Co-reporter:Chen Chen, Xuben Hou, Guohua Wang, Wenyan Pan, Xinying Yang, Yingkai Zhang, Hao Fang
European Journal of Medicinal Chemistry 2017 Volume 133(Volume 133) pp:
Publication Date(Web):16 June 2017
DOI:10.1016/j.ejmech.2017.03.064
•Novel quinoline-based N-hydroxycinnamamides were designed and synthesized.•Compound 4a showed better inhibitory activity for class I HDAC than Vorinostat.•Compound 4e exhibited excellent anti-proliferative activity against K562.•Compound 4a and 4e could promote cell apoptosis in vitro.Inhibition of histone deacetylase (HDAC) has been regarded as a potential therapeutic approach for treatment of multiple diseases including cancer. Based on pharmacophore model of HDAC inhibitors, a series of quinoline-based N-hydroxycinnamamides and N-hydroxybenzamides were designed and synthesized as potent HDAC inhibitors. All target compounds were evaluated for their in vitro HDAC inhibitory activities and anti-proliferative activities and the best compound 4a surpass Vorinostat in both enzymatic inhibitory activity and cellular anti-proliferative activity. In terms of HDAC isoforms selectivity, compounds 4a exhibited preferable inhibition for class I HDACs, especially for HDAC8, the IC50 value (442 nM) was much lower than that of Vorinostat (7468 nM). Subsequently, we performed class I & IIa HDACs whole cell enzyme assay to evaluate inhibitory activity in whole cell context. Compounds 4a and 4e displayed much better cellular activity for class I HDACs than that for class IIa HDACs, which indicated that 4a and 4e might be potent class I HDAC inhibitors. Meanwhile, flow cytometry analysis showed that compound 4a and 4e can promote cell apoptosis in vitro.Download high-res image (142KB)Download full-size image
Co-reporter:Guangsen Xu, Tingting Liu, Yi Zhou, Xinying Yang, Hao Fang
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.08.024
Bcl-2 proteins, such as B-cell lymphoma (Bcl-2) protein, myeloid cell leukemia sequence 1 (Mcl-1) protein, has been implicated in the progression and survival of multiple tumor types and become a validated and attractive target for cancer therapy. In this work, a series of 1-phenyl-1H-indole derivatives has been designed and synthesized. The preliminary biological studies (binding assay for Bcl-2 proteins and MTT assay) suggested that some active compounds showed potent inhibitory activities on Bcl-2/Mcl-1 without binding on Bcl-XL. Furthermore, Compound 9c and 9h showed better anti-proliferative activity than WL-276.Download high-res image (68KB)Download full-size image
Co-reporter:Renshuai Liu, Junhua Wang, Weiping Tang, Hao Fang
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 7) pp:1446-1454
Publication Date(Web):1 April 2016
DOI:10.1016/j.bmc.2016.02.005
Histone deacetylase inhibitors have been proved to be great potential for the treatment of cancer. Recently, we designed and modified a series of substituted purine hydroxamate analogs as potent HDAC inhibitors based on our previous studies. The target compounds were investigated for their in vitro HDAC inhibitory activities and anti-proliferative activities. Results indicated that these compounds could effectively inhibit HDAC and possess obvious anti-proliferative activity against tumor cells. Promisingly, target compounds 4m and 4n outperformed SAHA in both enzymatic inhibitory activity and cellular anti-proliferative activity assay.
Co-reporter:Lin Ge, Kang-shuai Li, Meng-meng Li, Peng Xiao, Xu-ben Hou, Xu Chen, Hong-da Liu, Amy Lin, Xiao Yu, Gui-jie Ren, Hao Fang, Jin-peng Sun
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 19) pp:4795-4798
Publication Date(Web):1 October 2016
DOI:10.1016/j.bmcl.2016.08.024
Protein tyrosine phosphatases (PTPs) play key roles in many physiological processes, including cell proliferation, differentiation, immune responses and neural activities. Inappropriate regulation of the PTP activity could lead to human diseases, such as cancer or diabetes. Functional studies of PTP can be greatly facilitated by chemical probes that covalently label the active site of a PTP through an activity-dependent chemical reaction. Here, we characterize compound E4 as a new class of PTP activity probes. Compound E4 inactivate STEP in a time- and concentration-dependent fashion. Further study showed that compound E4 inhibits a series of PTPs in a time dependent manner, whereas it shows little or no inhibition toward metal dependent protein phosphatases. Collectively, this new identified covalent inhibitor of PTPs has the potential to be developed to an active site Cys directed PTP probes to study the active properties of the PTPs in cell signaling.
Co-reporter:Tingting Liu;Yichao Wan;Renshuai Liu;Lin Ma
Chemical Research in Chinese Universities 2016 Volume 32( Issue 5) pp:768-774
Publication Date(Web):2016 October
DOI:10.1007/s40242-016-6159-6
Twenty-two novel 1,3,4-thiadiazole derivatives were synthesized using different aromatic acids as starting materials, followed by cyclization, coupling and deprotection reaction. The structures of all the target compounds were identified by means of 1H nuclear magnetic resonance(NMR), 13C NMR and high resolution mass spectrometer(HRMS). Further biological evaluations were performed for chronic myelogenous leukemia cell and breast cancer cell. The results suggest that most of the target compounds exhibit potent anti-proliferative activities. Especially, compound 5b shows better antiproliferative activities against MDA-MB-231 and K562 cell lines compared with gossypol.
Co-reporter:Xuben Hou; Kangshuai Li; Xiao Yu; Jin-peng Sun
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 9) pp:1973-1983
Publication Date(Web):September 11, 2015
DOI:10.1021/acs.jcim.5b00344
Incorporating protein flexibility is a major challenge for docking-based virtual screening. With an increasing number of available crystal structures, ensemble docking with multiple protein structures is an efficient approach to deal with protein flexibility. Herein, we report the successful application of a docking-based virtual screen using multiple crystal structures to discover novel inhibitors of lymphoid-specific tyrosine phosphatase (LYP), a potential drug target for autoimmune diseases. The appropriate use of multiple protein structures allowed a better enrichment than a single structure in the recovery of known inhibitors. Subsequently, an optimal ensemble of LYP structures was selected and used in docking-based virtual screening. Eight novel LYP inhibitors (IC50 ranging from 7.95 to 56.6 μM) were identified among 23 hit compounds. Further studies demonstrated that the most active compound B15 possessed some selectivity over other protein phosphatases and could effectively up-regulate TCR (T cell receptor)-mediated signaling in Jurkat T cells. These novel hits not only provided good starting points for the development of therapeutic agents useful in autoimmune diseases but also demonstrated the advantages of choosing an appropriate ensemble of protein structures in docking-based virtual screening over using a single protein conformation.
