Co-reporter:Timothy R. Valentic;David R. Jackson;Sean F. Brady
ACS Chemical Biology December 16, 2016 Volume 11(Issue 12) pp:3421-3430
Publication Date(Web):October 25, 2016
DOI:10.1021/acschembio.6b00658
Arixanthomycins are pentangular polyphenols (PP) with potent antiproliferative activities that were discovered through the heterologous expression of environmental DNA-derived gene clusters. The biosynthesis of arixanthomycin and other PPs is unusual because it requires several novel type II polyketide synthase (PKS) enzymes for its complete maturation. Most type II PKSs contain a ketoreductase (KR) that mediates the C7–C12 first ring cyclization and C-9 reduction. In contrast, based on previous studies of product analysis and genome mining, the arixanthomycin (ARX) gene cluster harbors a C-11 reducing KR (ARX 27), a C9–C14 first-ring aromatase/cyclase (ARX 19), and an unprecedented C-17 and C-19 reducing KR (ARX 21). While bioinformatics is useful for predicting novel enzymes, the functions of ARX 19, ARX 21, and ARX 27 have yet to be confirmed. Further, the structural features that predispose the ARX biosynthetic enzymes to process atypical poly-β-ketone scaffolds remain unknown. We report the crystal structure of ARX 21, the first structure of an enzyme involved in PP biosynthesis and likely a C-17 and C-19 reducing-KR, which is structurally similar to C-15 reducing KRs. Structural comparison of ARX 21 and other C-9 reducing KRs revealed a difference in the enzyme active site that may enlighten the molecular basis of KR substrate specificity. In addition, we report the successful in vitro reconstitution of ARX 19. The structural characterization of ARX 21 in conjunction with the in vitro results of ARX 19 lays the groundwork toward a complete in vitro and structural characterization of type II PKS enzymes involved in PP biogenesis.
Co-reporter:Jesus F. Barajas;Gaurav Shakya;Gabriel Moreno;David R. Jackson;Caitlyn L. Topper;Heriberto Rivera, Jr.;Anna L. Vagstad;Michael D. Burkart;James J. La Clair;Craig A. Townsend
PNAS 2017 Volume 114 (Issue 21 ) pp:E4142-E4148
Publication Date(Web):2017-05-23
DOI:10.1073/pnas.1609001114
Product template (PT) domains from fungal nonreducing polyketide synthases (NR-PKSs) are responsible for controlling the aldol
cyclizations of poly-β-ketone intermediates assembled during the catalytic cycle. Our ability to understand the high regioselective
control that PT domains exert is hindered by the inaccessibility of intrinsically unstable poly-β-ketones for in vitro studies.
We describe here the crystallographic application of “atom replacement” mimetics in which isoxazole rings linked by thioethers
mimic the alternating sites of carbonyls in the poly-β-ketone intermediates. We report the 1.8-Å cocrystal structure of the
PksA PT domain from aflatoxin biosynthesis with a heptaketide mimetic tethered to a stably modified 4′-phosphopantetheine,
which provides important empirical evidence for a previously proposed mechanism of PT-catalyzed cyclization. Key observations
support the proposed deprotonation at C4 of the nascent polyketide by the catalytic His1345 and the role of a protein-coordinated
water network to selectively activate the C9 carbonyl for nucleophilic addition. The importance of the 4′-phosphate at the
distal end of the pantetheine arm is demonstrated to both facilitate delivery of the heptaketide mimetic deep into the PT
active site and anchor one end of this linear array to precisely meter C4 into close proximity to the catalytic His1345. Additional
structural features, docking simulations, and mutational experiments characterize protein–substrate mimic interactions, which
likely play roles in orienting and stabilizing interactions during the native multistep catalytic cycle. These findings afford
a view of a polyketide “atom-replaced” mimetic in a NR-PKS active site that could prove general for other PKS domains.
Co-reporter:Alexander R. White, Brendan M. Duggan, Shiou-Chuan Tsai, and Christopher D. Vanderwal
Organic Letters 2016 Volume 18(Issue 5) pp:1124-1127
Publication Date(Web):February 18, 2016
DOI:10.1021/acs.orglett.6b00230
Many halogenases interchangeably incorporate chlorine and bromine into organic molecules. On the basis of an unsubstantiated report that the alga Ochromonas danica, a prodigious producer of chlorosulfolipids, was able to produce bromosulfolipids, we have investigated the promiscuity of its halogenases toward bromine incorporation. We have found that bromosulfolipids are produced with the exact positional and stereochemical selectivity as in the chlorosulfolipid danicalipin A when this alga is grown under modified conditions containing excess bromide ion.
Co-reporter:David R. Jackson, Stephanie S. Tu, MyChi Nguyen, Jesus F. Barajas, Andrew J. Schaub, Daniel Krug, Dominik Pistorius, Ray Luo, Rolf Müller, and Shiou-Chuan Tsai
ACS Chemical Biology 2016 Volume 11(Issue 1) pp:95
Publication Date(Web):October 16, 2015
DOI:10.1021/acschembio.5b00500
The incorporation of nonacetate starter units during type II polyketide biosynthesis helps diversify natural products. Currently, there are few enzymatic strategies for the incorporation of nonacetate starter units in type II polyketide synthase (PKS) pathways. Here we report the crystal structure of AuaEII, the anthranilate:CoA ligase responsible for the generation of anthraniloyl-CoA, which is used as a starter unit by a type II PKS in aurachin biosynthesis. We present structural and protein sequence comparisons to other aryl:CoA ligases. We also compare the AuaEII crystal structure to a model of a CoA ligase homologue, AuaE, which is present in the same gene cluster. AuaE is predicted to have the same fold as AuaEII, but instead of CoA ligation, AuaE catalyzes acyl transfer of anthranilate from anthraniloyl-CoA to the acyl carrier protein (ACP). Together, this work provides insight into the molecular basis for starter unit selection of anthranilate in type II PKS biosynthesis.
