Co-reporter:Charles P. Casey and Hairong Guan
Organometallics 2012 Volume 31(Issue 7) pp:2631-2638
Publication Date(Web):October 21, 2011
DOI:10.1021/om2007453
The bis(trimethylsilyl)-substituted hydroxycyclopentadienyl ruthenium hydride [2,5-(SiMe3)2-3,4-(CH2OCH2)(η5-C4COH)]Ru(CO)2H (10) is an efficient catalyst for hydrogenation of aldehydes and ketones. Because 10 transfers hydrogen rapidly to aldehydes and ketones and because it does not form an inactive bridging hydride during reaction, hydrogenation of aldehydes and ketones can be performed at room temperature under relatively low hydrogen pressure (3 atm); this is a significant improvement in comparison with previously developed Shvo type catalysts. Kinetic and 2H NMR spectroscopic studies of the stoichiometric reduction of aldehydes and ketones by 10 established a two-step process for the hydrogen transfer: (1) rapid and reversible hydrogen bond formation between OH of 10 and the oxygen of the aldehyde or ketone followed by (2) slow transfer of both proton and hydride from 10 to the aldehyde or ketone. The stoichiometric and catalytic activities of complex 10 are compared to those of other Shvo type ruthenium hydrides and related iron hydrides.
Co-reporter:Charles P. Casey, Jeffrey B. Johnson, Xiandong Jiao, Sharon E. Beetner and Steven W. Singer
Chemical Communications 2010 vol. 46(Issue 42) pp:7915-7917
Publication Date(Web):21 Sep 2010
DOI:10.1039/C0CC02875D
The ruthenium hydride of (Ar4CpOH)Ru(CO)2H exchanges cleanly and rapidly with D2 at room temperature to generate the ruthenium deuteride. A chain mechanism is proposed to explain the much more rapid exchange of RuH/D2 than RuCO exchange with 13CO.
Co-reporter: Charles P. Casey
ChemCatChem 2010 Volume 2( Issue 10) pp:1209-1211
Publication Date(Web):
DOI:10.1002/cctc.201000221
Co-reporter:Charles P. Casey, Xiangdong Jiao, and Ilia. A. Guzei
Organometallics 2010 Volume 29(Issue 21) pp:4829-4836
Publication Date(Web):April 7, 2010
DOI:10.1021/om100012j
The reaction of the dienone ruthenium dicarbonyl dimer {[2,5-Ph-3,4-Tol(η5-C4CO)]Ru(CO)2}2 (7) with propargyl alcohol at room temperature gave a high yield of β-ruthenium-enal (E)-[2,5-Ph-3,4-Tol(η5-C4COH)]Ru(CO)2(CH═CHCHO) (8E), which was characterized spectroscopically and by X-ray crystallography. Reaction of 7 with pent-2-yn-1-ol led to the kinetic formation of the E-isomer (E)-[2,5-Ph-3,4-Tol(η5-C4COH)]Ru(CO)2[C(CH2CH3)═CHCHO] (10E-Et), which isomerized to an equilibrium mixture of Z- and E-isomers upon heating. The intramolecular nature of the 1,2-hydrogen shifts involved in these reactions was established by the absence of crossover products in the reaction of 7 with a mixture of PhC≡CCH2OH and PhC≡CCD2OH. A primary deuterium isotope effect (kH/kD ≈ 11) was seen on the product-forming step in the reaction of 7 with PhC≡CCHDOH. The reaction of PhC≡CCH3 with 7 produced the alkyne complex [2,5-Ph2-3,4-Tol2(η5-C4COH)]Ru(CO)2(η2-PhC≡CCH3) (14). The key step in the mechanism of the reaction of 7 with propargyl alcohols is proposed to be an in-plane 1,2-hydrogen migration to an electrophilic carbon of a complexed alkyne.
Co-reporter:Charles P. Casey, Timothy M. Boller, Joseph S. M. Samec and John R. Reinert-Nash
Organometallics 2009 Volume 28(Issue 1) pp:123-131
Publication Date(Web):November 24, 2008
DOI:10.1021/om800739j
