Co-reporter:Gao-Lei Hou, Bo Chen, Wesley J. Transue, Zheng Yang, Hansjörg Grützmacher, Matthias Driess, Christopher C. Cummins, Weston Thatcher Borden, and Xue-Bin Wang
Journal of the American Chemical Society July 5, 2017 Volume 139(Issue 26) pp:8922-8922
Publication Date(Web):June 7, 2017
DOI:10.1021/jacs.7b02984
Three newly synthesized [Na+(221-Kryptofix)] salts containing AsCO–, PCO–, and PCS– anions were successfully electrosprayed into a vacuum, and these three ECX– anions were investigated by negative ion photoelectron spectroscopy (NIPES) along with high-resolution photoelectron imaging spectroscopy. For each ECX– anion, a well-resolved NIPE spectrum was obtained, in which every major peak is split into a doublet. The splittings are attributed to spin–orbit coupling (SOC) in the ECX• radicals. Vibrational progressions in the NIPE spectra of ECX– were assigned to the symmetric and the antisymmetric stretching modes in ECX• radicals. The electron affinities (EAs) and SO splittings of ECX• are determined from the NIPE spectra to be AsCO•: EA = 2.414 ± 0.002 eV, SO splitting = 988 cm–1; PCO•: EA = 2.670 ± 0.005 eV, SO splitting = 175 cm–1; PCS•: EA = 2.850 ± 0.005 eV, SO splitting = 300 cm–1. Calculations using the B3LYP, CASPT2, and CCSD(T) methods all predict linear geometries for both the anions and the neutral radicals. The calculated EAs and SO splittings for ECX• are in excellent agreement with the experimentally measured values. The simulated NIPE spectra, which are based on the calculated Franck–Condon factors, and the SO splittings nicely reproduce all of the observed spectral peaks, thus allowing unambiguous spectral assignments. The finding that PCS• has the greatest EA of the three triatomic molecules considered here is counterintuitive based upon simple electronegativity considerations, but this finding is understandable in terms of the movement of electron density from phosphorus in the HOMO of PCO– to sulfur in the HOMO of PCS–. Comparisons of the EAs of PCO• and PCS• with the previously measured EA values for NCO• and NCS• are made and discussed.
Co-reporter:Julia M. Stauber, Glen E. Alliger, Daniel G. Nocera, and Christopher C. Cummins
Inorganic Chemistry July 17, 2017 Volume 56(Issue 14) pp:7615-7615
Publication Date(Web):June 30, 2017
DOI:10.1021/acs.inorgchem.7b01335
The preparation of a selective turn-on colorimetric fluoride sensor was achieved through single cobalt(II) ion insertion into a macrobicyclic cryptand. Monometallic [Co(mBDCA-5t-H3)]− (1) and [Zn(mBDCA-5t-H3)]− (2) complexes were prepared in 74 and 84% yields, respectively. Structural characterization of 1 confirmed the presence of a proximal hydrogen-bonding network consisting of carboxamide N–H donors. The reaction of 1 with F– was accompanied by a distinct colorimetric turn-on response in mixed aqueous/organic media, and 1 was capable of selective fluoride sensing in the presence of large quantities of potentially competitive anions. Complex 1 represents a unique example of a fluoride sensor wherein selective F– binding takes place directly at a transition-metal center and induces a color change based upon metal-centered transitions. The metal(II) fluoride complexes [F⊂Co(mBDCA-5t-H3)]2– (3) and [F⊂Zn(mBDCA-5t-H3)]2– (4) were both fully characterized, including single crystal X-ray analyses. Fluoride binding is synergistic involving hydrogen-bond donors from the second-coordination sphere together with metal(II) ion complexation.
Co-reporter:Wesley J. Transue, Alexandra Velian, Matthew Nava, Cristina García-Iriepa, Manuel Temprado, and Christopher C. Cummins
Journal of the American Chemical Society August 9, 2017 Volume 139(Issue 31) pp:10822-10822
Publication Date(Web):July 13, 2017
DOI:10.1021/jacs.7b05464
Dibenzo-7-phosphanorbornadiene compounds, RPA (A = C14H10 or anthracene), are investigated as phosphinidene sources upon thermally induced (70–90 °C) anthracene elimination. Analysis of substituent effects reveals that π-donating dialkylamide groups are paramount to successful phosphinidene transfer; poorer π-donors give reduced or no transfer. Substituent steric bulk is also implicated in successful transfer. Molecular beam mass spectrometry (MBMS) studies of each derivative reveal dialkylamide derivatives to be promising precursors for further gas-phase spectroscopic studies of phosphinidenes; in particular, we present evidence of direct detection of the dimethylamide derivative, [Me2N═P]. Kinetic investigations of iPr2NPA thermolysis in 1,3-cyclohexadiene and/or benzene-d6 are consistent with a model of unimolecular fragmentation to yield free phosphinidene [iPr2N═P] as a transient reactive intermediate. This conclusion is probed by density functional theory (DFT) calculations, which favored a mechanistic model featuring free singlet aminophosphinidenes. The breadth of phosphinidene acceptors is expanded to unsaturated substrates beyond 1,3-dienes to include olefins and alkynes; this provides a new synthetic route to valuable amino-substituted phosphiranes and phosphirenes, respectively. Stereoselective phosphinidene transfer to olefins is consistent with singlet phosphinidene reactivity by analogy with the Skell hypothesis for singlet carbene addition to olefins.
Co-reporter:Julia M. Stauber and Christopher C. Cummins
Inorganic Chemistry 2017 Volume 56(Issue 5) pp:
Publication Date(Web):February 16, 2017
DOI:10.1021/acs.inorgchem.6b03149
The synthesis and characterization of tri- and tetrametaphosphate titanium(IV) oxo and peroxo complexes is described. Addition of 0.5 equiv of [OTi(acac)2]2 to dihydrogen tetrametaphosphate ([P4O12H2]2–) and monohydrogen trimetaphosphate ([P3O9H]2–) provided a bis(μ2,κ2,κ2) tetrametaphosphate titanyl dimer, [OTiP4O12]24– (1; 70% yield), and a trimetaphosphate titanyl acetylacetonate complex, [OTiP3O9(acac)]2– (2; 59% yield). Both 1 and 2 have been structurally characterized, crystallizing in the triclinic P1̅ and monoclinic P21 space groups, respectively. These complexes contain Ti≡O units with distances of 1.624(7) and 1.644(2) Å, respectively, and represent rare examples of structurally characterized terminal titanyls within an all-oxygen coordination environment. Complexes 1 and 2 react with hydrogen peroxide to produce the corresponding peroxotitanium(IV) metaphosphate complexes [O2TiP4O12]24–(3; 61% yield) and [O2TiP3O9(acac)]2– (4; 65% yield), respectively. Both 3 and 4 have been characterized by single-crystal X-ray diffraction studies, and their solid-state structures are presented. Complex 3 functions as an oxygen atom transfer (OAT) reagent capable of oxidizing phosphorus(III) compounds (P(OMe)3, PPh3) and SMe2 at ambient temperature to result in the corresponding organic oxide with regeneration of dimer 1.
Co-reporter:Shiyu Zhang;Matthew J. Nava;Gary K. Chow;Nazario Lopez;Gang Wu;David R. Britt;Daniel G. Nocera
Chemical Science (2010-Present) 2017 vol. 8(Issue 9) pp:6117-6122
Publication Date(Web):2017/08/21
DOI:10.1039/C7SC01230F
When solubilized in a hexacarboxamide cryptand anion receptor, the peroxide dianion reacts rapidly with CO2 in polar aprotic organic media to produce hydroperoxycarbonate (HOOCO2−) and peroxydicarbonate (−O2COOCO2−). Peroxydicarbonate is subject to thermal fragmentation into two equivalents of the highly reactive carbonate radical anion, which promotes hydrogen atom abstraction reactions responsible for the oxidative degradation of organic solvents. The activation and conversion of the peroxide dianion by CO2 is general. Exposure of solid lithium peroxide (Li2O2) to CO2 in polar aprotic organic media results in aggressive oxidation. These findings indicate that CO2 must not be introduced in conditions relevant to typical lithium–O2 cell configurations, as production of HOOCO2− and −O2COOCO2− during lithium–O2 cell cycling will lead to cell degradation via oxidation of organic electrolytes and other vulnerable cell components.
Co-reporter:Ioana Knopf;Daniel Tofan;Dirk Beetstra;Abdulaziz Al-Nezari;Khalid Al-Bahily
Chemical Science (2010-Present) 2017 vol. 8(Issue 2) pp:1463-1468
Publication Date(Web):2017/01/30
DOI:10.1039/C6SC03614G
A family of cis-macrocyclic diphosphines was prepared in just three steps from white phosphorus and commercial materials using a modular synthetic approach. Alkylation of bicyclic diphosphane 3,4,8,9-tetramethyl-1,6-diphosphabicyclo(4.4.0)deca-3,8-diene, or P2(dmb)2, produced phosphino-phosphonium salts [R-P2(dmb)2]X, where R is methyl, benzyl and isobutyl, in yields of 90–96%. Treatment of these salts with organolithium or Grignard reagents yielded symmetric and unsymmetric macrocyclic diphosphines of the form cis-1-R-6-R′-3,4,8,9-tetramethyl-2,5,7,10-tetrahydro-1,6-DiPhospheCine, or R,R′-DPC, in which R′ is methyl, cyclohexyl, phenyl or mesityl, in yields of 46–94%. Alternatively, symmetric diphosphine Cy2-DPC was synthesized in 74% yield from the dichlorodiphosphine Cl2P2(dmb)2. As a first application, these cis-macrocyclic diphosphines were used as ligands in the nickel-catalyzed synthesis of acrylate from CO2 and ethylene, for which they showed promising catalytic activity.
Co-reporter:Maximilian Joost;Wesley J. Transue
Chemical Communications 2017 vol. 53(Issue 77) pp:10731-10733
Publication Date(Web):2017/09/26
DOI:10.1039/C7CC06841G
Tungsten(IV) tetrakis(2,6-diisopropylphenoxide) (1) has been demonstrated to be a competent platform for decarbonylative formation of anionic terminal pnictide complexes upon treatment with pnictaethynolate anions: cyanate, 2-phosphaethynolate, and 2-arsaethynolate. These transformations constitute the first examples of terminal phosphide and arsenide complex formation at a transition metal center from OCP− and OCAs−, respectively. The phosphide and arsenide complexes are also the first to be isolated in a tetragonal, all-oxygen ligand environment. The scalar NMR coupling constants between tungsten-183 and nitrogen-15 or phosphorus-31 have been measured and contextualized using natural bond orbital (NBO) methods in terms of s orbital character in the σ bonding orbital and pnictide lone pair.
Co-reporter:Maximilian Joost;Matthew Nava;Wesley J. Transue
Chemical Communications 2017 vol. 53(Issue 83) pp:11500-11503
Publication Date(Web):2017/10/17
DOI:10.1039/C7CC06516G
Targeted as an example of a compound composed of a carbon atom together with two stable neutral leaving groups, 7-isocyano-7-azadibenzonorbornadiene, CN2A (1, A = C14H10 or anthracene) has been synthesized and spectroscopically and structurally characterized. The terminal C atom of 1 can be transferred: mesityl nitrile oxide reacts with 1 to produce carbon monoxide, likely via intermediacy of the N-isocyanate OCN2A. Reaction of 1 with [RuCl2(CO)(PCy3)2] leads to [RuCl2(CO)(1)(PCy3)2] which decomposes unselectively: in the product mixture, the carbide complex [RuCl2(C)(PCy3)2] was detected. Upon heating in the solid state or in solution, 1 decomposes to A, N2 and cyanogen (C2N2) as substantiated using molecular beam mass spectrometry, IR and NMR spectroscopy techniques.
Co-reporter:Wesley J. Transue; Alexandra Velian; Matthew Nava; Marie-Aline Martin-Drumel; Caroline C. Womack; Jun Jiang; Gao-Lei Hou; Xue-Bin Wang; Michael C. McCarthy; Robert W. Field
Journal of the American Chemical Society 2016 Volume 138(Issue 21) pp:6731-6734
Publication Date(Web):May 12, 2016
DOI:10.1021/jacs.6b03910
Dibenzo-7-phosphanorbornadiene Ph3PC(H)PA (1, A = C14H10, anthracene) is reported here as a molecular precursor to phosphaethyne (HC≡P), produced together with anthracene and triphenylphosphine. HCP generated by thermolysis of 1 has been observed by molecular beam mass spectrometry, laser-induced fluorescence, microwave spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy. In toluene, fragmentation of 1 has been found to proceed with activation parameters of ΔH⧧ = 25.5 kcal/mol and ΔS⧧ = −2.43 eu and is accompanied by formation of an orange insoluble precipitate. Results from computational studies of the mechanism of HCP generation are in good agreement with experimental data. This high-temperature method of HCP generation has pointed to new reaction chemistry with azide anion to produce the 1,2,3,4-phosphatriazolate anion, HCPN3–, for which structural data have been obtained in a single-crystal X-ray diffraction study. Negative-ion photoelectron spectroscopy has shown the adiabatic detachment energy for this anion to be 3.555(10) eV. The aromaticity of HCPN3– has been assessed using nucleus-independent chemical shift, quantum theory of atoms in molecules, and natural bond orbital methods.
Co-reporter:Marc-André Courtemanche, Wesley J. Transue, and Christopher C. Cummins
Journal of the American Chemical Society 2016 Volume 138(Issue 50) pp:16220-16223
Publication Date(Web):November 28, 2016
DOI:10.1021/jacs.6b10545
Toward the preparation of a coordination complex of the heterodiatomic molecule PN, P≡N-V(N[tBu]Ar)3 (1, Ar = 3,5-Me2C6H3), we report the use of ClPA (A = C14H10, anthracene) as a formal source of phosphorus(I) in its reaction with Na[NV(N[tBu]Ar)3] (Na[4]) to yield trimeric cyclo-triphosphane [PNV(N[tBu]Ar)3]3 (3) with a core composed exclusively of phosphorus and nitrogen. In the presence of NapS2 (peri-1,8-naphthalene disulfide), NapS2P-NV(N[tBu]Ar)3 (6) is instead generated in 80% yield, suggesting trapping of transient 1. Upon mild heating, 3 readily fragments into dimeric [PNV(N[tBu]Ar)3]2 (2), while in the presence of bis(trimethylsilyl)acetylene or cis-4-octene, the respective phosphirene (Ar[tBu]N)3VN-PC2(SiMe3)2 (7) or phosphirane (Ar[tBu]N)3VN-P(C8H16) (8) compounds are generated. Kinetic data were found to be consistent with unimolecular decay of 3, and [2+1]-cycloaddition with radical clocks ruled out a triplet intermediate, consistent with intermediate 1 reacting as a singlet phosphinidene. In addition, both 7 and 8 were shown to reversibly exchange cis-4-octene and bis(trimethylsilyl)acetylene, serving as formal sources of 1, a reactivity manifold traditionally reserved for transition metals.
Co-reporter:Gao-Lei Hou, Bo Chen, Wesley J. Transue, David A. Hrovat, Christopher C. Cummins, Weston Thatcher Borden and Xue-Bin Wang
Chemical Science 2016 vol. 7(Issue 7) pp:4667-4675
Publication Date(Web):19 Apr 2016
DOI:10.1039/C5SC04667J
We report here a negative ion photoelectron spectroscopy (NIPES) and ab initio study of the recently synthesized planar aromatic inorganic ion P2N3−, to investigate the electronic structures of P2N3− and its neutral P2N3˙ radical. The adiabatic detachment energy of P2N3− (electron affinity of P2N3˙) was determined to be 3.765 ± 0.010 eV, indicating high stability for the P2N3− anion. Ab initio electronic structure calculations reveal the existence of five, low-lying, electronic states in the neutral P2N3˙ radical. Calculation of the Franck–Condon factors (FCFs) for each anion-to-neutral electronic transition and comparison of the resulting simulated NIPE spectrum with the vibrational structure in the observed spectrum allows the first four excited states of P2N3˙ to be determined to lie 6.2, 6.7, 11.5, and 22.8 kcal mol−1 above the ground state of the radical, which is found to be a 6π-electron, 2A1, σ state.
Co-reporter:Khetpakorn Chakarawet, Ioana Knopf, Matthew Nava, Yanfeng Jiang, Julia M. Stauber, and Christopher C. Cummins
Inorganic Chemistry 2016 Volume 55(Issue 12) pp:6178-6185
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.inorgchem.6b00749
Metaphosphate acids cannot be thoroughly studied in aqueous media because their acidity is leveled by the solvent, and the resulting metaphosphates are susceptible to acid-catalyzed hydrolysis. Exploration of metaphosphate acid chemistry has now been made possible with the development of a general synthetic method for organic media soluble metaphosphate acids. Protonation of the [PPN]+ salts ([PPN]+ = [N(PPh3)2]+) of tri-, tetra-, and hexametaphosphates results in five new metaphosphate acids, [PPN]2[P3O9H] (2), [PPN]4[(P4O12)3H8] (3), [PPN]4[P6O18H2]·2H2O (4), [PPN]3[P6O18H3] (5), and [PPN]2[P6O18H2(H3O)2] (6), obtained in yields of 80, 71, 66, 88, and 76%, respectively. Additionally, our synthetic method can be extended to pyrophosphate to produce [PPN][P2O7H3] (7) in 77% yield. The structural configurations of these oxoacids are dictated by strong hydrogen bonds and the anticooperative effect. Intramolecular hydrogen bonds are observed in 2, 4, and 5 and the previously reported [PPN]2[P4O12H2] (1), while intermolecular hydrogen bonds are observed in 3, 6, and 7. The hydrogen bonds in 3–7 possess short distances and are classified as low-barrier hydrogen bonds. Gas-phase acidity computations reveal that the parent tri- and tetrametaphosphoric acids are superacids. Their remarkable acidity is attributable to the stabilization of their corresponding conjugate bases via intramolecular hydrogen bonding.