Co-reporter:David R. Jackson, Xia Yu, Guojung Wang, Avinash B. Patel, Jordi Calveras, Jesus F. Barajas, Eita Sasaki, Mikko Metsä-Ketelä, Hung-wen Liu, Jürgen Rohr, and Shiou-Chuan Tsai
ACS Chemical Biology 2016 Volume 11(Issue 4) pp:1137
Publication Date(Web):January 27, 2016
DOI:10.1021/acschembio.5b00913
Cores of aromatic polyketides are essential for their biological activities. Most type II polyketide synthases (PKSs) biosynthesize these core structures involving the minimal PKS, a PKS-associated ketoreductase (KR) and aromatases/cyclases (ARO/CYCs). Oxygenases (OXYs) are rarely involved. BE-7585A is an anticancer polyketide with an angucyclic core. 13C isotope labeling experiments suggest that its angucyclic core may arise from an oxidative rearrangement of a linear anthracyclinone. Here, we present the crystal structure and functional analysis of BexE, the oxygenase proposed to catalyze this key oxidative rearrangement step that generates the angucyclinone framework. Biochemical assays using various linear anthracyclinone model compounds combined with docking simulations narrowed down the substrate of BexE to be an immediate precursor of aklaviketone, possibly 12-deoxy-aklaviketone. The structural analysis, docking simulations, and biochemical assays provide insights into the role of BexE in BE-7585A biosynthesis and lay the groundwork for engineering such framework-modifying enzymes in type II PKSs.
Co-reporter:Heriberto Rivera Jr., Sachin Dhar, James J. La Clair, Shiou-Chuan Tsai, Michael D. Burkart
Tetrahedron 2016 Volume 72(Issue 25) pp:3605-3608
Publication Date(Web):23 June 2016
DOI:10.1016/j.tet.2016.01.062
Polyketide biosynthesis engages a series of well-timed biosynthetic operations to generate elaborate natural products from simple building blocks. Mimicry of these processes has offered practical means for total synthesis and provided a foundation for reaction discovery. We now report an unusual intramolecular trans-amidation reaction discovered while preparing stabilized probes for the study of actinorhodin biosynthesis. This rapid cyclization event offers insight into the natural cyclization process inherent to the biosynthesis of type II polyketide antibiotics.
Co-reporter:Jesus F. Barajas, Ryan M. Phelan, Andrew J. Schaub, Jaclyn T. Kliewer, Peter J. Kelly, David R. Jackson, Ray Luo, Jay D. Keasling, Shiou-Chuan Tsai
Chemistry & Biology 2015 Volume 22(Issue 8) pp:1018-1029
Publication Date(Web):20 August 2015
DOI:10.1016/j.chembiol.2015.06.022
•Highest resolution and first cofactor-bound structure of a terminal reductase domain•Computational modeling advances hypotheses made from the crystal structure•Biochemical analysis defines residues critical for substrate specificity and catalysis•Result-based engineering enabled improved reduction of highly reduced substratesThe terminal reductase (R) domain from the non-ribosomal peptide synthetase (NRPS) module MxaA in Stigmatella aurantiaca Sga15 catalyzes a non-processive four-electron reduction to produce the myxalamide family of secondary metabolites. Despite widespread use in nature, a lack of structural and mechanistic information concerning reductive release from polyketide synthase (PKS) and NRPS assembly lines principally limits our ability to redesign R domains with altered or improved activity. Here we report crystal structures for MxaA R, both in the absence and, for the first time, in the presence of the NADPH cofactor. Molecular dynamics simulations were employed to provide a deeper understanding of this domain and further identify residues critical for structural integrity, substrate binding, and catalysis. Aggregate computational and structural findings provided a basis for mechanistic investigations and, in the process, delivered a rationally altered variant with improved activity toward highly reduced substrates.Figure optionsDownload full-size imageDownload high-quality image (138 K)Download as PowerPoint slide
Co-reporter:Kara Finzel, Chi Nguyen, David R. Jackson, Aarushi Gupta, Shiou-Chuan Tsai, Michael D. Burkart
Chemistry & Biology 2015 Volume 22(Issue 11) pp:1453-1460
Publication Date(Web):19 November 2015
DOI:10.1016/j.chembiol.2015.09.009
•The application of mechanistic crosslinking to evaluate FabA specificity•Mutagenesis utilized to identify important residues for FabA substrate preference•First gain-of-function FabA mutant for shorter chain length fatty acidsMicrobial fatty acid biosynthetic enzymes are important targets for areas as diverse as antibiotic development to biofuel production. Elucidating the molecular basis of chain length control during fatty acid biosynthesis is crucial for the understanding of regulatory processes of this fundamental metabolic pathway. In Escherichia coli, the acyl carrier protein (AcpP) plays a central role by sequestering and shuttling the growing acyl chain between fatty acid biosynthetic enzymes. FabA, a β-hydroxyacyl-AcpP dehydratase, is an important enzyme in controlling fatty acid chain length and saturation levels. FabA-AcpP interactions are transient in nature and thus difficult to visualize. In this study, four mechanistic crosslinking probes mimicking varying acyl chain lengths were synthesized to systematically probe for modified chain length specificity of 14 FabA mutants. These studies provide evidence for the AcpP-interacting “positive patch,” FabA mutations that alter substrate specificity, and the roles that the FabA “gating residues” play in chain length control.Figure optionsDownload full-size imageDownload high-quality image (171 K)Download as PowerPoint slide
Co-reporter:Grace Caldara-Festin;David R. Jackson;Jesus F. Barajas;Avinash B. Patel;Stephanie Aguilar;Timothy R. Valentic;MyChi Nguyen;Michael Vo;Avinash Khanna;Eita Sasaki;Hung-wen Liu
PNAS 2015 Volume 112 (Issue 50 ) pp:E6844-E6851
Publication Date(Web):2015-12-15
DOI:10.1073/pnas.1512976112
Aromatic polyketides make up a large class of natural products with diverse bioactivity. During biosynthesis, linear poly-β-ketone
intermediates are regiospecifically cyclized, yielding molecules with defined cyclization patterns that are crucial for polyketide
bioactivity. The aromatase/cyclases (ARO/CYCs) are responsible for regiospecific cyclization of bacterial polyketides. The
