Co-reporter:Xinlei Zhang, Jingjing Lin, Shuangming Chen, Jia Yang, Li Song, Xiaojun Wu, and Hangxun Xu
ACS Applied Materials & Interfaces November 8, 2017 Volume 9(Issue 44) pp:38499-38499
Publication Date(Web):October 17, 2017
DOI:10.1021/acsami.7b11120
It is known that introducing metal nanoparticles (e.g., Fe and Co) into N-doped carbons can enhance the activity of N-doped carbons toward the oxygen reduction reaction (ORR). However, introducing metals into N-doped carbons inevitably causes the formation of multiple active sites. Thus, it is challenging to identify the active sites and unravel mechanisms responsible for enhanced ORR activity. Herein, by developing a new N-heterocyclic carbene (NHC)–Co complex as the nitrogen- and metal-containing precursor, we report the synthesis of N-doped carbon nanosheets embedded with Co nanoparticles as highly active ORR catalysts without direct metal–nitrogen bonding. Electrochemical measurements and X-ray absorption spectroscopy indicate that the carbon–nitrogen sites surrounding Co nanoparticles are responsible for the observed ORR activity and stability. Density functional theory calculations further reveal that Co nanoparticles could facilitate the protonation of O2 and thus promote the ORR activity. These results provide new prospects in the rational design and synthesis of heteroatom-doped carbon materials as non-precious-metal catalysts for various electrochemical reactions.Keywords: carbon nanosheets; cobalt nanoparticles; electrocatalysis; N-doping; oxygen reduction reaction;
Co-reporter:Xiuling Li, Haifeng Lv, Jun Dai, Liang Ma, Xiao Cheng Zeng, Xiaojun Wu, and Jinlong Yang
Journal of the American Chemical Society May 10, 2017 Volume 139(Issue 18) pp:6290-6290
Publication Date(Web):April 28, 2017
DOI:10.1021/jacs.7b01369
The development of one-dimensional (1D) molecular nanowires with high spin-polarization is important for both fundamental research and practical applications in nanoscale spintronics. Herein, we report new 1D metal trihydride molecular nanowires MH3 (M = Sc, Cr, Mn, and Co) with versatile magnetic properties on the basis of first-principles calculations and molecular assembly of their molecular motifs. Among the 1D nanowires considered, CrH3, MnH3, and CoH3 are either antiferromagnetic or ferromagnetic in their ground states. In particular, CoH3 nanowire is a half-metal, which ideally could provide 100% spin-polarized currents. Moreover, carrier doping in MnH3 nanowire can induce a transition from ferromagnetic metal to half-metal. Strong metal–metal interaction in 1D MH3 nanowires is responsible to versatile magnetic properties and high Curie temperature. Born–Oppenheimer molecular dynamics simulation indicates that these nanowires are stable at elevated temperature. In particular, the ScH3 nanowire appears to have the highest structural stability at temperature 1200 K. These novel properties of 1D MH3 nanowires render their potential applications in nanoscale spintronics.
Co-reporter:Yingjie Sun, Zhiwen Zhuo, Xiaojun Wu, and Jinlong Yang
Nano Letters May 10, 2017 Volume 17(Issue 5) pp:2771-2771
Publication Date(Web):April 25, 2017
DOI:10.1021/acs.nanolett.6b04884
Searching experimental feasible two-dimensional (2D) ferromagnetic crystals with large spin-polarization ratio, high Curie temperature and large magnetic anisotropic energy is one key to develop next-generation spintronic nanodevices. Here, 2D Fe2Si nanosheet, one counterpart of Hapkeite mineral discovered in meteorite with novel magnetism is proposed on the basis of first-principles calculations. The 2D Fe2Si crystal has a slightly buckled triangular lattice with planar hexacoordinated Si and Fe atoms. The spin-polarized calculations with hybrid HSE06 function method indicate that 2D Fe2Si is a ferromagnetic half metal at its ground state with 100% spin-polarization ratio at Fermi energy level. The phonon spectrum calculation and ab initio molecular dynamic simulation shows that 2D Fe2Si crystal has a high thermodynamic stability and its 2D lattice can be retained at the temperature up to 1200 K. Monte Carlo simulations based on the Ising model predict a Curie temperature over 780 K in 2D Fe2Si crystal, which can be further tuned by applying a biaxial strain. Moreover, 2D structure and strong in-plane Fe–Fe interaction endow Fe2Si nanosheet sizable magnetocrystalline anisotropy energy with the magnitude of at least two orders larger than those of Fe, Co and Ni bulks. These novel magnetic properties render the 2D Fe2Si crystal a very promising material for developing practical spintronic nanodevicesKeywords: ferromagnetism; First-principles calculations; two-dimensional Fe2Si;
Co-reporter:Keke Mao;Jinlong Yang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 6) pp:4435-4439
Publication Date(Web):2017/02/08
DOI:10.1039/C6CP07402B
Manipulation of the chemical reactivity of two-dimensional materials is a challenge for advancing various nanotechnologies, ranging from electronics to catalysis. In this study, on the basis of first-principles calculations, we demonstrated that the chemical reactivity of h-BN sheets towards O2 can be significantly enhanced via a metal substrate-mediated charge effect. The chemisorption of O2 molecule on the h-BN sheet deposited on Ni, Co, or Cu substrate were almost spontaneous with negligible energy barrier, distinctly different from that on the freestanding h-BN sheet, which has ultra-high chemical stability. In particular, the enhanced oxidation of h-BN sheet can be confined in the nanoscale region due to the localized electronic states in the h-BN sheet. These findings imply a pathway to selectively oxidize h-BN sheet by patterning the metal substrate.
Co-reporter:Ke Zhang;Rashid Khan;Hongyan Guo;Irfan Ali;Xiuling Li;Yunxiang Lin;Haiping Chen;Wensheng Yan;Li Song
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 3) pp:1735-1739
Publication Date(Web):2017/01/18
DOI:10.1039/C6CP07270D
A room-temperature ferromagnetic behavior was observed in a ternary layered-Cu2MoS4 nanosheet. Both the coercivity and magnetization saturation increased with a decrease in temperature. The electron paramagnetic resonance spectroscopy confirmed a high g value. Combined with atomic structural observations, our first principle calculations revealed that the ferromagnetism originated from the edged molybdenum atoms.
Co-reporter:Xiuling Li;Xiao Cheng Zeng
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 16) pp:10401-10405
Publication Date(Web):2017/04/19
DOI:10.1039/C7CP01020F
Developing freestanding silicene nanostructures with tunable electronic and magnetic properties is of particular importance for their applications in nanoelectronics, but still faces big challenges. On the basis of first-principles calculations, here we predict that embedded silicene nanoflakes and nanoribbons can be realized by partly dehydrogenating a freestanding polysilane (Si6H6) sheet. Born–Oppenheimer molecular dynamics simulations indicate that the embedded silicene nanostructures show good thermal stability at 500 K. In particular, the embedded silicene nanostructures exhibit similar electronics properties to those of isolated ones. These findings imply a practical solution to produce embedded silicene nanostructures from partly dehydrogenated freestanding polysilane.
