Co-reporter:Bernhard Hauer, Stefan Lutz
Current Opinion in Chemical Biology 2017 Volume 37(Volume 37) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.cbpa.2017.04.006
Co-reporter:Nico Kreß
BIOspektrum 2017 Volume 23( Issue 7) pp:836-838
Publication Date(Web):17 November 2017
DOI:10.1007/s12268-017-0878-1
Biocatalysis can serve as a basis for the synthesis of structurally challenging valuable compounds. The synergistic use of biological and chemical knowledge represents a fruitful approach for finding starting points in the development of novel enzymes. Several enzyme engineering techniques are nowadays available to evolve such starting activities towards tailored enzymes in artificial biochemical syntheses.
Co-reporter:Martin J. Weissenborn, Sandra Notonier, Sarah-Luise Lang, Konrad B. Otte, Susanne Herter, Nicholas J. Turner, Sabine L. Flitsch and Bernhard Hauer
Chemical Communications 2016 vol. 52(Issue 36) pp:6158-6161
Publication Date(Web):07 Apr 2016
DOI:10.1039/C6CC01749E
A readily available galactose oxidase (GOase) variant was used to develop a whole cell screening assay. This endpoint detection system was applied in a proof-of-concept approach by screening a focussed mutant library. This led to the discovery of the thus far most active P450 Marinobacter aquaeolei mutant catalysing the terminal hydroxylation of fatty acids.
Co-reporter:Melanie Melcher, Sandra J. Facey, Thorsten M. Henkes, Thomas Subkowski, and Bernhard Hauer
Biomacromolecules 2016 Volume 17(Issue 5) pp:
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.biomac.6b00135
Calcium phosphate mineralization is of particular interest in dental repair. A biomimetic approach using proteins or peptides is a highly promising way to reconstruct eroded teeth. In this study, the screening of several proteins is described for their binding and nucleating activities toward hydroxyapatite. Out of 27 tested candidates, only two hydrophobin fusion proteins showed binding abilities to hydroxyapatite in a mouthwash formulation and an increased nucleation in artificial saliva. Using a semirational approach, one of the two candidates (DEWA_5), a fusion protein consisting of a truncated section of the Bacillus subtilis synthase YaaD, the Aspergillus nidulans hydrophobin DEWA, and the rationally designed peptide P11-4 described in the literature, could be further engineered toward a faster mineral formation. The variants DEWA_5a (40aaYaaD-SDSDSD-DEWA) and DEWA_5b (40aaYaaD-RDRDRD-DEWA) were able to enhance the nucleation activity without losing the ability to form hydroxyapatite. In the case of variant DEWA_5b, an additional increase in the binding toward hydroxyapatite could be achieved. Especially with the variant DEWA_5a, the protein engineering of the rationally designed peptide sequence resulted in a resemblance of an amino acid motif that is found in nature. The engineered peptide resembles the amino acid motif in dentin phosphoprotein, one of the major proteins involved in dentinogenesis.
Co-reporter:Sebastian A. Löw;Isabell M. Löw;Dr. Martin J. Weissenborn;Dr. Bernhard Hauer
ChemCatChem 2016 Volume 8( Issue 5) pp:911-915
Publication Date(Web):
DOI:10.1002/cctc.201501230
Abstract
The reduction of activated C=C double bonds is an important reaction in synthetic chemistry owing to the potential formation of up to two new stereogenic centers. Artificial nicotinamide cofactors were recently presented as alternative suppliers of hydride equivalents needed for alkene reduction. To study the effect of cofactors on the reduction of activated alkenes, a set of N-substituted synthetic nicotinamide cofactors with differing oxidation potentials were synthesized and their electrochemical and kinetic behavior was studied. The effects of the synthetic cofactors on enzyme activity of four ene reductases are outlined in this study, where the cofactor mimic with an N-substituted 4-hydroxy-phenyl residue led to a sixfold higher vmax relative to the natural cofactor NADH.
Co-reporter:Sebastian A. Löw;Isabell M. Löw;Dr. Martin J. Weissenborn;Dr. Bernhard Hauer
ChemCatChem 2016 Volume 8( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/cctc.201600205
Co-reporter:Dr. Dennis Wetzl;Jennifer Bolsinger;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
ChemCatChem 2016 Volume 8( Issue 7) pp:1361-1366
Publication Date(Web):
DOI:10.1002/cctc.201501244
Abstract
Enzymes still have a limited application scope in synthetic organic chemistry. To expand this, different strategies exist that range from the de novo design of enzymes to the exploitation of the catalytic capabilities of known enzymes by converting different substrates; denoted as substrate promiscuity. We harnessed the synthetic potential offered by the taurine dioxygenase (TauD) from Escherichia coli (E. coli) by studying its promiscuous catalytic properties in the hydroxylation of carboxylic acid substrates. TauD showed high selectivities in the hydroxylation reaction but reduced levels of activity (26 % conversion, >96 % ee). We enhanced the enzyme substrate scope and improved the conversions for the tested substrates by introducing a point mutation at position 206 (F206Y). The conversions of the improved catalyst increased by at least 140 % compared to that of the wild-type enzyme. The number of carboxylic acids that accepted by the enzyme variant doubled from four to eight carboxylic acids.