Co-reporter:Alexandra Velian, Brandi M. Cossairt and Christopher C. Cummins
Dalton Transactions 2016 vol. 45(Issue 5) pp:1891-1895
Publication Date(Web):07 Oct 2015
DOI:10.1039/C5DT03383G
Complexes (THF)0–2E[P3Nb(ODipp)3]2 (E = Sn, Pb; Dipp = 2,6-iPr2C6H3) were isolated (>90%) from the salt metathesis of [Na(THF)3][P3Nb(ODipp)3] with E2+ salts. The reaction of (THF)Sn[P3Nb(ODipp)3]2 with pyridine-N-oxide was investigated as a method to deposit a new SnP6 phase. Additionally, the neutral complex P3Nb(ODipp)2(py)2 (py = pyridine) was prepared from [Na(THF)3][P3Nb(ODipp)3] in the presence of pyridine and salts of coordinating cations (Mg(II), Sn(II), Pb(II), Ge(II), Hg(II) and Ag(I)). P3Nb(ODipp)2(py)2 was found to successfully produce AsP3 upon treatment with AsCl3. The characterization of complexes (THF)0–1Sn[P3Nb(ODipp)3]2, (THF)2Pb[P3Nb(ODipp)3]2 and P3Nb(ODipp)2(py)2, including their solid state structures, is discussed.
Co-reporter:Gao-Lei Hou, Bo Chen, Wesley J. Transue, David A. Hrovat, Christopher C. Cummins, Weston Thatcher Borden, and Xue-Bin Wang
The Journal of Physical Chemistry A 2016 Volume 120(Issue 31) pp:6228-6235
Publication Date(Web):July 19, 2016
DOI:10.1021/acs.jpca.6b06343
We report here the results of a combined experimental and computational study of the negative ion photoelectron spectroscopy (NIPES) of the recently synthesized, planar, aromatic, HCPN3– ion. The adiabatic electron detachment energy of HCPN3– (electron affinity of HCPN3•) was measured to be 3.555 ± 0.010 eV, a value that is intermediate between the electron detachment energies of the closely related (CH)2N3– and P2N3– ions. High level electronic structure calculations and Franck–Condon factor (FCF) simulations reveal that transitions from the ground state of the anion to two nearly degenerate, low-lying, electronic states, of the neutral HCPN3• radical are responsible for the congested peaks at low binding energies in the NIPE spectrum. The best fit of the simulated NIPE spectrum to the experimental spectrum indicates that the ground state of HCPN3• is a 5π-electron 2A″ π radical state, with a 6π-electron, 2A′, σ radical state being at most 1.0 kcal/mol higher in energy.
Co-reporter:Julia M. Stauber; Eric D. Bloch; Konstantinos D. Vogiatzis; Shao-Liang Zheng; Ryan G. Hadt; Dugan Hayes; Lin X. Chen; Laura Gagliardi; Daniel G. Nocera
Journal of the American Chemical Society 2015 Volume 137(Issue 49) pp:15354-15357
Publication Date(Web):November 11, 2015
DOI:10.1021/jacs.5b09827
A dicobalt(II) complex, [Co2(mBDCA-5t)]2– (1), demonstrates a cofacial arrangement of trigonal monopyramidal Co(II) ions with an inter-metal separation of 6.2710(6) Å. Reaction of 1 with potassium superoxide generates an encapsulated Co–O–Co core in the dianionic complex, [Co2O(mBDCA-5t)]2– (2); to form the linear Co–O–Co core, the inter-metal distance has diminished to 3.994(3) Å. Co K-edge X-ray absorption spectroscopy data are consistent with a +2 oxidation state assignment for Co in both 1 and 2. Multireference complete active space calculations followed by second-order perturbation theory support this assignment, with hole equivalents residing on the bridging O-atom and on the cryptand ligand for the case of 2. Complex 2 acts as a 2-e– oxidant toward substrates including CO and H2, in both cases efficiently regenerating 1 in what represent net oxygen-atom-transfer reactions. This dicobalt system also functions as a catalase upon treatment with H2O2.
Co-reporter:Matthew Nava; Nazario Lopez; Peter Müller; Gang Wu; Daniel G. Nocera
Journal of the American Chemical Society 2015 Volume 137(Issue 46) pp:14562-14565
Publication Date(Web):October 14, 2015
DOI:10.1021/jacs.5b08495
The reactivity of peroxide dianion O22– has been scarcely explored in organic media due to the lack of soluble sources of this reduced oxygen species. We now report the finding that the encapsulated peroxide cryptate, [O2⊂mBDCA-5t-H6]2– (1), reacts with carbon monoxide in organic solvents at 40 °C to cleanly form an encapsulated carbonate. Characterization of the resulting hexacarboxamide carbonate cryptate by single crystal X-ray diffraction reveals that carbonate dianion forms nine complementary hydrogen bonds with the hexacarboxamide cryptand, [CO3⊂mBDCA-5t-H6]2– (2), a conclusion that is supported by spectroscopic data. Labeling studies and 17O solid-state NMR data confirm that two-thirds of the oxygen atoms in the encapsulated carbonate derive from peroxide dianion, while the carbon is derived from CO. Further evidence for the formation of a carbonate cryptate was obtained by three methods of independent synthesis: treatment of (i) free cryptand with K2CO3; (ii) monodeprotonated cryptand with PPN[HCO3]; and (iii) free cryptand with TBA[OH] and atmospheric CO2. This work demonstrates CO oxidation mediated by a hydrogen-bonding anion receptor, constituting an alternative to transition-metal catalysis.
Co-reporter:Heather A. Spinney, Christopher R. Clough and Christopher C. Cummins
Dalton Transactions 2015 vol. 44(Issue 15) pp:6784-6796
Publication Date(Web):16 Feb 2015
DOI:10.1039/C5DT00105F
This work explores the reduction of 4,4′-bipyridine using two equivalents of the titanium(III) complex Ti(N[tBu]Ar)3 resulting in formation of a black, crystalline complex, (4,4′-bipy){Ti(N[tBu]Ar)3}2, for which an X-ray structure determination is reported. The neutral, black, 4,4′-bipyridine-bridged bimetallic was found to be redox active, with mono- and di-anions being accessible electrochemically, and with the mono- and di-cations also being accessible chemically, and isolable, at least when using the weakly coordinating anion [B(C6F5)4]− as the counter-ion. It proved possible to crystallize the salt [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]2 for a single-crystal X-ray structure investigation; in this instance it was revealed that the aromaticity of the 4,4′-bipyridine ligand, that had been disrupted upon reduction, had been regained. A rare cationic d0 metal tris-amide complex, shown by X-ray crystallography to contain an intriguing pyramidal TiN3 core geometry, namely {Ti(N[tBu]Ar)3}+, could also be isolated when using [B(C6F5)4] as the essentially non-interacting counter-ion. This highly reactive cation should be considered as a potential intermediate in the plethora of reactions wherein Ti(N[tBu]Ar)3 has been shown to effect the reduction of substrates including halogenated organic molecules, carbonyl compounds, organic nitriles, and metal complexes.
Co-reporter:Paula L. Diaconescu and Christopher C. Cummins
Dalton Transactions 2015 vol. 44(Issue 6) pp:2676-2683
Publication Date(Web):05 Dec 2014
DOI:10.1039/C4DT02422B
The synthesis and characterization of (bipy)2U(N[t-Bu]Ar)2 (1-(bipy)2, bipy = 2,2′-bipyridyl, Ar = 3,5-C6H3Me2), (bipy)U(N[1Ad]Ar)3 (2-bipy), (bipy)2U(NC[t-Bu]Mes)3 (3-(bipy)2, Mes = 2,4,6-C6H2Me3), and IU(bipy)(NC[t-Bu]Mes)3 (3-I-bipy) are reported. X-ray crystallography studies indicate that bipy coordinates as a radical anion in 1-(bipy)2 and 2-bipy, and as a neutral ligand in 3-I-bipy. In 3-(bipy)2, one of the bipy ligands is best viewed as a radical anion, the other as a neutral ligand. The electronic structure assignments are supported by NMR spectroscopy studies of exchange experiments with 4,4′-dimethyl-2,2′-bipyridyl and also by optical spectroscopy. In all complexes, uranium was assigned a +4 formal oxidation state.
Co-reporter:Bryce L. Anderson, Andrew G. Maher, Matthew Nava, Nazario Lopez, Christopher C. Cummins, and Daniel G. Nocera
The Journal of Physical Chemistry B 2015 Volume 119(Issue 24) pp:7422-7429
Publication Date(Web):January 30, 2015
DOI:10.1021/jp5110505
The encapsulation of peroxide dianion by hexacarboxamide cryptand provides a platform for the study of electron transfer of isolated peroxide anion. Photoinitiated electron transfer (ET) between freely diffusing Ru(bpy)32+ and the peroxide dianion occurs with a rate constant of 2.0 × 1010 M–1 s–1. A competing electron transfer quenching pathway is observed within an ion pair. Picosecond transient spectroscopy furnishes a rate constant of 1.1 × 1010 s–1 for this first-order process. A driving force dependence for the ET rate within the ion pair using a series of Ru(bpy)32+ derivatives allows for the electronic coupling and reorganization energies to be assessed. The ET reaction is nonadiabatic and dominated by a large inner-sphere reorganization energy, in accordance with that expected for the change in bond distance accompanying the conversion of peroxide dianion to superoxide anion.
Co-reporter:Ioana Knopf and Christopher C. Cummins
Organometallics 2015 Volume 34(Issue 9) pp:1601-1603
Publication Date(Web):April 27, 2015
DOI:10.1021/acs.organomet.5b00190
The reduction of CO2 to formate using sodium borohydride was originally investigated in the 1950s. Despite this clue from the chemical literature, many recent publications describe catalytic CO2 hydroboration methods leading to formate or methoxide with more expensive and less reactive boranes such as pinacolborane. Herein we describe the uptake of 3 equiv of CO2 by NaBH4, along with full spectroscopic and crystallographic characterization of the resulting triformatoborohydride, Na[HB(OCHO)3]. Conducting the synthesis in acetonitrile under 300 psi of CO2 constitutes a new preparative procedure for generating Na[HB(OCHO)3]. This reaction does not require the presence of a strongly coordinating alkali metal cation, as evidenced by the analogous reactivity of [NEt4][BH4]. Even at 1 atm pressure and without using rigorously dry solvent, treatment of NaBH4 with CO2 and subsequent quenching gave formic acid (1.5 equiv based on B).
Co-reporter:Alexandra Velian, Wesley J. Transue, and Christopher C. Cummins
Organometallics 2015 Volume 34(Issue 19) pp:4644-4646
Publication Date(Web):August 10, 2015
DOI:10.1021/acs.organomet.5b00529
The dibenzo-7-dimethylgermanorbornadiene Me2GeA (A = C14H10) has been synthesized in one step by treatment of MgA·3THF with Me2GeCl2 in tetrahydrofuran (−35 °C) and isolated in 69% yield. The thermolysis of Me2GeA in toluene leads to the effective expansion of the bicyclic framework to the dibenzo-7,8-tetramethyldigermabicyclo[2.2.2]octadiene (Me2Ge)2A, isolated in 71% yield (based on germanium). The bicyclic compounds Me2GeA and (Me2Ge)2A have been characterized by single-crystal X-ray diffraction studies and their structures discussed.
Co-reporter:Alexandra Velian
Science 2015 Volume 348(Issue 6238) pp:1001-1004
Publication Date(Web):29 May 2015
DOI:10.1126/science.aab0204
An aromatic phosphorus and nitrogen ring
In chemistry, the term “aromatic” denotes the energy stabilization associated with electrons being shared among atoms in a ring. Benzene is the best-known aromatic compound, although numerous related hydrocarbons also manifest the property. Velian and Cummins now report a rare instance of an inorganic aromatic compound: a negatively charged pentagonal ring composed of three nitrogen and two phosphorus atoms.
Science, this issue p. 1001
Co-reporter:Yanfeng Jiang ; Khetpakorn Chakarawet ; Andrea Laura Kohout ; Matthew Nava ; Nadia Marino
Journal of the American Chemical Society 2014 Volume 136(Issue 34) pp:11894-11897
Publication Date(Web):August 7, 2014
DOI:10.1021/ja5058339
Dihydrogen tetrametaphosphate [P4O12H2]2– (1) can now be synthesized and isolated as its PPN salt ([PPN]+ = [N(PPh3)2]+) via treatment of [PPN]4[P4O12] with trifluoroacetic anhydride in wet acetone; this simple procedure affords the oxoacid salt in 94% yield. A pKa of 15.83 ± 0.11 in acetonitrile was determined. [P4O12H2]2– reacts with the dehydrating agent N,N′-dicyclohexylcarbodiimide to afford tetrametaphosphate anhydride [P4O11]2– (2) in 82% yield, also as the PPN salt. From 2 a monohydrogen tetrametaphosphate ester [P4O10(OH)(OMe)]2– (3, 96%) was derived by addition of methanol, illustrating that 2 can function as a reagent for chemical phosphorylation. Addition of water to 2 regenerates 1 quantitatively. Deprotonation of 1 by metal amides in the +2 oxidation state led to the unconventional monomeric tin(II) κ4 tetrametaphosphate [Sn(P4O12)]2– (4, 78%, a molecular analog of SnO) and binary dimeric chromium(II) bis(μ2,κ2,κ2) derivative [Cr2(P4O12)2]4– (5, 82%). Structural data stemming from single-crystal X-ray diffraction studies for the PPN salts of anions 1–5 are also reported.
Co-reporter:Alexandra Velian ; Matthew Nava ; Manuel Temprado ; Yan Zhou ; Robert W. Field
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13586-13589
Publication Date(Web):September 8, 2014
DOI:10.1021/ja507922x
The transannular diphosphorus bisanthracene adduct P2A2 (A = anthracene or C14H10) was synthesized from the 7-phosphadibenzonorbornadiene Me2NPA through a synthetic sequence involving chlorophosphine ClPA (28–35%) and the tetracyclic salt [P2A2Cl][AlCl4] (65%) as isolated intermediates. P2A2 was found to transfer P2 efficiently to 1,3-cyclohexadiene (CHD), 1,3-butadiene (BD), and (C2H4)Pt(PPh3)2 to form P2(CHD)2 (>90%), P2(BD)2 (69%), and (P2)[Pt(PPh3)2]2 (47%), respectively, and was characterized by X-ray diffraction as the complex [CpMo(CO)3(P2A2)][BF4]. Experimental and computational thermodynamic activation parameters for the thermolysis of P2A2 in a solution containing different amounts of CHD (0, 4.75, and 182 equiv) have been obtained and suggest that P2A2 thermally transfers P2 to CHD through two competitive routes: (i) an associative pathway in which reactive intermediate [P2A] adds the first molecule of CHD before departure of the second anthracene, and (ii) a dissociative pathway in which [P2A] fragments to P2 and A prior to addition of CHD. Additionally, a molecular beam mass spectrometry study on the thermolysis of solid P2A2 reveals the direct detection of molecular fragments of only P2 and anthracene, thus establishing a link between solution-phase P2-transfer chemistry and production of gas-phase P2 by mild thermal activation of a molecular precursor.
Co-reporter:Subhojit Majumdar, Julia M. Stauber, Taryn D. Palluccio, Xiaochen Cai, Alexandra Velian, Elena V. Rybak-Akimova, Manuel Temprado, Burjor Captain, Christopher C. Cummins, and Carl D. Hoff
Inorganic Chemistry 2014 Volume 53(Issue 20) pp:11185-11196
Publication Date(Web):October 3, 2014
DOI:10.1021/ic5017005
The enthalpy of oxygen atom transfer (OAT) to V[(Me3SiNCH2CH2)3N], 1, forming OV[(Me3SiNCH2CH2)3N], 1–O, and the enthalpies of sulfur atom transfer (SAT) to 1 and V(N[t-Bu]Ar)3, 2 (Ar = 3,5-C6H3Me2), forming the corresponding sulfides SV[(Me3SiNCH2CH2)3N], 1–S, and SV(N[t-Bu]Ar)3, 2–S, have been measured by solution calorimetry in toluene solution using dbabhNO (dbabhNO = 7-nitroso-2,3:5,6-dibenzo-7-azabicyclo[2.2.1]hepta-2,5-diene) and Ph3SbS as chalcogen atom transfer reagents. The V–O BDE in 1–O is 6.3 ± 3.2 kcal·mol–1 lower than the previously reported value for 2–O and the V–S BDE in 1–S is 3.3 ± 3.1 kcal·mol–1 lower than that in 2–S. These differences are attributed primarily to a weakening of the V–Naxial bond present in complexes of 1 upon oxidation. The rate of reaction of 1 with dbabhNO has been studied by low temperature stopped-flow kinetics. Rate constants for OAT are over 20 times greater than those reported for 2. Adamantyl isonitrile (AdNC) binds rapidly and quantitatively to both 1 and 2 forming high spin adducts of V(III). The enthalpies of ligand addition to 1 and 2 in toluene solution are −19.9 ± 0.6 and −17.1 ± 0.7 kcal·mol–1, respectively. The more exothermic ligand addition to 1 as compared to 2 is opposite to what was observed for OAT and SAT. This is attributed to less weakening of the V–Naxial bond in ligand binding as opposed to chalcogen atom transfer and is in keeping with structural data and computations. The structures of 1, 1–O, 1–S, 1–CNAd, and 2–CNAd have been determined by X-ray crystallography and are reported.
