Co-reporter:Lara E. Lemmerz, Valeri Leich, Daniel Martin, Thomas P. Spaniol, and Jun Okuda
Inorganic Chemistry December 18, 2017 Volume 56(Issue 24) pp:14979-14979
Publication Date(Web):December 1, 2017
DOI:10.1021/acs.inorgchem.7b02233
The magnesium triphenylsilyl complex [(Me3TACD)Mg(SiPh3)] (2) was synthesized from magnesium bis(triphenylsilyl) [Mg(SiPh3)2(THF)2]·THF (1; THF = tetrahydrofuran) and the NNNN-type macrocyclic amidotriamine proligand (Me3TACD)H ((Me3TACD)H = Me3[12]aneN4 = 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane). Treating 2 with AlR3 (R = Me, Et) gave the magnesium triphenylsilyl complexes with “blocked” amido function [(Me3TACD·AlR3)Mg(SiPh3)] (3a: R = Me; 3b: R = Et). Instead of forming a Mg–H bond upon reaction with dihydrogen or hydrosilanes, 2 and 3a,b underwent rapid silyl–silane exchange with hydrosilanes. Treating the ethyl complex [(Me3TACD·AlEt3)MgEt] with H3SiPh gave [(Me3TACD·AlEt3)MgH] (4), albeit not in a reproducible manner. The silyl–hydrosilane exchange allows access to other magnesium silyls of the type [(Me3TACD)Mg(SiR′3)] (5a: SiR′3 = SiH2Ph; 5b: SiR′3 = SiHPh2) and [(Me3TACD·AlR3)Mg(SiR′3)] (6a: SiR′3 = SiH2Ph, R = Me; 6b: SiR′3 = SiH2Ph, R = Et; 7a: SiR′3 = SiHPh2, R = Me; 7b: SiR′3 = SiHPh2, R = Et). The reaction of 2 with H2SiMePh in THF at room temperature resulted in an equilibrium (Keq ≈ 1). Protonolysis of 2 with Brønsted acids (HX) 2,5-di-tert-butylphenol, phenylacetylene, acetophenone, aniline, and triethylammonium chloride each gave a compound [(Me3TACD)Mg(X)] with the conjugated base coordinated at the magnesium along with HSiPh3. The magnesium silyls 1, 2, and 7b as well as the magnesium hydride 4 contain a distorted square-pyramidal magnesium center according to single-crystal X-ray diffraction.
Co-reporter:S. Schnitzler;P. Cui;T. P. Spaniol;J. Okuda
Dalton Transactions 2017 vol. 46(Issue 6) pp:1761-1765
Publication Date(Web):2017/02/14
DOI:10.1039/C6DT04654A
Molecular magnesium hydride with a terminal metal–hydrogen bond [Mg(iPr2TACN·AliBu3)H]2 supported by a monoanionic TACN-type ligand (TACN = 1,4-di-isopropyl-1,4,7-triazacyclononane) rearranges to form the dinuclear magnesium hydride [Mg(iPr2TACN·AlHiBu2)(μ-H)]2, exhibiting a rare Mg⋯H–Al interaction.
Co-reporter:Debabrata Mukherjee;Ann-Kristin Wiegand;Thomas P. Spaniol
Dalton Transactions 2017 vol. 46(Issue 19) pp:6183-6186
Publication Date(Web):2017/05/15
DOI:10.1039/C7DT01094J
The zinc hydridotriphenylborates [(L)Zn(TMDS)][HBPh3] and [(L)ZnX][HBPh3] (L = Me4TACD, Me4[12]aneN4; TMDS = N(SiHMe2)2; X = Cl, Br, I) were synthesized by BPh3-mediated β-SiH abstraction and salt metathesis with KHBPh3, respectively. CO2 is rapidly inserted into the B–H bonds. [(L)Zn(TMDS)][HBPh3] catalyzes the hydroboration of polar substrates including CO2.
Co-reporter:Debabrata Mukherjee;Hassan Osseili;Thomas P. Spaniol
Dalton Transactions 2017 vol. 46(Issue 25) pp:8017-8021
Publication Date(Web):2017/06/27
DOI:10.1039/C7DT01671A
The macrocyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane [LH = (Me3TACD)H] formed adducts with tetramethyl-silazides [M{N(SiHMe2)2}] (M = Li, Na, K) of light alkali metals. Upon heating, intramolecular dehydrocoupling occurred to give [M{(L)SiMe2N(SiHMe2)}]. BPh3 induced facile ring-opening of THF when reacted with [Li{(L)SiMe2N(SiHMe2)}].
Co-reporter:
Journal of Polymer Science Part A: Polymer Chemistry 2017 Volume 55(Issue 1) pp:175-180
Publication Date(Web):2017/01/01
DOI:10.1002/pola.28370
ABSTRACTPoly(styrene) microgels are known, but the influence of tacticity on particle formation and behavior has not been investigated yet. Isotactic poly(styrene) (iPS) with Mn = 15–120 kg/mol is synthesized by coordinate polymerization and cross-linked by Friedel–Crafts alkylation in a miniemulsion. Nuclear magnetic resonance (NMR) spectroscopy, light microscopy, cryogenic transmission electron microscopy, and wide-angle powder diffraction are applied to understand the structure of microgels obtained. Typically, spherical microgels with overall diameter of 40–500 nm are found. Isotacticity of the polymer is retained during microgel formation. Increase of cross-linker content leads to partial crystallinity inside the microgel. © 2016 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 175–180
Co-reporter:Danny Schuhknecht;Carolin Lhotzky;Dr. Thomas P. Spaniol; Dr. Laurent Maron; Dr. Jun Okuda
Angewandte Chemie 2017 Volume 129(Issue 40) pp:12539-12543
Publication Date(Web):2017/09/25
DOI:10.1002/ange.201706848
AbstractDie Reaktion des Dibenzylcalcium-Komplexes [Ca(Me4TACD)(CH2Ph)2], stabilisiert durch den neutralen, makrocyclischen Liganden 1,4,7,10-Tetramethyl-1,4,7,10-tetra<-azacyclododecan (Me4TACD), mit Triphenylsilan führte zu dem kationischen, zweikernigen Calciumhydrid [Ca2H2(Me4TACD)2](PhCHSiPh3)2, das mithilfe von NMR-Spektroskopie und Einkristallstrukturanalyse charakterisiert wurde. Das Kation kann als eine durch Liganden stabilisierte, dimere Form des hypothetischen [CaH]+ angesehen werden. Die Hydrierung des Benzylcalcium-Kations [Ca(Me4TACD)(CH2Ph)(thf)]+ führte zu dikationischen Calciumhydriden [Ca2H2(Me4TACD)2][BAr4]2 (Ar=C6H4-4-tBu; C6H3-3,5-Me2) mit schwach koordinierenden Anionen. In THF katalysierten die Komplexe den Isotopenaustausch zwischen H2 und D2 zu HD sowie die Hydrierung von 1-Alkenen.
Co-reporter:Debabrata Mukherjee;Thomas P. Spaniol
Dalton Transactions 2017 vol. 46(Issue 3) pp:651-655
Publication Date(Web):2017/01/17
DOI:10.1039/C6DT04379H
A macrocyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, (Me3TACD)H formed thermally stable adducts with MMe3 (M = Al, In). The reactions with cationic dialkyls [MR2]+ directly provided cationic monoalkyls [(Me3TACD)MR]+ under RH elimination. Reactions between [Li(Me3TACD)]2 and Me2MCl gave chloro-bridged heterobimetallic adducts.
Co-reporter:Debabrata Mukherjee;Satoru Shirase;Klaus Beckerle;Thomas P. Spaniol;Kazushi Mashima
Dalton Transactions 2017 vol. 46(Issue 26) pp:8451-8457
Publication Date(Web):2017/07/04
DOI:10.1039/C7DT01727H
Heteroleptic bis(silyl)amides of magnesium and calcium [(L)M{N(SiMe3)2}] [M = Mg, Ca; LH = 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane; (Me3TACD)H] were previously synthesized from LH and [M{N(SiMe3)2}2]. Strontium bis(silyl)amides [Sr{N(SiMe3)2}2(thf)2] and [Sr{N(SiHMe2)2}2(thf)2/3] reacted with LH to give different types of products, depending on the presence of the β-SiH function. While the former underwent protonolysis to give the amido-bridged dimer [(L)Sr{N(SiMe3)2}]2 (1), the latter gave the adduct [(LH)Sr{N(SiHMe2)2}2] (2) as a stable solid. 2 slowly underwent an intramolecular Si–H/H–N dehydrocoupling in solution to give [{(L)SiMe2N(SiHMe2)}Sr{N(SiHMe2)2}] (3) by liberating H2. The results of transamination of 1 with HN(SiHMe2)2 depended on the relative stoichiometric ratio. A 1 : 1 mixture in n-pentane gave [{(L)SiMe2N(SiHMe2)}Sr{N(SiMe3)2}] (4) and H2, while excess HN(SiHMe2)2 gave the adduct 2 under similar conditions. Compounds 2 and 3 exhibit Sr↼H–Si interactions according to X-ray crystallography, NMR, and IR spectroscopy. Lighter congeners of elusive [(L)Sr{N(SiHMe2)2}] were isolable for Mg (5) and Ca (6).
Co-reporter:Danny Schuhknecht;Carolin Lhotzky;Dr. Thomas P. Spaniol; Dr. Laurent Maron; Dr. Jun Okuda
Angewandte Chemie International Edition 2017 Volume 56(Issue 40) pp:12367-12371
Publication Date(Web):2017/09/25
DOI:10.1002/anie.201706848
AbstractReaction of dibenzyl calcium complex [Ca(Me4TACD)(CH2Ph)2], containing the neutral NNNN-type macrocyclic ligand Me4TACD (Me4TACD=1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane), with triphenylsilane gave the cationic dinuclear calcium hydride [Ca2H2(Me4TACD)2](PhCHSiPh3)2 which was characterized by NMR spectroscopy and single-crystal X-ray diffraction. The cation can be regarded as the ligand-stabilized dimeric form of hypothetical [CaH]+. Hydrogenolysis of benzyl calcium cation [Ca(Me4TACD)(CH2Ph)(thf)]+ gave dicationic calcium hydrides [Ca2H2(Me4TACD)2][BAr4]2 (Ar=C6H4-4-tBu; C6H3-3,5-Me2) containing weakly coordinating anions. In THF, they catalyzed the isotope exchange of H2 and D2 to give HD and the hydrogenation of unactivated 1-alkenes.
Co-reporter:Jun Okuda
Coordination Chemistry Reviews 2017 Volume 340(Volume 340) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.ccr.2016.09.009
•This review summarizes and classifies cationic rare-earth metal hydrides, a relatively recent addition to the growing body of molecular rare-earth metal hydrides.•The latter has been reviewed several times in the literature.•The introduction of cationic charges can be expected to reduce the nuclearity of rare-earth metal hydride fragments, at the same time to result in increase of Lewis acidity and electrophilicity.A survey of the literature on cationic rare-earth metal hydride complexes is presented. This overview follows systematics based on the nature of supporting ligands and provides comparisons between neutral and cationic hydrides with respect to their structure and reactivity.
Co-reporter:Debabrata Mukherjee;Hassan Osseili;Khai-Nghi Truong;Thomas P. Spaniol
Chemical Communications 2017 vol. 53(Issue 24) pp:3493-3496
Publication Date(Web):2017/03/21
DOI:10.1039/C7CC01159H
Molecular aluminum hydride [(L)AlH2] (L = Me3TACD) reacted with 2 equiv. of BPh3 in THF or THP to give the cationic alkoxides [(L)Al(OR)][HBPh3] (R = nBu, nPent) by facile ring-opening of the cyclic ethers. The Cα–O bond cleavage which involves the isolable intermediate [(L)AlH][HBPh3] is a result of hydride transfer to Cα from [HBPh3]−.
Co-reporter:Ann-Kristin Wiegand, Arnab Rit, Jun Okuda
Coordination Chemistry Reviews 2016 Volume 314() pp:71-82
Publication Date(Web):1 May 2016
DOI:10.1016/j.ccr.2015.08.010
•This review collates reports on molecular zinc hydrides.•Systematic follows ligands used to support zinc hydrides.•Synthetic procedures for molecular hydrides are collected.•NMR spectroscopic and crystallographic data are discussed.•Catalytic applications of zinc hydrides are discussed.An overview of structurally characterized molecular organozinc hydrides is given. The compounds are presented based on the type of supporting ligand. Their synthesis, structure, reactivity, and catalytic applications are summarized in this overview.
Co-reporter:Debabrata Mukherjee, Hassan Osseili, Thomas P. Spaniol, and Jun Okuda
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10790-10793
Publication Date(Web):August 11, 2016
DOI:10.1021/jacs.6b06319
Light alkali metal hydridotriphenylborates M[HBPh3] (M = Li, Na, K), characterized as tris{2-(dimethylamino)ethyl}amine (L) complexes [(L)M][HBPh3], act as efficient catalysts for the chemoselective hydroboration of a wide range of aldehydes and ketones using pinacolborane HBpin. The lithium derivative showed a remarkably high TOF of ≥17 s–1. These compounds also catalyze the hydroborative reduction of CO2 to give formoxyborane HCO2Bpin without any over-reduction.
Co-reporter:Silvia Schnitzler, Thomas P. Spaniol, and Jun Okuda
Inorganic Chemistry 2016 Volume 55(Issue 24) pp:12997-13006
Publication Date(Web):December 1, 2016
DOI:10.1021/acs.inorgchem.6b02509
The reactivity of the molecular magnesium hydride [Mg(Me3TACD·AliBu3)H] (1) featuring a terminal magnesium–hydrogen bond and an NNNN-type macrocyclic ligand, Me3TACD ((Me3TACD)H = Me3[12]aneN4 = 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane), can be grouped into protonolysis, oxidation, hydrometalation, (insertion), and hydride abstraction. Protonolysis of 1 with weak Brønsted acids HX such as terminal acetylenes, amines, silanols, and silanes gave the corresponding derivatives [Mg(Me3TACD·AliBu3)X] (X = C≡CPh, 3; HN(3,5-Me2-C6H3), 4; OSiMe3, 5; OSiPh3, 6; Cl, 7; Br, 8). Single-crystal X-ray diffraction of anilide 4 showed a square-pyramidal coordination geometry for magnesium. No correlation with the pKa values of the acids was detected. Oxidation of 1 with elemental iodine gave the iodide [Mg(Me3TACD·AliBu3)I] (9), and oxidation with nitrous oxide afforded the μ-oxo-bridged compound [{Mg(Me3TACD·AliBu3)}2(μ-O)] (10) with a linear Mg–O–Mg core, as characterized by single-crystal X-ray diffraction. The Mg–H bond reacted with benzaldehyde, benzophenone, fluorenone, and CO2 under insertion but not with the olefins 1,1,2-triphenylethylene, tert-butylethylene, and cyclopentene. The unstable formate, prepared also by salt metathesis of iodide 9 with potassium formate, revealed κO,κO′ coordination in the solid state. Hydride abstraction with triphenylborane gave the ion pair [Mg(Me3TACD·AliBu3)(thf)][HBPh3] (16), which catalyzed the hydroboration of polar substrates by pinacolborane.
Co-reporter:Catherine Hermans, Weifeng Rong, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2016 vol. 45(Issue 19) pp:8127-8133
Publication Date(Web):11 Apr 2016
DOI:10.1039/C6DT00272B
Lanthanum complexes [(L)LaX] (X = N(SiMe3)21, OiPr 2, BH43) supported by a ferrocene-based (OSSO)-type ligand LH2 were synthesized and characterized by elemental analysis, NMR spectroscopy and cyclic voltammetry. The structure of 1 was confirmed by single crystal X-ray diffraction. These complexes were highly active initiators for the ring-opening polymerization of rac-lactide (rac-LA). The activity depended on the initiating group in the order of 1 ≈ 2 > 3. The activities of 2 and 3 during polymerization were controlled in situ with external redox reagents by reversibly switching the oxidation state of the iron center.
Co-reporter:Tomoki Himiyama, Daniel F. Sauer, Akira Onoda, Thomas P. Spaniol, Jun Okuda, Takashi Hayashi
Journal of Inorganic Biochemistry 2016 Volume 158() pp:55-61
Publication Date(Web):May 2016
DOI:10.1016/j.jinorgbio.2015.12.026
•A terpyridine ligand is linked to nitrobindin within its hydrophobic cavity.•Cu2 +, Zn2 +, and Co2 + ions bind to the terpyridine in the cavity of nitrobindin.•A Cu2 +-bound NB–terpyridine hybrid catalyzes the Diels–Alder reaction.A hybrid biocatalyst containing a metal terpyridine (tpy) complex within a rigid β-barrel protein nitrobindin (NB) is constructed. A tpy ligand with a maleimide group, N-[2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethyl]maleimide (1), was covalently linked to Cys96 inside the cavity of NB to prepare a conjugate NB–1. Binding of Cu2 +, Zn2 +, or Co2 + ion to the tpy ligand in NB–1 was confirmed by UV–vis spectroscopy and ESI–TOF MS measurements. Cu2 +-bound NB–1 is found to catalyze a Diels–Alder reaction between azachalcone and cyclopentadiene in 22% yield, which is higher than that of the Cu2 +–tpy complex without the NB matrix. The results suggest that the hydrophobic cavity close to the copper active site within the NB scaffold supports the binding of the two substrates, dienophile and diene, to promote the reaction.Hybrid biocatalysts combining metal terpyridine complexes and β-barrel nitrobindin protein were prepared to catalyze a Diels–Alder reaction. The Cu2 +–terpyridine complex embedded within nitrobindin exhibits higher catalytic activity than the Cu2 +–terpyridine complex without a protein matrix. This demonstrates the importance of the outer coordination sphere provided by the protein cavity.
Co-reporter:Klaus Beckerle, Andreas Sauer, Thomas P. Spaniol, Jun Okuda
Polyhedron 2016 Volume 116() pp:105-110
Publication Date(Web):25 September 2016
DOI:10.1016/j.poly.2016.03.053
Bio-based polyols can be converted to olefins and furan derivatives in one step by combined reduction and dehydration (deoxydehydration, DODH). A series of octahedral complexes of hexavalent molybdenum containing an (OSSO)-type bis(phenolate) ligand were prepared and structurally characterized. These complexes were screened as catalyst precursors for the deoxydehydration of anhydroerythritol using 3-octanol as reducing agent. Microwave heating allows a lower reaction temperature.Octahedral complexes of hexavalent molybdenum containing an (OSSO)-type bis(phenolate) ligand were prepared and structurally characterized. These complexes were screened as catalysts for the deoxydehydration of anhydroerythritol using 3-octanol as reducing agent.
Co-reporter:Haruka Nishiyama, Keishi Yamamoto, Andreas Sauer, Hideaki Ikeda, Thomas P. Spaniol, Hayato Tsurugi, Kazushi Mashima, and Jun Okuda
Organometallics 2016 Volume 35(Issue 7) pp:932-935
Publication Date(Web):March 17, 2016
DOI:10.1021/acs.organomet.5b00855
A tungsten alkylidyne complex [{(OC6H2tBu2-4,6)2(SCH2CH2S)}W(≡CEt)X] (X = OtBu, 1a) with a bridged bis(phenolate) ligand was prepared from the alkoxy precursor (tBuO)3W≡CEt and the corresponding bis(phenol). The tert-butoxy ligand in 1a was substituted against chloride or trimethylsilylmethyl to give 1b (X = Cl) or 1c (X = CH2SiMe3). X-ray diffraction studies of 1a and 1c showed an octahedral cis α-configuration of the metal atom. Hydride abstraction from the β-position of the n-propylidyne ligand in 1a and 1c with [Ph3C][B(C6F5)4] gave high-valent cationic vinylidene complexes [{(OC6H2tBu2-4,6)2(SCH2CH2S)}W(═C═CHMe)X][B(C6F5)4] (2a: X = OtBu; 2c: X = CH2SiMe3), while protonation of 1a with [PhNMe2H][B(C6F5)4] gave the cationic n-propylidene complex [{(OC6H2tBu2-4,6)2(SCH2CH2S)}W(═CHEt)(OtBu)][B(C6F5)4] (3a). The alkylidyne complex 1a was regenerated from the alkylidene 3a by deprotonation with Ph3P═CH2, whereas 1c was regenerated from 2c by nucleophilic attack by a hydride at the β-carbon of the vinylidene moiety. The cationic tungsten alkylidyne complex [{(OC6H2tBu2-4,6)2(SCH2CH2S)}W(≡CEt)(PhNMe2)][B(C6F5)4] (4c) was obtained from [{(OC6H2tBu2-4,6)2(SCH2CH2S)}W(═CHEt)(CH2SiMe3)][B(C6F5)4] (3c) in chlorobenzene at 60 °C.
Co-reporter:Valeri Leich, Thomas P. Spaniol, and Jun Okuda
Organometallics 2016 Volume 35(Issue 9) pp:1179-1182
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.organomet.6b00160
The alkali-metal silyl [K(L)SiPh3] (1; L = 18-crown-6 ether) catalyzed the hydrosilylation of activated C═C double bonds. Isolation and characterization of an addition product is in agreement with the anti-Markovnikov selectivity. Second-order kinetics for the hydrosilylation of 1,1′-diphenylethylene and the kinetic isotope effect of kH/kD = 3.1 indicate that a silyl migration mechanism is operative.
Co-reporter:Valeri Leich;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie 2016 Volume 128( Issue 15) pp:4872-4876
Publication Date(Web):
DOI:10.1002/ange.201600552
Abstract
Die Hydrierung von Bis(triphenylsilyl)calcium mit dem neutralen makrocyclischen Aminliganden 1,4,7,10-Tetramethyl-1,4,7,10-tetraazacyclododecan (Me4TACD) vom NNNN-Typ, [Ca(Me4TACD)(SiPh3)2] (2), führte zu dem kationischen zweikernigen Calciumhydrid [Ca2H3(Me4TACD)2](SiPh3) (3), das über NMR-Spektroskopie, Einkristallstrukturanalyse und Dichtefunktionalrechnungen charakterisiert wurde. 3 reagierte mit Deuterium zu dem Deuterid [D3]-3.
Co-reporter:Dr. Debabrata Mukherjee;Daniel F. Sauer;Dr. Alessro Zanardi ;Dr. Jun Okuda
Chemistry - A European Journal 2016 Volume 22( Issue 23) pp:7730-7733
Publication Date(Web):
DOI:10.1002/chem.201601006
Abstract
Triphenylborane (BPh3) in highly polar, aprotic solvents catalyzes hydrosilylation of CO2 effectively under mild conditions to provide silyl formates with high chemoselectivity (>95 %) and without over-reduction. This system also promotes reductive hydrosilylation of tertiary amides as well as dehydrogenative coupling of silane with alcohols.
Co-reporter:Valeri Leich;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2016 Volume 55( Issue 15) pp:4794-4797
Publication Date(Web):
DOI:10.1002/anie.201600552
Abstract
Hydrogenolysis of bis(triphenylsilyl)calcium containing the neutral NNNN-type macrocyclic amine ligand Me4TACD [Ca(Me4TACD)(SiPh3)2] (2), gave the cationic dinuclear calcium hydride [Ca2H3(Me4TACD)2](SiPh3) (3), characterized by NMR spectroscopy, single-crystal X-ray analysis, and DFT calculations. Compound 3 reacted with deuterium to give the deuteride [D3]-3.
Co-reporter:Daniel F. Sauer, Tomoki Himiyama, Kengo Tachikawa, Kazuki Fukumoto, Akira Onoda, Eiichi Mizohata, Tsuyoshi Inoue, Marco Bocola, Ulrich Schwaneberg, Takashi Hayashi, and Jun Okuda
ACS Catalysis 2015 Volume 5(Issue 12) pp:7519
Publication Date(Web):November 16, 2015
DOI:10.1021/acscatal.5b01792
A series of Grubbs–Hoveyda type catalyst precursors for olefin metathesis containing a maleimide moiety in the backbone of the NHC ligand was covalently incorporated in the cavity of the β-barrel protein nitrobindin. By using two protein mutants with different cavity sizes and choosing the suitable spacer length, an artificial metalloenzyme for olefin metathesis reactions in water in the absence of any organic cosolvents was obtained. High efficiencies reaching TON > 9000 in the ROMP of a water-soluble 7-oxanorbornene derivative and TON > 100 in ring-closing metathesis (RCM) of 4,4-bis(hydroxymethyl)-1,6-heptadiene in water under relatively mild conditions (pH 6, T = 25–40 °C) were observed.Keywords: biohybrid catalysis; chemogenetic approach; metalloenzymes; metathesis; ruthenium
Co-reporter:V. Leich, T. P. Spaniol and J. Okuda
Chemical Communications 2015 vol. 51(Issue 79) pp:14772-14774
Publication Date(Web):17 Aug 2015
DOI:10.1039/C5CC06187C
Hydrogenation of easily accessible potassium triphenylsilyl [K(Me6TREN)SiPh3] gave the hydrogen storage material α-[KSiH3] in high yields by an unusual hydrogenolytic cleavage of silicon–phenyl bonds.
Co-reporter:Valeri Leich, Thomas P. Spaniol, and Jun Okuda
Inorganic Chemistry 2015 Volume 54(Issue 10) pp:4927-4933
Publication Date(Web):May 1, 2015
DOI:10.1021/acs.inorgchem.5b00527
Protonolysis of bis(triphenylsilyl)calcium [Ca(SiPh3)2(THF)4] (1; THF = tetrahydrofuran) with the NNNN-type macrocyclic amido triamine (Me3TACD)H (TACD = 1,4,7-triazacyclododecane) gave the heteroleptic calcium complex [Ca(Me3TACD)SiPh3] (2) in quantitative yield. Hydrogenolysis of 2 gave the cationic tricalcium dihydride cluster [Ca3H2(Me3TACD)3]+(SiPh3)−·2THF (4a) in high yield with concomitant formation of HSiPh3. In the crystal, 4a consists of a cluster cation and a free triphenylsilyl anion. 1H NMR spectroscopy and deuterium labeling experiments confirmed the selective cleavage of dihydrogen by the highly polar Ca–Si bond in 1.
Co-reporter:Marie Tschage;Seungwhan Jung;Thomas P. Spaniol
Macromolecular Rapid Communications 2015 Volume 36( Issue 2) pp:219-223
Publication Date(Web):
DOI:10.1002/marc.201400386
Co-reporter:Daniel F. Sauer;Dr. Marco Bocola;Claudio Broglia;Marcus Arlt;Dr. Lei-Lei Zhu;Dr. Melanie Brocker;Dr. Ulrich Schwaneberg;Dr. Jun Okuda
Chemistry – An Asian Journal 2015 Volume 10( Issue 1) pp:177-182
Publication Date(Web):
DOI:10.1002/asia.201403005
Abstract
A biohybrid ring-opening olefin metathesis polymerization catalyst based on the reengineered β-barrel protein FhuA ΔCVFtev was chemically modified with respect to the covalently anchored Grubbs–Hoveyda type catalyst. Shortening of the spacer (1,3-propanediyl to methylene) between the N-heterocyclic carbene ligand and the cysteine site 545 increased the ROMP activity toward a water-soluble 7-oxanorbornene derivative. The cis/trans ratio of the double bond in the polymer was influenced by the hybrid catalyst.
Co-reporter:Dr. Waldemar Fegler;Dr. Ajay Venugopal;Dr. Mathias Kramer;Dr. Jun Okuda
Angewandte Chemie International Edition 2015 Volume 54( Issue 6) pp:1724-1736
Publication Date(Web):
DOI:10.1002/anie.201406677
Abstract
Molecular hydrides of the rare-earth metals play an important role as homogeneous catalysts and as counterparts of solid-state interstitial hydrides. Structurally well-characterized non-metallocene-type hydride complexes allow the study of elementary reactions that occur at rare-earth-metal centers and of catalytic reactions involving bonds between rare-earth metals and hydrides. In addition to neutral hydrides, cationic derivatives have now become available.
Co-reporter:Daniel Martin;Dr. Klaus Beckerle;Silvia Schnitzler;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2015 Volume 54( Issue 13) pp:4115-4118
Publication Date(Web):
DOI:10.1002/anie.201411612
Abstract
Large magnesium hydride aggregates [Mg13(Me3TACD)6(μ2-H12)(μ3-H6)][A]2 ((Me3TACD)H=1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane; A=AlEt4, AlnBu4, B{3,5-(CF3)2C6H3}4) were synthesized stepwise from alkyl complexes [Mg2(Me3TACD)R3] (R=Et, nBu) and phenylsilane in the presence of additional MgII ions. The central magnesium atom is octahedrally coordinated by six hydrides as in solid α-MgH2 of the rutile type. Further coordination to six magnesium atoms leads to a substructure of seven edge-sharing octahedra as found in the hexagonal layer of brucite (Mg(OH)2). Upon protonolysis in the presence of 1,2-dimethoxyethane (DME), this cluster was degraded into a tetranuclear dication [Mg2(Me3TACD)(μ-H)2(DME)]2[A]2.
Co-reporter:Silvia Schnitzler;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Chemistry - A European Journal 2015 Volume 21( Issue 32) pp:11330-11334
Publication Date(Web):
DOI:10.1002/chem.201502232
Abstract
A complex featuring a terminal magnesium hydride bond supported by an NNNN macrocyclic ligand, [Mg{Me3TACD⋅Al(iBu)3}H] (3), was formed from its labile Al(iBu)3 adduct. Use of Al(iBu)3 to block the amido nitrogen of the NNNN macrocyclic ligand was essential to prevent aggregation. The structurally characterized compound 3 reacted with BH3 to give the BH4 derivative, whereas Me3SiCCH and PhSiH3 led to the corresponding acetylide and silyl derivative under H2 elimination. Pyridine is inserted into the MgH bond to give selectively the 1,4-dihydropyridinate.
Co-reporter:Dr. Waldemar Fegler;Dr. Ajay Venugopal;Dr. Mathias Kramer;Dr. Jun Okuda
Angewandte Chemie 2015 Volume 127( Issue 6) pp:1744-1757
Publication Date(Web):
DOI:10.1002/ange.201406677
Abstract
Molekulare Hydride der Seltenerdmetalle spielen eine wichtige Rolle als homogene Katalysatoren und als Modelle für Einlagerungshydride im Festkörper. Mit strukturell gut charakterisierten Hydridkomplexen vom Nichtmetallocen-Typ lassen sich Elementarreaktionen studieren, die an Seltenerdmetallzentren ablaufen. Untersuchen lassen sich auch katalytische Reaktionen, bei denen Seltenerdmetall-Hydrid-Bindungen beteiligt sind. Über neutrale Hydride hinaus sind nun auch kationische Derivate zugänglich.
Co-reporter:Daniel Martin;Dr. Klaus Beckerle;Silvia Schnitzler;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie 2015 Volume 127( Issue 13) pp:4188-4191
Publication Date(Web):
DOI:10.1002/ange.201411612
Abstract
Große Magnesiumhydrid-Aggregate [Mg13(Me3TACD)6(μ2-H12)(μ3-H6)][A]2 ((Me3TACD)H=1,4,7-Trimethyl-1,4,7,10-tetraazacyclododecan; A=AlEt4, AlnBu4, B{3,5-(CF3)2C6H3}4) wurden schrittweise aus Alkylkomplexen [Mg2(Me3TACD)R3] (R=Et, nBu) und Phenylsilan in Gegenwart von zusätzlichen MgII-Ionen synthetisiert. Das zentrale Magnesiumatom ist wie in festem α-MgH2 vom Rutil-Typ oktaedrisch von sechs Hydriden koordiniert. Weitere Koordination zu sechs Magnesiumatomen führt zu einer Unterstruktur mit sieben eckenverknüpften Oktaedern, ähnlich einer hexagonalen Schicht von Brucit (Mg(OH)2). Durch Protonolyse in Gegenwart von 1,2-Dimethoxyethan (DME) wurde dieser Cluster zu dem vierkernigen Dikation [Mg2(Me3TACD)(μ-H)2(DME)]2[A]2 abgebaut.
Co-reporter:Heiko Kulinna, Thomas P. Spaniol, and Jun Okuda
Organometallics 2015 Volume 34(Issue 11) pp:2160-2164
Publication Date(Web):October 3, 2014
DOI:10.1021/om500820k
