Co-reporter:Fanxun Zeng;Tiantian Qi;Chunyan Li;Tingfang Li;Honglin Li;Shiliang Li;Xiaoyong Xu
MedChemComm (2010-Present) 2017 vol. 8(Issue 6) pp:1297-1302
Publication Date(Web):2017/06/21
DOI:10.1039/C7MD00081B
A series of 4-thiazolidinone derivatives were synthesized and evaluated as novel human dihydroorotate dehydrogenase (hDHODH) inhibitors. Compounds 26 and 31 displayed IC50 values of 1.75 and 1.12 μM, respectively. The structure–activity relationship was summarized. Further binding mode analysis revealed that compound 31 could form a hydrogen bond with Tyr38 and a water-mediated hydrogen bond with Ala55, which may be necessary for maintaining the bioactivities of the compounds in this series. Further structural optimization of the para- or meta-position of the phenyl group at R will lead to the identification of more potent hDHODH inhibitors.
Co-reporter:Fanxun Zeng;Tiantian Qi;Chunyan Li;Tingfang Li;Honglin Li;Shiliang Li;Xiaoyong Xu
MedChemComm (2010-Present) 2017 vol. 8(Issue 6) pp:1297-1302
Publication Date(Web):2017/06/21
DOI:10.1039/C7MD00081B
A series of 4-thiazolidinone derivatives were synthesized and evaluated as novel human dihydroorotate dehydrogenase (hDHODH) inhibitors. Compounds 26 and 31 displayed IC50 values of 1.75 and 1.12 μM, respectively. The structure–activity relationship was summarized. Further binding mode analysis revealed that compound 31 could form a hydrogen bond with Tyr38 and a water-mediated hydrogen bond with Ala55, which may be necessary for maintaining the bioactivities of the compounds in this series. Further structural optimization of the para- or meta-position of the phenyl group at R will lead to the identification of more potent hDHODH inhibitors.
Co-reporter:Fangshu Wu, Junsheng Zhu, Honglin Li, Lili Zhu
Acta Pharmaceutica Sinica B 2017 Volume 7, Issue 3(Issue 3) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.apsb.2016.12.008
UbcH5c belongs to the ubiquitin-conjugating enzyme family and plays an important role in catalyzing ubiquitination during TNF-α--triggered NF-κB activation. Therefore, UbcH5c is a potent therapeutic target for the treatment of inflammatory and autoimmune diseases induced by aberrant activation of NF-κB. In this study, we established a stable expression system for recombinant UbcH5c and solved the crystal structure of UbcH5c belonging to space group P22121 with one molecule in the asymmetric unit. This study provides the basis for further study of UbcH5c including the design of UbcH5c inhibitors.A stable expression system for recombinant UbcH5c was established and one crystal structure of UbcH5c solved at a high resolution. This study provides the basis for further study of UbcH5c including the design of UbcH5c inhibitors.Download high-res image (317KB)Download full-size image
Co-reporter:Wenlin Song, Shiliang Li, Yi Tong, Jiawei Wang, Lina Quan, Zhuo Chen, Zhenjiang Zhao, Yufang Xu, Lili Zhu, Xuhong Qian and Honglin Li
MedChemComm 2016 vol. 7(Issue 7) pp:1441-1448
Publication Date(Web):13 Jun 2016
DOI:10.1039/C6MD00179C
It has been proven that inhibiting human dihydroorotate dehydrogenase (hDHODH) restricts the growth of rapidly proliferating cells, thus hDHODH can be developed as a promising target for the treatment of immunological disease and cancer. Here, a succession of substituted hydrazino-thiazole derivatives were designed, synthesized, and biologically evaluated through structure-based optimization, of which compound 22 was the most potent inhibitor of hDHODH with an IC50 value of 1.8 nM. Furthermore, 22 exhibited much better antiproliferative activity than brequinar, both in HCT-116 and BxPC-3 cancer cell lines. Flow cytometry analysis revealed that 22 induced S phase cell cycle arrest and promoted induction of apoptosis. All results established a proof that blocking the pyrimidine de novo synthesis pathway by inhibiting the rate-limiting enzyme hDHODH is an attractive therapy for cancer.
Co-reporter:Jiawei Wang, Yanyan Diao, Junsheng Zhu, Shiliang Li, Zhenjiang Zhao, Honglin Li and Lili Zhu
MedChemComm 2016 vol. 7(Issue 5) pp:853-858
Publication Date(Web):05 Feb 2016
DOI:10.1039/C6MD00024J