Co-reporter:Andrew M. Ullman, Xianru Sun, Daniel J. Graham, Nazario Lopez, Matthew Nava, Rebecca De Las Cuevas, Peter Müller, Elena V. Rybak-Akimova, Christopher C. Cummins, and Daniel G. Nocera
Inorganic Chemistry 2014 Volume 53(Issue 10) pp:5384-5391
Publication Date(Web):April 28, 2014
DOI:10.1021/ic500759g
A peroxide dianion (O22–) can be isolated within the cavity of hexacarboxamide cryptand, [(O2)⊂mBDCA-5t-H6]2–, stabilized by hydrogen bonding but otherwise free of proton or metal-ion association. This feature has allowed the electron-transfer (ET) kinetics of isolated peroxide to be examined chemically and electrochemically. The ET of [(O2)⊂mBDCA-5t-H6]2– with a series of seven quinones, with reduction potentials spanning 1 V, has been examined by stopped-flow spectroscopy. The kinetics of the homogeneous ET reaction has been correlated to heterogeneous ET kinetics as measured electrochemically to provide a unified description of ET between the Butler–Volmer and Marcus models. The chemical and electrochemical oxidation kinetics together indicate that the oxidative ET of O22– occurs by an outer-sphere mechanism that exhibits significant nonadiabatic character, suggesting that the highest occupied molecular orbital of O22– within the cryptand is sterically shielded from the oxidizing species. An understanding of the ET chemistry of a free peroxide dianion will be useful in studies of metal–air batteries and the use of [(O2)⊂mBDCA-5t-H6]2– as a chemical reagent.
Co-reporter:Christopher C. Cummins, Chao Huang, Tabitha J. Miller, Markus W. Reintinger, Julia M. Stauber, Isabelle Tannou, Daniel Tofan, Abouzar Toubaei, Alexandra Velian, and Gang Wu
Inorganic Chemistry 2014 Volume 53(Issue 7) pp:3678-3687
Publication Date(Web):March 12, 2014
DOI:10.1021/ic403178j
Treatment of P4 with in situ generated [Na][SnPh3] leads to the formation of the sodium monophosphide [Na][P(SnPh3)2] and the Zintl salt [Na]3[P7]. The former was isolated in 46% yield as the crystalline salt [Na(benzo-15-crown-5)][P(SnPh3)2] and used to prepare the homoleptic phosphine P(SnPh3)3, isolated in 67% yield, as well as the indium derivative (XL)2InP(SnPh3)2 (XL = S(CH2)2NMe2), isolated in 84% yield, and the gold complex (Ph3P)AuP(SnPh3)2. The compounds [Na(benzo-15-crown-5)][P(SnPh3)2], P(SnPh3)3, (XL)2InP(SnPh3)2, and (Ph3P)AuP(SnPh3)2 were characterized using multinuclear NMR spectroscopy and X-ray crystallography. The bonding in (Ph3P)AuP(SnPh3)2 was dissected using natural bond orbital (NBO) methods, in response to the observation from the X-ray crystal structure that the dative P:→Au bond is slightly shorter than the shared electron-pair P–Au bond. The bonding in (XL)2InP(SnPh3)2 was also interrogated using 31P and 13C solid-state NMR and computational methods. Co-product [Na]3[P7] was isolated in 57% yield as the stannyl heptaphosphide P7(SnPh3)3, following salt metathesis with ClSnPh3. Additionally, we report that treatment of P4 with sodium naphthalenide in dimethoxyethane at 22 °C is a convenient and selective method for the independent synthesis of Zintl ion [Na]3[P7]. The latter was isolated as the silylated heptaphosphide P7(SiMe3)3, in 67% yield, or as the stannyl heptaphosphide P7(SnPh3)3 in 65% yield by salt metathesis with ClSiMe3 or ClSnPh3, respectively.
Co-reporter:Anthony F. Cozzolino, Jared S. Silvia, Nazario Lopez and Christopher C. Cummins
Dalton Transactions 2014 vol. 43(Issue 12) pp:4639-4652
Publication Date(Web):03 Feb 2014
DOI:10.1039/C3DT52738G
An important challenge in the artificial fixation of N2 is to find atom efficient transformations that yield value-added products. Here we explore the coordination complex mediated conversion of ubiquitous species, CO and N2, into isocyanate. We have conceptually split the process into three steps: (1) the six-electron splitting of dinitrogen into terminal metal nitrido ligands, (2) the reduction of the complex by two electrons with CO to form an isocyanate linkage, and (3) the one electron reduction of the metal isocyanate complex to regenerate the starting metal complex and release the product. These steps are explored separately in an attempt to understand the limitations of each step and what is required of a coordination complex in order to facilitate a catalytic cycle. The possibility of this cyanate cycle was explored with both Mo and V complexes which have previously been shown to perform select steps in the sequence. Experimental results demonstrate the feasibility of some of the steps and DFT calculations suggest that, although the reduction of the terminal metal nitride complex by carbon monoxide should be thermodynamically favorable, there is a large kinetic barrier associated with the change in spin state which can be avoided in the case of the V complexes by an initial binding of the CO to the metal center followed by rearrangement. This mandates certain minimal design principles for the metal complex: the metal center should be sterically accessible for CO binding and the ligands should not readily succumb to CO insertion reactions.
Co-reporter:Cesar M. Manna, Mostafa Y. Nassar, Daniel Tofan, Khetpakorn Chakarawet and Christopher C. Cummins
Dalton Transactions 2014 vol. 43(Issue 4) pp:1509-1518
Publication Date(Web):07 Nov 2013
DOI:10.1039/C3DT52526K
We herein report the preparation of several mononuclear-metaphosphate complexes using simple techniques and mild conditions with yields ranging from 56% to 78%. Treatment of cyclo-tetrametaphosphate ([TBA]4[P4O12]·5H2O, TBA = tetra-n-butylammonium) with various metal sources including (CH3CN)3Mo(CO)3, (CH3CN)2Mo(CO)2(η3-C3H5)Cl, MoO2Cl2(OSMe2)2, and VOF3, leads to the clean and rapid formation of [TBA]4[(P4O12)Mo(CO)3]·2H2O, [TBA]3[(P4O12)Mo(CO)2(η3-C3H5)], [TBA]3[(P4O12)MoO2Cl] and [TBA]3[(P4O12)VOF2]·Et2O salts in isolated yields of 69, 56, 68, and 56% respectively. NMR spectroscopy, NMR simulations and single crystal X-ray studies reveal that the [P4O12]4− anion behaves as a tridentate ligand wherein one of the metaphosphate groups is not directly bound to the metal. cyclo-Trimetaphosphate-metal complexes were prepared using a similar procedure i.e., treatment of [PPN]3[P3O9]·H2O (PPN = bis(triphenylphosphine)iminium) with the metal sources (CH3CN)2Mo(CO)2(η3-C3H5)Cl, MoO2Cl2(OSMe2)2, MoOCl3, VOF3, WOCl4, and WO2Cl2(CH3CN)2 to produce the corresponding salts, [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)], [PPN]2[(P3O9)MoO2Cl], [PPN]2[(P3O9)MoOCl2], [PPN]2[(P3O9)VOF2]·2CH2Cl2, and [PPN]2[(P3O9)WO2Cl] in isolated yields of 78, 56, 75, 59, and 77% respectively. NMR spectroscopy, NMR simulations and single-crystal X-ray studies indicate that the trianionic ligand [P3O9]3− in these complexes also has κ3 connectivity.
Co-reporter:Jens M. Breunig;Daniel Tofan
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 10) pp:1605-1609
Publication Date(Web):
DOI:10.1002/ejic.201301140
Abstract
Reduction of mer-Cl3W(ODipp)3 with Na/Hg in the presence of P4 in toluene afforded the orange complex (η3-P3)W(ODipp)3 (δ = –156 ppm, 10 % isolated yield). Unlike the valence-isoelectronic anion [(η3-P3)Nb(ODipp)3]–, treatment with AsCl3 of the neutral (η3-P3)W(ODipp)3 did not transfer the cyclo-P3 ligand to generate AsP3. Instead, this complex underwent aryloxide ligand transfer to form the red complex (η3-P3)W(Cl)(ODipp)2(THF) (δ = –143 ppm, 32 % isolated yield).
Co-reporter:Daniel Tofan, Manuel Temprado, Subhojit Majumdar, Carl D. Hoff, and Christopher C. Cummins
Inorganic Chemistry 2013 Volume 52(Issue 15) pp:8851-8864
Publication Date(Web):July 10, 2013
DOI:10.1021/ic401052a
The 3,4,8,9-tetramethyl-1,6-diphospha-bicyclo-[4.4.0]deca-3,8-diene (P2(C6H10)2) framework containing a P–P bond has allowed for an unprecedented selectivity toward functionalization of a single phosphorus lone pair with reference to acyclic diphosphane molecules. Functionalization at the second phosphorus atom was found to proceed at a significantly slower rate, thus opening the pathway for obtaining mixed functional groups for a pair of P–P bonded λ5-phosphorus atoms. Reactivity with the chalcogen-atom donors MesCNO (Mes = 2,4,6-C6H2Me3) and SSbPh3 has allowed for the selective synthesis of the diphosphane chalcogenides OP2(C6H10)2 (87%), O2P2(C6H10)2 (94%), SP2(C6H10)2 (56%), and S2P2(C6H10)2 (87%). Computational studies indicate that the oxygen-atom transfer reactions involve penta-coordinated phosphorus intermediates that have four-membered {PONC} cycles. The P–E bond dissociation enthalpies in EP2(C6H10)2 were measured via calorimetric studies to be 134.7 ± 2.1 kcal/mol for P–O, and 93 ± 3 kcal/mol for P–S, respectively, in good agreement with the computed values. Additional reactivity with breaking of the P–P bond and formation of diphosphinate O3P2(C6H10)2 was only observed to occur upon heating of dimethylsulfoxide solutions of the precursor. Reactivity of diphosphane P2(C6H10)2 with azides allowed the isolation of monoiminophosphoranes (RN)P2(C6H10)2(R = Mes, CPh3, SiMe3), and treatment with additional MesN3 yielded symmetric and unsymmetric diiminodiphosphoranes (RN)(MesN)P2(C6H10)2 (91% for R = Mes). Metalation reactions with the bulky diiminodiphosphorane ligand (MesN)2P2(C6H10)2 (nppn) allowed for the isolation and characterization of (nppn)Mo(η3-C3H5)Cl(CO)2 (91%), (nppn)NiCl2 (76%), and [(nppn)Ni(η3-2-C3H4Me)][OTf] showing that these ligands provide an attractive preorganized binding pocket for both late and early transition metals.
Co-reporter:Lee-Ping Wang, Daniel Tofan, Jiahao Chen, Troy Van Voorhis and Christopher C. Cummins
RSC Advances 2013 vol. 3(Issue 45) pp:23166-23171
Publication Date(Web):25 Sep 2013
DOI:10.1039/C3RA43940B
We report a computational study of an energetically favorable pathway for the excited-state dissociation of a tetrahedral P4 molecule into two P2 molecules via the simultaneous breaking of four chemical bonds along a highly symmetric (D2d) reaction pathway. Along this pathway, a degeneracy occurs between the first excited state of P4 and the ground state of 2P2 at a lower total energy (ca. 4.7 eV) than the initial state, indicating that the initial photoexcitation provides sufficient energy for the dissociation without significant kinetic barriers. We also found that sequential dissociation of the four P–P bonds exhibits larger activation barriers thus making this a less viable dissociation pathway. Our computational investigation uncovers complicated photochemistry in elemental phosphorus, and suggests a likely mechanism for the environmentally friendly inclusion of phosphorus atoms into organic molecules.
Co-reporter:Bess Vlaisavljevich, Paula L. Diaconescu, Wayne L. Lukens Jr., Laura Gagliardi, and Christopher C. Cummins
Organometallics 2013 Volume 32(Issue 5) pp:1341-1352
Publication Date(Web):February 20, 2013
DOI:10.1021/om3010367
The electronic structure of the arene-bridged complex (μ-toluene)U2(N[tBu]Ar)4 (1a2-μ-toluene, Ar = 3,5-C6H3Me2) has been studied in relation to a variety of mononuclear uranium amide complexes, and their properties have been discussed comparatively. The syntheses, molecular structures (X-ray crystal structures and solution behavior based on variable-temperature NMR spectroscopic data), and corresponding spectroscopic (X-ray absorption near-edge structure and UV–vis–near-IR absorption) and magnetic properties are presented and interpreted with reference to results of density functional theory (DFT) and complete active space self-consistent field with corrections from second-order perturbation theory (CASSCF/CASPT2) calculations performed on model compounds. While the mononuclear compounds display expected electronic and magnetic properties for uranium complexes, 1a2-μ-toluene shows complicated properties in contrast. XANES spectroscopy, X-ray crystallography, and both density functional and CASSCF/CASPT2 results are consistent with the following electronic structure interpretation: f orbitals host the unpaired electrons, followed energetically by two δ bonds formed by filled uranium f orbitals and LUMOs of toluene.
Co-reporter:Alexandra Velian
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:13978-13981
Publication Date(Web):August 15, 2012
DOI:10.1021/ja306902j
Unprotected dibenzo-7λ3-phosphanorbornadiene derivatives RPA (A = C14H10 or anthracene; R = tBu, dbabh = NA, HMDS = (Me3Si)2N, iPr2N) are synthesized by treatment of the corresponding phosphorus dichloride RPCl2 with MgA·3THF, in cold THF (∼20% to 30% isolated yields). Anthracene and the corresponding cyclic phosphane (RP)n form as coproducts. Characteristic NMR features of the RPA derivatives include a doublet near 4 ppm in their 1H NMR spectra and a triplet peak in the 175–212 ppm region of the 31P NMR spectrum (2JPH ∼14 Hz). The X-ray structures of the AN–PA and (HMDS)PA derivatives are discussed. Thermolysis of RPA benzene-d6 solutions leads to anthracene extrusion. This process has a unimolecular kinetic profile for the iPr2NPA derivative. The 7-phosphanorbornene anti-iPr2NP(C6H8) could be synthesized (70% isolated yield) by thermolysis of iPr2NPA in 1,3-cyclohexadiene.
Co-reporter:Anthony F. Cozzolino ; Daniel Tofan ; Christopher C. Cummins ; Manuel Temprado ; Taryn D. Palluccio ; Elena V. Rybak-Akimova ; Subhojit Majumdar ; Xiaochen Cai ; Burjor Captain ;Carl D. Hoff
Journal of the American Chemical Society 2012 Volume 134(Issue 44) pp:18249-18252
Publication Date(Web):October 19, 2012
DOI:10.1021/ja309621h
Treatment of V(N[tBu]Ar)3 (1) (Ar = 3,5-Me2C6H3) with O2 was shown by stopped-flow kinetic studies to result in the rapid formation of (η1-O2)V(N[tBu]Ar)3 (2) (ΔH⧧ = 3.3 ± 0.2 kcal/mol and ΔS⧧ = −22 ± 1 cal mol–1 K–1), which subsequently isomerizes to (η2-O2)V(N[tBu]Ar)3 (3) (ΔH⧧ = 10.3 ± 0.9 kcal/mol and ΔS⧧ = −6 ± 4 cal mol–1 K–1). The enthalpy of binding of O2 to form 3 is −75.0 ± 2.0 kcal/mol, as measured by solution calorimetry. The reaction of 3 and 1 to form 2 equiv of O≡V(N[tBu]Ar)3 (4) occurs by initial isomerization of 3 to 2. The results of computational studies of this rearrangement (ΔH = 4.2 kcal/mol; ΔH⧧ = 16 kcal/mol) are in accord with experimental data (ΔH = 4 ± 3 kcal/mol; ΔH⧧ = 14 ± 3 kcal/mol). With the aim of suppressing the formation of 4, the reaction of O2 with 1 in the presence of tBuCN was studied. At −45 °C, the principal products of this reaction are 3 and tBuC(═O)N≡V(N[tBu]Ar)3 (5), in which the bound nitrile has been oxidized. Crystal structures of 3 and 5 are reported.
Co-reporter:Daniel Tofan and Christopher C. Cummins
Chemical Science 2012 vol. 3(Issue 8) pp:2474-2478
Publication Date(Web):22 May 2012
DOI:10.1039/C2SC20559A
Selective formation of bimetallic group 10 complexes using the Cs symmetric, bicyclic diphosphane P2C12H20 is reported herein. With its eclipsed lone pairs disposed at a relative angle of ca. 45°, the diphosphane framework is ideally suited to form multiple bridges between two metal centers. The complexes contain {M2P6} cages with three diphosphane bridges and a pair of trans-axial ligands such as EPh3 (E = P, As, Sb) or η1-P2C12H20. X-Ray crystallography experiments revealed that the cages have a pseudo-D3h symmetry, with metal–metal distances in the 3.9–4.1 Å range. The complexes were isolated in 48–91% yield as crystalline, bright yellow or orange powders. Substitution of the axial ligands with the {M2P6} cages remaining intact was also observed.