Co-reporter:Lei Wang;Yangyang Wan;Yanjun Ding;Yuchen Niu;Yujie Xiong;Hangxun Xu
Nanoscale (2009-Present) 2017 vol. 9(Issue 12) pp:4090-4096
Publication Date(Web):2017/03/23
DOI:10.1039/C7NR00534B
Nanostructured semiconducting polymers have emerged as a very promising class of metal-free photocatalytic materials for solar water splitting. However, they generally exhibit low efficiency and lack the ability to utilize long-wavelength photons in a photocatalytic oxygen evolution reaction (OER). Here, based on first-principles calculations, we reveal that the two-dimensional (2D) aza-fused conjugated microporous polymer (aza-CMP) with a honeycomb network is a semiconductor with novel layer-dependent electronic properties. The bandgap of the as-synthesized aza-CMP nanosheets is measured to be 1.22 eV, suggesting that they can effectively boost light absorption in the visible and near infrared (NIR) region. More importantly, aza-CMP also possesses a valence band margin suitable for a photocatalytic OER. Taking advantage of the 2D layered nanostructure, we further show that the exfoliated ultrathin aza-CMP nanosheets can exhibit a three-fold enhancement in the photocatalytic OER. After deposition of a Co(OH)2 cocatalyst, the hybrid Co(OH)2/aza-CMP photocatalyst exhibits a markedly improved performance for photocatalytic O2 evolution. Furthermore, first-principles calculations reveal that the photocatalytic O2 evolution reaction is energetically feasible for aza-CMP nanosheets under visible light irradiation. Our findings reveal that nanostructured polymers hold great potential for photocatalytic applications with efficient solar energy utilization.
Co-reporter:Zhi Jiang;Zhongti Sun;Yuqi Yang;Siyu Chen;Wenfeng Shangguan
Nanoscale (2009-Present) 2017 vol. 9(Issue 37) pp:14272-14279
Publication Date(Web):2017/09/28
DOI:10.1039/C7NR02913F
First-principles calculations and experiments with PtOx on TiO2 surfaces were performed together to understand the interactions of metal oxides during the calcination process and their influence on the growth pattern of Pt on the TiO2 surface. Our calculations indicate that PtOx with a high concentration of oxygen binds more strongly to rutile than to anatase, indicating the formation of stronger interactions between Pt oxide and the rutile surface compared with anatase during the calcination stage under an oxidative atmosphere. X-ray photoelectron spectra quantification analysis illustrates that higher amounts of Pt oxide are obtained when impregnation is performed with a rutile support after calcination in comparison with that of anatase. After reduction, the stronger interaction between PtOx and rutile leads to a larger amount of partially charged and highly dispersed Pt nanoclusters on the surface, while the weaker interaction between PtOx and anatase illustrates the dispersion and sintering of higher amounts of metal nanoparticles on the anatase surface. Furthermore, the photocatalytic oxygen evolution test highlights the importance of understanding the interaction between the metal oxide and anatase/rutile for targeted synthesis of the supported catalyst.
Co-reporter:Haiping Chen;Zhongti Sun;Chengming Wang;Xiuling Li;Xusheng Zheng;Youkui Zhang;Qun He;Li Song
Journal of Materials Chemistry C 2017 vol. 5(Issue 17) pp:4185-4189
Publication Date(Web):2017/05/04
DOI:10.1039/C7TC00336F
Two-dimensional layered structure of a single crystal is regarded as an ideal feature for physical and chemical fundamental studies. Herein, we demonstrated a high-quality (NH4)2V3O8 single crystal with a layered tetragonal structure prepared via a hydrothermal method. The lattice vibrational behavior and surface electronic state of (NH4)2V3O8 layers were systematically investigated via polarized Raman scattering spectroscopy and ultraviolet photoelectron spectroscopy (UPS), respectively. It was found that all Raman peaks of (NH4)2V3O8 could be clearly identified as four active Raman modes through parallel and perpendicular polarization configurations in the backscattering geometry for (001) crystal surface. The UPS results indicated that the valence band maximum of (NH4)2V3O8 was mainly composed of localized vanadium 3d states, which was further confirmed by the density functional theory calculations.
Co-reporter:Ruijie Huang;Zhongti Sun;Sheng Chen;Siyu Wu;Zeqi Shen;Jie Zeng
Chemical Communications 2017 vol. 53(Issue 51) pp:6922-6925
Publication Date(Web):2017/06/22
DOI:10.1039/C7CC03643D
Hierarchical metal nanostructures which exhibit an open structure and a high density of twin defects accessible to reactants hold great promise in catalysis. Here, we report a facile synthesis of Pt–Cu hierarchical quasi great dodecahedrons (HQGDs) which present 5-fold symmetry and are composed of multiple ordered branched units with a frame structure. HQGDs evolve from icosahedral seeds with multiple {111} twin planes, followed by the growth of higher-order branches. Owing to the unique frame structure associated with multiple twin defects, HQGDs showed much higher HER catalytic activity and better durability relative to commercial Pt/C.
Co-reporter:Ning Lu;Hongyan Guo;Wei Hu;Xiao Cheng Zeng
Journal of Materials Chemistry C 2017 vol. 5(Issue 12) pp:3121-3129
Publication Date(Web):2017/03/23
DOI:10.1039/C7TC00367F
We perform a comprehensive study of the effects of different types of line defects on the electronic and magnetic properties of ZnO nanoribbons and monolayer sheets by using first-principles computations. Our computations show that for zigzag ZnO nanoribbons, their metallic characteristics are unchanged by the line defects, although certain nanoribbons can exhibit much higher magnetic moments contributed by atoms within the line defects. For the armchair nanoribbons containing the 4–8 line defects, their semiconducting characteristics are the same as those of the defect-free nanoribbons. Besides the line defects, two large-angle grain boundaries are also considered for zigzag and armchair nanoribbons. In both cases, the ZnO nanoribbons show metallic characteristics. Finally, the effects of line defects on 2D ZnO monolayer sheets are also studied. It is found that the line defects in ZnO sheets can markedly enhance visible-light absorption.
Co-reporter:Laifa Shen;Haifeng Lv;Shuangqiang Chen;Peter Kopold;Peter A. van Aken;Joachim Maier;Yan Yu
Advanced Materials 2017 Volume 29(Issue 27) pp:
Publication Date(Web):2017/07/01
DOI:10.1002/adma.201700142
Lithium ion capacitors are new energy storage devices combining the complementary features of both electric double-layer capacitors and lithium ion batteries. A key limitation to this technology is the kinetic imbalance between the Faradaic insertion electrode and capacitive electrode. Here, we demonstrate that the Li3VO4 with low Li-ion insertion voltage and fast kinetics can be favorably used for lithium ion capacitors. N-doped carbon-encapsulated Li3VO4 nanowires are synthesized through a morphology-inheritance route, displaying a low insertion voltage between 0.2 and 1.0 V, a high reversible capacity of ≈400 mAh g−1 at 0.1 A g−1, excellent rate capability, and long-term cycling stability. Benefiting from the small nanoparticles, low energy diffusion barrier and highly localized charge-transfer, the Li3VO4/N-doped carbon nanowires exhibit a high-rate pseudocapacitive behavior. A lithium ion capacitor device based on these Li3VO4/N-doped carbon nanowires delivers a high energy density of 136.4 Wh kg−1 at a power density of 532 W kg−1, revealing the potential for application in high-performance and long life energy storage devices.
