Co-reporter:Hai Wang, Lu Su, Richen Li, Shiyi Zhang, Jingwei Fan, Fuwu Zhang, Tan P. Nguyen, and Karen L. Wooley
ACS Macro Letters March 21, 2017 Volume 6(Issue 3) pp:219-219
Publication Date(Web):February 16, 2017
DOI:10.1021/acsmacrolett.6b00966
The direct and facile synthesis of polyphosphoramidates (PPAs) with acid-labile phosphoramidate backbone linkages are reported, together with demonstration of their hydrolytic degradability, evaluated under acidic conditions. The introduction of acid-labile linkages along the polymer backbone led to rapid degradation of the polymer backbone dependent upon the environmental stimuli. An oxazaphospholidine monomer bearing a phosphoramidate linkage was designed and synthesized to afford the PPAs via organobase-catalyzed ring-opening polymerization in a controlled manner. The hydrolytic degradation of the PPAs was studied, revealing breakdown of the polymer backbone through cleavage of the phosphoramidate linkages under acidic conditions.
Co-reporter:Alexander T. Lonnecker, Young H. Lim, and Karen L. Wooley
ACS Macro Letters July 18, 2017 Volume 6(Issue 7) pp:748-748
Publication Date(Web):June 26, 2017
DOI:10.1021/acsmacrolett.7b00362
Herein, we demonstrate the synthesis of a bicyclic carbonate monomer of a d-glucal derivative, which originated from the natural product d-glucose, in an efficient three-step procedure and its ring-opening polymerization (ROP), initiated by 4-methylbenzyl alcohol, via organocatalysis. The ROP behavior was studied as a function of time, catalyst type, and catalyst concentration by using size exclusion chromatography (SEC) and nuclear magnetic resonance (NMR) spectroscopy. Using a cocatalyst system of 1,8-diazabicyclo[5.4.0]undec-7-ene and 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-2-thiourea (5 mol %) afforded poly(d-glucal-carbonate) (PGCC) with almost complete monomer conversion (ca. 99%) within 1 min, as analyzed by 1H NMR spectroscopy, and a monomodal SEC trace with dispersity of 1.13. The resulting PGCCs exhibited amorphous characteristics with a relatively high glass transition temperature at ca. 69 °C and onset decomposition temperature at ca. 190 °C, as analyzed by differential scanning calorimetry and thermogravimetric analysis, respectively. This new type of potentially degradable polymer system represents a reactive functional polymer architecture.
Co-reporter:Zhou Li, Jun Ma, Nam S. Lee, and Karen L. Wooley
Journal of the American Chemical Society February 9, 2011 Volume 133(Issue 5) pp:1228-1231
Publication Date(Web):January 4, 2011
DOI:10.1021/ja109191z
We have developed a hierarchical process that combines linear triblock copolymers into concentric globular subunits through strong chemical bonds and is followed by their supramolecular assembly via weak noncovalent interactions to afford one-dimensionally assembled, dynamic cylindrical nanostructures. The molecular brush architecture forces triblock copolymers to adopt intramolecular interactions within confined frameworks and then drives their intermolecular interactions in the mixtures of organic solvent and water. In contrast, the triblock copolymers, when not preconnected into the molecular brush architectures, organize only into globular assemblies.
Co-reporter:Yi-Yun Timothy Tsao and Karen L. Wooley
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5467-5467
Publication Date(Web):April 10, 2017
DOI:10.1021/jacs.7b01116
A grand challenge that crosses synthetic chemistry and biology is the scalable production of functional analogues of biomacromolecules. We have focused our attention on the use of deoxynucleoside building blocks bearing non-natural bases to develop a synthetic methodology that allows for the construction of high molecular weight deoxynucleotide polymers. Our six-membered cyclic phosphoester ring-opening polymerization strategy is demonstrated, herein, by an initial preparation of novel polyphosphoesters, comprised of butenyl-functionalized deoxyribonucleoside repeat units, connected via 3′,5′-backbone linkages. A thymidine-derived bicyclic monomer, 3′,5′-cyclic 3-(3-butenyl) thymidine ethylphosphate, was synthesized in two steps directly from thymidine, via butenylation and diastereoselective cyclization promoted by N,N-dimethyl-4-aminopyridine. Computational modeling of the six-membered 3′,5′-cyclic phosphoester ring derived from deoxyribose indicated strain energies at least 5.4 kcal/mol higher than those of the six-membered monocyclic phosphoester, 2-ethoxy-1,3,2-dioxaphosphinane 2-oxide. These calculations supported the hypothesis that the strained 3′,5′-cyclic monomer can promote ring-opening polymerization to afford the resulting poly(3′,5′-cyclic 3-(3-butenyl) thymidine ethylphosphate)s with low dispersities (Đ < 1.10). This advanced design combines the merits of natural product-derived materials and functional, degradable polymers to provide a new platform for functional, synthetically derived polydeoxyribonucleotide-analogue materials.
Co-reporter:Samantha L. Kristufek;Kevin T. Wacker;Yi-Yun Timothy Tsao;Lu Su
Natural Product Reports (1984-Present) 2017 vol. 34(Issue 4) pp:433-459
Publication Date(Web):2017/04/05
DOI:10.1039/C6NP00112B
Covering: 2010–Aug. 2016
In an effort towards enhancing function and sustainability, natural products have become of interest in the field of polymer chemistry. This review details the blending of chemistries developed through synthetic organic chemistry and polymer chemistry. Through synthetic organic chemical transformations, such as functional group interconversion, a protection/deprotection series, or installation of a functional group, various designs towards novel, synthetic, bio-based polymer systems are described. This review covers several classifications of natural products – oils and fatty acids, terpenes, lignin, and sugar derivatives – focusing on exploring monomers prepared by one or more synthetic steps.
Co-reporter:Jingwei Fan;Richen Li;Hai Wang;Xun He;Tan P. Nguyen;Rachel A. Letteri;Jiong Zou
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 24) pp:5145-5154
Publication Date(Web):2017/06/21
DOI:10.1039/C7OB00931C
A polypeptide-based hydrogel system, when prepared from a diblock polymer with a ternary copolypeptide as one block, exhibited thermo-, mechano- and enzyme-responsive properties, which enabled the encapsulation of naproxen (Npx) during the sol–gel transition and its release in the gel state. Statistical terpolymerizations of L-alanine (Ala), glycine (Gly) and L-isoleucine (Ile) NCAs at a 1 : 1 : 1 feed ratio initiated by monomethoxy monoamino-terminated poly(ethylene glycol) afforded a series of methoxy poly(ethylene glycol)-block-poly(L-alanine-co-glycine-co-L-isoleucine) (mPEG-b-P(A-G-I)) block polymers. β-Sheets were the dominant secondary structures within the polypeptide segments, which facilitated a heat-induced sol-to-gel transition, resulting from the supramolecular assembly of β-sheets into nanofibrils. Deconstruction of the three-dimensional networks by mechanical force (sonication) triggered the reverse gel-to-sol transition. Certain enzymes could accelerate the breakdown of the hydrogel, as determined by in vitro gel weight loss profiles. The hydrogels were able to encapsulate and release Npx over 6 days, demonstrating the potential application of these polypeptide hydrogels as an injectable local delivery system for small molecule drugs.
Co-reporter:Yue Song, Yingchao Chen, Lu Su, Richen Li, Rachel A. Letteri, Karen L. Wooley
Polymer 2017 Volume 122(Volume 122) pp:
Publication Date(Web):28 July 2017
DOI:10.1016/j.polymer.2017.06.065
•Fully natural product-based degradable amphiphilic polymers were synthesized.•Spherical, cylindrical and 2D bundled cylindrical micelles were formed.•The degradability of these micellar nanoparticles was demonstrated.•Crystallization-driven self assembly (CDSA) was broadened to zwitterionic polymers.Crystallization-driven self assembly (CDSA) was achieved with fully degradable amphiphilic block polymers derived from three natural products, l-lactide, l-cysteine and d-glucose, to afford spherical and cylindrical nanostructures. A series of functional l-cysteine-modified diblock copolymers, poly(l-lactide)-block-poly(α-d-glucose carbonate)s (PLLA-b-PDGC-cys), was synthesized by organocatalyzed sequential ring-opening polymerization (ROP) of l-lactide and an alkyne-substituted bicyclic α-d-glucose carbonate, followed by UV-initiated thiol-yne “click” reaction with l-cysteine to render the PDGC block hydrophilic. Incubation of the resulting amphiphilic diblock copolymers in water at 65 °C for 30 h, followed by cooling to room temperature yielded spherical, cylindrical and 2D platelet-like bundled cylinder micellar nanostructures, depending on the PLLA weight percentage in the block copolymer, as revealed by transmission electron microscopy (TEM) and atomic force microscopy (AFM). 1H NMR spectroscopy was employed to monitor the degradation of the materials over 100 d in aqueous solution at pH 1 and 10 at 37 °C, which allowed for characterization of the stability of the micelles, and for determination of the hydrolytic degradability of the polymer backbone and cleavage of the side chain moieties. Electrospray ionization (ESI) and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry were used to identify the hydrolytic degradation products of the copolymers. Overall, this work broadens the scope of CDSA to functional, natural-product based degradable block copolymers (BCPs), and the polymeric nanomaterials synthesized in this work hold promise in drug and antimicrobial delivery applications, among others.Download high-res image (231KB)Download full-size image
Co-reporter:Lu Su;Sarosh Khan;Jingwei Fan;Yen-Nan Lin;Hai Wang;Tiffany P. Gustafson;Fuwu Zhang
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 10) pp:1699-1707
Publication Date(Web):2017/03/07
DOI:10.1039/C6PY01978A
Fundamental synthetic methodology was advanced to allow for the preparation of a reactive glucose-based block copolycarbonate, which was conveniently transformed into a series of amphiphilic block copolymers that underwent aqueous assembly into functional nanoparticle morphologies having practical utility in biomedical and other applications. Two degradable D-glucose carbonate monomers, with one carrying alkyne functionality, were designed and synthesized to access well-defined block polycarbonates (Đ < 1.1) via sequential organocatalytic ring opening polymerizations (ROPs). Kinetic studies of the organocatalyzed sequential ROPs showed a linear relationship between the monomer conversion and the polymer molecular weight, which indicated the controlled fashion during each polymerization. The pendant alkyne groups underwent two classic click reactions, copper-catalyzed azide–alkyne dipolar cycloaddition (CuAAC) and thiol–yne addition reactions, which were employed to render hydrophilicity for the alkyne-containing block and to provide a variety of amphiphilic diblock poly(D-glucose carbonate)s (PGCs). The resulting amphiphilic PGCs were further assembled into a family of nanostructures with different sizes, morphologies, surface charges and functionalities. These non-ionic and anionic nanoparticles showed low cytotoxicity in RAW 264.7 mouse macrophage cells and MC3T3 healthy mouse osteoblast precursor cells, while the cationic nanoparticles exhibited significantly higher IC50 (162 μg mL−1 in RAW 264.7; 199 μg mL−1 in MC3T3) compared to the commercially available cationic lipid-based formulation, Lipofectamine (IC50 = 31 μg mL−1), making these nanomaterials of interest for biomedical applications.
Co-reporter:Fuwu Zhang;Sarosh Khan;Richen Li;Justin A. Smolen;Shiyi Zhang;Guizhi Zhu;Lu Su;Ashlee A. Jahnke;Mahmoud Elsabahy;Xiaoyuan Chen
Nanoscale (2009-Present) 2017 vol. 9(Issue 41) pp:15773-15777
Publication Date(Web):2017/10/26
DOI:10.1039/C7NR05935C
Multifunctional polyphosphoester-based nanoparticles capable of loading paclitaxel (PTX) both chemically and physically were prepared, achieving an ultrahigh equivalent PTX aqueous concentration of 25.30 mg mL−1. The dual-loaded nanoparticles were effective in killing cancer cells, which has the potential to minimize the amount of nanocarriers needed for clinical applications, due to their ultrahigh loading capacity.
Co-reporter:Jennifer S. Zigmond, Adriana Pavía-Sanders, Joel D. Russell, and Karen L. Wooley
Chemistry of Materials 2016 Volume 28(Issue 15) pp:5471
Publication Date(Web):July 18, 2016
DOI:10.1021/acs.chemmater.6b02013
Amphiphilic hyperbranched fluoropolymer coatings incorporating liquid crystalline moieties and poly(ethylene glycol) cross-linkers were found to demonstrate noteworthy anti-icing properties. A series of amphiphilic networks was synthesized through variation of the polymer molecular weights and hydrophilic/hydrophobic component ratios. These innovative materials show a remarkable reduction in the free water melting transition (Tm) temperature (−10 °C), measured by differential scanning calorimetry, and an increase in water contact angle for dry and water-swollen systems. The addition of this ordered parameter generated a unique coating topography, which can be visualized via polarized optical microscopy and 3D optical microscopy, while maintaining an overall macroscopic homogeneity.
Co-reporter:Xun He, Jingwei Fan, Jiong Zou and Karen L. Wooley
Chemical Communications 2016 vol. 52(Issue 54) pp:8455-8458
Publication Date(Web):16 Jun 2016
DOI:10.1039/C6CC03579E
A strategy for reversible patterning of soft conductive materials is described, based upon a combination of peptide-based block copolymer hydrogelators and photo-thermally-active carbon nanotubes. This composite displays photo-responsive gelation at application-relevant timescales (<10 s), allowing for rapid and spatially-defined construction of conductive patterns (>100 S m−1), which, additionally, hold the capability to revert to sol upon sonication for reprocessing.
Co-reporter:Tyler S. Kristufek, Samantha L. Kristufek, Lauren A. Link, Andrew C. Weems, Sarosh Khan, Soon-Mi Lim, Alexander T. Lonnecker, Jeffery E. Raymond, Duncan J. Maitland and Karen L. Wooley
Polymer Chemistry 2016 vol. 7(Issue 15) pp:2639-2644
Publication Date(Web):29 Mar 2016
DOI:10.1039/C5PY01659B
The rapid synthesis of an optically-transparent, flexible elastomer was performed utilizing the naturally-derived source, isosorbide. A novel monomer based on isosorbide (isosorbide dialloc, IDA) was prepared by installing carbonate functionalities along with external olefins for use in thiol–ene click chemistry. Cross-linked networks were created using the commercially-available cross-linker, trimethylolpropane tris(3-mercaptopropionate) (TMPTMP) and resulted in IDA-co-TMPTMP, an optically-transparent elastomer. Systematically, IDA-co-TMPTMP networks were synthesized using a photoinitiator, a UV cure time of one minute and varied post cure times (0–24 h, 125 mm Hg) at 100 °C to observe effects on mechanical, thermal and surface alterations. The mechanical properties also had limited changes with post cure time, including a modulus at 25 °C of 1.9–2.8 MPa and an elongation of 220–344%. The thermal decomposition temperatures of the networks were consistent, ca. 320 °C, while the glass transition temperature remained below room temperature for all samples. A cell viability assay and fluorescence imaging with adherent cells are also reported in this study to show the potential of the material as a biomedical substrate. A degradation study for 60 days resulted in 8.3 ± 3.5% and 97.7 ± 0.3% mass remaining under accelerated (1 M NaOH, 60 °C) and biological conditions (pH 7.4 PBS at 37 °C), respectively. This quickly-synthesized material has the potential to hydrolytically degrade into biologically-benign and environmentally-friendly by-products and may be utilized in renewable plastics and/or bioelastomer applications.
Co-reporter:Jennifer S. Zigmond, Rachel A. Letteri, and Karen L. Wooley
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 49) pp:
Publication Date(Web):December 5, 2016
DOI:10.1021/acsami.6b11112
Linear and hyperbranched poly(ethylene glycol)-cross-linked amphiphilic fluoropolymer networks comprised of different liquid crystalline comonomers were developed and evaluated as functional coatings in extreme weather-challenging conditions. Through variation of the liquid-crystalline comonomer and hydrophilic:hydrophobic component ratios, several series of coatings were synthesized and underwent a variety of analyses including differential scanning calorimetry, water contact angle measurements and solution stability studies in aqueous media. These materials maintained an unprecedented reduction in the free water melting transition (Tm) temperature across the hyperbranched and linear versions. The coatings synthesized from hyperbranched fluoropolymers preserved the liquid crystalline character of the mesogenic components, as seen by polarized optical microscopy, and demonstrated stability in saltwater aqueous environments and in cold weather conditions.Keywords: amphiphilic; anti-icing; dynamic coating; hyperbranched; liquid crystalline;
Co-reporter:Alexander T. Lonnecker, Young H. Lim, Simcha E. Felder, Céline J. Besset, and Karen L. Wooley
Macromolecules 2016 Volume 49(Issue 20) pp:7857-7867
Publication Date(Web):October 7, 2016
DOI:10.1021/acs.macromol.6b00591
Strategies for the preparation of polycarbonates, derived from the natural product d-glucose, which have the potential to degrade back into their bioresorbable starting material and CO2, were developed. By employing established carbohydrate protection/deprotection chemistries, two d-glucose derivatives, methyl 4,6-O-benzylidene-α-d-glucopyranoside or methyl α-d-glucopyranoside, were converted into four different regioisomeric diol monomers, i.e., 1,4-, 1,6-, 2,6-, or 3,6-diols, as confirmed by nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, and mass spectrometry. Each type of regioisomeric monomer was then employed in a condensation polymerization with phosgene, generated in situ from triphosgene, as a comonomer, in the presence of pyridine, to produce four types of polycarbonates with different backbone regio-connectivity, as characterized by size exclusion chromatography, NMR spectroscopy, and IR spectroscopy. Interestingly, their thermal properties, i.e., glass transition temperature (Tg) and thermal degradation behavior, were tunable by changing the topological composition of the monomeric unit. That is, polycarbonates with 2,6- and 3,6-backbone connectivity resulted in significantly higher Tg of ca. 85 and 83 °C, respectively, as compared to those with 1,4- and 1,6-backbone connectivity, showing a Tg of ca. 33 °C, as measured by differential scanning calorimetry. Furthermore, when the thermal decomposition temperature was measured by thermogravimetric analysis, the nonanomeric carbon backbone-based polycarbonates (2,6- and 3,6-) exhibited higher thermal stability and a sharper decomposition profile, with onset decomposition temperature (Td,onset) at 363 or 336 °C, as compared with those polymers containing the anomeric carbon in the carbonate linkage (1,4- and 1,6-), having Td,onset at 171 and 163 °C.
Co-reporter:Xun He;Jingwei Fan ; Karen L. Wooley
Chemistry – An Asian Journal 2016 Volume 11( Issue 4) pp:437-447
Publication Date(Web):
DOI:10.1002/asia.201500957
Abstract
The past decade has witnessed significantly increased interest in the development of smart polypeptide-based organo- and hydrogel systems with stimuli responsiveness, especially those that exhibit sol–gel phase-transition properties, with an anticipation of their utility in the construction of adaptive materials, sensor designs, and controlled release systems, among other applications. Such developments have been facilitated by dramatic progress in controlled polymerizations of α-amino acid N-carboxyanhydrides (NCAs), together with advanced orthogonal functionalization techniques, which have enabled economical and practical syntheses of well-defined polypeptides and peptide hybrid polymeric materials. One-dimensional stacking of polypeptides or peptide aggregations in the forms of certain ordered conformations, such as α helices and β sheets, in combination with further physical or chemical cross-linking, result in the construction of three-dimensional matrices of polypeptide gel systems. The macroscopic sol–gel transitions, resulting from the construction or deconstruction of gel networks and the conformational changes between secondary structures, can be triggered by external stimuli, including environmental factors, electromagnetic fields, and (bio)chemical species. Herein, the most recent advances in polypeptide gel systems are described, covering synthetic strategies, gelation mechanisms, and stimuli-triggered sol–gel transitions, with the aim of demonstrating the relationships between chemical compositions, supramolecular structures, and responsive properties of polypeptide-based organo- and hydrogels.
Co-reporter:Jennifer S. Zigmond;Kevin A. Pollack;Sarah Smedley;Jeffery E. Raymond;Lauren A. Link;Adriana Pavía-Sers;Michael A. Hickner
Journal of Polymer Science Part A: Polymer Chemistry 2016 Volume 54( Issue 2) pp:238-244
Publication Date(Web):
DOI:10.1002/pola.27800
Co-reporter:Mahmoud Elsabahy, Gyu Seong Heo, Soon-Mi Lim, Guorong Sun, and Karen L. Wooley
Chemical Reviews 2015 Volume 115(Issue 19) pp:10967
Publication Date(Web):August 4, 2015
DOI:10.1021/acs.chemrev.5b00135
Co-reporter:Mahmoud Elsabahy and Karen L. Wooley
Accounts of Chemical Research 2015 Volume 48(Issue 6) pp:1620
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.accounts.5b00066
The potential immunotoxicity of nanoparticles that are currently being approved, in different phases of clinical trials, or undergoing rigorous in vitro and in vivo characterizations in several laboratories has recently raised special attention. Products with no apparent in vitro or in vivo toxicity may still trigger various components of the immune system unintentionally and lead to serious adverse reactions. Cytokines are one of the useful biomarkers for predicting the effect of biotherapeutics on modulation of the immune system and for screening the immunotoxicity of nanoparticles both in vitro and in vivo, and they were recently found to partially predict the in vivo pharmacokinetics and biodistribution of nanomaterials. Control of polymer chemistry and supramolecular assembly provides a great opportunity for the construction of biocompatible nanoparticles for biomedical clinical applications. However, the sources of data collected regarding immunotoxicities of nanomaterials are diverse, and experiments are usually conducted using different assays under specific conditions. As a result, making direct comparisons nearly impossible, and thus, tailoring the properties of nanomaterials on the basis of the available data is challenging. In this Account, the effects of chemical structure, cross-linking, degradability, morphology, concentration, and surface chemistry on the immunotoxicity of an expansive array of polymeric nanomaterials will be highlighted, with a focus on assays conducted using the same in vitro and in vivo models and experimental conditions. Furthermore, numerical descriptive values have been utilized uniquely to stand for induction of cytokines by nanoparticles. This treatment of available data provides a simple way to compare the immunotoxicities of various nanomaterials, and the values were found to correlate well with published data. On the basis of the polymeric systems investigated in this study, valuable information has been collected that will aid in the future design of nanomaterials for biomedical applications, including the following: (a) the immunotoxicity of nanomaterials is concentration- and dose-dependent; (b) the synthesis of degradable nanoparticles is essential to decrease toxicity; (c) cross-linking minimizes the release of free polymeric chains and maintains high stability of the nanoparticles, thereby lowering their immunotoxicity; (d) lowering the amine density for cationic polymers that are being utilized for delivery of nucleic acids lowers the toxicity of the nanoparticles; (e) among neutral, zwitterionic, anionic, and cationic nanomaterials, neutral and cationic nanoparticles usually have the lowest and highest immunotoxicities, respectively; and (f) morphology, dimension, and surface chemistry have a great influence on the ability of nanomaterials to interact with the various components of the biological system and to modulate the immune system.
Co-reporter:Fuwu Zhang; Shiyi Zhang; Stephanie F. Pollack; Richen Li; Amelia M. Gonzalez; Jingwei Fan; Jiong Zou; Sarah E. Leininger; Adriana Pavía-Sanders; Rachel Johnson; Laura D. Nelson; Jeffery E. Raymond; Mahmoud Elsabahy; Dennis M. P. Hughes; Mark W. Lenox; Tiffany P. Gustafson
Journal of the American Chemical Society 2015 Volume 137(Issue 5) pp:2056-2066
Publication Date(Web):January 28, 2015
DOI:10.1021/ja512616s
Nanomaterials have great potential to offer effective treatment against devastating diseases by providing sustained release of high concentrations of therapeutic agents locally, especially when the route of administration allows for direct access to the diseased tissues. Biodegradable polyphosphoester-based polymeric micelles and shell cross-linked knedel-like nanoparticles (SCKs) have been designed from amphiphilic block-graft terpolymers, PEBP-b-PBYP-g-PEG, which effectively incorporate high concentrations of paclitaxel (PTX). Well-dispersed nanoparticles physically loaded with PTX were prepared, exhibiting desirable physiochemical characteristics. Encapsulation of 10 wt% PTX, into either micelles or SCKs, allowed for aqueous suspension of PTX at concentrations up to 4.8 mg/mL, as compared to <2.0 μg/mL for the aqueous solubility of the drug alone. Drug release studies indicated that PTX released from these nanostructures was defined through a structure–function relationship, whereby the half-life of sustained PTX release was doubled through cross-linking of the micellar structure to form SCKs. In vitro, physically loaded micellar and SCK nanotherapeutics demonstrated IC50 values against osteosarcoma cell lines, known to metastasize to the lungs (CCH-OS-O and SJSA), similar to the pharmaceutical Taxol formulation. Evaluation of these materials in vivo has provided an understanding of the effects of nanoparticle structure–function relationships on intratracheal delivery and related biodistribution and pharmacokinetics. Overall, we have demonstrated the potential of these novel nanotherapeutics toward future sustained release treatments via administration directly to the sites of lung metastases of osteosarcoma.
Co-reporter:Jeniree A. Flores, Adriana Pavía-Sanders, Yingchao Chen, Darrin J. Pochan, and Karen L. Wooley
Chemistry of Materials 2015 Volume 27(Issue 10) pp:3775
Publication Date(Web):May 7, 2015
DOI:10.1021/acs.chemmater.5b01523