Co-reporter:Alexandra Velian and Christopher C. Cummins
Chemical Science 2012 vol. 3(Issue 4) pp:1003-1006
Publication Date(Web):20 Jan 2012
DOI:10.1039/C2SC00931E
Dimer [P2Nb(ODipp)3]2 (Dipp = 2,6-iPr2C6H3) has been obtained via a novel “2(3−1)” synthetic strategy. The mononuclear diphosphorus complex P2Nb(ODipp)3 targeted for generation by formal P− abstraction from previously reported [Na(THF)3][P3Nb(ODipp)3] ostensibly undergoes irreversible dimerization to form the [P2Nb(ODipp)3]2 complex, and is alternatively trapped reversibly by 1,3-cyclohexadiene with in situ formation of C6H8P2Nb(ODipp)3. The molecular structure of [P2Nb(ODipp)3]2 has been determined by X-ray crystallography. Computational studies provide further insights into the bonding and reactivity of P2Nb(ODipp)3, [P2Nb(ODipp)3]2, and C6H8P2Nb(ODipp)3.
Co-reporter:Alexander R. Fox and Christopher C. Cummins
Chemical Communications 2012 vol. 48(Issue 25) pp:3061-3063
Publication Date(Web):23 Jan 2012
DOI:10.1039/C2CC17212G
The synthesis, structure, and spectroscopic features of a bimetallic cyanogen complex obtained from the reductive coupling of cyanide by a niobium(IV) precursor are described, and a mechanism for the coupling reaction is proposed based on DFT calculations.
Co-reporter:Paula L. Diaconescu
Inorganic Chemistry 2012 Volume 51(Issue 5) pp:2902-2916
Publication Date(Web):February 16, 2012
DOI:10.1021/ic202163m
Diuranium μ-η6,η6-arene complexes supported by ketimide ligands were synthesized and characterized. Disodium or dipotassium salts of the formula M2(μ-η6,η6-arene)[U(NCtBuMes)3]2 (M = Na or K, Mes = 2,4,6-C6H2Me3) and monopotassium salts of the formula K(μ-η6,η6-arene)[U(NCtBuMes)3]2 (arene = naphthalene, biphenyl, trans-stilbene, or p-terphenyl) were both observed. Two different salts of the monoanionic, toluene-bridged complexes are also described. Density functional theory calculations have been employed to illuminate the electronic structure of the μ-η6,η6-arene diuranium complexes and to facilitate the comparison with related transition-metal systems, in particular (μ-η6,η6-C6H6)[VCp]2. It was found that the μ-η6,η6-arene diuranium complexes were isolobal with (μ-η6,η6-C6H6)[VCp]2 and that the principal arene-binding interaction was a pair of δ bonds (total of 4e) involving both metals and the arene lowest unoccupied molecular orbital. Reactivity studies have been carried out with the mono- and dianionic μ-η6,η6-arene diuranium complexes, revealing contrasting modes of redox chemistry as a function of the system’s state of charge.
Co-reporter:Matthew A. Rankin and Christopher C. Cummins
Dalton Transactions 2012 vol. 41(Issue 32) pp:9615-9618
Publication Date(Web):27 Jun 2012
DOI:10.1039/C2DT31082A
Terminal, 4-coordinate phosphinidenes of Ta supported by bulky anilide ligands are prepared by an apparent reaction sequence involving metallaziridine phosphanide complexes.
Co-reporter:Christopher R. Clough, Jared S. Silvia, Peter Müller, Christopher C. Cummins
Inorganica Chimica Acta 2012 Volume 382() pp:195-198
Publication Date(Web):15 March 2012
DOI:10.1016/j.ica.2011.11.060
Treatment of (CH3CN)3Mo(CO)3(CH3CN)3Mo(CO)3 with the trimetaphosphate salt [PPN]3[P3O9]·H2O([PPN]+=[Ph3PN-PPh3]+) (acetonitrile, 25°C) gave the trimetaphosphate-molybdenum complex [(P3O9)Mo(CO)3]3-[(P3O9)Mo(CO)3]3- in 91% isolated yield. [PPN]3[(P3O9)Mo(CO)3][PPN]3[(P3O9)Mo(CO)3] was also obtained in 77% isolated yield by directly treating Mo(CO)6Mo(CO)6 with [PPN]3[P3O9]·H2O[PPN]3[P3O9]·H2O in refluxing acetonitrile, presumably by generating (CH3CN)3Mo(CO)3(CH3CN)3Mo(CO)3in situ . Reported herein is the full characterization and structural determination of [PPN]3[(P3O9)Mo(CO)3][PPN]3[(P3O9)Mo(CO)3] and comparison to synthesized and calculated [(P3O9)M(CO)3]n-[(P3O9)M(CO)3]n- (M = group 6–9) complexes.Graphical abstractTrimetaphosphate molybdenum tricarbonyl trianion was obtained from molybdenum hexacarbonyl upon refluxing in acetonitrile.Highlights► Presented herein is the first characterized example of a molybdenum trimetaphosphate complex. ► The synthesis involves mixing a trimetaphosphate salt with molybdenum hexacarbonyl in refluxing acetonitrile. ► Experimental data from [(P3O9)Mo(CO)3]3− are compared to previous theoretical predictions.
Co-reporter:Nazario Lopez;Daniel J. Graham;Robert McGuire Jr.;Glen E. Alliger;Yang Shao-Horn;Daniel G. Nocera
Science 2012 Volume 335(Issue 6067) pp:450-453
Publication Date(Web):27 Jan 2012
DOI:10.1126/science.1212678
Co-reporter:Ivo Krummenacher, Christopher C. Cummins
Polyhedron 2012 Volume 32(Issue 1) pp:10-13
Publication Date(Web):20 January 2012
DOI:10.1016/j.poly.2011.07.016
A novel borane-capped niobium phosphide anion ([B(C6F5)3-1]) has been prepared in 87% yield by reaction of [PNb(N[Np]Ar)3]− (1, Ar = 3,5-C6H3Me2 and Np = neopentyl) with the Lewis acidic borane B(C6F5)3. Room-temperature reaction of this adduct with carbon dioxide readily yields the OCP− ion 3, isolated as its sodium salt, [Na(dme)2][OCP] (dme = dimethoxyethane), in 70% yield, with concomitant formation of a borane-substituted niobium oxo complex [B(C6F5)3-2] (2 = ONb(N[Np]Ar)3).Graphical abstractThe niobium phosphide anion complex [PNb(N[Np]Ar)3]−, where Np = neopentyl and Ar = 3,5-C6H3Me2, was converted to its fluorinated borane adduct, [(F5C6)3BPNb(N[Np]Ar)3]−. Upon reaction of the latter anion with CO2, formation of phosphaethynolate, [OCP]−, was confirmed, together with formation of borane-capped niobium oxo, (F5C6)3BONb(N[Np]Ar)3.Highlights► A niobium phosphide anion complex was complexed with perfluorotriphenylborane. ► Carbon dioxide was transformed into the sodium salt of phosphaethynolate. ► Sodium phosphaethynolate was formed in good yield and isolated in pure form.
Co-reporter:Sidney E. Creutz, Ivo Krummenacher, Christopher R. Clough and Christopher C. Cummins
Chemical Science 2011 vol. 2(Issue 11) pp:2166-2172
Publication Date(Web):16 Aug 2011
DOI:10.1039/C1SC00375E
Treatment of terminal phosphide anion [PNb(N[Np]Ar)3]− ([Na(OEt2)]+ salt, Ar = 3,5-Me2C6H3) with two diphenylketene equivalents led to isolation of anion [P(C{CPh2}O)2Nb(N[Np]Ar)3]− in 78% yield as its [Na(THF)]+ salt. The latter reacts with diphenylketene (1 equiv) to provide the triple diphenylketene addition product [P(C{CPh2}O)3Nb(N[Np]Ar)3]− in 88% yield as its [Na(THF)]+ salt; the same material is obtained alternatively in 93% yield by reaction of diphenylketene (3 equiv) directly with niobium phosphide [Na(OEt2)][PNb(N[Np]Ar)3]. The anion [P(C{CPh2}O)3Nb(N[Np]Ar)3]− was also crystallized as its ion-separated [Na(THF)6]+ salt as illuminated by a single-crystal X-ray diffraction study, which also revealed the three-fold symmetric nature of the highly hindered tertiary phosphine comprising the anionic component. Coinage metal monocations of the new, anionic phosphine were prepared via salt elimination; structural studies on the zwitterionic complexes (py)M[P(C{CPh2}O)3Nb(N[Np]Ar)3] (M = Cu, Ag, and Au) showed them to be isostructural in the space groupP21/c while illustrating the ensconcement of a two-coordinate coinage metal in a deep binding pocket flanked by three phenyl residues. Rhodium(I) incorporation into the anion's binding pocket illustrated versatility of the latter: in contrast to structural observations for the M(I) complexes (M = Cu, Ag, and Au), in the case of Rh[P(C{CPh2}O)3Nb(N[Np]Ar)3] an X-ray study reveals strong interactions (η6 and η2, respectively) with two of the three phenyl residues that are proximal to Rh(I) when connected to the phosphine lone pair of the anion.
Co-reporter:Jared S. Silvia and Christopher C. Cummins
Chemical Science 2011 vol. 2(Issue 8) pp:1474-1479
Publication Date(Web):26 May 2011
DOI:10.1039/C1SC00215E
The titanium oxo anion complex [(Et2O)2Li][OTi(N[tBu]3,5-Me2C6H3)3] ([(Et2O)2Li][1]) reacts with CO2 in diethyl ether to form the carbonate complex ([Li][O2COTi(N[tBu]3,5-Me2C6H3)3])6 ([Li][2]). The solid-state structure of complex [Li][2] is a hexamer with a hexagonal prismatic core comprised of six lithium cations bridged by the carbonate functionality. In the monomeric subunits of [Li][2], the carbonate ligand is bound κ1- to the titanium metal center and pseudo κ2- to the lithium countercation. The hexameric structure persists in benzene solutions as determined by 1H DOSY NMR techniques. The binding of CO2 in complex [Li][2] is reversible and can be effected by the introduction of the lithium sequestration reagent 12-crown-4 to diethyl ether solutions of [Li][2]. Complex [Li][2] is readily functionalized with Me3SiOS(O)2CF3 to yield the silyl carbonate complex Me3SiOC(O)OTi(N[tBu]3,5-Me2C6H3)3 (3), the solid-state structure of which is presented. Functionalization with pivaloyl chloride results in the rapid loss of CO2 and formation of the pivalate complex tBuC(O)OTi(N[tBu]3,5-Me2C6H3)3 (4).
Co-reporter:Michael Montag, Christopher R. Clough, Peter Müller and Christopher C. Cummins
Chemical Communications 2011 vol. 47(Issue 2) pp:662-664
Publication Date(Web):29 Nov 2010
DOI:10.1039/C0CC04342G
Examination of cyclotriphosphate and cyclotetraphosphate as ligands for Co(III) in aqueous solutions revealed that cyclotetraphosphate affords stable complexes as a hemilabile ligand, while cyclotriphosphate exhibits facile hydrolysis.
Co-reporter:Glen E. Alliger, Peter Müller, Loi H. Do, Christopher C. Cummins, and Daniel G. Nocera
Inorganic Chemistry 2011 Volume 50(Issue 9) pp:4107-4115
Publication Date(Web):March 29, 2011
DOI:10.1021/ic200143b
A series of coordination compounds has been prepared comprising manganese, iron, nickel, and zinc bound by a hexaanionic cryptand where carboxamides are anionic N-donors. The metal complexes have been investigated by X-ray crystallography, and possess metal centers in trigonal monopyramidal geometries with intermetallic distances spanning dMn,avg = 6.080 Å to dNi,avg = 6.495 Å. All complexes featuring trigonal monopyramidal metal(II) ions crystallize in Cc, and feature extended three-dimensional networks composed of cryptate anions linked by bridging potassium countercations. We also report the first solid state structure of the free cryptand ligand, which features no guest in its cavity and which possesses an extended hydrogen-bonding network. SQuID magnetometry data of the metal complexes reveal weak antiferromagnetic coupling of the metal centers. Only the diiron(II) complex exhibits reversible electrochemistry, and correspondingly, its chemical oxidation yields a powder formulated as the diiron(III) congener. The insertion of cyanide into the intermetallic cleft of the diiron(II) complex has been achieved, and comparisons of its solid state structure to the recently reported dicobalt(II) analogue are made. The antiferromagnetic coupling between the diiron(II) and the dicobalt(II) centers when bridged by cyanide does not increase significantly relative to the unbridged congeners. A one-site model satisfactorily fits Mössbauer spectra of unbridged diiron(II) and diiron(III) complexes whereas a two site fit was needed to model the iron(II) centers that are bridged by cyanide.
Co-reporter:Diego Solis-Ibarra, Jared S. Silvia, Vojtech Jancik, and Christopher C. Cummins
Inorganic Chemistry 2011 Volume 50(Issue 20) pp:9980-9984
Publication Date(Web):September 21, 2011
DOI:10.1021/ic200510y
Crystallization of Na2VOP2O7 from its aqueous solution results in formation of a one-dimensional inorganic polymer {Na2VO(H2O)P2O7·7H2O}n (1). When this polymer is dehydrated at elevated temperatures this polymer undergoes a phase transition to form the two-dimensional framework β-Na2VOP2O7, which although previously reported had been difficult to access. Exchanging lithium for sodium via ion-exchange chromatography results in formation of a discrete, cyclic, tetramer species, Li8[VOP2O7(H2O)·4H2O]4 (2). Isolation of crystalline β-Li2VOP2O7 using a dehydration procedure analogous to the one employed for the sodium derivative was unsuccessful. In contrast, we show that β-K2VOP2O7 can be obtained from the amorphous phase K2VOP2O7·nH2O (n = 0–7) upon thermal dehydration.
Co-reporter:Daniel Tofan, Brandi M. Cossairt, and Christopher C. Cummins
Inorganic Chemistry 2011 Volume 50(Issue 24) pp:12349-12358
Publication Date(Web):September 6, 2011
DOI:10.1021/ic2014607
The Nb–P triple bond in [P≡Nb(N[Np]Ar)3]− (Np = CH2tBu; Ar = 3,5-Me2C6H3) has produced the first case of P4 activation by a metal–ligand multiple bond. Treatment of P4 with the sodium salt of the niobium phosphide complex in weakly coordinating solvents led to formation of the cyclo-P3 anion [(P3)Nb(N[Np]Ar)3]−. Treatment in tetrahydrofuran (THF) led to the formation of a cyclo-P5 anion [(Ar[Np]N)(η4-P5)Nb(N[Np]Ar)2]−, which represents a rare example of a substituted pentaphosphacyclopentadienyl ligand. The P4 activation pathway was shown to depend on the dimer–monomer equilibrium of the niobium phosphide reagent, which, in turn, depends on the solvent used for the reaction. The pathway leading to the cyclo-P3 product was shown to require a 2:1 ratio of the phosphide anion to P4, while the cyclo-P5 formation requires a 1:1 ratio. The cyclo-P3 salt has been isolated in 56% yield as orange crystals of the [Na(THF)]2[(P3)Nb(N[Np]Ar)3]2 dimer or in 83% yield as an orange powder of [Na(12-crown-4)2][(P3)Nb(N[Np]Ar)3]. A solid-state X-ray diffraction experiment on the former salt revealed that each Nb–P3 unit exhibits pseudo-C3 symmetry, while 31P NMR spectroscopy showed a sharp signal at −223 ppm that splits into a doublet–triplet pair below −50 °C. It was demonstrated that this salt can serve as a P33– source upon treatment with AsCl3, albeit with modest yield of AsP3. The cyclo-P5 salt was isolated in 71% yield and structurally characterized from red crystals of [Na(THF)6][(Ar[Np]N)(η4-P5)Nb(N[Np]Ar)2]. The anion in this salt can be interpreted as the product of trapping of an intermediate pentaphosphacycplopentadienyl structure through migration of one anilide ligand onto the P5 ring. The W(CO)5-capped cyclo-P3 salt was also isolated in 60% yield as [Na(THF)][(OC)5W(P3)Nb(N[Np]Ar)3] from the activation of 0.5 equiv of P4 with the sodium salt of the tungsten pentacarbonyl adduct of the niobium phosphide anion.
Co-reporter:John J. Curley, Anthony F. Cozzolino and Christopher C. Cummins
Dalton Transactions 2011 vol. 40(Issue 11) pp:2429-2432
Publication Date(Web):04 Feb 2011
DOI:10.1039/C0DT01326A
Facile methoxymethylation of N2-derived nitride NMo(N[tBu]Ar)3 provided the imido cation [MeOCH2NMo(N[tBu]Ar)3]+ as its triflate salt in 88% yield. Treatment of the latter with LiN(SiMe3)2 provided blue methoxyketimide complex MeO(H)CNMo(N[tBu]Ar)3 in 95% yield. Conversion of the latter to the terminal cyanide complex NCMo(N[tBu]Ar)3, which was the subject of a single-crystal X-ray diffraction study, was accomplished in 51% yield upon treatment with a combination of SnCl2 and Me2NSiMe3.