Co-reporter:Yuqiao Guo;Haitao Deng;Xu Sun;Xiuling Li;Jiyin Zhao;Junchi Wu;Wangsheng Chu;Sijia Zhang;Haibin Pan;Xusheng Zheng;Changqing Jin;Changzheng Wu;Yi Xie
Advanced Materials 2017 Volume 29(Issue 29) pp:
Publication Date(Web):2017/08/01
DOI:10.1002/adma.201700715
2D transition-metal dichalcogenides (TMDCs) are currently the key to the development of nanoelectronics. However, TMDCs are predominantly nonmagnetic, greatly hindering the advancement of their spintronic applications. Here, an experimental realization of intrinsic magnetic ordering in a pristine TMDC lattice is reported, bringing a new class of ferromagnetic semiconductors among TMDCs. Through van der Waals (vdW) interaction engineering of 2D vanadium disulfide (VS2), dual regulation of spin properties and bandgap brings about intrinsic ferromagnetism along with a small bandgap, unravelling the decisive role of vdW gaps in determining the electronic states in 2D VS2. An overall control of the electronic states of VS2 is also demonstrated: bond-enlarging triggering a metal-to-semiconductor electronic transition and bond-compression inducing metallization in 2D VS2. The pristine VS2 lattice thus provides a new platform for precise manipulation of both charge and spin degrees of freedom in 2D TMDCs availing spintronic applications.
Co-reporter:Zhiwen Zhuo; Xiaojun Wu;Jinlong Yang
Journal of the American Chemical Society 2016 Volume 138(Issue 22) pp:7091-7098
Publication Date(Web):May 12, 2016
DOI:10.1021/jacs.6b02964
Exploring stable two-dimensional (2D) crystalline structures of phosphorus with tunable properties is of considerable importance partly due to the novel anisotropic behavior in phosphorene and potential applications in high-performance devices. Here, 21 new 2D phosphorus allotropes with porous structure are reported based on topological modeling method and first-principles calculations. We establish that stable 2D phosphorus crystals can be obtained by topologically assembling selected phosphorus monomer, dimer, trimer, tetramer, and hexamer. Nine of reported structures are predicted to be more stable than white phosphorus. Their dynamic and thermal stabilities are confirmed by the calculated vibration spectra and Born–Oppenheimer molecular dynamic simulation at temperatures up to 1500 K. These phosphorus porous polymorphs have isotropic mechanic properties that are significantly softer than phosphorene. The electronic band structures calculated with the HSE06 method indicate that new structures are semiconductors with band gaps ranging widely from 0.15 to 3.42 eV, which are tuned by the basic units assembled in the network. Of particular importance is that the position of both conduction and valence band edges of some allotropes matches well with the chemical reaction potential of H2/H+ and O2/H2O, which can be used as element photocatalysts for visible-light-driven water splitting.
Co-reporter:Zhiwen Zhuo, Xiaojun Wu, and Jinlong Yang
The Journal of Physical Chemistry C 2016 Volume 120(Issue 46) pp:26453-26458
Publication Date(Web):November 1, 2016
DOI:10.1021/acs.jpcc.6b10873
The discovery of new phosphorus allotropes has attracted continuous attention over recent decades, partly due to the importance of phosphorus in life and their fantastic structural diversity. Generally, phosphorus allotropes consist of covalently linked substructures, stacked together with van der Waals interactions, and a few phosphorus allotropes possess three-dimensional covalently linked structures only at high pressure. On the basis of first-principles calculations, five new phosphorus allotropes with three-dimensional covalently linked structures are predicted by assembling phosphorus units at ambient pressure, which are energetically more favorable than white phosphorus. Particularly, three of them share the same structures as those of previously reported three-dimensional nitrogen allotropes. These new allotropes are semiconductors with band gaps ranging from 0.52 to 2.39 eV, and the Young’s modulus varies from 39 to 72 GPa. The structural stability of the new phosphorus allotropes are confirmed with a phonon spectrum and Born–Oppenheimer molecular dynamic simulation at temperatures up to 700 K. Our findings enrich the phosphorus allotrope family with three-dimensional covalently linked structures at ambient pressure and versatile electronic properties.
Co-reporter:Qin Liu;Xiuling Li;Zhangru Xiao;Yu Zhou;Haipin Chen;Adnan Khalil;Ting Xiang;Junqing Xu;Wangsheng Chu;Jinlong Yang;Chengming Wang;Yujie Xiong;Chuanhong Jin;Pulickel M. Ajayan;Li Song
Advanced Materials 2015 Volume 27( Issue 33) pp:4837-4844
Publication Date(Web):
DOI:10.1002/adma.201502134
Co-reporter:Qiudi Yue, Yangyang Wan, Zijun Sun, Xiaojun Wu, Yupeng Yuan and Pingwu Du
Journal of Materials Chemistry A 2015 vol. 3(Issue 33) pp:16941-16947
Publication Date(Web):21 Jul 2015
DOI:10.1039/C5TA03949E
The generation of hydrogen (H2) through photocatalytic water splitting by employing various cocatalysts has attracted much attention. Herein we report for the first time that metallic molybdenum phosphide (MoP), as a highly active cocatalyst, can significantly enhance photocatalytic H2 production from water. A series of MoP/CdS nanorod (NR) hybrids were facilely prepared. The optimal amount of MoP led to a maximal H2 evolution rate of 163.2 μmol h−1 mg−1 under visible light illumination (λ > 420 nm), which is more than 20 times higher than that of freshly prepared CdS NRs. This work demonstrated that the suitable Fermi level alignment of MoP and CdS is responsible for the high photocatalytic activity of H2 production under visible light in the present system, as evidenced by both experimental and theoretical results.