Hydrogenation of bis(trimethylsilylmethyl) zirconium complex [Zr(Me2TACD)(CH2SiMe3)2] (1), prepared by reacting [Zr(CH2SiMe3)4] with 1,7-dimethyl-1,4,7,10-tetraazacyclododecane (Me2TACD)H2, gave dinuclear alkyl hydride complex [Zr(Me2TACD)(CH2SiMe3)2(μ-H)2Zr(Me2TACD)] (2). According to NMR spectroscopic and single-crystal X-ray diffraction studies, 2 exhibits a Cs-symmetrical structure with two distinct zirconium centers, one eight- and the other seven-coordinate, bridged by an amido donor and two hydrides. Abstraction of the trimethylsilylmethyl groups by the weak Brønsted acid [NEt3H][B(3,5-C6H3Cl2)4] gave the monocationic mono(alkyl) dihydride [Zr(Me2TACD)(CH2SiMe3)(μ-H)2Zr(Me2TACD)][B(3,5-C6H3Cl2)4] (3) and the dicationic hydride complex [Zr(Me2TACD)(THF)2(μ-H)2Zr(Me2TACD)][B(3,5-C6H3Cl2)4]2 (4). X-ray crystallography of the cationic complexes 3 and 4 revealed a Cs-symmetrical dinuclear structure derived from that of 2.
Co-reporter:Stefan Arndt, Mathias U. Kramer, Waldemar Fegler, Yumiko Nakajima, Iker Del Rosal, Romuald Poteau, Thomas P. Spaniol, Laurent Maron, and Jun Okuda
Organometallics 2015 Volume 34(Issue 15) pp:3739-3747
Publication Date(Web):July 17, 2015
DOI:10.1021/acs.organomet.5b00422
Monocationic bis(hydrocarbyl)yttrium complexes [YR2(THF)2][A] (R = CH2SiMe3, CH2C6H4-o-NMe2; A = weakly coordinating anion) underwent hydrogenolysis using dihydrogen or phenylsilane to give a mixture of polynuclear solvent-stabilized dihydride cations [YH2(THF)2]n[A]n. The mixture composition as well as the nuclearity n depended on the starting material, solvent, and reaction conditions. NMR spectroscopic data in solution and X-ray diffraction data suggested that the main product was tetranuclear, although conclusive structural data were not obtained. DFT calculations supported a closo-type tetrahedral [YH2(THF)2]44+ core. The hydridic character of these cations was revealed by their reaction with benzophenone to give the bis(diphenylmethoxy) cation [Y(OCHPh2)2(THF)4][AlR4]. The reaction of this cluster with the (NNNN)-type macrocycle Me4TACD under dihydrogen gave the dinuclear tetrahydride dication with quadruply bridging hydride ligands, [Y2(μ-H)4(Me4TACD)2][A]2, analogous to the previously characterized lutetium derivative. NH-acidic (Me3TACD)H gave the dinuclear dihydride dication [Y2(μ-H)2(Me3TACD)2(THF)2][A]2.
Co-reporter:Valeri Leich, Thomas P. Spaniol, Laurent Maron and Jun Okuda
Chemical Communications 2014 vol. 50(Issue 18) pp:2311-2314
Publication Date(Web):07 Jan 2014
DOI:10.1039/C3CC49308C
Bis(triphenylsilyl)calcium [Ca(SiPh3)2(thf)] obtained in high yield as a crystalline ether adduct catalyzes the hydrosilylation of activated C–C double bonds efficiently and regioselectively.
Co-reporter:Peng Cui, Thomas P. Spaniol, Laurent Maron and Jun Okuda
Chemical Communications 2014 vol. 50(Issue 4) pp:424-426
Publication Date(Web):01 Nov 2013
DOI:10.1039/C3CC47805J
A scandium hydride supported by a (NNNN)-type macrocycle consists of a trinuclear cation and a trinuclear anion. The anion shows significantly higher reactivity.
Co-reporter:Valeri Leich, Kevin Lamberts, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2014 vol. 43(Issue 38) pp:14315-14321
Publication Date(Web):07 May 2014
DOI:10.1039/C4DT00916A
Alkali metal triphenylsilyls [Li(12-crown-4)SiPh3]·(thf)0.5 (2), [Na(15-crown-5)SiPh3]·(thf)0.5 (3) and [K(18-crown-6)SiPh3(thf)] (4) were synthesized using 1,1,1-trimethyl-2,2,2-triphenyldisilane (Ph3SiSiMe3) and isolated in high yields. Solid state structures were determined by single crystal X-ray diffraction. These alkali metal silyls catalyzed the regioselective hydrosilylation of 1,1-diphenylethylene to give the anti-Markovnikov product. The presence of crown ethers enhanced the reactivity of the metal silyls in hydrosilylation catalysis.
Co-reporter:Christiane Hohberger;Thomas P. Spaniol
Macromolecular Chemistry and Physics 2014 Volume 215( Issue 20) pp:2001-2006
Publication Date(Web):
DOI:10.1002/macp.201400169
Co-reporter:Dr. Arnab Rit;Dr. Alessro Zanardi;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13273-13277
Publication Date(Web):
DOI:10.1002/anie.201408346
Abstract
The trinuclear cationic zinc hydride cluster [(IMes)3Zn3H4(THF)](BPh4)2 (1) was obtained either by protonation of the neutral zinc dihydride [(IMes)ZnH2]2 with a Brønsted acid or by addition of the putative zinc dication [(IMes)Zn(THF)]2+. A triply bridged thiophenolato complex 2 was formed upon oxidation of 1 with PhSSPh. Protonolysis of 1 by methanol or water gave the corresponding trinuclear dicationic derivatives. At ambient temperature, 1 catalyzed the hydrosilylation of aldehydes, ketones, and nitriles. Carbon dioxide was also hydrosilylated under forcing conditions when using (EtO)3SiH, giving silylformate as the main product.
Co-reporter:Dr. Arnab Rit;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Chemistry – An Asian Journal 2014 Volume 9( Issue 2) pp:612-619
Publication Date(Web):
DOI:10.1002/asia.201301268
Abstract
Methylene-linked bis(N,N′-di-tert-butylimidazol-2-ylidene) 1 reacted with diethylzinc to give dinuclear zinc ethyl compound 2, which contains a formally anionic bis(carbene) ligand as a result of deprotonation of the methylene bridge. The reaction of 2 with PhSiH3 gave the phenylsilyl compound 3. The zinc hydride 4 was obtained by the reaction of 2 with LiAlH4 or Ph3SiOH followed by treatment with PhSiH3. X-ray diffraction studies show that compounds 2, 3, and 4 all have a similar dimeric structure with D2h symmetry. The reaction of hydride 4 with carbon dioxide and N,N′-diisopropylcarbodiimide gave formato (5) and formamidinato (7) derivatives as a result of the insertion of the heterocumulene into both ZnH bonds. Reaction with Ph2CO gave the diphenylmethoxy compound 6. Hydride 4 shows catalytic activity in the hydrosilylation of 1,1-diphenylethylene and methanolysis of silanes.
Co-reporter:Arnab Rit, Thomas P. Spaniol, Laurent Maron, and Jun Okuda
Organometallics 2014 Volume 33(Issue 8) pp:2039-2047
Publication Date(Web):April 11, 2014
DOI:10.1021/om500190c
The (NNNN)-type macrocycle 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane (Me3TACD, 1,4,7-Me3[12]aneN4) reacted with 1 equiv of ZnEt2 under ethane elimination to give the mononuclear ethyl complex [(Me3TACD)ZnEt] (1). Upon treatment of (Me3TACD)H with 2 equiv of ZnEt2, the dinuclear complex [(Me3TACD)(ZnEt)(ZnEt2)] (2) was formed, which was converted with an additional 1 equiv of (Me3TACD)H to 1. Reaction of 1 with PhSiH3 led to the formation of a tetranuclear ethyl hydrido complex [{(Me3TACD)ZnEt}2(ZnEtH)2] (3). Single-crystal X-ray diffraction study revealed 3 to be a centrosymmetric dimer featuring two [(Me3TACD)ZnEt] units coordinated to a [Zn(μ-H)2Zn] core via amido nitrogen atoms of the Me3TACD ligands. Substitution of the two [(Me3TACD)ZnEt] units in 3 by N-heterocyclic carbene IMes [1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene] gave [(IMes)ZnEtH]2 (4b). The mixed alkyl hydrido complexes [(IMes)ZnRH]2 (R = Me, 4a; Et, 4b) were alternatively synthesized in quantitative yield by reacting [(IMes)ZnR2] (R = Me, Et) with [(IMes)ZnH2]2 in 2:1 ratio. Methyl complex 4a reacted with CO2 (p(CO2) = 0.5 bar) under facile insertion of CO2 into Zn–H bonds to give dinuclear formate complex [(IMes)ZnMe(O2CH)]2 (5a). Treatment of 4b with CO2 (p(CO2) = 0.5 bar) afforded a mixture of di- and trinuclear formate complexes [(IMes)ZnEt(O2CH)]2 (5b) and [(IMes)2Zn3Et3(O2CH)3] (6) under elimination of one IMes as CO2 adduct IMes·CO2.
Co-reporter:Ajay Venugopal, Floriana Tuna, Thomas P. Spaniol, Liviu Ungur, Liviu F. Chibotaru, Jun Okuda and Richard A. Layfield
Chemical Communications 2013 vol. 49(Issue 9) pp:901-903
Publication Date(Web):11 Dec 2012
DOI:10.1039/C2CC38036F
An experimental and ab initio computational study of an unsymmetrical, hydride-bridged di-dysprosium single-molecule magnet is reported.
Co-reporter:Andreas Sauer, Andreas Kapelski, Christophe Fliedel, Samuel Dagorne, Moshe Kol and Jun Okuda
Dalton Transactions 2013 vol. 42(Issue 25) pp:9007-9023
Publication Date(Web):11 Mar 2013
DOI:10.1039/C3DT00010A
Polylactide (PLA) is an attractive polymeric material due to its origin from annually renewable resources and its biodegradability. The ring-opening polymerization (ROP) of lactide initiated by Lewis acidic and oxophilic metal-based catalysts constitutes the method of choice to access PLA in a controlled and stereoselective manner. The design and synthesis of ligand-supported metal complexes to act as effective ROP initiators of lactide monomers have been the subject of numerous investigations over the past decades. In view of their oxophilic nature, well-defined group 4 metal complexes supported by polydentate supporting ligands have appeared as active initiators for lactide ROP. This perspective summarizes various classes of structurally well-defined group 4 metal initiators developed for lactide ROP. It also provides observed trends regarding their catalytic performance. Whenever appropriate and possible, catalyst structure–ROP performance (i.e. activity, control and stereoselectivity) relationships are rationalized.
Co-reporter:Daniel Martin;Julian Kleemann;Elise Abinet;Thomas P. Spaniol;Laurent Maron
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 22-23) pp:3987-3992
Publication Date(Web):
DOI:10.1002/ejic.201300167
Abstract
Hydrogenation of the neutral bis(allyl) complexes of the early lanthanides [Ln(Me3TACD)(η3-C3H5)2] [(Me3TACD)H = 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, Me3[12]aneN4] with phenylsilane gave the tetranuclear octahydrido complexes [Ln(Me3TACD)(μ-H)2]4 [Ln = Ce (1-Ce), Pr (2-Pr)] or the dinuclear allyl/hydrido complexes [Ln(Me3TACD)(η3-C3H5)(μ-H)]2 [Ln = Nd (3-Nd), Sm (4-Sm)], which were isolated and characterized. The structures of 1-Ce and 2-Pr are constructed of a tetrahedral Ln4H8 core. Single-crystal X-ray diffraction analyses of 3-Nd and 4-Sm revealed a C2 symmetric planar Ln2H2 core. The experimental structures agreed with the results of DFT calculations, which predict that the nuclearity of the dihydrido complexes depend on the ionic radius of the metal. Compounds 1-Ce, 2-Pr, 3-Nd and 4-Sm were tested as catalysts in the copolymerization of cyclohexene oxide with CO2 to give highly carbonate-linked copolymers with moderate activities.
Co-reporter:Dipl.-Chem. Crispin Lichtenberg ;Dr. Jun Okuda
Angewandte Chemie 2013 Volume 125( Issue 20) pp:5336-5354
Publication Date(Web):
DOI:10.1002/ange.201208942
Abstract
Metallorganische Allylverbindungen sind von Bedeutung als Allylierungsreagentien in der organischen Synthese, als Polymerisationskatalysatoren und als flüchtige Metallvorstufen in den Materialwissenschaften. Während die Allylchemie der Lanthanoide und syntheserelevanter Übergangsmetalle wie Palladium gut etabliert ist, bestand für die Allylchemie der Hauptgruppenmetalle Nachholbedarf. Jüngste Erkenntnisse über die Allylkomplexe der Gruppen 1, 2 und 12–16 zeichnen nun ein vollständigeres Bild. Dies beruht auf dem grundlegenden Verständnis der Metall-Allyl-Bindungswechselwirkungen im Festkörper und in Lösung. Außerdem wurden Reaktivitätstrends rational erklärt und neue, allylspezifische Reaktivitätsmuster aufgedeckt. Schlüsselerkenntnisse umfassen 1) die Nutzung unterschiedlicher Metall-Allyl-Wechselwirkungen (stark ionisch bis vorwiegend kovalent), 2) das Nutzen synergetischer Effekte in Heterodimetallverbindungen und 3) die Regulierung der Lewis-Acidität durch Variation der Ladung von Allylkomplexen.
Co-reporter:Dr. Arnab Rit;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2013 Volume 52( Issue 17) pp:4664-4667
Publication Date(Web):
DOI:10.1002/anie.201300749
Co-reporter:Waldemar Fegler;Dr. Ajay Venugopal;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie 2013 Volume 125( Issue 31) pp:8134-8138
Publication Date(Web):
DOI:10.1002/ange.201303958
Co-reporter:Heiko Kulinna, Thomas P. Spaniol, Jun Okuda
Journal of Organometallic Chemistry 2013 744() pp: 49-52
Publication Date(Web):
DOI:10.1016/j.jorganchem.2013.04.047
Co-reporter:Andreas Kapelski
Journal of Polymer Science Part A: Polymer Chemistry 2013 Volume 51( Issue 23) pp:4983-4991
Publication Date(Web):
DOI:10.1002/pola.26925
ABSTRACT
Ring-opening polymerization of rac- and meso-lactide initiated by indium bis(phenolate) isopropoxides {1,4-dithiabutanediylbis(4,6-di-tert-butylphenolate)}(isopropoxy)indium (1) and {1,4-dithiabutanediylbis(4,6-di(2-phenyl-2-propyl)phenolate)}(isopropoxy)indium (2) is found to follow first-order kinetics for monomer conversion. Activation parameters ΔH‡ and ΔS‡ suggest an ordered transition state. Initiators 1 and 2 polymerize meso-lactide faster than rac-lactide. In general, compound 2 with the more bulky cumyl ortho-substituents in the phenolate moiety shows higher polymerization activity than 1 with tert-butyl substituents. meso-Lactide is polymerized to syndiotactic poly(meso-lactides) in THF, while polymerization of rac-lactide in THF gives atactic poly(rac-lactides) with solvent-dependent preferences for heterotactic (THF) or isotactic (CH2Cl2) sequences. Indium bis(phenolate) compound rac-(1,2-cyclohexanedithio-2,2′-bis{4,6-di(2-phenyl-2-propyl)phenolato}(isopropoxy)indium (3) polymerizes meso-lactide to give syndiotactic poly(meso-lactide) with narrow molecular weight distributions and rac-lactide in THF to give heterotactically enriched poly(rac-lactides). © 2013 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 4983–4991
Co-reporter:Dipl.-Chem. Crispin Lichtenberg ;Dr. Jun Okuda
Angewandte Chemie International Edition 2013 Volume 52( Issue 20) pp:5228-5246
Publication Date(Web):
DOI:10.1002/anie.201208942
Abstract
Organometallic allyl compounds are important as allylation reagents in organic synthesis, as polymerization catalysts, and as volatile metal precursors in material science. Whereas the allyl chemistry of synthetically relevant transition metals such as palladium and of the lanthanoids is well-established, that of main group metals has been lagging behind. Recent progress on allyl complexes of Groups 1, 2, and 12–16 now provides a more complete picture. This is based on a fundamental understanding of metal–allyl bonding interactions in solution and in the solid state. Furthermore, reactivity trends have been rationalized and new types of allyl-specific reactivity patterns have been uncovered. Key features include 1) the exploitation of the different types of metal–allyl bonding (highly ionic to predominantly covalent), 2) the use of synergistic effects in heterobimetallic compounds, and 3) the adjustment of Lewis acidity by variation of the charge of allyl compounds.
Co-reporter:Waldemar Fegler;Dr. Ajay Venugopal;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2013 Volume 52( Issue 31) pp:7976-7980
Publication Date(Web):
DOI:10.1002/anie.201303958
Co-reporter:Peng Cui, Thomas P. Spaniol, and Jun Okuda
Organometallics 2013 Volume 32(Issue 5) pp:1176-1182
Publication Date(Web):December 10, 2012
DOI:10.1021/om300986w
The macrocyclic diamino diamine (1,7-Me2TACD)H2 (1,7-Me2TACD = 1,7-dimethyl-1,4,7,10-tetraazacyclododecane, 1,7-Me2[12]aneN4), reacted under propylene elimination with [Ln(η3-C3H5)3(diox)] (Ln = Y, La) to give the mono(allyl) complexes [(1,7-Me2TACD)Ln(η3-C3H5)]2 (Ln = Y (1a), La (1b)). A single-crystal X-ray diffraction study shows 1b to be a centrosymmetric dimer with lanthanum atoms bridged by one of the two amido nitrogen atoms. Complexes 1a,b were treated with 2 equiv of the potassium allyl KC3H5 to give the corresponding heterometallic allyl complexes [(1,7-Me2TACD)Ln(η3-C3H5)2K(THF)]n (Ln = Y (2a), La (2b)). A single-crystal X-ray diffraction study revealed that 2a,b are polymeric in the solid state with allyl ligands bridging the metal centers in addition to the presence of μ2-amido functions of the 1,7-Me2TACD ligand. Hydrogenolysis of the yttrium compound 2a with 1 bar of H2 led to the formation of the heterometallic Y4K2 hydrido complex [(1,7-Me2TACD)2Y2H3K(THF)2]2 (3a), which can also be synthesized from a 1:1 mixture of 1a and KC3H5 with 1 bar of H2. A single-crystal X-ray diffraction study of 3a revealed a dimer of heterotrinuclear Y2K trihydride aggregate. Treatment of 2b with 1 bar of H2 afforded the heptanuclear La3K4 heptahydrido complex [(1,7-Me2TACD)3La3H7K4(THF)7] (3b).
Co-reporter:Heiko Kulinna;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Chemistry - A European Journal 2013 Volume 19( Issue 29) pp:9468-9471
Publication Date(Web):
DOI:10.1002/chem.201301202
Co-reporter:Dr. Peng Cui;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Chemistry - A European Journal 2013 Volume 19( Issue 40) pp:13437-13444
Publication Date(Web):
DOI:10.1002/chem.201301732
Abstract
The rare-earth-metal hydride complexes [{(1,7-Me2TACD)LnH}4] (Ln=La 1 a, Y 1 b; (1,7-Me2TACD)H2=1,7-dimethyl-1,4,7,10-tetraazacyclododecane, 1,7-Me2[12]aneN4) were synthesized by hydrogenolysis of [{(1,7-Me2TACD)Ln(η3-C3H5)}2] with 1 bar H2. The tetrameric structures were confirmed by 1H NMR spectroscopy and single-crystal X-ray diffraction of compound 1 a. Both complexes catalyze the dehydrogenation of secondary amineborane Me2NH⋅BH3 to afford the cyclic dimer (Me2NBH2)2 and (Me2N)2BH under mild conditions. Whilst the complete conversion of Me2NH⋅BH3 was observed within 2 h with lanthanumhydride 1 a, the yttrium homologue 1 b required 48 h to reach 95 % conversion. Further reactions of compound 1 a with Me2NH⋅BH3 in various stoichiometric ratios gave a series of intermediate products, [{(1,7-Me2TACD)LaH}4](Me2NBH2)2 (2 a), [(1,7-Me2TACDH)La(Me2NBH3)2] (3 a), [(1,7-Me2TACD)(Me2NBH2)La(Me2NBH3)] (4 a), and [(1,7-Me2TACD)(Me2NBH2)2La(Me2NBH3)] (5 a). Complexes 2 a, 3 a, and 5 a were isolated and characterized by multinuclear NMR spectroscopy and single-crystal X-ray diffraction studies. These intermediates revealed the activation and coordination modes of “Me2NH⋅BH3” fragments that were trapped within the coordination sphere of a rare-earth-metal center.
Co-reporter:Dr. Freddi Philippart;Marcus Arlt;Steve Gotzen;Dr. Stefanie-Joana Tenne;Dr. Marco Bocola;Dr. Hsui-Hui Chen;Dr. Leilei Zhu; Ulrich Schwaneberg; Jun Okuda
Chemistry - A European Journal 2013 Volume 19( Issue 41) pp:13865-13871
Publication Date(Web):
DOI:10.1002/chem.201301515
Abstract
A β-barrel protein hybrid catalyst was prepared by covalently anchoring a Grubbs–Hoveyda type olefin metathesis catalyst at a single accessible cysteine amino acid in the barrel interior of a variant of β-barrel transmembrane protein ferric hydroxamate uptake protein component A (FhuA). Activity of this hybrid catalyst type was demonstrated by ring-opening metathesis polymerization of a 7-oxanorbornene derivative in aqueous solution.
Co-reporter:Andreas Sauer;Jean-Charles Buffet;Thomas P. Spaniol;Haruki Nagae; Kazushi Mashima; Jun Okuda
ChemCatChem 2013 Volume 5( Issue 5) pp:1088-1091
Publication Date(Web):
DOI:10.1002/cctc.201200705
Co-reporter:Crispin Lichtenberg ; Thomas P. Spaniol ; Ilja Peckermann ; Timothy P. Hanusa
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:811-821
Publication Date(Web):December 14, 2012
DOI:10.1021/ja310112e
Starting from bis(allyl)magnesium [Mg(C3H5)2], a set of cationic, neutral, anionic, and dianionic allyl magnesium compounds has been isolated and characterized, including [Mg(C3H5)(THF)5][B(C6F5)4] (3), [Mg(C3H5)2(1,4-dioxane)(THF)] (2), [KMg(C3H5)3(THF)] (6), and [MMg(C3H5)4] (8: M = K2; 9: M = Ca). In solution, the allyl ligands of the compounds display fluxional behavior, even at low temperatures. Single crystal X-ray analysis reveals unusual μ2-η1:η3- and unprecedented μ3-η1:η3:η3-coordination modes in the heterobimetallic compounds 6 and [8·(THF)2]. Density functional theory calculations confirm that these metal–allyl conformations are energetically stable. The magnesium compounds have been investigated as initiators for butadiene polymerization and ethylene oligomerization. The heterobimetallic compounds display initiation properties, including higher reaction rates, that are distinctively different from those of the monometallic species. Reactivity trends depend on the formal charge of the magnesium compounds (dianionic, higher-order magnesiate > monoanionic, lower-order magnesiate) and on the nature of the counterion (K+ > Ca2+).
Co-reporter:Crispin Lichtenberg ; Julien Engel ; Thomas P. Spaniol ; Ulli Englert ; Gerhard Raabe
Journal of the American Chemical Society 2012 Volume 134(Issue 23) pp:9805-9811
Publication Date(Web):May 16, 2012
DOI:10.1021/ja303480a
The reinvestigation of two allyl zinc compounds, parent bis(allyl)zinc [Zn(C3H5)2] (1) and 2-methallyl chloro zinc [Zn(C4H7)Cl] (2), revealed two new coordination modes in the solid state for the allyl ligand, viz cis- and trans-μ2-η1:η1. These results call for modification of the conventional interpretation of zinc–allyl interactions. Computational results indicate that the classical η3-bonding mode of the allyl ligand is not favored in zinc compounds. A rare case of a zinc–olefin interaction in the dimer of [Zn(η1-C3H5)(OC(C3H5)Ph2)] was found in the monoinsertion product of 1 with benzophenone.
Co-reporter:Crispin Lichtenberg ; Thomas P. Spaniol
Inorganic Chemistry 2012 Volume 51(Issue 4) pp:2254-2262
Publication Date(Web):January 23, 2012
DOI:10.1021/ic202293t
A series of cationic, neutral, and anionic allylgallium complexes has been isolated and fully characterized. It includes neutral [Ga(η1-C3H5)3(L)] (1, L = THF; 2, L = OPPh3), cationic [Ga(η1-C3H5)2(THF)2]+[A]− (3, [A]− = [B(C6F5)4]−; 4, [A]− = [B(C6H3Cl2)4]−), as well as anionic [Cat]+[Ga(η1-C3H5)4]− (5, [Cat]+ = K+; 6, [Cat]+ = [K(dibenzo-18-c-6]+; 7, [Cat]+ = [PPh4]+). Binding modes of the allyl ligand in solution and in the solid state have been studied comparatively. Single crystal X-ray analyses revealed a four-coordinate neutral gallium center in 2, a five-coordinate cationic gallium center in 4 and [4·THF], and a four-coordinate anionic gallium center with a bridging μ2-η1:η2 coordination mode of the allyl ligand in 6. The reactivity of this series of allylgallium complexes toward benzophenone and N-heteroaromatics has been investigated. Counterion effects have also been studied. Reactions of 1 and 5 with isoquinoline revealed the first examples of organogallium complexes reacting under 1,2-insertion with pyridine derivatives.
Co-reporter:Andreas Sauer ; Jean-Charles Buffet ; Thomas P. Spaniol ; Haruki Nagae ; Kazushi Mashima
Inorganic Chemistry 2012 Volume 51(Issue 10) pp:5764-5770
Publication Date(Web):May 9, 2012
DOI:10.1021/ic300271h
A series of group 4 metal complexes Zr-(1)2, Zr-(2)2, Zr-(3)2, Zr-(4)2, Zr-(5)2, Hf-(1)2, and Hf-(4)2 containing two bridged bis(phenolate) ligands of the (OSSO)-type were prepared by the reaction of the corresponding bis(phenol) and group 4 metal precursor MX4 (X = OiPr, CH2Ph) and isolated as robust, colorless crystals. NMR spectra indicate D2 symmetry, in agreement with the solid state structure determined by single crystal X-ray diffraction study of the complexes Zr-(1)2, Hf-(1)2, Zr-(3)2, Zr-(4)2, and Zr-(5)2. The complexes with the 1,4-dithiabutanediyl bridged ligands exhibit a highly symmetric coordination around the metal center. The introduction of the rigid trans-1,2-cyclohexanediyl bridged ligands led to a distorted coordination around the metal center in Zr-(4)2 and Zr-(5)2 when the ortho substituent is tert-butyl and the para substituent is larger than methyl. The complexes Zr-(1)2, Zr-(2)2, Zr-(3)2, Zr-(4)2 as well as Hf-(1)2 and Hf-(4)2 initiated the ring-opening polymerization of meso-lactide at 100 °C to give heterotactic polylactide with pronounced heterotacticity (>70%) and varying polydispersity (1.05 < Mw/Mn < 1.61). As shown by kinetic studies, zirconium complex Zr-(1)2 polymerized meso-lactide faster than the homologous hafnium complex Hf-(1)2.
Co-reporter:Heiko Kulinna, Thomas P. Spaniol, Laurent Maron, and Jun Okuda
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12462-12472
Publication Date(Web):October 31, 2012
DOI:10.1021/ic301880z
An air- and light-sensitive, but thermally stable tris[(trimethylsilyl)methyl]zirconium complex containing an NNNN-type macrocyclic ligand [Zr(Me3TACD)(CH2SiMe3)3] (1; Me3TACD = Me3[12]aneN4: 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane) was prepared by reacting [Zr(CH2SiMe3)4] with (Me3TACD)H. Reaction of the zirconium tris(alkyl) 1 with a Lewis or Brønsted acid gave a dialkyl cation with a weakly coordinating anion [Zr(Me3TACD)(CH2SiMe3)2][A] [A = Al{OC(CF3)3}4 (2a), B{3,5-C6H3(CF3)2}4 (2b), B(3,5-C6H3Cl2)4 (2c), and BPh4) (2d)]. Hydrogenolysis of 2a–2c resulted in the formation of the dinuclear tetrahydride dication [{Zr(Me3TACD)(μ-H)2}2][A]2 (3a–3c). Compounds 1–3 were characterized by multinuclear NMR spectroscopy, and the solid-state structures of 1, 2c, and 3b were established by single-crystal X-ray diffraction studies. The dinuclear hydride complex 3b exhibits a quadruply bridged {Zr2(μ-H)4} core in solution and in the solid state with a relatively short Zr···Zr distance of 2.8752(11) Å. Density functional theory computations at the B3PW91 level reproduced this structure (Zr···Zr distance of 2.900 Å). The cationic hydride complex 3b reacted with excess carbon monoxide in tetrahydrofuran at room temperature to give ethylene in 25% yield based on 3b. Upon analysis of 13C NMR spectra of the reaction mixture using 13CO, oxymethylene and enolate complexes were detected as intermediates among other complexes.
Co-reporter:Julien P. Davin, Jean-Charles Buffet, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2012 vol. 41(Issue 40) pp:12612-12618
Publication Date(Web):21 Aug 2012
DOI:10.1039/C2DT31309J
A chiral, tetradentate polyether ligand with a trans-1,2-cyclohexanediyl backbone, bis(methoxyethoxy)-trans-1,2-cyclohexane (5), was synthesized as both a racemate and the (S,S) enantiomer. 5 was found to form stable adducts with alkaline earth metal amides [M{N(SiMe3)2}2(thf)x] (M = Mg (x = 0), Ca (x = 2) and Sr (x = 2/3)), [Ca{N(SiHMe2)2}2(thf)] as well as with hydrocarbyl compounds [Mg(CH2SiMe3)2] and [Ca(η3-C3H5)2]. X-ray diffraction study of the bis(amide) [((S,S)-5)Ca{N(SiMe3)2}2] and of the bis(allyl) [(rac-5)Ca(η3-C3H5)2] was performed. The complexes obtained were tested as initiators for the ring-opening polymerization of meso-, racemic and L-lactide.
Co-reporter:Phillip Jochmann, Julien P. Davin, Stefanie Maslek, Thomas P. Spaniol, Yann Sarazin, Jean-Francois Carpentier and Jun Okuda
Dalton Transactions 2012 vol. 41(Issue 30) pp:9176-9181
Publication Date(Web):10 May 2012
DOI:10.1039/C2DT30743J
The synthesis and attempted isolation of neutral bis(allyl)strontium [Sr(C3H5)2] (1) resulted in the isolation of potassium tris(allyl)strontiate K[Sr(C3H5)3] (2). In situ generated 1 shows a pronounced Brønsted basicity, inducing polymerisation of THF. Ate complex 2 crystallises as [K(THF)2{Sr(C3H5)3}(THF)]∞ (2·(THF)3). The salt-like solid state structure of 2·(THF)3 comprises a two-dimensional network of (μ2-η3:η3-C3H5)− bridged potassium and strontium centres. Synthesis of allyl complexes 1 and 2 utilised SrI2, [Sr(TMDS)2] (3) (TMDS = tetramethyldisilazanide), and [Sr(HMDS)2] (HMDS = hexamethyldisilazanide) as strontium precursors. The solid state structure of previously reported [Sr(TMDS)2] (3) was established by X-ray single crystal analysis as a dissymmetric dimer of [Sr2(TMDS)4(THF)3] (3·(THF)3) with multiple Si–H⋯Sr agostic interactions. The presence of ether ligands (THF, 18-crown-6) influenced the Si–H⋯Sr resonances in the NMR spectra of the amido complex 3.
Co-reporter:Dr. Phillip Jochmann;Julien P. Davin;Dr. Thomas P. Spaniol;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2012 Volume 51( Issue 18) pp:4452-4455
Publication Date(Web):
DOI:10.1002/anie.201200690
Co-reporter:Dipl.-Chem. Andreas Kapelski;Dr. Jean-Charles Buffet;Dr. Thomas P. Spaniol; Jun Okuda
Chemistry – An Asian Journal 2012 Volume 7( Issue 6) pp:1320-1330
Publication Date(Web):
DOI:10.1002/asia.201100826
Abstract