Co-reporter:Brandi M. Cossairt, Nicholas A. Piro and Christopher C. Cummins
Chemical Reviews 2010 Volume 110(Issue 7) pp:4164
Publication Date(Web):February 22, 2010
DOI:10.1021/cr9003709
Co-reporter:Jared S. Silvia
Journal of the American Chemical Society 2010 Volume 132(Issue 7) pp:2169-2171
Publication Date(Web):February 1, 2010
DOI:10.1021/ja910445r
The terminal nitride anion complex [Na][N≡Nb(N[tBu]Ar)3] ([Na][1], Ar = 3,5-Me2C6H3) reacts quantitatively with CO2 to give the carbamate complex [Na][O2CN≡Nb(N[tBu]Ar)3] ([Na][O2C-1]). The structure of [Na][O2C-1] as the bis-12-crown-4 solvate, as determined by X-ray crystallography, displays a unique N-bound carbamate ligand without any metal−oxygen interactions. When treated with organic acid anhydrides or acid chlorides, complex [Na][O2C-1] reacts via salt elimination to give the five-coordinate complexes (RC(O)O)(OCN)Nb(N[tBu]Ar)3 (R-2, R = Me, tBu, F3C). We show that complexes R-2 yield the starting complex [Na][1] with concomitant release of CO upon two-electron reduction. This series of reactions constitutes a closed cycle for the conversion of CO2 to CO mediated by a terminal nitride anion complex.
Co-reporter:Alexander R. Fox ; Polly L. Arnold
Journal of the American Chemical Society 2010 Volume 132(Issue 10) pp:3250-3251
Publication Date(Web):February 24, 2010
DOI:10.1021/ja910364u
Reaction of the uranium(III) tris(anilide) complex (THF)U(N[t-Bu]Ar)3 (1, THF = tetrahydrofuran; Ar = 3,5-Me2C6H3) with MN3 (M = Na, [N(n-Bu)4]) results in the formation of the bimetallic diuranium(IV/IV) complexes M[(μ-N)(U(N[t-Bu]Ar)3)2] (M[3]), which feature a single nitride ligand engaged as a linear, symmetric bridge between two uranium centers. The stability of the U═N═U core across multiple charge states is illustrated by stepwise chemical oxidation of Na[3] to the diuranium(IV/V) complex (μ-N)(U(N[t-Bu]Ar)3)2 (3) and the diuranium(V/V) complex [(μ-N)(U(N[t-Bu]Ar)3)2][B(ArF)4] {[3][B(ArF)4]; ArF = 3,5-(CF3)2C6H3}. M[3], 3, and [3][B(ArF)4] were characterized by NMR spectroscopy, single-crystal X-ray diffraction, and elemental analysis. The cyclic voltammogram of 3 reveals two clean, reversible one-electron electrochemical events at E1/2 = −1.69 and −0.67 V, assigned to the [3]−/3 and 3/[3]+ redox couples, respectively. The X-ray crystal structures of [N(n-Bu)4][3], 3, and [3][B(ArF)4] reveal a linear U═N═U core that contracts by only ∼0.03 Å across the [3]n (n = −1, 0, +1) series, an effect that is rationalized as being primarily electrostatic in origin. [3][B(ArF)4] reacts with NaCN, eliminating Na[B(ArF)4] and forming the known diuranium(IV/IV) cyanoimide complex (μ-NCN)(U(N[t-Bu]Ar)3)2, suggesting that the U═N═U core has metallonitrene-like character.
Co-reporter:Brandi M. Cossairt ; Christopher C. Cummins ; Ashley R. Head ; Dennis L. Lichtenberger ; Raphael J. F. Berger ; Stuart A. Hayes ; Norbert W. Mitzel ;Gang Wu
Journal of the American Chemical Society 2010 Volume 132(Issue 24) pp:8459-8465
Publication Date(Web):June 1, 2010
DOI:10.1021/ja102580d
The molecular and electronic structures of AsP3 and P4 have been investigated. Gas-phase electron diffraction studies of AsP3 have provided rg bond lengths of 2.3041(12) and 2.1949(28) Å for the As−P interatomic distances and the P−P interatomic distances, respectively. The gas-phase electron diffraction structure of P4 has been redetermined and provides an updated value of 2.1994(3) Å for the P−P interatomic distances, reconciling conflicting literature values. Gas-phase photoelectron spectroscopy provides experimental values for the energies of ionizations from the valence molecular orbitals of AsP3 and P4 and shows that electronically AsP3 and P4 are quite similar. Solid-state 75As and 31P NMR spectroscopy demonstrate the plastic nature of AsP3 and P4 as solids, and an extreme upfield 75As chemical shift has been confirmed for the As atom in AsP3. Finally, quantum chemical gauge-including magnetically induced current calculations show that AsP3 and P4 can accurately be described as strongly aromatic. Together these data provide a cohesive description of the molecular and electronic properties of these two tetraatomic molecules.
Co-reporter:Matthew A. Rankin
Journal of the American Chemical Society 2010 Volume 132(Issue 29) pp:10021-10023
Publication Date(Web):July 2, 2010
DOI:10.1021/ja104761n
The preparation of tantalaziridine−hydride complex (Ar[tBuCH2]N)2(η2-tBu(H)CNAr)TaH (1) is reported (Ar = 3,5-Me2C6H3). While stable for months in the solid state at −35 °C, in solution this complex undergoes partial conversion to isomeric hydride (Ar[tBuCH2]N)2(κ2-CH2C(Me)2CH2NAr)TaH (2). Although 1 and 2 exist in equilibrium in benzene solution, complex 2 can be isolated cleanly from 1 by selective precipitation using cold n-pentane; solid-state structures for both 1 and 2 are presented. Exposure of 1 to ca. 1 atm of CO2 results in the production of methylene diolate complex {(Ar[tBuCH2]N)2(η2-tBu(H)CNAr)Ta}2(μ-OCH2O) as a mixture of rac and meso diastereomers (3r,m). Similar reactivity for the Nb congener of 1 is reported herein. Further-more, methylene diolate complex {(Ar[tBuCH2]N)2(κ2-CH2C(Me)2CH2NAr)Ta}2(μ-OCH2O) (4) is produced from 2 upon treatment with CO2 and was characterized crystallographically. Complexes 3r,m (and the Nb analogues) as well as 4 establish the feasibility of the formation of methylene diolate products from CO2 and two terminal transition-metal hydrides. Reaction of formate (Ar[tBu]N)3TiOC(O)H with 1 generates the related, structurally characterized heterobimetallic complex (Ar[tBuCH2]N)2(η2-tBu(H)CNAr)TaOCH2OTi(N[tBu]Ar)3 (5), which further contributes to the class of complexes reported herein that effectively stabilizes an unusual H2CO22− ligand between two metal centers.
Co-reporter:Glen E. Alliger ; Peter Müller ; Christopher C. Cummins ;Daniel G. Nocera
Inorganic Chemistry 2010 Volume 49(Issue 8) pp:3697-3699
Publication Date(Web):March 25, 2010
DOI:10.1021/ic100395a
A hexacarboxamide cryptand featuring appended polyether moieties is used as a binucleating ligand for two CoII centers, marking the first time cryptands have been used as hexaanionic N donors for metal coordination. A synthesis for the hexacarboxamide cryptand, culminating in a 23% yield high-dilution step and proceeding in 8% overall yield, is reported. The ligand is metalated using cobalt(II) acetate, and a solid-state structure is presented, revealing an intermetallic void over 6.4 Å in length. The reactivity of this new type of cryptate is also probed; treatment of the dicobalt cryptate with potassium cyanide at elevated temperature results in a bridging cyanide complex.
Co-reporter:Alexander R. Fox, Sidney E. Creutz and Christopher C. Cummins
Dalton Transactions 2010 vol. 39(Issue 29) pp:6632-6634
Publication Date(Web):25 Jun 2010
DOI:10.1039/C0DT00419G
The synthesis, spectroscopy, structure, and bonding of the molecular uranium dicarbide complex (μ,η1:η1-C2)[U(N[t-Bu]Ar)3]2 (Ar = 3,5-Me2C6H3) is described.
Co-reporter:Brandi M. Cossairt and Christopher C. Cummins
New Journal of Chemistry 2010 vol. 34(Issue 8) pp:1533-1536
Publication Date(Web):13 Apr 2010
DOI:10.1039/C0NJ00124D
A reaction scheme has been devised according to 3 RX + 3 Ti(III) + 0.25 P4 → PR3 + 3 XTi(IV), wherein RX = PhBr, CyBr, Me3SiI or Ph3SnCl, with contrasting results in the case of more hindered RX. The scheme accomplishes the direct radical functionalization of white phosphorus without the intermediacy of PCl3.
Co-reporter:Bri M. Cossairt ; Christopher C. Cummins
Chemistry - A European Journal 2010 Volume 16( Issue 42) pp:12603-12608
Publication Date(Web):
DOI:10.1002/chem.201001819
Abstract
Treatment of AsP3 with 0.75 equivalents of [{GaC(SiMe3)3}4] resulted in selective insertion of three equivalents of {GaC(SiMe3)3} into the three AsP bonds to give [As{GaC(SiMe3)3}3P3] (1-As) with an intact cyclo-P3 ring. This yellow compound has been characterized by NMR spectroscopy, combustion analysis, single-crystal X-ray diffraction, UV/Vis spectroscopy, Raman spectroscopy, and cyclic voltammetry (THF, 0.2 M [TBA][B(C6F5)4]; TBA=tetrabutyl ammonium). Computational models of 1-As and the isomeric [P{GaC(SiMe3)3}3AsP2] (1-P) have been investigated as well, revealing several interesting electronic features of these cage molecules. Following from the cyclic voltammetry studies of 1-As that highlight an irreversible two-electron reduction at −2.2 V versus Fc/Fc+, treatment with one equivalent of [Mg(C14H10)(thf)3] resulted in two-electron reduction to provide [As{GaC(SiMe3)3}3P3Mg(thf)3] (2), in which the Mg2+ ion has inserted into one of the PP bonds of the cyclo-P3 ring. It was also found that treatment of AsP3 or P4 with one equivalent of [{GaC(SiMe3)3}4] resulted in formation of the quadruple insertion products [As{GaC(SiMe3)3}4P3] (3) and [P{GaC(SiMe3)3}4P3] (4), respectively.
Co-reporter:Joshua S. Figueroa, Nicholas A. Piro, Daniel J. Mindiola, Michael G. Fickes, and Christopher C. Cummins
Organometallics 2010 Volume 29(Issue 21) pp:5215-5229
Publication Date(Web):October 15, 2010
DOI:10.1021/om100522p
Presented herein are synthetic, structural, and reactivity studies delineating the characteristics of the niobaziridine hydride functional group as it pertains to the stabilization of trisanilide niobium complexes of the type Nb(N[R]Ar)3 (1R, Ar = 3,5-Me2C6H3). Utilization of the N-isopropyl anilide ligand, N[i-Pr]Ar, results in the niobaziridine hydride dimer [Nb(H)(η2-Me2C═NAr)(N[i-Pr]Ar)2]2 ([2i-Pr-H]2). Dimer [2i-Pr-H]2 is thermally unstable at room temperature and decomposes via ortho-metalation and i-Pr radical ejection to a species containing a Nb−Nb bond. The ligand variant N[Np]Ar (Np = neopentyl) provides the room-temperature-stable niobaziridine hydride monomer Nb(H)(η2-t-Bu(H)C═NAr)(N[Np]Ar)2 (2Np-H). Thermal decomposition of 2Np-H at elevated temperature (75 °C) provides the neopentyl imido complex Nb(NNp)(Ar)(N[Np]Ar)2 (5Np). H/D isotopic labeling studies provide evidence for reversible β-H elimination interconverting 2Np-H and its trisanilide tautomer [Nb(N[Np]Ar)3] (1Np), with the latter thereby implicated as an intermediate during the 2Np-H → 5Np conversion. Reactivity studies between 2Np-H and certain small-molecule substrates confirm that the niobaziridine hydride group can effectively mask a reactive d2 Nb(III) trisanilide center. However, 2Np-H exhibits insertion chemistry when treated with a variety of unsaturated organic substrates, thus demonstrating a pronounced tendency to additionally function as a Lewis acidic, early transition metal hydride species. A general mechanism accounting for the divergent reactivity of 2Np-H is proposed. Niobaziridine hydride complexes derived from the amido ligands N[CH2Ad]Ar, N[Cy]Ar, and NCy2 (Ad = 1-adamantyl, Cy = cyclohexyl) are also presented, and their thermal behavior and reaction chemistry are compared with those of 2Np-H. In addition, the radical anion of 2Np-H is reported and compared with the neutral d1 molybdaziridine hydride complex Mo(H)(η2-Me2C═NAr)(N[i-Pr]Ar)2 (3i-Pr-H), to which it is isoelectronic.
Co-reporter:BriM. Cossairt ;ChristopherC. Cummins
Angewandte Chemie International Edition 2010 Volume 49( Issue 9) pp:1595-1598
Publication Date(Web):
DOI:10.1002/anie.200906633
Co-reporter:Daniel Tofan ; Christopher C. Cummins
Angewandte Chemie International Edition 2010 Volume 49( Issue 41) pp:7516-7518
Publication Date(Web):
DOI:10.1002/anie.201004385
Co-reporter:Daniel Tofan ; Christopher C. Cummins
Angewandte Chemie 2010 Volume 122( Issue 41) pp:7678-7680
Publication Date(Web):
DOI:10.1002/ange.201004385
Co-reporter:BriM. Cossairt ;ChristopherC. Cummins
Angewandte Chemie 2010 Volume 122( Issue 9) pp:1639-1642
Publication Date(Web):
DOI:10.1002/ange.200906633
Co-reporter:Brandi M. Cossairt;Mariam-Céline Diawara
Science 2009 Vol 323(5914) pp:602
Publication Date(Web):30 Jan 2009
DOI:10.1126/science.1168260
Abstract
The elements phosphorus and arsenic form tetrahedral P4 and As4 molecules, of which the former is a commodity chemical and the latter unstable. Previously, information on molecules of intermediate composition was limited to a spectroscopic study involving hot gas-phase mixtures of phosphorus and arsenic. The AsP3 molecule has yielded to chemical synthesis and was isolated in solid form as a pure substance with a melting point from 71° to 73°C. Physical properties and spectroscopic characterization data for AsP3 are described, and its structure was determined as a ligand in a coordination complex. Soluble in organic solvents, AsP3 represents an attractive starting point for any synthesis in which the target molecule or material contains an exact 1:3 ratio of arsenic to phosphorus.
Co-reporter:Nicholas A. Piro
Journal of the American Chemical Society 2009 Volume 131(Issue 25) pp:8764-8765
Publication Date(Web):June 8, 2009
DOI:10.1021/ja903860k
The terminal phosphorus monoxide complex (OP)Mo(N[tBu]Ar)3, 1 (Ar = 3,5-Me2C6H3), undergoes an O-for-PSiR3 metathesis reaction with the niobium phosphinidene complex iPr3SiPNb(N[CH2tBu]Ar)3, 2, to generate the oxoniobium complex ONb(N[CH2tBu]Ar)3, 3, and the diphosphenido complex iPr3SiPPMo(N[tBu]Ar)3, 4. The structure of 4, as determined by X-ray crystallography, contains a “singly bent” diphosphenido moiety, suggesting that the diphosphenido ligand serves as a 3e− donor to a formally d2 metal center. This bonding characterization was supported by DFT calculations and is unique among known diphosphenido complexes. Diphosphenido 4 was found to react over time to produce products consistent with a bimolecular degradation pathway where the terminal phosphide complex PMo(N[tBu]Ar)3, 5, serves as a stable leaving group. Mixtures of 4 and PPh3 were observed to set up an equilibrium (Keq = 0.7) between 4, PPh3, and the products of phosphinidene transfer, 5 and iPr3SiP═PPh3.
Co-reporter:Brandi M. Cossairt
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15501-15511
Publication Date(Web):October 2, 2009
DOI:10.1021/ja906294m
Facile synthetic access to the isolable, thermally robust AsP3 molecule has allowed for a thorough study of its physical properties and reaction chemistry with a variety of transition-metal and organic fragments. The electronic properties of AsP3 in comparison with P4 are revealed by DFT and atoms in molecules (AIM) approaches and are discussed in relation to the observed electrochemical profiles and the phosphorus NMR properties of the two molecules. An investigation of the nucleus independent chemical shifts revealed that AsP3 retains spherical aromaticity. The thermodynamic properties of AsP3 and P4 are described. The reaction types explored in this study include the thermal decomposition of the AsP3 tetrahedron to its elements, the synthesis and structural characterization of [(AsP3)FeCp*(dppe)][BPh4] (dppe = 1,2-bis(diphenylphosphino)ethane), 1, selective single As-P bond cleavage reactions, including the synthesis and structural characterization of AsP3(P(N(iPr)2)N(SiMe3)2)2, 2, and activations of AsP3 by reactive early transition-metal fragments including Nb(H)(η2-tBu(H)C═NAr)(N[CH2tBu]Ar)2 and Mo(N[tBu]Ar)3 (Ar = 3,5-Me2C6H3). In the presence of reducing equivalents, AsP3 was found to allow access to [Na][E3Nb(ODipp)3] (Dipp = 2,6-diisopropylphenyl) complexes (E = As or P) which themselves allow access to mixtures of AsnP4−n (n = 1−4).