two most common cyclization patterns are C7–C12 and C9–C14 cyclizations. We have previously characterized three monodomain
ARO/CYCs: ZhuI, TcmN, and WhiE. The last remaining uncharacterized class of ARO/CYCs is the di-domain ARO/CYCs, which catalyze
C7–C12 cyclization and/or aromatization. Di-domain ARO/CYCs can further be separated into two subclasses: “nonreducing” ARO/CYCs,
which act on nonreduced poly-β-ketones, and “reducing” ARO/CYCs, which act on cyclized C9 reduced poly-β-ketones. For years,
the functional role of each domain in cyclization and aromatization for di-domain ARO/CYCs has remained a mystery. Here we
present what is to our knowledge the first structural and functional analysis, along with an in-depth comparison, of the nonreducing
(StfQ) and reducing (BexL) di-domain ARO/CYCs. This work completes the structural and functional characterization of mono-
and di-domain ARO/CYCs in bacterial type II polyketide synthases and lays the groundwork for engineered biosynthesis of new
bioactive polyketides.
Co-reporter:Gaurav Shakya ; Heriberto Rivera ; Jr.; D. John Lee ; Matt J. Jaremko ; James J. La Clair ; Daniel T. Fox ; Robert W. Haushalter ; Andrew J. Schaub ; Joel Bruegger ; Jesus F. Barajas ; Alexander R. White ; Parminder Kaur ; Emily R. Gwozdziowski ; Fiona Wong ; Shiou-Chuan Tsai ;Michael D. Burkart
Journal of the American Chemical Society 2014 Volume 136(Issue 48) pp:16792-16799
Publication Date(Web):November 19, 2014
DOI:10.1021/ja5064857
The mechanistic details of many polyketide synthases (PKSs) remain elusive due to the instability of transient intermediates that are not accessible via conventional methods. Here we report an atom replacement strategy that enables the rapid preparation of polyketone surrogates by selective atom replacement, thereby providing key substrate mimetics for detailed mechanistic evaluations. Polyketone mimetics are positioned on the actinorhodin acyl carrier protein (actACP) to probe the underpinnings of substrate association upon nascent chain elongation and processivity. Protein NMR is used to visualize substrate interaction with the actACP, where a tetraketide substrate is shown not to bind within the protein, while heptaketide and octaketide substrates show strong association between helix II and IV. To examine the later cyclization stages, we extended this strategy to prepare stabilized cyclic intermediates and evaluate their binding by the actACP. Elongated monocyclic mimics show much longer residence time within actACP than shortened analogs. Taken together, these observations suggest ACP-substrate association occurs both before and after ketoreductase action upon the fully elongated polyketone, indicating a key role played by the ACP within PKS timing and processivity. These atom replacement mimetics offer new tools to study protein and substrate interactions and are applicable to a wide variety of PKSs.
Co-reporter:Gregory L. Challis and Shiou-Chuan Tsai
Natural Product Reports 2014 vol. 31(Issue 10) pp:1241-1241
Publication Date(Web):03 Sep 2014
DOI:10.1039/C4NP90033B
Co-reporter:Joel Bruegger, Bob Haushalter, Anna Vagstad, Gaurav Shakya, Nathan Mih, Craig A. Townsend, Michael D. Burkart, Shiou-Chuan Tsai
Chemistry & Biology 2013 Volume 20(Issue 9) pp:1135-1146
Publication Date(Web):19 September 2013
DOI:10.1016/j.chembiol.2013.07.012
•An activity-based crosslinker successfully detects PKS interdomain interactions•The crosslinking efficiency is correlated with starter unit specificity of KSs•The ACPs and KSs from NR-PKSs are interchangeable for crosslinking•Mutations identify KS surface residues important for ACP⋅KS interactionsProtein⋅protein interactions, which often involve interactions among an acyl carrier protein (ACP) and ACP partner enzymes, are important for coordinating polyketide biosynthesis. However, the nature of such interactions is not well understood, especially in the fungal nonreducing polyketide synthases (NR-PKSs) that biosynthesize toxic and pharmaceutically important polyketides. Here, we employ mechanism-based crosslinkers to successfully probe ACP and ketosynthase (KS) domain interactions in NR-PKSs. We found that crosslinking efficiency is closely correlated with the strength of ACP⋅KS interactions and that KS demonstrates strong starter unit selectivity. We further identified positively charged surface residues by KS mutagenesis, which mediates key interactions with the negatively charged ACP surface. Such complementary/matching contact pairs can serve as “adapter surfaces” for future efforts to generate new polyketides using NR-PKSs.Figure optionsDownload full-size imageDownload high-quality image (245 K)Download as PowerPoint slide
Co-reporter:Pouya Javidpour, Joel Bruegger, Supawadee Srithahan, Tyler P. Korman, Matthew P. Crump, John Crosby, Michael D. Burkart, Shiou-Chuan Tsai
Chemistry & Biology 2013 Volume 20(Issue 10) pp:1225-1234
Publication Date(Web):24 October 2013
DOI:10.1016/j.chembiol.2013.07.016
•ActKR surface arginines are important for ACP-binding and activity toward polyketides•In contrast to the S-specific P94L actKR, mutant V151L displays R-stereospecificity•ActKR is proposed to mediate C7–C12 polyketide cyclization prior to C9-ketoreductionIn the actinorhodin type II polyketide synthase, the first polyketide modification is a regiospecific C9-carbonyl reduction, catalyzed by the ketoreductase (actKR). Our previous studies identified the actKR 94-PGG-96 motif as a determinant of stereospecificity. The molecular basis for reduction regiospecificity is, however, not well understood. In this study, we examined the activities of 20 actKR mutants through a combination of kinetic studies, PKS reconstitution, and structural analyses. Residues have been identified that are necessary for substrate interaction, and these observations have suggested a structural model for this reaction. Polyketides dock at the KR surface and are steered into the enzyme pocket where C7–C12 cyclization is mediated by the KR before C9-ketoreduction can occur. These molecular features can potentially serve as engineering targets for the biosynthesis of novel, reduced polyketides.