PMe3 adds selectively to the central carbon of the η3-propargyl complex [C5Me5(CO)2Re(η3-CH2C≡CCMe3)][BF4] (1-t-Bu) to form the metallacyclobutene [C5Me5(CO)2Re(CH2C(PMe3)═CCMe3)][BF4] (7). The rate of rearrangement of the metallacyclobutene 7 to η2-alkyne complex [C5Me5(CO)2Re(η2-Me3PCH2C≡CCMe3)][BF4] (8) is independent of phosphine concentration, consistent with a dissociative mechanism proceeding via η3-propargyl complex 1-t-Bu. The rate of this rearrangement is 480 times slower than the rate of exchange of PMe3 with the labeled metallacyclobutene 7-d9. This rate ratio provides an indirect measurement of the regioselectivity for addition of PMe3 to the central carbon of η3-propargyl complex 1-t-Bu to give 7 compared to addition to a terminal carbon to give 8. The addition of PPh3 to 1-t-Bu gives the metallacyclobutene [C5Me5(CO)2Re(CH2C(PPh3)═CCMe3)][BF4] (11). Low-temperature 1H NMR spectra provide evidence for an equilibrium between metallacyclobutene 11 and η3-propargyl complex 1-t-Bu (Keq ≈ 44 M−1 at −46 °C and ΔG°(0 °C) = −1.2 ± 0.2 kcal mol−1).
Co-reporter:Charles P. Casey, Trevor L. Dzwiniel, Stefan Kraft, Michael A. Kozee, Douglas R. Powell
Inorganica Chimica Acta 2003 Volume 345() pp:320-326
Publication Date(Web):10 March 2003
DOI:10.1016/S0020-1693(02)01273-2
Dimerization of the alkynylcarbene complex (C5H4Me)(CO)2MnC(Tol)CCTol (10) occurred at 65 °C to give a mixture of E- and Z-enediyne complexes [(C5H4Me)(OC)2Mn]2[η2,η2-TolCC(Tol)CC(Tol)CCTol] (13). Thermolysis of alkynye complexes 10 at higher (100 °C) temperature led to the formation of manganese-free E- and Z-enediynes TolCC(Tol)CC(Tol)CCTol (14-E and 14-Z). Attempted synthesis of the tethered bis-(alkynylcarbene) complex Cp(OC)2MnC(Ph)CCCH2CH2CH2CCC(Ph)Mn(CO)2Cp led instead to the cyclic enediyne complex [Cp(OC)2Mn]2[η2,η2-PhCCC(CH2CH2CH2)CCCPh] (19), from which the metal-free enediyne 1,2-bis(phenylethynyl)cyclopentene (20) was released by thermolysis at 90 °C.Manganese alkynylcarbene complexes dimerize to enediyne manganese complexes at 65 °C. The free enediynes are released upon heating to 100 °C. Attempted synthesis of a tethered bis-(alkynylcarbene) complex led directly to a cyclic enediyne complex from which the metal-free enediyne was released at 90 °C.
Co-reporter:Charles P Casey, Steven Chung
Inorganica Chimica Acta 2002 Volume 334() pp:283-293
Publication Date(Web):30 May 2002
DOI:10.1016/S0020-1693(02)00740-5
Protonation of η2-enyne rhenium complex C5Me5(CO)2Re(η2-3,4-trans-PhCCCHCHPh) (24) occurred at the coordinated alkyne to produce 1-metallacyclopropene complex C5Me5(CO)2Re[η2-trans-(PhHCCH)CCHPh]+BF4 − (26), instead of protonation at the non-coordinated alkene to produce an η3-propargyl complex. Protonation of the alkene coordinated η2-enyne rhenium complex C5Me5(CO)2Re(η2-1,2-cis-PhHCCHCCPh] (25) occurred at the non-coordinated alkyne to produce exo-alkylidene η3-allyl complex C5Me5(CO)2Re(η3-exo,anti-CHPhCHCCHPh)+BF4 − (27), instead of protonation at the coordinated alkene to produce an η3-propargyl complex. Protonation of η2-diyne complex C5Me5(CO)2Re(η2-CH3CCCCCH3) (32) at −78 °C produced the rhenium hydride complex trans-C5Me5(CO)2ReH(η2-CH3CCCCCH3)+BF4 − (33), which was converted to exo-alkylidene η3-propargyl complexes C5Me5(CO)2Re[η3-(CH3CH)CCCCH3]+BF4 − (34a and 34b) at 0 °C.Protonation of η2-diyne complex C5Me5(CO)2Re(η2-CH3CCCCCH3) produced the exo-alkylidene η3-propargyl complex C5Me5(CO)2Re[η3-(CH3CH)CCCCH3]+BF4 −.
Co-reporter:Charles P. Casey, Jeffrey B. Johnson, Xiandong Jiao, Sharon E. Beetner and Steven W. Singer
Chemical Communications 2010 - vol. 46(Issue 42) pp:NaN7917-7917
Publication Date(Web):2010/09/21
DOI:10.1039/C0CC02875D
The ruthenium hydride of (Ar4CpOH)Ru(CO)2H exchanges cleanly and rapidly with D2 at room temperature to generate the ruthenium deuteride. A chain mechanism is proposed to explain the much more rapid exchange of RuH/D2 than RuCO exchange with 13CO.