Hybrid inorganic/organic composite materials have been synthesized from the coupling of amine-functionalized iron oxide nanoparticles (amine-IONs) and pre-established shell cross-linked knedel-like (SCK) polymer nanoconstructs. The magnetically active hybrid networks (MHNs), composed of several interconnected SCKs bound to magnetically active amine-IONs, were designed for their application in the sequestration of hydrophobic contaminants from polluted environments. Initial assessment of the ability of the MHNs to capture complex pollutants, such as crude oil (oil), determined a loading capacity in the range of 3.5–4.5 mg of oil sequestered/mg of MHNs. The magnetic component of the hybrid nanoconstructs was exploited as a facile method of manipulation of the loaded networks, which allowed for the recovery of ca. 93% of the MHNs deployed in water, as well as ca. 90% recovery of the oil originally sequestered. Reutilization of these materials exhibited comparable efficiency after three cycles of remediation, which involved deployment, magnetic recovery, and organic washes to remove the cargo. The multiple characteristics of these materials could be exploited in the cleaning of water contaminated during the process of drilling, extraction, and transport of crude oil, among other applications.
Co-reporter:Fuwu Zhang, Justin A. Smolen, Shiyi Zhang, Richen Li, Parth N. Shah, Sangho Cho, Hai Wang, Jeffery E. Raymond, Carolyn L. Cannon and Karen L. Wooley
Nanoscale 2015 vol. 7(Issue 6) pp:2265-2270
Publication Date(Web):09 Jan 2015
DOI:10.1039/C4NR07103D
In this study, a new type of degradable polyphosphoester-based polymeric nanoparticle, capable of carrying silver cations via interactions with alkyne groups, has been developed as a potentially effective and safe treatment for lung infections. It was found that up to 15% (w/w) silver loading into the nanoparticles could be achieved, consuming most of the pendant alkyne groups along the backbone, as revealed by Raman spectroscopy. The well-defined Ag-loaded nanoparticles released silver in a controlled and sustained manner over 5 days, and displayed enhanced in vitro antibacterial activities against cystic fibrosis-associated pathogens and decreased cytotoxicity to human bronchial epithelial cells, in comparison to silver acetate.
Co-reporter:Kellie Seetho, Shiyi Zhang, Kevin A. Pollack, Jiong Zou, Jeffery E. Raymond, Edgar Martinez, and Karen L. Wooley
ACS Macro Letters 2015 Volume 4(Issue 5) pp:505
Publication Date(Web):April 17, 2015
DOI:10.1021/mz500818c
An antibiofouling polymer coating, combined with both zwitterionic and amphiphilic features, is engineered by a two-step modification of a commodity polymer. The surface properties of the resultant polymer coating can be easily tuned by varying the extent of cross-linking in the network. Higher antibiofouling efficiency was observed for these surfaces vs. an elastomeric polydimethylsiloxane standard (Sylgard 184) against the adsorption of biomacromolecules and a marine fouling organism (Ulva zoospores) has been demonstrated. This design establishes a platform for the achievement of functionalized amphiphilic zwitterionic copolymers from relatively inexpensive starting materials via simple chemical manipulations.
Co-reporter:Amandine Noel, Yannick P. Borguet, and Karen L. Wooley
ACS Macro Letters 2015 Volume 4(Issue 6) pp:645
Publication Date(Web):June 2, 2015
DOI:10.1021/acsmacrolett.5b00227
A series of hydrolytically degradable fluorescent poly(ferulic acid-co-tyrosine)-g-mPEG graft copolymers were synthesized and shown to undergo self-assembly in aqueous media to yield fluorescent micelles. The polymers and their micellar assemblies exhibited greater fluorescence emission intensity than did their small molecular building blocks, which provides a self-reporting character that has potential for monitoring the polymer integrity and also for performing in theranostics applications. The amphiphilic graft-copolymers were synthesized by Cu-assisted azide–alkyne “click” addition of azido-functionalized mPEG polymers onto fluorescent degradable hydrophobic copolymers displaying randomly distributed alkyne side-chain groups along their biorenewably derived poly(ferulic acid-co-tyrosine) backbones. The morphologies and photophysical properties of the supramolecular assemblies generated in aqueous solutions were evaluated by DLS, TEM, AFM, and steady-state optical spectroscopies. The 15–30 nm sized micelles behaved as broad-band emitters in the 350–600 nm range, which highlights their potential as self-reporting nanomaterials for in vitro studies.
Co-reporter:Guorong Sun;Sangho Cho;Fan Yang;Xun He;Adriana Pavía-Sers;Corrie Clark;Jeffery E. Raymond;Stanislav V. Verkhoturov;Emile A. Schweikert;James W. Thackeray;Peter Trefonas
Journal of Polymer Science Part A: Polymer Chemistry 2015 Volume 53( Issue 2) pp:193-199
Publication Date(Web):
DOI:10.1002/pola.27362
Co-reporter:Sangho Cho, Gyu Seong Heo, Sarosh Khan, Amelia M. Gonzalez, Mahmoud Elsabahy, and Karen L. Wooley
Macromolecules 2015 Volume 48(Issue 24) pp:8797-8805
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.macromol.5b01974
Drawbacks of poly(ethylene glycol) (PEG), the most widely used water-soluble polymer in nanomedicines, have stimulated development of alternative hydrophilic polymers. Among the substitutes, poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) exhibits water solubility, minimal toxicity, and the possibility to introduce functionalities through pendant hydroxyl groups; however, nondegradability may cause long-term health and environmental issues. Alternatively, polycarbonates based on bis-MPA derivatives, which are well-known to be biocompatible, biodegradable, and of low toxicity in vivo, could be utilized as degradable equivalents to polymethacrylates. Therefore, we developed a polycarbonate-based PHPMA analogue, poly(5-methyl-5-(2-hydroxypropyl)aminocarbonyl-1,3-dioxan-2-one) (PMHPAC), by amidation of carboxylic acid-functional polycarbonates with 1-amino-2-propanol. The resulting PMHPAC was highly water-soluble, with low cyto-/immuno-toxicities, and readily functionalizable. These characteristics make PMHPAC a promising candidate as a degradable alternative to PEG and PHPMA. Furthermore, a fully degradable PMHPAC block copolymer was synthesized to demonstrate synthetic versatility and formation of nanostructures in aqueous solution for potential biomedical applications.
Co-reporter:Young H. Lim, Kristin M. Tiemann, Gyu Seong Heo, Patrick O. Wagers, Yohannes H. Rezenom, Shiyi Zhang, Fuwu Zhang, Wiley J. Youngs, David A. Hunstad, and Karen L. Wooley
ACS Nano 2015 Volume 9(Issue 2) pp:1995
Publication Date(Web):January 26, 2015
DOI:10.1021/nn507046h
The development of well-defined polymeric nanoparticles (NPs) as delivery carriers for antimicrobials targeting human infectious diseases requires rational design of the polymer template, an efficient synthetic approach, and fundamental understanding of the developed NPs, e.g., drug loading/release, particle stability, and other characteristics. Herein, we developed and evaluated the in vitro antimicrobial activity of silver-bearing, fully biodegradable and functional polymeric NPs. A series of degradable polymeric nanoparticles (dNPs), composed of phosphoester and l-lactide and designed specifically for silver loading into the hydrophilic shell and/or the hydrophobic core, were prepared as potential delivery carriers for three different types of silver-based antimicrobials–silver acetate or one of two silver carbene complexes (SCCs). Silver-loading capacities of the dNPs were not influenced by the hydrophilic block chain length, loading site (i.e., core or shell), or type of silver compound, but optimization of the silver feed ratio was crucial to maximize the silver loading capacity of dNPs, up to ca. 12% (w/w). The release kinetics of silver-bearing dNPs revealed 50% release at ca. 2.5–5.5 h depending on the type of silver compound. In addition, we undertook a comprehensive evaluation of the rates of hydrolytic or enzymatic degradability and performed structural characterization of the degradation products. Interestingly, packaging of the SCCs in the dNP-based delivery system improved minimum inhibitory concentrations up to 70%, compared with the SCCs alone, as measured in vitro against 10 contemporary epidemic strains of Staphylococcus aureus and eight uropathogenic strains of Escherichia coli. We conclude that these dNP-based delivery systems may be beneficial for direct epithelial treatment and/or prevention of ubiquitous bacterial infections, including those of the skin and urinary tract.Keywords: (bio)degradable polymeric nanoparticles; Escherichia coli; functional polymeric nanoparticles; in vitro antimicrobial efficacy; nanoparticle-based antimicrobial delivery system; silver carbene complexes; Staphylococcus aureus;
Co-reporter:Jingwei Fan, Jiong Zou, Xun He, Fuwu Zhang, Shiyi Zhang, Jeffery E. Raymond and Karen L. Wooley
Chemical Science 2014 vol. 5(Issue 1) pp:141-150
Publication Date(Web):18 Oct 2013
DOI:10.1039/C3SC52504J
The simple copolymerization of N-carboxyanhydride (NCA) monomers is utilized to generate copolypeptides having a combination of α-helix and β-sheet sub-structures that, when grown from a solvophilic synthetic polymer block segment, are capable of driving mechano-responsive supramolecular sol-to-gel-to-sol and sol-to-gel-to-gel transitions reversibly, which allow also for injection-based processing and self-healing behaviors. A new type of polypeptide-based organogelator, methoxy poly(ethylene glycol)-block-poly(γ-benzyl-L-glutamate-co-glycine) (mPEG-b-P(BLG-co-Gly)), is facilely synthesized by statistical ring-opening copolymerizations (ROPs) of γ-benzyl-L-glutamate (BLG) and glycine (Gly) NCAs initiated by mPEG-amine. These systems exhibit tunable secondary structures and result in sonication stimulus responsiveness of the organogels with the polypeptide segment variation, controlled by varying the ratio of BLG NCA to Gly NCA during the copolymerizations. Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) studies indicate the α-helical component decreases while the β-sheet content increases systematically with a higher mole fraction of Gly in the polypeptide segment. The supramolecular assembly of β-sheet nanofibrils, having a tunable width over the range of 10.4–14.5 nm with varied BLG to Gly ratio, are characterized by transmission electron microscopy (TEM). The further self-assembly of these nanostructures into 3-D gel networks within N,N-dimethylformamide (DMF) occurs at low critical gelation concentrations (CGC) (lowest ca. 0.6 wt%). Increased BLG to Gly ratios lead to an increase of the α-helical component in the secondary structures of the polypeptide segments, resulting in wider and more flexible nanofibrils. The presence of α-helical component in the polymers enhances the stability of the organogels against sonication, and instantaneous gel-to-gel transitions are observed as in situ reconstruction of networks occurs within the gelled materials after sonication. In marked contrast, the β-sheet-rich gel, prepared from mPEG-b-PGly, exhibits an instant gel-to-sol transition after sonication is applied. The CGC concentration and stiffness of this mPEG-b-P(BLG-co-Gly) organogel system can be tuned by simply varying the percentages of α-helix and β-sheet in the secondary structures through control of the BLG to Gly ratio during synthesis. The mechanical properties of these organogels are studied by dynamic mechanical analyses (DMA), having storage moduli of ca. 12.1 kPa at room temperature. The injectability and self-healing capabilities are demonstrated by direct observation of the macroscopic self-healing behavior experiment.
Co-reporter:Xun He, Jingwei Fan, Fuwu Zhang, Richen Li, Kevin A. Pollack, Jeffery E. Raymond, Jiong Zou and Karen L. Wooley
Journal of Materials Chemistry A 2014 vol. 2(Issue 46) pp:8123-8130
Publication Date(Web):23 Jul 2014
DOI:10.1039/C4TB00909F
A multi-responsive triblock hydrogelator oligo(DL-allylglycine)-block-poly(ethylene glycol)-block-oligo(DL-allylglycine) (ODLAG-b-PEG-b-ODLAG) was synthesized facilely by ring-opening polymerization (ROP) of DLAG N-carboxyanhydride (NCA) with a diamino-terminated PEG as the macroinitiator. This system exhibited heat-induced sol-to-gel transitions and either sonication- or enzyme-induced gel-to-sol transitions. The β-sheeting of the oligopeptide segments was confirmed by attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) and wide-angle X-ray scattering (WAXS). The β-sheets further displayed tertiary ordering into fibrillar structures that, in turn generated a porous and interconnected hydrogel matrix, as observed via transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The reversible macroscopic sol-to-gel transitions triggered by heat and gel-to-sol transitions triggered by sonication were correlated with the transformation of nanostructural morphologies, with fibrillar structures observed in gel and spherical aggregates in sol, respectively. The enzymatic breakdown of the hydrogels was also investigated. This allyl-functionalized hydrogelator can serve as a platform for the design of smart hydrogels, appropriate for expansion into biological systems as bio-functional and bio-responsive materials.
Co-reporter:Kevin A. Pollack, Philip M. Imbesi, Jeffery E. Raymond, and Karen L. Wooley
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 21) pp:19265
Publication Date(Web):October 20, 2014
DOI:10.1021/am505296n
Synthesis of terpolymer coatings composed of hyperbranched fluoropolymers cross-linked with bisamino-propyl poly(ethylene glycol) and bisamino-propyl polydimethylsiloxane (PDMS) was performed to generate antibiofouling surfaces. Nanoscale imaging and surface spectroscopy confirmed that this system possessed complex surface topographies and chemical compositions. Surface complexity was determined to be due to molecular interactions, phase segregation, and compositional gradients arising between the three components. A clear difference in surface behavior was observable before and after exposure to water. Antibiofouling characteristics were investigated by bovine serum albumin (BSA) adsorption studies; the terpolymer coating displayed a 60% greater resistance to protein adsorption in comparison to the fouling of a commercial antibiofouling silicone coating. The unique surface topography, topology, and chemical heterogeneity expressed at a variety of scales provide a robust regime for the generation of hardy, complex surfaces known to incorporate characteristics appropriate for antibiofouling applications. Thorough assessment of thermal responses and mechanical properties in relevant environments demonstrated a formulation platform immediately appropriate for consideration in marine and in vivo applications.Keywords: antibiofouling; coatings; cross-linked networks; hyperbranched fluoropolymer
Co-reporter:Sandani Samarajeewa, Ryan P. Zentay, Nema D. Jhurry, Ang Li, Kellie Seetho, Jiong Zou and Karen L. Wooley
Chemical Communications 2014 vol. 50(Issue 8) pp:968-970
Publication Date(Web):04 Dec 2013
DOI:10.1039/C3CC46013D
Electrostatic interaction-mediated enzymatic-hydrolysis of poly(lactide)-containing nanoscale assemblies is described. At physiological pH, degradable core–shell morphologies with charged shells can readily attract or repel enzymes carrying opposite or similar charges, respectively.
Co-reporter:Lauren A. Link, Alexander T. Lonnecker, Keith Hearon, Cameron A. Maher, Jeffery E. Raymond, and Karen L. Wooley
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 20) pp:17370
Publication Date(Web):October 7, 2014
DOI:10.1021/am506087e
Polycarbonate networks derived from the natural product quinic acid that can potentially return to their natural building blocks upon hydrolytic degradation are described herein. Solvent-free thiol–ene chemistry was utilized in the copolymerization of tris(alloc)quinic acid and a variety of multifunctional thiol monomers to obtain poly(thioether-co-carbonate) networks with a wide range of achievable thermomechanical properties including glass transition temperatures from −18 to +65 °C and rubbery moduli from 3.8 to 20 MPa. The network containing 1,2-ethanedithiol expressed an average toughness at 25 and 63 °C of 1.08 and 2.35 MJ/m3, respectively, and an order-of-magnitude increase in the average toughness at 37 °C of 15.56 MJ/m3.Keywords: photo-cross-linking; polycarbonates; quinic acid; renewable polymers; thiol−ene chemistry
Co-reporter:Gyu Seong Heo, Sangho Cho and Karen L. Wooley
Polymer Chemistry 2014 vol. 5(Issue 11) pp:3555-3558
Publication Date(Web):23 Apr 2014
DOI:10.1039/C4PY00456F
Ozonolysis of allyl-functional polycarbonates provides aldehyde-functional polycarbonates that have potential to be reactive platforms for transformation into diverse active materials.
Co-reporter:Jiong Zou;Fuwu Zhang;Shiyi Zhang;Stephanie F. Pollack;Mahmoud Elsabahy;Jingwei Fan
Advanced Healthcare Materials 2014 Volume 3( Issue 3) pp:441-448
Publication Date(Web):
DOI:10.1002/adhm.201300235
There has been an increasing interest to develop new types of stimuli-responsive drug delivery vehicles with high drug loading and controlled release properties for chemotherapeutics. An acid-labile poly(ethylene oxide)-block-polyphosphoester-graft-PTX drug conjugate (PEO-b-PPE-g-PTX G2) degradable, polymeric paclitaxel (PTX) conjugate containing ultra-high levels of PTX loading is improved significantly, in this second-generation development, which involves connection of each PTX molecule to the polymer backbone via a pH-sensitive β-thiopropionate linkage. The PEO-b-PPE-g-PTX G2 forms well-defined nanoparticles in an aqueous solution, by direct dissolution into water, with a number-averaged hydrodynamic diameter of 114 ± 31 nm, and exhibits a PTX loading capacity as high as 53 wt%, with a maximum PTX concentration of 0.68 mg mL−1 in water (vs 1.7 μg mL−1 for free PTX). The PEO-b-PPE-g-PTX G2 shows accelerated drug release under acidic conditions (≈50 wt% PTX released in 8 d) compared with neutral conditions (≈20 wt% PTX released in 8 d). Compared to previously reported polyphosphoester-based PTX drug conjugates, PEO-b-PPE-g-PTX G1 without the β-thiopropionate linker, the PEO-b-PPE-g-PTX G2 shows pH-triggered drug release property and 5- to 8-fold enhanced in vitro cytotoxicity against two cancer cell lines.
Co-reporter:Jingwei Fan, Richen Li, Xun He, Kellie Seetho, Fuwu Zhang, Jiong Zou and Karen L. Wooley
Polymer Chemistry 2014 vol. 5(Issue 13) pp:3977-3981
Publication Date(Web):27 May 2014
DOI:10.1039/C4PY00628C
Sequential polymerization of N-carboxyanhydrides accelerated by nitrogen flow is utilized to generate a novel well-defined diblock copolypeptide (PDI = 1.08), with incorporation of alkyne-functionalized side-chain groups allowing for rapid and efficient thiol–yne click-type modifications, followed by self-assembly into nanopure water to construct a helical polypeptide-based versatile and functional nanoparticle platform.
Co-reporter:Sangho Cho;Fan Yang;Guorong Sun;Michael J. Eller;Corrie Clark;Emile A. Schweikert;James W. Thackeray;Peter Trefonas
Macromolecular Rapid Communications 2014 Volume 35( Issue 4) pp:437-441
Publication Date(Web):
DOI:10.1002/marc.201300845
Co-reporter:Dr. Jiong Zou;Xun He;Jingwei Fan;Dr. Jeffery E. Raymond ;Dr. Karen L. Wooley
Chemistry - A European Journal 2014 Volume 20( Issue 29) pp:8842-8847
Publication Date(Web):
DOI:10.1002/chem.201403027
Abstract
A facile polymerization of an allyl-functionalized N-carboxyanhydride (NCA) monomer is utilized to construct an A-B-A-type triblock structure containing β-sheet-rich oligomeric peptide segments tethered by a poly(ethylene oxide) chain, which are capable of dispersing and gelating single-walled carbon nanotubes (SWCNTs) noncovalently in organic solvents, resulting in significant enhancement of the mechanical properties of polypeptide-based organogels.
Co-reporter:Young H. Lim, Gyu Seong Heo, Yohannes H. Rezenom, Stephanie Pollack, Jeffery E. Raymond, Mahmoud Elsabahy, and Karen L. Wooley
Macromolecules 2014 Volume 47(Issue 14) pp:4634-4644
Publication Date(Web):July 2, 2014
DOI:10.1021/ma402480a
A novel polyphosphoester (PPE) with vinyl ether side chain functionality was developed as a versatile template for postpolymerization modifications, and its degradability and biocompatibility were evaluated. An organocatalyzed ring-opening polymerization of ethylene glycol vinyl ether-pendant cyclic phosphotriester monomer allowed for construction of poly(ethylene glycol vinyl ether phosphotriester) (PEVEP). This vinyl ether-functionalized PPE scaffold was coupled with hydroxyl- or thiol-containing model small molecules via three different types of conjugation chemistries—thiol–ene “click” reaction, acetalization, or thio-acetalization reaction—to afford modified polymers that accommodated either stable thio–ether or hydrolytically labile acetal or thio–acetal linkages. Amphiphilic diblock copolymers of poly(ethylene glycol) and PEVEP formed well-defined micelles with a narrow and monomodal size distribution in water, as confirmed by dynamic light scattering (DLS), transmission electron microscopy, and atomic force microscopy. The stability of the micelles and the hydrolytic degradability of the backbone and side chains of the PEVEP block segment were assessed by DLS and nuclear magnetic resonance spectroscopy (1H and 31P), respectively, in aqueous buffer solutions at pH values of 5.0 and 7.4 and at temperatures of 25 and 37 °C. The hydrolytic degradation products of the PEVEP segments of the block copolymers were then identified by electrospray ionization, gas chromatography, and matrix-assisted laser desorption/ionization mass spectrometry. The parent micelles and their degradation products were found to be non-cytotoxic at concentrations up to 3 mg/mL, when evaluated with RAW 264.7 mouse macrophages and OVCAR-3 human ovarian adenocarcinoma cells.
Co-reporter:Amandine Noel, Yannick P. Borguet, Jeffery E. Raymond, and Karen L. Wooley
Macromolecules 2014 Volume 47(Issue 20) pp:7109-7117
Publication Date(Web):October 14, 2014
DOI:10.1021/ma5015534
The photophysical and mechanical properties of novel poly(carbonate-amide)s derived from two biorenewable resources, ferulic acid (FA) and l-tyrosine ethyl ester, were evaluated in detail. From these two bio-based precursors, a series of four monomers were generated (having amide and/or carbonate coupling units with remaining functionalities to allow for carbonate formation) and transformed to a series of four poly(carbonate-amide)s. The simplest monomer, which was biphenolic and was obtained in a single amidation synthetic step, displayed bright, visible fluorescence that was twice brighter than FA. Multidimensional fluorescence spectroscopy of the polymers in solution highlighted the strong influence that regioselectivity and the degree of polymerization have on their photophysical properties. The regiochemistry of the system had little effect on the wettability, surface free energy, and Young’s modulus (ca. 2.5 GPa) in the solid state. Confocal imaging of solvent-cast films of each polymer revealed microscopically flat surfaces with fluorescent emission deep into the visible region. Fortuitously, one of the two regiorandom polymers (obtainable from the biphenolic monomer in only an overall two synthetic steps from FA and l-tyrosine ethyl ester) displayed the most promising fluorescent properties both in the solid state and in solution, allowing for the possibility of translating this system as a self-reporting or imaging agent in future applications. To further evaluate the potential of this polymer as a biodegradable material, hydrolytic degradation studies at different pH values and temperatures were investigated. Additionally, the antioxidant properties of the degradation products of this polymer were compared with its biphenolic monomer and FA.
Co-reporter:Amandine Noel, Yannick P. Borguet, Jeffery E. Raymond, and Karen L. Wooley
Macromolecules 2014 Volume 47(Issue 9) pp:2974-2983
Publication Date(Web):April 16, 2014
DOI:10.1021/ma500454f
Ferulic acid (FA), a bio-based resource found in fruits and vegetables, was coupled with a hydroxyl-amino acid to generate a new class of monomers to afford poly(carbonate–amide)s with potential to degrade into natural products. l-Serine was first selected as the hydroxyl-amino partner for FA, from which the activated p-nitrophenyl carbonate monomer was synthesized. Unfortunately, polymerizations were unsuccessful, and the elimination product was systematically obtained. To avoid elimination, we revised our strategy and used l-tyrosine ethyl ester, which lacks an acidic proton on the α position of the ethyl ester. Four new monomers were synthesized and converted into the corresponding poly(carbonate–amide)s with specific regioselectivities. The polymers were fully characterized through thermal and spectroscopic analyses. Preliminary fluorescent studies revealed interesting photophysical properties for the monomers and their corresponding poly(carbonate–amide)s, beyond the fluorescence characteristics of l-tyrosine and FA, making these materials potentially viable for sensing and/or imaging applications, in addition to their attractiveness as engineering materials derived from renewable resources.
Co-reporter:Tiffany P. Gustafson, Young H. Lim, Jeniree A. Flores, Gyu Seong Heo, Fuwu Zhang, Shiyi Zhang, Sandani Samarajeewa, Jeffery E. Raymond, and Karen L. Wooley
Langmuir 2014 Volume 30(Issue 2) pp:631-641
Publication Date(Web):2017-2-22
DOI:10.1021/la403943w
The successful development of degradable polymeric nanostructures as optical probes for use in nanotheranostic applications requires the intelligent design of materials such that their surface response, degradation, drug delivery, and imaging properties are all optimized. In the case of imaging, optimization must result in materials that allow differentiation between unbound optical contrast agents and labeled polymeric materials as they undergo degradation. In this study, we have shown that use of traditional electrophoretic gel-plate assays for the determination of the purity of dye-conjugated degradable nanoparticles is limited by polymer degradation characteristics. To overcome these limitations, we have outlined a holistic approach to evaluating dye and peptide–polymer nanoparticle conjugation by utilizing steady-state fluorescence, anisotropy, and emission and anisotropy lifetime decay profiles, through which nanoparticle–dye binding can be assessed independently of perturbations, such as those presented during the execution of electrolyte gel-based assays. This approach has been demonstrated to provide an overall understanding of the spectral signature–structure–function relationship, ascertaining key information on interactions between the fluorophore, polymer, and solvent components that have a direct and measurable impact on the emissive properties of the optical probe. The use of these powerful techniques provides feedback that can be utilized to improve nanotheranostics by evaluating dye emissivity in degradable nanotheranostic systems, which has become increasingly important as modern platforms transition to architectures intentionally reliant on degradation and built-in environmental responses.
Co-reporter:Mahmoud Elsabahy and Karen L. Wooley
Chemical Society Reviews 2013 vol. 42(Issue 12) pp:5552-5576
Publication Date(Web):03 Apr 2013
DOI:10.1039/C3CS60064E