Co-reporter:Shiou-Chuan Tsai
Chemistry & Biology 2012 Volume 19(Issue 7) pp:787-788
Publication Date(Web):27 July 2012
DOI:10.1016/j.chembiol.2012.07.006
Enzymes involved in natural product biosynthesis employ a variety of cofactors, reaction mechanisms, and substrate preferences to achieve remarkable chemical diversity found in nature. In this issue of Chemistry & Biology, Goldman and colleagues show how cofactor (FAD) binding affinity impacts the reaction mechanism and outcome of two related proteins, RebC and StaC, involved in indolocarbazoles biosynthesis.
Co-reporter:Ming-Yue Lee, Brian D. Ames, and Shiou-Chuan Tsai
Biochemistry 2012 Volume 51(Issue 14) pp:
Publication Date(Web):March 20, 2012
DOI:10.1021/bi201705q
Aromatic polyketides are biologically active natural products. Many important pharmaceuticals are derived from aromatic polyketides. Especially important in aromatic polyketide biosynthesis is the regiospecific cyclization of a linear, preassembled polyketide chain catalyzed by aromatase/cyclase (ARO/CYC), which serves as a key control point in aromatic ring formation. How different ARO/CYCs promote different cyclization patterns is not well understood. The whiE locus of Streptomyces coelicolor A3(2) is responsible for the biosynthesis of an aromatic polyketide precursor to the gray spore pigment. The WhiE ARO/CYC catalyzes the regiospecific C9–C14 and C7–C16 cyclization and aromatization of a 24-carbon polyketide chain. WhiE ARO/CYC shares a high degree of similarity to another nonreducing PKS ARO/CYC, TcmN ARO/CYC. This paper presents the apo crystal structure of WhiE ARO/CYC, and cocrystal structures of WhiE and TcmN ARO/CYCs bound with polycyclic aromatic compounds that mimic the respective ARO/CYC products. Site-directed mutagenesis coupled with in vitro PKS reconstitution assays was used to characterize the interior pocket residues of WhiE ARO/CYC. The results confirmed that the interior pocket of ARO/CYCs is a critical determinant of polyketide cyclization specificity. A unified ARO/CYC-mediated cyclization mechanism is proposed on the basis of these structural and functional results.
Co-reporter:Pouya Javidpour, Tyler Paz Korman, Gaurav Shakya, and Shiou-Chuan Tsai
Biochemistry 2011 Volume 50(Issue 21) pp:
Publication Date(Web):April 20, 2011
DOI:10.1021/bi200335f
Type II polyketides include antibiotics such as tetracycline and chemotherapeutics such as daunorubicin. Type II polyketides are biosynthesized by the type II polyketide synthase (PKS) that consists of 5–10 stand-alone domains. In many type II PKSs, the type II ketoreductase (KR) specifically reduces the C9-carbonyl group. How the type II KR achieves such a high regiospecificity and the nature of stereospecificity are not well understood. Sequence alignment of KRs led to a hypothesis that a well-conserved 94-XGG-96 motif may be involved in controlling the stereochemistry. The stereospecificity of single-, double-, and triple-mutant combinations of P94L, G95D, and G96D were analyzed in vitro and in vivo for the actinorhodin KR (actKR). The P94L mutation is sufficient to change the stereospecificity of actKR. Binary and ternary crystal structures of both wild-type and P94L actKR were determined. Together with assay results, docking simulations, and cocrystal structures, a model for stereochemical control is presented herein that elucidates how type II polyketides are introduced into the substrate pocket such that the C9-carbonyl can be reduced with high regio- and stereospecificities. The molecular features of actKR important for regio- and stereospecificities can potentially be applied in biosynthesizing new polyketides via protein engineering that rationally controls polyketide keto reduction.