Human dihydroorotate dehydrogenase (hDHODH) is an enzyme that catalyzes the fourth step in de novo pyrimidine biosynthesis, and its inhibitors restrict the growth of rapidly proliferating cells. Therefore, hDHODH has been reported as an attractive target for the treatment of cancer and autoimmune diseases. In this study, several quinoline derivatives were identified as potent inhibitors against hDHODH, among which compound A9 was the most potent one with an IC50 value of 9.7 nM. We further verified by thermal shift assay (TSA), surface plasmon resonance (SPR) and X-ray crystallography that A9 could directly bind to the target hDHODH. The crystal structure of hDHODH in complex with compound A9 was refined to 1.90 Å and the binding mode of compound A9 was summarized. Moreover, structure–activity relationship (SAR) analysis of quinoline derivatives with hDHODH indicated that quinoline derivatives with a carboxyl group in R1, a bromine atom in R2 and a para-alkyl-substituted phenyl group in R5, were beneficial for potency against hDHODH, which plays an important role in inhibitor design and optimization.
Co-reporter:Junsheng Zhu; Le Han; Yanyan Diao; Xiaoli Ren; Minghao Xu; Liuxin Xu; Shiliang Li; Qiang Li; Dong Dong; Jin Huang; Xiaofeng Liu; Zhenjiang Zhao; Rui Wang; Lili Zhu; Yufang Xu; Xuhong Qian;Honglin Li
Journal of Medicinal Chemistry 2015 Volume 58(Issue 3) pp:1123-1139
Publication Date(Web):January 12, 2015
DOI:10.1021/jm501127s
Human dihydroorotate dehydrogenase (HsDHODH) is a flavin-dependent mitochondrial enzyme that has been certified as a potential therapeutic target for the treatment of rheumatoid arthritis and other autoimmune diseases. On the basis of lead compound 4, which was previously identified as potential HsDHODH inhibitor, a novel series of thiazole derivatives were designed and synthesized. The X-ray complex structures of the promising analogues 12 and 33 confirmed that these inhibitors bind at the putative ubiquinone binding tunnel and guided us to explore more potent inhibitors, such as compounds 44, 46, and 47 which showed double digit nanomolar activities of 26, 18, and 29 nM, respectively. Moreover, 44 presented considerable anti-inflammation effect in vivo and significantly alleviated foot swelling in a dose-dependent manner, which disclosed that thiazole-scaffold analogues can be developed into the drug candidates for the treatment of rheumatoid arthritis by suppressing the bioactivity of HsDHODH.
Co-reporter:Minghao Xu ; Junsheng Zhu ; Yanyan Diao ; Hongchang Zhou ; Xiaoli Ren ; Deheng Sun ; Jin Huang ; Dongmei Han ; Zhenjiang Zhao ; Lili Zhu ; Yufang Xu ;Honglin Li
Journal of Medicinal Chemistry 2013 Volume 56(Issue 20) pp:7911-7924
Publication Date(Web):September 27, 2013
DOI:10.1021/jm400938g
Taking the emergence of drug resistance and lack of effective antimalarial vaccines into consideration, it is of significant importance to develop novel antimalarial agents for the treatment of malaria. Herein, we elucidated the discovery and structure–activity relationships of a series of dihydrothiophenone derivatives as novel specific inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH). The most promising compound, 50, selectively inhibited PfDHODH (IC50 = 6 nM, with >14 000-fold species-selectivity over hDHODH) and parasite growth in vitro (IC50 = 15 and 18 nM against 3D7 and Dd2 cells, respectively). Moreover, an oral bioavailability of 40% for compound 50 was determined from in vivo pharmacokinetic studies. These results further indicate that PfDHODH is an effective target for antimalarial chemotherapy, and the novel scaffolds reported in this work might lead to the discovery of new antimalarial agents.
Co-reporter:Yuwei Song, Huangtao Jin, Xiaofeng Liu, Lili Zhu, Jin Huang, Honglin Li
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 7) pp:2078-2082
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmcl.2013.01.128
Plasmepsin II (PM II) is an attractive target for anti-malaria drug discovery, which involves in host hemoglobin degradation in the acidic food vacuole. In this study, we demonstrated the successful use of structure-based virtual screening to identify inhibitors of PM II from two chemical database. Five novel non-peptide inhibitors were identified and revealed moderate inhibitory potencies with IC50 ranged from 4.62 ± 0.39 to 9.47 ± 0.71 μM. The detailed analysis of binding modes using docking simulations for five inhibitors showed that the inhibitors could be stabilized by forming multiple hydrogen bonds with catalytic residues (Asp 34 and Asp 214) and also with other key residues.