Nanoscale objects, whether of biologic origin or synthetically created, are being developed into devices for a variety of bionanotechnology diagnostic and pharmaceutical applications. However, the potential immunotoxicity of these nanomaterials and mechanisms by which they may induce adverse reactions have not received sufficient attention. Nanomaterials, depending on their characteristics and compositions, can interact with the immune system in several ways and either enhance or suppress immune system function. Cytokines perform pleiotropic functions to mediate and regulate the immune response and are generally recognized as biomarkers of immunotoxicity. While the specificity and validity of certain cytokines as markers of adverse immune response has been established for chemicals, small and macromolecular drugs, research on their applicability for predicting and monitoring the immunotoxicity of engineered nanomaterials is still ongoing. The goal of this review is to provide guidelines as to important cytokines that can be utilized for evaluating the immunotoxicity of nanomaterials and to highlight the role of those cytokines in mediating adverse reactions, which is of particular importance for the clinical development of nanopharmaceuticals and other nanotechnology-based products. Importantly, the rational design of nanomaterials of low immunotoxicity will be discussed, focusing on synthetic nanodevices, with emphasis on both the nanoparticle-forming materials and the embedded cargoes.
Co-reporter:Yuefei Shen;Shiyi Zhang;Fuwu Zhang;Alexer Loftis;Adriana Pavía-Sers;Jiong Zou;Jingwei Fan;John-Stephen A. Taylor
Advanced Materials 2013 Volume 25( Issue 39) pp:5609-5614
Publication Date(Web):
DOI:10.1002/adma.201302842
Co-reporter:Mahmoud Elsabahy, Ritu Shrestha, Corrie Clark, Sara Taylor, Jeffrey Leonard, and Karen L. Wooley
Nano Letters 2013 Volume 13(Issue 5) pp:2172-2181
Publication Date(Web):April 10, 2013
DOI:10.1021/nl4006645
Development of multifunctional nanostructures that can be tuned to codeliver multiple drugs and diagnostic agents to diseased tissues is of great importance. Hierarchically assembled theranostic (HAT) nanostructures based on anionic cylindrical shell cross-linked nanoparticles and cationic shell cross-linked knedel-like nanoparticles (cSCKs) have recently been developed by our group to deliver siRNA intracellularly and to undergo radiolabeling. In the current study, paclitaxel, a hydrophobic anticancer drug, and siRNA have been successfully loaded into the cylindrical and spherical components of the hierarchical assemblies, respectively. Cytotoxicity, immunotoxicity, and intracellular delivery mechanism of the HAT nanostructures and their individual components have been investigated. Decoration of nanoparticles with F3-tumor homing peptide was shown to enhance the selective cellular uptake of the spherical particles, whereas the HAT nanoassemblies underwent an interesting disassembly process in contact with either OVCAR-3 or RAW 264.7 cell lines. The HAT nanostructures were found to “stick” to the cell membrane and “trigger” the release of spherical cSCKs templated onto their surfaces intracellularly, while retaining the cylindrical part on the cell surface. Combination of paclitaxel and cell-death siRNA (siRNA that induces cell death) into the HAT nanostructures resulted in greater reduction in cell viability than siRNA complexed with Lipofectamine and the assemblies loaded with the individual drugs. In addition, a shape-dependent immunotoxicity was observed for both spherical and cylindrical nanoparticles with the latter being highly immunotoxic. Supramolecular assembly of the two nanoparticles into the HAT nanostructures significantly reduced the immunotoxicity of both cSCKs and cylinders. HAT nanostructures decorated with targeting moieties, loaded with nucleic acids, hydrophobic drugs, radiolabels, and fluorophores, with control over their toxicity, immunotoxicity, and intracellular delivery might have great potential for biomedical delivery applications.
Co-reporter:Koichiro Mikami ; Alexander T. Lonnecker ; Tiffany P. Gustafson ; Nathanael F. Zinnel ; Pei-Jing Pai ; David H. Russell
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6826-6829
Publication Date(Web):April 30, 2013
DOI:10.1021/ja402319m
An organocatalyzed ring-opening polymerization methodology was developed for the preparation of polycarbonates derived from glucose as a natural product starting material. The cyclic 4,6-carbonate monomer of glucose having the 1, 2, and 3 positions methyl-protected was prepared in three steps from a commercially available glucose derivative, and the structure was confirmed by means of NMR and IR spectroscopies, electrospray ionization mass spectrometry (MS), and single-crystal X-ray analysis. Polymerization of the monomer, initiated by 4-methylbenzyl alcohol in the presence of 1,5,7-triazabicyclo[4.4.0]dec-5-ene as the organocatalyst, proceeded effectively in a controlled fashion to afford the polycarbonate with a tunable degree of polymerization, narrow molecular weight distribution, and well-defined end groups, as confirmed by a combination of NMR spectroscopy, gel-permeation chromatography, and MALDI-TOF MS. A distribution of head-to-head, head-to-tail, and tail-to-tail regiochemistries was determined by NMR spectroscopy and tandem MS analysis by electron transfer dissociation. These polycarbonates are of interest as engineering materials because of their origination from renewable resources combined with their amorphous character and relatively high glass transition temperatures as determined by X-ray diffraction and differential scanning calorimetry studies.
Co-reporter:Jiahua Zhu;Shiyi Zhang;Fuwu Zhang;Darrin J. Pochan
Advanced Functional Materials 2013 Volume 23( Issue 14) pp:1767-1773
Publication Date(Web):
DOI:10.1002/adfm.201202323
Abstract
A challenging aim in both materials physics and chemistry is the construction of complex and functional superstructures from designed nanoscale building units. Block copolymer nanoparticles with morphological variety and compositional complexity have been made with solution-based assembly. However, routine ability to build hierarchical superstructures by inter-nanoparticle association is not yet possible. A hierarchical assembly strategy of organizing pre-formed spherical block copolymer nanoparticles into superstructures, including linear, circular, and close-packed arrays, via tunable interparticle interactions is presented. Solution-state mixtures are made of two amphiphilic diblock copolymers, poly(acrylic acid)-block-poly(methyl methacrylate) (PAA-b-PMMA) and poly(acrylic acid)-block-polybutadiene (PAA-b-PB) with additional crown ether functionalities grafted onto 40 mol% of the AA repeat units on the PAA-b-PMMA diblock copolymer. Through kinetic control of the solution assembly process in aqueous/N,N-dimethylformamide (DMF) mixtures (4:1 water:DMF), spherical nanoparticles with compositional complexity confined in both the core and shell are obtained. Benefiting from host-guest chemistry, interparticle association is triggered and tuned by the addition of di-functional organoamines due to amine-crown ether complexation. The resultant multiparticle superstructures contain well-defined multicompartments within individual, constituent nanoparticles due to the local separation of unlike PB and PMMA hydrophobic blocks within the cores of the individual particles. Through competitive complexation with potassium ions, the superstructures are disassembled into individual multicomparment nanoparticles.
Co-reporter:Shiyi Zhang, Jiong Zou, Mahmoud Elsabahy, Amolkumar Karwa, Ang Li, Dennis A. Moore, Richard B. Dorshow and Karen L. Wooley
Chemical Science 2013 vol. 4(Issue 5) pp:2122-2126
Publication Date(Web):06 Mar 2013
DOI:10.1039/C3SC50252J
A new type of degradable, nanoscopic polymer assembly containing ultra-high levels of drug loading via covalent attachment within amphiphilic core–shell nanoparticle morphology has been generated as a potentially effective and safe anti-cancer agent. Poly(ethylene oxide)-block-polyphosphoester-based paclitaxel drug conjugates (PEO-b-PPE-g-PTX) were synthesized by a rapid, scalable and versatile approach that involves only two steps: organocatalyst-promoted ring-opening-polymerization followed by click reaction-based conjugation of a PTX prodrug. Variations in the polymer-to-PTX stoichiometries allowed for optimization of the conjugation efficiency, the PTX drug loading and the resulting water solubilities of the entire polymer and the PTX content. The PEO-b-PPE-g-PTX formed well-defined micelles in aqueous solution, with a PTX loading capacity as high as 65 wt%, and a maximum PTX concentration of 6.2 mg mL−1 in water, which is 25000-fold higher than the aqueous solubility of free PTX. The positive cell-killing activity of PEO-b-PPE-g-PTX against several cancer cell lines is demonstrated, and the presence of pendant reactive functionality provides a powerful platform for future work to involve conjugation of multiple drugs and imaging agents to achieve chemotherapy and bioimaging.
Co-reporter:Fuwu Zhang, Mahmoud Elsabahy, Shiyi Zhang, Lily Yun Lin, Jiong Zou and Karen L. Wooley
Nanoscale 2013 vol. 5(Issue 8) pp:3220-3225
Publication Date(Web):11 Mar 2013
DOI:10.1039/C3NR34320K
Polymeric micelles and shell crosslinked knedel-like (SCK) nanoparticles were loaded with up to 48% (w/w) cisplatin. These spherical cisplatin-loaded nanoparticles displayed sustained platinum release over 5 days in PBS, enhanced stability over free cisplatin in aqueous milieu, and significant antitumor activity in vitro against two cancer cell lines.
Co-reporter:Mahmoud Elsabahy, Sandani Samarajeewa, Jeffery E. Raymond, Corrie Clark and Karen L. Wooley
Journal of Materials Chemistry A 2013 vol. 1(Issue 39) pp:5241-5255
Publication Date(Web):18 Jun 2013
DOI:10.1039/C3TB20668H
The development of stable nanoparticles that can withstand the changing conditions experienced in a biological setting and also be of low toxicity and immunogenicity is of particular importance to address the problems associated with currently utilized nanotechnology-based therapeutics and diagnostics. The use of crosslinked nanoparticles continues to receive special impetus, due to their robust structure and high kinetic stability, and they have recently been shown to induce lower cytotoxicity than their non-crosslinked micellar counterparts. In the current study, poly(acrylamidoethylamine)-block-poly(DL-lactide) (PAEA90-b-PDLLA40) copolymers were synthesized, self-assembled in water to yield nanoscopic polymeric micelles, and the effects of decorating the micellar surface with poly(ethylene glycol) (i.e. PEGylation) and crosslinking the PAEA layer to varying extents on the physicochemical characteristics, cytotoxicity and immunotoxicity of the nanoparticles were studied. Herein, we report for the first time that crosslinking can efficiently reduce the immunotoxicity of polymeric nanomaterials. In addition, increasing the degree of crosslinking further reduced the accessibility of biomolecules to the core of the nanoparticles and decreased their cytotoxicity and immunotoxicity. It is also highlighted that crosslinking can be more efficient than PEGylation in reducing the immunotoxicity of nanomaterials. Shell-crosslinking of block copolymer micelles, therefore, is expected to advance their clinical development beyond the earlier known effects, and to broaden the implications in the field of nanomedicine.
Co-reporter:Young H. Lim, Gyu Seong Heo, Sangho Cho, and Karen L. Wooley
ACS Macro Letters 2013 Volume 2(Issue 9) pp:785
Publication Date(Web):August 19, 2013
DOI:10.1021/mz400229m
The development of a diblock copolymer, polyphosphoester-block-poly(l-lactide), which has potential for being fully degradable and biocompatible, was achieved by one-pot sequential ring-opening polymerizations (ROPs) of two cyclic monomers: alkyne-functionalized phospholane and l-lactide (LLA). A kinetic study of the polymerization in each step was investigated in a detailed manner by nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC), revealing living/controlled characteristics with narrow molecular weight distributions and a linear increase of molecular weights vs monomer conversion and time. Subsequently, photoinduced thiol-yne “click” reactions with small-molecule thiols bearing either carboxylic acid or amino groups afforded amphiphilic diblock copolymers with carboxylate or amino side-chain functionalities along the polyphosphoester segment of the diblock copolymer backbone. Finally, direct dissolution of the two different types of amphiphilic diblock copolymers in aqueous solutions yielded well-defined spherical micelles with corresponding negative or positive surface charges, respectively, as confirmed by transmission electron microscopy (TEM), dynamic light scattering (DLS), and zeta potential analyses.
Co-reporter:Sandani Samarajeewa, Ritu Shrestha, Mahmoud Elsabahy, Amolkumar Karwa, Ang Li, Ryan P. Zentay, James G. Kostelc, Richard B. Dorshow, and Karen L. Wooley
Molecular Pharmaceutics 2013 Volume 10(Issue 3) pp:1092-1099
Publication Date(Web):February 19, 2013
DOI:10.1021/mp3005897
Paclitaxel-loaded shell cross-linked polymeric nanoparticles having an enzymatically and hydrolytically degradable poly(lactic acid) core and a glutathione-responsive disulfide cross-linked poly(oligoethylene glycol)-containing corona were constructed in aqueous solution and investigated for their stimuli-responsive release of the embedded therapeutics and in vitro cytotoxicity. Paclitaxel release from the nanoparticles in PBS buffer was accelerated in the presence of glutathione at both pH 5.5 and pH 7.4, reaching ca. 65% cumulative drug release after 8 d, whereas only ca. 50% and 35% extents of release were observed in the absence of glutathione at pH 5.5 and pH 7.4, respectively. Enzyme-catalyzed hydrolysis of the nanoparticle core resulted in the degradation of ca. 30% of the poly(lactic acid) core to lactic acid within 12 h, with coincidently triggered paclitaxel release of ca. 37%, as opposed to only ca. 17% release from the uncatalyzed nanoparticles at pH 7.4. While empty nanoparticles did not show any inherent cytotoxicity at the highest tested concentrations, paclitaxel-loaded nanoparticles showed IC50 values that were similar to those of free paclitaxel at 72 h incubation with KB cells and were more efficacious at ca. 3-fold lower IC50 value (0.031 μM vs 0.085 μM) at 2 h of incubation. Against human ovarian adenocarcinoma cells, the paclitaxel-loaded nanoparticles exhibited a remarkable ca. 11-fold lower IC50 than a Taxol-mimicking formulation (0.0007 μM vs 0.008 μM) at 72 h of incubation. These tunable dual-responsive degradable nanoparticles show great promise for delivery of paclitaxel to tumor tissues, given their superior in vitro efficacies compared to that of free paclitaxel and Taxol-mimicking formulations.Keywords: cell viability; degradable; disulfide cross-linker; paclitaxel; poly(dl-lactic acid); poly(dl-lactide); polymeric nanoparticles;
Co-reporter:Parth N. Shah, Lily Yun Lin, Justin A. Smolen, Jasur A. Tagaev, Sean P. Gunsten, Daniel S. Han, Gyu Seong Heo, Yali Li, Fuwu Zhang, Shiyi Zhang, Brian D. Wright, Matthew J. Panzner, Wiley J. Youngs, Steven L. Brody, Karen L. Wooley, and Carolyn L. Cannon
ACS Nano 2013 Volume 7(Issue 6) pp:4977
Publication Date(Web):May 29, 2013
DOI:10.1021/nn400322f
The use of nebulizable, nanoparticle-based antimicrobial delivery systems can improve efficacy and reduce toxicity for treatment of multi-drug-resistant bacteria in the chronically infected lungs of cystic fibrosis patients. Nanoparticle vehicles are particularly useful for applying broad-spectrum silver-based antimicrobials, for instance, to improve the residence time of small-molecule silver carbene complexes (SCCs) within the lung. Therefore, we have synthesized multifunctional, shell cross-linked knedel-like polymeric nanoparticles (SCK NPs) and capitalized on the ability to independently load the shell and core with silver-based antimicrobial agents. We formulated three silver-loaded variants of SCK NPs: shell-loaded with silver cations, core-loaded with SCC10, and combined loading of shell silver cations and core SCC10. All three formulations provided a sustained delivery of silver over the course of at least 2–4 days. The two SCK NP formulations with SCC10 loaded in the core each exhibited excellent antimicrobial activity and efficacy in vivo in a mouse model of Pseudomonas aeruginosa pneumonia. SCK NPs with shell silver cation-load only, while efficacious in vitro, failed to demonstrate efficacy in vivo. However, a single dose of core SCC10-loaded SCK NPs (0.74 ± 0.16 mg Ag) provided a 28% survival advantage over sham treatment, and administration of two doses (0.88 mg Ag) improved survival to 60%. In contrast, a total of 14.5 mg of Ag+ delivered over 5 doses at 12 h intervals was necessary to achieve a 60% survival advantage with a free-drug (SCC1) formulation. Thus, SCK NPs show promise for clinical impact by greatly reducing antimicrobial dosage and dosing frequency, which could minimize toxicity and improve patient adherence.Keywords: cystic fibrosis; multi-drug-resistant bacteria; nebulizable nanoparticles; Pseudomonas aeruginosa pneumonia; shell cross-linked knedel-like polymeric nanoparticles; silver carbene complexes
Co-reporter:Adriana Pavía-Sanders, Shiyi Zhang, Jeniree A. Flores, Jonathan E. Sanders, Jeffery E. Raymond, and Karen L. Wooley
ACS Nano 2013 Volume 7(Issue 9) pp:7552
Publication Date(Web):August 29, 2013
DOI:10.1021/nn401541e
Well-defined, magnetic shell cross-linked knedel-like nanoparticles (MSCKs) with hydrodynamic diameters ca. 70 nm were constructed through the co-assembly of amphiphilic block copolymers of PAA20-b-PS280 and oleic acid-stabilized magnetic iron oxide nanoparticles using tetrahydrofuran, N,N-dimethylformamide, and water, ultimately transitioning to a fully aqueous system. These hybrid nanomaterials were designed for application as sequestering agents for hydrocarbons present in crude oil, based upon their combination of amphiphilic organic domains, for aqueous solution dispersibility and capture of hydrophobic guest molecules, with inorganic core particles for magnetic responsivity. The employment of these MSCKs in a contaminated aqueous environment resulted in the successful removal of the hydrophobic contaminants at a ratio of 10 mg of oil per 1 mg of MSCK. Once loaded, the crude oil-sorbed nanoparticles were easily isolated via the introduction of an external magnetic field. The recovery and reusability of these MSCKs were also investigated. These results suggest that deployment of hybrid nanocomposites, such as these, could aid in environmental remediation efforts, including at oil spill sites, in particular, following the bulk recovery phase.Keywords: hybrid organic−inorganic nanoparticles; magnetic nanoparticles; oil recovery
Co-reporter:Shiyi Zhang, Hai Wang, Yuefei Shen, Fuwu Zhang, Kellie Seetho, Jiong Zou, John-Stephen A. Taylor, Andrew P. Dove, and Karen L. Wooley
Macromolecules 2013 Volume 46(Issue 13) pp:5141-5149
Publication Date(Web):June 27, 2013
DOI:10.1021/ma400675m
The direct synthesis of an acid-labile polyphosphoramidate by organobase-catalyzed ring-opening polymerization and an overall two-step preparation of polyphosphodiester ionomers (PPEI) by acid-assisted cleavage of the phosphoramidate bonds along the backbone of the polyphosphoramidate were developed in this study. The ultrafast organobase-catalyzed ring-opening polymerization of a cyclic phospholane methoxyethyl amidate monomer initiated by benzyl alcohol allowed for the preparation of well-defined polyphosphoramidates (PPA) with predictable molecular weights, narrow molecular weight distributions (PDI < 1.10), and well-defined chain ends. Cleavage of the acid-labile phosphoramidate bonds on the polyphosphoramidate repeat units was evaluated under acidic conditions over a pH range of 1–5, and the complete hydrolysis produced polyphosphodiesters. The thermal properties of the resulting polyphosphoester ionomer acid and polyphosphoester ionomer sodium salt exhibited significant thermal stability. The parent PPA and both forms of the PPEIs showed low cytotoxicities toward HeLa cells and RAW 264.7 mouse macrophage cells. The synthetic methodology developed here has enriched the family of water-soluble polymers prepared by rapid and convenient organobase-catalyzed ring-opening polymerizations and straightforward chemical medication reactions, which are designed to be hydrolytically degradable and have promise for numerous biomedical and other applications.
Co-reporter:Tiffany P. Gustafson, Alexander T. Lonnecker, Gyu Seong Heo, Shiyi Zhang, Andrew P. Dove, and Karen L. Wooley
Biomacromolecules 2013 Volume 14(Issue 9) pp:
Publication Date(Web):August 19, 2013
DOI:10.1021/bm4010832
A natural product-based polymer platform, having the characteristics of being derived from renewable materials and capable of breaking down, ultimately, into natural byproducts, has been prepared through the ring-opening polymerization (ROP) of a glucose-based bicyclic carbonate monomer. ROP was carried out via chain extension of a polyphosphoester (PPE) macroinitiator in the presence of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) organocatalyst to afford the PPE-b-poly(d-glucose carbonate) (PDGC) block copolymer. This new copolymer represents a functional architecture that can be rapidly transformed through thiol-yne reactions along the PPE segment into a diverse variety of amphiphilic polymers, which interestingly display stimuli-sensitive phase behavior in the form of a lower critical solution temperature (LCST). Below the LCST, they undergo self-assembly to form spherical core–shell nanostructures that display a poorly defined core–shell morphology. It is expected that hydrophobic patches are exposed within the micellar corona, reminiscent of the surface complexity of proteins, making these materials of interest for triggered and reversible assembly disassembly processes.
Co-reporter:Sandani Samarajeewa, Aida Ibricevic, Sean P. Gunsten, Ritu Shrestha, Mahmoud Elsabahy, Steven L. Brody, and Karen L. Wooley
Biomacromolecules 2013 Volume 14(Issue 4) pp:
Publication Date(Web):March 19, 2013
DOI:10.1021/bm3018774
In this work, degradable cationic shell cross-linked knedel-like (deg-cSCK) nanoparticles were developed as an alternative platform to replace similar nondegradable cSCK nanoparticles that have been utilized for nucleic acids delivery. An amphiphilic diblock copolymer poly(acrylamidoethylamine)90-block-poly(dl-lactide)40 (PAEA90-b-PDLLA40) was synthesized, self-assembled in aqueous solution, and shell cross-linked using a hydrolyzable cross-linker to afford deg-cSCKs with an average core diameter of 45 ± 7 nm. These nanoparticles were fluorescently labeled for in vitro tracking. The enzymatic- and hydrolytic-degradability, siRNA binding affinity, cell uptake and cytotoxicity of the deg-cSCKs were evaluated. Esterase-catalyzed hydrolysis of the nanoparticles resulted in the degradation of ca. 24% of the PDLLA core into lactic acid within 5 d, as opposed to only ca. 9% degradation from aqueous solutions of the deg-cSCK nanoparticles in the absence of enzyme. Cellular uptake of deg-cSCKs was efficient, while exhibiting low cytotoxicity with LD50 values of ca. 90 and 30 μg/mL in RAW 264.7 mouse macrophages and MLE 12 cell lines, respectively, ca. 5- to 6-fold lower than the cytotoxicity observed for nondegradable cSCK analogs. Additionally, deg-cSCKs were able to complex siRNA at an N/P ratio as low as 2, and were efficiently able to facilitate cellular uptake of the complexed nucleic acids.
Co-reporter:Jiong Zou, Jingwei Fan, Xun He, Shiyi Zhang, Hai Wang, and Karen L. Wooley
Macromolecules 2013 Volume 46(Issue 10) pp:4223-4226
Publication Date(Web):May 2, 2013
DOI:10.1021/ma4007939
Co-reporter:Mahmoud Elsabahy and Karen L. Wooley
Chemical Society Reviews 2012 vol. 41(Issue 7) pp:2545-2561
Publication Date(Web):14 Feb 2012
DOI:10.1039/C2CS15327K
Polymeric nanoparticles-based therapeutics show great promise in the treatment of a wide range of diseases, due to the flexibility in which their structures can be modified, with intricate definition over their compositions, structures and properties. Advances in polymerization chemistries and the application of reactive, efficient and orthogonal chemical modification reactions have enabled the engineering of multifunctional polymeric nanoparticles with precise control over the architectures of the individual polymer components, to direct their assembly and subsequent transformations into nanoparticles of selective overall shapes, sizes, internal morphologies, external surface charges and functionalizations. In addition, incorporation of certain functionalities can modulate the responsiveness of these nanostructures to specific stimuli through the use of remote activation. Furthermore, they can be equipped with smart components to allow their delivery beyond certain biological barriers, such as skin, mucus, blood, extracellular matrix, cellular and subcellular organelles. This tutorial review highlights the importance of well-defined chemistries, with detailed ties to specific biological hurdles and opportunities, in the design of nanostructures for various biomedical delivery applications.
Co-reporter:Shiyi Zhang ; Jiong Zou ; Fuwu Zhang ; Mahmoud Elsabahy ; Simcha E. Felder ; Jiahua Zhu ; Darrin J. Pochan
Journal of the American Chemical Society 2012 Volume 134(Issue 44) pp:18467-18474
Publication Date(Web):October 23, 2012
DOI:10.1021/ja309037m
A rapid and efficient approach for the preparation and modification of a versatile class of functional polymer nanoparticles has been developed, for which the entire engineering process from small molecules to polymers to nanoparticles bypasses typical slow and inefficient procedures and rather employs a series of steps that capture fully the “click” chemistry concepts that have greatly facilitated the preparation of complex polymer materials over the past decade. The construction of various nanoparticles with functional complexity from a versatile platform is a challenging aim to provide materials for fundamental studies and also optimization toward a diverse range of applications. In this paper, we demonstrate the rapid and facile preparation of a family of nanoparticles with different surface charges and functionalities based on a biodegradable polyphosphoester block copolymer system. From a retrosynthetic point of view, the nonionic, anionic, cationic, and zwitterionic micelles with hydrodynamic diameters between 13 and 21 nm and great size uniformity were quickly formed by suspending, independently, four amphiphilic diblock polyphosphoesters into water, which were functionalized from the same parental hydrophobic-functional AB diblock polyphosphoester by click-type thiol–yne reactions. The well-defined (PDI < 1.2) hydrophobic-functional AB diblock polyphosphoester was synthesized by an ultrafast (<5 min) organocatalyzed ring-opening polymerization in a two-step, one-pot manner with the quantitative conversions of two kinds of cyclic phospholane monomers. The whole programmable process starting from small molecules to nanoparticles could be completed within 6 h, as the most rapid approach for the anionic and nonionic nanoparticles, although the cationic and zwitterionic nanoparticles required ca. 2 days due to purification by dialysis. The micelles showed high biocompatibility, with even the cationic micelles exhibiting a 6-fold lower cytotoxicity toward RAW 264.7 mouse macrophage cells, as compared to the commercial transfection agent Lipofectamine.
Co-reporter:Ritu Shrestha ; Mahmoud Elsabahy ; Hannah Luehmann ; Sandani Samarajeewa ; Stephanie Florez-Malaver ; Nam S. Lee ; Michael J. Welch ; Yongjian Liu