Co-reporter:Pouya Javidpour, Abhirup Das, Chaitan Khosla, and Shiou-Chuan Tsai
Biochemistry 2011 Volume 50(Issue 34) pp:
Publication Date(Web):July 21, 2011
DOI:10.1021/bi2006866
Bacterial aromatic polyketides that include many antibiotic and antitumor therapeutics are biosynthesized by the type II polyketide synthase (PKS), which consists of 5–10 stand-alone enzymatic domains. Hedamycin, an antitumor antibiotic polyketide, is uniquely primed with a hexadienyl group generated by a type I PKS followed by coupling to a downstream type II PKS to biosynthesize a 24-carbon polyketide, whose C9 position is reduced by hedamycin type II ketoreductase (hedKR). HedKR is homologous to the actinorhodin KR (actKR), for which we have conducted extensive structural studies previously. How hedKR can accommodate a longer polyketide substrate than the actKR, and the molecular basis of its regio- and stereospecificities, is not well understood. Here we present a detailed study of hedKR that sheds light on its specificity. Sequence alignment of KRs predicts that hedKR is less active than actKR, with significant differences in substrate/inhibitor recognition. In vitro and in vivo assays of hedKR confirmed this hypothesis. The hedKR crystal structure further provides the molecular basis for the observed differences between hedKR and actKR in the recognition of substrates and inhibitors. Instead of the 94-PGG-96 motif observed in actKR, hedKR has the 92-NGG-94 motif, leading to S-dominant stereospecificity, whose molecular basis can be explained by the crystal structure. Together with mutations, assay results, docking simulations, and the hedKR crystal structure, a model for the observed regio- and stereospecificities is presented herein that elucidates how different type II KRs recognize substrates with different chain lengths, yet precisely reduce only the C9-carbonyl group. The molecular features of hedKR important for regio- and stereospecificities can potentially be applied to biosynthesize new polyketides via protein engineering that rationally controls polyketide ketoreduction.
Co-reporter:Brian D. Ames, Ming-Yue Lee, Colleen Moody, Wenjun Zhang, Yi Tang, and Shiou-Chuan Tsai
Biochemistry 2011 Volume 50(Issue 39) pp:
Publication Date(Web):August 26, 2011
DOI:10.1021/bi200593m
Aromatic polyketides comprise an important class of natural products that possess a wide range of biological activities. The cyclization of the polyketide chain is a critical control point in the biosynthesis of aromatic polyketides. The aromatase/cyclases (ARO/CYCs) are an important component of the type II polyketide synthase (PKS) and help fold the polyketide for regiospecific cyclizations of the first ring and/or aromatization, promoting two commonly observed first-ring cyclization patterns for the bacterial type II PKSs: C7–C12 and C9–C14. We had previously reported the crystal structure and enzymological analyses of the TcmN ARO/CYC, which promotes C9–C14 first-ring cyclization. However, how C7–C12 first-ring cyclization is controlled remains unresolved. In this work, we present the 2.4 Å crystal structure of ZhuI, a C7–C12-specific first-ring ARO/CYC from the type II PKS pathway responsible for the production of the R1128 polyketides. Though ZhuI possesses a helix-grip fold shared by TcmN ARO/CYC, there are substantial differences in overall structure and pocket residue composition that may be important for directing C7–C12 (rather than C9–C14) cyclization. Docking studies and site-directed mutagenesis coupled to an in vitro activity assay demonstrate that ZhuI pocket residues R66, H109, and D146 are important for enzyme function. The ZhuI crystal structure helps visualize the structure and putative dehydratase function of the didomain ARO/CYCs from KR-containing type II PKSs. The sequence–structure–function analysis described for ZhuI elucidates the molecular mechanisms that control C7–C12 first-ring polyketide cyclization and builds a foundation for future endeavors into directing cyclization patterns for engineered biosynthesis of aromatic polyketides.
Co-reporter:Ana Arabolaza, Mary Elizabeth Shillito, Ting-Wan Lin, Lautaro Diacovich, Melrose Melgar, Huy Pham, Deborah Amick, Hugo Gramajo and Shiou-Chuan Tsai
Biochemistry 2010 Volume 49(Issue 34) pp:
Publication Date(Web):July 22, 2010
DOI:10.1021/bi1005305
The first committed step of fatty acid and polyketides biosynthesis, the biotin-dependent carboxylation of an acyl-CoA, is catalyzed by acyl-CoA carboxylases (ACCases) such as acetyl-CoA carboxylase (ACC) and propionyl-CoA carboxylase (PCC). ACC and PCC in Streptomyces coelicolor are homologue multisubunit complexes that can carboxylate different short chain acyl-CoAs. While ACC is able to carboxylate acetyl-, propionyl-, or butyryl-CoA with approximately the same specificity, PCC only recognizes propionyl- and butyryl-CoA as substrates. How ACC and PCC have such different specificities toward these substrates is only partially understood. To further understand the molecular basis of how the active site residues can modulate the substrate recognition, we mutated D422, N80, R456, and R457 of PccB, the catalytic beta subunit of PCC. The crystal structures of six PccB mutants and the wild type crystal structure were compared systematically to establish the sequence−structure−function relationship that correlates the observed substrate specificity toward acetyl-, propionyl-, and butyryl-CoA with active site geometry. The experimental data confirmed that D422 is a key determinant of substrate specificity, influencing not only the active site properties but further altering protein stability and causing long-range conformational changes. Mutations of N80, R456, and R457 lead to variations in the quaternary structure of the beta subunit and to a concomitant loss of enzyme activity, indicating the importance of these residues in maintaining the active protein conformation as well as a critical role in substrate binding.