Co-reporter:Hongyan Guo
The Journal of Physical Chemistry C 2015 Volume 119(Issue 12) pp:6912-6917
Publication Date(Web):March 9, 2015
DOI:10.1021/acs.jpcc.5b00681
Co-reporter:Wenqiang Yang, Zhenbin Wang, Zhiquan Wang, Zhenghui Yang, Changrong Xia, Ranran Peng, Xiaojun Wu, and Yalin Lu
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 23) pp:21051
Publication Date(Web):November 7, 2014
DOI:10.1021/am505900g
Using the first-principles calculation and the electronic conductivity relaxation (ECR) experimental technique, we investigated the adsorption and dissociation behaviors of O2 on Pt-modified La0.625Sr0.375Co0.25Fe0.75O3−δ (LSCF) surface. Toward the O2 reduction, the calculation results show that the perfect LSCF (100) surface is catalytically less active than both the defective (100) surface and the perfect (110) surface. O2 molecule can weakly adsorb on the perfect LSCF (100) surface with a small adsorption energy of about −0.30 eV, but the dissociation energy barrier of the O2 molecule is about 1.33–1.43 eV. Doping of Pt cluster on the LSCF (100) surface can remarkably enhance its catalytic activity. The adsorption energies of O2 molecules become −1.16 and −1.89 eV for the interfacial Feint site and the Ptbri bridge site of Pt4-cluster, respectively. Meanwhile, the dissociation energy barriers are reduced to 0.37 and 0.53 eV, respectively. The migration energy barrier of the dissociated oxygen from the interfacial Pt to the LSCF surface is 0.66 eV, and it is 2.58 eV from the top site of the Pt cluster to the interfacial Pt site, suggesting that it is extremely difficult for oxygen to migrate over the Pt cluster. The Bader charge analysis results further indicate that the charges transferring from Pt cluster to LSCF surface promote the adsorption and dissociation of O2 molecules. Experimentally, a dramatic decrease of the surface oxygen exchange relaxation time was observed on Pt-modified LSCF cathode, with a chemical surface exchange coefficient increased from 6.05 × 10–5 cm/s of the bare LSCF cathode to 4.04 × 10–4 cm/s of the Pt-modified LSCF cathode, agreeing very well with our theoretical predictions.Keywords: density functional theory; LSCF; oxygen reduction; Pt modification; solid oxide fuel cells
Co-reporter:Hongyan Guo ; Ning Lu ; Lu Wang ; Xiaojun Wu ;Xiao Cheng Zeng
The Journal of Physical Chemistry C 2014 Volume 118(Issue 13) pp:7242-7249
Publication Date(Web):February 25, 2014
DOI:10.1021/jp501734s
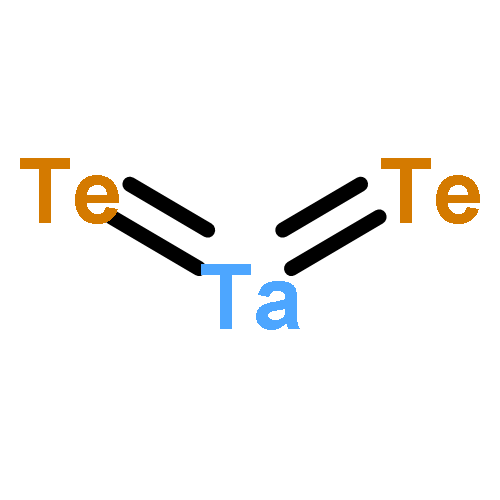
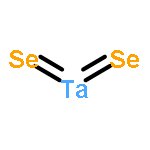
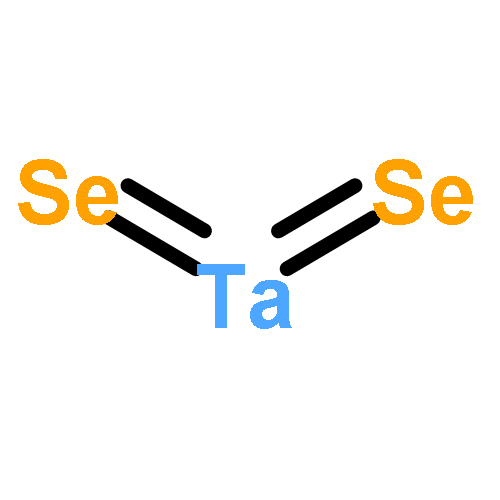
We have performed a systematic first-principles study of the effect of tensile strains on the electronic properties of early transition-metal dichalcogenide (TMDC) monolayers MX2 (M = Sc, Ti, Zr, Hf, Ta, Cr; X = S, Se, Te). Our density functional theory calculations suggest that the tensile strain can significantly affect the electronic properties of many early TMDCs in general and the electronic bandgap in particular. For group IVB TMDCs (TiX2, ZrX2, HfX2), the bandgap increases with the tensile strain, but for ZrX2 and HfX2 (X = S, Se), the bandgap starts to decrease at strain 6–8%. For the group VB TMDCs (TaX2), the tensile strain can either induce the ferromagnetism or enhance the existing ferromagnetism. For the group VIB TMDCs (CrX2), the direct-to-indirect bandgap transition is seen upon application of the tensile strain, except CrTe2 whose bandgap decreases with the tensile strain even though the direct character of its bandgap is retained. Lastly, for the group IIIB TMDCs (ScX2) in the T metallic phase, we find that the tensile strain has little effect on their electronic and magnetic properties. Our study suggests that strain engineering is an effective approach to modify electronic and magnetic properties of most early TMDC monolayers, thereby opening an alternative way for future optoelectronic and spintronic applications.
Co-reporter:Ran Long;Keke Mao;Ming Gong;Shan Zhou;Jiahua Hu;Min Zhi;Yang You;Song Bai; Jun Jiang; Qun Zhang; Xiaojun Wu; Yujie Xiong
Angewandte Chemie International Edition 2014 Volume 53( Issue 12) pp:3205-3209
Publication Date(Web):
DOI:10.1002/anie.201309660
Abstract
The charge state of the Pd surface is a critical parameter in terms of the ability of Pd nanocrystals to activate O2 to generate a species that behaves like singlet O2 both chemically and physically. Motivated by this finding, we designed a metal–semiconductor hybrid system in which Pd nanocrystals enclosed by {100} facets are deposited on TiO2 supports. Driven by the Schottky junction, the TiO2 supports can provide electrons for metal catalysts under illumination by appropriate light. Further examination by ultrafast spectroscopy revealed that the plasmonics of Pd may force a large number of electrons to undergo reverse migration from Pd to the conduction band of TiO2 under strong illumination, thus lowering the electron density of the Pd surface as a side effect. We were therefore able to rationally tailor the charge state of the metal surface and thus modulate the function of Pd nanocrystals in O2 activation and organic oxidation reactions by simply altering the intensity of light shed on Pd–TiO2 hybrid structures.
Co-reporter:Hongyan Guo ; Ning Lu ; Jun Dai ; Xiaojun Wu ;Xiao Cheng Zeng
The Journal of Physical Chemistry C 2014 Volume 118(Issue 25) pp:14051-14059
Publication Date(Web):June 6, 2014
DOI:10.1021/jp505257g
We perform a comprehensive first-principles study of the electronic properties of phosphorene nanoribbons, phosphorus nanotubes, multilayer phosphorene sheets, and heterobilayers of phosphorene and two-dimensional (2D) transition-metal dichalcogenide (TMDC) monolayer. The tensile strain and electric-field effects on electronic properties of low-dimensional phosphorene nanostructures are also investigated. Our calculations show that the bare zigzag phosphorene nanoribbons (z-PNRs) are metals regardless of the ribbon width, whereas the bare armchair phosphorene nanoribbons (a-PNRs) are semiconductors with indirect bandgaps and the bandgaps decrease with increasing ribbon width. We find that compressive (or tensile) strains can reduce (or enlarge) the bandgap of the bare a-PNRs while an in-plane electric field can significantly reduce the bandgap of the bare a-PNRs, leading to the semiconductor-to-metal transition beyond certain electric field. For edge-passivated PNR by hydrogen, z-PNRs become semiconductor with nearly direct bandgaps and a-PNRs are still semiconductor but with direct bandgaps. The response to tensile strain and electric field for the edge-passivated PNRs is similar to that for the edge-unpassivated (bare) a-PNRs. For single-walled phosphorus nanotubes, both armchair and zigzag nanotubes are semiconductors with direct bandgaps. With either tensile strains or transverse electric field, behavior of bandgap modulation similar to that for a-PNRs can arise. It is known that multilayer phosphorene sheets are semiconductors whose bandgaps decrease with an increase in the number of multilayers. In the presence of a vertical electric field, the bandgaps of multilayer phosphorene sheets decrease with increasing electric field and the bandgap modulation is more significant with more layers. Lastly, heterobilayers of phosphorene (p-type) with an n-type TMDC (MoS2 or WS2) monolayer are still semiconductors while their bandgaps can be reduced by applying a vertical electric field as well. We also show that the combined phosphorene/MoS2 heterolayers can be an effective solar cell material. Our estimated power conversion efficiency for the phosphorene/MoS2 heterobilayer has a theoretical maximum value of 17.5%.