Co-reporter:Xuben Hou, Jintong Du, Renshuai Liu, Yi Zhou, Minyong Li, Wenfang Xu, and Hao Fang
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 4) pp:861-871
Publication Date(Web):March 10, 2015
DOI:10.1021/ci500762z
As key regulators of epigenetic regulation, human histone deacetylases (HDACs) have been identified as drug targets for the treatment of several cancers. The proper recognition of zinc-binding groups (ZBGs) will help improve the accuracy of virtual screening for novel HDAC inhibitors. Here, we developed a high-specificity ZBG-based pharmacophore model for HDAC8 inhibitors by incorporating customized ZBG features. Subsequently, pharmacophore-based virtual screening led to the discovery of three novel HDAC8 inhibitors with low micromole IC50 values (1.8–1.9 μM). Further studies demonstrated that compound H8-A5 was selective for HDAC8 over HDAC 1/4 and showed antiproliferation activity in MDA-MB-231 cancer cells. Molecular docking and molecular dynamic studies suggested a possible binding mode for H8-A5, which provides a good starting point for the development of HDAC8 inhibitors in cancer treatment.
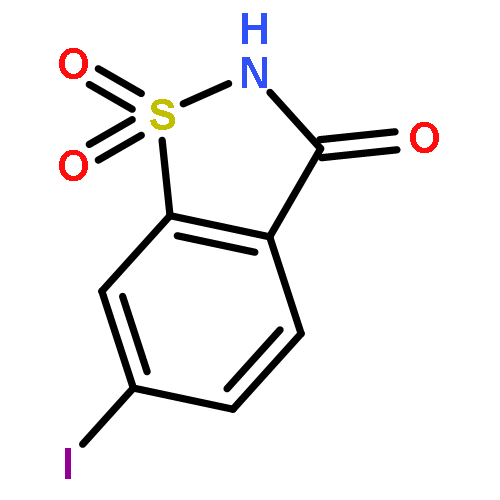
Co-reporter:Huansheng Fu, Leiqiang Han, Xuben Hou, Yanyan Dun, Lei Wang, Xiaowei Gong, Hao Fang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 17) pp:5774-5781
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmc.2015.07.008
We report the development of a novel series of saccharin-based N-hydroxybenzamides as histone deacetylases inhibitors. Among them, 6j exhibited potent HDACs inhibitory activity against Hela nuclear extract. Further biological evaluation found 6i showed similar antiproliferative activities in vitro compared with the approved SAHA.
Co-reporter:Yichao Wan, Junhua Wang, Feng’e Sun, Minglu Chen, Xuben Hou, Hao Fang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 24) pp:7685-7693
Publication Date(Web):15 December 2015
DOI:10.1016/j.bmc.2015.11.014
Anti-apoptotic proteins, such as B-cell lymphoma (Bcl-2) protein, myeloid cell leukemia sequence 1 (Mcl-1) protein, are potential targets for cancer treatment. In the studies, a series of pyrrolidine derivatives were developed as potent Mcl-1 inhibitors. The preliminary biological studies suggested that most of target compounds exhibit good abilities for targeting Mcl-1 protein. Among them, compound 21 (Ki = 0.53 μM) exhibited equal inhibitory activities towards Mcl-1 protein compared to positive control gossypol (Ki = 0.39 μM). This compound also possessed good antiproliferative activities against MDA-MB-231 and PC-3 cancer cells.
Co-reporter:Yichao Wan, Shaolei Wu, Guizhi Xiao, Tingting Liu, Xuben Hou, Chen Chen, Peng Guan, Xinying Yang, Hao Fang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 9) pp:1994-2003
Publication Date(Web):1 May 2015
DOI:10.1016/j.bmc.2015.03.024
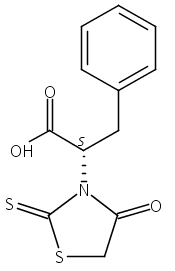
The B-cell lymphoma-2 (Bcl-2) protein is a promising target for cancer therapy. In the present study, a series of 2-thioxo-4-thiazolidinone derivatives were designed and synthesized as Bcl-2 inhibitors. Most of them possessed decent inhibitory activity for anti-apoptotic Bcl-2 proteins. Among them, compound 31 has similar growth inhibition towards K562 compared to (R)-Gossypol. In addition, it inhibits the myeloid cell leukemia sequence 1 (Mcl-1) protein with a Ki value of 74 nM.
Co-reporter:Yu Chen, Li Su, Xinying Yang, Wenyan Pan, Hao Fang
Tetrahedron 2015 Volume 71(Issue 49) pp:9234-9239
Publication Date(Web):9 December 2015
DOI:10.1016/j.tet.2015.10.041
A mild method to convert optically pure amino acid thiourea and urea derivatives to thiohydantoins and hydantoins, respectively, is described. It provides an efficient way to realize enantioselective synthesis of thiohydantoins and hydantoins with good to high isolated yields and enantiomeric purities. We found that the enantiomeric purities were highly dependent on the reaction conditions including bases, solvents, and temperature.
Co-reporter:Huansheng Fu, Xuben Hou, Lei Wang, Yanyan Dun, Xinying Yang, Hao Fang
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5265-5269
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.09.051
Co-reporter:Lei Wang, Xuben Hou, Huansheng Fu, Xiaole Pan, Wenfang Xu, Weiping Tang, Hao Fang
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4364-4374
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.024
Co-reporter:Xuben Hou ; Rong Li ; Kangshuai Li ; Xiao Yu ; Jin-Peng Sun
Journal of Medicinal Chemistry 2014 Volume 57(Issue 22) pp:9309-9322
Publication Date(Web):November 5, 2014
DOI:10.1021/jm500692u
Lymphoid-specific tyrosine phosphatase (Lyp), a critical signaling regulator of immune cells, is associated with various autoimmune diseases, including type 1 diabetes, rheumatoid arthritis, and systemic lupus erythematosus. Recent research suggests that Lyp is a potential drug target for autoimmune diseases. Herein, we applied a target–ligand interaction-based virtual screening method to identify novel Lyp inhibitors. Nine Lyp inhibitors with novel scaffolds were identified with eight reversible inhibitors (Ki values ranged from 2.87 to 28.03 μM) and one covalent inhibitor (Ki = 40.98 ± 13.19 μM). The top four compounds (A2, A15, A19, and A26) displayed selectivity over other phosphatases in preliminary experiments, and kinetic analysis indicated that these compounds are competitive inhibitors of Lyp. Compounds A15 and A19 up-regulated TCR (T cell receptor) mediated signaling and transcriptional activation through inhibition of Lyp activity in T cells. The new chemotypes of Lyp selective inhibitors identified through the target–ligand interaction-based virtual screening may provide new leads for Lyp targeted therapeutic development.
Co-reporter:Xinying Yang, Li Su, Xuben Hou, Shengyong Ding, Wenfang Xu, Binghe Wang, Hao Fang
Journal of Chromatography A 2014 Volume 1355() pp:291-295
Publication Date(Web):15 August 2014
DOI:10.1016/j.chroma.2014.06.022