A series of 1,ω-dithiaalkanediyl-bridged bis(phenols) of the general type [OSSO]H2 with variable steric properties and various bridges were prepared. The stoichiometric reaction of the bis(phenols) 1,3-dithiapropanediyl-2,2′-bis(4,6-di-tert-butylphenol), 1,3-dithiapropanediyl-2,2′-bis[4,6-di(2-phenyl-2-propyl)phenol], rac-2,3-trans-propanediyl-1,4-dithiabutanediyl-2,2′-bis[4,6-di(2-phenyl-2-propyl)phenol], rac-2,3-trans-butanediyl-1,4-dithiabutane diyl-2,2′-bis[4,6-di(2-phenyl-2-propyl)phenol], rac-2,3-trans-hexanediyl-1,4-dithiabutanediyl-2,2′-bis[4,6-di(2-phenyl-2-propyl)phenol], 1,3-dithiapropanediyl-2,2′-bis[6-(1-methylcyclohexyl)-4-methylphenol] (C1, R=1-methylcyclohexyl), and 1,4-dithiabutanediyl-2,2′-bis[6-(1-methylcyclohexyl)-4-methylphenol] with rare-earth metal silylamido precursors [Ln{N(SiHMe2)2}3(thf)x] (Ln=Sc, x=1 or Ln=Y, x=2; thf=tetrahydrofuran) afforded the corresponding scandium and yttrium bis(phenolate) silylamido complexes [Ln(OSSO){N(SiHMe2)2}(thf)] in moderate to good yields. The monomeric nature of these complexes was shown by an X-ray diffraction study of one of the yttrium complexes. The complexes efficiently initiated the ring-opening polymerization of rac- and meso-lactide to give heterotactic-biased poly(rac-lactides) and highly syndiotactic poly(meso-lactides). Variation of the ligand backbone and the steric properties of the ortho substituents affected the level of tacticity in the polylactides.
Co-reporter:Dipl.-Chem. Crispin Lichtenberg;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Angewandte Chemie 2012 Volume 124( Issue 32) pp:8225-8229
Publication Date(Web):
DOI:10.1002/ange.201203698
Co-reporter:Dipl.-Chem. Crispin Lichtenberg;M.Sc. Fangfang Pan;Dr. Thomas P. Spaniol;Dr. Ulli Englert ;Dr. Jun Okuda
Angewandte Chemie 2012 Volume 124( Issue 52) pp:13186-13190
Publication Date(Web):
DOI:10.1002/ange.201206782
Co-reporter:Crispin Lichtenberg;Fangfang Pan;Dr. Thomas P. Spaniol;Dr. Ulli Englert ;Dr. Jun Okuda
Angewandte Chemie International Edition 2012 Volume 51( Issue 52) pp:13011-13015
Publication Date(Web):
DOI:10.1002/anie.201206782
Co-reporter:Dipl.-Chem. Crispin Lichtenberg;Dr. Thomas P. Spaniol;Dr. Lionel Perrin;Dr. Laurent Maron;Dr. Jun Okuda
Chemistry - A European Journal 2012 Volume 18( Issue 21) pp:6448-6452
Publication Date(Web):
DOI:10.1002/chem.201200808
Co-reporter:Dipl.-Chem. Crispin Lichtenberg;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Angewandte Chemie International Edition 2012 Volume 51( Issue 32) pp:8101-8105
Publication Date(Web):
DOI:10.1002/anie.201203698
Co-reporter:Klaus Beckerle, Jun Okuda
Journal of Molecular Catalysis A: Chemical 2012 Volume 356() pp:158-164
Publication Date(Web):April 2012
DOI:10.1016/j.molcata.2012.01.008
d-Glucose and cellobiose were converted into 5-hydroxymethylfurfural (HMF) by rare earth metal chlorides LnCl3 (Ln = Sc, Y, La) in N,N′-dimethylacetamide (DMA). Both conversion and selectivity strongly depend on the ionic radii of the rare earth metal center. Conversion of fructose into HMF proceeds significantly faster and with higher selectivity than of glucose, suggesting a mechanism that involves the transformation of glucose into fructose as a crucial, rate determining step.Graphical abstractHighlights► Rare earth metal chlorides can be applied for the dehydration of carbohydrates in DMA. ► Scandium is considerably more active than yttrium or lanthanum. ► Data suggest a mechanism involving the transformation of glucose into fructose. ► Cellobiose can be transformed into HMF in a one-pot approach.
Co-reporter:Ajay Venugopal ; Waldemar Fegler ; Thomas P. Spaniol ; Laurent Maron
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17574-17577
Publication Date(Web):October 7, 2011
DOI:10.1021/ja207180x
The dinuclear lutetium dihydride dication supported by metalated tripodal ligands undergoes facile hydrogenolysis with H2 to form a trihydride dication. Molecular orbital analysis shows that the LUMO is a bonding Lu···Lu orbital that is poised to activate dihydrogen.
Co-reporter:Sabine Standfuss, Elise Abinet, Thomas P. Spaniol and Jun Okuda
Chemical Communications 2011 vol. 47(Issue 41) pp:11441-11443
Publication Date(Web):23 Sep 2011
DOI:10.1039/C1CC14180E
Neutral, cationic and anionic allyl compounds of scandium contain highly fluxional allyl ligands in solution, whilst in the solid state both η1- and η3-binding modes are detected.
Co-reporter:Ilja Peckermann, Gerhard Raabe, Thomas P. Spaniol and Jun Okuda
Chemical Communications 2011 vol. 47(Issue 17) pp:5061-5063
Publication Date(Web):24 Mar 2011
DOI:10.1039/C1CC00039J
Allyl and 2-methylallyl indium compounds were prepared by salt metathesis starting from indium trichloride and a Grignard reagent. They are highly fluxional in solution and reveal coordination numbers of the indium atoms of four and five in the solid state.
Co-reporter:Jean-Charles Buffet and Jun Okuda
Chemical Communications 2011 vol. 47(Issue 16) pp:4796-4798
Publication Date(Web):15 Mar 2011
DOI:10.1039/C1CC10149H
Group 4 metal initiators with a tetradentate bis(phenolato) ligand polymerized meso-lactide efficiently under ring-opening to give syndiotactic polylactide. L-Lactide was converted faster than rac-lactide and meso-lactide.
Co-reporter:Jean-Charles Buffet and Jun Okuda
Polymer Chemistry 2011 vol. 2(Issue 12) pp:2758-2763
Publication Date(Web):18 Aug 2011
DOI:10.1039/C1PY00206F
Poly(lactides) (PLAs), or poly(lactic acid)s, are among the first commercial biodegradable polymers that have the potential to become commodity plastics. meso-Lactide, a by-product of L-lactide production, will become more easily available. Highly stereoregular poly(meso-lactides) should be crystalline and thus interesting as polymeric material. In this short review, initiators capable of inducing the stereoselective ring-opening polymerization of meso-lactide to ideally give syndiotactic or heterotactic PLAs will be discussed. Mechanistic discussions with regard to understanding the reactivity differences between the various lactide monomers are included.
Co-reporter:Jean-Charles Buffet, Ashley N. Martin, Moshe Kol and Jun Okuda
Polymer Chemistry 2011 vol. 2(Issue 10) pp:2378-2384
Publication Date(Web):15 Aug 2011
DOI:10.1039/C1PY00266J
Group 4 metal complexes Zr-1 to Zr-4, Ti-4 and Ti-4a that contain an (OSSO)-type tetradentate bis(phenolate) ligand were found to initiate the ring-opening polymerization of meso-, rac-, and L-lactides in toluene solution. The polymerizations were controlled with initiator efficiency values around one and gave polymers with low polydispersity (Mw/Mn < 1.2). The polylactides were atactic when rac-lactide was used as the monomer and heterotactic biased when meso-lactide was used. Kinetic measurements showed that meso-lactide was polymerized generally faster than rac- and L-lactide.
Co-reporter:Jean-Charles Buffet and Jun Okuda
Dalton Transactions 2011 vol. 40(Issue 30) pp:7748-7754
Publication Date(Web):11 Apr 2011
DOI:10.1039/C1DT10075K
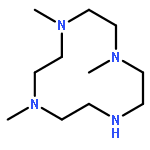
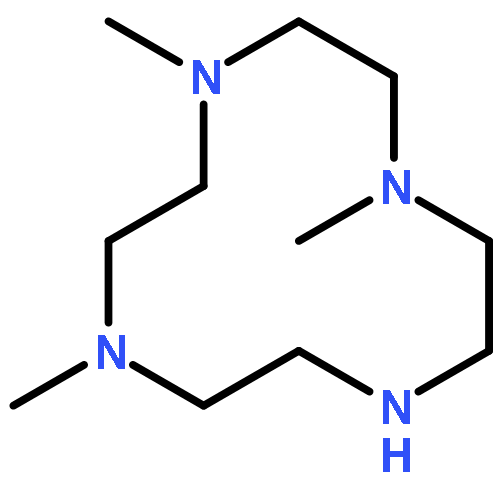
Cyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, (Me3TACD)H (= Me3[12]aneN4), reacted with [K{N(SiHMe2)2}] in benzene-d6 to give [K{(Me3TACD)SiMe2N(SiHMe2)}] (1) under hydrogen evolution. Single-crystal X-ray diffraction of 1 shows a dinuclear structure in the solid state, featuring a bridging μ-amido and a weak β-agostic Si–H bond. 1,7-Dimethyl-1,4,7,10-tetraazacyclododecane (Me2TACD)H2 (= Me2[12]aneN4) and (Me3TACD)H were reacted with [Sc{N(SiHMe2)2}3(thf)] in benzene-d6 to give [{(Me2TACD)SiMe2N(SiHMe2)}Sc{N(SiHMe2)2}] (2) and [(Me3TACD)Sc{N(SiHMe2)2}2SiMe2] (3), respectively. Both compounds are monomeric in solution and X-ray diffraction studies showed the scandium metal centers to be six-coordinate. The scandium alkyl complex [Sc(Me3TACD)(CH2SiMe3)2] (4) was obtained by reacting (Me3TACD)H with [Sc(CH2SiMe3)3(thf)] in benzene-d6. The scandium amide complexes 2 and 3 catalyzed the ring-opening polymerization (ROP) of meso-lactide to give syndiotactic polylactides.
Co-reporter:Jean-Charles Buffet, Julien P. Davin, Thomas P. Spaniol and Jun Okuda
New Journal of Chemistry 2011 vol. 35(Issue 10) pp:2253-2257
Publication Date(Web):01 Jul 2011
DOI:10.1039/C1NJ20259F
Cyclic tetra-amine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, (Me3TACD)H (= Me3[12]aneN4) was reacted with [M{N(SiMe3)2}2(thf)x] (M = Mg, x = 0; M = Ca, x = 2) in n-pentane to give thf-free complexes [{Me3TACD}M{N(SiMe3)2}] (M = Mg (1); M = Ca (2)). Both compounds are monomeric in solution and X-ray diffraction studies showed the metal centers to be five-coordinated. The proligand (Me3TACD)H reacted with [Ca{N(SiHMe2)2}2(thf)2] in benzene-d6 to give [{(Me3TACD)SiMe2N(SiHMe2)}Ca{N(SiHMe2)2}] (3) with hydrogen evolution. Single-crystal X-ray diffraction showed 3 to be monomeric with a six-coordinated calcium center in the solid state. The alkaline earth metal amide complexes 1 to 3 catalyzed the ring-opening polymerization of lactide monomers to give syndiotactic-rich polylactides from meso-lactide and isotactic-rich polylactides from rac- and L-lactide.
Co-reporter:Nicolas Susperregui, Mathias U. Kramer, Jun Okuda, and Laurent Maron
Organometallics 2011 Volume 30(Issue 6) pp:1326-1333
Publication Date(Web):February 17, 2011
DOI:10.1021/om100606p
The mechanism of the ring-opening polymerization (ROP) of ε-caprolactone by the rare-earth borohydride cations [YMe(BH4)(THF)5]+ has been investigated at the DFT level. The reaction with [YMe(BH4)(THF)5]+ is predicted to occur either on the borohydride or on the methyl group. This also indicates the presence of some trans effect that is further demonstrated by replacing the borohydride with a dimethylamido ligand, [YMe(NMe2)(THF)5]+.
Co-reporter:Phillip Jochmann, Stefanie Maslek, Thomas P. Spaniol, and Jun Okuda
Organometallics 2011 Volume 30(Issue 7) pp:1991-1997
Publication Date(Web):March 9, 2011
DOI:10.1021/om200012k
Allyl calcium compounds of different chain lengths [Ca(R)2(THF)x] (x = 0.15−0.25 (1-Ca, 3-Ca), 0.25−0.75 (2-Ca)) were synthesized by salt metathesis of CaI2 with allylpotassium reagents [K(R)] (R = n-butenyl (1-K), isobutenyl (2-K), n-hexenyl (3-K)), prepared from the corresponding α-olefin and Schlosser base. The new calcium derivatives were obtained in nearly quantitative yields. 1-Ca and 2-Ca could be crystallized as triglyme adducts (triglyme: tris(ethylene glycol)dimethyl ether) and structurally characterized by single-crystal X-ray diffraction. All potassium precursors [K(R)] were also isolated and characterized by 1H and 13C NMR spectroscopy. The solution properties in THF are in agreement with an η3-coordination mode of the allyl moiety for all isolated compounds. For the potassium reagents 1-K and 3-K, endo/exo equilibrium distributions of >99:<1 and 85:15 were observed, whereas for the calcium compounds 1-Ca and 3-Ca, distributions of 60:40 and 42:58 were found at 25 °C, respectively. This distribution pattern is discussed in the context of the behavior found for alkali metal analogues.
Co-reporter:Crispin Lichtenberg, Thomas P. Spaniol, and Jun Okuda
Organometallics 2011 Volume 30(Issue 16) pp:4409-4417
Publication Date(Web):July 25, 2011
DOI:10.1021/om200492f
Tris(allyl)aluminum as its THF adduct, [Al(η1-C3H5)3(THF)] (1), reacted with pyridine either under substitution to give [Al(η1-C3H5)3(py)] (7) (py = pyridine) or under carbometalation to give N-metalated 2-allyldihydropyridine depending on the solvent. The substituent pattern of the pyridine substrate and the aluminum center’s electrophilicity in [Al(η1-C3H5)3(L)] (L = neutral ligand) influenced the outcome of the reaction. Reactions of one N-metalated dihydropyridine with electrophiles have been studied. A crystalline derivative of tris(allyl)aluminum, [Al(η1-C3H5)3(OPPh3)] (2), and tetrakis(allyl)aluminate in the ion pair [K(15-crown-5)2][Al(η1-C3H5)4] (4) were characterized by single-crystal X-ray diffraction and shown to contain four-coordinate aluminum centers.
Co-reporter:Phillip Jochmann, Thomas P. Spaniol, Stephen C. Chmely, Timothy P. Hanusa, and Jun Okuda
Organometallics 2011 Volume 30(Issue 19) pp:5291-5296
Publication Date(Web):September 14, 2011
DOI:10.1021/om200749f
The synthesis, characterization, and decomposition pathway of the 18-crown-6 adduct of bis(allyl)calcium [Ca(η1-C3H5)(η3-C3H5)(18-crown-6)] (2) are reported. The solid-state structure of adduct 2 features one σ- and one π-bonded allyl ligand at the metal center. 2 is the first structurally characterized example of a mononuclear calcium complex bearing a purely σ-bound allyl ligand. DFT calculations indicate that [Ca(η1-C3H5)(η3-C3H5)(18-crown-6)] and [Ca(η1-C3H5)2(18-crown-6)] conformations are close in energy. In THF solution, η3-allyl ligands are observed exclusively at both ambient temperature and −95 °C. 2 undergoes rapid cleavage of the crown ether to give vinyl-terminated alcoholates of different chain length with a half-life of t1/2 = 2.5 h.
Co-reporter:Phillip Jochmann;Valeri Leich;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Chemistry - A European Journal 2011 Volume 17( Issue 43) pp:12115-12122
Publication Date(Web):
DOI:10.1002/chem.201101489
Abstract
A facile and general synthetic pathway for the production of dearomatized, allylated, and CH bond activated pyridine derivatives is presented. Reaction of the corresponding derivative with the previously reported reagent bis(allyl)calcium, [Ca(C3H5)2] (1), cleanly affords the product in high yield. The range of N-heterocyclic compounds studied comprised 2-picoline (2), 4-picoline (3), 2,6-lutidine (4), 4-tert-butylpyridine (5), 2,2′-bipyridine (6), acridine (7), quinoline (8), and isoquinoline (9). Depending on the substitution pattern of the pyridine derivative, either carbometalation or CH bond activation products are obtained. In the absence of methyl groups ortho or para to the nitrogen atom, carbometalation leads to dearomatized products. C(sp3)H bond activation occurs at ortho and para situated methyl groups. Steric shielding of the 4-position in pyridine yields the ring-metalated product through C(sp2)H bond activation instead. The isolated compounds [Ca(2-CH2-C5H4N)2(THF)] (2 b⋅(THF)), [Ca(4-CH2-C5H4N)2(THF)2] (3 b⋅(THF)2), [Ca(2-CH2-C5H3N-6-CH3)2(THF)n] (4 b⋅(THF)n; n=0, 0.75), [Ca{2-C5H3N-4-C(CH3)3}2(THF)2] (5 c⋅(THF)2), [Ca{4,4′-(C3H5)2-(C10H8N2)}(THF)] (6 a⋅(THF)), [Ca(NC13H9-9-C3H5)2(THF)] (7 a⋅(THF)), [Ca(4-C3H5-C9H7N)2(THF)] (8 b⋅(THF)), and [Ca(1-C3H5-C9H7N)2(THF)3] (9 a⋅(THF)3) have been characterized by NMR spectroscopy and metal analysis. 9 a⋅(THF)4 and 4 b⋅(THF)3 were additionally characterized in the solid state by X-ray diffraction experiments. 4 b⋅(THF)3 shows an aza-allyl coordination mode in the solid state. Based on the results, mechanistic aspects are discussed in the context of previous findings.
Co-reporter:Dipl.-Chem. Elise Abinet;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Chemistry – An Asian Journal 2011 Volume 6( Issue 2) pp:389-391
Publication Date(Web):
DOI:10.1002/asia.201000598
Co-reporter:Crispin Lichtenberg;Phillip Jochmann;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Angewandte Chemie International Edition 2011 Volume 50( Issue 25) pp:5753-5756
Publication Date(Web):
DOI:10.1002/anie.201100073
Co-reporter:Dipl.-Chem. Elise Abinet;Dipl.-Chem. Daniel Martin;Dipl.-Chem. Sabine Stfuss;Dipl.-Chem. Heiko Kulinna;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Chemistry - A European Journal 2011 Volume 17( Issue 52) pp:15014-15026
Publication Date(Web):
DOI:10.1002/chem.201102145
Abstract
The preparation and characterization of a series of neutral rare-earth metal complexes [Ln(Me3TACD)(η3-C3H5)2] (Ln=Y, La, Ce, Pr, Nd, Sm) supported by the 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane anion (Me3TACD−) are reported. Upon treatment of the neutral allyl complexes [Ln(Me3TACD)(η3-C3H5)2] with Brønsted acids, monocationic allyl complexes [Ln(Me3TACD)(η3-C3H5)(thf)2][B(C6X5)4] (Ln=La, Ce, Nd, X=H, F) were isolated and characterized. Hydrogenolysis gave the hydride complexes [Ln(Me3TACD)H2]n (Ln=Y, n=3; La, n=4; Sm). X-ray crystallography showed the lanthanum hydride to be tetranuclear. Reactivity studies of [Ln(Me3TACD)R2]n (R=η3-C3H5, n=0; R=H, n=3,4) towards furan derivatives includes hydrosilylation and deoxygenation under ring-opening conditions.
Co-reporter:Crispin Lichtenberg;Phillip Jochmann;Dr. Thomas P. Spaniol ;Dr. Jun Okuda
Angewandte Chemie 2011 Volume 123( Issue 25) pp:5872-5875
Publication Date(Web):
DOI:10.1002/ange.201100073
Co-reporter:Christiane Hohberger, Klaus Beckerle and Jun Okuda
Polymer Chemistry 2010 vol. 1(Issue 4) pp:534-539
Publication Date(Web):21 Jan 2010
DOI:10.1039/B9PY00286C
p-(2,2′-Diphenylethyl)styrene (DPES) was polymerized in an atactic, syndiotactic, and isospecific fashion. Atactic polymerization was initiated by 2,2′-azobis(2-methylpropionitrile) (AIBN). Syndiotactic polymer was obtained using the catalyst system TiCp*Cl3/MAO. Isospecific polymerization was performed with the homochiral postmetallocene catalyst dichloro{trans-1,2-dithiocyclohexanediyl-2,2′-bis(4,6-di-tert-butylphenolato)}titanium/MAO. Optically active isotactic polymers were obtained by a controlled reduction of the molecular weight, employing two different chain transfer methodologies. In addition to 1-hexene as a chain transfer agent (CTA), the use of diethylzinc as a CTA for DPES oligomerization was introduced. Polymers with molecular weights below Mn of 50,000 g mol−1 showed specific rotation values ([α]23D) between ±0.2 and 2.2.
Co-reporter:Waldemar Fegler, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2010 vol. 39(Issue 29) pp:6774-6779
Publication Date(Web):28 Apr 2010
DOI:10.1039/C001699C
Tris(trimethylsilylmethyl) complexes of yttrium and lutetium [LnR3(THF)2] (R = CH2SiMe3) were treated with sterically bulky N-heterocyclic carbenes (NHC) 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) and 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes). IPr gave labile mono-adducts [LnR3(NHC)], isolated as thermally robust crystals and fully characterized by NMR spectroscopy and X-ray diffraction. IMes gave a similar lutetium mono-adduct [LuR3(IMes)] with the lutetium alkyl [LuR3(THF)2], whereas the yttrium alkyl [YR3(THF)2] resulted in the formation of an ortho-metalated product. This compound, isolated as a crystalline bis(THF) adduct, contains a strained six-membered chelate ring that has been formed by the C–H bond activation of one of the ortho-methyl groups of the mesityl group. In contrast [LuR3(IMes)] only slowly underwent a similar C–H bond activation.
Co-reporter:Sabine Stfuss;Thomas P. Spaniol
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 19) pp:2987-2991
Publication Date(Web):
DOI:10.1002/ejic.201000199
Abstract
Cyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, (Me3TACD)H (1), was metalated with n-butyllithium in pentane to give [Li2(Me3TACD)2] (2). The structure of this compound is dimeric in the solid state as shown by single-crystal X-ray diffraction. With an excess of nBuLi, nBuLi is incorporated into the product. Depending on the stoichiometry, the compounds [Li3(nBu)(Me3TACD)2] (3) or [Li4(nBu)2(Me3TACD)2] (4) are formed. As shown by single-crystal X-ray diffraction, both molecular structures show a ladder motif. (Me3TACD)H reacted with NaI/Na2CO3 in acetonitrile to give benzene-soluble [NaI(Me3TACD)H] (5).
Co-reporter:Phillip Jochmann;Thomas S. Dols;Dr. Thomas P. Spaniol;Dr. Lionel Perrin;Dr. Laurent Maron;Dr. Jun Okuda
Angewandte Chemie International Edition 2010 Volume 49( Issue 42) pp:7795-7798
Publication Date(Web):
DOI:10.1002/anie.201003704
Co-reporter:Crispin Lichtenberg, Dominique Robert, Thomas P. Spaniol, and Jun Okuda
Organometallics 2010 Volume 29(Issue 21) pp:5714-5721
Publication Date(Web):October 8, 2010
DOI:10.1021/om100809h
Cationic, neutral, and anionic aluminum allyl compounds were synthesized, and their reactivity toward electrophiles was studied. The THF adduct of the previously elusive tris(allyl)aluminum, [Al(η1-C3H5)3(THF)] (1), was isolated as an oil. Protonolysis of one allyl ligand in 1 using [NEt3H][BPh4] gave the cationic bis(allyl)aluminum, a fragment of the crystalline [Al(η1-C3H5)2(THF)3−n]+[BPh4]−·(n+1)THF (n = 0, 1) (2). Single-crystal X-ray diffraction of [Al(η1-C3H5)2(THF)2]+[BPh4]− (2a) revealed a tetrahedral aluminum center, while [Al(η1-C3H5)2(THF)3]+[BPh4]− (2b) contains a trigonal-bipyramidal aluminum center with both allyl ligands in the equatorial plane. The tetrakis(allyl)aluminate K+[Al(η1-C3H5)4]− (3) was also synthesized from the reaction of 1 with K(C3H5). Reactions of the allyl compounds 1−3 with (i) benzophenone, (ii) allyl halides C3H5X (X = Cl, Br, I), and (iii) halogen X2 (X = Br, I) showed considerable difference with respect to the ionic charge of the aluminum allyl.
Co-reporter:Ilja Peckermann, Thomas S. Dols, Thomas P. Spaniol, Jun Okuda
Journal of Organometallic Chemistry 2010 695(21) pp: 2325-2328
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.06.027
Co-reporter:Waldemar Fegler, Teruhiko Saito, Kazushi Mashima, Thomas P. Spaniol, Jun Okuda
Journal of Organometallic Chemistry 2010 695(25–26) pp: 2794-2797
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.08.021
Co-reporter:Tsuneomi Kawasaki, Christiane Hohberger, Yuko Araki, Kunihiko Hatase, Klaus Beckerle, Jun Okuda and Kenso Soai
Chemical Communications 2009 (Issue 37) pp:5621-5623
Publication Date(Web):21 Aug 2009
DOI:10.1039/B912813A
Chiral isotactic polystyrenes induce the enantioselective addition of diisopropylzinc to pyrimidine-5-carbaldehyde, affording the enantiomerically enriched pyrimidyl alkanol with the corresponding absolute configuration to that of cryptochiral polystyrenes in conjunction with asymmetric autocatalysis.
Co-reporter:Ilja Peckermann, Andreas Kapelski, Thomas P. Spaniol and Jun Okuda
Inorganic Chemistry 2009 Volume 48(Issue 12) pp:5526-5534
Publication Date(Web):May 8, 2009
DOI:10.1021/ic900161w
Indium bis(phenolato) complexes [In(L)R(THF)n] (L = 1,4-dithiabutanediylbis(4,6-di-tert-butylphenolato) (etbbp), R = Cl, n = 0, 1; L = 1,3-dithiapropanediylbis(6-tert-butyl-4-methylphenolato) (mtbmp), R = Me, n = 1, 2; L = 1,4-dithiabutanediyl-bis(6-tert-butyl-4-methyl-phenolato) (etbmp), R = Me, n = 0, 3; L = etbbp, R = CH2SiMe3, n = 0, 4; L = 1,4-dithiabutanediylbis{4,6-di(2-phenyl-2-propyl)phenolato} (etccp), R = CH2SiMe3, n = 0, 5) were prepared from indium trichloride or from the corresponding tris(alkyl) complexes and 1 equiv of tetradentate 1,ω-dithiaalkanediyl-bridged bis(phenol) LH2. The monomeric nature of the alkyl indium complexes was shown by X-ray diffraction studies of the complexes [In(mtbmp)Me(THF)] (2), [In(etbbp)(CH2SiMe3)] (4), and [In(etccp)(CH2SiMe3)] (5). Pseudo-octahedral configuration was found for 2, while square pyramidal structure was observed for 4 and 5. The isopropoxy complexes [In(L)(OiPr)] (L = etbbp, 6; etccp, 7) were synthesized starting with indium tris(isopropoxide). Complex 6 crystallized as homochiral dimer of pseudo-octahedral fragments with bridging μ2-alkoxide ligands, but in solution two diastereomers were observed. The isopropoxy complexes efficiently initiated the ring-opening polymerization of l-lactide in toluene to give isotactic poly(l-lactides) with narrow molecular weight distribution (Mw/Mn = 1.03−1.18).
Co-reporter:Geert-Jan M. Meppelder, Hong-Tao Fan, Thomas P. Spaniol and Jun Okuda
Inorganic Chemistry 2009 Volume 48(Issue 15) pp:7378-7388
Publication Date(Web):July 2, 2009
DOI:10.1021/ic900903b
A series of chiral linear tetradentate bis(phenols) that contain both sulfur and nitrogen donors of the type [2,2′-(HOC6H2-6-tBu-4-R1)2SC6H10NR2] [R1 = Me (a), tBu (b); R2 = H (1), Me (2)], [2,2′-(HOC6H2-4,6-tBu2)2SC6H10N═CH] (3), and [2,2′-(HOC6H2-4,6-tBu2)2SC6H10NHCH2] (4) were synthesized. The reaction of these bis(phenols) with TiX4 (X = Cl, OiPr) afforded the corresponding C1-symmetric titanium complexes [Ti{2,2′-(OC6H2-6-tBu-4-R1)2SC6H10NR2}X2] [R1 = Me (a), tBu (b); R2 = H, X = Cl (5a, 5b), OiPr (6a); R2 = Me, X = Cl (7a, 7b), OiPr (8a)], [Ti{2,2′-(OC6H2-4,6-tBu2)2SC6H10N═CH}Cl2] (9), and [Ti{2,2′-(OC6H2-4,6-tBu2)2SC6H10NHCH2}Cl2] (10). The formation of titanium complexes 5−8 proceeded diastereoselectively, but a mixture of two isomers (a and b) was obtained for 9 and 10. The configuration of the ligand around the metal center was determined by a combination of NMR spectroscopy and single-crystal X-ray diffraction studies of 5b, 7b, 8a, 9b, 10a, and 10b. All titanium complexes were configurationally stable in solution up to 100 °C. For compounds 5−8, cis-α and cis-β2 coordination modes of the ligand were observed, depending on the nitrogen substituent and the auxiliary ligand. In compounds 9 and 10, both configurations coexist and do not interconvert at elevated temperatures, but HCl catalyzes the isomerization of 10a to 10b. Upon activation with methylaluminoxane, [Ti{OSNO}X2] complexes show moderate activity in the polymerization of styrene and trace activity in the polymerization of 1-hexene.
Co-reporter:Bing Lian, Haiyan Ma, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2009 (Issue 41) pp:9033-9042
Publication Date(Web):25 Aug 2009
DOI:10.1039/B909287K
Reaction of trimethylaluminium with the linked bis(phenol) trans-1,4-dithiocyclohexanediyl-2,2′-bis(4,6-di-tert-butylphenol) (cydtbpH2, 1a) led to the chiral methyl aluminium complex [Al(cydtbp)Me] (2a) as both racemate and resolved enantiomers, which were characterized by multinuclear NMR spectroscopy and elemental analysis. Rac-{trans-1,2-dithiocyclohexanediyl-2,2′-bis(6-tert-butyl-4-methylphenol)} (cytbmpH2, rac-1b) similarly gave rac-2b. Single-crystal X-ray diffraction of (S,S)-2a and rac-2b showed a strongly distorted trigonal bipyramidal geometry with significantly differing Al–S distances. Reaction of rac-2b with (−)-borneol gave the alkoxy complex 5 as a mixture of two diastereomers. Methyl abstraction from the neutral metal complex rac-2a using B(C6F5)3 gave the cationic complex [rac-Al(cydtbp)(THF)2]+[MeB(C6F5)3]− (rac-6·(THF)2) that according to X-ray crystallography adopts an octahedral coordination geometry. Upon reacting trimethylaluminium with a bis(phenol) with a longer link, 3,4-trans-butanediyl-1,6-dithiahexanediyl)-2,2′-bis(6-tert-butyl-4-methylphenol), (cmtbmpH2, 3, a dinuclear compound 4 was obtained that contains two square pyramidal aluminium centers with two “fly-over” bis(phenolate) ligands. The neutral methyl aluminium complexes are active in the polymerization of methyl methacrylate (MMA) and in the ring-polymerization of rac-lactide (LA), producing PMMAs and PLAs in a controlled fashion. The cationic aluminium complexes were active in the cationic polymerization of isoprene and benzofuran.
Co-reporter:Bing Lian;Klaus Beckerle;Thomas P. Spaniol
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 2) pp:311-316
Publication Date(Web):
DOI:10.1002/ejic.200800810
Abstract
Reaction of group 4 metal tetrabenzyl complexes [M(CH2Ph)4] (M = Ti, Zr, Hf) with 1 or 2 equiv. of a thioether-functionalized phenol 2,4-tBu2-6-(PhSCH2)C6H2OH afforded dibenzyl complexes [M(L)2(CH2Ph)2] (M = Ti, 1; M = Zr, 2; M = Hf, 3) and the tribenzyl zirconium complex [Zr(L)(CH2Ph)3] (4). Benzyl abstraction with a Lewis acid gave the zirconium monobenzyl cation [Zr(L)2CH2Ph]+[PhCH2B(C6F5)3]– (5) and the dibenzyl cation [Zr(L)(CH2Ph)2]+[PhCH2B(C6F5)3]– (6). The monobenzyl cation 5 oligomerized 1-hexene to low molecular-weight atactic oligo(1-hexene) (Mn = 560–820), while the dibenzyl cation 6 polymerized 1-hexene isospecifically ([mmmm] > 90 %). (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Bing Lian;Thomas P. Spaniol;Patricia Horrillo-Martínez;Kai C. Hultzsch
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 3) pp:429-434
Publication Date(Web):
DOI:10.1002/ejic.200800977
Abstract