Co-reporter:Zhenbin Wang, Ranran Peng, Wenhua Zhang, Xiaojun Wu, Changrong Xia and Yalin Lu
Journal of Materials Chemistry A 2013 vol. 1(Issue 41) pp:12932-12940
Publication Date(Web):20 Aug 2013
DOI:10.1039/C3TA11554B
Oxygen reduction and successive migration on a cathode are key steps in solid oxide fuel cells. In this work, we have systematically studied the adsorption, dissociation, incorporation, and successive diffusion of oxygen species on the La1−xSrxCo1−yFeyO3 (LSCF) cathode on the basis of density-functional theory calculation. We found that the O2 molecule prefers to be adsorbed on the transition metal atoms at the B site (Fe or Co) than those at the A site (La or Sr). The oxygen molecule forms either superoxide (O2−) or peroxide (O22−) species on the surface transition metal atoms, and the isomerisation energy barrier energies between them are less than 0.14 eV. The SrCo-terminated surface has the smallest oxygen vacancy formation energy, and the existence of surface oxygen vacancy promotes the oxygen dissociation on the B-site atom without an energy barrier. Instead, without the surface oxygen vacancy, the oxygen dissociation on the Co site needs to overcome an energy barrier of 0.30 eV, while that on the Fe site is about 0.14 eV. The calculated minimum energy pathways indicate that the energy barrier of oxygen migration on the surface is much higher than that in the bulk which contains the oxygen vacancy. Moreover, increasing the concentration of Co will effectively facilitate the formation of oxygen vacancy, greatly enhancing the oxygen bulk transport. Our study presents a comprehensive understanding of the mechanism of oxygen reduction and migration on the LSCF cathode.
Co-reporter:Jun Dai ; Yu Zhao ; Xiaojun Wu ; Xiao Cheng Zeng ;Jinlong Yang
The Journal of Physical Chemistry C 2013 Volume 117(Issue 42) pp:22156-22161
Publication Date(Web):September 26, 2013
DOI:10.1021/jp408347w
Motivated from experimentally realized Cr-chemisorbed graphene, we have systematically explored the electronic properties of organometallic complexes of graphene with covalent monohexahapto-TM (TM = Cr, Fe, Ni) bonds using density-functional theory (DFT) calculations. We show that besides Cr, Fe and Ni can also bind strongly with the graphene. At the experimentally suggested coverage ratio (TM:C = 1:18, TM = Cr, Fe, Ni), our calculations suggest that the computed band gap of perfectly arranged networks of (η6-graphene)-Cr(CO)3, (η6-graphene)-Fe(CO)2, and (η6-graphene)-NiCO can be enlarged to 1.08, 0.61, and 0.29 eV, respectively. The inconsistency between the computed gap (1.08 eV) and the experimental gap (∼10 meV) for the (η6-graphene)-Cr(CO)3 is explained, which is possibly due to the existence of regions with relatively lower coverage ratio, in view of the much smaller band gap for (η6-graphene)-Cr(CO)3 with Cr:C = 1:32 (54 meV) and with Cr:C = 1:50 (20 meV), respectively. Both band gap values are much closer to the measured band gap (∼10 meV). Yet, the functionalized graphene shows little structural distortion from its original planar structure. The notable features of the direct band gaps along with the planar structure render the TM-functionalized graphenes quite appealing for applications not only in nanoelectronics but also in optoelectronics such as the infrared detector and solar cell photoanode.
Co-reporter:Xiaojun Wu, Jun Dai, Yu Zhao, Zhiwen Zhuo, Jinlong Yang, and Xiao Cheng Zeng
ACS Nano 2013 Volume 7(Issue 2) pp:880
Publication Date(Web):February 26, 2013
DOI:10.1021/nn305981j
Co-reporter:Jun Dai, Xiaojun Wu, Jinlong Yang, and Xiao Cheng Zeng
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 20) pp:3484-3488
Publication Date(Web):September 30, 2013
DOI:10.1021/jz4018877
Two metallic zeolite-like microporous BN crystals with all-sp2 bonding networks are predicted from an unbiased structure search based on the particle-swarm optimization (PSO) algorithm in combination with first-principles density functional theory (DFT) calculations. The stabilities of both microporous structures are confirmed via the phonon spectrum analysis and Born–Oppenheimer molecular dynamics simulations with temperature control at 1000 K. The unusual metallicity for the microporous BN allotropes stems from the delocalized p electrons along the axial direction of the micropores. Both microporous BN structures entail large surface areas, ranging from 3200 to 3400 m2/g. Moreover, the microporous BN structures show a preference toward organic molecule adsorption (e.g., the computed adsorption energy for CH3CH2OH is much more negative than that of H2O). This preferential adsorption can be exploited for water cleaning, as demonstrated recently using porous boron BN nanosheets ( Nat. Commun. 2013, 4, 1777).Keywords: anisotropic metallicity; global structure search; large surface areas; metallic boron nitride network;
Co-reporter:Jun Dai, Yu Zhao, Xiaojun Wu, Jinlong Yang, and Xiao Cheng Zeng
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 4) pp:561-567
Publication Date(Web):January 31, 2013
DOI:10.1021/jz302000q
The most stable structures of two-dimensional (2D) boron–silicon (B–Si) compounds containing planar sp2-bonding silicon (sp2-Si) are explored using the first-principles calculation-based particle-swarm optimization method. Among 10 B–Si compounds considered, we find that for BSi4, BSi3, BSi, B2Si, B3Si, B5Si, and B6Si, each Si atom is bonded with three B or Si atoms within the same plane, representing a preference of planar sp2-Si structure in B–Si compounds. For BSi2 and B4Si, the predicted lowest-energy structures entail a small out-of-plane buckling. Furthermore, a planar-tetracoordinated Si (ptSi) atom bonded with four B atoms within the same plane is observed in the lowest-energy structure of B7Si compound. Dynamical stabilities of the predicted 10 2D B–Si compounds are confirmed via phonon-spectrum calculation. The lowest-energy 2D B–Si compounds are all metals, regardless of the B–Si stoichiometry considered in this study.Keywords: density functional theory; electronic properties; phonon spectra; sp2-bonding silicon; structure search;
Co-reporter:Zhi Jiang, Hongyan Guo, Zheng Jiang, Guosheng Chen, Longfei Xia, Wenfeng Shangguan and Xiaojun Wu
Chemical Communications 2012 vol. 48(Issue 77) pp:9598-9600
Publication Date(Web):18 Jul 2012
DOI:10.1039/C2CC34437H
Clean Pt/TiO2 with highly dispersed controlled Pt nanocrystals was prepared by a novel approach combining an in situ polyol process with light induced photocatalysis oxidation. The interaction between TiO2 and Pt under the assistance of surfactants hinders the agglomeration. The preference to form three dimensional clusters as the cluster size increases enables the formation of controlled Pt nanocrystals.
Co-reporter:Yu Zhao, Xiaojun Wu, Jinlong Yang and Xiao Cheng Zeng
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 16) pp:5545-5550
Publication Date(Web):17 Feb 2012
DOI:10.1039/C2CP40081B
Two-dimensional (2D) hexagonal boron-nitride oxide (h-BNO) is a structural analogue of graphene oxide. Motivated by recent experimental studies of graphene oxide, we have investigated the chemical oxidation of 2D h-BN sheet and the associated electronic properties of h-BNO. Particular emphasis has been placed on the most favorable site(s) for chemisorption of atomic oxygen, and on the migration barrier for an oxygen atom hopping to the top, bridge, or hollow site on the h-BN surface, as well as the most likely pathway for the dissociation of an oxygen molecule on the h-BN surface. We find that when an oxygen atom migrates on the h-BN surface, it is most likely to be over an N atom, but confined by three neighbor B atoms (forming a triangle ring). In general, chemisorption of an oxygen atom will stretch the B–N bond, and under certain conditions may even break the B–N bond. Depending on the initial location of the first chemisorbed O atom, subsequent oxidation tends to form an O domain or O chain on the h-BN sheet. The latter may lead to a synthetic strategy for the unzipping of the h-BN sheet along a zigzag direction. A better understanding of the oxidation of h-BN sheet has important implications for tailoring the properties of the h-BN sheet for applications.