![Benzenamine, 4-[2-(dimethylamino)ethoxy]-2-methoxy-](http://img.cochemist.com/ccimg/927700/927672-74-0.png)
![Benzenamine, 4-[2-(dimethylamino)ethoxy]-2-methoxy-](http://img.cochemist.com/ccimg/927700/927672-74-0_b.png)


![2-METHOXY-4-[4-(4-METHYLPIPERAZIN-1-YL)PIPERIDIN-1-YL]ANILINE](http://img.cochemist.com/ccimg/761500/761440-75-9.png)
![2-METHOXY-4-[4-(4-METHYLPIPERAZIN-1-YL)PIPERIDIN-1-YL]ANILINE](http://img.cochemist.com/ccimg/761500/761440-75-9_b.png)






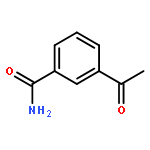

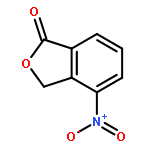

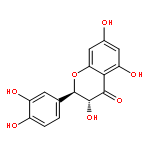
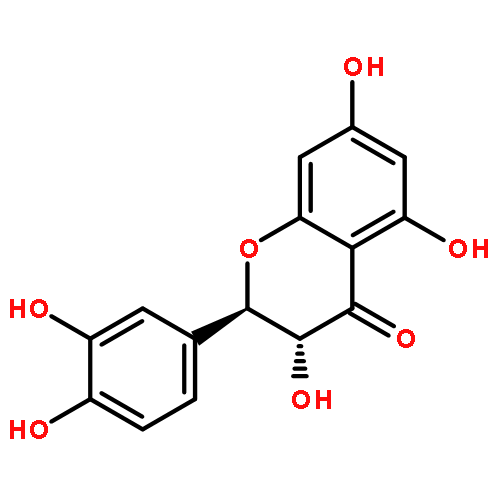
![Benzoic acid, 3-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/61500/61471-37-2.png)
![Benzoic acid, 3-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/61500/61471-37-2_b.png)






![Benzoic acid, 4-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/200/122-92-9.png)
![Benzoic acid, 4-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/200/122-92-9_b.png)


![Benzene, 1-chloro-2-[(2-chloro-4-nitrophenoxy)methyl]-3-fluoro-](http://img.cochemist.com/ccimg/867300/867288-06-0.png)
![Benzene, 1-chloro-2-[(2-chloro-4-nitrophenoxy)methyl]-3-fluoro-](http://img.cochemist.com/ccimg/867300/867288-06-0_b.png)
![Benzenamine, 3-chloro-4-[(2-chloro-6-fluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/867300/867288-04-8.png)
![Benzenamine, 3-chloro-4-[(2-chloro-6-fluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/867300/867288-04-8_b.png)
![[1,1'-Biphenyl]-4-amine, 3,5-difluoro-3'-methoxy-](http://img.cochemist.com/ccimg/867300/867288-00-4.png)
![[1,1'-Biphenyl]-4-amine, 3,5-difluoro-3'-methoxy-](http://img.cochemist.com/ccimg/867300/867288-00-4_b.png)