Salt metathesis reaction of the imido complex [Ti(NR)Cl2(NC5H5)3] (R = tBu, C6H3iPr2-2,6) with 1 equiv. of the lithium salt of the corresponding [OSSO]-type bis(phenol) [edtbpH2: (HOC6H2-tBu2-4,6)2(SCH2CH2S); rac-(cydtbp)H2: (HOC6H2-tBu2-4,6)2(S2C6H10-1,2)] afforded imido titanium complexes [Ti(edtbp)(NtBu)(NC5H5)] (1), [Ti(edtbp)(NC6H3iPr2-2,6)(NC5H5)] (2), and [Ti{rac-(cydtbp)}(NtBu)(NC5H5)] (3). The bis(dimethylamido)titanium complex [Ti(edtbp)(NMe2)2] (4) was synthesized by protonolysis of [Ti(NMe2)4] with bis(phenol) edtbpH2. Reaction of [Ti(NMe2)Cl3] with the lithium salt of the bis(phenol) gave the chloro dimethylamido complex [Ti(edtbp)(NMe2)Cl] (5) in high yield. All complexes were characterized by NMR spectroscopy and elemental analysis. Additionally, complexes 1 and 5 were studied by X-ray diffraction analysis. Imido titanium complex 1 shows moderate activity in the intramolecular hydroamination reaction of 5-phenylpent-4-ynylamine. Complexes 1–3 catalyze the intramolecular hydroamination of aminoalkenes. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Phillip Jochmann;ThomasS. Dols;ThomasP. Spaniol Dr.;Lionel Perrin Dr.;Laurent Maron Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 31) pp:5715-5719
Publication Date(Web):
DOI:10.1002/anie.200901743
Co-reporter:Geert-Jan M. Meppelder, Tobias S. Halbach, Thomas P. Spaniol, Rolf Mülhaupt, Jun Okuda
Journal of Organometallic Chemistry 2009 694(7–8) pp: 1235-1237
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.11.008
Co-reporter:Geert-Jan M. Meppelder, Hong-Tao Fan, Thomas P. Spaniol and Jun Okuda
Organometallics 2009 Volume 28(Issue 17) pp:5159-5165
Publication Date(Web):August 5, 2009
DOI:10.1021/om9004274
Reaction of 1,4-diazabutanediyl-bridged bis(phenols) [(HOC6H2-4,6-tBu2)2NR(CH2)2NR] (R = H, 1; Me, 2) with the appropriate metal precursors MX4 (M = Ti, Zr, Hf; X = Cl, OiPr) gave a series of group 4 metal dichloro and di(isopropoxy) complexes [M{2,2′-(OC6H2-4,6-tBu2)2NR(CH2)2NR}X2] (R = H, X = Cl, M = Ti, 3; R = Me, X = OiPr, M = Ti, 4; R = Me, X = Cl, M = Ti, 5; Zr, 6; Hf, 7). While for complexes 3, 4, 6, and 7 single isomers were observed, titanium complex 5 formed as a mixture of cis-α (5a) and cis-β (5b) isomers. Variable-temperature NMR spectroscopy shows that titanium complexes 3 and 5b are unstable and favor a cis-α ligand configuration at elevated temperatures, whereas 4 and 5a are configurationally stable. Complexes 6 and 7 show rigid C2-symmetric structures in solution up to 100 °C. Upon activation with methylaluminoxane, 3−7 show good (M = Ti) to excellent (M = Zr, Hf) activity in the polymerization of 1-hexene, giving low molecular weight atactic polymers. Styrene is polymerized with modest activity.
Co-reporter:Dominique Robert Dr.;Elise Abinet Dipl.-Chem.;ThomasP. Spaniol Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 44) pp:11937-11947
Publication Date(Web):
DOI:10.1002/chem.200901616
Abstract
Monocationic bis-allyl complexes [Ln(η3-C3H5)2(thf)3]+[B(C6X5)4]− (Ln=Y, La, Nd; X=H, F) and dicationic mono-allyl complexes of yttrium and the early lanthanides [Ln(η3-C3H5)(thf)6]2+[BPh4]2− (Ln=La, Nd) were prepared by protonolysis of the tris-allyl complexes [Ln(η3-C3H5)3(diox)] (Ln=Y, La, Ce, Pr, Nd, Sm; diox=1,4-dioxane) isolated as a 1,4-dioxane-bridged dimer (Ln=Ce) or THF adducts [Ln(η3-C3H5)3(thf)2] (Ln=Ce, Pr). Allyl abstraction from the neutral tris-allyl complex by a Lewis acid, ER3 (Al(CH2SiMe3)3, BPh3) gave the ion pair [Ln(η3-C3H5)2(thf)3]+[ER3(η1-CH2CHCH2)]− (Ln=Y, La; ER3=Al(CH2SiMe3)3, BPh3). Benzophenone inserts into the LaCallyl bond of [La(η3-C3H5)2(thf)3]+[BPh4]− to form the alkoxy complex [La{OCPh2(CH2CHCH2)}2(thf)3]+[BPh4]−. The monocationic half-sandwich complexes [Ln(η5-C5Me4SiMe3)(η3-C3H5)(thf)2]+[B(C6X5)4]− (Ln=Y, La; X=H, F) were synthesized from the neutral precursors [Ln(η5-C5Me4SiMe3)(η3-C3H5)2(thf)] by protonolysis. For 1,3-butadiene polymerization catalysis, the yttrium-based systems were more active than the corresponding lanthanum or neodymium homologues, giving polybutadiene with approximately 90 % 1,4-cis stereoselectivity.
Co-reporter:Haiyan Ma ; Thomas P. Spaniol
Inorganic Chemistry 2008 Volume 47(Issue 8) pp:3328-3339
Publication Date(Web):March 4, 2008
DOI:10.1021/ic702312b
Monomeric yttrium and lutetium bis(phenolato) complexes [Ln(OSSO){N(SiHMe2)2}(THF)] (Ln = Y, Lu) were prepared from the reaction of silylamido complexes [Ln{N(SiHMe2)2}3(THF)2] with 1 equiv of tetradentate 1,ω-dithiaalkanediyl-bridged bis(phenol) (OSSO)H2 1–9 in moderate to high yields. In contrast to the rigid configuration of scandium analogues, the yttrium complexes 2b and 3b and the lutetium complex 3c that contain a C2 bridge between the two sulfur donors of the ligand are symmetric in solution. The monomeric nature of these complexes was indicated by an X-ray diffraction study of the yttrium complex 6b. The yttrium center in 6b is coordinated to the tetradentate [OSSO]-type ligand, one silylamido group and one THF ligand with the two oxygen donors of the [OSSO]-type ligand located trans. Corresponding bis(phenolato) silylamido complexes of larger rare-earth metals could not be obtained from similar reactions: Reaction of [La{N(SiHMe2)2}3(THF)2] with 1,2-xylylene-linked bis(phenol) gave a dinuclear lanthanum complex 6d of the formula [La2(OSSO)3] with two inequivalent eight-coordinate metal centers. The yttrium and lutetium complexes efficiently initiated the ring-opening polymerization (ROP) of lactides in THF. The heteroselectivity during the ROP of rac-lactide was enhanced when the steric demand of the bis(phenolato) ligand was increased, either by extending the bridge length or by introducing bulky ortho-substituents in the phenoxy units. A C3 bridge within the ligand backbone is essential to allow configurational interconversion of the active site between Λ and Δ configuration during polymerization, allowing accommodation of both enantiomers of the monomer in an alternating fashion.
Co-reporter:Mathias U. Kramer ; Dominique Robert ; Stefan Arndt ; Peter M. Zeimentz ; Thomas P. Spaniol ; Ahmed Yahia ; Laurent Maron ; Odile Eisenstein
Inorganic Chemistry 2008 Volume 47(Issue 20) pp:9265-9278
Publication Date(Web):September 25, 2008
DOI:10.1021/ic801259n
Synthesis, structure, and reactivity of two families of rare-earth metal complexes containing discrete methyl cations [LnMe(2−x)(thf)n](1+x)+ (x = 0, 1; thf = tetrahydrofuran) have been studied. As a synthetic equivalent for the elusive trimethyl complex [LnMe3], lithium methylates of the approximate composition [Li3LnMe6(thf)n] were prepared by treating rare-earth metal trichlorides [LnCl3(thf)n] with 6 equiv of methyllithium in diethyl ether. Heteronuclear complexes of the formula [Li3Ln2Me9Ln] (Ln = Sc, Y, Tb; L = Et2O, thf) were isolated by crystallization from diethyl ether. Single crystal X-ray diffraction studies revealed a heterometallic aggregate of composition [Li3Ln2Me9(thf)n(Et2O)m] with a [LiLn2Me9]2− core (Ln = Sc, Y, Tb). When tris(tetramethylaluminate) [Ln(AlMe4)3] (Ln = Y, Lu) was reacted with less than 1 equiv of [NR3H][BPh4], the dimethyl cations [LnMe2(thf)n][BPh4] were obtained. The coordination number as well as cis/trans isomer preference was studied by crystallographic and computational methods. Dicationic methyl complexes of the rare-earth metals of the formula [LnMe(thf)n][BAr4]2 (Ln = Sc, Y, La−Nd, Sm, Gd−Lu; Ar = Ph, C6H4F-4) were synthesized, by protonolysis of either the ate complex [Li3LnMe6(thf)n] (Ln = Sc, Y, Gd−Lu) or the tris(tetramethylaluminate) [Ln(AlMe4)3] (Ln = La−Nd, Sm, Dy, Gd) with ammonium borates [NR3H][BAr4] in thf. The number of coordinated thf ligands varied from n = 5 (Ln = Sc, Tm) to n = 6 (Ln = La, Y, Sm, Dy, Ho). The configuration of representative examples was determined by X-ray diffraction studies and confirmed by density-functional theory calculations. The highly polarized bonding between the methyl group and the rare-earth metal center results in the reactivity pattern dominated by the carbanionic character and the pronounced Lewis acidity: The dicationic methyl complex [YMe(thf)6]2+ inserted benzophenone as an electrophile to give the alkoxy complex [Y(OCMePh2)(thf)5]2+. Nucleophilic addition of the soft anion X− (X− = I−, BH4−) led to the monocationic methyl complexes [YMe(X)(thf)5]+.
Co-reporter:Dominique Robert, Małgorzata Kondracka and Jun Okuda
Dalton Transactions 2008 (Issue 20) pp:2667-2669
Publication Date(Web):27 Mar 2008
DOI:10.1039/B801030G
Rare-earth metal tris(borohydrides) [Ln(BH4)3(thf)3] (Ln = Y, La, Nd, Sm) are converted with one equivalent of the Brønsted acid [NEt3H]+[BPh4]− in thf into the monocationic bis(borohydride) complexes [Ln(BH4)2(thf)5]+[BPh4]−. They efficiently initiate the ring-opening polymerisation of ε-caprolactone.
Co-reporter:Dominique Robert;Peter Voth;Thomas P. Spaniol
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 18) pp:2810-2819
Publication Date(Web):
DOI:10.1002/ejic.200800216
Abstract
Trimethylsilylmethyl complexes of the type [Ln(η5-C5Me4CH2SiMe2NC6H4R-4-κN)(CH2SiMe3)(thf)n] containing a dianionic ligand [C5Me4CH2SiMe2NC6H4R-4]2– with a para-substituted anilido group and a CH2SiMe2 link were prepared. The yttrium complex [Y(η5-C5Me4CH2SiMe2NPh-κN)(CH2SiMe3)(thf)2] (2a) reacts with H2 to generate the corresponding dimeric hydride [Y(η5-C5Me4CH2SiMe2NPh-κN)(μ-H)(thf)]2 (5a). Pyridine inserts into the Y–H bond in a 1,2-fashion to afford the stable 2-hydropyridyl complex [Y(η5-C5Me4CH2SiMe2NPh-κN)(η1-NC5H6)(py)2] (6a). Upon reaction with tBuC≡CH, protonolysis takes place to give the dimeric alkynyl complex [Y(η5-C5Me4CH2SiMe2NPh-κN)(μ-C≡CtBu)(thf)]2 (7a).(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Dominique Robert;Thomas P. Spaniol
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 18) pp:2801-2809
Publication Date(Web):
DOI:10.1002/ejic.200800040
Abstract
Half-sandwich rare-earth metal tetramethylaluminate complexes [Ln(η5-C5Me4SiMe3){(μ-Me)2(AlMe2)}2] (Ln = Y, La, Nd, Sm, Gd, Lu) were obtained by reaction of the neutral homoleptic tetramethylaluminate complex [Ln{(μ-Me)2(AlMe2)}3] with tetramethyl(trimethylsilyl)cyclopentadiene, (C5Me4H)SiMe3. Protonolysis reaction of the neutral mono(cyclopentadienyl) complexes with the Brønsted acid [NEt3H]+[BPh4]– in thf led to the formation of the monocationic methyl complexes [Ln(η5-C5Me4SiMe3)Me(thf)3]+[BPh4]– (Ln = Y, La, Nd, Sm, Lu). Single-crystal X-ray diffraction study on the Y, Sm, and Lu derivatives showed a four-legged piano-stool configuration. Upon activation with [Ph3C]+[B(C6F5)4]–, the neutral half-sandwich tetramethylaluminate complex [La(η5-C5Me4SiMe3){(μ-Me)2(AlMe2)}2] catalyzed the polymerization of butadiene in the presence of [AliBu3] to give trans-1,4-polybutadiene with narrow polydispersities (Mn/Mw = 1.05–1.09).(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Dominique Robert;Thomas P. Spaniol
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 18) pp:
Publication Date(Web):
DOI:10.1002/ejic.200890047
Abstract
The cover picture shows the reactivity of half-sandwich bis(aluminate) complexes of the rare-earth metals that can be converted either into cationic methyl species by protonolysis or into dimeric dimethyl species by donor-induced cleavage of the aluminate moieties. Details are discussed in the article by J. Okuda et al. on p. 2801 ff.
Co-reporter:Geert-Jan M. Meppelder;Klaus Beckerle Dr.;Ramanujachary Manivannan Dr.;Bing Lian Dr.;Gerhard Raabe Dr.;Thomas P. Spaniol Dr. Dr.
Chemistry – An Asian Journal 2008 Volume 3( Issue 8-9) pp:1312-1323
Publication Date(Web):
DOI:10.1002/asia.200800064
Abstract
Chiral 1,2-trans-dithiocyclohexanediyl-bridged bis(phenols) of the type [2,2′-{HOC6H2-6-R1-4-R2}2S2C6H10] ([OSSO]H2, R1=tBu, iPr, H; R2=tBu, iPr, Me) could be conveniently and selectively synthesized in three steps, starting from cyclohexene oxide and arene thiolate. The racemic bis(phenols) could be resolved using an enantiopure (S)-camphorsulfonic ester auxiliary or by (chiral) HPLC. Complexation of the racemic bis(phenols) to TiX4 (X=Cl, OiPr) proceeds in a diastereoselective fashion to give only the Λ,R,R and Δ,S,S enantiomers. Racemic [Ti{(OC6H2-6-tBu-4-Me)2S2C6H10}Cl2] reacts with benzyl magnesium bromide to afford the crystallographically characterized dibenzyl complex. The benzyl cation formed using B(C6F5)3 in C6D5Br slowly decomposes at temperatures above +10 °C. When treated with methylaluminoxane, the dichloro complexes [Ti{OSSO}Cl2] polymerize styrene with activities up to 146 kg (mol catalyst)−1 [styrene (mol L−1)]−1 h−1; diisopropoxy complexes [Ti{OSSO}(OiPr)2] show mere trace activity. With 1-hexene as a chain-transfer agent, activated enantiopure titanium complexes give low-molecular-weight homochiral isotactic oligostyrenes, terminated by one to five 1-hexene units with Mn values as low as 750 g mol−1 for R=tBu and 1290 g mol−1 for R=Me. Below Mn≈5000 these oligostyrenes show optical activity.
Co-reporter:Marcin Konkol ; Malgorzata Kondracka ; Peter Voth ; Thomas P. Spaniol
Organometallics 2008 Volume 27(Issue 15) pp:3774-3784
Publication Date(Web):July 9, 2008
DOI:10.1021/om800161u
Rare-earth metal alkyl complexes with tridentate [OSO]-type and tetradentate [OSSO]-type bis(phenolato) ligands, [Ln(L)(CH2SiMe3)(THF)n] (LH2 = 2,2′-thiobis(6-tert-butyl-4-methylphenol) (tbmpH2), 1,3-dithiapropanediylbis(6-tert-butyl-4-methylphenol) (mtbmpH2), 1,4-dithiabutanediylbis(6-tert-butyl-4-methylphenol) (etbmpH2); Ln = Y (1−3), Sc (4, 5), Lu (7), Ho (9, 10)), were synthesized from the reactions of the tris(alkyl) complexes [Ln(CH2SiMe3)3(THF)2] with the corresponding bis(phenol) via alkane elimination. The alkyl complexes were characterized by NMR spectroscopy (Y, Sc, Lu) and elemental analysis as well as by X-ray crystal structure analysis (5, 7). The reaction of [Lu(CH2SiMe3)3(THF)2] with H2etbmp in a 1:2 ratio led to the formation of the bis(phenolato)-bridged dinuclear complex [Lu2(etbmp)3(THF)2] (8). The reaction of the holmium alkyl complexes 9 and 10 with PhSiH3 resulted in the formation of the corresponding hydrido complexes [Ho(L)(μ-H)(THF)n]2 (L = tbmp, n = 3, 11; L = etbmp, n = 2, 12). The formation of the yttrium analogues could be observed by NMR spectroscopy. Complexes 2, 4, and 5 were tested in the hydrosilylation of a wide variety of aliphatic and aromatic 1-alkenes and 1,5-hexadiene with various silanes (PhSiH3, nBuSiH3, and Ph2SiH2). In the case of terminal aliphatic alkenes an anti-Markovnikov (1,2) addition takes place with 80−99% regioselectivity. The hydrosilylation of styrene afforded the Markovnikov (2,1) addition product PhHC(SiH2Ph)Me with 97% regioselectivity. The hydrosilylation of 1,5-hexadiene by PhSiH3 catalyzed by 2 resulted in the formation of a linear product, 1,6-bis(phenylsilyl)hexane (ca. 90%), and a cyclic product, (phenylsilylmethyl)cyclopentane (ca. 10%), whereas with nBuSiH3 84% of the cyclic product was obtained.
Co-reporter:Ilja Peckermann, Dominique Robert, Ulli Englert, Thomas P. Spaniol and Jun Okuda
Organometallics 2008 Volume 27(Issue 18) pp:4817-4820
Publication Date(Web):August 27, 2008
DOI:10.1021/om800562z
The trimethylsilylmethyl indium complex [In(CH2SiMe3)3] (1) was studied by single-crystal X-ray structure analysis at low temperature and shown to be weakly associated as dimeric units with a long indium−carbon bond in the bridges. The bis(trimethylsilylmethyl) cation [In(CH2SiMe3)2(THF)3][B(C6F5)4] (2) was prepared by protonolysis of the neutral complex [In(CH2SiMe3)3] with [NPhMe2H][B(C6F5)4] in THF. Crystal structure analysis revealed that mononuclear 2 adopts a square-pyramidal geometry.
Co-reporter:Mathias U. Kramer;Dominique Robert;Yumiko Nakajima;Ulli Englert;Thomas P. Spaniol
European Journal of Inorganic Chemistry 2007 Volume 2007(Issue 5) pp:
Publication Date(Web):4 JAN 2007
DOI:10.1002/ejic.200601025
The alkyl abstraction reaction of [YR3(thf)2] (R = CH2SiMe3) with group-13 trialkyl complexes [(ER3)n] (E = B, Ga, In: n = 1; E = Al: n = 2) forming cationic yttrium species [YR2(thf)4]+[ER4]– (E = Al, Ga, In) shows a strong dependence on the Lewis acidic metal centre E and on the solvent basicity. Whilst the boron compound does not react with [YR3(thf)2], the heavier homologues form the ion pairs [YR2(thf)4]+[ER4]– (E = Al, Ga, In) which dissociate to give the neutral compounds in apolar solvents such as benzene. Single-crystal structure analysis of the gallate [YR2(thf)4]+[GaR4]– shows the presence of an ion pair with cis-arranged alkyl ligands in the octahedral yttrium cation and a tetrahedral gallate anion. Group-13 trialkyl compounds [(ER3)n] (E = B, Al, Ga), all liquids at room temperature, and [Li(12-crown-4)2]+[AlR4]– were characterised by single-crystal X-ray diffraction and NMR spectroscopy. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Bing Lian, Thomas P. Spaniol and Jun Okuda
Organometallics 2007 Volume 26(Issue 26) pp:6653-6660
Publication Date(Web):November 20, 2007
DOI:10.1021/om700785c
Reaction of the titanium dichloro complex containing a [OSSO]-type bis(phenolate) ligand (edtbp)TiCl2 (1) (edtbpH2 = (HOC6H2-tBu2-4,6)2(SCH2CH2S)) with 2 equiv of AlMe3 produces the methylchloro complex (edtbp)TiMeCl (2) in 77% yield, alternatively prepared from comproportionation of 1 and the dimethyl complex (edtbp)TiMe2 (3). The titanium ester enolate complex (edtbp)TiMe{O(iPrO)C=CMe2} (4) was synthesized by reaction of 2 with lithium isopropylisobutyrate, LiO(iPrO)C=CMe2, in a 1:1 molar ratio. This complex reacted with alcohols ROH (R = iPr, Ph, Ph3C) to give alkoxy complexes (edtbp)TiMe(OR) (6−8) and with acetone under C−C coupling to afford an aldolate complex (edtbp)TiMe(OCMe2CMe2CO2iPr) (9). Methyl abstraction from the neutral complexes 4, 7, and 9 using with B(C6F5)3 gave the corresponding cationic species [(edtbp)Ti(OR)(THF)n][MeB(C6F5)3]. All complexes have been characterized by multinuclear NMR spectroscopy and elemental analysis. Single-crystal X-ray diffraction studies were performed for complexes 2, 4, and 9. Cationic titanium enolate species initiate the polymerization of methyl methacrylate (MMA) at room temperature, producing syndiotactic-enriched poly(methyl methacrylates) (PMMAs). In the presence of the neutral enolate complex, MMA polymerization proceeds in a living fashion. The neutral titanium enolate complex 4 was found to be moderately active in n-butyl acrylate (n-BA) polymerization.
Co-reporter:Bing Lian Dr.;Klaus Beckerle Dr.;Thomas P. Spaniol Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 44) pp:
Publication Date(Web):27 SEP 2007
DOI:10.1002/ange.200703218
Wechselhafte Katalyse: Kationische Bis(phenolato)-Metallkomplexe der Gruppe 4 katalysieren effizient die Oligomerisation von 1-Hexen. Während mit dem Titankomplex Oligo(1-hexene) entstehen, die aus einer 2,1-Insertion des 1-Hexens resultieren, katalysieren die Zirconium- und Hafniumkomplexe eine 1,2-Insertion (siehe Schema).
Co-reporter:Klaus Beckerle Dr.;Ramanujachary Manivannan Dr.;Bing Lian Dr.;Geert-Jan M. Meppelder;Gerhard Raabe Dr.;Thomas P. Spaniol Dr.;Henner Ebeling Dr.;Frederic Pelascini Dr.;Rolf Mülhaupt Dr. Dr.
Angewandte Chemie 2007 Volume 119(Issue 25) pp:
Publication Date(Web):29 MAY 2007
DOI:10.1002/ange.200700783
Im Gleichtakt: Titankatalysatorvorstufen in beiden enantiomeren Formen wurden diastereoselektiv hergestellt und nach MAO-Aktivierung zur stereospezifischen Styrololigomerisation eingesetzt (siehe Schema). Die isotaktischen Polystyrole zeigen bis zu einem Polymerisationsgrad von 45 messbare optische Aktivität, was bestätigt, dass bei diesen Homogenkatalysatoren eine stereochemische Steuerung durch das Metallzentrum stattfindet.
Co-reporter:Bing Lian Dr.;Klaus Beckerle Dr.;Thomas P. Spaniol Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 44) pp:
Publication Date(Web):27 SEP 2007
DOI:10.1002/anie.200703218
A cat of two tales: Cationic Group 4 metal catalysts that contain a linked bis(phenolato) ligand efficiently catalyze the oligomerization of 1-hexene. Whereas oligo(1-hexene)s arising from 2,1-insertion of 1-hexene are observed for the titanium complexes, those prepared using zirconium and hafnium complexes are formed by 1,2-insertion.
Co-reporter:Klaus Beckerle Dr.;Ramanujachary Manivannan Dr.;Bing Lian Dr.;Geert-Jan M. Meppelder;Gerhard Raabe Dr.;Thomas P. Spaniol Dr.;Henner Ebeling Dr.;Frederic Pelascini Dr.;Rolf Mülhaupt Dr. Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 25) pp:
Publication Date(Web):29 MAY 2007
DOI:10.1002/anie.200700783
Tactful: Titanium catalyst precursors in both optically active, enantiomeric forms have been obtained diastereoselectively, and after activation with methylaluminoxane (MAO) they were used for the stereospecific oligomerization of styrene. The isotactic polystyrenes with a degree of polymerization of up to 45 show measurable optical activity, thus corroborating that enantiomorphic site control is operating in these homogeneous catalysts.
Co-reporter:Benjamin R. Elvidge, Stefan Arndt, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2006 (Issue 7) pp:890-901
Publication Date(Web):06 Dec 2005
DOI:10.1039/B512285F
Rare-earth metal alkyl tri(tert-butoxy)silanolate complexes [Ln{μ,η2-OSi(OtBu)3}(CH2SiMe3)2]2 (Ln = Y (1), Tb (2), Lu (3)) were prepared via protonolysis of the appropriate tris(alkyl) complex [Ln(CH2SiMe3)3(thf)2] with tri(tert-butoxy)silanol in pentane. Crystal structure analysis revealed a dinuclear structure for 1 with square pyramidal geometry at the yttrium centre. The silanolate ligand coordinates in an η2-bridging coordination mode giving a 4-rung truncated ladder and non-crystallographic inversion centre. Addition of two equiv. of 12-crown-4 to a pentane solution of 1 or 3 respectively gave [Ln{OSi(OtBu)3}(CH2SiMe3)2(12-crown-4)]·12-crown-4 (Ln = Y (4), Lu (5)). Crystal structure analysis of 5 showed a slightly distorted octahedral geometry at the lutetium centre. The silanolate ligand adopts an η1-terminal coordination mode, whilst the crown ether unit coordinates in an unusual κ3-fashion. Reaction of 1–3 with [NEt3H]+[BPh4]− in thf yielded the cationic derivatives [Ln{OSi(OtBu)3}(CH2SiMe3)(thf)4]+[BPh4]− (Ln = Y (6), Tb (7) and Lu (8)); coordination of crown ether led to compounds of the form [Ln{OSi(OtBu)3}(CH2SiMe3)(L)(thf)n]+[BPh4]− (Ln = Y, Lu, L = 12-crown-4, n = 1 (9, 10); Ln = Y, Lu, L = 15-crown-5, n = 0 (11, 12)). Reaction of 1 with [NMe2PhH]+[B(C6F5)4]−, [Al(CH2SiMe3)3] or BPh3 in thf gave the ion pairs [Y{OSi(OtBu)3}(CH2SiMe3)(thf)4]+[A]− ([A]− = [B(C6F5)4]− (13), [Al(CH2SiMe3)4]− (14), [BPh3(CH2SiMe3)]− (15)), whilst two equiv. [NMe2PhH]+[BPh4]− with 1 in thf produced the dicationic ion triple [Y{OSi(OtBu)3}(thf)6]2+[BPh4]−2 (16). Crystal structure analysis revealed that 16 is mononuclear with pentagonal bipyramidal geometry at the yttrium centre. The silanolate ligand coordinates in an η1-terminal fashion. All diamagnetic compounds have been characterized by NMR spectroscopy. 1, 3, 4, 6 and 13 were tested as olefin hydrosilylation pre-catalysts with a variety of substrates; 1 was found to be highly active in 1-decene hydrosilylation.
Co-reporter:Peter M. Zeimentz, Thomas P. Spaniol, Jun Okuda
Inorganica Chimica Acta 2006 Volume 359(Issue 15) pp:4769-4773
Publication Date(Web):1 December 2006
DOI:10.1016/j.ica.2006.04.018
Neutral tris(trimethylsilylmethyl) complexes [Ln(CH2SiMe3)3(L)] (Ln = Sc (1), Lu (2)) and cationic bis(trimethylsilylmethyl) complexes [Ln(CH2SiMe3)2(L)(THF)]+[BPh4]−, (Ln = Sc (3), Lu (4)) that contain bis(2-methoxyethyl)(trimethylsilyl)amine (L = Me3SiN(CH2CH2OMe)2) as a neutral, tridentate ligand were synthesized and characterized by NMR spectroscopy. X-ray structural analysis was performed for the scandium complex 1 and exhibited a distorted octahedral coordination geometry with a facially arranged ligand at the neutral scandium center. NMR spectroscopy corroborated the coordination of the tertiary amine function of the ligand to the metal. Complexes 3 and 4 expand the still limited range of cationic rare-earth metal alkyl complexes with known neutral, multidentate ligands.Neutral tris(trimethylsilylmethyl) complexes [Ln(CH2SiMe3)3(L)] and cationic bis(trimethylsilylmethyl) complexes [Ln(CH2SiMe3)2(L)(THF)]+ [BPh4]−, (Ln = Sc, Lu) that contain bis(2-methoxyethyl)(trimethylsilyl)amine (Me3SiN(CH2CH2OMe)2) as a neutral, tridentate ligand L, were synthesized and characterized by NMR spectroscopy. X-ray structural analysis was performed for the neutral scandium complex and exhibited a distorted octahedral coordination geometry with a facially arranged ligand at the neutral scandium center.
Co-reporter:Haiyan Ma Dr.;Thomas P. Spaniol Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 46) pp:
Publication Date(Web):26 OCT 2006
DOI:10.1002/anie.200603178
Tactical response: High heterotactic stereocontrol was observed from the ring-opening polymerization of rac-lactide on using a series of bis(phenolato)scandium complexes (see scheme). This stereocontrol appears to involve a dynamic monomer-recognition process as a result of the interconversion of the configuration from Λ into Δ.
Co-reporter:Haiyan Ma Dr.;Thomas P. Spaniol Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 46) pp:
Publication Date(Web):26 OCT 2006
DOI:10.1002/ange.200603178
Die richtige Taktik: Eine ausgeprägte heterotaktische Stereokontrolle kennzeichnet die Ringöffnungspolymerisation von rac-Lactid mit einer Reihe von Bis(phenolato)scandium-Komplexen (siehe Schema). An dieser Stereokontrolle scheint eine dynamische Monomererkennung als Folge der Umwandlung der Konfiguration von Λ zu Δ beteiligt zu sein.
Co-reporter:Stefan Arndt
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 2-3) pp:
Publication Date(Web):14 FEB 2005
DOI:10.1002/adsc.200404269
Cationic alkyl complexes of the rare-earth metals [LnRm(L)n](3–m)+ (R=alkyl; m=1, 2; L=Lewis base) were virtually unknown species until recently. Because of their increased Lewis acidity/electrophilicity they should have considerable potential as homogeneous catalysts in olefin polymerization and in organic transformations. They can be generated by treating the neutral rare-earth metal precursors containing at least two alkyl groups R with suitable Lewis or Brønsted acids in the presence of weakly coordinating anions. Not only monocationic but also dicationic alkyl derivatives have been shown to be accessible. In the context of modeling homogeneous ethylene polymerization using a mixture consisting of LnR3/AlR3/[NMe2HPh][B(C6F5)4], such dications were discovered. Some thermally robust examples of mono- and dicationic alkyl complexes have been structurally characterized as solvent-separated ion pairs. Neutral and monoanionic macrocycles such as crown ethers or aza-crown ethers as well as amidinato, β-diketiminato, and substituted cyclopentadienyl ligands are suitable ancillary ligands to stabilize the cationic alkyl fragments.
Co-reporter:Haiyan Ma, Gianluca Melillo, Leone Oliva, Thomas P. Spaniol, Ulli Englert and Jun Okuda
Dalton Transactions 2005 (Issue 4) pp:721-727
Publication Date(Web):12 Jan 2005
DOI:10.1039/B416875E
Aluminium alkyl complexes [(OSSO)AlR]
(1–3: R = Me, Et) were isolated in good yields from the protonolysis reaction of AlR3 with the corresponding tetradentate 1,ω-dithiaalkanediyl-bridged bisphenols (1,4-dithiabutanediyl-bis(6-tert-butyl-4-methylphenol), etbmpH2; ortho-xylylenedithio-bis(6-tert-butyl-4-methylphenol), xytbmpH2). The monomeric structures of all three complexes were confirmed by X-ray diffraction studies. Complexes 1 and 2 have an isotypic packing arrangement. The aluminium center is coordinated by the etbmp ligand and one alkyl group with distorted trigonal bipyramidal geometry. Complex 3 shows Cs symmetry with square pyramidal geometry around the metal center. Substitution reaction of complex 1 with trityl alcohol gave the monomeric alkoxide complex [(etbmp)Al(OCPh3)]