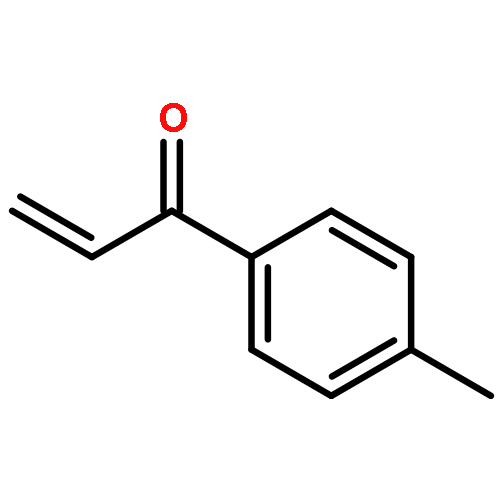
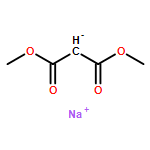
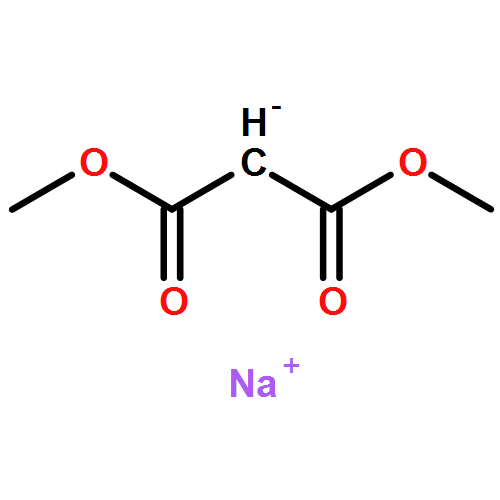