Co-reporter:Hongyan Guo, Yu Zhao, Ning Lu, Erjun Kan, Xiao Cheng Zeng, Xiaojun Wu, and Jinlong Yang
The Journal of Physical Chemistry C 2012 Volume 116(Issue 20) pp:11336-11342
Publication Date(Web):May 3, 2012
DOI:10.1021/jp2125069
We have studied structural, electronic, and magnetic properties of the graphene-like ZnO monolayer doped with nonmetal species using the first-principles calculations. Particular attention has been placed on the ZnO monolayer with one or two oxygen atoms per supercell substituted by carbon, boron, or nitrogen atoms. We find that the ZnO monolayer with one oxygen atom per supercell substituted by a carbon or boron atom is ferromagnetic (FM) half metal (HM), while that with a nitrogen atom per supercell is a FM semiconductor. Upon the ZnO monolayer with two oxygen atoms per supercell substituted by carbon or boron, the magnetic properties vary, depending on the distance between two impurities. Two neighboring carbon or boron atoms in the ZnO monolayer form dimer pairs, which convert the ZnO monolayer into an n-type semiconductor with a nonmagnetic (NM) ground state. As the distance between two carbon or boron atoms increases, the doped ZnO monolayer undergoes both NM–AFM–FM and semiconductor–HM transitions. However, the ZnO monolayer with two N atoms per supercell is a p-type semiconductor with the antiferromagnetic (AFM) ground state, regardless of the distance between N atoms. The negligible energy difference between AFM and FM states of the N-doped ZnO monolayer implies it exhibits paramagnetic behavior at room temperature. Our study demonstrates that nonmetal-doped ZnO monolayers possess tunable magnetic and electronic properties, suitable for applications in electronics and spintronics at nanoscale.
Co-reporter:Xiuling Li, Xiaojun Wu, Xiao Cheng Zeng, and Jinlong Yang
ACS Nano 2012 Volume 6(Issue 5) pp:4104
Publication Date(Web):April 8, 2012
DOI:10.1021/nn300495t
We perform a comprehensive study of the effects of line defects on electronic and magnetic properties of monolayer boron-nitride (BN) sheets, nanoribbons, and single-walled BN nanotubes using first-principles calculations and Born–Oppenheimer quantum molecular dynamic simulation. Although line defects divide the BN sheet (or nanotube) into domains, we show that certain line defects can lead to tailor-made edges on BN sheets (or imperfect nanotube) that can significantly reduce the band gap of the BN sheet or nanotube. In particular, we find that the line-defect-embedded zigzag BN nanoribbons (LD-zBNNRs) with chemically homogeneous edges such as B- or N-terminated edges can be realized by introducing a B2, N2, or C2 pentagon–octagon–pentagon (5–8–5) line defect or through the creation of the antisite line defect. The LD-zBNNRs with only B-terminated edges are predicted to be antiferromagnetic semiconductors at the ground state, whereas the LD-zBNNRs with only N-terminated edges are metallic with degenerated antiferromagnetic and ferromagnetic states. In addition, we find that the hydrogen-passivated LD-zBNNRs as well as line-defect-embedded BN sheets (and nanotubes) are nonmagnetic semiconductors with markedly reduced band gap. The band gap reduction is attributed to the line-defect-induced impurity states. Potential applications of line-defect-embedded BN nanomaterials include nanoelectronic and spintronic devices.Keywords: band gap reduction; h-BN sheet; line defect; nanoribbon; nanotube
Co-reporter:Xiaojun Wu, Jun Dai, Yu Zhao, Zhiwen Zhuo, Jinlong Yang, and Xiao Cheng Zeng
ACS Nano 2012 Volume 6(Issue 8) pp:7443
Publication Date(Web):July 20, 2012
DOI:10.1021/nn302696v
Boron, a nearest-neighbor of carbon, is possibly the second element that can possess free-standing flat monolayer structures, evidenced by recent successful synthesis of single-walled and multiwalled boron nanotubes (MWBNTs). From an extensive structural search using the first-principles particle-swarm optimization (PSO) global algorithm, two boron monolayers (α1- and β1-sheet) are predicted to be the most stable α- and β-types of boron sheets, respectively. Both boron sheets possess greater cohesive energies than the state-of-the-art two-dimensional boron structures (by more than 60 meV/atom based on density functional theory calculation using PBE0 hybrid functional), that is, the α-sheet previously predicted by Tang and Ismail-Beigi and the g1/8- and g2/15-sheets (both belonging to the β-type) recently reported by Yakobson and co-workers. Moreover, the PBE0 calculation predicts that the α-sheet is a semiconductor, while the α1-, β1-, g1/8-, and g2/15-sheets are all metals. When two α1 monolayers are stacked on top each other, the bilayer α1-sheet remains flat with an optimal interlayer distance of ∼3.62 Å, which is close to the measured interlayer distance (∼3.2 Å) in MWBNTs.Keywords: boron monolayer sheet; double-walled boron nanotube; hybrid density functional; interlayer distance
Co-reporter:Yan Zhang ; Xiaojun Wu ; Qunxiang Li ;Jinlong Yang
The Journal of Physical Chemistry C 2012 Volume 116(Issue 16) pp:9356-9359
Publication Date(Web):April 10, 2012
DOI:10.1021/jp301691z
The band-gap modulation of graphane nanoribbons under uniaxial elastic strain is investigated with the density functional theory method. Our results predict that the band gap of graphane nanoribbons can be tuned linearly with strain regardless of their widths or edge structures. The band gap increases remarkably from 2.49 to 4.11 eV and 2.04 to 4.21 eV for 13-armchair and 6-zigzag graphane nanoribbons when the strain changes from −10.0% to 10.0%, respectively. Moreover, the band gap of the graphane nanoribbon is more sensitive to the compressive than tensile deformation, which mainly originates from the shift of its valence band edge under strain. Our results imply the great potential of graphane nanoribbons in the pressure sensor and optical electronics applications at nanoscale.
Co-reporter:Fang Wu, Erjun Kan and Xiaojun Wu
Nanoscale 2011 vol. 3(Issue 9) pp:3620-3622
Publication Date(Web):15 Aug 2011
DOI:10.1039/C1NR10569H
The properties of dopant-related defects in silicon nanowires are key characteristics in semiconductive devices. Our first-principles calculations predicted that the preferred doping sites of B and P atoms in hydrogen-passivated silicon nanowires have opposite distribution behavior under electric field, suggesting a steady intrinsic p-n junction can be spontaneously formed in (B and P) codoped silicon nanowires.