•Separation of eleven chiral 3,5-disubstituted hydantoins were achieved on one given CSP with the same eluent.•The temperature effects on retention are well described by the Van’t Hoff model.•Mechanistic aspects of the chiral recognition and separation are discussed with respect to the structures of the analytes.•Both temperature-induced and solvent-induced reversals of the elution order were observed.Enantioseparations were achieved for eleven 3,5-disubstituted hydantoins in HPLC under the normal phase mode using Chiralpak IA. The effects of polar alcoholic modifier and column temperature on retention and enantioseparation were determined. Importantly, we found two kinds of enantiomer elution order (EEO) reversals, which include solvent-induced EEO reversal for compound 9 and temperature-induced EEO reversals for compound 3 and compound 6. The phenomena of these EEO reversals were described for the first time in present work, which is helpful to elucidate the chiral separation mechanism of these hydantoins.
Co-reporter:Peng Guan, Lei Wang, Xuben Hou, Yichao Wan, Wenfang Xu, Weiping Tang, Hao Fang
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:5766-5775
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.09.039
A series of 1,3,4-thiadiazole-containing hydroxamic acids, in accord with the common pharmacophore of histone deacetylase (HDAC) inhibitors (a Zn2+ binding moiety–a linker–a surface recognition motif), was identified as submicromolar HDAC inhibitors by our group. In this study, we continued our efforts to develop 1,3,4-thiadiazole bearing hydroxamate analogues by modifying the surface recognition motif. We found that 1,3,4-thiadiazoles having a heteroaromatic substituent showed better HDAC inhibitory activity in enzymatic assay and higher antiproliferative potency in cellular assay compared to SAHA.
Co-reporter:Junhua Wang, Feng'e Sun, Leiqiang Han, Xuben Hou, Xiaole Pan, Renshuai Liu, Weiping Tang and Hao Fang
MedChemComm 2014 vol. 5(Issue 12) pp:1887-1891
Publication Date(Web):15 Sep 2014
DOI:10.1039/C4MD00203B
Histone deacetylase (HDAC) is a clinically validated target for anti-tumor therapy. In order to increase HDAC inhibition and efficiency, we developed a series of novel substituted purine hydroxamic acids as potent HDAC inhibitors. The biological evaluation suggests that compound 5r (IC50 = 0.075 μmol L−1) exhibits better HDAC1 and 2 inhibitory activity compared to the approved drug SAHA (IC50 = 0.14 μmol L−1). Further biological evaluation indicated that compounds 5r, 5w, and 5x have potent anti-proliferative activities against eight tumor cells, including MDA-MB231, KG1, PC3, U937 and so on.
Co-reporter:Leiqiang Han, Lei Wang, Xuben Hou, Huansheng Fu, Weiguo Song, Weiping Tang, Hao Fang
Bioorganic & Medicinal Chemistry 2014 22(5) pp: 1529-1538
Publication Date(Web):
DOI:10.1016/j.bmc.2014.01.045
Co-reporter:Xuben Hou, Jintong Du, Jian Zhang, Lupei Du, Hao Fang, and Minyong Li
Journal of Chemical Information and Modeling 2013 Volume 53(Issue 1) pp:188-200
Publication Date(Web):December 17, 2012
DOI:10.1021/ci300417y
Molecular docking, which is the indispensable emphasis in predicting binding conformations and energies of ligands to receptors, constructs the high-throughput virtual screening available. So far, increasingly numerous molecular docking programs have been released, and among them, AutoDock 4.2 is a widely used docking program with exceptional accuracy. It has heretofore been substantiated that the calculation of partial charge is very fundamental for the accurate conformation search and binding energy estimation. However, no systematic comparison of the significances of electrostatic potentials on docking accuracy of AutoDock 4.2 has been determined. In this paper, nine different charge-assigning methods, including AM1-BCC, Del-Re, formal, Gasteiger–Hückel, Gasteiger–Marsili, Hückel, Merck molecular force field (MMFF), and Pullman, as well as the ab initio Hartree–Fock charge, were sufficiently explored for their molecular docking performance by using AutoDock4.2. The results clearly demonstrated that the empirical Gasteiger–Hückel charge is the most applicable in virtual screening for large database; meanwhile, the semiempirical AM1-BCC charge is practicable in lead compound optimization as well as accurate virtual screening for small databases.
Co-reporter:Junhua Wang;Que Wang;Liangren Zhang
Chinese Journal of Chemistry 2013 Volume 31( Issue 9) pp:1181-1191
Publication Date(Web):
DOI:10.1002/cjoc.201300420
Abstract
Novel purine-2,6-diamine derivatives were designed and synthesized as cyclin-dependent kinase (CDK) inhibitors. According to the preliminary biological evaluation, most of the compounds show good inhibitory activities in CDK1 enzyme assay and potent antiproliferative activities in some tumor cell lines. Especially, compound 11a (IC50=0.35 µmol/L for CDK1/cyclin B and IC50=0.023 µmol/L for CDK2/cyclin A) possessed better inhibitory effect compared with Roscovitine (IC50=2.54 (mol/L for CDK1/cyclin B and IC50=0.092 µmol/L for CDK2/cyclin A).
Co-reporter:Li Su, Jiangying Cao, Yuping Jia, Xiaonan Zhang, Hao Fang, and Wenfang Xu
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 12) pp:959
Publication Date(Web):September 21, 2012
DOI:10.1021/ml3000758
Cancer metastasis is a major barrier to its treatment and an important cause of patient death. Antimetastatic agents hold promise for patients with advanced metastatic tumors. Aminopeptidase N/CD13 (APN) is being pursued by many as an important target against cancer metastasis and angiogenesis, but there are few reports on the in vivo evaluation of synthetic APN inhibitors. Herein, a series of compounds targeting APN were synthesized and evaluated for their antimetastasis and antiangiogenesis potency both in vitro and in vivo. Excitingly, compounds 4m, 4t, and 4cc, with the most potent APN inhibitory activities, displayed significant antimetastasis and antiangiogenesis effects in vitro and in vivo, suggesting that those synthetic APN inhibitors have the potential to overcome cancer metastasis and angiogenesis.Keywords: angiogenesis; anticancer; APN/CD13; inhibitors; metastasis
Co-reporter:Peng Guan, Feng’e Sun, Xuben Hou, Feng Wang, Fan Yi, Wenfang Xu, Hao Fang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 12) pp:3865-3872
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmc.2012.04.032
Histone deacetylase (HDAC) inhibitors have emerged as a new class of anticancer agents, targeting the biological processes including cell cycle, apoptosis and differentiation. In the present study, a series of 1,3,4-thiadiazole based hydroxamic acids were developed as potent HDAC inhibitors. Some of them showed good inhibitory activity in HDAC enzyme assay and potent growth inhibition in some tumor cell lines. Among them, compound 6i (IC50 = 0.089 μM), exhibited better inhibitory effect compared with SAHA (IC50 = 0.15 μM).
Co-reporter:Zhongyu Wu, Minyong Li, Hao Fang, Binghe Wang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7179-7182
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.060