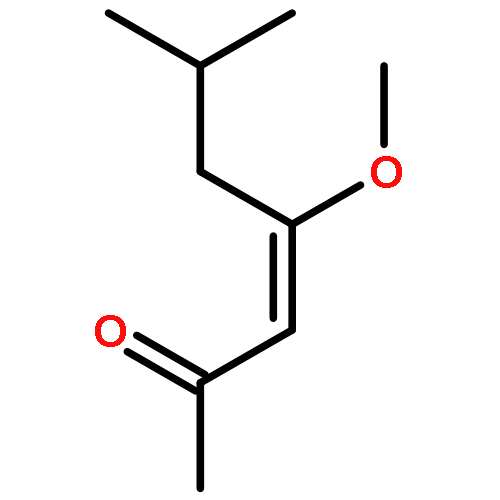


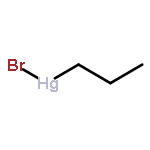
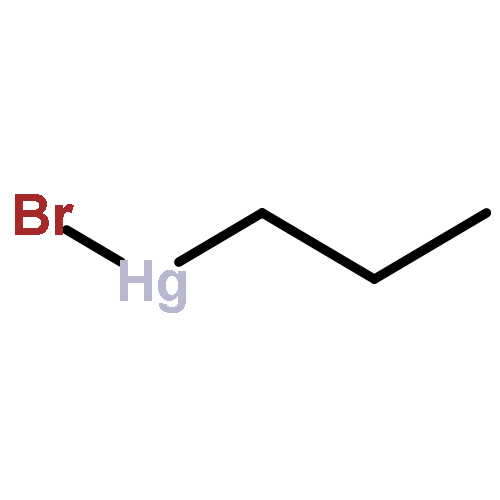


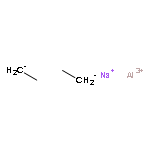
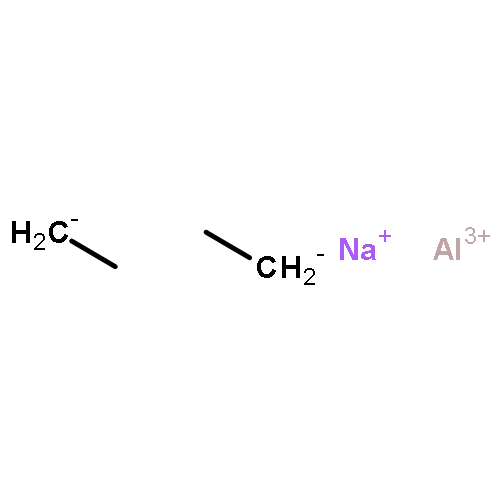
















![Benzene,1,1'-[(1R,2S)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-73-0.png)
![Benzene,1,1'-[(1R,2S)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-73-0_b.png)
![Benzene,1,1'-[(1R,2R)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-72-9.png)
![Benzene,1,1'-[(1R,2R)-1-methyl-1,2-cyclopropanediyl]bis-, rel- (9CI)](http://img.cochemist.com/ccimg/14200/14161-72-9_b.png)




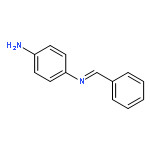
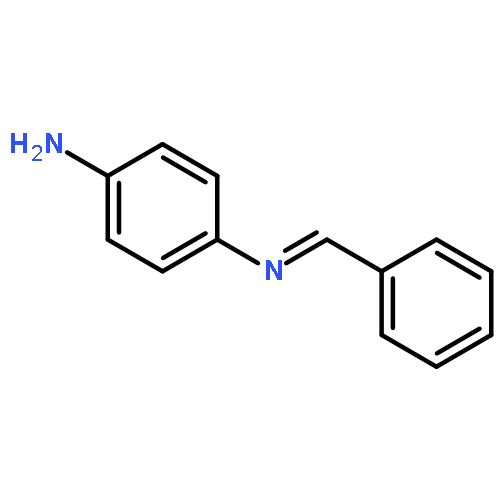
![N-[(E)-phenylmethylidene]biphenyl-4-amine](http://img.cochemist.com/ccimg/14000/13924-28-2.png)
![N-[(E)-phenylmethylidene]biphenyl-4-amine](http://img.cochemist.com/ccimg/14000/13924-28-2_b.png)










![Tungsten, [(2,3,5,6-h)-bicyclo[2.2.1]hepta-2,5-diene]tetracarbonyl-](http://img.cochemist.com/ccimg/12200/12129-25-8.png)
![Tungsten, [(2,3,5,6-h)-bicyclo[2.2.1]hepta-2,5-diene]tetracarbonyl-](http://img.cochemist.com/ccimg/12200/12129-25-8_b.png)