4, which has a similar trigonal bipyramidal geometry around the aluminium atom as complex 1. In the presence of isopropanol, complexes 1–3 initiated the living ring-opening polymerization of rac-lactide (PDI = 1.03–1.06, Mw/Mn). The ligand structure influenced the tacticity of the obtained polymer, with complex 3 giving heterotactic-enriched polylactides.
Co-reporter:Stefan Arndt Dr.;Klaus Beckerle Dr.;Peter M. Zeimentz Dipl.-Chem.;Thomas P. Spaniol Dr. Dr.
Angewandte Chemie 2005 Volume 117(Issue 45) pp:
Publication Date(Web):25 OCT 2005
DOI:10.1002/ange.200502915
Strukturell charakterisierte mono- und dikationische Yttrium-Methyl-Komplexe 3 und 4 sind durch Protonolyse der bekannten Hexamethylat-Komplexe 1 und 2 zugänglich (siehe Schema) und dienen als Modelle für homogene Katalysatoren zur 1,4-cis-selektiven Polymerisation von 1,3-Dienen durch industriell verwendete Mehrkomponenten-Katalysatoren.
Co-reporter:Stefan Arndt, Klaus Beckerle, Peter M. Zeimentz, Thomas P. Spaniol,Jun Okuda
Angewandte Chemie International Edition 2005 44(45) pp:7473-7477
Publication Date(Web):
DOI:10.1002/anie.200502915
Co-reporter:Alexander A. Trifonov, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2004 (Issue 15) pp:2245-2250
Publication Date(Web):28 Jun 2004
DOI:10.1039/B406071G
The dimeric hydrido complex [Y(L)(THF)(μ-H)]2
(2) containing the CH2SiMe2-linked amido-cyclopentadienyl ligand L = C5Me4CH2SiMe2NCMe32− catalyzed the hydrosilylation of 1,5-hexadiene, 1,7-octadiene and vinylcyclohexene by PhSiH3. As demonstrated for 1,7-octadiene, the product distribution of the hydrosilylation strongly depends on the molar ratio of the reagents. In the absence of PhSiH3, the stoichiometric reaction of 2 with 1,5-hexadiene gave the isolable crystalline cyclopentylmethyl complex [Y(L){CH2CH(CH2)4}(THF)]
(3). Internal olefins such as trans-stilbene and alkynes such as tert-butylacetylene were not hydrosilylated by 2. trans-Stilbene was inserted into the yttrium–hydride bond of 2 to give the 1,2-diphenylethyl complex [Y(L){CH(CH2Ph)Ph}(THF)]
(4). tert-Butylacetylene reacted with 2 to give the dimeric acetylide [Y(L)(CCCMe3)]2
(5). In an attempt to detect the monomeric hydrido species as a DME adduct [Y(L)H(DME)], complex 2 was reacted with DME to form the sparingly soluble, dimeric 2-methoxyethoxy complex [Y(L)(μ-OCH2CH2OMe-κO)]2
(6) under C–O splitting.
Co-reporter:Maria D'Acunzi;Oriana Motta;Leone Oliva;Antonio Proto;Carmine Capacchione
Macromolecular Chemistry and Physics 2004 Volume 205(Issue 3) pp:370-373
Publication Date(Web):9 FEB 2004
DOI:10.1002/macp.200300115
Summary: Copolymerization of styrene with small amounts of ethylene using the catalyst system dichloro{1,4-dithiabutanediyl-2,2′-bis(4,6-di-tert-butyl-phenoxy)}titanium/methylaluminoxane results in the unprecedented formation of isotactic polystyrene containing isolated ethylene units. 13C NMR spectroscopic analysis of the copolymer indicates that an “enantiomorphic site” control mechanism for isospecific propagation is operating. DSC measurements also indicate the presence of isolated ethylene units which modify the crystallization behavior of the isotactic polystyrene.
Co-reporter:Shigekazu Matsui, Yasunori Yoshida, Yukihiro Takagi, Thomas P. Spaniol, Jun Okuda
Journal of Organometallic Chemistry 2004 Volume 689(Issue 7) pp:1155-1164
Publication Date(Web):1 April 2004
DOI:10.1016/j.jorganchem.2003.11.041
A series of pyrrolyl-imines HL1–6 was prepared by the condensation of pyrrole-2-carboxyaldehyde with different amines. The reaction of 2 equiv of pyrrolyl-imine with tetrabenzyl complexes of hafnium and zirconium M(CH2Ph)4 (M=Hf or Zr) gave dibenzyl complexes (L3–6)2M(CH2Ph)2, which were characterized by NMR spectroscopy and crystal structure analysis. NMR spectra of the complexes with secondary alkyl substituents at the imine nitrogen (isopropyl: 3a, 4-tert-butylcyclohexyl: 4a and 4b) suggest that rapid racemization between Δ and Λ configurations occurs in solution on the NMR time scale. The complexes with pyrrolide-imine ligands with a tertiary alkyl group such as tert-butyl (5a and 5b) or 1-adamantyl (6a and 6b) at the imine nitrogen possess cis-configured benzyl groups. Hafnium complexes 5a and 6a react with B(C6F5)3 in bromobenzene-d5 to give the corresponding cationic benzyl complexes, which exhibit high activity for ethylene polymerization (5a: 2242 kg-polymer/ mol-Hf h bar, 6a: 2096 kg-polymer/ mol-Hf h bar). Zirconium complexes 5b and 6b display a remarkably high ethylene polymerization activity when activated with methylaluminoxane (5b: 17,952 kg-polymer/mol-Zr h bar, 6b: 22,944 kg-polymer/mol-Zr h bar).The reaction of pyrrolyl-imine with a tertiary alkyl group at the imine nitrogen LH with M(CH2Ph)4 (M=Hf or Zr) gave dibenzyl hafnium and zirconium complexes (L)2M(CH2Ph)2. The complexes possess cis-configured benzyl groups and exhibit high activity for ethylene polymerization of up to 2 × 104 kg-polymer/mol-M h bar, when activated with B(C6F5)3 or methylaluminoxane.
Co-reporter:Klaus Beckerle, Carmine Capacchione, Henner Ebeling, R. Manivannan, Rolf Mülhaupt, Antonio Proto, Thomas P. Spaniol, Jun Okuda
Journal of Organometallic Chemistry 2004 Volume 689(Issue 24) pp:4636-4641
Publication Date(Web):29 November 2004
DOI:10.1016/j.jorganchem.2004.08.019
In the context of developing single-site stereoselective post-metallocene catalysts, the case for isospecific styrene polymerization catalysts based on methylaluminoxane-activated group 4 metal bis(phenolato) complexes is summarized. Ligands derived from the 1,4-dithiabutanediyl-linked bis(phenol)s have been found to induce stereochemical rigidity by the presence of the hemi-labile sulfide donor functions. Isospecific styrene polymerization was achieved using easily accessible catalyst precursors of the type [MX2(OC6H2-tBu2-4,6)2{S(CH2)2S}] (M = Ti, Zr, Hf; X = Cl, OiPr, CH2Ph). Activating the dibenzyl titanium complex [Ti(CH2Ph)2(OC6H2-tBu2-4,6)2{S(CH2)2S}] with B(C6F5)3 and AliBu3, controlled isotactic polymerization became possible at lower temperatures. A remarkable dependence of both the activity and stereoselectivity on the ligand substitution pattern was observed. Analogous precursors with the 1,5-dithiapentanediyl-linked bis(phenolato) ligand gave syndiotactic polystyrene with lower activity.In the context of developing single-site stereoselective post-metallocene catalysts, the case for isospecific styrene polymerization catalysts based on methylaluminoxane-activated group 4 metal bis(phenolato) complexes is summarized.
Co-reporter:Michael W. Neiser Dr.;Sra Muth;Ute Kolb Dr.;J. Robin Harris Dr.;Manfred Schmidt Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 24) pp:
Publication Date(Web):9 JUN 2004
DOI:10.1002/anie.200353259
Giant rod–coil amphiphilic block copolymers were prepared by metallocene-catalyzed polymerization. The hydrophilic coiled block consists of polymethacrylic acid and the rod component of a hydrophobic cylindrical brush polymer. The high solubility of the stiff block results in the micelles being formed with the stiff block forming the solubilizing corona.
Co-reporter:Michael W. Neiser Dr.;Sra Muth;Ute Kolb Dr.;J. Robin Harris Dr.;Manfred Schmidt Dr.
Angewandte Chemie 2004 Volume 116(Issue 24) pp:
Publication Date(Web):9 JUN 2004
DOI:10.1002/ange.200353259
Gigantisch: Riesige amphiphile Knäuel-Stäbchen-Blockcopolymere wurden durch metallocenkatalysierte Polymerisation synthetisiert. Der hydrophile Knäuelblock besteht aus Polymethacrylsäure, die Stäbchenkomponente aus einem zylindrischen Bürstenpolymer. Wegen der ausgezeichneten Löslichkeit der kettensteifen Komponente konnten Micellen gebildet werden (siehe Schema), deren solubilisierende Schale aus dem Stäbchenblock besteht.
Co-reporter:Carmine Capacchione, Antonio Proto, Henner Ebeling, Rolf Mülhaupt, Klaus Möller, R Manivannan, Thomas P Spaniol, Jun Okuda
Journal of Molecular Catalysis A: Chemical 2004 Volume 213(Issue 1) pp:137-140
Publication Date(Web):13 April 2004
DOI:10.1016/j.molcata.2003.10.057
The development of an isospecific styrene polymerization catalysts system based on methylaluminoxane (MAO)-activated group 4 metal non-metallocene complexes is reported. The ligand derived from the 1,4-dithiabutanediyl-linked bis(phenol)s has been designed in order to ensure stereochemical rigidity utilizing the potentially hemi-labile function of the sulfur donors. Isospecificity during styrene polymerization was achieved using easily accessible catalyst precursors of the type (M = Ti, Zr, Hf; X = Cl, CH2Ph). A remarkable dependence of both the activity and stereoselectivity on the ligand backbone was observed, as analogous precursors with 1,5-dithiapentanediyl-linked bis(phenolato) ligand gave syndiotactic polystyrene with lower activity.Graphic
Co-reporter:Albert Paparo, Jun Okuda
Coordination Chemistry Reviews (1 March 2017) Volume 334() pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.ccr.2016.06.005
•The literature on CO2 metal complexes is critically surveyed.•The CO2 complexes are classified by the covalent bond classification (CBC) method.•The CBC method reveals the existence of bonding modes that have not been recognized so far.•A generalized summary of all known synthetic methods is provided.•An overview of metal centered reductive transformations of CO2 is given.A critical survey on the literature on CO2 metal complexes is provided. A re-evaluation of all known CO2 complexes reveals the existence of several bonding modes, which have not been recognized as separate classes so far. This review further provides suggestions on alternative interpretations of the bonding in reported complexes. The accurate classification of the bonding situation as well as the oxidation states of carbon and the metals involved in binding CO2 allowed for establishing a generalized summary of all known synthetic methods. Finally, a short overview of reductive carbon dioxide transformations at metal centers is given.
Co-reporter:Waldemar Fegler, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2010 - vol. 39(Issue 29) pp:NaN6779-6779
Publication Date(Web):2010/04/28
DOI:10.1039/C001699C
Tris(trimethylsilylmethyl) complexes of yttrium and lutetium [LnR3(THF)2] (R = CH2SiMe3) were treated with sterically bulky N-heterocyclic carbenes (NHC) 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) and 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes). IPr gave labile mono-adducts [LnR3(NHC)], isolated as thermally robust crystals and fully characterized by NMR spectroscopy and X-ray diffraction. IMes gave a similar lutetium mono-adduct [LuR3(IMes)] with the lutetium alkyl [LuR3(THF)2], whereas the yttrium alkyl [YR3(THF)2] resulted in the formation of an ortho-metalated product. This compound, isolated as a crystalline bis(THF) adduct, contains a strained six-membered chelate ring that has been formed by the C–H bond activation of one of the ortho-methyl groups of the mesityl group. In contrast [LuR3(IMes)] only slowly underwent a similar C–H bond activation.
Co-reporter:Jean-Charles Buffet and Jun Okuda
Dalton Transactions 2011 - vol. 40(Issue 30) pp:NaN7754-7754
Publication Date(Web):2011/04/11
DOI:10.1039/C1DT10075K
Cyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, (Me3TACD)H (= Me3[12]aneN4), reacted with [K{N(SiHMe2)2}] in benzene-d6 to give [K{(Me3TACD)SiMe2N(SiHMe2)}] (1) under hydrogen evolution. Single-crystal X-ray diffraction of 1 shows a dinuclear structure in the solid state, featuring a bridging μ-amido and a weak β-agostic Si–H bond. 1,7-Dimethyl-1,4,7,10-tetraazacyclododecane (Me2TACD)H2 (= Me2[12]aneN4) and (Me3TACD)H were reacted with [Sc{N(SiHMe2)2}3(thf)] in benzene-d6 to give [{(Me2TACD)SiMe2N(SiHMe2)}Sc{N(SiHMe2)2}] (2) and [(Me3TACD)Sc{N(SiHMe2)2}2SiMe2] (3), respectively. Both compounds are monomeric in solution and X-ray diffraction studies showed the scandium metal centers to be six-coordinate. The scandium alkyl complex [Sc(Me3TACD)(CH2SiMe3)2] (4) was obtained by reacting (Me3TACD)H with [Sc(CH2SiMe3)3(thf)] in benzene-d6. The scandium amide complexes 2 and 3 catalyzed the ring-opening polymerization (ROP) of meso-lactide to give syndiotactic polylactides.
Co-reporter:Phillip Jochmann, Julien P. Davin, Stefanie Maslek, Thomas P. Spaniol, Yann Sarazin, Jean-Francois Carpentier and Jun Okuda
Dalton Transactions 2012 - vol. 41(Issue 30) pp:NaN9181-9181
Publication Date(Web):2012/05/10
DOI:10.1039/C2DT30743J
The synthesis and attempted isolation of neutral bis(allyl)strontium [Sr(C3H5)2] (1) resulted in the isolation of potassium tris(allyl)strontiate K[Sr(C3H5)3] (2). In situ generated 1 shows a pronounced Brønsted basicity, inducing polymerisation of THF. Ate complex 2 crystallises as [K(THF)2{Sr(C3H5)3}(THF)]∞ (2·(THF)3). The salt-like solid state structure of 2·(THF)3 comprises a two-dimensional network of (μ2-η3:η3-C3H5)− bridged potassium and strontium centres. Synthesis of allyl complexes 1 and 2 utilised SrI2, [Sr(TMDS)2] (3) (TMDS = tetramethyldisilazanide), and [Sr(HMDS)2] (HMDS = hexamethyldisilazanide) as strontium precursors. The solid state structure of previously reported [Sr(TMDS)2] (3) was established by X-ray single crystal analysis as a dissymmetric dimer of [Sr2(TMDS)4(THF)3] (3·(THF)3) with multiple Si–H⋯Sr agostic interactions. The presence of ether ligands (THF, 18-crown-6) influenced the Si–H⋯Sr resonances in the NMR spectra of the amido complex 3.
Co-reporter:Andreas Sauer, Andreas Kapelski, Christophe Fliedel, Samuel Dagorne, Moshe Kol and Jun Okuda
Dalton Transactions 2013 - vol. 42(Issue 25) pp:NaN9023-9023
Publication Date(Web):2013/03/11
DOI:10.1039/C3DT00010A
Polylactide (PLA) is an attractive polymeric material due to its origin from annually renewable resources and its biodegradability. The ring-opening polymerization (ROP) of lactide initiated by Lewis acidic and oxophilic metal-based catalysts constitutes the method of choice to access PLA in a controlled and stereoselective manner. The design and synthesis of ligand-supported metal complexes to act as effective ROP initiators of lactide monomers have been the subject of numerous investigations over the past decades. In view of their oxophilic nature, well-defined group 4 metal complexes supported by polydentate supporting ligands have appeared as active initiators for lactide ROP. This perspective summarizes various classes of structurally well-defined group 4 metal initiators developed for lactide ROP. It also provides observed trends regarding their catalytic performance. Whenever appropriate and possible, catalyst structure–ROP performance (i.e. activity, control and stereoselectivity) relationships are rationalized.
Co-reporter:Valeri Leich, Kevin Lamberts, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2014 - vol. 43(Issue 38) pp:NaN14321-14321
Publication Date(Web):2014/05/07
DOI:10.1039/C4DT00916A
Alkali metal triphenylsilyls [Li(12-crown-4)SiPh3]·(thf)0.5 (2), [Na(15-crown-5)SiPh3]·(thf)0.5 (3) and [K(18-crown-6)SiPh3(thf)] (4) were synthesized using 1,1,1-trimethyl-2,2,2-triphenyldisilane (Ph3SiSiMe3) and isolated in high yields. Solid state structures were determined by single crystal X-ray diffraction. These alkali metal silyls catalyzed the regioselective hydrosilylation of 1,1-diphenylethylene to give the anti-Markovnikov product. The presence of crown ethers enhanced the reactivity of the metal silyls in hydrosilylation catalysis.
Co-reporter:Catherine Hermans, Weifeng Rong, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2016 - vol. 45(Issue 19) pp:NaN8133-8133
Publication Date(Web):2016/04/11
DOI:10.1039/C6DT00272B
Lanthanum complexes [(L)LaX] (X = N(SiMe3)21, OiPr 2, BH43) supported by a ferrocene-based (OSSO)-type ligand LH2 were synthesized and characterized by elemental analysis, NMR spectroscopy and cyclic voltammetry. The structure of 1 was confirmed by single crystal X-ray diffraction. These complexes were highly active initiators for the ring-opening polymerization of rac-lactide (rac-LA). The activity depended on the initiating group in the order of 1 ≈ 2 > 3. The activities of 2 and 3 during polymerization were controlled in situ with external redox reagents by reversibly switching the oxidation state of the iron center.
Co-reporter:Sabine Standfuss, Elise Abinet, Thomas P. Spaniol and Jun Okuda
Chemical Communications 2011 - vol. 47(Issue 41) pp:NaN11443-11443
Publication Date(Web):2011/09/23
DOI:10.1039/C1CC14180E
Neutral, cationic and anionic allyl compounds of scandium contain highly fluxional allyl ligands in solution, whilst in the solid state both η1- and η3-binding modes are detected.
Co-reporter:Jean-Charles Buffet and Jun Okuda
Chemical Communications 2011 - vol. 47(Issue 16) pp:NaN4798-4798
Publication Date(Web):2011/03/15
DOI:10.1039/C1CC10149H
Group 4 metal initiators with a tetradentate bis(phenolato) ligand polymerized meso-lactide efficiently under ring-opening to give syndiotactic polylactide. L-Lactide was converted faster than rac-lactide and meso-lactide.
Co-reporter:Valeri Leich, Thomas P. Spaniol, Laurent Maron and Jun Okuda
Chemical Communications 2014 - vol. 50(Issue 18) pp:NaN2314-2314
Publication Date(Web):2014/01/07
DOI:10.1039/C3CC49308C
Bis(triphenylsilyl)calcium [Ca(SiPh3)2(thf)] obtained in high yield as a crystalline ether adduct catalyzes the hydrosilylation of activated C–C double bonds efficiently and regioselectively.
Co-reporter:V. Leich, T. P. Spaniol and J. Okuda
Chemical Communications 2015 - vol. 51(Issue 79) pp:NaN14774-14774
Publication Date(Web):2015/08/17
DOI:10.1039/C5CC06187C
Hydrogenation of easily accessible potassium triphenylsilyl [K(Me6TREN)SiPh3] gave the hydrogen storage material α-[KSiH3] in high yields by an unusual hydrogenolytic cleavage of silicon–phenyl bonds.
Co-reporter:Debabrata Mukherjee, Ann-Kristin Wiegand, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2017 - vol. 46(Issue 19) pp:NaN6186-6186
Publication Date(Web):2017/04/18
DOI:10.1039/C7DT01094J
The zinc hydridotriphenylborates [(L)Zn(TMDS)][HBPh3] and [(L)ZnX][HBPh3] (L = Me4TACD, Me4[12]aneN4; TMDS = N(SiHMe2)2; X = Cl, Br, I) were synthesized by BPh3-mediated β-SiH abstraction and salt metathesis with KHBPh3, respectively. CO2 is rapidly inserted into the B–H bonds. [(L)Zn(TMDS)][HBPh3] catalyzes the hydroboration of polar substrates including CO2.
Co-reporter:S. Schnitzler, P. Cui, T. P. Spaniol and J. Okuda
Dalton Transactions 2017 - vol. 46(Issue 6) pp:NaN1765-1765
Publication Date(Web):2017/01/11
DOI:10.1039/C6DT04654A
Molecular magnesium hydride with a terminal metal–hydrogen bond [Mg(iPr2TACN·AliBu3)H]2 supported by a monoanionic TACN-type ligand (TACN = 1,4-di-isopropyl-1,4,7-triazacyclononane) rearranges to form the dinuclear magnesium hydride [Mg(iPr2TACN·AlHiBu2)(μ-H)]2, exhibiting a rare Mg⋯H–Al interaction.
Co-reporter:Debabrata Mukherjee, Satoru Shirase, Thomas P. Spaniol, Kazushi Mashima and Jun Okuda
Chemical Communications 2016 - vol. 52(Issue 89) pp:NaN13158-13158
Publication Date(Web):2016/10/13
DOI:10.1039/C6CC06805G
Magnesium bis(hydridotriphenylborate), isolated as a solvent-separated ion pair [Mg(thf)6][HBPh3]2, effectively catalyzed the hydroboration of several unsaturated substrates including CO2.
Co-reporter:D. F. Sauer, S. Gotzen and J. Okuda
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 39) pp:NaN9183-9183
Publication Date(Web):2016/08/15
DOI:10.1039/C6OB01475E
The incorporation of organometallic catalyst precursors in proteins results in so-called artificial metalloenzymes. The protein structure will control activity, selectivity and stability of the organometallic site in aqueous medium and allow non-natural reactions in biological settings. Grubbs-Hoveyda type ruthenium catalysts with an N-heterocyclic carbene (NHC) as ancillary ligand, known to be active in olefin metathesis, have recently been incorporated in various proteins. An overview of these artificial metalloproteins and their potential application in olefin metathesis is given.
Co-reporter:Debabrata Mukherjee, Hassan Osseili, Khai-Nghi Truong, Thomas P. Spaniol and Jun Okuda
Chemical Communications 2017 - vol. 53(Issue 24) pp:NaN3496-3496
Publication Date(Web):2017/03/03
DOI:10.1039/C7CC01159H
Molecular aluminum hydride [(L)AlH2] (L = Me3TACD) reacted with 2 equiv. of BPh3 in THF or THP to give the cationic alkoxides [(L)Al(OR)][HBPh3] (R = nBu, nPent) by facile ring-opening of the cyclic ethers. The Cα–O bond cleavage which involves the isolable intermediate [(L)AlH][HBPh3] is a result of hydride transfer to Cα from [HBPh3]−.
Co-reporter:Debabrata Mukherjee, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2017 - vol. 46(Issue 3) pp:NaN655-655
Publication Date(Web):2016/12/02
DOI:10.1039/C6DT04379H
A macrocyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane, (Me3TACD)H formed thermally stable adducts with MMe3 (M = Al, In). The reactions with cationic dialkyls [MR2]+ directly provided cationic monoalkyls [(Me3TACD)MR]+ under RH elimination. Reactions between [Li(Me3TACD)]2 and Me2MCl gave chloro-bridged heterobimetallic adducts.
Co-reporter:Debabrata Mukherjee, Hassan Osseili, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2017 - vol. 46(Issue 25) pp:NaN8021-8021
Publication Date(Web):2017/05/31
DOI:10.1039/C7DT01671A
The macrocyclic polyamine 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane [LH = (Me3TACD)H] formed adducts with tetramethyl-silazides [M{N(SiHMe2)2}] (M = Li, Na, K) of light alkali metals. Upon heating, intramolecular dehydrocoupling occurred to give [M{(L)SiMe2N(SiHMe2)}]. BPh3 induced facile ring-opening of THF when reacted with [Li{(L)SiMe2N(SiHMe2)}].
Co-reporter:Debabrata Mukherjee, Satoru Shirase, Klaus Beckerle, Thomas P. Spaniol, Kazushi Mashima and Jun Okuda
Dalton Transactions 2017 - vol. 46(Issue 26) pp:NaN8457-8457
Publication Date(Web):2017/06/02
DOI:10.1039/C7DT01727H
Heteroleptic bis(silyl)amides of magnesium and calcium [(L)M{N(SiMe3)2}] [M = Mg, Ca; LH = 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane; (Me3TACD)H] were previously synthesized from LH and [M{N(SiMe3)2}2]. Strontium bis(silyl)amides [Sr{N(SiMe3)2}2(thf)2] and [Sr{N(SiHMe2)2}2(thf)2/3] reacted with LH to give different types of products, depending on the presence of the β-SiH function. While the former underwent protonolysis to give the amido-bridged dimer [(L)Sr{N(SiMe3)2}]2 (1), the latter gave the adduct [(LH)Sr{N(SiHMe2)2}2] (2) as a stable solid. 2 slowly underwent an intramolecular Si–H/H–N dehydrocoupling in solution to give [{(L)SiMe2N(SiHMe2)}Sr{N(SiHMe2)2}] (3) by liberating H2. The results of transamination of 1 with HN(SiHMe2)2 depended on the relative stoichiometric ratio. A 1:1 mixture in n-pentane gave [{(L)SiMe2N(SiHMe2)}Sr{N(SiMe3)2}] (4) and H2, while excess HN(SiHMe2)2 gave the adduct 2 under similar conditions. Compounds 2 and 3 exhibit Sr↼H–Si interactions according to X-ray crystallography, NMR, and IR spectroscopy. Lighter congeners of elusive [(L)Sr{N(SiHMe2)2}] were isolable for Mg (5) and Ca (6).
Co-reporter:Peng Cui, Thomas P. Spaniol, Laurent Maron and Jun Okuda
Chemical Communications 2014 - vol. 50(Issue 4) pp:NaN426-426
Publication Date(Web):2013/11/01
DOI:10.1039/C3CC47805J
A scandium hydride supported by a (NNNN)-type macrocycle consists of a trinuclear cation and a trinuclear anion. The anion shows significantly higher reactivity.
Co-reporter:Ajay Venugopal, Floriana Tuna, Thomas P. Spaniol, Liviu Ungur, Liviu F. Chibotaru, Jun Okuda and Richard A. Layfield
Chemical Communications 2013 - vol. 49(Issue 9) pp:NaN903-903
Publication Date(Web):2012/12/11
DOI:10.1039/C2CC38036F
An experimental and ab initio computational study of an unsymmetrical, hydride-bridged di-dysprosium single-molecule magnet is reported.
Co-reporter:Ilja Peckermann, Gerhard Raabe, Thomas P. Spaniol and Jun Okuda
Chemical Communications 2011 - vol. 47(Issue 17) pp:NaN5063-5063
Publication Date(Web):2011/03/24
DOI:10.1039/C1CC00039J
Allyl and 2-methylallyl indium compounds were prepared by salt metathesis starting from indium trichloride and a Grignard reagent. They are highly fluxional in solution and reveal coordination numbers of the indium atoms of four and five in the solid state.
Co-reporter:Tsuneomi Kawasaki, Christiane Hohberger, Yuko Araki, Kunihiko Hatase, Klaus Beckerle, Jun Okuda and Kenso Soai
Chemical Communications 2009(Issue 37) pp:NaN5623-5623
Publication Date(Web):2009/08/21
DOI:10.1039/B912813A
Chiral isotactic polystyrenes induce the enantioselective addition of diisopropylzinc to pyrimidine-5-carbaldehyde, affording the enantiomerically enriched pyrimidyl alkanol with the corresponding absolute configuration to that of cryptochiral polystyrenes in conjunction with asymmetric autocatalysis.
Co-reporter:Bing Lian, Haiyan Ma, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2009(Issue 41) pp:NaN9042-9042
Publication Date(Web):2009/08/25
DOI:10.1039/B909287K
Reaction of trimethylaluminium with the linked bis(phenol) trans-1,4-dithiocyclohexanediyl-2,2′-bis(4,6-di-tert-butylphenol) (cydtbpH2, 1a) led to the chiral methyl aluminium complex [Al(cydtbp)Me] (2a) as both racemate and resolved enantiomers, which were characterized by multinuclear NMR spectroscopy and elemental analysis. Rac-{trans-1,2-dithiocyclohexanediyl-2,2′-bis(6-tert-butyl-4-methylphenol)} (cytbmpH2, rac-1b) similarly gave rac-2b. Single-crystal X-ray diffraction of (S,S)-2a and rac-2b showed a strongly distorted trigonal bipyramidal geometry with significantly differing Al–S distances. Reaction of rac-2b with (−)-borneol gave the alkoxy complex 5 as a mixture of two diastereomers. Methyl abstraction from the neutral metal complex rac-2a using B(C6F5)3 gave the cationic complex [rac-Al(cydtbp)(THF)2]+[MeB(C6F5)3]− (rac-6·(THF)2) that according to X-ray crystallography adopts an octahedral coordination geometry. Upon reacting trimethylaluminium with a bis(phenol) with a longer link, 3,4-trans-butanediyl-1,6-dithiahexanediyl)-2,2′-bis(6-tert-butyl-4-methylphenol), (cmtbmpH2, 3, a dinuclear compound 4 was obtained that contains two square pyramidal aluminium centers with two “fly-over” bis(phenolate) ligands. The neutral methyl aluminium complexes are active in the polymerization of methyl methacrylate (MMA) and in the ring-polymerization of rac-lactide (LA), producing PMMAs and PLAs in a controlled fashion. The cationic aluminium complexes were active in the cationic polymerization of isoprene and benzofuran.
Co-reporter:Dominique Robert, Małgorzata Kondracka and Jun Okuda
Dalton Transactions 2008(Issue 20) pp:NaN2669-2669
Publication Date(Web):2008/03/27
DOI:10.1039/B801030G
Rare-earth metal tris(borohydrides) [Ln(BH4)3(thf)3] (Ln = Y, La, Nd, Sm) are converted with one equivalent of the Brønsted acid [NEt3H]+[BPh4]− in thf into the monocationic bis(borohydride) complexes [Ln(BH4)2(thf)5]+[BPh4]−. They efficiently initiate the ring-opening polymerisation of ε-caprolactone.
Co-reporter:Julien P. Davin, Jean-Charles Buffet, Thomas P. Spaniol and Jun Okuda
Dalton Transactions 2012 - vol. 41(Issue 40) pp:NaN12618-12618
Publication Date(Web):2012/08/21
DOI:10.1039/C2DT31309J
A chiral, tetradentate polyether ligand with a trans-1,2-cyclohexanediyl backbone, bis(methoxyethoxy)-trans-1,2-cyclohexane (5), was synthesized as both a racemate and the (S,S) enantiomer. 5 was found to form stable adducts with alkaline earth metal amides [M{N(SiMe3)2}2(thf)x] (M = Mg (x = 0), Ca (x = 2) and Sr (x = 2/3)), [Ca{N(SiHMe2)2}2(thf)] as well as with hydrocarbyl compounds [Mg(CH2SiMe3)2] and [Ca(η3-C3H5)2]. X-ray diffraction study of the bis(amide) [((S,S)-5)Ca{N(SiMe3)2}2] and of the bis(allyl) [(rac-5)Ca(η3-C3H5)2] was performed. The complexes obtained were tested as initiators for the ring-opening polymerization of meso-, racemic and L-lactide.






