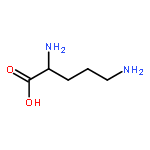
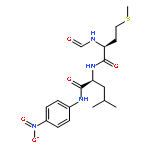
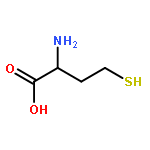
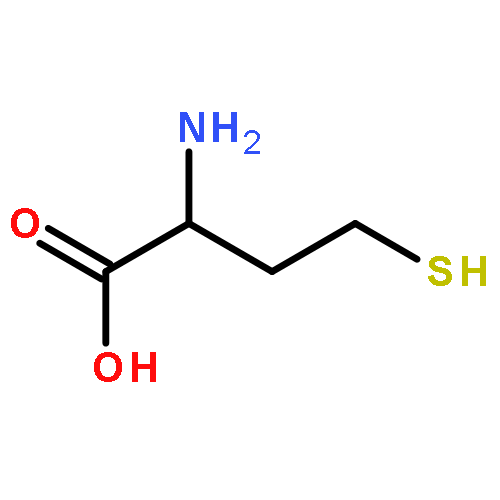
Co-reporter:Walaa Bedewy;Hui Liao;Nageh A. Abou-Taleb;Sherif F. Hammad;Tamer Nasr
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 21) pp:4540-4543
Publication Date(Web):2017/05/31
DOI:10.1039/C7OB00430C
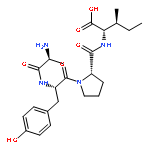
Cyclic peptides are capable of binding and modulating challenging drug targets including protein–protein interactions. However, their lack of membrane permeability prevents their application against intracellular targets. In this study, we show that it is possible to design a cell-permeable and biologically active cycloheptapeptide inhibitor against the intracellular enzyme peptidyl–prolyl isomerase Pin1 by integrating cell-penetrating and target-binding sequences.
Co-reporter:Hui Liao
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 45) pp:9595-9598
Publication Date(Web):2017/11/22
DOI:10.1039/C7OB02562A
Protein tyrosine phosphatases (PTPs) have been challenging targets for inhibitor design, because all PTPs share a highly conserved active site structure, which is positively charged and requires negatively charged moieties for tight binding. In this study, we developed cell-permeable bicyclic peptidyl inhibitors against T-cell PTP (TCPTP), which feature a cell-penetrating motif in one ring and a target-binding sequence in the second ring.
Co-reporter:Ziqing Qian, Patrick G Dougherty, Dehua Pei
Current Opinion in Chemical Biology 2017 Volume 38(Volume 38) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.cbpa.2017.03.011
•Cyclic peptides are an emerging class of drug modality for PPI inhibition.•Rational design and library screening are established to generate lead peptides.•Membrane permeability can be enhanced by various techniques.•Integration of membrane permeability and binding affinity is essential.Intracellular protein–protein interactions (PPIs) are challenging targets for conventional drug modalities, because small molecules generally do not bind to their large, flat binding sites with high affinity, whereas monoclonal antibodies cannot cross the cell membrane to reach the targets. Cyclic peptides in the 700–2000 molecular-weight range have the sufficient size and a balanced conformational flexibility/rigidity for binding to flat PPI interfaces with antibody-like affinity and specificity. Several powerful cyclic peptide library technologies were developed over the past decade to rapidly discover potent, specific cyclic peptide ligands against proteins of interest including those involved in PPIs. Methods are also being developed to enhance the membrane permeability of cyclic peptides through both passive diffusion and active transport mechanisms. Integration of the permeability-enhancing elements into cyclic peptide design has led to an increasing number of cell-permeable and biologically active cyclic peptides against intracellular PPIs. In this account, we review the recent developments in the design and synthesis of cell-permeable cyclic peptides.Download high-res image (108KB)Download full-size image
Co-reporter:Ziqing Qian, Agnieszka Martyna, Ryan L. Hard, Jiang Wang, George Appiah-Kubi, Christopher Coss, Mitch A. Phelps, Jeremy S. Rossman, and Dehua Pei
Biochemistry 2016 Volume 55(Issue 18) pp:2601-2612
Publication Date(Web):April 18, 2016
DOI:10.1021/acs.biochem.6b00226