Co-reporter:Heather A. Spinney ; Nicholas A. Piro
Journal of the American Chemical Society 2009 Volume 131(Issue 44) pp:16233-16243
Publication Date(Web):October 20, 2009
DOI:10.1021/ja906550h
While P4 is the stable molecular form of phosphorus, a recent study illustrated the possibility of P2 generation for reactions in organic media under mild conditions. The heavier group 15 element arsenic can exist as As4 molecules, but As4 cannot be stored as a pure substance because it is both light-sensitive and reverts thermally to its stable, metallic gray form. Herein we report As4 activation giving rise to a μ-As2 diniobium complex, serving in turn as precursor to a terminal arsenide anion complex of niobium. Functionalization of the latter provides the new AsPNMes* ligand, which when complexed with tungsten pentacarbonyl elicits extrusion of the (AsP)W(CO)5 molecule as a reactive intermediate. Trapping reactions of the latter with organic dienes are found to furnish double Diels-Alder adducts in which the AsP unit is embedded in a polycyclic organic framework. Thermal generation of (AsP)W(CO)5 in the presence of the neutral terminal phosphide complex P≡Mo(N[iPr]Ar)3 leads to the cyclo-AsP2 complex (OC)5W(cyclo-AsP2)Mo(N[iPr]Ar)3. The (AsP)W(CO)5 trapping products were crystallized and characterized by X-ray diffraction methods, and computational methods were applied for analysis of the As−As and As−P bonds in the complexes.
Co-reporter:John J. Curley, Nicholas A. Piro and Christopher C. Cummins
Inorganic Chemistry 2009 Volume 48(Issue 20) pp:9599-9601
Publication Date(Web):September 18, 2009
DOI:10.1021/ic9016068
A terminal molybdenum arsenide complex is synthesized in one step from the reactive As4 molecule. The properties of this complex with its arsenic atom ligand are discussed in relation to the analogous nitride and phosphide complexes.
Co-reporter:John J. Curley, Tetsuro Murahashi and Christopher C. Cummins
Inorganic Chemistry 2009 Volume 48(Issue 15) pp:7181-7193
Publication Date(Web):July 6, 2009
DOI:10.1021/ic900495s
A series of cationic diazoalkane complexes [4-RC6H4C(H)NNMo(N[t-Bu]Ar)3][AlCl4], [1-R][AlCl4] (R = NMe2, Me, H, Br, CN; Ar = 3,5-C6H3Me2) has been prepared by treatment of the N2-derived diazenido complex Me3SiNNMo(N[t-Bu]Ar)3 with 4-RC6H4CHO and 2 equiv of AlCl3. The structures of [1-H][AlCl4] and [1-NMe2][AlCl4] were determined by X-ray crystallography. The C−N and N−N stretching modes were identified by a combined IR and Raman spectroscopy study, and other physical properties are discussed in detail. The electrochemical reduction potential for [1-R][AlCl4] was shown to be linear with the Hammett σ parameter. This reduction process forms the C−C bonded dimer, μ-(4-RC6H4C(H)NN)2[Mo(N[t-Bu]Ar)3]2, that was characterized by X-ray crystallography for R = H. Possible mechanisms for the formation of this dimer are presented. Both electrochemical investigations and quantum chemical calculations are used to describe the odd-electron complex 4-RC6H4C(H)NNMo(N[t-Bu]Ar)3, 1-R, that is an intermediate in the formation of [1-R]2. The C−C bond in [1-R]2 is redox-noninnocent and is broken upon oxidation. This reaction was used to prepare [1-H][A] (A = PF6−, OTf−), and possible uses of this property in charge-storage devices are discussed.
Co-reporter:NicholasA. Piro ;ChristopherC. Cummins Dr.
Angewandte Chemie 2009 Volume 121( Issue 5) pp:952-956
Publication Date(Web):
DOI:10.1002/ange.200804432
Co-reporter:NicholasA. Piro ;ChristopherC. Cummins Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 5) pp:934-938
Publication Date(Web):
DOI:10.1002/anie.200804432
Co-reporter:James E. McDonough ; Arjun Mendiratta ; John J. Curley ; George C. Fortman ; Serena Fantasia ; Christopher C. Cummins ; Elena V. Rybak-Akimova ; Steven P. Nolan ;Carl D. Hoff
Inorganic Chemistry 2008 Volume 47(Issue 6) pp:2133-2141
Publication Date(Web):February 9, 2008
DOI:10.1021/ic701611p
Enthalpies of chalcogen atom transfer to Mo(N[t-Bu]Ar)3, where Ar = 3,5-C6H3Me2, and to IPr (defined as bis-(2,6-isopropylphenyl)imidazol-2-ylidene) have been measured by solution calorimetry leading to bond energy estimates (kcal/mol) for EMo(N[t-Bu]Ar)3 (E = S, 115; Se, 87; Te, 64) and EIPr (E = S, 102; Se, 77; Te, 53). The enthalpy of S-atom transfer to PMo(N[t-Bu]Ar)3 generating SPMo(N[t-Bu]Ar)3 has been measured, yielding a value of only 78 kcal/mol. The kinetics of combination of Mo(N[t-Bu]Ar)3 with SMo(N[t-Bu]Ar)3 yielding (μ-S)[Mo(N[t-Bu]Ar)3]2 have been studied, and yield activation parameters ΔH‡ = 4.7 ± 1 kcal/mol and ΔS‡ = −33 ± 5 eu. Equilibrium studies for the same reaction yielded thermochemical parameters ΔH° = −18.6 ± 3.2 kcal/mol and ΔS° = −56.2 ± 10.5 eu. The large negative entropy of formation of (μ-S)[Mo(N[t-Bu]Ar)3]2 is interpreted in terms of the crowded molecular structure of this complex as revealed by X-ray crystallography. The crystal structure of Te-atom transfer agent TePCy3 is also reported. Quantum chemical calculations were used to make bond energy predictions as well as to probe terminal chalcogen bonding in terms of an energy partitioning analysis.
Co-reporter:Manuel Temprado ; James Eric McDonough ; Arjun Mendiratta ; Yi-Chou Tsai ; George C. Fortman ; Christopher C. Cummins ; Elena V. Rybak-Akimova ;Carl D. Hoff
Inorganic Chemistry 2008 Volume 47(Issue 20) pp:9380-9389
Publication Date(Web):September 13, 2008
DOI:10.1021/ic800945m
Synthetic studies are reported that show that the reaction of either H2SnR2 (R = Ph, n-Bu) or HMo(CO)3(Cp) (1-H, Cp = η5-C5H5) with Mo(N[t-Bu]Ar)3 (2, Ar = 3,5-C6H3Me2) produce HMo(N[t-Bu]Ar)3 (2-H). The benzonitrile adduct (PhCN)Mo(N[t-Bu]Ar)3 (2-NCPh) reacts rapidly with H2SnR2 or 1-H to produce the ketimide complex (Ph(H)C═N)Mo(N[t-Bu]Ar)3 (2-NC(H)Ph). The X-ray crystal structures of both 2-H and 2-NC(H)Ph are reported. The enthalpy of reaction of 1-H and 2 in toluene solution has been measured by solution calorimetry (ΔH = −13.1 ± 0.7 kcal mol−1) and used to estimate the Mo−H bond dissociation enthalpy (BDE) in 2-H as 62 kcal mol−1. The enthalpy of reaction of 1-H and 2-NCPh in toluene solution was determined calorimetrically as ΔH = −35.1 ± 2.1 kcal mol−1. This value combined with the enthalpy of hydrogenation of [Mo(CO)3(Cp)]2 (12) gives an estimated value of 90 kcal mol−1 for the BDE of the ketimide C−H of 2-NC(H)Ph. These data led to the prediction that formation of 2-NC(H)Ph via nitrile insertion into 2-H would be exothermic by ∼36 kcal mol−1, and this reaction was observed experimentally. Stopped flow kinetic studies of the rapid reaction of 1-H with 2-NCPh yielded ΔH⧧ = 11.9 ± 0.4 kcal mol−1, ΔS⧧ = −2.7 ± 1.2 cal K−1 mol−1. Corresponding studies with DMo(CO)3(Cp) (1-D) showed a normal kinetic isotope effect with kH/kD ≈ 1.6, ΔH⧧ = 13.1 ± 0.4 kcal mol−1 and ΔS⧧ = 1.1 ± 1.6 cal K−1 mol−1. Spectroscopic studies of the much slower reaction of 1-H and 2 yielding 2-H and 1/212 showed generation of variable amounts of a complex proposed to be (Ar[t-Bu]N)3Mo−Mo(CO)3(Cp) (1−2). Complex 1−2 can also be formed in small equilibrium amounts by direct reaction of excess 2 and 12. The presence of 1−2 complicates the kinetic picture; however, in the presence of excess 2, the second-order rate constant for H atom transfer from 1-H has been measured: 0.09 ± 0.01 M−1 s−1 at 1.3 °C and 0.26 ± 0.04 M−1 s−1 at 17 °C. Study of the rate of reaction of 1-D yielded kH/kD = 1.00 ± 0.05 consistent with an early transition state in which formation of the adduct (Ar[t-Bu]N)3Mo···HMo(CO)3(Cp) is rate limiting.
Co-reporter:Brandi M. Cossairt
Inorganic Chemistry 2008 Volume 47(Issue 20) pp:9363-9371
Publication Date(Web):September 19, 2008
DOI:10.1021/ic8009282
The diniobium octaphosphorus complex (P8)[Nb(OC[2Ad]Mes)3]2 (1) (Ad = adamantylidene, Mes = 2,4,6-Me3C6H2) contains a reactive niobium phosphinidene moiety that can be exploited for metathetical scission of the Nb═P bond. When 1 is treated with aryl ketones, loss of ONb(OC[2Ad]Mes)3(OEt2) (2) is observed along with concomitant formation of the corresponding phosphaalkene (RC6H4)2C═PP7Nb(OC[2Ad]Mes)3 (3-R). Complexes 3-R rearrange to incorporate the (RC6H4)2C═P unit into the phosphorus cage, thereby generating a saturated organo-phosphorus cluster complexed to the niobium tris-enolate platform, (RC6H4)2CP8Nb(OC[2Ad]Mes)3 (4-R). The structure of one such rearranged cluster 4-H, as determined by X-ray crystallography, is briefly discussed. An Eyring analysis of the first-order rearrangement of 3-H to 4-H gives activation parameters of ΔH⧧ = 16.7 kcal/mol and ΔS⧧ = −20.4 eu. A Hammett analysis of the phosphaalkene rearrangement, 3-R to 4-R, with substitution varying at the para positions of the aryl rings, reveals a linear relationship between the σ values and the rearrangement rate constants. A concerted, asynchronous mechanism for the least-motion rearrangement of 3-H to 4-H is presented. When 1 is treated with alkyl ketones, similar loss of 2 and formation of the corresponding phosphaalkene is observed; however, the phosphaalkene complexes have considerably greater stability and are readily isolated.
Co-reporter:BriM. Cossairt ;ChristopherC. Cummins Dr.
Angewandte Chemie 2008 Volume 120( Issue 46) pp:8995-8998
Publication Date(Web):
DOI:10.1002/ange.200803971
Co-reporter:BriM. Cossairt ;ChristopherC. Cummins Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 46) pp:8863-8866
Publication Date(Web):
DOI:10.1002/anie.200803971
Co-reporter:BriM. Cossairt ;ChristopherC. Cummins Dr.
Angewandte Chemie International Edition 2007 Volume 47( Issue 1) pp:169-172
Publication Date(Web):
DOI:10.1002/anie.200704354
Co-reporter:BriM. Cossairt ;ChristopherC. Cummins Dr.
Angewandte Chemie 2007 Volume 120( Issue 1) pp:175-178
Publication Date(Web):
DOI:10.1002/ange.200704354
Co-reporter:Paresh Agarwal;Nicholas A. Piro;Karsten Meyer Dr.;Peter Müller Dr.;Christopher C. Cummins Dr.
Angewandte Chemie 2007 Volume 119(Issue 17) pp:
Publication Date(Web):12 APR 2007
DOI:10.1002/ange.200790068
Große redoxaktive Metalloliganden stabilisieren das Phosphorradikal [.P{NV[N(Np)Ar]3}2] (Np=Neopentyl) so stark, dass es selbst im festen Zustand als Monomer vorliegt. Aus dem EPR-Spektrum des Radikals (siehe Bild; P orange, N blau, V gelb) leiten C. C. Cummins und Mitarbeiter in ihrer Zuschrift auf S. 3171 ff. eine Delokalisierung des ungepaarten Elektrons über beide Vanadiumzentren ab. Radikalische Bindungsbildungen am Phosphoratom führen zu diamagnetischen Produkten.
Co-reporter:Paresh Agarwal;Nicholas A. Piro;Karsten Meyer Dr.;Peter Müller Dr.;Christopher C. Cummins Dr.
Angewandte Chemie 2007 Volume 119(Issue 17) pp:
Publication Date(Web):12 MAR 2007
DOI:10.1002/ange.200700059
Unterstützung für ein Radikal: Das neutrale Radikal [.P{NV[N(Np)Ar]3}2] (Np=Neopentyl, Ar=3,5-Me2C6H3) ist durch zwei Nitridovanadiumtrisanilid-Liganden über einen VIV/VV-Redoxzyklus resonanzstabilisiert (siehe EPR-Spektrum und berechnetes SOMO für [.P{NV[N(Me)Ph]3}2]). Die Verbindung liegt im Festkörper als Monomer vor und geht durch radikalische Reaktionen am Phosphorzentrum in diamagnetische Produkte über.
Co-reporter:Alexer R. Fox;Christopher R. Clough;Nicholas A. Piro;Christopher C. Cummins
Angewandte Chemie 2007 Volume 119(Issue 6) pp:
Publication Date(Web):9 JAN 2007
DOI:10.1002/ange.200604736
Ligand verloren und gewonnen: Ein Wolframkomplex mit endständigem Nitridliganden wird in einer Eintopfsequenz von Atomtransferreaktionen (siehe Schema) in den Komplex mit endständigem Phosphidliganden umgewandelt. Der Phosphidkomplex wird anschließend durch Behandlung mit einem Phosphorelektrophil funktionalisiert. Der dabei gebildete phosphorreiche Komplex weist P-P-Mehrfachbindungscharakter auf.
Co-reporter:Paresh Agarwal;Nicholas A. Piro;Karsten Meyer Dr.;Peter Müller Dr.;Christopher C. Cummins Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 17) pp:
Publication Date(Web):12 APR 2007
DOI:10.1002/anie.200790068
Bulky redox-active metalloligands stabilize the phosphorus radical [.P{NV[N(Np)Ar]3}2] (Np=neopentyl) to the extent that it exists as a monomer even in the solid state. C. C. Cummins and co-workers describe in their Communication on page 3111 ff. the EPR spectrum of the radical (see picture; P orange, N blue, V yellow), which reveals delocalization of the unpaired electron onto both vanadium centers. Radical bond formation at the phosphorus atom leads to diamagnetic products.
Co-reporter:Paresh Agarwal;Nicholas A. Piro;Karsten Meyer Dr.;Peter Müller Dr.;Christopher C. Cummins Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 17) pp:
Publication Date(Web):12 MAR 2007
DOI:10.1002/anie.200700059
Aiding and abetting a wanted radical: The neutral phosphorus radical [.P{NV[N(Np)Ar]3}2] (Np=neopentyl, Ar=3,5-Me2C6H3) exists as a monomer in the solid state and is resonance-stabilized by two nitridovanadium trisanilide metalloligands through the VIV/VV redox couple (see EPR trace and calculated SOMO of [.P{NV[N(Me)Ph]3}2]). The compound undergoes radical reactions at its phosphorus center to form diamagnetic compounds.
Co-reporter:Alexer R. Fox;Christopher R. Clough;Nicholas A. Piro;Christopher C. Cummins
Angewandte Chemie International Edition 2007 Volume 46(Issue 6) pp:
Publication Date(Web):9 JAN 2007
DOI:10.1002/anie.200604736
Ligands lost, ligands gained: A terminal tungsten nitride is converted into the corresponding terminal tungsten phosphide by a one-pot sequence of atom-transfer reactions (see scheme). The phosphide complex is subsequently functionalized by treatment with a phosphorus-based electrophile. The resulting phosphorus-rich complex displays PP multiple-bonding character.
Co-reporter:Nicholas A. Piro;Joshua S. Figueroa;Jessica T. McKellar
Science 2006 Vol 313(5791) pp:1276-1279
Publication Date(Web):01 Sep 2006
DOI:10.1126/science.1129630
Abstract
We report a mild method for generating the diphosphorus molecule or its synthetic equivalent in homogeneous solution; the P2 allotrope of the element phosphorus is normally obtained only under extreme conditions (for example, from P4 at 1100 kelvin). Diphosphorus is extruded from a niobium complex designed for this purpose and can be trapped efficiently by two equivalents of an organic diene to produce an organodiphosphorus compound. Diphosphorus stabilized by coordination to tungsten pentacarbonyl can be generated similarly at 25°C, and in this stabilized form it still efficiently consumes two organic diene molecules for every diphosphorus unit.