Catechol skeleton widely exists in natural products and bioactive substances. Fluorescent reporters which could recognize catechol are very promising for the construction of chemosensors to detect catechol and its derivatives in biological environment. Herein, we reported a novel catechol reporter, 2-(4-boronophenyl)quinoline-4-carboxylic acid, which exhibits significant fluorescent property changes upon binding catechol containing molecules in an aqueous solution.
Co-reporter:Yingjie Zhang ; Jinhong Feng ; Yuping Jia ; Xuejian Wang ; Lei Zhang ; Chunxi Liu ; Hao Fang ;Wenfang Xu
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:2823-2838
Publication Date(Web):April 5, 2011
DOI:10.1021/jm101605z
Inhibition of histone deacetylase (HDAC) results in growth arrest, differentiation, and apoptosis in nearly all tumor cell lines, promoting HDACs as promising targets for antitumor therapy. In our previous study we developed a novel series of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives as HDAC inhibitors (HDACi), among which compound 7d exhibited promising HDAC8 inhibitory and antiproliferative activities. Herein, we report the design and development of a new class of tetrahydroisoquinoline-bearing hydroxamic acid analogues as potential HDACi and anticancer agents. In vitro biological evaluation of these compounds showed improved HDAC8 inhibition (compounds 31a and 31b exhibited mid-nM IC50 values against HDAC8) and potent growth inhibition in multiple tumor cell lines. Most importantly, compounds 25e, 34a, and 34b exhibited excellent in vivo anticancer activities in a human breast carcinoma (MDA-MB-231) xenograft model compared with suberoylanilide hydroxamic acid (SAHA), an approved HDACi. Collectively, our results indicate that tetrahydroisoquinoline bearing a hydroxamic acid is an excellent template to develop novel HDACi as potential anticancer agents.
Co-reporter:Li Su, Hao Fang, Kanghui Yang, Yingying Xu, Wenfang Xu
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 2) pp:900-906
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmc.2010.11.066
As the exopeptidase over-expressed in the cell surface of endothelial cells, aminopeptidase N (APN/CD13) is an essential target for tumor angiogenesis and metastasis. Based on the previous work of l-lysine amide derivatives in our laboratory, we designed and synthesized two series of l-lysine ureido derivatives as APN inhibitors. Within these compounds, one compound, 5d (IC50 = 4.51 μM), showed similar inhibitory effect compared with Bestatin (IC50 = 5.87 μM).
Co-reporter:Kanghui Yang, Qiang Wang, Li Su, Hao Fang, Xuejian Wang, Jianzhi Gong, Binghe Wang, Wenfang Xu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 11) pp:3810-3817
Publication Date(Web):1 June 2009
DOI:10.1016/j.bmc.2009.04.038
Herein we report a series of novel chloramphenicol amine derivatives as aminopeptidase N (APN)/CD13 inhibitors. All compounds were synthesized starting from commercially available (1S,2S)-2-amino-1-(4-nitrophenyl) propane-1,3-diol. The preliminary biological screening showed that some compounds exhibited potent inhibitory activity against APN. It should be noted that one compound, 13b (IC50 = 7.1 μM), possess similar APN inhibitory activity compared with Bestatin (IC50 = 3.0 μM).The compound 13b was built and docked into the active site of APN (PDB code: 2DQM) using sybyl7.0. The docking result of 13b is showed by Ligplot.
Co-reporter:Yan Ling Li, Hao Fang, Wen Fang Xu, Bing He Wang
Chinese Chemical Letters 2008 Volume 19(Issue 5) pp:541-543
Publication Date(Web):May 2008
DOI:10.1016/j.cclet.2008.03.025
As an important intermediate to study cyclin-dependent kinase (CDK) inhibitors, 2-aryl-8-(piperidin-4-yl)-5,7-dimethoxy-4H-chromen-4-one derivatives were prepared using β-diketone route with low yield. In our study, chalcone route has been investigated and the result suggested that the benzaldehydes substituted with electron-donating group give much better yield than β-diketone route. This new method will be an efficient way to start further research on new anticancer flavonoids.
Co-reporter:Hao Fang, Min Yong Li, Lin Xia
Chinese Chemical Letters 2007 Volume 18(Issue 1) pp:41-44
Publication Date(Web):January 2007
DOI:10.1016/j.cclet.2006.11.022
A series of arylpiperazinesquinazoline-2,4-diamine compounds were designed and synthesized based on pharmacophore for uro-selective α1-adrenoceptor antagonists and 3D chemical database searching. The in vitro functional analysis showed that compounds 9 and 14 showed better and similar α1-AR antagonistic activity compared with prazosin.
Co-reporter:Tingting Liu, Yichao Wan, Renshuai Liu, Lin Ma, Minyong Li, Hao Fang
Bioorganic & Medicinal Chemistry (15 March 2017) Volume 25(Issue 6) pp:
Publication Date(Web):15 March 2017
DOI:10.1016/j.bmc.2017.02.014
The B-cell lymphoma-2 (Bcl-2) family proteins are attractive targets for cancer therapy. In our previous work, the structure-activity relationship of WL-276 was studied. According to the results, rhodanine derivatives show potent binding affinity for Bcl-2 and Mcl-1 protein and show weaker activity against Bcl-XL protein. Based on the previous results, a new class of indole-3-carboxylic acid-based derivatives were designed and synthesized as Bcl-2/Mcl-1 dual inhibitors. Among them, compound 17 has a Ki value of 0.26 μM for Bcl-2 protein and is better than WL-276. Furthermore, it inhibits the myeloid cell leukemia sequence 1 (Mcl-1) protein with a Ki value of 72 nM. Especially, compound 31 can selectively acting on Bcl-2 and Mcl-1 protein but not Bcl-XL protein, which has great significance for developing dual inhibitors targeting Bcl-2 and Mcl-1 protein, as well as specific antitumor abilities in cells.
Co-reporter:Yichao Wan, Tingting Liu, Xiaoxian Li, Chen Chen, Hao Fang
Bioorganic & Medicinal Chemistry (1 January 2017) Volume 25(Issue 1) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.bmc.2016.10.020
•A series of pyrrolidine derivatives as Mcl-1 inhibitors were synthesized.•Some of them exhibited remarkably inhibitory activities for Mcl-1 protein.•Compound 40 possessed good anti-proliferative activities against PC-3 cells.As an important member of anti-apoptotic Bcl-2 protein, myeloid cell leukemia sequence 1 (Mcl-1) protein is an attractive target for cancer therapy. In this study, a new series of pyrrolidine derivatives as Mcl-1 inhibitors were developed by mainly modifying the amino acid side chain of compound 1. Among them, compound 18 (Ki = 0.077 μM) exhibited better potent inhibitory activities towards Mcl-1 protein compared to positive control Gossypol (Ki = 0.18 μM). In addition, compound 40 possessed good antiproliferative activities against PC-3 cells (Ki = 8.45 μM), which was the same as positive control Gossypol (Ki = 7.54 μM).