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17362-17365
Publication Date(Web):October 10, 2012
DOI:10.1021/ja306616n
Dual functional hierarchically assembled nanostructures, with two unique functions of carrying therapeutic cargo electrostatically and maintaining radiolabeled imaging agents covalently within separate component building blocks, have been developed via the supramolecular assembly of several spherical cationic shell cross-linked nanoparticles clustered around a central anionic shell cross-linked cylinder. The shells of the cationic nanoparticles and the hydrophobic core domain of the anionic central cylindrical nanostructure of the assemblies were utilized to complex negatively charged nucleic acids (siRNA) and to undergo radiolabeling, respectively, for potential theranostic applications. The assemblies exhibited exceptional cell transfection and radiolabeling efficiencies, providing an overall advantage over the individual components, which could each facilitate only one or the other of the functions.
Co-reporter:Lily Yun Lin ; Kristin M. Tiemann ; Yali Li ; Jerome S. Pinkner ; Jennifer N. Walker ; Scott J. Hultgren ; David A. Hunstad
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:3938-3941
Publication Date(Web):February 23, 2012
DOI:10.1021/ja2091917
Amphiphilic block copolymer nanoparticles are conjugated with uropathogenic Escherichia coli type 1 pilus adhesin FimHA through amidation chemistry to enable bladder epithelial cell binding and internalization of the nanoparticles in vitro.
Co-reporter:Philip M. Imbesi, Jeffery E. Raymond, Bryan S. Tucker and Karen L. Wooley
Journal of Materials Chemistry A 2012 vol. 22(Issue 37) pp:19462-19473
Publication Date(Web):01 Jun 2012
DOI:10.1039/C2JM32005C
The synthesis of heterogeneous, amphiphilic crosslinked networks from photo-initiated thiol-ene chemistry and full characterization of the physicochemical, anti-biofouling, mechanical, and thermal properties of this system are reported. Although interest in coatings that present heterogeneous surface features is increasing, anti-biofouling performance is typically compared to homogeneous biocidal- or non-toxic polydimethylsiloxane-based paints, for the advancement of commercial and novel systems, which leaves a need for a benchmark to compare highly complex, heterogeneous composites with similar complexities. This system has been generated and rigorously analyzed to understand how microscopic and nanoscopic disorder, resistance to protein adsorption and surface mechanical properties can be fine-tuned and optimized through oligomer selection, blend ratios and process conditions. Solution-state studies of individual and blended constituents probed the relative fluorescence intensities based on concentration and neighboring species that were used to identify microscopic disorder on the surface. Incubation of a fluorescently labelled protein on the benchmark surface showed 42% and 72% less adsorption than on model surfaces that largely expressed a single component. The extent of reaction and the identification of unconsumed functionalities were found through infrared spectroscopy. The benchmark surface had a Young's modulus of approximately 0.5 GPa, 7 to 35 times higher than model surfaces, with 50 times the variation in modulus. Nanoscale surface adhesion force variation and bulk wettability and bulk thermal stability are also reported. This study provides an extensive list of metrics to be used for the development of complex, heterogeneous, anti-biofouling coatings, appropriate to both surface optimization and mechanical tunability, for implementation in real-world applications.
Co-reporter:Philip M. Imbesi, John A. Finlay, Nick Aldred, Michael J. Eller, Simcha E. Felder, Kevin A. Pollack, Alexander T. Lonnecker, Jeffery E. Raymond, Michael E. Mackay, Emile A. Schweikert, Anthony S. Clare, James A. Callow, Maureen E. Callow and Karen L. Wooley
Polymer Chemistry 2012 vol. 3(Issue 11) pp:3121-3131
Publication Date(Web):31 May 2012
DOI:10.1039/C2PY20317K
A two-dimensional array of amphiphilic crosslinked networks was prepared by systematic alteration of both the composition of hyperbranched fluoropolymers (HBFPs) and the relative stoichiometries upon crosslinking with poly(ethylene glycol) (PEG). Results of physicochemical, mechanical, surface and biofouling assessment are described in full. The materials were designed to present complex surface topographies, morphologies, and chemical features over nano- and microscopic dimensions to explicitly inhibit microorganism settlement and adhesion. A multi-dimensional, tunable matrix was generated to understand and optimize the composition–structure–property relationships. The thermal properties of the crosslinked networks were analyzed by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), where the onset of thermal degradation, overall thermal stability and phase transition temperatures could be controlled based on the formulation. Investigation of the mechanical properties of the coatings in the water-swollen state found that the Young's modulus, ranging between 10.0 and 121 MPa, was dependent on both the wt% of PEG crosslinker and chemical composition of the HBFP. This result, on average, gives Young's moduli an order of magnitude larger than that previously reported for HBFP–PEG networks. Use of atomic force microscopy (AFM) provided insight into the nanoscale topography of the networks. Interestingly, it was observed that for all networks, surface roughness increased with water swelling going from an average of 115 ± 49 nm (dry) to 214 ± 106 nm (swelled) RMS roughness. Probing the surface chemistry by optical tensiometry revealed an increase in the static water contact angle by as much as 40° after water swelling. These two findings display a counter-intuitive increase of the surface hydrophobicity. Secondary ion mass spectrometry (SIMS) confirmed migration of hydrophobic fluorocarbon units to the surface by quantifying a 24% increase in fluorine species ejected from a dry versus water-swollen surface. Selected formulations of HBFP–PEG that demonstrated complex surface features and an overall high mechanical strength were tested in biological assays and all surfaces (3 formulations × 12 replicates) completely resisted the settlement of barnacle cyprids (Balanus amphitrite). Diatoms (Navicula incerta) were two- to three-times more easily removed from the HBFP–PEG surfaces compared to a homogeneous polydimethylsiloxane elastomer (PDMSe) standard surface. In contrast, algal spores (Ulva linza) were able to colonize the surfaces and were more difficult to remove in comparison to the PDMSe standard, pointing to the challenges associated with the development of a single material that is capable of broad anti-biofouling performance.
Co-reporter:Jiong Zou, Shiyi Zhang, Ritu Shrestha, Kellie Seetho, Carrie L. Donley and Karen L. Wooley
Polymer Chemistry 2012 vol. 3(Issue 11) pp:3146-3156
Publication Date(Web):28 Jun 2012
DOI:10.1039/C2PY20324C
Functionally-responsive amphiphilic core–shell nanoscopic objects, capable of either complete or partial inversion processes, were produced by the supramolecular assembly of pH-responsive block copolymers, without or with covalent crosslinking of the shell layer, respectively. A new type of well-defined, dual-functionalized boronic acid- and amino-based diblock copolymer poly(3-acrylamidophenylboronic acid)30-block-poly(acrylamidoethylamine)25 (PAPBA30-b-PAEA25) was synthesized by sequential reversible addition–fragmentation chain transfer (RAFT) polymerization and then assembled into cationic micelles in aqueous solution at pH 5.5. The micelles were further cross-linked throughout the shell domain comprised of poly(acrylamidoethylamine) by reaction with a bis-activated ester of 4,15-dioxo-8,11-dioxa-5,14-diazaoctadecane-1,18-dioic acid, upon increase of the pH to 7, to different cross-linking densities (2%, 5% and 10%), forming well-defined shell cross-linked nanoparticles (SCKs) with hydrodynamic diameters of ca. 50 nm. These smart micelles and SCKs presented switchable cationic, zwitterionic and anionic properties, and existed as stable nanoparticles with high positive surface charge at low pH (pH = 2, zeta potential ∼ +40 mV) and strong negative surface charge at high pH (pH = 12, zeta potential ∼ −35 mV). 1H NMR spectroscopy, X-ray photoelectron spectroscopy (XPS), dynamic light scattering (DLS), transmission electron microscopy (TEM), atomic force microscopy (AFM), and zeta potential, were used to characterize the chemical compositions, particle sizes, morphologies and surface charges. Precipitation occurred near the isoelectric points (IEP) of the polymer/particle solutions, and the IEP values could be tuned by changing the shell cross-linking density. The block copolymer micelles were capable of full reversible morphological inversion as a function of pH, by orthogonal protonation of the PAEA and hydroxide association with the PAPBA units, whereas the SCKs underwent only reptation of the PAPBA chain segments through the crosslinked shell of PAEA as the pH was elevated. Further, these nanomaterials also showed D-glucose-responsive properties.
Co-reporter:Shiyi Zhang, Ang Li, Jiong Zou, Lily Yun Lin, and Karen L. Wooley
ACS Macro Letters 2012 Volume 1(Issue 2) pp:328
Publication Date(Web):February 3, 2012
DOI:10.1021/mz200226m
“Click” chemistry is a library of efficient and reliable reactions, which have been used to functionalize various classes of bio- and synthetic macromolecular systems for the incorporation of designed properties and functions. In this report, azide–alkyne Huisgen cycloaddition and thiol-yne reactions, two classical “click” chemistries, were employed to functionalize biodegradable, clickable polyphosphoester homopolymers, and their water-soluble copolymers. A stable alkyne-functionalized phospholane monomer was synthesized, its organocatalyzed polymerization kinetics were evaluated, and the resulting (co)polymers were utilized to develop this facile method that provides the synthesis of clickable, water-soluble, and degradable polyphosphoesters, which can be adapted for various applications.
Co-reporter:Philip M. Imbesi, Christopher Fidge, Jeffery E. Raymond, Solène I. Cauët, and Karen L. Wooley
ACS Macro Letters 2012 Volume 1(Issue 4) pp:473
Publication Date(Web):March 20, 2012
DOI:10.1021/mz200137m
Diels–Alder (DA) chemistry was used in the construction of amphiphilic cross-linked polymer networks comprised of furan-functionalized hyperbranched fluoropolymers and maleimide-functionalized linear poly(ethylene glycol)s, which were designed as antibiofouling coatings capable of repair. Discrete molecules and a linear polymer analog were studied as model systems to understand the nature of the thermally reversible [4 + 2] cycloaddition reaction involving a tetrafluorobenzylfuranyl ether unit, which was part of the structure for the incorporation of the DA functionalities into the composite network materials. Atomic force microscopy confirmed the complex, nanoscopically resolved topography needed for antibiofouling. Bright field and fluorescence imaging monitored healing at damage sites as well as the ability of the coatings to resist protein adsorption.
Co-reporter:Ritu Shrestha, Yuefei Shen, Kevin A. Pollack, John-Stephen A. Taylor, and Karen L. Wooley
Bioconjugate Chemistry 2012 Volume 23(Issue 3) pp:574
Publication Date(Web):February 28, 2012
DOI:10.1021/bc200629f
In this work, multifunctional biosynthetic hybrid nanostructures were prepared and studied for their potential utility in the recognition and inhibition of mRNA sequences for inducible nitric oxide synthase (iNOS), which are overexpressed at sites of inflammation, such as in cases of acute lung injury. Shell cross-linked knedel-like polymer nanoparticles (SCKs) that present peptide nucleic acids, for binding to complementary mRNAs, and cell penetrating peptides (CPPs), to gain cell entry, along with fluorescent labels and sites for radiolabeling, were prepared by a series of robust, efficient, and versatile synthetic steps that proceeded from monomers to polymers to functional nanoparticles. Amphiphilic block graft copolymers having combinations of methoxy- and thioacetyl-terminated poly(ethylene glycol) (PEG) and DOTA-lysine units grafted from the backbone of poly(acrylic acid) (PAA) and extending with a backbone segment of poly(octadecyl acrylate-co-decyl acrylate) (P(ODA-co-DA)) were prepared by a combination of reversible addition–fragmentation chain transfer (RAFT) polymerization and chemical modification reactions, which were then used as the building blocks for the formation of well-defined SCKs decorated with reactive thiols accessible to the surface. Fluorescent labeling with Alexa Fluor 633 hydrazide was then accomplished by amidation with residual acrylic acid residues within the SCK shells. Finally, the PNAs and CPP units were covalently conjugated to the SCKs via Michael addition of thiols on the SCKs to maleimide units on the termini of PNAs and CPPs. Confirmation of the ability of the PNAs to bind selectively to the target iNOS mRNAs when tethered to the SCK nanoparticles was determined by in vitro competition experiments. When attached to the SCKs having a hydrodynamic diameter of 60 ± 16 nm, the Kd values of the PNAs were ca. an order of magnitude greater than the free PNAs, while the mismatched PNA showed no significant binding.
Co-reporter:Lily Yun Lin, Amolkumar Karwa, James G. Kostelc, Nam S. Lee, Richard B. Dorshow, and Karen L. Wooley
Molecular Pharmaceutics 2012 Volume 9(Issue 8) pp:2248-2255
Publication Date(Web):June 28, 2012
DOI:10.1021/mp3000887
Block copolymer nanoparticles having two different hydrodynamic diameters (120 nm vs 50 nm) and core diameters (60 nm vs 20 nm) with variable paclitaxel loading (5 to 20 wt % with respect to polymer weight, 4.4 μg/mL to 21.7 μg/mL paclitaxel concentrations in ultrapure water) were prepared for their in vitro cytotoxicity evaluation. Empty nanoparticles did not show any inherent cytotoxicity even at their highest concentration, whereas paclitaxel-loaded nanoparticles resulted in IC50 values that were better than free paclitaxel at 2 h (0.021 μM vs 0.046 μM) incubation periods, and approximately equal to free paclitaxel at 72 h (0.004 μM vs 0.003 μM) continuous incubation. Confocal fluorescence microscopy images demonstrated that the drug-loaded nanoparticles internalized into KB cells within 2 h and released their payload, resulting in cytotoxicity as evident from the fragmented nuclei present. Functionalization of the nanoparticle surfaces with poly(ethylene oxide) (2 kDa PEO, 5 PEO per block copolymer chain) did not affect the loading of paclitaxel or cell kill ability. No free paclitaxel was found in these nanoparticle formulations indicated by analytical assays.Keywords: cell internalization; cell viability; chemotherapy; paclitaxel; polymeric nanoparticles;
Co-reporter:Ritu Shrestha, Mahmoud Elsabahy, Stephanie Florez-Malaver, Sandani Samarajeewa, Karen L. Wooley
Biomaterials 2012 33(33) pp: 8557-8568
Publication Date(Web):
DOI:10.1016/j.biomaterials.2012.07.054
Co-reporter:Andreas M. Nyström and Karen L. Wooley
Accounts of Chemical Research 2011 Volume 44(Issue 10) pp:969
Publication Date(Web):June 15, 2011
DOI:10.1021/ar200097k
Nanomedicine is a rapidly evolving field, for which polymer building blocks are proving useful for the construction of sophisticated devices that provide enhanced diagnostic imaging and treatment of disease, known as theranostics. These well-defined nanoscopic objects have high loading capacities, can protect embedded therapeutic cargo, and offer control over the conditions and rates of release. Theranostics also offer external surface area for the conjugation of ligands to impart stealth characteristics and/or direct their interactions with biological receptors and provide a framework for conjugation of imaging agents to track delivery to diseased site(s). The nanoscopic dimensions allow for extensive biological circulation. The incorporation of such multiple functions is complicated, requiring exquisite chemical control during production and rigorous characterization studies to confirm the compositions, structures, properties, and performance.We are particularly interested in the study of nanoscopic objects designed for treatment of lung infections and acute lung injury, urinary tract infections, and cancer. This Account highlights our work over several years to tune the assembly of unique nanostructures. We provide examples of how the composition, structure, dimensions, and morphology of theranostic devices can tune their performance as drug delivery agents for the treatment of infectious diseases and cancer.The evolution of nanostructured materials from relatively simple overall shapes and internal morphologies to those of increasing complexity is driving the development of synthetic methodologies for the preparation of increasingly complex nanomedicine devices. Our nanomedicine devices are derived from macromolecules that have well-defined compositions, structures, and topologies, which provide a framework for their programmed assembly into nanostructures with controlled sizes, shapes, and morphologies. The inclusion of functional units within selective compartments/domains allows us to create (multi)functional materials. We employ combinations of controlled radical and ring-opening polymerizations, chemical transformations, and supramolecular assembly to construct such materials as functional entities. The use of multifunctional monomers with selective polymerization chemistries affords regiochemically functionalized polymers. Further supramolecular assembly processes in water with further chemical transformations provide discrete nanoscopic objects within aqueous solutions. This approach echoes processes in nature, whereby small molecules (amino acids, nucleic acids, saccharides) are linked into polymers (proteins, DNA/RNA, polysaccharides, respectively) and then those polymers fold into three-dimensional conformations that can lead to nanoscopic functional entities.
Co-reporter:Sandani Samarajeewa ; Ritu Shrestha ; Yali Li
Journal of the American Chemical Society 2011 Volume 134(Issue 2) pp:1235-1242
Publication Date(Web):December 13, 2011
DOI:10.1021/ja2095602
Comparative studies of bulk samples of hydrolytically degradable poly(lactic acid) (PLA) vs core–shell block copolymer micelles having PLA cores revealed remarkable acceleration in the proteinase K enzymatic hydrolysis of the nanoparticulate forms and demonstrated that even with amidation-based shell cross-linking the core domain remained accessible. Kinetic analyses by 1H NMR spectroscopy showed less than 20% lactic acid released from enzymatically catalyzed hydrolysis of poly(l-lactic acid) in bulk, whereas ca. 70% of the core degraded within 48 h for block copolymer micelles of poly(N-(acryloyloxy)succinimide-copolymer-N-acryloylmorpholine)-block-poly(L-lactic acid) (P(NAS-co-NAM)-b-PLLA), with only a slight reduction to ca. 50% for the shell cross-linked derivatives. Rigorous characterization measurements by NMR spectroscopy, fluorescence spectroscopy, dynamic light scattering, atomic force microscopy, and transmission electron microscopy were employed to confirm core excavation. These studies provide important fundamental understanding of the effects of nanoscopic dimensions on protein–polymer interactions and polymer degradability, which will guide the development of these degradable nanoconstructs to reach their potential for controlled release of therapeutics and biological clearance.
Co-reporter:Guorong Sun, Honggang Cui, Lily Yun Lin, Nam S. Lee, Chao Yang, William L. Neumann, John N. Freskos, Jeng J. Shieh, Richard B. Dorshow, and Karen L. Wooley
Journal of the American Chemical Society 2011 Volume 133(Issue 22) pp:8534-8543
Publication Date(Web):May 16, 2011
DOI:10.1021/ja200182t
Pyrazine-labeled multicompartment nanostructures are shown to exhibit enhanced pH-responsive blue-shifted fluorescence emission intensities compared to their simpler core–shell spherical analogs. An amphiphilic linear triblock terpolymer of ethylene oxide, N-acryloxysuccinimide, and styrene, PEO45-b-PNAS105-b-PS45, which lacks significant incompatibility for the hydrophobic block segments and undergoes gradual hydrolysis of the NAS units, underwent supramolecular assembly in mixtures of organic solvent and water to afford multicompartment micelles (MCMs) with a narrow size distribution. The assembly process was followed over time and found to evolve from individual polymer nanodroplets containing internally phase segregated domains, of increasing definition, and ultimately to dissociate into discrete micelles. Upon covalent cross-linking of the MCMs with pH-insensitive pyrazine-based diamino cross-linkers, pH-responsive, photonic multicompartment nanostructures (MCNs) were produced. These MCNs exhibited significant enhancement of overall structural stability, in comparison with the MCMs, and internal structural tunability through the cross-linking chemistry. Meanwhile, the complex compartmentalized morphology exerted unique pH-responsive fluorescence dual-emission properties, indicating promise in ratiometric pH-sensing applications.
Co-reporter:Shiyi Zhang ; Zhou Li ; Sandani Samarajeewa ; Guorong Sun ; Chao Yang
Journal of the American Chemical Society 2011 Volume 133(Issue 29) pp:11046-11049
Publication Date(Web):July 6, 2011
DOI:10.1021/ja203133h
Synthetic asymmetrical systems, Janus particles and patchy particles, are capable of undergoing hierarchical assembly processes that mimic those of Nature, to serve as switchable devices, optical probes, phase-transfer catalysts, and multifunctional drug carriers, each of which benefits from opposing surface patterns that behave differently. Production of nanometer-sized Janus particles that are equipped with efficient chemistries remains a challenge. A robust Janus-faced polymer nanoparticle framework that presents two orthogonally click-reactive surface chemistries has been generated by a recyclable strategy that involves reactive functional group transfer by templating against gold nanoparticle substrates. This anisotropic functionalization approach is compatible with a wide range of soft materials, providing Janus nanoparticles for the construction of dual-functionalized devices by accurately controlling chemical functionality at the nanoscopic level.
Co-reporter:Nam S. Lee, Guorong Sun, Lily Yun Lin, William L. Neumann, John N. Freskos, Amolkumar Karwa, Jeng J. Shieh, Richard B. Dorshow and Karen L. Wooley
Journal of Materials Chemistry A 2011 vol. 21(Issue 37) pp:14193-14202
Publication Date(Web):27 Jun 2011
DOI:10.1039/C1JM11854D
Dual-emitting photonic nano-objects that can sense changes in the environmental pH are designed based on shell-crosslinked micelles assembled from amphiphilic block copolymers and crosslinked with pH-insensitive chromophores. The chromophoric crosslinkers are tetra-functionalized pyrazine molecules that bear a set of terminal aliphatic amine groups and a set of anilino amine groups, which demonstrate morphology-dependent reactivities towards the poly(acrylic acid) shell domain of the nano-objects. The extent to which the anilino amine groups react with the nano-object shell is shown to affect the hypsochromic shift (blue-shift). The ratio of fluorescence intensity at 496 nm over that of 560 nm is dependent upon the solution pH. We report, herein, observations on the pH-sensitive dual-emission photophysical properties of rod-shaped or spherical nano-objects, whose shell domains offer two distinct platforms for amidation reactions to occur—through formation of activated esters upon addition of carbodiimide or pre-installation of activated ester groups. We demonstrate that physical manipulations (changes in morphology or particle dimensions) or chemical manipulations of the crosslinking reaction (the order of installation of activated esters) lead to fine tuning of dual-emission over ca. 60 nm in a physiologically relevant pH range. Rod-shaped shell-crosslinked nanostructures with poly(p-hydroxystyrene) core show blue-shift as a function of increasing pH while spherical shell-crosslinked nanostructures with polystyrene core and poly(ethylene oxide) corona exhibit blue-shift as a function of decreasing pH.
Co-reporter:Jennifer L. Sorrells, Ritu Shrestha, William L. Neumann and Karen L. Wooley
Journal of Materials Chemistry A 2011 vol. 21(Issue 25) pp:8983-8986
Publication Date(Web):27 May 2011
DOI:10.1039/C0JM04507A
Inclusion of porphyrins as crosslinkers within aqueous block copolymer assemblies imparts water solubility to the hydrophobic macrocycle, provides critical structural information about the particle interior, gives unique pH-independent photophysical properties, and has the potential for future development of SOD mimics.
Co-reporter:Jeremy W. Bartels, Philip M. Imbesi, John A. Finlay, Christopher Fidge, Jun Ma, Jonathan E. Seppala, Andreas M. Nystrom, Michael E. Mackay, James A. Callow, Maureen E. Callow, and Karen L. Wooley
ACS Applied Materials & Interfaces 2011 Volume 3(Issue 6) pp:2118
Publication Date(Web):June 6, 2011
DOI:10.1021/am200337q
A series of thiol-ene generated amphiphilic cross-linked networks was prepared by reaction of alkene-modified Boltorn polyesters (Boltorn-ene) with varying weight percent of 4-armed poly(ethylene glycol) (PEG) tetrathiol (0–25 wt %) and varying equivalents of pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) (0–64 wt %). These materials were designed to present complex surface topographies and morphologies, with heterogeneity of surface composition and properties and robust mechanical properties, to serve as nontoxic antibiofouling coatings that are amenable to large-scale production for application in the marine environment. Therefore, a two-dimensional matrix of materials compositions was prepared to study the physical and mechanical properties, over which the compositions spanned from 0 to 25 wt % PEG tetrathiol and 0–64 wt % PETMP (the overall thiol/alkene (SH/ene) ratios ranged from 0.00 to 1.00 equiv), with both cross-linker weight percentages calculated with respect to the weight of Boltorn-ene. The Boltorn-ene components were prepared through the esterification of commercially available Boltorn H30 with 3-butenoic acid. The subsequent cross-linking of the Boltorn-PEG-PETMP films was monitored using IR spectroscopy, where it was found that near-complete consumption of both thiol and alkene groups occurred when the stoichiometry was ca. 48 wt % PETMP (0.75 equiv SH/ene, independent of PEG amount). The thermal properties of the films showed an increase in Tg with an increase in 4-armed PEG-tetrathiol wt %, regardless of the PETMP concentration. Investigation of the bulk mechanical properties in dry and wet states found that the Young’s modulus was the greatest at 48 wt % PETMP (0.75 equiv of SH/ene). The ultimate tensile strength increased when PETMP was constant and the PEG concentration was increased. The Young’s modulus was slightly lower for wet films at constant PEG or constant PETMP amounts, than for the dry samples. The nanoscopic surface features were probed using atomic force microscopy (AFM), where it was observed that the surface of the amphiphilic films became increasingly rough with increasing PEG wt %. On the basis of the physicochemical data from the diverse sample matrix, a focused compositional profile was then investigated further to determine the antifouling performance of the cross-linked Boltorn-PEG-PETMP networks. For these studies, a low, constant PETMP concentration of 16 wt % was maintained with variation in the PEG wt % (0–35 wt %). Antifouling and fouling-release activities were tested against the marine alga Ulva. Spore settlement densities were low on these films, compared to that on standards of polydimethylsiloxane and glass.Keywords: Boltorn; cross-linking; marine fouling; poly(ethylene glycol); thiol-ene; Ulva