Previous cell-penetrating peptides (CPPs) generally have low cytosolic delivery efficiencies, because of inefficient endosomal escape. In this study, a family of small, amphipathic cyclic peptides was found to be highly efficient CPPs, with cytosolic delivery efficiencies of up to 120% (compared to 2.0% for Tat). These cyclic CPPs bind directly to the plasma membrane phospholipids and enter mammalian cells via endocytosis, followed by efficient release from the endosome. Their total cellular uptake efficiency correlates positively with the binding affinity for the plasma membrane, whereas their endosomal escape efficiency increases with the endosomal membrane-binding affinity. The cyclic CPPs induce membrane curvature on giant unilamellar vesicles and budding of small vesicles, which subsequently collapse into amorphous lipid/peptide aggregates. These data suggest that cyclic CPPs exit the endosome by binding to the endosomal membrane and inducing CPP-enriched lipid domains to bud off as small vesicles. Together with their high proteolytic stability, low cytotoxicity, and oral bioavailability, these cyclic CPPs should provide a powerful system for intracellular delivery of therapeutic agents and chemical probes.
Co-reporter:Thi B. Trinh, Punit Upadhyaya, Ziqing Qian, and Dehua Pei
ACS Combinatorial Science 2016 Volume 18(Issue 1) pp:75
Publication Date(Web):December 8, 2015
DOI:10.1021/acscombsci.5b00164
Cyclic peptides have great potential as therapeutic agents and research tools. However, their applications against intracellular targets have been limited, because cyclic peptides are generally impermeable to the cell membrane. It was previously shown that fusion of cyclic peptides with a cyclic cell-penetrating peptide resulted in cell-permeable bicyclic peptides that are proteolytically stable and biologically active in cellular assays. In this work, we tested the generality of the bicyclic approach by synthesizing a combinatorial library of 5.7 × 106 bicyclic peptides featuring a degenerate sequence in the first ring and an invariant cell-penetrating peptide in the second ring. Screening of the library against oncoprotein K-Ras G12V followed by hit optimization produced a moderately potent and cell-permeable K-Ras inhibitor, which physically blocks the Ras-effector interactions in vitro, inhibits the signaling events downstream of Ras in cancer cells, and induces apoptosis of the cancer cells. Our approach should be generally applicable to developing cell-permeable bicyclic peptide inhibitors against other intracellular proteins.Keywords: anticancer agent; bicyclic peptide; cell-penetrating peptide; peptide library; Ras inhibitor
Co-reporter:Ziqing Qian, Patrick G. Dougherty and Dehua Pei
Chemical Communications 2015 vol. 51(Issue 11) pp:2162-2165
Publication Date(Web):17 Dec 2014
DOI:10.1039/C4CC09441G
We report a simple, effective method to assess the cytosolic delivery efficiency and kinetics of cell-penetrating peptides using a pH-sensitive fluorescent probe, naphthofluorescein.
Co-reporter:Bisheng Jiang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 15) pp:6306-6312
Publication Date(Web):July 21, 2015
DOI:10.1021/acs.jmedchem.5b00411
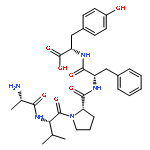
Pin1 regulates the levels and functions of phosphoproteins by catalyzing phosphorylation-dependent cis/trans isomerization of peptidyl–prolyl bonds. Previous Pin1 inhibitors contained phosphoamino acids, which are metabolically unstable and have poor membrane permeability. In this work, we report a cell-permeable and metabolically stable nonphosphorylated bicyclic peptide as a potent and selective Pin1 inhibitor, which inhibited the intracellular Pin1 activity in cultured mammalian cells but had little effect on other isomerases such as Pin4, FKBP12, or cyclophilin A.