![4'-Chloro-[1,1'-biphenyl]-2-carbaldehyde](http://img.cochemist.com/ccimg/153900/153850-83-0.png)
![4'-Chloro-[1,1'-biphenyl]-2-carbaldehyde](http://img.cochemist.com/ccimg/153900/153850-83-0_b.png)



![1,3,4-Thiadiazol-2-amine, 5-([1,1'-biphenyl]-4-ylmethyl)-](http://img.cochemist.com/ccimg/87600/87527-45-5.png)
![1,3,4-Thiadiazol-2-amine, 5-([1,1'-biphenyl]-4-ylmethyl)-](http://img.cochemist.com/ccimg/87600/87527-45-5_b.png)
![L-Tyrosine, N-[(1,1-dimethylethoxy)carbonyl]-O-(1-methylethyl)-](http://img.cochemist.com/ccimg/76800/76757-94-3.png)
![L-Tyrosine, N-[(1,1-dimethylethoxy)carbonyl]-O-(1-methylethyl)-](http://img.cochemist.com/ccimg/76800/76757-94-3_b.png)
![L-TYROSINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-O-PROPYL-](http://img.cochemist.com/ccimg/76800/76757-93-2.png)
![L-TYROSINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-O-PROPYL-](http://img.cochemist.com/ccimg/76800/76757-93-2_b.png)