Co-reporter:Sara M. Hoffmann;Dr. Martin J. Weissenborn;&x141;ukasz Gricman;Sra Notonier;Dr. Jürgen Pleiss ;Dr. Bernhard Hauer
ChemCatChem 2016 Volume 8( Issue 8) pp:1591-1597
Publication Date(Web):
DOI:10.1002/cctc.201501397
Abstract
Three different reductases have been fused to CYP153 monooxygenase from Marinobacter aquaeolei. The most promising candidate has been analysed in terms of its linker part, which connects the reductase with the haem domain through sequence alignment of the corresponding reductase family CYP116B. To improve the artificial fusion construct, the linker length has been varied, thereby only altering the non-conserved middle part of the linker. This way seven artificial fusion constructs have been engineered, which varied in linker length between 11 and 32 amino acids (“natural” is 16). These variations showed a substantial impact on the fusion construct. The best mutant, extended by two amino acids, showed an improved activity (67 %), higher stability (67 % more active haem domain after 2 h) and a coupling efficiency of 94 % (55 % higher than before). Presented in this paper is an approach to find and optimise artificial fusion constructs for P450 monooxygenases.
Co-reporter:Dr. Martin J. Weissenborn;Dr. Sebastian A. Löw;Niels Borlinghaus;Miriam Kuhn;Stefanie Kummer;Fabian Rami;Dr. Bernd Plietker;Dr. Bernhard Hauer
ChemCatChem 2016 Volume 8( Issue 9) pp:1636-1640
Publication Date(Web):
DOI:10.1002/cctc.201600227
Abstract
The Wittig-type carbonyl olefination reaction has no biocatalytic equivalent. To build complex molecular scaffolds, however, C−C bond-forming reactions are pivotal for biobased economy and synthetic biology. The heme-containing E. coli protein YfeX was found to catalyze carbonyl olefination by reaction of benzaldehyde with ethyl diazoacetate under aerobic conditions in the absence of a triphenylphosphine oxophile. The reaction was performed in whole cells and showed a product formation of 440 mg L−1 in 1 h. It was, moreover, shown that the reaction could be performed under Wittig-analogue conditions in the presence of triphenylphosphine or triphenylarsine.
Co-reporter:Dr. Martin J. Weissenborn;Dr. Sebastian A. Löw;Niels Borlinghaus;Miriam Kuhn;Stefanie Kummer;Fabian Rami;Dr. Bernd Plietker;Dr. Bernhard Hauer
ChemCatChem 2016 Volume 8( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cctc.201600483
Co-reporter:Dr. Sabrina Reich;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
ChemBioChem 2016 Volume 17( Issue 7) pp:561-565
Publication Date(Web):
DOI:10.1002/cbic.201500604
Abstract
The enzymatic reduction of C=C bonds in allylic alcohols with Old Yellow Enzymes represents a challenging task, due to insufficient activation through the hydroxy group. In our work, we coupled an alcohol dehydrogenase with three wild-type ene reductases—namely nicotinamide-dependent cyclohex-2-en-1-one reductase (NCR) from Zymomonas mobilis, OYE1 from Saccharomyces pastorianus and morphinone reductase (MR) from Pseudomonas putida M10—and four rationally designed β/α loop variants of NCR in the bienzymatic cascade hydrogenation of allylic alcohols. Remarkably, the wild type of NCR was not able to catalyse the cascade reaction whereas MR and OYE1 demonstrated high to excellent activities. Through the rational loop grafting of two intrinsic β/α surface loop regions near the entrance of the active site of NCR with the corresponding loops from OYE1 or MR we successfully transferred the cascade reduction activity from one family member to another. Further we observed that loop grafting revealed certain influences on the interaction with the nicotinamide cofactor.
Co-reporter:Dr. Sra Notonier;Dr. &x141;ukasz Gricman;Dr. Jürgen Pleiss ;Dr. Bernhard Hauer
ChemBioChem 2016 Volume 17( Issue 16) pp:1550-1557
Publication Date(Web):
DOI:10.1002/cbic.201600207
Abstract
The regioselective terminal hydroxylation of alkanes and fatty acids is of great interest in a variety of industrial applications, such as in cosmetics, in fine chemicals, and in the fragrance industry. The chemically challenging activation and oxidation of non-activated C−H bonds can be achieved with cytochrome P450 enzymes. CYP153AM.aq.-CPRBM3 is an artificial fusion construct consisting of the heme domain from Marinobacter aquaeolei and the reductase domain of CYP102A1 from Bacillus megaterium. It has the ability to hydroxylate medium- and long-chain fatty acids selectively at their terminal positions. However, the activity of this interesting P450 construct needs to be improved for applications in industrial processes. For this purpose, the design of mutant libraries including two consecutive steps of mutagenesis is demonstrated. Targeted positions and residues chosen for substitution were based on semi-rational protein design after creation of a homology model of the heme domain of CYP153AM.aq., sequence alignments, and docking studies. Site-directed mutagenesis was the preferred method employed to address positions within the binding pocket, whereas diversity was created with the aid of a degenerate codon for amino acids located at the substrate entrance channel. Combining the successful variants led to the identification of a double variant—G307A/S233G—that showed alterations of one position within the binding pocket and one position located in the substrate access channel. This double variant showed twofold increased activity relative to the wild type for the terminal hydroxylation of medium-chain-length fatty acids. This variant furthermore showed improved activity towards short- and long-chain fatty acids and enhanced stability in the presence of higher concentrations of fatty acids.