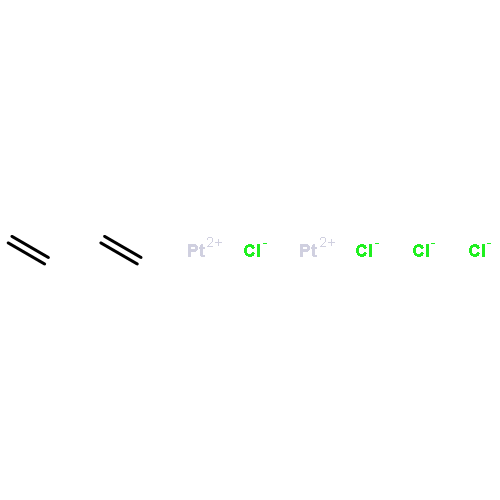







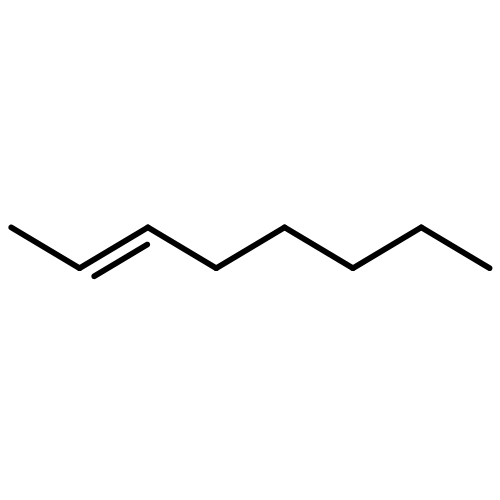

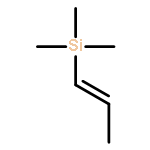
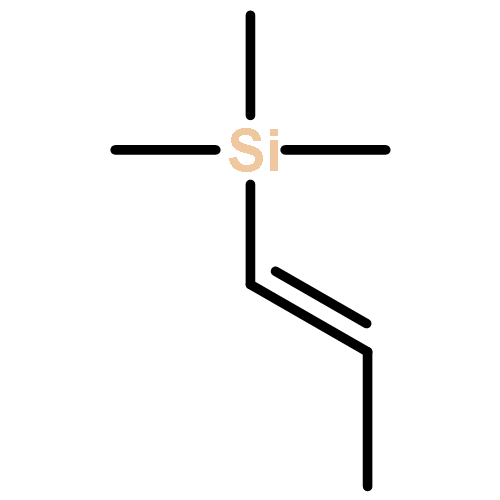
![Silane, trimethyl[(3-methyl-1-methylene-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/6700/6651-46-3.png)
![Silane, trimethyl[(3-methyl-1-methylene-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/6700/6651-46-3_b.png)
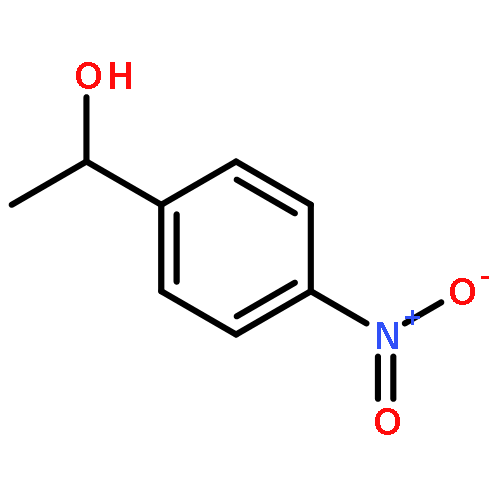
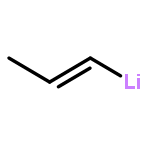
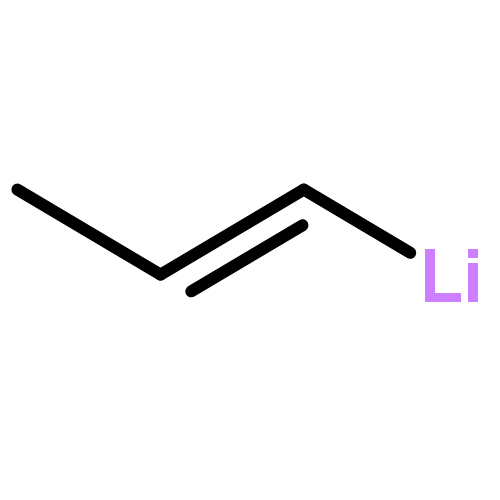
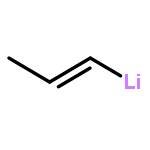