![5-(4-Methoxy-benzyl)-[1,3,4]thiadiazol-2-ylamine](http://img.cochemist.com/ccimg/63700/63617-18-5.png)
![5-(4-Methoxy-benzyl)-[1,3,4]thiadiazol-2-ylamine](http://img.cochemist.com/ccimg/63700/63617-18-5_b.png)


![5-(4-CHLORO-BENZYL)-[1,3,4]THIADIAZOL-2-YLAMINE](http://img.cochemist.com/ccimg/39200/39181-43-6.png)
![5-(4-CHLORO-BENZYL)-[1,3,4]THIADIAZOL-2-YLAMINE](http://img.cochemist.com/ccimg/39200/39181-43-6_b.png)















![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0.png)
![2-Propenamide, N-hydroxy-3-[3-[(phenylamino)sulfonyl]phenyl]-, (2E)-](http://img.cochemist.com/ccimg/866400/866323-14-0_b.png)


![4-[(1,1-dioxido-3-oxo-1,2-benzisothiazol-2(3H)-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/694500/694473-93-3.png)
![4-[(1,1-dioxido-3-oxo-1,2-benzisothiazol-2(3H)-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/694500/694473-93-3_b.png)


![4-oxo-4-{[4-(pyridin-4-ylmethyl)phenyl]amino}butanoic acid](http://img.cochemist.com/ccimg/676600/676599-90-9.png)
![4-oxo-4-{[4-(pyridin-4-ylmethyl)phenyl]amino}butanoic acid](http://img.cochemist.com/ccimg/676600/676599-90-9_b.png)






![ETHANONE, 2,2,2-TRIFLUORO-1-[1-(2-METHYLPHENYL)-1H-1,2,3-TRIAZOL-4-YL]-](http://img.cochemist.com/ccimg/478400/478358-87-1.png)
![ETHANONE, 2,2,2-TRIFLUORO-1-[1-(2-METHYLPHENYL)-1H-1,2,3-TRIAZOL-4-YL]-](http://img.cochemist.com/ccimg/478400/478358-87-1_b.png)
![Ethanone, 1-[1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl]-2,2,2-trifluoro-](http://img.cochemist.com/ccimg/478400/478358-86-0.png)
![Ethanone, 1-[1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl]-2,2,2-trifluoro-](http://img.cochemist.com/ccimg/478400/478358-86-0_b.png)






















![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)