Co-reporter:Joshua S. Figueroa and Christopher C. Cummins
Dalton Transactions 2006 (Issue 18) pp:2161-2168
Publication Date(Web):03 Apr 2006
DOI:10.1039/B602530G
This short review describes a breakthrough embodied by the synthesis of a niobaziridine hydride complex. This reactive entity reacts directly with white phosphorus to provide a bridging diphosphorus diniobium complex that upon reduction splits to afford a terminal niobium phosphide anion, isolated as its sodium salt. Reactions of the latter with acid chlorides constitute a new synthesis of phosphaalkynes, while treatment with chlorodiorganophosphanes leads to complexed 1,1-diorganophosphanylphosphinidene systems. Additionally, reactions of the sodium salt of the niobium phosphide anion with divalent main group element salts (E = Ge, Sn, or Pb) provide complexed triatomic EP2 triangles. Dinitrogen cleavage was realized via reduction of a heterodinuclear niobium/molybdenum dinitrogen complex, and this provided an entry to a nitrogen-15 labeled terminal nitride anion of niobium as its sodium salt. In a fashion analogous to the aforementioned phosphaalkyne synthesis, acid chlorides are transformed upon reaction with the niobium nitride anion into corresponding nitrogen-15 labeled organic nitriles. Complete synthetic cycles are achieved in both the phosphaalkyne and the organic nitrile syntheses, as the oxoniobium(V) byproduct can be recycled in high yield to the title niobaziridine hydride complex.
Co-reporter:Christopher C. Cummins Dr.
Angewandte Chemie 2006 Volume 118(Issue 6) pp:
Publication Date(Web):30 DEC 2005
DOI:10.1002/ange.200503327
Anionische Komplexe mit terminalen einatomigen Nitrid-, Phosphid- und Carbidliganden sind ausgezeichnete Ausgangsmaterialien für die Synthese von Liganden mit niederkoordinierten Phosphorzentren in der schützenden Koordinationssphäre eines Metallkomplexes. Salzeliminierungsreaktionen mit Chlorphosphanen führen zu Phosphaisocyanid-, Iminophosphinimid- und Diorganophosphanylphosphiniden-Komplexen, in denen die ungewöhnlichen Phosphorliganden durch Koordination stabilisiert sind. Röntgenstrukturanalysen und Dichtefunktionalrechnungen erhellen die Bindungsverhältnisse in diesen Verbindungen.
Co-reporter:Christopher C. Cummins
Angewandte Chemie International Edition 2006 45(6) pp:862-870
Publication Date(Web):
DOI:10.1002/anie.200503327
Co-reporter:Arjun Mendiratta, Joshua S. Figueroa and Christopher C. Cummins
Chemical Communications 2005 (Issue 27) pp:3403-3405
Publication Date(Web):09 Jun 2005
DOI:10.1039/B504492H
Deprotonation of the titanium formate complex [Ar(t-Bu)N]3TiOC(O)H with LiN(i-Pr)2 resulted in the release of free CO and the formation of a titanium(IV) oxoanion complex, isolated as its lithium salt.
Co-reporter:Joshua S. Figueroa Dr.
Angewandte Chemie 2005 Volume 117(Issue 29) pp:
Publication Date(Web):30 JUN 2005
DOI:10.1002/ange.200500707
Salze von zweiwertigen Gruppe-14-Elementen reagieren mit dem [Na(thf)x]+-Derivat des terminalen Niobphosphid-Anions [PNb{N(Np)Ar}3]− (Np=Neopentyl, Ar=3,5-Me2C6H3) zu Komplexen der Form [(μ2:η3,η3-cyclo-EP2){Nb[N(Np)Ar]3}2] (E=Ge, Sn, Pb; siehe Struktur). Die verbrückenden {cyclo-EP2}-Einheiten dieser Komplexe können als neutrale dreigliedrige 2π-Elektronenringe gesehen werden, die isolobal zum Cyclopropenium-Ion sind.
Co-reporter:Tetsuro Murahashi Dr.;Christopher R. Clough;Joshua S. Figueroa Dr.
Angewandte Chemie 2005 Volume 117(Issue 17) pp:
Publication Date(Web):22 MAR 2005
DOI:10.1002/ange.200463057
Distickstoff zwischen Molybdän und Phosphor: Im abgebildeten kationischen Komplex bildet der Diazo(triorgano)phosphoran-Ligand Ph2MePN2 , der aus koordiniertem N2 am Metallzentrum erzeugt wurde, mit seinem N-Ende eine Mehrfachbindung zum Molybdän. Die N-N-Bindung in diesem Komplex ist Rechnungen zufolge stark aktiviert, bei Einelektronenreduktion wird jedoch Zerfall unter N2-Verlust beobachtet.
Co-reporter:Joshua S. Figueroa Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 29) pp:
Publication Date(Web):30 JUN 2005
DOI:10.1002/anie.200500707
Divalent Group 14 salts react with the [Na(thf)x]+ derivative of the terminal niobium phosphide anion [PNb{N(Np)Ar}3]− (Np=neopentyl, Ar=3,5-Me2C6H3) to give complexes of the form [(μ2:η3,η3-cyclo-EP2){Nb[N(Np)Ar]3}2] (E=Ge, Sn, and Pb; see structure). The bridging {cyclo-EP2} units in these complexes can be considered as neutral 2π-electron three-membered rings that are isolobal to the cyclopropenium ion.
Co-reporter:Tetsuro Murahashi Dr.;Christopher R. Clough;Joshua S. Figueroa Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 17) pp:
Publication Date(Web):22 MAR 2005
DOI:10.1002/anie.200463057
Dinitrogen connects molybdenum and phosphorus: The diazo(triorgano)phosphorane ligand Ph2MePN2 is multiply bonded through its N-terminus to molybdenum in the depicted cationic complex. The Ph2MePN2 ligand was synthesized within molybdenum's coordination sphere by rational assembly from a precursor N2 complex. Computational studies reveal the NN bond to be highly activated in the complex, which nonetheless readily fragments (with N2 loss) upon one-electron reduction.
Co-reporter:Joshua S. Figueroa Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 8) pp:
Publication Date(Web):11 FEB 2004
DOI:10.1002/anie.200352779
Reductive cleavage of a bridging diphosphide complex with sodium amalgam affords the sodium salt of the terminal niobium phosphide anion [PNb(N[Np]Ar)3]− (1; Np=neopentyl, Ar=3,5-Me2C6H3), which is best formulated as containing an NbP triple bond. The phosphorus atom of 1 is nucleophilic. Treatment of 1 with ClP(tBu)2 or ClP(Ph)2 provides η2-phosphanylphosphinidene complexes, which are the first examples of such complexed ligands bound to an early-transition-metal fragment.
Co-reporter:Justin K. Brask, Víctor Durà-Vilà, Paula L. Diaconescu and Christopher C. Cummins
Chemical Communications 2002 (Issue 8) pp:902-903
Publication Date(Web):26 Mar 2002
DOI:10.1039/B111550M
A dramatic difference in behavior is observed for the dithiocarbamate and carbamate complexes [Ar(But)N]3V(NCE2)Na(THF)2 (E = S or O, respectively), prepared from the corresponding nitride species {[Ar(But)N]3VNNa}2 by way of a nucleophilic addition reaction involving carbon disulfide or dioxide, and is rationalized with the aid of DFT calculations.
Co-reporter:Justin K. Brask, Michael G. Fickes, Preeyanuch Sangtrirutnugul, Víctor Durà-Vilà, Aaron L. Odom and Christopher C. Cummins
Chemical Communications 2001 (Issue 17) pp:1676-1677
Publication Date(Web):16 Aug 2001
DOI:10.1039/B105584B
The iminophosphinimide complexes
[Ar(R)N]3M(NPNBut) (M = V, Nb) were prepared from the
corresponding anionic nitride species
{[Ar(R)N]3MNNa}2 by way of a
four-step synthetic strategy.
Co-reporter:Daniel J. Mindiola, Yi-Chou Tsai, Ryuichiro Hara, Qinghao Chen, Karsten Meyer and Christopher C. Cummins
Chemical Communications 2001 (Issue 1) pp:125-126
Publication Date(Web):18 Dec 2000
DOI:10.1039/B006517J
A cyanoimide [NCN] transfer reagent has been developed and
applied to the synthesis of the μ-NCN systems
(μ2;η1,η1-NCN)
{M[N(R)Ar]3}2 (Ar =
C6H3Me2-3,5; M = V or U, R =
But; M = Mo, R = Pri).
Co-reporter:Karsten Meyer;Daniel J. Mindiola;Thomas A. Baker;William M. Davis;Christopher C. Cummins Dr.
Angewandte Chemie 2000 Volume 112(Issue 17) pp:
Publication Date(Web):4 SEP 2000
DOI:10.1002/1521-3757(20000901)112:17<3191::AID-ANGE3191>3.0.CO;2-0
Co-reporter:Daniel J. Mindiola
Angewandte Chemie International Edition 1998 Volume 37(Issue 7) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980420)37:7<945::AID-ANIE945>3.0.CO;2-X
Instead of azides, which can be explosive and poorly soluble in nonpolar solvents, azabicycloheptadienes can be used to synthesize nitrides. The reaction of diiodide 1 with the lithium amide 3 provides the red nitride 2 in 60 % yield with loss of lithium iodide and anthracene. The remaining iodo ligand in 2 can undergo an exchange reaction with lithium amide 3, but no more anthracene is released.
Co-reporter:Daniel J. Mindiola;Christopher C. Cummins
Angewandte Chemie 1998 Volume 110(Issue 7) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980403)110:7<983::AID-ANGE983>3.0.CO;2-P
Die häufig explosiven und in organischen Solventien schwerlöslichen Azide lassen sich als Vorstufen für Nitride vermeiden, wenn als N-Quelle ein Azabicycloheptadien verwendet wird. So ergibt die Umsetzung des Diiodids 1 mit dem Amid 3 unter Abspaltung von Lithiumiodid und Anthracen das rote Nitrid 2 in 60% Ausbeute. Der verbliebene Iodoligand in 2 läßt sich mit dem Lithiumamid 3 noch austauschen, allerdings wird dann kein Anthracen mehr abgespalten.
Co-reporter:Catalina E. Laplaza;Dr. William M. Davis; Dr. Christopher C. Cummins
Angewandte Chemie 1995 Volume 107(Issue 18) pp:
Publication Date(Web):21 JAN 2006
DOI:10.1002/ange.19951071819
Co-reporter:Julia M. Stauber, Peter Müller, Yizhe Dai, Gang Wu, Daniel G. Nocera and Christopher C. Cummins
Chemical Science (2010-Present) 2016 - vol. 7(Issue 12) pp:NaN6933-6933
Publication Date(Web):2016/07/06
DOI:10.1039/C6SC01754A
Cofacial bimetallic tin(II) ([Sn2(mBDCA-5t)]2−, 1) and lead(II) ([Pb2(mBDCA-5t)]2−, 2) complexes have been prepared by hexadeprotonation of hexacarboxamide cryptand mBDCA-5t-H6 together with double Sn(II) or Pb(II) insertion. Reaction of 1 with elemental sulfur or selenium generates di-tin polychalcogenide complexes containing μ-E and bridging μ-E5 ligands where E = S or Se, and the Sn(II) centers have both been oxidized to Sn(IV). Solution and solid-state UV-Vis spectra of [(μ-S5)Sn2(μ-S)(mBDCA-5t)]2− (4) indicate that the complex acts reversibly as a source of S3˙− in DMF solution with a Keq = 0.012 ± 0.002. Reductive removal of all six chalcogen atoms is achieved through treatment of [(μ-E5)Sn2(μ-E)(mBDCA-5t)]2− with PR3 (R = tBu, Ph, OiPr) to produce six equiv. of the corresponding EPR3 compound with regeneration of di-tin(II) cryptand complex 1.
Co-reporter:Ioana Knopf, Daniel Tofan, Dirk Beetstra, Abdulaziz Al-Nezari, Khalid Al-Bahily and Christopher C. Cummins
Chemical Science (2010-Present) 2017 - vol. 8(Issue 2) pp:NaN1468-1468
Publication Date(Web):2016/10/11
DOI:10.1039/C6SC03614G
A family of cis-macrocyclic diphosphines was prepared in just three steps from white phosphorus and commercial materials using a modular synthetic approach. Alkylation of bicyclic diphosphane 3,4,8,9-tetramethyl-1,6-diphosphabicyclo(4.4.0)deca-3,8-diene, or P2(dmb)2, produced phosphino-phosphonium salts [R-P2(dmb)2]X, where R is methyl, benzyl and isobutyl, in yields of 90–96%. Treatment of these salts with organolithium or Grignard reagents yielded symmetric and unsymmetric macrocyclic diphosphines of the form cis-1-R-6-R′-3,4,8,9-tetramethyl-2,5,7,10-tetrahydro-1,6-DiPhospheCine, or R,R′-DPC, in which R′ is methyl, cyclohexyl, phenyl or mesityl, in yields of 46–94%. Alternatively, symmetric diphosphine Cy2-DPC was synthesized in 74% yield from the dichlorodiphosphine Cl2P2(dmb)2. As a first application, these cis-macrocyclic diphosphines were used as ligands in the nickel-catalyzed synthesis of acrylate from CO2 and ethylene, for which they showed promising catalytic activity.
Co-reporter:Alexander R. Fox, Sidney E. Creutz and Christopher C. Cummins
Dalton Transactions 2010 - vol. 39(Issue 29) pp:NaN6634-6634
Publication Date(Web):2010/06/25
DOI:10.1039/C0DT00419G
The synthesis, spectroscopy, structure, and bonding of the molecular uranium dicarbide complex (μ,η1:η1-C2)[U(N[t-Bu]Ar)3]2 (Ar = 3,5-Me2C6H3) is described.
Co-reporter:John J. Curley, Anthony F. Cozzolino and Christopher C. Cummins
Dalton Transactions 2011 - vol. 40(Issue 11) pp:NaN2432-2432
Publication Date(Web):2011/02/04
DOI:10.1039/C0DT01326A
Facile methoxymethylation of N2-derived nitride NMo(N[tBu]Ar)3 provided the imido cation [MeOCH2NMo(N[tBu]Ar)3]+ as its triflate salt in 88% yield. Treatment of the latter with LiN(SiMe3)2 provided blue methoxyketimide complex MeO(H)CNMo(N[tBu]Ar)3 in 95% yield. Conversion of the latter to the terminal cyanide complex NCMo(N[tBu]Ar)3, which was the subject of a single-crystal X-ray diffraction study, was accomplished in 51% yield upon treatment with a combination of SnCl2 and Me2NSiMe3.
Co-reporter:Anthony F. Cozzolino, Jared S. Silvia, Nazario Lopez and Christopher C. Cummins
Dalton Transactions 2014 - vol. 43(Issue 12) pp:NaN4652-4652
Publication Date(Web):2014/02/03
DOI:10.1039/C3DT52738G
An important challenge in the artificial fixation of N2 is to find atom efficient transformations that yield value-added products. Here we explore the coordination complex mediated conversion of ubiquitous species, CO and N2, into isocyanate. We have conceptually split the process into three steps: (1) the six-electron splitting of dinitrogen into terminal metal nitrido ligands, (2) the reduction of the complex by two electrons with CO to form an isocyanate linkage, and (3) the one electron reduction of the metal isocyanate complex to regenerate the starting metal complex and release the product. These steps are explored separately in an attempt to understand the limitations of each step and what is required of a coordination complex in order to facilitate a catalytic cycle. The possibility of this cyanate cycle was explored with both Mo and V complexes which have previously been shown to perform select steps in the sequence. Experimental results demonstrate the feasibility of some of the steps and DFT calculations suggest that, although the reduction of the terminal metal nitride complex by carbon monoxide should be thermodynamically favorable, there is a large kinetic barrier associated with the change in spin state which can be avoided in the case of the V complexes by an initial binding of the CO to the metal center followed by rearrangement. This mandates certain minimal design principles for the metal complex: the metal center should be sterically accessible for CO binding and the ligands should not readily succumb to CO insertion reactions.
Co-reporter:Cesar M. Manna, Mostafa Y. Nassar, Daniel Tofan, Khetpakorn Chakarawet and Christopher C. Cummins
Dalton Transactions 2014 - vol. 43(Issue 4) pp:NaN1518-1518
Publication Date(Web):2013/11/07
DOI:10.1039/C3DT52526K
We herein report the preparation of several mononuclear-metaphosphate complexes using simple techniques and mild conditions with yields ranging from 56% to 78%. Treatment of cyclo-tetrametaphosphate ([TBA]4[P4O12]·5H2O, TBA = tetra-n-butylammonium) with various metal sources including (CH3CN)3Mo(CO)3, (CH3CN)2Mo(CO)2(η3-C3H5)Cl, MoO2Cl2(OSMe2)2, and VOF3, leads to the clean and rapid formation of [TBA]4[(P4O12)Mo(CO)3]·2H2O, [TBA]3[(P4O12)Mo(CO)2(η3-C3H5)], [TBA]3[(P4O12)MoO2Cl] and [TBA]3[(P4O12)VOF2]·Et2O salts in isolated yields of 69, 56, 68, and 56% respectively. NMR spectroscopy, NMR simulations and single crystal X-ray studies reveal that the [P4O12]4− anion behaves as a tridentate ligand wherein one of the metaphosphate groups is not directly bound to the metal. cyclo-Trimetaphosphate-metal complexes were prepared using a similar procedure i.e., treatment of [PPN]3[P3O9]·H2O (PPN = bis(triphenylphosphine)iminium) with the metal sources (CH3CN)2Mo(CO)2(η3-C3H5)Cl, MoO2Cl2(OSMe2)2, MoOCl3, VOF3, WOCl4, and WO2Cl2(CH3CN)2 to produce the corresponding salts, [PPN]2[(P3O9)Mo(CO)2(η3-C3H5)], [PPN]2[(P3O9)MoO2Cl], [PPN]2[(P3O9)MoOCl2], [PPN]2[(P3O9)VOF2]·2CH2Cl2, and [PPN]2[(P3O9)WO2Cl] in isolated yields of 78, 56, 75, 59, and 77% respectively. NMR spectroscopy, NMR simulations and single-crystal X-ray studies indicate that the trianionic ligand [P3O9]3− in these complexes also has κ3 connectivity.