Co-reporter:Dr. Ziqing Qian;Dr. Xiaohua Xu;Dr. Jeanine F. Amacher;Dr. Dean R. Madden;Dr. Estelle Cormet-Boyaka;Dr. Dehua Pei
Angewandte Chemie International Edition 2015 Volume 54( Issue 20) pp:5874-5878
Publication Date(Web):
DOI:10.1002/anie.201411594
Abstract
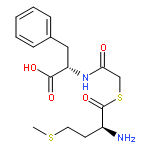
A general strategy was developed for the intracellular delivery of linear peptidyl ligands through fusion to a cell-penetrating peptide and cyclization of the fusion peptides via a disulfide bond. The resulting cyclic peptides are cell permeable and have improved proteolytic stability. Once inside the cell, the disulfide bond is reduced to produce linear biologically active peptides. This strategy was applied to generate a cell-permeable peptide substrate for real-time detection of intracellular caspase activities during apoptosis and an inhibitor for the CFTR-associated ligand (CAL) PDZ domain as a potential treatment for cystic fibrosis.
Co-reporter:Dr. Ziqing Qian;Dr. Xiaohua Xu;Dr. Jeanine F. Amacher;Dr. Dean R. Madden;Dr. Estelle Cormet-Boyaka;Dr. Dehua Pei
Angewandte Chemie 2015 Volume 127( Issue 20) pp:5972-5976
Publication Date(Web):
DOI:10.1002/ange.201411594
Abstract
A general strategy was developed for the intracellular delivery of linear peptidyl ligands through fusion to a cell-penetrating peptide and cyclization of the fusion peptides via a disulfide bond. The resulting cyclic peptides are cell permeable and have improved proteolytic stability. Once inside the cell, the disulfide bond is reduced to produce linear biologically active peptides. This strategy was applied to generate a cell-permeable peptide substrate for real-time detection of intracellular caspase activities during apoptosis and an inhibitor for the CFTR-associated ligand (CAL) PDZ domain as a potential treatment for cystic fibrosis.
Co-reporter:Wenlong Lian ; Bisheng Jiang ; Ziqing Qian
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9830-9833
Publication Date(Web):June 27, 2014
DOI:10.1021/ja503710n
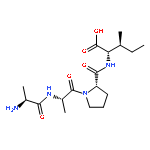
Cyclic peptides have great potential as therapeutic agents and research tools but are generally impermeable to the cell membrane. Fusion of cyclic peptides with a cyclic cell-penetrating peptide produces bicyclic peptides that are cell-permeable and retain the ability to recognize specific intracellular targets. Application of this strategy to protein tyrosine phosphatase 1B and a peptidyl-prolyl cis−trans isomerase (Pin1) isomerase resulted in potent, selective, proteolytically stable, and biologically active inhibitors against the enzymes.
Co-reporter:Ziqing Qian ; Patrick G. Dougherty ; Tao Liu ; Shameema Oottikkal ; Patrick G. Hogan ; Christopher M. Hadad
Journal of Medicinal Chemistry 2014 Volume 57(Issue 18) pp:7792-7797
Publication Date(Web):August 27, 2014
DOI:10.1021/jm500743t
Calcineurin inhibitors such as cyclosporine A and FK506 are effective immunosuppressants but produce severe side effects. Rational modification of a previously reported peptide inhibitor, GPHPVIVITGPHEE (KD ∼ 500 nM), by replacing the two valine residues with tert-leucine and the C-terminal proline with a cis-proline analogue, gave an improved inhibitor ZIZIT-cisPro, which binds to calcineurin with a KD value of 2.6 nM and is more resistant to proteolysis.