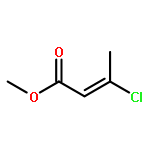

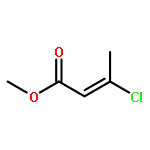
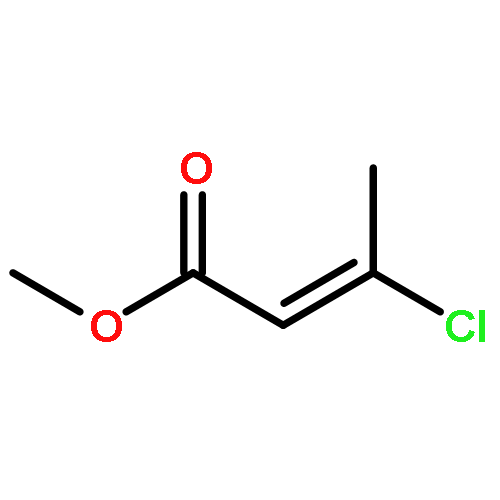






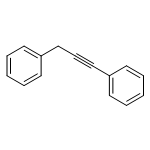













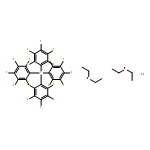
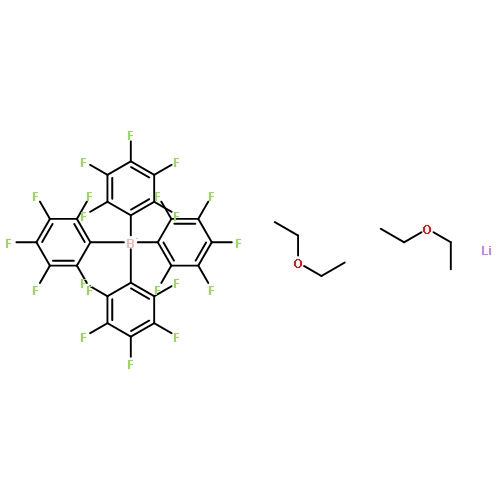
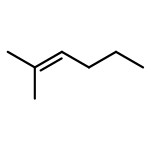
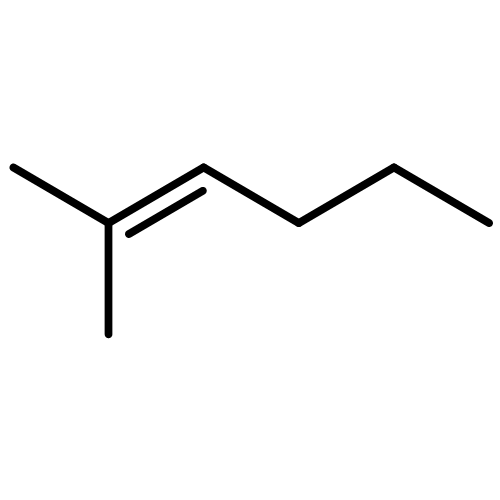