![5-(2-Bromo-phenyl)-[1,3,4]thiadiazol-2-ylamine](http://img.cochemist.com/ccimg/108700/108656-64-0.png)
![5-(2-Bromo-phenyl)-[1,3,4]thiadiazol-2-ylamine](http://img.cochemist.com/ccimg/108700/108656-64-0_b.png)
![4'-Nitro-[1,1'-biphenyl]-4-carbaldehyde](http://img.cochemist.com/ccimg/98700/98648-23-8.png)
![4'-Nitro-[1,1'-biphenyl]-4-carbaldehyde](http://img.cochemist.com/ccimg/98700/98648-23-8_b.png)
![[2,2'-Binaphthalene]-8,8'-dicarboxaldehyde,1,1',6,6',7,7'-hexahydroxy-3,3'-dimethyl-5,5'-bis(1-methylethyl)-, (2R)-](http://img.cochemist.com/ccimg/90200/90141-22-3.png)
![[2,2'-Binaphthalene]-8,8'-dicarboxaldehyde,1,1',6,6',7,7'-hexahydroxy-3,3'-dimethyl-5,5'-bis(1-methylethyl)-, (2R)-](http://img.cochemist.com/ccimg/90200/90141-22-3_b.png)




![3-(1,1-Dioxido-3-oxobenzo[d]isothiazol-2(3H)-yl)propanoic acid](http://img.cochemist.com/ccimg/83800/83747-21-1.png)
![3-(1,1-Dioxido-3-oxobenzo[d]isothiazol-2(3H)-yl)propanoic acid](http://img.cochemist.com/ccimg/83800/83747-21-1_b.png)








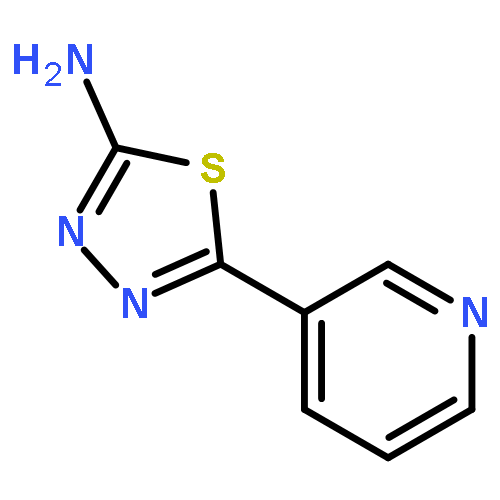





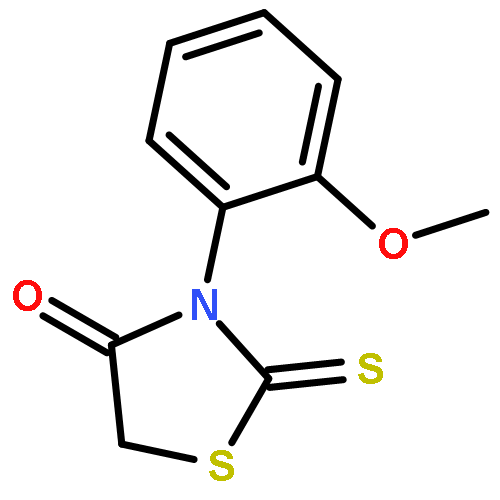
![2-(1,1-Dioxido-3-oxobenzo[d]isothiazol-2(3H)-yl)acetic acid](http://img.cochemist.com/ccimg/52200/52188-11-1.png)
![2-(1,1-Dioxido-3-oxobenzo[d]isothiazol-2(3H)-yl)acetic acid](http://img.cochemist.com/ccimg/52200/52188-11-1_b.png)
![Benzoic acid, 4-[(bromoacetyl)amino]-](http://img.cochemist.com/ccimg/52200/52134-28-8.png)
![Benzoic acid, 4-[(bromoacetyl)amino]-](http://img.cochemist.com/ccimg/52200/52134-28-8_b.png)





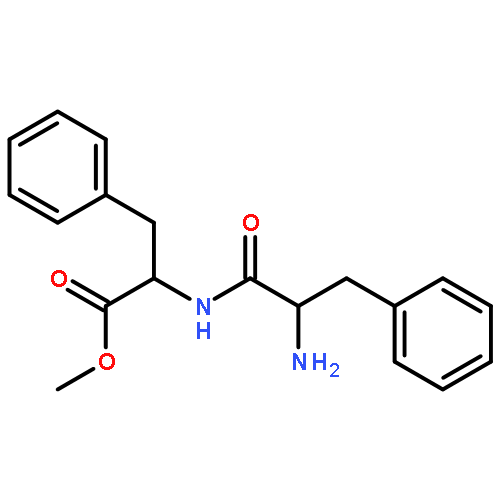
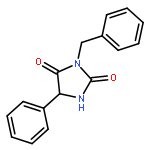


![Benzenesulfonamide,4-methyl-N-[(2-thiazolylamino)carbonyl]-](http://img.cochemist.com/ccimg/24600/24535-63-5.png)
![Benzenesulfonamide,4-methyl-N-[(2-thiazolylamino)carbonyl]-](http://img.cochemist.com/ccimg/24600/24535-63-5_b.png)
![L-Phenylalanine, N-[(phenylamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/23700/23631-66-5.png)
![L-Phenylalanine, N-[(phenylamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/23700/23631-66-5_b.png)


![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-phenylalanyl-, methyl ester](/data/chemimg/1085000/13122-89-9.png)
![L-Phenylalanine, N-[(1,1-dimethylethoxy)carbonyl]-L-phenylalanyl-, methyl ester](/data/chemimg/1085000/13122-89-9_b.png)