Co-reporter:Céline J. Besset, Alexander T. Lonnecker, Jennifer M. Streff, and Karen L. Wooley
Biomacromolecules 2011 Volume 12(Issue 7) pp:
Publication Date(Web):June 6, 2011
DOI:10.1021/bm2003048
Strategies for the preparation of polycarbonates, derived from natural polyhydroxy monomeric repeat units, were developed for biosourced polycarbonates based on quinic acid. The design and synthesis of regioselectively tert-butyldimethylsilyloxy (TBS)-protected 1,4- and 1,5-diol monomers of quinic acid were followed by optimization of their copolymerizations with phosgene, generated in situ from trichloromethyl chloroformate, to yield protected poly(1,4-quinic acid carbonate) and poly(1,5-quinic acid carbonate). The molecular weights reached ca. 7.6 kDa, corresponding to degrees of polymerization of ca. 24, with polydispersities ranging from 2.0 to 3.5, as measured by SEC using tetrahydrofuran as the eluent and with polystyrene calibration standards. Partially because of the presence of the bicyclic backbone, each regioisomeric poly(quinic acid carbonate) exhibited relatively high glass-transition temperatures, 209 °C for poly(1,4-quinic acid carbonate) and 229 °C for poly(1,5-quinic acid carbonate). Removal of the TBS-protecting groups was studied under mild conditions to achieve control over potential competing reactions involving polymer degradation, which could include cleavage of lactones within the repeat units, carbonate linkages, or both between the repeat units. Full deprotection was not achieved without some degree of polymer degradation. The regiochemistry of the monomer showed significant impact on the reactivity during deprotection and also on the thermal properties, with the 1,5-regioisomeric polymer having lower reactivity and giving higher Tg values, in comparison with the 1,4-regioisomer. Each regioisomer underwent a 10–20 °C increase in Tg upon partial removal of the TBS-protecting groups. As the extent of deprotection increased, the solubility decreased. Ultimately, at long deprotection reaction times, the solubility increased and the Tg decreased because of significant degradation of the polymers.
Co-reporter:Simon Harrisson, Glenna L. Drisko, Eva Malmström, Anders Hult, and Karen L. Wooley
Biomacromolecules 2011 Volume 12(Issue 4) pp:
Publication Date(Web):March 7, 2011
DOI:10.1021/bm101506j
Rigid nanoscale polymer rods were prepared by grafting preformed amine-terminated poly(styrene) and poly(tert-butyl acrylate) onto oxidized cellulose microcrystals. Low polydispersity polymers, grown using atom transfer radical polymerization, were characterized and purified prior to cellulose attachment. Oxidation of the cellulose microcrystal led to the formation of carboxylic acids on the surface of the microcrystals. Covalent attachment of the polymers onto the cellulose microcrystals was achieved via a carbodiimide-mediated amidation reaction. The length and diameter of the polymer-cellulose composites increased upon surface modification. Typically, polymer-cellulose composites are synthesized by a grafting-from method because it can be difficult to obtain sufficient graft density using a grafting-to preparation. However, the composites reported here comprised 60−64% grafted polymer by mass. This degree of grafting-to allowed the composite to form stable suspensions in organic solvents.
Co-reporter:Jennifer L. Sorrells;Ying-Hsin Tsai
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 20) pp:4465-4472
Publication Date(Web):
DOI:10.1002/pola.24237
Abstract
In the effort towards making nanoscale objects and assemblies feasible for use as functional materials, it is imperative to obtain control over the fundamental architectures and essential to understand what experimental conditions cause the manifestation of specific morphologies. A number of factors are known to influence the shape during the self-assembly of amphiphilic block copolymers in solution, including solvent composition, polymer length, hydrophobicity versus hydrophilicity, as well as the addition of additives that can interact with segments of the block copolymers. This research, focused on developing an understanding of the micellar architectures accessed by the amphiphilic triblock copolymer of acrylic acid, methyl acrylate, and styrene, PAA85-b-PMA40-b-PS35, as a function of the stirring rate, together with other factors, when undergoing coassembly with ethylenediamine or diethylenetriamine in water/tetrahydrofuran solutions. The work demonstrates that the rate at which the polymer solution was stirred impacts the shape of the solution-state assemblies formed by the triblock copolymer. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2010
Co-reporter:Wenjun Du;Yali Li;Andreas M. Nyström;Chong Cheng
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 15) pp:3487-3496
Publication Date(Web):
DOI:10.1002/pola.24149
Abstract
Complex amphiphilic polymers were synthesized via core-first polymerization followed by alkylation-based grafting of poly(ethylene oxide) (PEO). Inimer 1-(4′-(bromomethyl)benzyloxy)-2,3,5,6-tetrafluoro-4-vinylbenzene was synthesized and subjected to atom transfer radical self-condensing vinyl polymerization to afford hyperbranched fluoropolymer (HBFP) as the hydrophobic core component with a number-averaged molecular weight of 29 kDa and polydispersity index of 2.1. The alkyl halide chain ends on the HBFP were allowed to undergo reaction with monomethoxy-terminated poly(ethylene oxide) amine (PEOx-NH2) at different grafting numbers and PEO chain lengths to afford PEO-functionalized HBFPs [(PEOx)y-HBFPs], with x = 15 while y = 16, 22, or 29, x = 44 while y = 16, and x = 112 while y = 16. The amphiphilic, grafted block copolymers were found to aggregate in aqueous solution to give micelles with number-averaged diameters (Dav) of 12–28 nm, as measured by transmission electron microscopy (TEM). An increase of the PEO:HBFP ratio, by increase in either the grafting densities (y values) or the chain lengths (x values), led to decreased TEM-measured diameters. These complex, amphiphilic (PEOx)y-HBFPs, with tunable sizes, might find potential applications as nanoscopic biomedical devices, such as drug delivery vehicles and 19F magnetic resonance imaging agents. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 3487–3496, 2010
Co-reporter:Rhiannon K. Iha;Brooke A. van Horn
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 16) pp:3553-3563
Publication Date(Web):
DOI:10.1002/pola.24132
Abstract
Degradable, amphiphilic graft copolymers of poly(ε-caprolactone)-graft-poly(ethylene oxide), PCL-g-PEO, were synthesized via a grafting onto strategy taking advantage of the ketones presented along the backbone of the statistical copolymer poly(ε-caprolactone)-co-(2-oxepane-1,5-dione), (PCL-co-OPD). Through the formation of stable ketoxime ether linkages, 3 kDa PEO grafts and p-methoxybenzyl side chains were incorporated onto the polyester backbone with a high degree of fidelity and efficiency, as verified by NMR spectroscopies and GPC analysis (90% grafting efficiency in some cases). The resulting block graft copolymers displayed significant thermal differences, specifically a depression in the observed melting transition temperature, Tm, in comparison with the parent PCL and PEO polymers. These amphiphilic block graft copolymers undergo self-assembly in aqueous solution with the P(CL-co-OPD-co-(OPD-g-PEO)) polymer forming spherical micelles and a P(CL-co-OPD-co-(OPD-g-PEO)-co-(OPD-g-pMeOBn)) forming cylindrical or rod-like micelles, as observed by transmission electron microscopy and atomic force microscopy. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 3553–3563, 2010
Co-reporter:Zhou Li;Ke Zhang;Jun Ma;Chong Cheng
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 20) pp:5557-5563
Publication Date(Web):
DOI:10.1002/pola.23626
Co-reporter:Zhou Li ; Jun Ma ; Nam S. Lee
Journal of the American Chemical Society () pp:
Publication Date(Web):January 4, 2011
DOI:10.1021/ja109191z
We have developed a hierarchical process that combines linear triblock copolymers into concentric globular subunits through strong chemical bonds and is followed by their supramolecular assembly via weak noncovalent interactions to afford one-dimensionally assembled, dynamic cylindrical nanostructures. The molecular brush architecture forces triblock copolymers to adopt intramolecular interactions within confined frameworks and then drives their intermolecular interactions in the mixtures of organic solvent and water. In contrast, the triblock copolymers, when not preconnected into the molecular brush architectures, organize only into globular assemblies.
Co-reporter:Jingwei Fan, Jiong Zou, Xun He, Fuwu Zhang, Shiyi Zhang, Jeffery E. Raymond and Karen L. Wooley
Chemical Science (2010-Present) 2014 - vol. 5(Issue 1) pp:NaN150-150
Publication Date(Web):2013/10/18
DOI:10.1039/C3SC52504J
The simple copolymerization of N-carboxyanhydride (NCA) monomers is utilized to generate copolypeptides having a combination of α-helix and β-sheet sub-structures that, when grown from a solvophilic synthetic polymer block segment, are capable of driving mechano-responsive supramolecular sol-to-gel-to-sol and sol-to-gel-to-gel transitions reversibly, which allow also for injection-based processing and self-healing behaviors. A new type of polypeptide-based organogelator, methoxy poly(ethylene glycol)-block-poly(γ-benzyl-L-glutamate-co-glycine) (mPEG-b-P(BLG-co-Gly)), is facilely synthesized by statistical ring-opening copolymerizations (ROPs) of γ-benzyl-L-glutamate (BLG) and glycine (Gly) NCAs initiated by mPEG-amine. These systems exhibit tunable secondary structures and result in sonication stimulus responsiveness of the organogels with the polypeptide segment variation, controlled by varying the ratio of BLG NCA to Gly NCA during the copolymerizations. Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) studies indicate the α-helical component decreases while the β-sheet content increases systematically with a higher mole fraction of Gly in the polypeptide segment. The supramolecular assembly of β-sheet nanofibrils, having a tunable width over the range of 10.4–14.5 nm with varied BLG to Gly ratio, are characterized by transmission electron microscopy (TEM). The further self-assembly of these nanostructures into 3-D gel networks within N,N-dimethylformamide (DMF) occurs at low critical gelation concentrations (CGC) (lowest ca. 0.6 wt%). Increased BLG to Gly ratios lead to an increase of the α-helical component in the secondary structures of the polypeptide segments, resulting in wider and more flexible nanofibrils. The presence of α-helical component in the polymers enhances the stability of the organogels against sonication, and instantaneous gel-to-gel transitions are observed as in situ reconstruction of networks occurs within the gelled materials after sonication. In marked contrast, the β-sheet-rich gel, prepared from mPEG-b-PGly, exhibits an instant gel-to-sol transition after sonication is applied. The CGC concentration and stiffness of this mPEG-b-P(BLG-co-Gly) organogel system can be tuned by simply varying the percentages of α-helix and β-sheet in the secondary structures through control of the BLG to Gly ratio during synthesis. The mechanical properties of these organogels are studied by dynamic mechanical analyses (DMA), having storage moduli of ca. 12.1 kPa at room temperature. The injectability and self-healing capabilities are demonstrated by direct observation of the macroscopic self-healing behavior experiment.
Co-reporter:Nam S. Lee, Guorong Sun, Lily Yun Lin, William L. Neumann, John N. Freskos, Amolkumar Karwa, Jeng J. Shieh, Richard B. Dorshow and Karen L. Wooley
Journal of Materials Chemistry A 2011 - vol. 21(Issue 37) pp:NaN14202-14202
Publication Date(Web):2011/06/27
DOI:10.1039/C1JM11854D
Dual-emitting photonic nano-objects that can sense changes in the environmental pH are designed based on shell-crosslinked micelles assembled from amphiphilic block copolymers and crosslinked with pH-insensitive chromophores. The chromophoric crosslinkers are tetra-functionalized pyrazine molecules that bear a set of terminal aliphatic amine groups and a set of anilino amine groups, which demonstrate morphology-dependent reactivities towards the poly(acrylic acid) shell domain of the nano-objects. The extent to which the anilino amine groups react with the nano-object shell is shown to affect the hypsochromic shift (blue-shift). The ratio of fluorescence intensity at 496 nm over that of 560 nm is dependent upon the solution pH. We report, herein, observations on the pH-sensitive dual-emission photophysical properties of rod-shaped or spherical nano-objects, whose shell domains offer two distinct platforms for amidation reactions to occur—through formation of activated esters upon addition of carbodiimide or pre-installation of activated ester groups. We demonstrate that physical manipulations (changes in morphology or particle dimensions) or chemical manipulations of the crosslinking reaction (the order of installation of activated esters) lead to fine tuning of dual-emission over ca. 60 nm in a physiologically relevant pH range. Rod-shaped shell-crosslinked nanostructures with poly(p-hydroxystyrene) core show blue-shift as a function of increasing pH while spherical shell-crosslinked nanostructures with polystyrene core and poly(ethylene oxide) corona exhibit blue-shift as a function of decreasing pH.
Co-reporter:Jennifer L. Sorrells, Ritu Shrestha, William L. Neumann and Karen L. Wooley
Journal of Materials Chemistry A 2011 - vol. 21(Issue 25) pp:NaN8986-8986
Publication Date(Web):2011/05/27
DOI:10.1039/C0JM04507A
Inclusion of porphyrins as crosslinkers within aqueous block copolymer assemblies imparts water solubility to the hydrophobic macrocycle, provides critical structural information about the particle interior, gives unique pH-independent photophysical properties, and has the potential for future development of SOD mimics.
Co-reporter:Philip M. Imbesi, Jeffery E. Raymond, Bryan S. Tucker and Karen L. Wooley
Journal of Materials Chemistry A 2012 - vol. 22(Issue 37) pp:NaN19473-19473
Publication Date(Web):2012/06/01
DOI:10.1039/C2JM32005C
The synthesis of heterogeneous, amphiphilic crosslinked networks from photo-initiated thiol-ene chemistry and full characterization of the physicochemical, anti-biofouling, mechanical, and thermal properties of this system are reported. Although interest in coatings that present heterogeneous surface features is increasing, anti-biofouling performance is typically compared to homogeneous biocidal- or non-toxic polydimethylsiloxane-based paints, for the advancement of commercial and novel systems, which leaves a need for a benchmark to compare highly complex, heterogeneous composites with similar complexities. This system has been generated and rigorously analyzed to understand how microscopic and nanoscopic disorder, resistance to protein adsorption and surface mechanical properties can be fine-tuned and optimized through oligomer selection, blend ratios and process conditions. Solution-state studies of individual and blended constituents probed the relative fluorescence intensities based on concentration and neighboring species that were used to identify microscopic disorder on the surface. Incubation of a fluorescently labelled protein on the benchmark surface showed 42% and 72% less adsorption than on model surfaces that largely expressed a single component. The extent of reaction and the identification of unconsumed functionalities were found through infrared spectroscopy. The benchmark surface had a Young's modulus of approximately 0.5 GPa, 7 to 35 times higher than model surfaces, with 50 times the variation in modulus. Nanoscale surface adhesion force variation and bulk wettability and bulk thermal stability are also reported. This study provides an extensive list of metrics to be used for the development of complex, heterogeneous, anti-biofouling coatings, appropriate to both surface optimization and mechanical tunability, for implementation in real-world applications.
Co-reporter:Xun He, Jingwei Fan, Fuwu Zhang, Richen Li, Kevin A. Pollack, Jeffery E. Raymond, Jiong Zou and Karen L. Wooley
Journal of Materials Chemistry A 2014 - vol. 2(Issue 46) pp:NaN8130-8130
Publication Date(Web):2014/07/23
DOI:10.1039/C4TB00909F
A multi-responsive triblock hydrogelator oligo(DL-allylglycine)-block-poly(ethylene glycol)-block-oligo(DL-allylglycine) (ODLAG-b-PEG-b-ODLAG) was synthesized facilely by ring-opening polymerization (ROP) of DLAG N-carboxyanhydride (NCA) with a diamino-terminated PEG as the macroinitiator. This system exhibited heat-induced sol-to-gel transitions and either sonication- or enzyme-induced gel-to-sol transitions. The β-sheeting of the oligopeptide segments was confirmed by attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) and wide-angle X-ray scattering (WAXS). The β-sheets further displayed tertiary ordering into fibrillar structures that, in turn generated a porous and interconnected hydrogel matrix, as observed via transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The reversible macroscopic sol-to-gel transitions triggered by heat and gel-to-sol transitions triggered by sonication were correlated with the transformation of nanostructural morphologies, with fibrillar structures observed in gel and spherical aggregates in sol, respectively. The enzymatic breakdown of the hydrogels was also investigated. This allyl-functionalized hydrogelator can serve as a platform for the design of smart hydrogels, appropriate for expansion into biological systems as bio-functional and bio-responsive materials.
Co-reporter:Mahmoud Elsabahy and Karen L. Wooley
Chemical Society Reviews 2012 - vol. 41(Issue 7) pp:NaN2561-2561
Publication Date(Web):2012/02/14
DOI:10.1039/C2CS15327K
Polymeric nanoparticles-based therapeutics show great promise in the treatment of a wide range of diseases, due to the flexibility in which their structures can be modified, with intricate definition over their compositions, structures and properties. Advances in polymerization chemistries and the application of reactive, efficient and orthogonal chemical modification reactions have enabled the engineering of multifunctional polymeric nanoparticles with precise control over the architectures of the individual polymer components, to direct their assembly and subsequent transformations into nanoparticles of selective overall shapes, sizes, internal morphologies, external surface charges and functionalizations. In addition, incorporation of certain functionalities can modulate the responsiveness of these nanostructures to specific stimuli through the use of remote activation. Furthermore, they can be equipped with smart components to allow their delivery beyond certain biological barriers, such as skin, mucus, blood, extracellular matrix, cellular and subcellular organelles. This tutorial review highlights the importance of well-defined chemistries, with detailed ties to specific biological hurdles and opportunities, in the design of nanostructures for various biomedical delivery applications.
Co-reporter:Mahmoud Elsabahy and Karen L. Wooley
Chemical Society Reviews 2013 - vol. 42(Issue 12) pp:NaN5576-5576
Publication Date(Web):2013/04/03
DOI:10.1039/C3CS60064E
Nanoscale objects, whether of biologic origin or synthetically created, are being developed into devices for a variety of bionanotechnology diagnostic and pharmaceutical applications. However, the potential immunotoxicity of these nanomaterials and mechanisms by which they may induce adverse reactions have not received sufficient attention. Nanomaterials, depending on their characteristics and compositions, can interact with the immune system in several ways and either enhance or suppress immune system function. Cytokines perform pleiotropic functions to mediate and regulate the immune response and are generally recognized as biomarkers of immunotoxicity. While the specificity and validity of certain cytokines as markers of adverse immune response has been established for chemicals, small and macromolecular drugs, research on their applicability for predicting and monitoring the immunotoxicity of engineered nanomaterials is still ongoing. The goal of this review is to provide guidelines as to important cytokines that can be utilized for evaluating the immunotoxicity of nanomaterials and to highlight the role of those cytokines in mediating adverse reactions, which is of particular importance for the clinical development of nanopharmaceuticals and other nanotechnology-based products. Importantly, the rational design of nanomaterials of low immunotoxicity will be discussed, focusing on synthetic nanodevices, with emphasis on both the nanoparticle-forming materials and the embedded cargoes.
Co-reporter:Shiyi Zhang, Jiong Zou, Mahmoud Elsabahy, Amolkumar Karwa, Ang Li, Dennis A. Moore, Richard B. Dorshow and Karen L. Wooley
Chemical Science (2010-Present) 2013 - vol. 4(Issue 5) pp:NaN2126-2126
Publication Date(Web):2013/03/06
DOI:10.1039/C3SC50252J
A new type of degradable, nanoscopic polymer assembly containing ultra-high levels of drug loading via covalent attachment within amphiphilic core–shell nanoparticle morphology has been generated as a potentially effective and safe anti-cancer agent. Poly(ethylene oxide)-block-polyphosphoester-based paclitaxel drug conjugates (PEO-b-PPE-g-PTX) were synthesized by a rapid, scalable and versatile approach that involves only two steps: organocatalyst-promoted ring-opening-polymerization followed by click reaction-based conjugation of a PTX prodrug. Variations in the polymer-to-PTX stoichiometries allowed for optimization of the conjugation efficiency, the PTX drug loading and the resulting water solubilities of the entire polymer and the PTX content. The PEO-b-PPE-g-PTX formed well-defined micelles in aqueous solution, with a PTX loading capacity as high as 65 wt%, and a maximum PTX concentration of 6.2 mg mL−1 in water, which is 25000-fold higher than the aqueous solubility of free PTX. The positive cell-killing activity of PEO-b-PPE-g-PTX against several cancer cell lines is demonstrated, and the presence of pendant reactive functionality provides a powerful platform for future work to involve conjugation of multiple drugs and imaging agents to achieve chemotherapy and bioimaging.
Co-reporter:Xun He, Jingwei Fan, Jiong Zou and Karen L. Wooley
Chemical Communications 2016 - vol. 52(Issue 54) pp:NaN8458-8458
Publication Date(Web):2016/06/16
DOI:10.1039/C6CC03579E
A strategy for reversible patterning of soft conductive materials is described, based upon a combination of peptide-based block copolymer hydrogelators and photo-thermally-active carbon nanotubes. This composite displays photo-responsive gelation at application-relevant timescales (<10 s), allowing for rapid and spatially-defined construction of conductive patterns (>100 S m−1), which, additionally, hold the capability to revert to sol upon sonication for reprocessing.
Co-reporter:Sandani Samarajeewa, Ryan P. Zentay, Nema D. Jhurry, Ang Li, Kellie Seetho, Jiong Zou and Karen L. Wooley
Chemical Communications 2014 - vol. 50(Issue 8) pp:NaN970-970
Publication Date(Web):2013/12/04
DOI:10.1039/C3CC46013D
Electrostatic interaction-mediated enzymatic-hydrolysis of poly(lactide)-containing nanoscale assemblies is described. At physiological pH, degradable core–shell morphologies with charged shells can readily attract or repel enzymes carrying opposite or similar charges, respectively.
Co-reporter:Mahmoud Elsabahy, Sandani Samarajeewa, Jeffery E. Raymond, Corrie Clark and Karen L. Wooley
Journal of Materials Chemistry A 2013 - vol. 1(Issue 39) pp:NaN5255-5255
Publication Date(Web):2013/06/18
DOI:10.1039/C3TB20668H
The development of stable nanoparticles that can withstand the changing conditions experienced in a biological setting and also be of low toxicity and immunogenicity is of particular importance to address the problems associated with currently utilized nanotechnology-based therapeutics and diagnostics. The use of crosslinked nanoparticles continues to receive special impetus, due to their robust structure and high kinetic stability, and they have recently been shown to induce lower cytotoxicity than their non-crosslinked micellar counterparts. In the current study, poly(acrylamidoethylamine)-block-poly(DL-lactide) (PAEA90-b-PDLLA40) copolymers were synthesized, self-assembled in water to yield nanoscopic polymeric micelles, and the effects of decorating the micellar surface with poly(ethylene glycol) (i.e. PEGylation) and crosslinking the PAEA layer to varying extents on the physicochemical characteristics, cytotoxicity and immunotoxicity of the nanoparticles were studied. Herein, we report for the first time that crosslinking can efficiently reduce the immunotoxicity of polymeric nanomaterials. In addition, increasing the degree of crosslinking further reduced the accessibility of biomolecules to the core of the nanoparticles and decreased their cytotoxicity and immunotoxicity. It is also highlighted that crosslinking can be more efficient than PEGylation in reducing the immunotoxicity of nanomaterials. Shell-crosslinking of block copolymer micelles, therefore, is expected to advance their clinical development beyond the earlier known effects, and to broaden the implications in the field of nanomedicine.
Co-reporter:Jingwei Fan, Richen Li, Hai Wang, Xun He, Tan P. Nguyen, Rachel A. Letteri, Jiong Zou and Karen L. Wooley
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 24) pp:NaN5154-5154
Publication Date(Web):2017/06/02
DOI:10.1039/C7OB00931C
A polypeptide-based hydrogel system, when prepared from a diblock polymer with a ternary copolypeptide as one block, exhibited thermo-, mechano- and enzyme-responsive properties, which enabled the encapsulation of naproxen (Npx) during the sol–gel transition and its release in the gel state. Statistical terpolymerizations of L-alanine (Ala), glycine (Gly) and L-isoleucine (Ile) NCAs at a 1:1:1 feed ratio initiated by monomethoxy monoamino-terminated poly(ethylene glycol) afforded a series of methoxy poly(ethylene glycol)-block-poly(L-alanine-co-glycine-co-L-isoleucine) (mPEG-b-P(A-G-I)) block polymers. β-Sheets were the dominant secondary structures within the polypeptide segments, which facilitated a heat-induced sol-to-gel transition, resulting from the supramolecular assembly of β-sheets into nanofibrils. Deconstruction of the three-dimensional networks by mechanical force (sonication) triggered the reverse gel-to-sol transition. Certain enzymes could accelerate the breakdown of the hydrogel, as determined by in vitro gel weight loss profiles. The hydrogels were able to encapsulate and release Npx over 6 days, demonstrating the potential application of these polypeptide hydrogels as an injectable local delivery system for small molecule drugs.