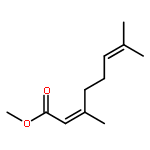







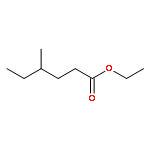

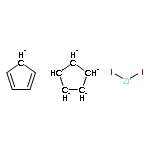



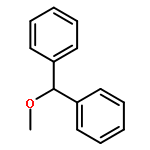










![Phosphine, bis[3,5-bis(trifluoromethyl)phenyl]ethenyl-](http://img.cochemist.com/ccimg/199400/199342-63-7.png)
![Phosphine, bis[3,5-bis(trifluoromethyl)phenyl]ethenyl-](http://img.cochemist.com/ccimg/199400/199342-63-7_b.png)
![1,2-BIS[BIS(3,5-DITRIFLUOROMETHYLPHENYL)PHOSPHINO]ETHANE](http://img.cochemist.com/ccimg/199400/199342-62-6.png)
![1,2-BIS[BIS(3,5-DITRIFLUOROMETHYLPHENYL)PHOSPHINO]ETHANE](http://img.cochemist.com/ccimg/199400/199342-62-6_b.png)

![Benzene, [(1R,2S)-2-(2-methyl-1-propenyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/89500/89486-57-7.png)
![Benzene, [(1R,2S)-2-(2-methyl-1-propenyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/89500/89486-57-7_b.png)
![Benzene, [(1R,2R)-2-(2-methyl-1-propenyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/89500/89486-56-6.png)
![Benzene, [(1R,2R)-2-(2-methyl-1-propenyl)cyclopropyl]-, rel-](http://img.cochemist.com/ccimg/89500/89486-56-6_b.png)
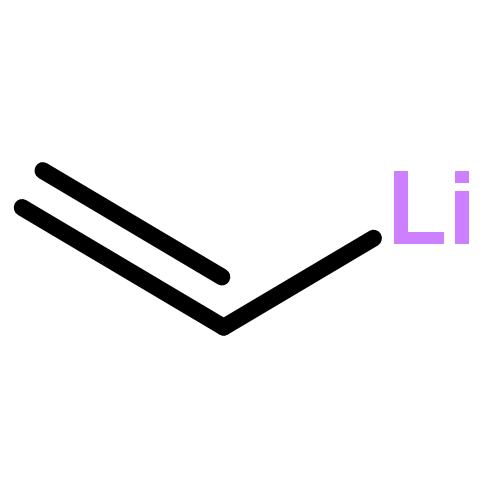
![Lithium, [1-(diphenylphosphino)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/83300/83272-80-4.png)
![Lithium, [1-(diphenylphosphino)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/83300/83272-80-4_b.png)


![2-Oxabicyclo[4.1.0]heptane, 1-(4-methylphenyl)-](http://img.cochemist.com/ccimg/77400/77352-25-1.png)
![2-Oxabicyclo[4.1.0]heptane, 1-(4-methylphenyl)-](http://img.cochemist.com/ccimg/77400/77352-25-1_b.png)


![Benzene, [2-(1,1-dimethylethyl)cyclopropyl]-, trans-](/data/chemimg/1287600/64433-16-5.png)
![Benzene, [2-(1,1-dimethylethyl)cyclopropyl]-, trans-](/data/chemimg/1287600/64433-16-5_b.png)
![Benzene, [2-(1,1-dimethylethyl)cyclopropyl]-, cis-](/data/chemimg/1284100/64433-15-4.png)
![Benzene, [2-(1,1-dimethylethyl)cyclopropyl]-, cis-](/data/chemimg/1284100/64433-15-4_b.png)




![Lithium, [(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/53000/52999-17-4.png)
![Lithium, [(4-methylphenyl)ethynyl]-](http://img.cochemist.com/ccimg/53000/52999-17-4_b.png)