Co-reporter:Sebastian A. Löw, Bettina M. Nestl, Martin J. Weissenborn, Ferdinand Zepeck, and Bernhard Hauer
Organic Process Research & Development 2015 Volume 19(Issue 11) pp:1544-1547
Publication Date(Web):April 21, 2015
DOI:10.1021/acs.oprd.5b00070
The facile synthesis of rifamycin S from rifamycin B, a member of the ansamycin family of antibiotics, via the oxidation of rifamycin B was developed. Currently on an industrial scale, this oxidation is performed using harsh pH conditions and chlorinated solvents. With the development of a suitable buffer/methanol system, a similar yield and space-time-yield in comparison to the current process can be obtained renouncing chlorinated solvents. Employment of methanol as a reaction medium in this process is crucial for attaining high yields under mild reaction conditions. With this method a space-time-yield of 189 g L–1 h–1 of rifamycin S was achieved in one step.
Co-reporter:Christine Gally;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
Angewandte Chemie International Edition 2015 Volume 54( Issue 44) pp:12952-12956
Publication Date(Web):
DOI:10.1002/anie.201506527
Abstract
The asymmetric dihydroxylation of olefins is of special interest due to the facile transformation of the chiral diol products into valuable derivatives. Rieske non-heme iron oxygenases (ROs) represent promising biocatalysts for this reaction as they can be engineered to efficiently catalyze the selective mono- and dihydroxylation of various olefins. The introduction of a single point mutation improved selectivities (≥95 %) and conversions (>99 %) towards selected alkenes. By modifying the size of one active site amino acid side chain, we were able to modulate the regio- and stereoselectivity of these enzymes. For distinct substrates, mutants displayed altered regioselectivities or even favored opposite enantiomers compared to the wild-type ROs, offering a sustainable approach for the oxyfunctionalization of a wide variety of structurally different olefins.
Co-reporter:Christine Gally;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
Angewandte Chemie 2015 Volume 127( Issue 44) pp:13144-13148
Publication Date(Web):
DOI:10.1002/ange.201506527
Abstract
The asymmetric dihydroxylation of olefins is of special interest due to the facile transformation of the chiral diol products into valuable derivatives. Rieske non-heme iron oxygenases (ROs) represent promising biocatalysts for this reaction as they can be engineered to efficiently catalyze the selective mono- and dihydroxylation of various olefins. The introduction of a single point mutation improved selectivities (≥95 %) and conversions (>99 %) towards selected alkenes. By modifying the size of one active site amino acid side chain, we were able to modulate the regio- and stereoselectivity of these enzymes. For distinct substrates, mutants displayed altered regioselectivities or even favored opposite enantiomers compared to the wild-type ROs, offering a sustainable approach for the oxyfunctionalization of a wide variety of structurally different olefins.
Co-reporter:Bettina M. Nestl and Bernhard Hauer
ACS Catalysis 2014 Volume 4(Issue 9) pp:3201
Publication Date(Web):August 8, 2014
DOI:10.1021/cs500325p
Co-reporter:Philipp N. Scheller;Silvia Fademrecht;Sebastian Hofelzer; Dr. Jürgen Pleiss;Dr. Friedemann Leipold; Dr. Nicholas J. Turner;Dr. Bettina M. Nestl; Dr. Bernhard Hauer
ChemBioChem 2014 Volume 15( Issue 15) pp:2201-2204
Publication Date(Web):
DOI:10.1002/cbic.201402213
Abstract
Reducing reactions are among the most useful transformations for the generation of chiral compounds in the fine-chemical industry. Because of their exquisite selectivities, enzymatic approaches have emerged as the method of choice for the reduction of CO and activated CC bonds. However, stereoselective enzymatic reduction of CN bonds is still in its infancy—it was only recently described after the discovery of enzymes capable of imine reduction. In our work, we increased the spectrum of imine-reducing enzymes by database analysis. By combining the currently available knowledge about the function of imine reductases with the experimentally uncharacterized diversity stored in protein sequence databases, three novel imine reductases with complementary enantiopreference were identified along with amino acids important for catalysis. Furthermore, their reducing capability was demonstrated by the reduction of the pharmaceutically relevant prochiral imine 2-methylpyrroline. These novel enzymes exhibited comparable to higher catalytic efficiencies than previously described enzymes, and their biosynthetic potential is highlighted by the full conversion of 2-methylpyrroline in whole cells with excellent selectivities.
Co-reporter:Dr. Bettina M. Nestl;Dipl.-Chem. Stephan C. Hammer;Dr. Bernd A. Nebel ;Dr. Bernhard Hauer
Angewandte Chemie 2014 Volume 126( Issue 12) pp:3132-3158
Publication Date(Web):
DOI:10.1002/ange.201302195
Abstract
Die Verwendung von Enzymen als Katalysatoren zur Synthese neuartiger Verbindungen ist in den letzten Jahren zunehmend in den Fokus gerückt. Entsprechend hohe Erwartungen werden an die Entdeckung und Identifizierung neuer Biokatalysatoren für die organische Synthese gestellt. Dies spiegelt sich auch in der Komplexität der adressierten Reaktionen wider. Der Anwendungsbereich von Biokatalysatoren umfasst die Synthese wichtiger Zwischenprodukte für die pharmazeutische und chemische Industrie sowie die Entwicklung neuer enzymatischer Techniken und Prozesse. Enzyme sind ein wichtiger Teil des Spektrums an Katalysatoren, die der Synthesechemie zur Verfügung stehen. Die Vorteile und Anwendungsmöglichkeiten der neuesten und höchst vielversprechenden Generation von Biokatalysatoren – Reduktasen, Transaminasen, Ammoniak-Lyasen, Epoxidhydrolasen und Dehalogenasen – werden hier vorgestellt und anhand der Synthese von Schlüsselmolekülen beispielhaft diskutiert.