![Acetamide, 2-azido-N-(3',6'-dihydroxy-3-oxospiro[isobenzofuran-1(3H),9'-[9H]xanthen]-5-yl)-](/data/chemimg/179000/1185289-36-4.png)
![Acetamide, 2-azido-N-(3',6'-dihydroxy-3-oxospiro[isobenzofuran-1(3H),9'-[9H]xanthen]-5-yl)-](/data/chemimg/179000/1185289-36-4_b.png)


![1-(5-AMINOPENTYL)-3-(3',6'-DIHYDROXY-3-OXOSPIRO[2-BENZOFURAN-1,9'-XANTHENE]-5-YL)THIOUREA](http://img.cochemist.com/ccimg/786800/786705-84-8.png)
![1-(5-AMINOPENTYL)-3-(3',6'-DIHYDROXY-3-OXOSPIRO[2-BENZOFURAN-1,9'-XANTHENE]-5-YL)THIOUREA](http://img.cochemist.com/ccimg/786800/786705-84-8_b.png)
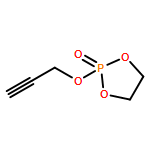
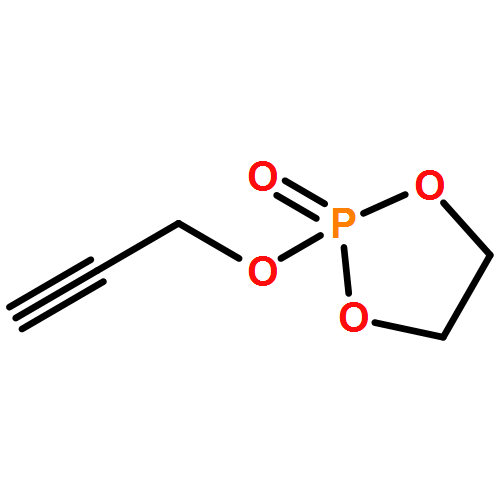