Co-reporter:Nicholas G. Selner, Rinrada Luechapanichkul, Xianwen Chen, Benjamin G. Neel, Zhong-Yin Zhang, Stefan Knapp, Charles E. Bell, and Dehua Pei
Biochemistry 2014 Volume 53(Issue 2) pp:
Publication Date(Web):December 20, 2013
DOI:10.1021/bi401223r
The sequence selectivity of 14 classical protein-tyrosine phosphatases (PTPs) (PTPRA, PTPRB, PTPRC, PTPRD, PTPRO, PTP1B, SHP-1, SHP-2, HePTP, PTP-PEST, TCPTP, PTPH1, PTPD1, and PTPD2) was systematically profiled by screening their catalytic domains against combinatorial peptide libraries. All of the PTPs exhibit similar preference for pY peptides rich in acidic amino acids and disfavor positively charged sequences but differ vastly in their degrees of preference/disfavor. Some PTPs (PTP-PEST, SHP-1, and SHP-2) are highly selective for acidic over basic (or neutral) peptides (by >105-fold), whereas others (PTPRA and PTPRD) show no to little sequence selectivity. PTPs also have diverse intrinsic catalytic efficiencies (kcat/KM values against optimal substrates), which differ by >105-fold due to different kcat and/or KM values. Moreover, PTPs show little positional preference for the acidic residues relative to the pY residue. Mutation of Arg47 of PTP1B, which is located near the pY-1 and pY-2 residues of a bound substrate, decreased the enzymatic activity by 3–18-fold toward all pY substrates containing acidic residues anywhere within the pY-6 to pY+5 region. Similarly, mutation of Arg24, which is situated near the C-terminus of a bound substrate, adversely affected the kinetic activity of all acidic substrates. A cocrystal structure of PTP1B bound with a nephrin pY1193 peptide suggests that Arg24 engages in electrostatic interactions with acidic residues at the pY+1, pY+2, and likely other positions. These results suggest that long-range electrostatic interactions between positively charged residues near the PTP active site and acidic residues on pY substrates allow a PTP to bind acidic substrates with similar affinities, and the varying levels of preference for acidic sequences by different PTPs are likely caused by the different electrostatic potentials near their active sites. The implications of the varying sequence selectivity and intrinsic catalytic activities with respect to PTP in vivo substrate specificity and biological functions are discussed.
Co-reporter:Ziqing Qian, Jonathan R. LaRochelle, Bisheng Jiang, Wenlong Lian, Ryan L. Hard, Nicholas G. Selner, Rinrada Luechapanichkul, Amy M. Barrios, and Dehua Pei
Biochemistry 2014 Volume 53(Issue 24) pp:
Publication Date(Web):June 4, 2014
DOI:10.1021/bi5004102
Cyclic heptapeptide cyclo(FΦRRRRQ) (cFΦR4, where Φ is l-2-naphthylalanine) was recently found to be efficiently internalized by mammalian cells. In this study, its mechanism of internalization was investigated by perturbing various endocytic events through the introduction of pharmacologic agents and genetic mutations. The results show that cFΦR4 binds directly to membrane phospholipids, is internalized into human cancer cells through endocytosis, and escapes from early endosomes into the cytoplasm. Its cargo capacity was examined with a wide variety of molecules, including small-molecule dyes, linear and cyclic peptides of various charged states, and proteins. Depending on the nature of the cargos, they may be delivered by endocyclic (insertion of cargo into the cFΦR4 ring), exocyclic (attachment of cargo to the Gln side chain), or bicyclic approaches (fusion of cFΦR4 and cyclic cargo rings). The overall delivery efficiency (i.e., delivery of cargo into the cytoplasm and nucleus) of cFΦR4 was 4–12-fold higher than those of nonaarginine, HIV Tat-derived peptide, or penetratin. The higher delivery efficiency, coupled with superior serum stability, minimal toxicity, and synthetic accessibility, renders cFΦR4 a useful transporter for intracellular cargo delivery and a suitable system for investigating the mechanism of endosomal escape.