Co-reporter:Bettina M. Nestl;Stephan C. Hammer;Bernd A. Nebel ;Dr. Bernhard Hauer
Angewandte Chemie International Edition 2014 Volume 53( Issue 12) pp:3070-3095
Publication Date(Web):
DOI:10.1002/anie.201302195
Abstract
The use of enzymes as catalysts for the preparation of novel compounds has received steadily increasing attention over the past few years. High demands are placed on the identification of new biocatalysts for organic synthesis. The catalysis of more ambitious reactions reflects the high expectations of this field of research. Enzymes play an increasingly important role as biocatalysts in the synthesis of key intermediates for the pharmaceutical and chemical industry, and new enzymatic technologies and processes have been established. Enzymes are an important part of the spectrum of catalysts available for synthetic chemistry. The advantages and applications of the most recent and attractive biocatalysts—reductases, transaminases, ammonia lyases, epoxide hydrolases, and dehalogenases—will be discussed herein and exemplified by the syntheses of interesting compounds.
Co-reporter:Konrad B. Otte;Jens Kittelberger;Marko Kirtz;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
ChemCatChem 2014 Volume 6( Issue 4) pp:1003-1009
Publication Date(Web):
DOI:10.1002/cctc.201300787
Abstract
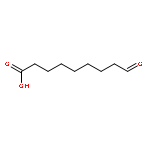
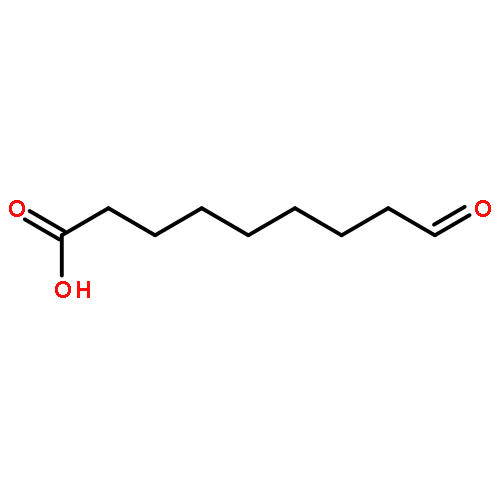
Polymers benefit from the use of biogenic resources such as fatty acids. They enable easy access to valuable monomeric building blocks, which, in comparison to their exclusively fossil counterparts, lead to products with improved physicochemical properties. Monomers of special interest are medium-chain dicarboxylic acids, which are not easy to obtain by traditional chemical means. Previously, we established an in vitro pathway that combined a 9-lipoxygenase and a 9/13-hydroperoxide lyase, which enabled the conversion of linoleic acid via a hydroperoxy intermediate into 9-oxononanoic acid, the precursor of azelaic acid. Herein, we aimed for the further development of the multi-enzyme cascade, which included the oxidation of 9-oxononanoic acid and the establishment of a suitable whole-cell catalyst. A detailed investigation of the simultaneous in vitro reaction setup revealed that both lipoxygenase activation and the subsequent hydroperoxide lyase reaction depend on the hydroperoxide reaction intermediate. For the activation of lipoxygenase, the hydroperoxide lyase activity, therefore, has to be significantly reduced. In accordance with these observations, we established a suitable dual-expression system and we further demonstrated that endogenous E. coli redox enzymes are feasible to oxidize 9-oxononanoic acid to azelaic acid. The resulting whole-cell catalyst is, therefore, able to perform the direct bioconversion of linoleic acid into azelaic acid. The use of organic solvent as the second phase improved the overall performance of the E. coli host strain. The developed one-pot, single-step process afforded 29 mg L−1 of azelaic acid within 8 h with a substrate conversion of 34 % and a selectivity of 47 %.
Co-reporter:Konrad B. Otte;Jens Kittelberger;Marko Kirtz;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
ChemCatChem 2014 Volume 6( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/cctc.201400103
Abstract
The front cover artwork for Issue 04/2014 is provided by the Institute of Technical Biochemistry, University of Stuttgart. The image shows the depiction of an Escherichia coli whole cell factory, which converts linoleic acid directly into azelaic acid. The three reaction steps are indicated in blue, green, and orange in the zoom-in. See the Full Paper itself at http://dx.doi.org/10.1002/cctc.201300787.