![Silane, trimethyl[(phenylsilyl)methyl]-](http://img.cochemist.com/ccimg/882000/881995-18-2.png)
![Silane, trimethyl[(phenylsilyl)methyl]-](http://img.cochemist.com/ccimg/882000/881995-18-2_b.png)
![Phenol, 2,2'-[1,3-propanediylbis(thio)]bis[4,6-bis(2-methylpropyl)-](http://img.cochemist.com/ccimg/827400/827308-18-9.png)
![Phenol, 2,2'-[1,3-propanediylbis(thio)]bis[4,6-bis(2-methylpropyl)-](http://img.cochemist.com/ccimg/827400/827308-18-9_b.png)
![Phenol, 2,2'-[1,2-ethanediylbis(thio)]bis[4,6-bis(2-methylpropyl)-](http://img.cochemist.com/ccimg/827400/827308-17-8.png)
![Phenol, 2,2'-[1,2-ethanediylbis(thio)]bis[4,6-bis(2-methylpropyl)-](http://img.cochemist.com/ccimg/827400/827308-17-8_b.png)




![2-Azaspiro[4.5]decane, 3-methyl-](http://img.cochemist.com/ccimg/592500/592478-37-0.png)
![2-Azaspiro[4.5]decane, 3-methyl-](http://img.cochemist.com/ccimg/592500/592478-37-0_b.png)