Co-reporter:Yu Zhao, Xiaojun Wu, Jinlong Yang and Xiao Cheng Zeng
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 24) pp:11766-11772
Publication Date(Web):20 May 2011
DOI:10.1039/C1CP20534J
We have studied non-covalent functionalization of boron nitride nanotubes (BNNTs) with benzene molecule and with seven other different heterocyclic aromatic rings (furan, thiophene, pyrrole, pyridine, pyrazine, pyrimidine, and pyridazine, respectively). A hybrid density functional theory (DFT) method with the inclusion of dispersion correction is employed. The structural and electronic properties of the functionalized BNNTs are obtained. The DFT calculation shows that upon adsorption to the BNNT, the center of aromatic rings tend to locate on top of the nitrogen site. The trend of adsorption energy for the aromatic rings on the BNNTs shows marked dependence on different intermolecular interactions, including the dispersion interaction (area of the delocalized π bond), the dipole–dipole interaction (polarization), and the electrostatic repulsion (lone pair electrons). The DFT calculation also shows that non-covalent functionalization of BNNTs with aromatic rings can give rise to new impurity states within the band gap of pristine BNNTs, suggesting possible carrier doping of BNNTs via selective adsorption of aromatic rings.
Co-reporter:Yuling Liu ; Xiaojun Wu ; Yu Zhao ; Xiao Cheng Zeng ;Jinlong Yang
The Journal of Physical Chemistry C 2011 Volume 115(Issue 19) pp:9442-9450
Publication Date(Web):April 28, 2011
DOI:10.1021/jp201350e
Motivated by successful fabrication of monolayer materials consisting of hybrid graphene and boron nitride domains (Ci, L.; et al. Nat. Mater. 2010, 9, 430–435), we report a first-principles study of hybrid graphene/boron nitride (C-BN) nanoribbons with dihydrogenated edge(s). The first-principles study suggests that hybrid C-BN nanoribbons can possess half-metallicity with a certain range of widths for the graphene and BN sections. In general, the hybrid C-BN nanoribbons, either in HC1HB2–(C2)m(BN)n or HC2HB2–(C2)m(BN)n form, can undergo the semiconductor-to-half-metal-to-metal transitions as the width of both graphene and BN nanoribbons increases. The calculated electronic structures of the hybrid C-BN nanoribbons suggest that dihydrogenation of the boron edge can induce localized edge states around the Fermi level, and the interaction among the localized edge states can lead to the semiconductor-to-half-metal-to-metal transitions.
Co-reporter:Zhi Jiang ; Wenhua Zhang ; Wenfeng Shangguan ; Xiaojun Wu ;Yasutake Teraoka
The Journal of Physical Chemistry C 2011 Volume 115(Issue 26) pp:13035-13040
Publication Date(Web):May 30, 2011
DOI:10.1021/jp203492j
The electronic properties of spinel-type CuFe2O4 material and the adsorption behavior of NO molecule on CuFe2O4 (100) surface were studied by using density functional theory method with on-site correction for Coulomb interaction (DFT+U). Our studies suggest that the ground state of CuFe2O4 bulk has an inverse spinel structure, which is a magnetic semiconductor. On the inverse spinel-type CuFe2O4 (100) surface, NO molecule prefers to adsorb on the top site of surface Fe atom with the formed N–Fe bond. The adsorption energy, electronic properties, and structures were investigated to provide an initial understanding to the catalysis of NO molecule over CuFe2O4 surface.
Co-reporter:Ruijie Huang, Zhongti Sun, Sheng Chen, Siyu Wu, Zeqi Shen, Xiaojun Wu and Jie Zeng
Chemical Communications 2017 - vol. 53(Issue 51) pp:NaN6925-6925
Publication Date(Web):2017/06/06
DOI:10.1039/C7CC03643D
Hierarchical metal nanostructures which exhibit an open structure and a high density of twin defects accessible to reactants hold great promise in catalysis. Here, we report a facile synthesis of Pt–Cu hierarchical quasi great dodecahedrons (HQGDs) which present 5-fold symmetry and are composed of multiple ordered branched units with a frame structure. HQGDs evolve from icosahedral seeds with multiple {111} twin planes, followed by the growth of higher-order branches. Owing to the unique frame structure associated with multiple twin defects, HQGDs showed much higher HER catalytic activity and better durability relative to commercial Pt/C.
Co-reporter:Xiuling Li, Xiao Cheng Zeng and Xiaojun Wu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 16) pp:NaN10405-10405
Publication Date(Web):2017/03/24
DOI:10.1039/C7CP01020F
Developing freestanding silicene nanostructures with tunable electronic and magnetic properties is of particular importance for their applications in nanoelectronics, but still faces big challenges. On the basis of first-principles calculations, here we predict that embedded silicene nanoflakes and nanoribbons can be realized by partly dehydrogenating a freestanding polysilane (Si6H6) sheet. Born–Oppenheimer molecular dynamics simulations indicate that the embedded silicene nanostructures show good thermal stability at 500 K. In particular, the embedded silicene nanostructures exhibit similar electronics properties to those of isolated ones. These findings imply a practical solution to produce embedded silicene nanostructures from partly dehydrogenated freestanding polysilane.
Co-reporter:Keke Mao, Xiaojun Wu and Jinlong Yang
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 6) pp:NaN4439-4439
Publication Date(Web):2017/01/12
DOI:10.1039/C6CP07402B
Manipulation of the chemical reactivity of two-dimensional materials is a challenge for advancing various nanotechnologies, ranging from electronics to catalysis. In this study, on the basis of first-principles calculations, we demonstrated that the chemical reactivity of h-BN sheets towards O2 can be significantly enhanced via a metal substrate-mediated charge effect. The chemisorption of O2 molecule on the h-BN sheet deposited on Ni, Co, or Cu substrate were almost spontaneous with negligible energy barrier, distinctly different from that on the freestanding h-BN sheet, which has ultra-high chemical stability. In particular, the enhanced oxidation of h-BN sheet can be confined in the nanoscale region due to the localized electronic states in the h-BN sheet. These findings imply a pathway to selectively oxidize h-BN sheet by patterning the metal substrate.
Co-reporter:Ke Zhang, Rashid Khan, Hongyan Guo, Irfan Ali, Xiuling Li, Yunxiang Lin, Haiping Chen, Wensheng Yan, Xiaojun Wu and Li Song
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 3) pp:NaN1739-1739
Publication Date(Web):2016/12/12
DOI:10.1039/C6CP07270D
A room-temperature ferromagnetic behavior was observed in a ternary layered-Cu2MoS4 nanosheet. Both the coercivity and magnetization saturation increased with a decrease in temperature. The electron paramagnetic resonance spectroscopy confirmed a high g value. Combined with atomic structural observations, our first principle calculations revealed that the ferromagnetism originated from the edged molybdenum atoms.
Co-reporter:Ning Lu, Hongyan Guo, Wei Hu, Xiaojun Wu and Xiao Cheng Zeng
Journal of Materials Chemistry A 2017 - vol. 5(Issue 12) pp:NaN3129-3129
Publication Date(Web):2017/02/23
DOI:10.1039/C7TC00367F
We perform a comprehensive study of the effects of different types of line defects on the electronic and magnetic properties of ZnO nanoribbons and monolayer sheets by using first-principles computations. Our computations show that for zigzag ZnO nanoribbons, their metallic characteristics are unchanged by the line defects, although certain nanoribbons can exhibit much higher magnetic moments contributed by atoms within the line defects. For the armchair nanoribbons containing the 4–8 line defects, their semiconducting characteristics are the same as those of the defect-free nanoribbons. Besides the line defects, two large-angle grain boundaries are also considered for zigzag and armchair nanoribbons. In both cases, the ZnO nanoribbons show metallic characteristics. Finally, the effects of line defects on 2D ZnO monolayer sheets are also studied. It is found that the line defects in ZnO sheets can markedly enhance visible-light absorption.