Co-reporter:Punit Upadhyaya, Ziqing Qian, Nurlaila A.A. Habir, Dehua Pei
Tetrahedron 2014 70(42) pp: 7714-7720
Publication Date(Web):
DOI:10.1016/j.tet.2014.05.113
Co-reporter:Qing Xiao ; Rinrada Luechapanichkul ; Yujing Zhai
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9760-9767
Publication Date(Web):June 12, 2013
DOI:10.1021/ja401692t
A combinatorial library method was developed to systematically profile the substrate specificity of protein phosphatases toward phosphoseryl (pS) and phosphothreonyl (pT) peptides. Application of this method and a previously reported phosphotyrosyl (pY) library screening technique to dual-specificity phosphatase (DUSP) VH1 of vaccinia virus revealed that VH1 is highly active toward both pS/pT and pY peptides. VH1 exhibits different and more stringent sequence specificity toward pS/pT than pY substrates. Unlike previously characterized protein tyrosine phosphatases (PTPs), the activity and specificity of VH1 are primarily determined by the amino acid residues C-terminal to the pS, pT, or pY residue. In contrast, the mammalian VH1-related (VHR) DUSP has intrinsically low catalytic activity toward pS and pT substrates, suggesting that its primary physiological function is to dephosphorylate pY residues in substrate proteins. This method is applicable to other DUSPs and protein-serine/threonine phosphatases, and the substrate specificity data will be useful for identifying the physiological substrates of these enzymes.
Co-reporter:Wenlong Lian, Punit Upadhyaya, Curran A. Rhodes, Yusen Liu, and Dehua Pei
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:11990-11995
Publication Date(Web):July 18, 2013
DOI:10.1021/ja405106u
Protein–protein interactions represent a new class of exciting but challenging drug targets, because their large, flat binding sites lack well-defined pockets for small molecules to bind. We report here a methodology for chemical synthesis and screening of large combinatorial libraries of bicyclic peptides displayed on rigid small-molecule scaffolds. With planar trimesic acid as the scaffold, the resulting bicyclic peptides are effective for binding to protein surfaces such as the interfaces of protein–protein interactions. Screening of a bicyclic peptide library against tumor necrosis factor-α (TNFα) identified a potent antagonist that inhibits the TNFα–TNFα receptor interaction and protects cells from TNFα-induced cell death. Bicyclic peptides of this type may provide a general solution for inhibition of protein–protein interactions.
Co-reporter:Ziqing Qian, Tao Liu, Yu-Yu Liu, Roger Briesewitz, Amy M. Barrios, Sissy M. Jhiang, and Dehua Pei
ACS Chemical Biology 2013 Volume 8(Issue 2) pp:423
Publication Date(Web):November 6, 2012
DOI:10.1021/cb3005275
Cyclic peptides hold great potential as therapeutic agents and research tools, but their broad application has been limited by poor membrane permeability. Here, we report a potentially general approach for intracellular delivery of cyclic peptides. Short peptide motifs rich in arginine and hydrophobic residues (e.g., FΦRRRR, where Φ is l-2-naphthylalanine), when embedded into small- to medium-sized cyclic peptides (7–13 amino acids), bound to the plasma membrane of mammalian cultured cells and were subsequently internalized by the cells. Confocal microscopy and a newly developed peptide internalization assay demonstrated that cyclic peptides containing these transporter motifs were translocated into the cytoplasm and nucleus at efficiencies 2–5-fold higher than that of nonaarginine (R9). Furthermore, incorporation of the FΦRRRR motif into a cyclic peptide containing a phosphocoumaryl aminopropionic acid (pCAP) residue generated a cell permeable, fluorogenic probe for detecting intracellular protein tyrosine phosphatase activities.