![Potassium,[1,2,3,4-tetramethyl-5-(trimethylsilyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/385900/385828-50-2.png)
![Potassium,[1,2,3,4-tetramethyl-5-(trimethylsilyl)-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/385900/385828-50-2_b.png)
![3,6,9-Triazabicyclo[9.3.1]pentadeca-1(15),11,13-triene](http://img.cochemist.com/ccimg/374900/374806-99-2.png)
![3,6,9-Triazabicyclo[9.3.1]pentadeca-1(15),11,13-triene](http://img.cochemist.com/ccimg/374900/374806-99-2_b.png)




![1,4,7,10-Tetraazacyclododecane, 1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/289800/289719-40-0.png)
![1,4,7,10-Tetraazacyclododecane, 1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/289800/289719-40-0_b.png)
















































![Ethanone, 1-[4-[(dimethylamino)methyl]phenyl]-](http://img.cochemist.com/ccimg/91300/91245-82-8.png)
![Ethanone, 1-[4-[(dimethylamino)methyl]phenyl]-](http://img.cochemist.com/ccimg/91300/91245-82-8_b.png)


![[1-(HYDROXYMETHYL)CYCLOPENT-3-EN-1-YL]METHANOL](http://img.cochemist.com/ccimg/77000/76910-02-6.png)
![[1-(HYDROXYMETHYL)CYCLOPENT-3-EN-1-YL]METHANOL](http://img.cochemist.com/ccimg/77000/76910-02-6_b.png)