Co-reporter:Tyler Paz Korman;Jason M. Crawford;Jason W. Labonte;Adam G. Newman;Justin Wong;Craig A. Townsend;
Proceedings of the National Academy of Sciences 2010 107(14) pp:6246-6251
Publication Date(Web):March 23, 2010
DOI:10.1073/pnas.0913531107
Polyketide natural products possess diverse architectures and biological functions and share a subset of biosynthetic steps
with fatty acid synthesis. The final transformation catalyzed by both polyketide synthases (PKSs) and fatty acid synthases
is most often carried out by a thioesterase (TE). The synthetic versatility of TE domains in fungal nonreducing, iterative
PKSs (NR-PKSs) has been shown to extend to Claisen cyclase (CLC) chemistry by catalyzing C–C ring closure reactions as opposed
to thioester hydrolysis or O–C/N–C macrocyclization observed in previously reported TE structures. Catalysis of C–C bond formation
as a product release mechanism dramatically expands the synthetic potential of PKSs, but how this activity was acquired has
remained a mystery. We report the biochemical and structural analyses of the TE/CLC domain in polyketide synthase A, the multidomain
PKS central to the biosynthesis of aflatoxin B1, a potent environmental carcinogen. Mutagenesis experiments confirm the predicted identity of the catalytic triad and its
role in catalyzing the final Claisen-type cyclization to the aflatoxin precursor, norsolorinic acid anthrone. The 1.7 Å crystal
structure displays an α/β-hydrolase fold in the catalytic closed form with a distinct hydrophobic substrate-binding chamber.
We propose that a key rotation of the substrate side chain coupled to a protein conformational change from the open to closed
form spatially governs substrate positioning and C–C cyclization. The biochemical studies, the 1.7 Å crystal structure of
the TE/CLC domain, and intermediate modeling afford the first mechanistic insights into this widely distributed C–C bond-forming
class of TEs.
Co-reporter:Peter Smith, Bingsen Zhou, Nam Ho, Yate-Ching Yuan, Leila Su, Shiou-Chuan Tsai and Yun Yen
Biochemistry 2009 Volume 48(Issue 46) pp:
Publication Date(Web):September 3, 2009
DOI:10.1021/bi9001425
Human p53R2 (hp53R2) is a 351-residue p53-inducible ribonucleotide reductase (RNR) small subunit. It shares >80% sequence identity with hRRM2, the small RNR subunit responsible for normal maintenance of the deoxyribonucleotide (dNTP) pool used for DNA replication, which is active during the S phase in a cell cycle-dependent fashion. But rather than cyclic dNTP synthesis, hp53R2 has been shown to supply dNTPs for DNA repair to cells in G0-G1 in a p53-dependent fashion. The first X-ray crystal structure of hp53R2 is determined to 2.6 Å, in which monomers A and B exhibit mono- and binuclear iron occupancy, respectively. The pronounced structural differences at three regions between hp53R2 and hRRM2 highlight the possible regulatory role in iron assimilation and help explain previously observed physical and biochemical differences in the mobility and accessibility of the radical iron center, as well as radical transfer pathways between the two enzymes. The sequence−structure−function correlations that differentiate hp53R2 and hRRM2 are revealed for the first time. Insight gained from this structural work will be used in the identification of biological function, regulation mechanism, and inhibitor selection in RNR small subunits.
Co-reporter:Jason M. Crawford,
Tyler P. Korman,
Jason W. Labonte,
Anna L. Vagstad,
Eric A. Hill,
Oliver Kamari-Bidkorpeh,
Shiou-Chuan Tsai
&
Craig A. Townsend
Nature 2009 461(7267) pp:1139
Publication Date(Web):2009-10-22
DOI:10.1038/nature08475
Regiospecific cyclizations of reactive poly--keto intermediates are known to lead to the structural variability of aromatic products of fungal nonreducing, multidomain iterative polyketide synthases (NR-PKS group of IPKSs), but questions about the process remain. The crystal structure and mutational studies of a dissected product template monodomain from PksA, the NR-PKS that initiates the biosynethesis of the hepatocarcinogen aflatoxin B1, are now presented.
Co-reporter:Peter Smith, Ping-Hui Szu, Cynthia Bui, Hung-wen Liu and Shiou-Chuan Tsai
Biochemistry 2008 Volume 47(Issue 24) pp:
Publication Date(Web):May 21, 2008
DOI:10.1021/bi702449p
Pyridoxal 5′-phosphate (PLP) and pyridoxamine 5′-phosphate (PMP) are highly versatile coenzymes whose importance is well recognized. The capability of PLP/PMP-dependent enzymes to catalyze a diverse array of chemical reactions is attributed to fine-tuning of the cofactor−substrate interactions in the active site. CDP-6-deoxy-l-threo-d-glycero-4-hexulose 3-dehydrase (E1), along with its reductase (E3), catalyzes the C-3 deoxygenation of CDP-4-keto-6-deoxy-d-glucose to form the dehydrated product, CDP-4-keto-3,6-dideoxy-d-glucose, in the ascarylose biosynthetic pathway. This product is the progenitor to most 3,6-dideoxyhexoses, which are the major antigenic determinants of many Gram-negative pathogens. The dimeric [2Fe-2S] protein, E1, cloned from Yersinia pseudotuberculosis, is the only known enzyme whose catalysis involves the direct participation of PMP in one-electron redox chemistry. E1 also contains an unusual [2Fe-2S] cluster with a previously unknown binding motif (C-X57-C-X1-C-X7-C). Herein we report the first X-ray crystal structure of E1, which exhibits an aspartate aminotransferase (AAT) fold. A comparison of the E1 active site architecture with homologous structures uncovers residues critical for the dehydration versus transamination activity. Site-directed mutagenesis of four E1 residues, D194H, Y217H, H220K, and F345H, converted E1 from a PMP-dependent dehydrase to a PLP/glutamate-dependent aminotransferase. The E1 quadruple mutant, having been conferred this altered enzyme activity, can transaminate the natural substrate to CDP-4,6-dideoxy-4-amino-d-galactose without E3. Taken together, these results provide the molecular basis of the functional switch of E1 toward dehydration, epimerization, and transamination. The insights gained from these studies can be used for the development of inhibitors of disease-relevant PLP/PMP-dependent enzymes.