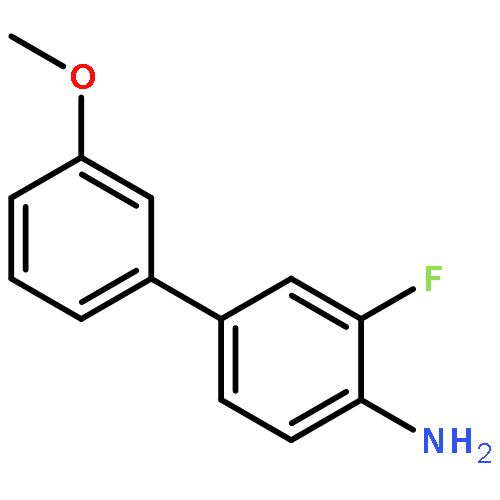




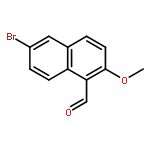


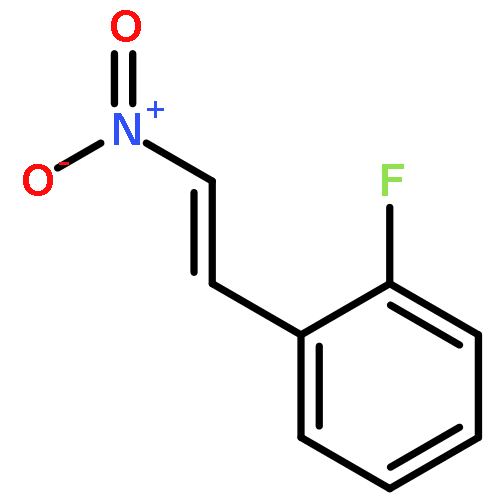
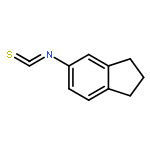



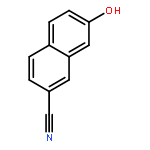
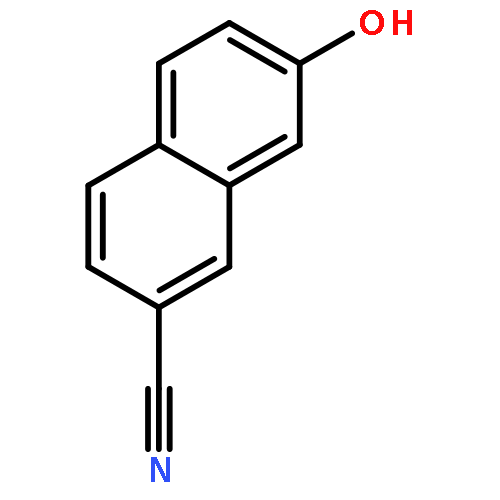





![4H-1-Benzopyran-4-one, 2-(4-hydroxyphenyl)-7-methoxy-5-[(6-O-β-D-xylopyranosyl-β-D-glucopyranosyl)oxy]-](/data/chemimg/1972600/77099-20-8.png)
![4H-1-Benzopyran-4-one, 2-(4-hydroxyphenyl)-7-methoxy-5-[(6-O-β-D-xylopyranosyl-β-D-glucopyranosyl)oxy]-](/data/chemimg/1972600/77099-20-8_b.png)
![[1,1'-Biphenyl]-4-amine, 3-fluoro-](http://img.cochemist.com/ccimg/76400/76302-56-2.png)
![[1,1'-Biphenyl]-4-amine, 3-fluoro-](http://img.cochemist.com/ccimg/76400/76302-56-2_b.png)








![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6.png)
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6_b.png)
![Thiourea,N-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/19300/19250-03-4.png)
![Thiourea,N-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/19300/19250-03-4_b.png)








![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)