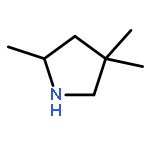
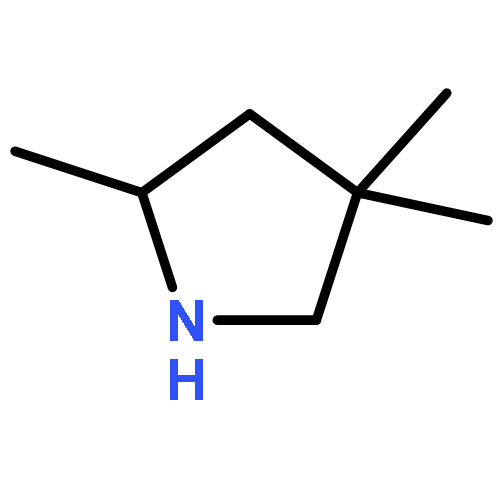
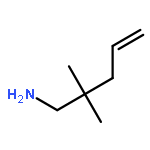
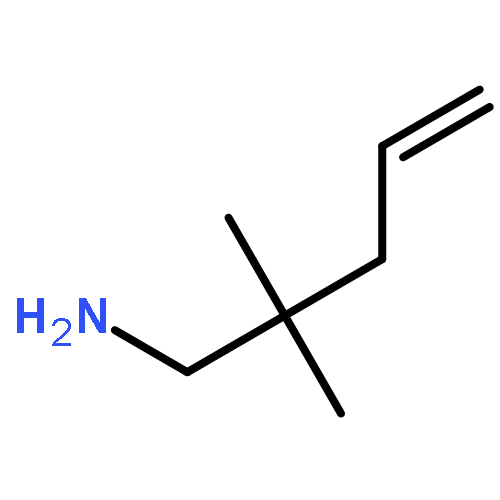




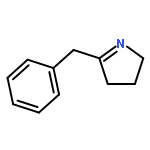
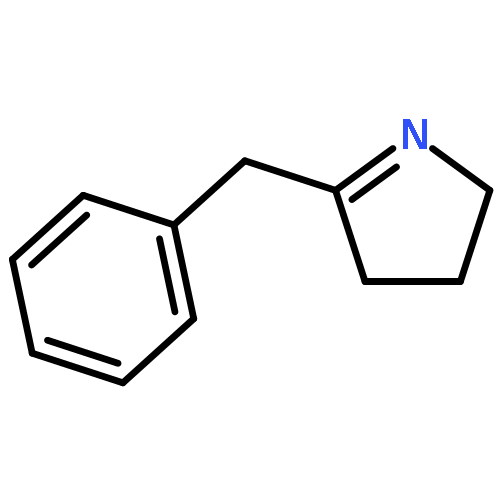






![Silane, [2-(3-cyclohexen-1-yl)ethyl]phenyl-](http://img.cochemist.com/ccimg/60200/60171-22-4.png)
![Silane, [2-(3-cyclohexen-1-yl)ethyl]phenyl-](http://img.cochemist.com/ccimg/60200/60171-22-4_b.png)


![Magnesium, bis[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/51400/51329-17-0.png)
![Magnesium, bis[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/51400/51329-17-0_b.png)
![Propanedinitrile, 2-[2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2138200/51325-91-8.png)
![Propanedinitrile, 2-[2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2138200/51325-91-8_b.png)


![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9.png)
![Lanthanum Tris[bis(trimethylsilyl)amide]](http://img.cochemist.com/ccimg/35800/35788-99-9_b.png)

















![Benzoic acid, 4-[(dimethylamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/18200/18153-53-2.png)
![Benzoic acid, 4-[(dimethylamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/18200/18153-53-2_b.png)



























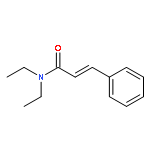

















![Dimethyl (1r,2s,3r,4s)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate](http://img.cochemist.com/ccimg/2000/1984-43-6.png)
![Dimethyl (1r,2s,3r,4s)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate](http://img.cochemist.com/ccimg/2000/1984-43-6_b.png)









![7-Azabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/300/286-18-0.png)
![7-Azabicyclo[4.1.0]heptane](http://img.cochemist.com/ccimg/300/286-18-0_b.png)


![1,3-DIOXANE-5-METHANOL, 5,5'-[OXYBIS(METHYLENE)]BIS[2,2-DIMETHYL-](http://img.cochemist.com/ccimg/881400/881399-31-1.png)
![1,3-DIOXANE-5-METHANOL, 5,5'-[OXYBIS(METHYLENE)]BIS[2,2-DIMETHYL-](http://img.cochemist.com/ccimg/881400/881399-31-1_b.png)
![PHENOL, 2,2'-[1,3-PROPANEDIYLBIS(THIO)]BIS[6-(1,1-DIMETHYLETHYL)-4-METHYL-](http://img.cochemist.com/ccimg/664000/663910-60-9.png)
![PHENOL, 2,2'-[1,3-PROPANEDIYLBIS(THIO)]BIS[6-(1,1-DIMETHYLETHYL)-4-METHYL-](http://img.cochemist.com/ccimg/664000/663910-60-9_b.png)











![Poly[[tetrahydro-3,4-bis(methoxymethyl)-2,5-furandiyl]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/117000/116910-52-2.png)
![Poly[[tetrahydro-3,4-bis(methoxymethyl)-2,5-furandiyl]-1,2-ethenediyl]](http://img.cochemist.com/ccimg/117000/116910-52-2_b.png)
![2-[2-(2-HYDROXYPHENYL)SULFANYLETHYLSULFANYL]PHENOL](http://img.cochemist.com/ccimg/107800/107777-29-7.png)
![2-[2-(2-HYDROXYPHENYL)SULFANYLETHYLSULFANYL]PHENOL](http://img.cochemist.com/ccimg/107800/107777-29-7_b.png)








![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)














![Phenol, 2,2'-[1,3-propanediylbis(thio)]bis[4,6-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/664000/663910-61-0.png)
![Phenol, 2,2'-[1,3-propanediylbis(thio)]bis[4,6-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/664000/663910-61-0_b.png)
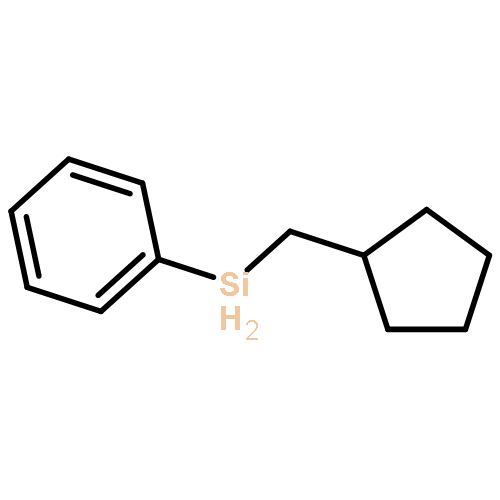
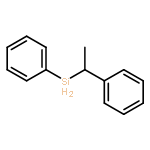
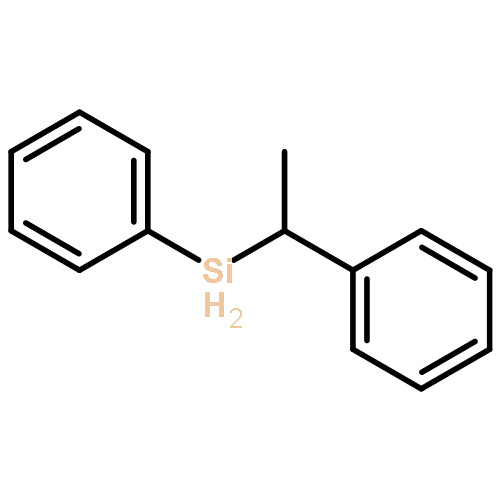






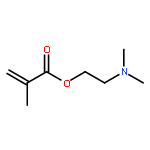
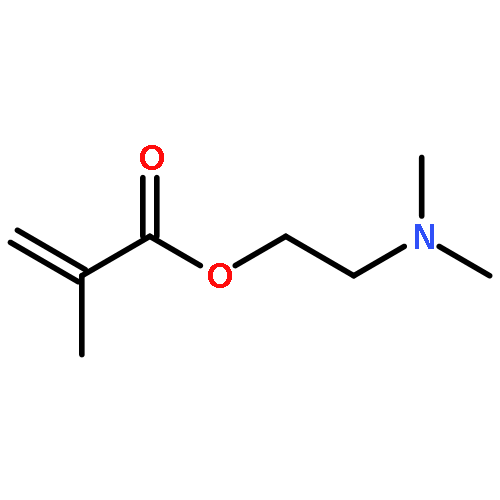







![Indium, tris[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/69900/69833-15-4.png)
![Indium, tris[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/69900/69833-15-4_b.png)
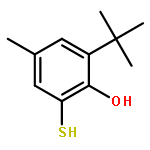
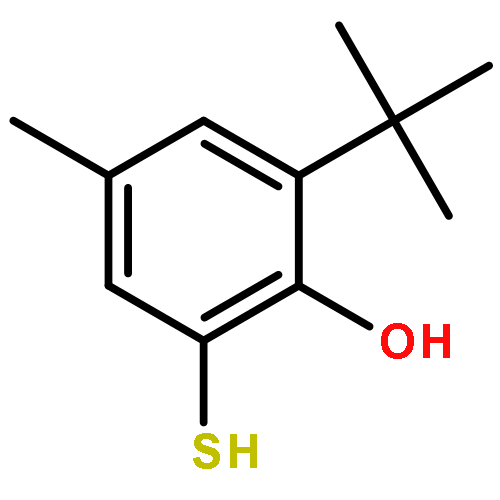





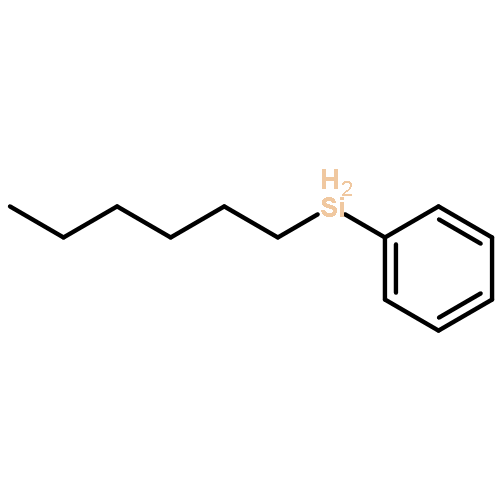




![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2.png)
![Poly[oxy[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/26200/26161-42-2_b.png)
![Phenol, 2,2'-[1,2-ethanediylbis(thio)]bis[4,6-bis(1-methyl-1-phenylethyl)-](http://img.cochemist.com/ccimg/856100/856014-29-4.png)
![Phenol, 2,2'-[1,2-ethanediylbis(thio)]bis[4,6-bis(1-methyl-1-phenylethyl)-](http://img.cochemist.com/ccimg/856100/856014-29-4_b.png)
![Phenol, 2,2'-[1,2-ethanediylbis(thio)]bis[6-(1,1-dimethylethyl)-4-methyl-](http://img.cochemist.com/ccimg/664000/663910-59-6.png)
![Phenol, 2,2'-[1,2-ethanediylbis(thio)]bis[6-(1,1-dimethylethyl)-4-methyl-](http://img.cochemist.com/ccimg/664000/663910-59-6_b.png)


![PHENOL, 2,2'-[1,2-ETHANEDIYLBIS(THIO)]BIS[4,6-BIS(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/443300/443287-30-7.png)
![PHENOL, 2,2'-[1,2-ETHANEDIYLBIS(THIO)]BIS[4,6-BIS(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/443300/443287-30-7_b.png)