Co-reporter:Brian Douglas Ames;Tyler Paz Korman;Wenjun Zhang;Peter Smith;Thanh Vu;Yi Tang
PNAS 2008 105 (14 ) pp:5349-5354
Publication Date(Web):2008-04-08
DOI:10.1073/pnas.0709223105
Polyketides are a class of natural products with highly diverse chemical structures and pharmaceutical activities. Polyketide
cyclization, promoted by the aromatase/cyclase (ARO/CYC), helps diversify aromatic polyketides. How the ARO/CYC promotes highly
specific cyclization is not well understood because of the lack of a first-ring ARO/CYC structure. The 1.9 Å crystal structure
of Tcm ARO/CYC reveals that the enzyme belongs to the Bet v1-like superfamily (or STAR domain family) with a helix–grip fold,
and contains a highly conserved interior pocket. Docking, mutagenesis, and an in vivo assay show that the size, shape, and composition of the pocket are important to orient and specifically fold the polyketide
chain for C9-C14 first-ring and C7-C16 second-ring cyclizations. Two pocket residues, R69 and Y35, were found to be essential
for promoting first- and second-ring cyclization specificity. Different pocket residue mutations affected the polyketide product
distribution. A mechanism is proposed based on the structure-mutation-docking results. These results strongly suggest that
the regiospecific cyclizations of the first two rings and subsequent aromatizations take place in the interior pocket. The
chemical insights gleaned from this work pave the foundation toward defining the molecular rules for the ARO/CYC cyclization
specificity, whose rational control will be important for future endeavors in the engineered biosynthesis of novel anticancer
and antibiotic aromatic polyketides.
Co-reporter:Stuart Smith and Shiou-Chuan Tsai
Natural Product Reports 2007 vol. 24(Issue 5) pp:1041-1072
Publication Date(Web):02 Jul 2007
DOI:10.1039/B603600G
Covering: up to the end of 2006
Co-reporter:Ting-Wan Lin;Melrose M. Melgar;Gabriela Gago;Hugo Gramajo;Pierre Baldi;Daniel Kurth;S. Joshua Swamidass;Teresa Tseng;John Purdon
PNAS 2006 Volume 103 (Issue 9 ) pp:3072-3077
Publication Date(Web):2006-02-28
DOI:10.1073/pnas.0510580103
Mycolic acids and multimethyl-branched fatty acids are found uniquely in the cell envelope of pathogenic mycobacteria. These
unusually long fatty acids are essential for the survival, virulence, and antibiotic resistance of Mycobacterium tuberculosis. Acyl-CoA carboxylases (ACCases) commit acyl-CoAs to the biosynthesis of these unique fatty acids. Unlike other organisms
such as Escherichia coli or humans that have only one or two ACCases, M. tuberculosis contains six ACCase carboxyltransferase domains, AccD1–6, whose specific roles in the pathogen are not well defined. Previous
studies indicate that AccD4, AccD5, and AccD6 are important for cell envelope lipid biosynthesis and that its disruption leads
to pathogen death. We have determined the 2.9-Å crystal structure of AccD5, whose sequence, structure, and active site are
highly conserved with respect to the carboxyltransferase domain of the Streptomyces coelicolor propionyl-CoA carboxylase. Contrary to the previous proposal that AccD4–5 accept long-chain acyl-CoAs as their substrates,
both crystal structure and kinetic assay indicate that AccD5 prefers propionyl-CoA as its substrate and produces methylmalonyl-CoA,
the substrate for the biosyntheses of multimethyl-branched fatty acids such as mycocerosic, phthioceranic, hydroxyphthioceranic,
mycosanoic, and mycolipenic acids. Extensive in silico screening of National Cancer Institute compounds and the University of California, Irvine, ChemDB database resulted in the
identification of one inhibitor with a K
i of 13.1 μM. Our results pave the way toward understanding the biological roles of key ACCases that commit acyl-CoAs to the
biosynthesis of cell envelope fatty acids, in addition to providing a target for structure-based development of antituberculosis
therapeutics.