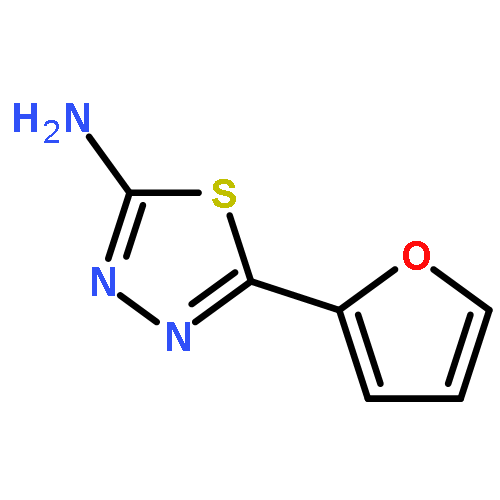


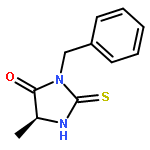
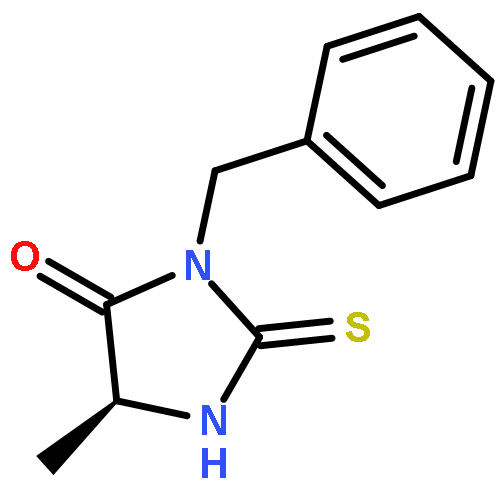
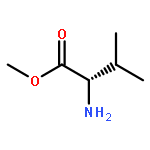
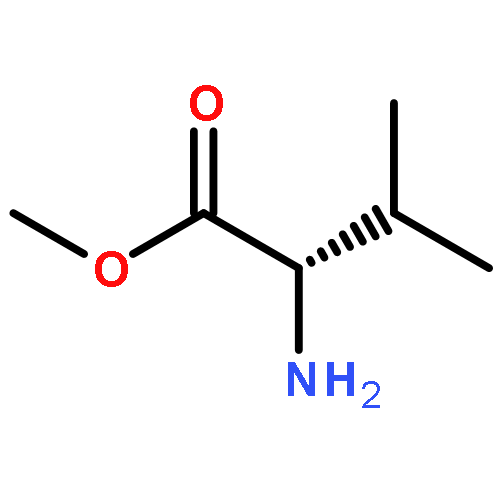

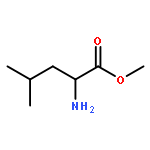
![1,3,4-Thiadiazol-2-amine, 5-[4-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/3200/3121-63-9.png)
![1,3,4-Thiadiazol-2-amine, 5-[4-(dimethylamino)phenyl]-](http://img.cochemist.com/ccimg/3200/3121-63-9_b.png)
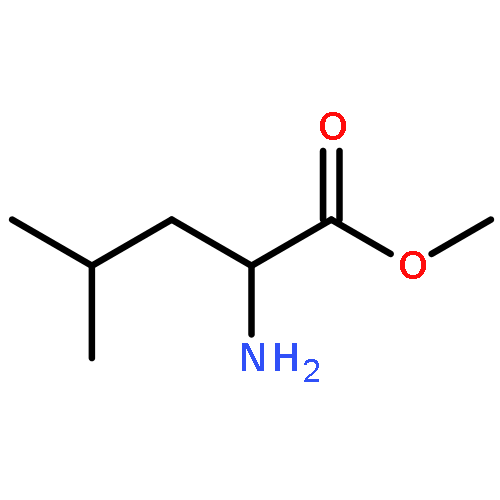





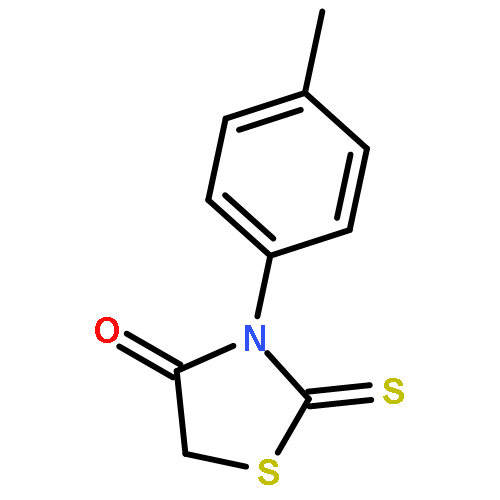
![4-Thiazolidinone,3-[(4-fluorophenyl)methyl]-2-thioxo-](http://img.cochemist.com/ccimg/500/453-69-0.png)
![4-Thiazolidinone,3-[(4-fluorophenyl)methyl]-2-thioxo-](http://img.cochemist.com/ccimg/500/453-69-0_b.png)

![4-Thiazolidinone,2-thioxo-3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/400/315-08-2.png)
![4-Thiazolidinone,2-thioxo-3-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/400/315-08-2_b.png)




![4-[(4-Formylphenoxy)methyl]benzoic acid](http://img.cochemist.com/ccimg/428500/428468-34-2.png)
![4-[(4-Formylphenoxy)methyl]benzoic acid](http://img.cochemist.com/ccimg/428500/428468-34-2_b.png)