![L-TYROSINE, N-[(2E)-3-(4-HYDROXY-3-METHOXYPHENYL)-1-OXO-2-PROPENYL]-](http://img.cochemist.com/ccimg/731800/731773-64-1.png)
![L-TYROSINE, N-[(2E)-3-(4-HYDROXY-3-METHOXYPHENYL)-1-OXO-2-PROPENYL]-](http://img.cochemist.com/ccimg/731800/731773-64-1_b.png)




![POLY[OXYCARBONYLOXY[2-METHYL-2-[(PHENYLMETHOXY)CARBONYL]-1,3-PROPANEDIYL]]](http://img.cochemist.com/ccimg/247200/247142-75-2.png)
![POLY[OXYCARBONYLOXY[2-METHYL-2-[(PHENYLMETHOXY)CARBONYL]-1,3-PROPANEDIYL]]](http://img.cochemist.com/ccimg/247200/247142-75-2_b.png)




![Carbamic acid, N-[2-[(2-methyl-1-oxo-2-propen-1-yl)amino]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/1788700/206554-56-5.png)
![Carbamic acid, N-[2-[(2-methyl-1-oxo-2-propen-1-yl)amino]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/1788700/206554-56-5_b.png)


![Benzenemethanol, 3,5-bis[(pentafluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/188200/188111-77-5.png)
![Benzenemethanol, 3,5-bis[(pentafluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/188200/188111-77-5_b.png)
![Glycine,N-[[2-[[(diphenylmethoxy)carbonyl]amino]-1,6-dihydro-6-oxo-9H-purin-9-yl]acetyl]-N-[2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/186100/186046-83-3.png)
![Glycine,N-[[2-[[(diphenylmethoxy)carbonyl]amino]-1,6-dihydro-6-oxo-9H-purin-9-yl]acetyl]-N-[2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/186100/186046-83-3_b.png)
![Glycine,N-[[6-[[(diphenylmethoxy)carbonyl]amino]-9H-purin-9-yl]acetyl]-N-[2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/186100/186046-82-2.png)
![Glycine,N-[[6-[[(diphenylmethoxy)carbonyl]amino]-9H-purin-9-yl]acetyl]-N-[2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/186100/186046-82-2_b.png)
![Glycine, N-[2-(3,4-dihydro-5-Methyl-2,4-dioxo-1(2H)-pyriMidinyl)acetyl]-N-[2-[[(9H-fluoren-9-ylMethoxy)carbonyl]aMino]ethyl]-](http://img.cochemist.com/ccimg/169400/169396-92-3.png)
![Glycine, N-[2-(3,4-dihydro-5-Methyl-2,4-dioxo-1(2H)-pyriMidinyl)acetyl]-N-[2-[[(9H-fluoren-9-ylMethoxy)carbonyl]aMino]ethyl]-](http://img.cochemist.com/ccimg/169400/169396-92-3_b.png)
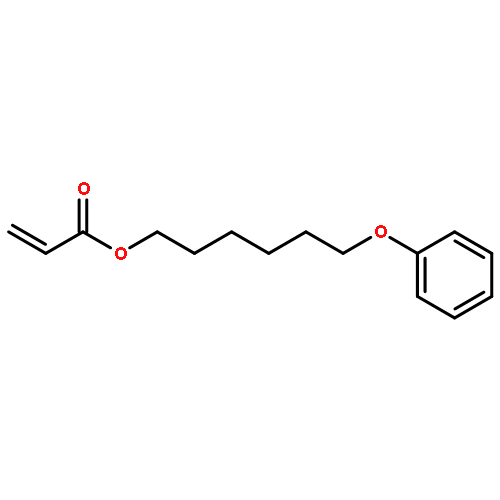