Co-reporter:Paula L. Diaconescu and Christopher C. Cummins
Dalton Transactions 2015 - vol. 44(Issue 6) pp:NaN2683-2683
Publication Date(Web):2014/12/05
DOI:10.1039/C4DT02422B
The synthesis and characterization of (bipy)2U(N[t-Bu]Ar)2 (1-(bipy)2, bipy = 2,2′-bipyridyl, Ar = 3,5-C6H3Me2), (bipy)U(N[1Ad]Ar)3 (2-bipy), (bipy)2U(NC[t-Bu]Mes)3 (3-(bipy)2, Mes = 2,4,6-C6H2Me3), and IU(bipy)(NC[t-Bu]Mes)3 (3-I-bipy) are reported. X-ray crystallography studies indicate that bipy coordinates as a radical anion in 1-(bipy)2 and 2-bipy, and as a neutral ligand in 3-I-bipy. In 3-(bipy)2, one of the bipy ligands is best viewed as a radical anion, the other as a neutral ligand. The electronic structure assignments are supported by NMR spectroscopy studies of exchange experiments with 4,4′-dimethyl-2,2′-bipyridyl and also by optical spectroscopy. In all complexes, uranium was assigned a +4 formal oxidation state.
Co-reporter:Heather A. Spinney, Christopher R. Clough and Christopher C. Cummins
Dalton Transactions 2015 - vol. 44(Issue 15) pp:NaN6796-6796
Publication Date(Web):2015/02/16
DOI:10.1039/C5DT00105F
This work explores the reduction of 4,4′-bipyridine using two equivalents of the titanium(III) complex Ti(N[tBu]Ar)3 resulting in formation of a black, crystalline complex, (4,4′-bipy){Ti(N[tBu]Ar)3}2, for which an X-ray structure determination is reported. The neutral, black, 4,4′-bipyridine-bridged bimetallic was found to be redox active, with mono- and di-anions being accessible electrochemically, and with the mono- and di-cations also being accessible chemically, and isolable, at least when using the weakly coordinating anion [B(C6F5)4]− as the counter-ion. It proved possible to crystallize the salt [(4,4′-bipy){Ti(N[tBu]Ar)3}2][B(C6F5)4]2 for a single-crystal X-ray structure investigation; in this instance it was revealed that the aromaticity of the 4,4′-bipyridine ligand, that had been disrupted upon reduction, had been regained. A rare cationic d0 metal tris-amide complex, shown by X-ray crystallography to contain an intriguing pyramidal TiN3 core geometry, namely {Ti(N[tBu]Ar)3}+, could also be isolated when using [B(C6F5)4] as the essentially non-interacting counter-ion. This highly reactive cation should be considered as a potential intermediate in the plethora of reactions wherein Ti(N[tBu]Ar)3 has been shown to effect the reduction of substrates including halogenated organic molecules, carbonyl compounds, organic nitriles, and metal complexes.
Co-reporter:Alexandra Velian, Brandi M. Cossairt and Christopher C. Cummins
Dalton Transactions 2016 - vol. 45(Issue 5) pp:NaN1895-1895
Publication Date(Web):2015/10/07
DOI:10.1039/C5DT03383G
Complexes (THF)0–2E[P3Nb(ODipp)3]2 (E = Sn, Pb; Dipp = 2,6-iPr2C6H3) were isolated (>90%) from the salt metathesis of [Na(THF)3][P3Nb(ODipp)3] with E2+ salts. The reaction of (THF)Sn[P3Nb(ODipp)3]2 with pyridine-N-oxide was investigated as a method to deposit a new SnP6 phase. Additionally, the neutral complex P3Nb(ODipp)2(py)2 (py = pyridine) was prepared from [Na(THF)3][P3Nb(ODipp)3] in the presence of pyridine and salts of coordinating cations (Mg(II), Sn(II), Pb(II), Ge(II), Hg(II) and Ag(I)). P3Nb(ODipp)2(py)2 was found to successfully produce AsP3 upon treatment with AsCl3. The characterization of complexes (THF)0–1Sn[P3Nb(ODipp)3]2, (THF)2Pb[P3Nb(ODipp)3]2 and P3Nb(ODipp)2(py)2, including their solid state structures, is discussed.
Co-reporter:Daniel Tofan and Christopher C. Cummins
Chemical Science (2010-Present) 2012 - vol. 3(Issue 8) pp:NaN2478-2478
Publication Date(Web):2012/05/22
DOI:10.1039/C2SC20559A
Selective formation of bimetallic group 10 complexes using the Cs symmetric, bicyclic diphosphane P2C12H20 is reported herein. With its eclipsed lone pairs disposed at a relative angle of ca. 45°, the diphosphane framework is ideally suited to form multiple bridges between two metal centers. The complexes contain {M2P6} cages with three diphosphane bridges and a pair of trans-axial ligands such as EPh3 (E = P, As, Sb) or η1-P2C12H20. X-Ray crystallography experiments revealed that the cages have a pseudo-D3h symmetry, with metal–metal distances in the 3.9–4.1 Å range. The complexes were isolated in 48–91% yield as crystalline, bright yellow or orange powders. Substitution of the axial ligands with the {M2P6} cages remaining intact was also observed.
Co-reporter:Jared S. Silvia and Christopher C. Cummins
Chemical Science (2010-Present) 2011 - vol. 2(Issue 8) pp:NaN1479-1479
Publication Date(Web):2011/05/26
DOI:10.1039/C1SC00215E
The titanium oxo anion complex [(Et2O)2Li][OTi(N[tBu]3,5-Me2C6H3)3] ([(Et2O)2Li][1]) reacts with CO2 in diethyl ether to form the carbonate complex ([Li][O2COTi(N[tBu]3,5-Me2C6H3)3])6 ([Li][2]). The solid-state structure of complex [Li][2] is a hexamer with a hexagonal prismatic core comprised of six lithium cations bridged by the carbonate functionality. In the monomeric subunits of [Li][2], the carbonate ligand is bound κ1- to the titanium metal center and pseudo κ2- to the lithium countercation. The hexameric structure persists in benzene solutions as determined by 1H DOSY NMR techniques. The binding of CO2 in complex [Li][2] is reversible and can be effected by the introduction of the lithium sequestration reagent 12-crown-4 to diethyl ether solutions of [Li][2]. Complex [Li][2] is readily functionalized with Me3SiOS(O)2CF3 to yield the silyl carbonate complex Me3SiOC(O)OTi(N[tBu]3,5-Me2C6H3)3 (3), the solid-state structure of which is presented. Functionalization with pivaloyl chloride results in the rapid loss of CO2 and formation of the pivalate complex tBuC(O)OTi(N[tBu]3,5-Me2C6H3)3 (4).
Co-reporter:Alexandra Velian and Christopher C. Cummins
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1006-1006
Publication Date(Web):2012/01/20
DOI:10.1039/C2SC00931E
Dimer [P2Nb(ODipp)3]2 (Dipp = 2,6-iPr2C6H3) has been obtained via a novel “2(3−1)” synthetic strategy. The mononuclear diphosphorus complex P2Nb(ODipp)3 targeted for generation by formal P− abstraction from previously reported [Na(THF)3][P3Nb(ODipp)3] ostensibly undergoes irreversible dimerization to form the [P2Nb(ODipp)3]2 complex, and is alternatively trapped reversibly by 1,3-cyclohexadiene with in situ formation of C6H8P2Nb(ODipp)3. The molecular structure of [P2Nb(ODipp)3]2 has been determined by X-ray crystallography. Computational studies provide further insights into the bonding and reactivity of P2Nb(ODipp)3, [P2Nb(ODipp)3]2, and C6H8P2Nb(ODipp)3.
Co-reporter:Alexander R. Fox and Christopher C. Cummins
Chemical Communications 2012 - vol. 48(Issue 25) pp:NaN3063-3063
Publication Date(Web):2012/01/23
DOI:10.1039/C2CC17212G
The synthesis, structure, and spectroscopic features of a bimetallic cyanogen complex obtained from the reductive coupling of cyanide by a niobium(IV) precursor are described, and a mechanism for the coupling reaction is proposed based on DFT calculations.
Co-reporter:Michael Montag, Christopher R. Clough, Peter Müller and Christopher C. Cummins
Chemical Communications 2011 - vol. 47(Issue 2) pp:NaN664-664
Publication Date(Web):2010/11/29
DOI:10.1039/C0CC04342G
Examination of cyclotriphosphate and cyclotetraphosphate as ligands for Co(III) in aqueous solutions revealed that cyclotetraphosphate affords stable complexes as a hemilabile ligand, while cyclotriphosphate exhibits facile hydrolysis.
Co-reporter:Gao-Lei Hou, Bo Chen, Wesley J. Transue, David A. Hrovat, Christopher C. Cummins, Weston Thatcher Borden and Xue-Bin Wang
Chemical Science (2010-Present) 2016 - vol. 7(Issue 7) pp:NaN4675-4675
Publication Date(Web):2016/04/19
DOI:10.1039/C5SC04667J
We report here a negative ion photoelectron spectroscopy (NIPES) and ab initio study of the recently synthesized planar aromatic inorganic ion P2N3−, to investigate the electronic structures of P2N3− and its neutral P2N3˙ radical. The adiabatic detachment energy of P2N3− (electron affinity of P2N3˙) was determined to be 3.765 ± 0.010 eV, indicating high stability for the P2N3− anion. Ab initio electronic structure calculations reveal the existence of five, low-lying, electronic states in the neutral P2N3˙ radical. Calculation of the Franck–Condon factors (FCFs) for each anion-to-neutral electronic transition and comparison of the resulting simulated NIPE spectrum with the vibrational structure in the observed spectrum allows the first four excited states of P2N3˙ to be determined to lie 6.2, 6.7, 11.5, and 22.8 kcal mol−1 above the ground state of the radical, which is found to be a 6π-electron, 2A1, σ state.
Co-reporter:Sidney E. Creutz, Ivo Krummenacher, Christopher R. Clough and Christopher C. Cummins
Chemical Science (2010-Present) 2011 - vol. 2(Issue 11) pp:NaN2172-2172
Publication Date(Web):2011/08/16
DOI:10.1039/C1SC00375E
Treatment of terminal phosphide anion [PNb(N[Np]Ar)3]− ([Na(OEt2)]+ salt, Ar = 3,5-Me2C6H3) with two diphenylketene equivalents led to isolation of anion [P(C{CPh2}O)2Nb(N[Np]Ar)3]− in 78% yield as its [Na(THF)]+ salt. The latter reacts with diphenylketene (1 equiv) to provide the triple diphenylketene addition product [P(C{CPh2}O)3Nb(N[Np]Ar)3]− in 88% yield as its [Na(THF)]+ salt; the same material is obtained alternatively in 93% yield by reaction of diphenylketene (3 equiv) directly with niobium phosphide [Na(OEt2)][PNb(N[Np]Ar)3]. The anion [P(C{CPh2}O)3Nb(N[Np]Ar)3]− was also crystallized as its ion-separated [Na(THF)6]+ salt as illuminated by a single-crystal X-ray diffraction study, which also revealed the three-fold symmetric nature of the highly hindered tertiary phosphine comprising the anionic component. Coinage metal monocations of the new, anionic phosphine were prepared via salt elimination; structural studies on the zwitterionic complexes (py)M[P(C{CPh2}O)3Nb(N[Np]Ar)3] (M = Cu, Ag, and Au) showed them to be isostructural in the space groupP21/c while illustrating the ensconcement of a two-coordinate coinage metal in a deep binding pocket flanked by three phenyl residues. Rhodium(I) incorporation into the anion's binding pocket illustrated versatility of the latter: in contrast to structural observations for the M(I) complexes (M = Cu, Ag, and Au), in the case of Rh[P(C{CPh2}O)3Nb(N[Np]Ar)3] an X-ray study reveals strong interactions (η6 and η2, respectively) with two of the three phenyl residues that are proximal to Rh(I) when connected to the phosphine lone pair of the anion.
Co-reporter:Matthew A. Rankin and Christopher C. Cummins
Dalton Transactions 2012 - vol. 41(Issue 32) pp:NaN9618-9618
Publication Date(Web):2012/06/27
DOI:10.1039/C2DT31082A
Terminal, 4-coordinate phosphinidenes of Ta supported by bulky anilide ligands are prepared by an apparent reaction sequence involving metallaziridine phosphanide complexes.
.jpg)
![Phosphine,(tricyclo[3.3.1.13,7]dec-1-ylmethylidyne)-](http://img.cochemist.com/ccimg/101100/101055-70-3.png)
![Phosphine,(tricyclo[3.3.1.13,7]dec-1-ylmethylidyne)-](http://img.cochemist.com/ccimg/101100/101055-70-3_b.png)
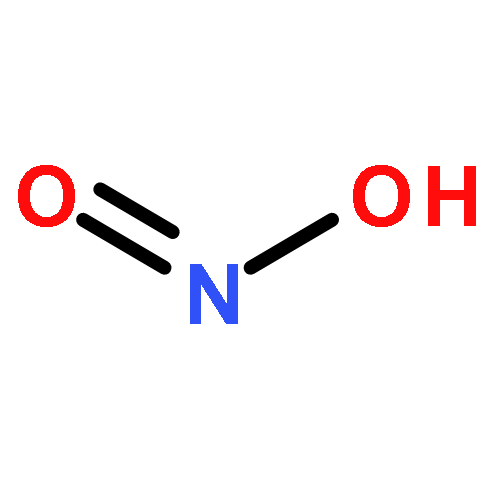
![Naphtho[1,8-cd]-1,2-dithiole](http://img.cochemist.com/ccimg/300/209-22-3.png)
![Naphtho[1,8-cd]-1,2-dithiole](http://img.cochemist.com/ccimg/300/209-22-3_b.png)


![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8.png)
![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8_b.png)
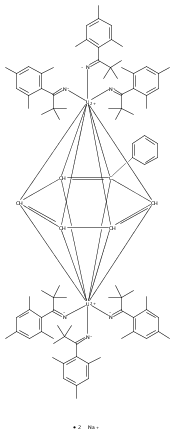
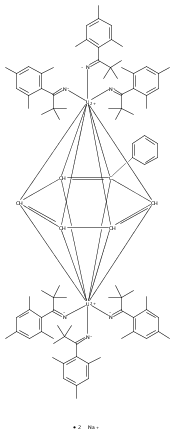






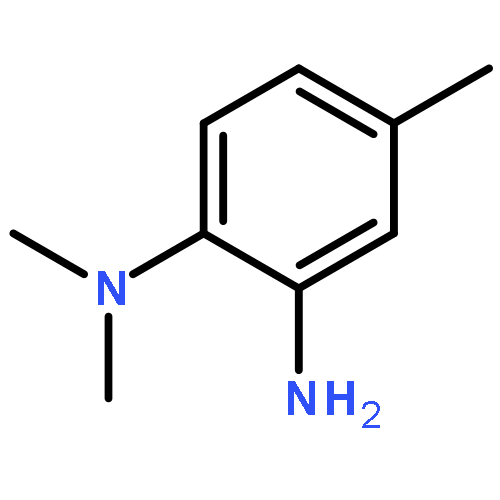


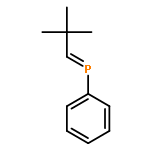




![Heptaphosphatricyclo[2.2.1.02,6]heptane, tris(triphenylstannyl)-, (±)-](http://img.cochemist.com/ccimg/110300/110298-19-6.png)
![Heptaphosphatricyclo[2.2.1.02,6]heptane, tris(triphenylstannyl)-, (±)-](http://img.cochemist.com/ccimg/110300/110298-19-6_b.png)




![Heptaphosphatricyclo[2.2.1.02,6]heptane, tris(trimethylsilyl)-](http://img.cochemist.com/ccimg/58000/57990-97-3.png)
![Heptaphosphatricyclo[2.2.1.02,6]heptane, tris(trimethylsilyl)-](http://img.cochemist.com/ccimg/58000/57990-97-3_b.png)
![[[DICHLOROPHOSPHANYL(TRIMETHYLSILYL)AMINO]-DIMETHYLSILYL]METHANE](http://img.cochemist.com/ccimg/54100/54036-90-7.png)
![[[DICHLOROPHOSPHANYL(TRIMETHYLSILYL)AMINO]-DIMETHYLSILYL]METHANE](http://img.cochemist.com/ccimg/54100/54036-90-7_b.png)











![Nickel, di-m-chlorobis[(1,2,3-h)-2-methyl-2-propen-1-yl]di-](http://img.cochemist.com/ccimg/12200/12145-60-7.png)
![Nickel, di-m-chlorobis[(1,2,3-h)-2-methyl-2-propen-1-yl]di-](http://img.cochemist.com/ccimg/12200/12145-60-7_b.png)