Co-reporter:Thi B. Trinh, Qing Xiao, and Dehua Pei
Biochemistry 2013 Volume 52(Issue 33) pp:
Publication Date(Web):July 15, 2013
DOI:10.1021/bi4008947
A robust, high-throughput method has been developed to screen one-bead–one-compound peptide libraries to systematically profile the sequence specificity of protein kinases. Its ability to provide individual sequences of the preferred substrates permits the identification of sequence contextual effects and nonpermissive residues. Application of the library method to kinases Pim1, MKK6, and Csk revealed that Pim1 and Csk are highly active toward peptide substrates and recognize specific sequence motifs, whereas MKK6 has little activity or sequence selectivity against peptide substrates. Pim1 recognizes peptide substrates of the consensus RXR(H/R)X(S/T); it accepts essentially any amino acid at the S/T–2 and S/T+1 positions, but strongly disfavors acidic residues (Asp or Glu) at the S/T–2 position and a proline residue at the S/T+1 position. The selected Csk substrates show strong sequence covariance and fall into two classes with the consensus sequences of (D/E)EPIYϕXϕ and (D/E)(E/D)S(E/D/I)YϕXϕ (where X is any amino acid and ϕ is a hydrophobic amino acid). Database searches and in vitro kinase assays identified phosphatase PTP-PEST as a Pim1 substrate and phosphatase SHP-1 as a potential Csk substrate. Our results demonstrate that the sequence specificity of protein kinases is defined not only by favorable interactions between permissive residue(s) on the substrate and their cognate binding site(s) on the kinase but also by repulsive interactions between the kinase and nonpermissive residue(s).
Co-reporter:Amit Thakkar, Thi Ba Trinh, and Dehua Pei
ACS Combinatorial Science 2013 Volume 15(Issue 2) pp:120
Publication Date(Web):December 22, 2012
DOI:10.1021/co300136j
Cyclic peptides are of considerable interest in drug discovery and nanotechnology. However, macrocyclization of peptides and other compounds has often been perceived as synthetically challenging and the cyclization yields are affected by several factors including the ring size, peptide sequence, and the reaction conditions. Through the screening of combinatorial peptide libraries, we analyzed the cyclization efficiency of >2 million peptide sequences to determine the effect of ring size, peptide sequence, and solvent on the backbone (N-to-C) cyclization of peptides. Our results show that on-resin cyclization of medium- and large-sized rings (cyclohexapeptides and above) with PyBOP is essentially quantitative for ≥99.96% of the sequences, with small amounts of dimer formation observed for <4% of these sequences. Cyclization of small rings (cyclotetrapeptides and cyclopentapeptides) is considerably more difficult and accompanied by significant cyclic dimer formation. Peptides that are difficult to cyclize are generally rich in Lys(Boc) and Arg(Pbf) residues as well as sterically hindered residues [e.g., Thr(tBu)] at the N-terminus. The majority of these difficult sequences can be cyclized to completion by the addition of aqueous additives to the cyclization reaction.Keywords: combinatorial chemistry; cyclic peptides; macrocyclization; one-bead-two-compound library; partial Edman degradation; peptide sequencing
Co-reporter:Xianghong Wu, Punit Upadhyaya, Miguel A. Villalona-Calero, Roger Briesewitz and Dehua Pei
MedChemComm 2013 vol. 4(Issue 2) pp:378-382
Publication Date(Web):27 Nov 2012
DOI:10.1039/C2MD20329D
A combinatorial library of 6 × 106 cyclic peptides was synthesized in the one bead-two compound format, with each bead displaying a unique cyclic peptide on its surface and a linear peptide encoding tag in its interior. Screening of the library against K-Ras identified compounds that bound K-Ras with submicromolar affinity and disrupted its interaction with effector proteins.