Co-reporter:Konrad B. Otte;Jens Kittelberger;Marko Kirtz;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
ChemCatChem 2014 Volume 6( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/cctc.201490020
Co-reporter:Konrad B. Otte;Marko Kirtz;Dr. Bettina M. Nestl ;Dr. Bernhard Hauer
ChemSusChem 2013 Volume 6( Issue 11) pp:2149-2156
Publication Date(Web):
DOI:10.1002/cssc.201300183
Abstract
Polymers based on renewable resources have become increasingly important. The natural functionalization of fats and oils enables an easy access to interesting monomeric building blocks, which in turn transform the derivative biopolymers into high-performance materials. Unfortunately, interesting building blocks of medium-chain length are difficult to obtain by traditional chemical means. Herein, a biotechnological pathway is established that could provide an environmentally suitable and sustainable alternative. A multiple enzyme two-step one-pot process efficiently catalyzed by a coupled 9S-lipoxygenase (St-LOX1, Solanum tuberosum) and 9/13-hydroperoxide lyase (Cm-9/13HPL, Cucumis melo) cascade reaction is proposed as a potential route for the conversion of linoleic acid into 9-oxononanoic acid, which is a precursor for biopolymers. Lipoxygenase catalyzes the insertion of oxygen into linoleic acid through a radical mechanism to give 9S-hydroperoxy-octadecadienoic acid (9S-HPODE) as a cascade intermediate, which is subsequently cleaved by the action of Cm-9/13HPL. This one-pot process afforded a yield of 73 % combined with high selectivity. The best reaction performance was achieved when lipoxygenase and hydroperoxide lyase were applied in a successive rather than a simultaneous manner. Green leaf volatiles, which are desired flavor and fragrance products, are formed as by-products in this reaction cascade. Furthermore, we have investigated the enantioselectivity of 9/13-HPLs, which exhibited a strong preference for 9S-HPODE over 9R-HPODE.
Co-reporter:Miriam Seitz;Dr. Per-Olof Syrén;Lisa Steiner;Dr. Janosch Klebensberger;Dr. Bettina M. Nestl ; Dr. Bernhard Hauer
ChemBioChem 2013 Volume 14( Issue 4) pp:436-439
Publication Date(Web):
DOI:10.1002/cbic.201300018
Co-reporter:Stephan C. Hammer;Bettina M. Nestl
BIOspektrum 2013 Volume 19( Issue 5) pp:574-576
Publication Date(Web):2013 September
DOI:10.1007/s12268-013-0355-4
Enzymes are highly attractive catalysts for organic synthesis. Traditionally, new enzymatic function has been identified by screening large libraries of mutants and microorganisms. Recently, the field has developed to use small, functionally rich libraries combined with a chemical-based enzyme engineering approach. Key advances derive from bioinformatics along with a better understanding of the mechanisms of natural protein evolution, all this in combination with chemical intuition.
Co-reporter:Christian Kazenwadel;Janosch Klebensberger
Applied Microbiology and Biotechnology 2013 Volume 97( Issue 16) pp:7215-7227
Publication Date(Web):2013 August
DOI:10.1007/s00253-012-4579-x
Phenoxy radical coupling reactions are involved in the biosynthesis of lignans in planta. Interestingly, the reaction can be guided by dirigent proteins, which mediate the stereoselective formation of either (+) or (−)-pinoresinol from coniferyl alcohol. So far, the mechanism is poorly understood, and for detailed mechanistic studies, a heterologous expression platform which allows the cost-effective, fast, and robust expression in high yields is needed. We established a reliable, high-yield fed-batch fermentation process with Pichia pastoris resulting in 47 mg L−1 of the dirigent protein AtDIR6, which represents a more than 250-fold increase compared to previous studies. Biochemical characterization of AtDIR6 produced with P. pastoris showed an overall agreement in protein structure, N-glycosylation sites, and dirigent activity compared to AtDIR6 produced by plant cell cultures of Solanum peruvianum. CD spectroscopy verified the β-barrel structure proposed by earlier studies and bioconversion experiments revealed similar activities to plant-derived protein, validating P. pastoris as a suitable expression system for dirigent proteins. Compared to the complex glycan structures of most plant cells, proteins produced with P. pastoris have the advantage that they can be enzymatically deglycosylated under non-denaturating conditions. With this study, we demonstrate that the glycan structures of AtDIR6 are essential for structure, solubility, and function of the protein as deglycosylation induced conformational changes leading to the complete loss in dirigent activity and subsequent protein aggregation.
Co-reporter:Sumire Honda Malca, Daniel Scheps, Lisa Kühnel, Elena Venegas-Venegas, Alexander Seifert, Bettina M. Nestl and Bernhard Hauer
Chemical Communications 2012 vol. 48(Issue 42) pp:5115-5117
Publication Date(Web):26 Mar 2012
DOI:10.1039/C2CC18103G
CYP153A from Marinobacter aquaeolei has been identified as a fatty acid ω-hydroxylase with a broad substrate range. Two hotspots predicted to influence substrate specificity and selectivity were exchanged. Mutant G307A is 2- to 20-fold more active towards fatty acids than the wild-type. Residue L354 is determinant for the enzyme ω-regioselectivity.
