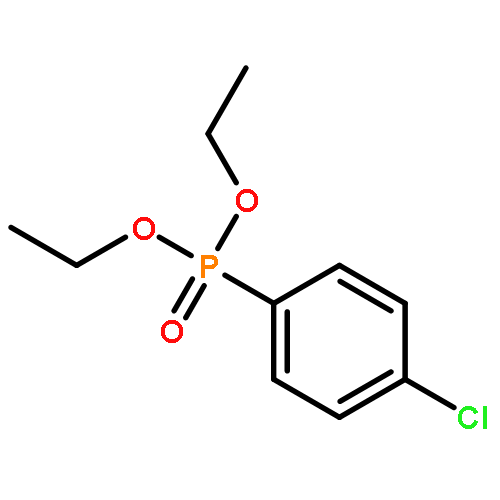



![Benzene, 1-methyl-3-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/21900/21895-16-9.png)
![Benzene, 1-methyl-3-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/21900/21895-16-9_b.png)
![[1,1'-Biphenyl]-2-amine,2'-methyl-](http://img.cochemist.com/ccimg/1300/1203-41-4.png)
![[1,1'-Biphenyl]-2-amine,2'-methyl-](http://img.cochemist.com/ccimg/1300/1203-41-4_b.png)
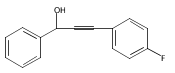
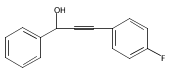
![Benzenemethanol, a-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/109700/109643-11-0.png)
![Benzenemethanol, a-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/109700/109643-11-0_b.png)





































![4-Methyl-[1,1'-biphenyl]-2-amine](http://img.cochemist.com/ccimg/42400/42308-28-1.png)
![4-Methyl-[1,1'-biphenyl]-2-amine](http://img.cochemist.com/ccimg/42400/42308-28-1_b.png)





![Benzenesulfonamide, 4-methyl-N-[2-(2-phenylethynyl)phenyl]-](/data/chemimg/65200/442155-91-1.png)
![Benzenesulfonamide, 4-methyl-N-[2-(2-phenylethynyl)phenyl]-](/data/chemimg/65200/442155-91-1_b.png)










![Phosphonic acid, [(cyclohexylamino)phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/184000/183964-51-4.png)
![Phosphonic acid, [(cyclohexylamino)phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/184000/183964-51-4_b.png)


![Phosphonic acid, [(butylamino)phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/157900/157841-12-8.png)
![Phosphonic acid, [(butylamino)phenylmethyl]-, diethyl ester](http://img.cochemist.com/ccimg/157900/157841-12-8_b.png)
![Phosphine oxide, [(1E)-2-cyclohexylethenyl]diphenyl-](/data/chemimg/120600/112863-51-1.png)
![Phosphine oxide, [(1E)-2-cyclohexylethenyl]diphenyl-](/data/chemimg/120600/112863-51-1_b.png)




![[1,1'-Biphenyl]-2-amine, 4'-methyl-](/data/chemimg/370200/1204-43-9.png)
![[1,1'-Biphenyl]-2-amine, 4'-methyl-](/data/chemimg/370200/1204-43-9_b.png)


![Phosphonic acid, [phenyl[(phenylmethyl)amino]methyl]-, dimethyl ester](http://img.cochemist.com/ccimg/192900/192868-43-2.png)
![Phosphonic acid, [phenyl[(phenylmethyl)amino]methyl]-, dimethyl ester](http://img.cochemist.com/ccimg/192900/192868-43-2_b.png)










![Hydrazine,[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/2300/2217-77-8.png)
![Hydrazine,[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/2300/2217-77-8_b.png)


![Benzenesulfonamide, 4-methyl-N-[4-methyl-2-(2-phenylethynyl)phenyl]-](/data/chemimg/107200/946161-99-5.png)
![Benzenesulfonamide, 4-methyl-N-[4-methyl-2-(2-phenylethynyl)phenyl]-](/data/chemimg/107200/946161-99-5_b.png)