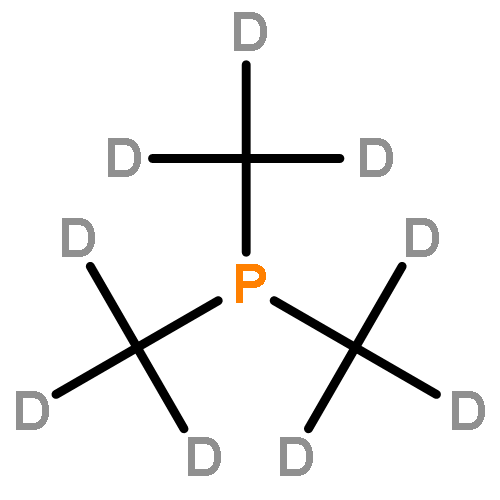


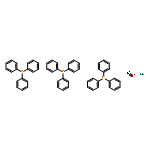





![Lithium, [(phenylthio)methyl]-](http://img.cochemist.com/ccimg/13400/13307-75-0.png)
![Lithium, [(phenylthio)methyl]-](http://img.cochemist.com/ccimg/13400/13307-75-0_b.png)




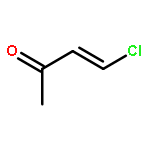
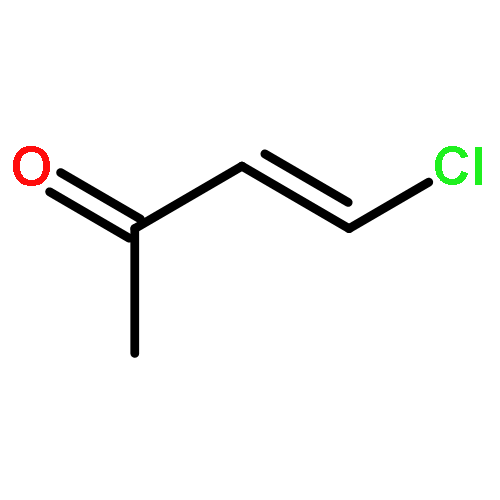



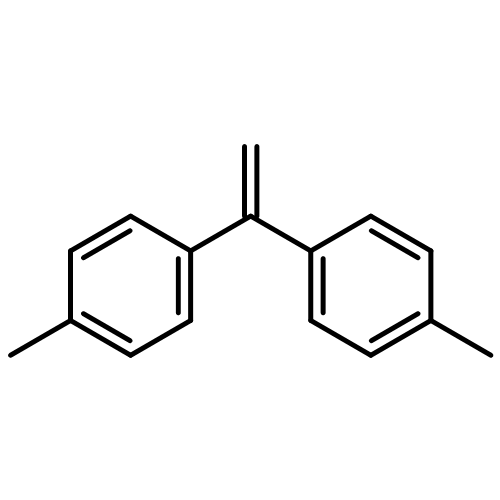
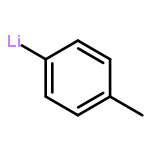
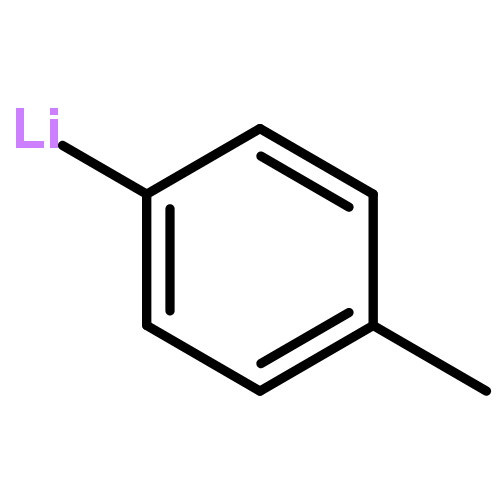
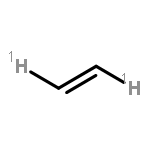




![2-OXABICYCLO[3.1.0]HEXANE, 1-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/73200/73184-81-3.png)
![2-OXABICYCLO[3.1.0]HEXANE, 1-(4-METHYLPHENYL)-](http://img.cochemist.com/ccimg/73200/73184-81-3_b.png)



























![1,6-DIMETHYLBICYCLO[4.1.0]HEPT-3-ENE](http://img.cochemist.com/ccimg/38800/38749-44-9.png)
![1,6-DIMETHYLBICYCLO[4.1.0]HEPT-3-ENE](http://img.cochemist.com/ccimg/38800/38749-44-9_b.png)