Co-reporter:Sabrina Reich;Dr. Hans Wolfgang Hoeffken;Dr. Bettina Rosche;Dr. Bettina M. Nestl; Dr. Bernhard Hauer
ChemBioChem 2012 Volume 13( Issue 16) pp:2400-2407
Publication Date(Web):
DOI:10.1002/cbic.201200404
Abstract
The crystal structure of the “ene” nicotinamide-dependent cyclohexenone reductase (NCR) from Zymomonas mobilis (PDB ID: 4A3U) has been determined in complex with acetate ion, FMN, and nicotinamide, to a resolution of 1.95 Å. To study the activity and enantioselectivity of this enzyme in the bioreduction of activated α,β-unsaturated alkenes, the rational design methods site- and loop-directed mutagenesis were applied. Based on a multiple sequence alignment of various members of the Old Yellow Enzyme family, eight single-residue variants were generated and investigated in asymmetric bioreduction. Furthermore, a structural alignment of various ene reductases predicted four surface loop regions that are located near the entrance of the active site. Four NCR loop variants, derived from loop-swapping experiments with OYE1 from Saccharomyces pastorianus, were analysed for bioreduction. The three enzyme variants, P245Q, D337Y and F314Y, displayed increased activity compared to wild-type NCR towards the set of substrates tested. The active-site mutation Y177A demonstrated a clear influence on the enantioselectivity. The loop-swapping variants retained reduction efficiency, but demonstrated decreased enzyme activity compared with the wild-type NCR ene reductase enzyme.
Co-reporter:Stephan C. Hammer, Jörg M. Dominicus, Per-Olof Syrén, Bettina M. Nestl, Bernhard Hauer
Tetrahedron 2012 68(37) pp: 7624-7629
Publication Date(Web):
DOI:10.1016/j.tet.2012.06.041
Co-reporter:Daniel Scheps, Sumire Honda Malca, Helen Hoffmann, Bettina M. Nestl and Bernhard Hauer
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 19) pp:6727-6733
Publication Date(Web):27 Jun 2011
DOI:10.1039/C1OB05565H
The oxofunctionalization of saturated hydrocarbons is an important goal in basic and applied chemistry. Biocatalysts like cytochrome P450 enzymes can introduce oxygen into a wide variety of molecules in a very selective manner, which can be used for the synthesis of fine and bulk chemicals. Cytochrome P450 enzymes from the CYP153A subfamily have been described as alkane hydroxylases with high terminal regioselectivity. Here we report the product yields resulting from C5–C12alkane and alcohol oxidation catalyzed by CYP153A enzymes from Mycobacterium marinum (CYP153A16) and Polaromonas sp. (CYP153A P. sp.). For all reactions, byproduct formation is described in detail. Following cloning and expression in Escherichia coli, the activity of the purified monooxygenases was reconstituted with putidaredoxin (CamA) and putidaredoxin reductase (CamB). Although both enzyme systems yielded primary alcohols and α,ω-alkanediols, each one displayed a different oxidation pattern towards alkanes. For CYP153A P. sp. a predominant ω-hydroxylation activity was observed, while CYP153A16 possessed the ability to catalyze both ω-hydroxylation and α,ω-dihydroxylation reactions.
Co-reporter:Mina Lalli;Dr. Sra J. Facey ; Dr. Bernhard Hauer
ChemBioChem 2011 Volume 12( Issue 10) pp:1519-1521
Publication Date(Web):
DOI:10.1002/cbic.201100210
Co-reporter:Danni Liu;Peter Trodler;Sabine Eiben;Katja Koschorreck;Monika Müller;Jürgen Pleiss ;Steffen C. Maurer;Cecilia Branneby;Rolf D. Schmid
ChemBioChem 2010 Volume 11( Issue 6) pp:789-795
Publication Date(Web):
DOI:10.1002/cbic.200900776
Abstract
Pseudozyma antarctica lipase B (CALB) shows activity in the acrylation of hydroxypropylcarbamate, a racemic mixture of enantiomers of primary and secondary alcohols. However, full conversion is hampered by the slowly reacting S enantiomer of the secondary alcohol. The same is true for a wide range of secondary alcohols, for example, octan-2- and -3-ol. In order to get high conversion in these reactions in a short time, the stereospecificity pocket of CALB was redesigned by using predictions from molecular modeling. Positions 278, 104, and 47 were targeted, and a library for two-site saturation mutagenesis at positions 104 and 278 was constructed. The library was then screened for hydrolysis of acrylated hydroxypropylcarbamates. The best mutants L278A, L278V, L278A/W104F, and L278A/W104F/S47A showed an increased conversion in hydrolysis and transesterification of more than 30 %. While the wild-type showed only 73 % conversion in the acrylation of hydroxypropylcarbamate after 6 h, 97 % conversion was achieved by L278A in this time. Besides this, L278A/W104F reached >96 % conversion in the acrylation of octan-2- and -3-ol within 48 h and showed a significant decrease in stereoselectivity, while the wild-type reached only 68 and 59 % conversion, respectively. Thus the new biocatalysts can be used for efficient transformation of racemic alcohols and esters with high activity when the high stereoselectivity of the wild-type hampers complete conversion of racemic substrates in a short time.
Co-reporter:Marcus Schallmey, Peter Jekel, Lixia Tang, Maja Majerić Elenkov, Hans Wolfgang Höffken, Bernhard Hauer, Dick B. Janssen
Enzyme and Microbial Technology (March 2015) Volume 70() pp:50-57
Publication Date(Web):1 March 2015
DOI:10.1016/j.enzmictec.2014.12.009
•Halohydrin dehalogenase catalyzes epoxide-ring opening with cyanide.•A single point mutation increases the rate of β-hydroxynitrile synthesis.•Crystal structures indicate that proton transfer to the epoxide is accelerated.The cyanide-mediated ring opening of epoxides catalyzed by halohydrin dehalogenases yields β-hydroxynitriles that are of high interest for synthetic chemistry. The best studied halohydrin dehalogenase to date is the enzyme from Agrobacterium radiobacter, but this enzyme (HheC) exhibits only low cyanolysis activities. Sequence comparison between a pair of related halohydrin dehalogenases from Corynebacterium and Mycobacterium suggested that substitution of a threonine that interacts with the active site might be responsible for the higher cyanolytic activity of the former enzyme. Here we report that a variant of HheC in which this substitution (T134A) is adopted displays an up to 11-fold higher activity in cyanide-mediated epoxide ring-opening. The mutation causes removal of the hydrogen bond between residue 134 and the side chain O of the active site serine 132, which donates a hydrogen bond to the substrate oxygen. The mutation also increases dehalogenase rates with various substrates. Structural analysis revealed that the anion-binding site of the mutant enzyme remained unaltered, showing that the enhanced activity is due to altered interactions with the substrate oxygen rather than changes in the nucleophile binding site.