Co-reporter:Varun Dewan, Tao Liu, Kuan-Ming Chen, Ziqing Qian, Yong Xiao, Lawrence Kleiman, Kiran V. Mahasenan, Chenglong Li, Hiroshi Matsuo, Dehua Pei, and Karin Musier-Forsyth
ACS Chemical Biology 2012 Volume 7(Issue 4) pp:761
Publication Date(Web):January 25, 2012
DOI:10.1021/cb200450w
The human immunodeficiency virus type 1 (HIV-1) capsid protein (CA) plays a critical role in the viral life cycle. The C-terminal domain (CTD) of CA binds to human lysyl-tRNA synthetase (hLysRS), and this interaction facilitates packaging of host cell tRNALys,3, which serves as the primer for reverse transcription. Here, we report the library synthesis, high-throughput screening, and identification of cyclic peptides (CPs) that bind HIV-1 CA. Scrambling or single-residue changes of the selected peptide sequences eliminated binding, suggesting a sequence-specific mode of interaction. Two peptides (CP2 and CP4) subjected to detailed analysis also inhibited hLysRS/CA interaction in vitro. Nuclear magnetic resonance spectroscopy and mutagenesis studies revealed that both CPs bind to a site proximal to helix 4 of the CA-CTD, which is the known site of hLysRS interaction. These results extend the current repertoire of CA-binding molecules to a new class of peptides targeting a novel site with potential for development into novel antiviral agents.
Co-reporter:Yanyan Zhang, Jinjin Zhang, Chunhua Yuan, Ryan L. Hard, In-Hee Park, Chenglong Li, Charles Bell, and Dehua Pei
Biochemistry 2011 Volume 50(Issue 35) pp:
Publication Date(Web):July 30, 2011
DOI:10.1021/bi200439v
Src homology 2 (SH2) domains mediate protein–protein interactions by recognizing phosphotyrosine (pY)-containing sequences of target proteins. In all of the SH2 domain–pY peptide interactions described to date, the SH2 domain binds to a single pY peptide. Here, determination of the cocrystal structure of the N-terminal SH2 domain of phosphatase SHP-2 bound to a class IV peptide (VIpYFVP) revealed a noncanonical 1:2 (protein–peptide) complex. The first peptide binds in a canonical manner with its pY side chain inserted in the usual binding pocket, while the second pairs up with the first to form two antiparallel β-strands that extend the central β-sheet of the SH2 domain. This unprecedented binding mode was confirmed in the solution phase by NMR experiments and shown to be adopted by pY peptides derived from cellular proteins. Site-directed mutagenesis and surface plasmon resonance studies revealed that the binding of the first peptide is pY-dependent, but phosphorylation is not required for the second peptide. Our findings suggest a potential new function for the SH2 domain as a molecular clamp to promote dimerization of signaling proteins.
Co-reporter:Tao Liu, Ziqing Qian, Qing Xiao, and Dehua Pei
ACS Combinatorial Science 2011 Volume 13(Issue 5) pp:537
Publication Date(Web):August 16, 2011
DOI:10.1021/co200101w
One-bead-one-compound (OBOC) libraries provide a powerful tool for drug discovery as well as biomedical research. However, screening a large number of beads/compounds (>1 million) and rank ordering the initial hits (which are covalently attached to a solid support) according to their potencies still post significant technical challenges. In this work, we have integrated some of the latest technical advances from our own as well as other laboratories to develop a general methodology for rapidly screening large OBOC libraries. The methodology has been applied to synthesize and screen a cyclic peptide library that features: (1) spatially segregated beads containing cyclic peptides on the surface layer and linear encoding peptides in their interior; (2) rapid on-bead screening of the library (>1 million) by a multistage procedure (magnetic bead sorting, enzyme-linked assay, and fluorescence based screening); (3) selective release of cyclic peptides from single positive beads for solution-phase determination of their binding affinities; and (4) hit identification by partial Edman degradation/mass spectrometry (PED/MS). Screening of the library against protein phosphatase calcineurin (Cn) identified a series of cyclic peptides that bind to the substrate-docking site for nuclear factor of activated T cells (NFAT) with KD values of ∼1 μM. Further improvement of the affinity and specificity of these compounds may lead to a new class of immunosuppressive agents that are more selective and therefore less toxic than cyclosporine A and FK506.Keywords: calcineurin; combinatorial library; cyclic peptides; high-throughput screening; protein−protein interaction