Co-reporter:Zhi Jiang, Hongyan Guo, Zheng Jiang, Guosheng Chen, Longfei Xia, Wenfeng Shangguan and Xiaojun Wu
Chemical Communications 2012 - vol. 48(Issue 77) pp:NaN9600-9600
Publication Date(Web):2012/07/18
DOI:10.1039/C2CC34437H
Clean Pt/TiO2 with highly dispersed controlled Pt nanocrystals was prepared by a novel approach combining an in situ polyol process with light induced photocatalysis oxidation. The interaction between TiO2 and Pt under the assistance of surfactants hinders the agglomeration. The preference to form three dimensional clusters as the cluster size increases enables the formation of controlled Pt nanocrystals.
Co-reporter:Qiudi Yue, Yangyang Wan, Zijun Sun, Xiaojun Wu, Yupeng Yuan and Pingwu Du
Journal of Materials Chemistry A 2015 - vol. 3(Issue 33) pp:NaN16947-16947
Publication Date(Web):2015/07/21
DOI:10.1039/C5TA03949E
The generation of hydrogen (H2) through photocatalytic water splitting by employing various cocatalysts has attracted much attention. Herein we report for the first time that metallic molybdenum phosphide (MoP), as a highly active cocatalyst, can significantly enhance photocatalytic H2 production from water. A series of MoP/CdS nanorod (NR) hybrids were facilely prepared. The optimal amount of MoP led to a maximal H2 evolution rate of 163.2 μmol h−1 mg−1 under visible light illumination (λ > 420 nm), which is more than 20 times higher than that of freshly prepared CdS NRs. This work demonstrated that the suitable Fermi level alignment of MoP and CdS is responsible for the high photocatalytic activity of H2 production under visible light in the present system, as evidenced by both experimental and theoretical results.
Co-reporter:Yu Zhao, Xiaojun Wu, Jinlong Yang and Xiao Cheng Zeng
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 24) pp:NaN11772-11772
Publication Date(Web):2011/05/20
DOI:10.1039/C1CP20534J
We have studied non-covalent functionalization of boron nitride nanotubes (BNNTs) with benzene molecule and with seven other different heterocyclic aromatic rings (furan, thiophene, pyrrole, pyridine, pyrazine, pyrimidine, and pyridazine, respectively). A hybrid density functional theory (DFT) method with the inclusion of dispersion correction is employed. The structural and electronic properties of the functionalized BNNTs are obtained. The DFT calculation shows that upon adsorption to the BNNT, the center of aromatic rings tend to locate on top of the nitrogen site. The trend of adsorption energy for the aromatic rings on the BNNTs shows marked dependence on different intermolecular interactions, including the dispersion interaction (area of the delocalized π bond), the dipole–dipole interaction (polarization), and the electrostatic repulsion (lone pair electrons). The DFT calculation also shows that non-covalent functionalization of BNNTs with aromatic rings can give rise to new impurity states within the band gap of pristine BNNTs, suggesting possible carrier doping of BNNTs via selective adsorption of aromatic rings.
Co-reporter:Yu Zhao, Xiaojun Wu, Jinlong Yang and Xiao Cheng Zeng
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 16) pp:NaN5550-5550
Publication Date(Web):2012/02/17
DOI:10.1039/C2CP40081B
Two-dimensional (2D) hexagonal boron-nitride oxide (h-BNO) is a structural analogue of graphene oxide. Motivated by recent experimental studies of graphene oxide, we have investigated the chemical oxidation of 2D h-BN sheet and the associated electronic properties of h-BNO. Particular emphasis has been placed on the most favorable site(s) for chemisorption of atomic oxygen, and on the migration barrier for an oxygen atom hopping to the top, bridge, or hollow site on the h-BN surface, as well as the most likely pathway for the dissociation of an oxygen molecule on the h-BN surface. We find that when an oxygen atom migrates on the h-BN surface, it is most likely to be over an N atom, but confined by three neighbor B atoms (forming a triangle ring). In general, chemisorption of an oxygen atom will stretch the B–N bond, and under certain conditions may even break the B–N bond. Depending on the initial location of the first chemisorbed O atom, subsequent oxidation tends to form an O domain or O chain on the h-BN sheet. The latter may lead to a synthetic strategy for the unzipping of the h-BN sheet along a zigzag direction. A better understanding of the oxidation of h-BN sheet has important implications for tailoring the properties of the h-BN sheet for applications.
Co-reporter:Zhenbin Wang, Ranran Peng, Wenhua Zhang, Xiaojun Wu, Changrong Xia and Yalin Lu
Journal of Materials Chemistry A 2013 - vol. 1(Issue 41) pp:NaN12940-12940
Publication Date(Web):2013/08/20
DOI:10.1039/C3TA11554B
Oxygen reduction and successive migration on a cathode are key steps in solid oxide fuel cells. In this work, we have systematically studied the adsorption, dissociation, incorporation, and successive diffusion of oxygen species on the La1−xSrxCo1−yFeyO3 (LSCF) cathode on the basis of density-functional theory calculation. We found that the O2 molecule prefers to be adsorbed on the transition metal atoms at the B site (Fe or Co) than those at the A site (La or Sr). The oxygen molecule forms either superoxide (O2−) or peroxide (O22−) species on the surface transition metal atoms, and the isomerisation energy barrier energies between them are less than 0.14 eV. The SrCo-terminated surface has the smallest oxygen vacancy formation energy, and the existence of surface oxygen vacancy promotes the oxygen dissociation on the B-site atom without an energy barrier. Instead, without the surface oxygen vacancy, the oxygen dissociation on the Co site needs to overcome an energy barrier of 0.30 eV, while that on the Fe site is about 0.14 eV. The calculated minimum energy pathways indicate that the energy barrier of oxygen migration on the surface is much higher than that in the bulk which contains the oxygen vacancy. Moreover, increasing the concentration of Co will effectively facilitate the formation of oxygen vacancy, greatly enhancing the oxygen bulk transport. Our study presents a comprehensive understanding of the mechanism of oxygen reduction and migration on the LSCF cathode.
Co-reporter:Haiping Chen, Zhongti Sun, Chengming Wang, Xiuling Li, Xusheng Zheng, Youkui Zhang, Qun He, Xiaojun Wu and Li Song
Journal of Materials Chemistry A 2017 - vol. 5(Issue 17) pp:NaN4189-4189
Publication Date(Web):2017/03/29
DOI:10.1039/C7TC00336F
Two-dimensional layered structure of a single crystal is regarded as an ideal feature for physical and chemical fundamental studies. Herein, we demonstrated a high-quality (NH4)2V3O8 single crystal with a layered tetragonal structure prepared via a hydrothermal method. The lattice vibrational behavior and surface electronic state of (NH4)2V3O8 layers were systematically investigated via polarized Raman scattering spectroscopy and ultraviolet photoelectron spectroscopy (UPS), respectively. It was found that all Raman peaks of (NH4)2V3O8 could be clearly identified as four active Raman modes through parallel and perpendicular polarization configurations in the backscattering geometry for (001) crystal surface. The UPS results indicated that the valence band maximum of (NH4)2V3O8 was mainly composed of localized vanadium 3d states, which was further confirmed by the density functional theory calculations.