Co-reporter:Sabrina Reich, Nico Kress, Bettina M. Nestl, Bernhard Hauer
Journal of Structural Biology (February 2014) Volume 185(Issue 2) pp:228-233
Publication Date(Web):1 February 2014
DOI:10.1016/j.jsb.2013.04.004
The engineering of protein stability is of major importance for the application of enzymes in a wide range of industrial applications. Here we study the determinants of the thermo- and solvent stability of the Zymomonas mobilis ene reductase NCR using a rational protein engineering approach based on analyses of structural and sequence data. We designed and created two loop mutants with the aim to increase their overall stability. They all retained catalytic activity but exhibited altered thermostability relative to the wild-type enzyme. The modulation of one specific loop segment near the active site of NCR showed an increased tolerance to organic solvents along with an enhanced thermostability.
Co-reporter:Miriam Seitz, Janosch Klebensberger, Sascha Siebenhaller, Michael Breuer, Gabriele Siedenburg, Dieter Jendrossek, Bernhard Hauer
Journal of Molecular Catalysis B: Enzymatic (December 2012) Volume 84() pp:72-77
Publication Date(Web):1 December 2012
DOI:10.1016/j.molcatb.2012.02.007
Squalene-hopene cyclases (SHC; EC 5.4.99.17) catalyze the cyclization of triterpenoids via cationic intermediates in one of the most complex reactions known in biochemistry. In this study, we report the functional expression of a novel SHC from the ethanol producing bacterium Zymomonas mobilis (ZmoSHC1; YP_163283.1). Biochemical characterization of ZmoSHC1 uncovered unique substrate activity patterns compared to the previously reported AacSHC from Alicyclobacillus acidocaldarius and ZmoSHC2, the second squalene-hopene cyclase from Z. mobilis. ZmoSHC1 showed cyclization of the non-natural substrates homofarnesol (C16) and citronellal (C10) in addition to hopene formation from squalene (C30). Moreover, ZmoSHC1 turned out to reveal high biocatalytic stability during long-term incubations. Remarkably, ZmoSHC1 exhibited a shift of activity towards substrates of shorter chain lengths, displaying over 50-fold higher conversion of homofarnesol and more than 2-fold higher conversion of citronellal in comparison to squalene conversion.Graphical abstractDownload full-size imageHighlights► We cloned and heterologously expressed a novel squalene-hopene cyclase (SHC) of the ethanol producing bacterium Zymomonas mobilis (ZmoSHC1). ► We established biotransformation setups for three model reactions using three SHCs as biocatalysts. ► Unexpected activity of ZmoSHC1 towards substrates of shorter chain lengths as well as high biochemical stability could be shown.
Co-reporter:Marko Kirtz, Janosch Klebensberger, Konrad B. Otte, Sven M. Richter, Bernhard Hauer
Journal of Biotechnology (20 July 2016) Volume 230() pp:30-33
Publication Date(Web):20 July 2016
DOI:10.1016/j.jbiotec.2016.05.017
•E. coli as a bacterial host for the de novo production of mid-chain fatty acids.•Engineering E. coli for the increased production of octanoic acid.•Establishing a whole-cell biocatalyst for the production of ω-hydroxy octanoic acid.The present proof-of-concept study reports the construction of a whole-cell biocatalyst for the de novo production of ω-hydroxy octanoic acid. This was achieved by hijacking the natural fatty acid cycle and subsequent hydroxylation using a specific monooxygenase without the need for the additional feed of alkene-like precursors. For this, we used the model organism Escherichia coli and increased primarily the release of the octanoic acid precursors by overexpressing the plant thioesterase FatB2 from Cuphea hookeriana in a β-oxidation deficient strain, which lead to the production of 2.32 mM (8.38 mg gcww−1) octanoic acid in 24 h. In order to produce the corresponding ω-hydroxy derivative, we additionally expressed the engineered self-sufficient monooxygenase fusion protein CYP153AMaq(G307A)-CPRBM3 within the octanoic acid producing strain. With this, we finally produced 234 μM (0.95 mg gcww−1) ω-hydroxy octanoic acid in a 20 h fed-batch set-up.
Co-reporter:Sumire Honda Malca, Daniel Scheps, Lisa Kühnel, Elena Venegas-Venegas, Alexander Seifert, Bettina M. Nestl and Bernhard Hauer
Chemical Communications 2012 - vol. 48(Issue 42) pp:NaN5117-5117
Publication Date(Web):2012/03/26
DOI:10.1039/C2CC18103G
CYP153A from Marinobacter aquaeolei has been identified as a fatty acid ω-hydroxylase with a broad substrate range. Two hotspots predicted to influence substrate specificity and selectivity were exchanged. Mutant G307A is 2- to 20-fold more active towards fatty acids than the wild-type. Residue L354 is determinant for the enzyme ω-regioselectivity.