![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)




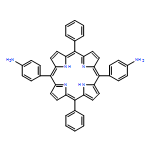
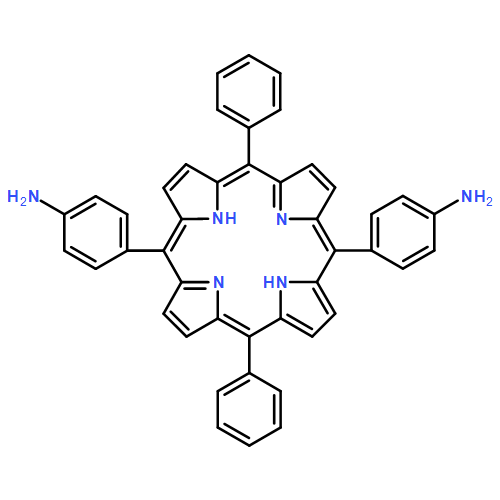
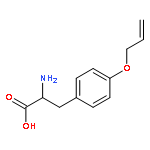



![Acetamide,N-(3',6'-dihydroxy-3-oxospiro[isobenzofuran-1(3H),9'-[9H]xanthen]-5-yl)-2-iodo-](/data/chemimg/941800/63368-54-7.png)
![Acetamide,N-(3',6'-dihydroxy-3-oxospiro[isobenzofuran-1(3H),9'-[9H]xanthen]-5-yl)-2-iodo-](/data/chemimg/941800/63368-54-7_b.png)
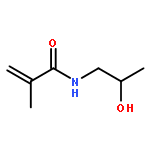
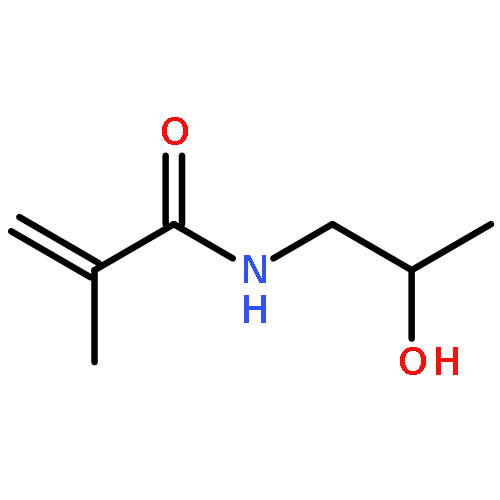
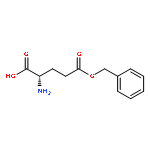
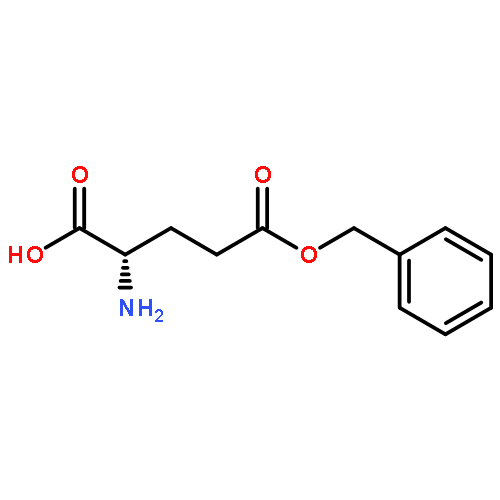













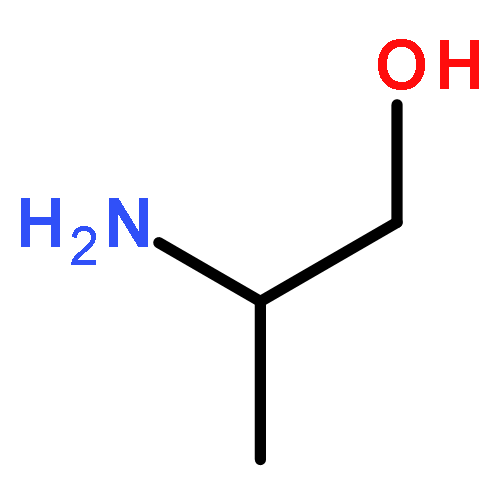
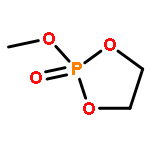
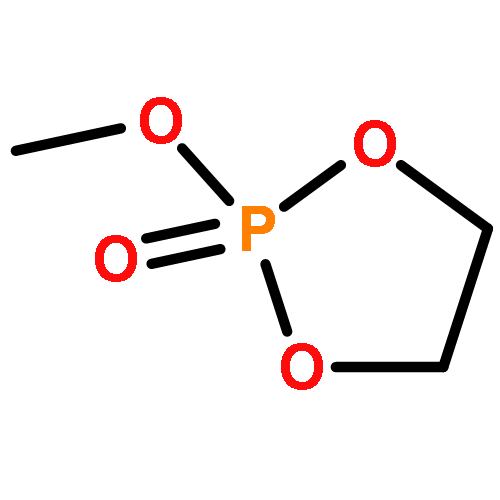






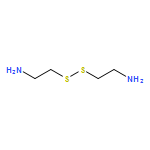
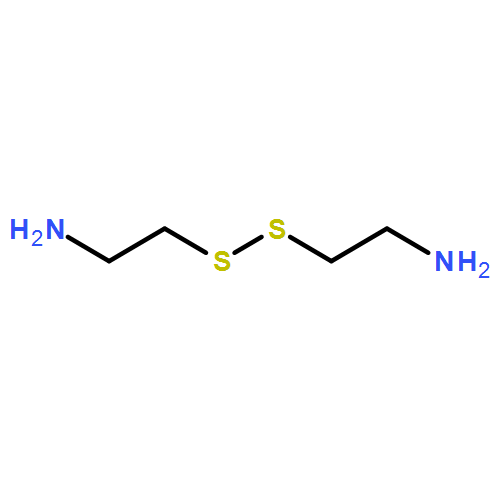
![1H-1,2,3-Triazole-1-acetic acid, 4-[[2-[[(dodecylthio)thioxomethyl]thio]-2-methyl-1-oxopropoxy]methyl]-α-methyl-, 2-[2-[2-(4-ethenyl-2,3,5,6-tetrafluorophenoxy)ethoxy]ethoxy]ethyl ester](/data/chemimg/3721900/1964482-96-9.png)
![1H-1,2,3-Triazole-1-acetic acid, 4-[[2-[[(dodecylthio)thioxomethyl]thio]-2-methyl-1-oxopropoxy]methyl]-α-methyl-, 2-[2-[2-(4-ethenyl-2,3,5,6-tetrafluorophenoxy)ethoxy]ethoxy]ethyl ester](/data/chemimg/3721900/1964482-96-9_b.png)
![Propanoic acid, 2-azido-, 2-[2-[2-(4-ethenyl-2,3,5,6-tetrafluorophenoxy)ethoxy]ethoxy]ethyl ester](/data/chemimg/3723200/1964482-95-8.png)
![Propanoic acid, 2-azido-, 2-[2-[2-(4-ethenyl-2,3,5,6-tetrafluorophenoxy)ethoxy]ethoxy]ethyl ester](/data/chemimg/3723200/1964482-95-8_b.png)
![Propanoic acid, 2-[[(dodecylthio)thioxomethyl]thio]-2-methyl-, 2-propyn-1-yl ester](/data/chemimg/104300/949151-11-5.png)
![Propanoic acid, 2-[[(dodecylthio)thioxomethyl]thio]-2-methyl-, 2-propyn-1-yl ester](/data/chemimg/104300/949151-11-5_b.png)

![Poly[oxycarbonyloxy[2-methyl-2-[(2-propenyloxy)carbonyl]-1,3-propaned
iyl]]](http://img.cochemist.com/ccimg/532500/532424-78-5.png)
![Poly[oxycarbonyloxy[2-methyl-2-[(2-propenyloxy)carbonyl]-1,3-propaned
iyl]]](http://img.cochemist.com/ccimg/532500/532424-78-5_b.png)




![2-Propenoic acid, 6-([1,1'-biphenyl]-4-yloxy)hexyl ester](http://img.cochemist.com/ccimg/73600/73586-35-3.png)
![2-Propenoic acid, 6-([1,1'-biphenyl]-4-yloxy)hexyl ester](http://img.cochemist.com/ccimg/73600/73586-35-3_b.png)
![1-HEXANOL, 6-([1,1'-BIPHENYL]-4-YLOXY)-](http://img.cochemist.com/ccimg/73600/73586-33-1.png)
![1-HEXANOL, 6-([1,1'-BIPHENYL]-4-YLOXY)-](http://img.cochemist.com/ccimg/73600/73586-33-1_b.png)


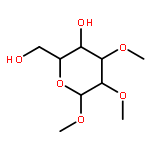
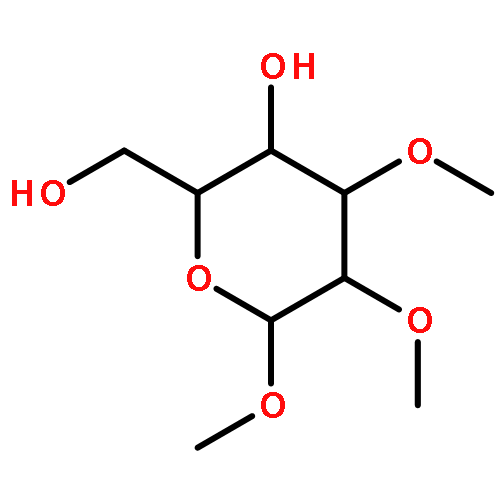
![6-Oxabicyclo[3.2.1]octan-7-one, 1,3,4-trihydroxy-, (1S,3R,4R,5R)-](http://img.cochemist.com/ccimg/700/665-27-0.png)
![6-Oxabicyclo[3.2.1]octan-7-one, 1,3,4-trihydroxy-, (1S,3R,4R,5R)-](http://img.cochemist.com/ccimg/700/665-27-0_b.png)


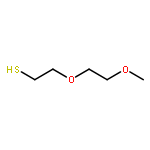



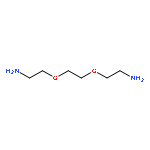
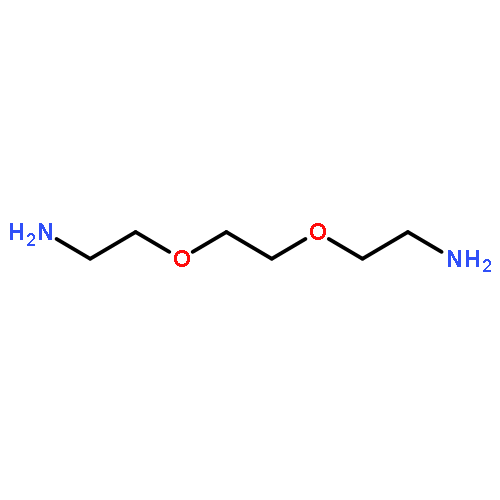
![Carbamic acid, [2-[(1-oxo-2-propenyl)amino]ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/165200/165196-44-1.png)
![Carbamic acid, [2-[(1-oxo-2-propenyl)amino]ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/165200/165196-44-1_b.png)