 Tsai-UC Irvine(4).jpg)





















![2-Naphthalenesulfonic acid,6-hydroxy-5-[[2-methoxy-4-[(4-sulfophenyl)azo]phenyl]azo]-](http://img.cochemist.com/ccimg/556800/556795-95-0.png)
![2-Naphthalenesulfonic acid,6-hydroxy-5-[[2-methoxy-4-[(4-sulfophenyl)azo]phenyl]azo]-](http://img.cochemist.com/ccimg/556800/556795-95-0_b.png)




![2,4-Quinazolinediamine, 5-[[4-(trifluoromethyl)phenyl]methoxy]-](http://img.cochemist.com/ccimg/216000/215925-78-3.png)
![2,4-Quinazolinediamine, 5-[[4-(trifluoromethyl)phenyl]methoxy]-](http://img.cochemist.com/ccimg/216000/215925-78-3_b.png)



![2,4-Quinazolinediamine, 5-[(4-chlorophenyl)thio]-](http://img.cochemist.com/ccimg/169000/168910-48-3.png)
![2,4-Quinazolinediamine, 5-[(4-chlorophenyl)thio]-](http://img.cochemist.com/ccimg/169000/168910-48-3_b.png)
![4H-1-Benzopyran-4-one,2,3-dihydro-2,7-dihydroxy-2-[(6-hydroxy-4-oxo-4H-pyran-2-yl)methyl]-5-methyl-](http://img.cochemist.com/ccimg/162200/162112-40-5.png)
![4H-1-Benzopyran-4-one,2,3-dihydro-2,7-dihydroxy-2-[(6-hydroxy-4-oxo-4H-pyran-2-yl)methyl]-5-methyl-](http://img.cochemist.com/ccimg/162200/162112-40-5_b.png)












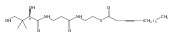
![2-BUTENETHIOIC ACID, S-[2-(ACETYLAMINO)ETHYL] ESTER, (E)-](http://img.cochemist.com/ccimg/67100/67024-18-4.png)
![2-BUTENETHIOIC ACID, S-[2-(ACETYLAMINO)ETHYL] ESTER, (E)-](http://img.cochemist.com/ccimg/67100/67024-18-4_b.png)

















![[4-(aminomethyl)-5-hydroxy-6-methylpyridin-3-yl]methyl Dihydrogen Phosphate](http://img.cochemist.com/ccimg/600/529-96-4.png)
![[4-(aminomethyl)-5-hydroxy-6-methylpyridin-3-yl]methyl Dihydrogen Phosphate](http://img.cochemist.com/ccimg/600/529-96-4_b.png)
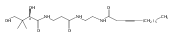
![(R)-2,4-dihydroxy-N-[3-[(2-mercaptoethyl)amino]-3-oxopropyl]-3,3-dimethylbutyramide](http://img.cochemist.com/ccimg/500/496-65-1.png)
![(R)-2,4-dihydroxy-N-[3-[(2-mercaptoethyl)amino]-3-oxopropyl]-3,3-dimethylbutyramide](http://img.cochemist.com/ccimg/500/496-65-1_b.png)


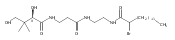


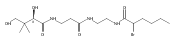
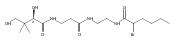


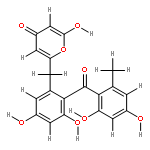
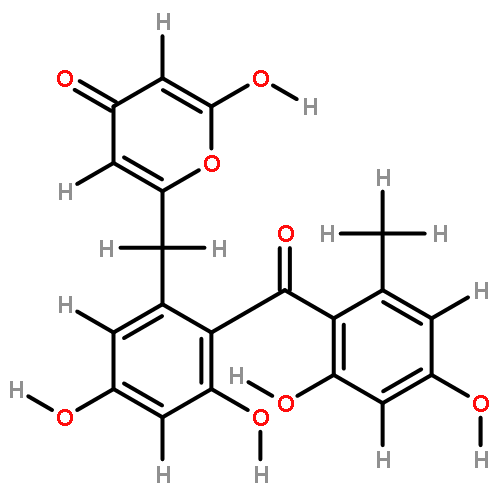
![D-Glucitol,2,3:4,5-dianhydro-1,6-dideoxy-5-C-[7,12-dihydro-11-hydroxy-5-methyl-4,7,12-trioxo-8-[2,3,6-trideoxy-3-(dimethylamino)-b-D-arabino-hexopyranosyl]-10-[2,3,6-trideoxy-3-(dimethylamino)-3-C-methyl-a-L-lyxo-hexopyranosyl]-4H-anthra[1,2-b]pyran-2-yl]-](http://img.cochemist.com/ccimg/11100/11048-97-8.png)
![D-Glucitol,2,3:4,5-dianhydro-1,6-dideoxy-5-C-[7,12-dihydro-11-hydroxy-5-methyl-4,7,12-trioxo-8-[2,3,6-trideoxy-3-(dimethylamino)-b-D-arabino-hexopyranosyl]-10-[2,3,6-trideoxy-3-(dimethylamino)-3-C-methyl-a-L-lyxo-hexopyranosyl]-4H-anthra[1,2-b]pyran-2-yl]-](http://img.cochemist.com/ccimg/11100/11048-97-8_b.png)








![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)
![4H-1-Benzopyran-4-one,2,3-dihydro-2,7-dihydroxy-2-[(4-hydroxy-2-oxo-2H-pyran-6-yl)methyl]-5-methyl-](http://img.cochemist.com/ccimg/625200/625120-84-5.png)
![4H-1-Benzopyran-4-one,2,3-dihydro-2,7-dihydroxy-2-[(4-hydroxy-2-oxo-2H-pyran-6-yl)methyl]-5-methyl-](http://img.cochemist.com/ccimg/625200/625120-84-5_b.png)


![[8,8'-Bi-1H-naphtho[2,3-c]pyran]-3,3'-diaceticacid,3,3',4,4',5,5',10,10'-octahydro-6,6',9,9'-tetrahydroxy-1,1'-dimethyl-5,5',10,10'-tetraoxo-,(1R,1'R,3S,3'S)-](http://img.cochemist.com/ccimg/1400/1397-77-9.png)
![[8,8'-Bi-1H-naphtho[2,3-c]pyran]-3,3'-diaceticacid,3,3',4,4',5,5',10,10'-octahydro-6,6',9,9'-tetrahydroxy-1,1'-dimethyl-5,5',10,10'-tetraoxo-,(1R,1'R,3S,3'S)-](http://img.cochemist.com/ccimg/1400/1397-77-9_b.png)


![4H-1-Benzopyran-4-one,2,3-dihydro-2,7-dihydroxy-5-[(4-hydroxy-2-oxo-2H-pyran-6-yl)methyl]-2-methyl-](http://img.cochemist.com/ccimg/625200/625120-83-4.png)
![4H-1-Benzopyran-4-one,2,3-dihydro-2,7-dihydroxy-5-[(4-hydroxy-2-oxo-2H-pyran-6-yl)methyl]-2-methyl-](http://img.cochemist.com/ccimg/625200/625120-83-4_b.png)