Co-reporter:Martin J. Weissenborn, Sandra Notonier, Sarah-Luise Lang, Konrad B. Otte, Susanne Herter, Nicholas J. Turner, Sabine L. Flitsch and Bernhard Hauer
Chemical Communications 2016 - vol. 52(Issue 36) pp:NaN6161-6161
Publication Date(Web):2016/04/07
DOI:10.1039/C6CC01749E
A readily available galactose oxidase (GOase) variant was used to develop a whole cell screening assay. This endpoint detection system was applied in a proof-of-concept approach by screening a focussed mutant library. This led to the discovery of the thus far most active P450 Marinobacter aquaeolei mutant catalysing the terminal hydroxylation of fatty acids.
Co-reporter:Daniel Scheps, Sumire Honda Malca, Helen Hoffmann, Bettina M. Nestl and Bernhard Hauer
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 19) pp:NaN6733-6733
Publication Date(Web):2011/06/27
DOI:10.1039/C1OB05565H
The oxofunctionalization of saturated hydrocarbons is an important goal in basic and applied chemistry. Biocatalysts like cytochrome P450 enzymes can introduce oxygen into a wide variety of molecules in a very selective manner, which can be used for the synthesis of fine and bulk chemicals. Cytochrome P450 enzymes from the CYP153A subfamily have been described as alkane hydroxylases with high terminal regioselectivity. Here we report the product yields resulting from C5–C12alkane and alcohol oxidation catalyzed by CYP153A enzymes from Mycobacterium marinum (CYP153A16) and Polaromonas sp. (CYP153A P. sp.). For all reactions, byproduct formation is described in detail. Following cloning and expression in Escherichia coli, the activity of the purified monooxygenases was reconstituted with putidaredoxin (CamA) and putidaredoxin reductase (CamB). Although both enzyme systems yielded primary alcohols and α,ω-alkanediols, each one displayed a different oxidation pattern towards alkanes. For CYP153A P. sp. a predominant ω-hydroxylation activity was observed, while CYP153A16 possessed the ability to catalyze both ω-hydroxylation and α,ω-dihydroxylation reactions.







![Oxirane, 2-methyl-2-[(1R)-4-methyl-3-cyclohexen-1-yl]-](http://img.cochemist.com/ccimg/184500/184488-93-5.png)
![Oxirane, 2-methyl-2-[(1R)-4-methyl-3-cyclohexen-1-yl]-](http://img.cochemist.com/ccimg/184500/184488-93-5_b.png)


















































![Benzene, [(3,7-dimethyl-2,6-octadienyl)oxy]-, (E)-](/data/chemimg/1204200/35266-82-1.png)
![Benzene, [(3,7-dimethyl-2,6-octadienyl)oxy]-, (E)-](/data/chemimg/1204200/35266-82-1_b.png)


























![Benzene, [(3-methyl-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/14400/14309-15-0.png)
![Benzene, [(3-methyl-2-butenyl)oxy]-](http://img.cochemist.com/ccimg/14400/14309-15-0_b.png)








































![Phenol,4,4'-[(1S,3aR,4S,6aR)-tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl]bis[2-methoxy-](http://img.cochemist.com/ccimg/500/487-36-5.png)
![Phenol,4,4'-[(1S,3aR,4S,6aR)-tetrahydro-1H,3H-furo[3,4-c]furan-1,4-diyl]bis[2-methoxy-](http://img.cochemist.com/ccimg/500/487-36-5_b.png)








![S-[(5-methoxy-2-oxo-1,3,4-thiadiazol-3(2H)-yl)methyl] O,O-dimethyl phosphorothioate](http://img.cochemist.com/ccimg/39900/39856-16-1.png)
![S-[(5-methoxy-2-oxo-1,3,4-thiadiazol-3(2H)-yl)methyl] O,O-dimethyl phosphorothioate](http://img.cochemist.com/ccimg/39900/39856-16-1_b.png)












![(3s,3as,5ar,5br,7as,11as,11br,13ar,13bs)-5a,5b,8,8,11a,13b-hexamethyl-3-prop-1-en-2-yl-1,2,3,3a,4,5,6,7,7a,9,10,11,11b,12,13,13a-hexadecahydrocyclopenta[a]chrysene](http://img.cochemist.com/ccimg/1700/1615-91-4.png)
![(3s,3as,5ar,5br,7as,11as,11br,13ar,13bs)-5a,5b,8,8,11a,13b-hexamethyl-3-prop-1-en-2-yl-1,2,3,3a,4,5,6,7,7a,9,10,11,11b,12,13,13a-hexadecahydrocyclopenta[a]chrysene](http://img.cochemist.com/ccimg/1700/1615-91-4_b.png)