Co-reporter:Xianghong Wu, Lisheng Wang, Yaohua Han, Nicholas Regan, Pui-Kai Li, Miguel A. Villalona, Xiche Hu, Roger Briesewitz, and Dehua Pei
ACS Combinatorial Science 2011 Volume 13(Issue 5) pp:486
Publication Date(Web):July 19, 2011
DOI:10.1021/co200057n
FK506 and rapamycin are immunosuppressive drugs with a unique mode of action. Prior to binding to their protein targets, these drugs form a complex with an endogenous chaperone FK506-binding protein 12 (FKBP12). The resulting composite FK506-FKBP and rapamycin-FKBP binding surfaces recognize the relatively flat target surfaces of calcineurin and mTOR, respectively, with high affinity and specificity. To test whether this mode of action may be generalized to inhibit other protein targets, especially those that are challenging to inhibit by conventional small molecules, we have developed a parallel synthesis method to generate a 200-member library of bifunctional cyclic peptides as FK506 and rapamycin analogues, which were referred to as “rapalogs”. Each rapalog consists of a common FKBP-binding moiety and a variable effector domain. The rapalogs were tested for binding to FKBP12 by a fluorescence polarization competition assay. Our results show that FKBP12 binds to most of the rapalogs with high affinity (KI values in the nanomolar to low micromolar range), creating a large repertoire of composite surfaces for potential recognition of macromolecular targets such as proteins.Keywords: cyclic peptides; FK506; FKBP; rapamycin; structure-activity relationship
Co-reporter:Lige Ren, Xianwen Chen, Rinrada Luechapanichkul, Nicholas G. Selner, Tiffany M. Meyer, Anne-Sophie Wavreille, Richard Chan, Caterina Iorio, Xiang Zhou, Benjamin G. Neel, and Dehua Pei
Biochemistry 2011 Volume 50(Issue 12) pp:
Publication Date(Web):February 3, 2011
DOI:10.1021/bi1014453
We determined the substrate specificities of the protein tyrosine phosphatases (PTPs) PTP1B, RPTPα, SHP-1, and SHP-2 by on-bead screening of combinatorial peptide libraries and solution-phase kinetic analysis of individually synthesized phosphotyrosyl (pY) peptides. These PTPs exhibit different levels of sequence specificity and catalytic efficiency. The catalytic domain of RPTPα has very weak sequence specificity and is approximately 2 orders of magnitude less active than the other three PTPs. The PTP1B catalytic domain has modest preference for acidic residues on both sides of pY, is highly active toward multiply phosphorylated peptides, but disfavors basic residues at any position, a Gly at the pY−1 position, or a Pro at the pY+1 position. By contrast, SHP-1 and SHP-2 share similar but much narrower substrate specificities, with a strong preference for acidic and aromatic hydrophobic amino acids on both sides of the pY residue. An efficient SHP-1/2 substrate generally contains two or more acidic residues on the N-terminal side and one or more acidic residues on the C-terminal side of pY but no basic residues. Subtle differences exist between SHP-1 and SHP-2 in that SHP-1 has a stronger preference for acidic residues at the pY−1 and pY+1 positions and the two SHPs prefer acidic residues at different positions N-terminal to pY. A survey of the known protein substrates of PTP1B, SHP-1, and SHP-2 shows an excellent agreement between the in vivo dephosphorylation pattern and the in vitro specificity profiles derived from library screening. These results suggest that different PTPs have distinct sequence specificity profiles and the intrinsic activity/specificity of the PTP domain is an important determinant of the enzyme’s in vivo substrate specificity.
Co-reporter:Dehua Pei
Chemistry & Biology 2010 Volume 17(Issue 1) pp:3-4
Publication Date(Web):29 January 2010
DOI:10.1016/j.chembiol.2010.01.003
In this issue of Chemistry & Biology, Astle et al. report the rapid identification of the most active hits from a large one-bead-one-compound peptoid library by magnetic sorting and without the need for labor-intensive resynthesis of the hits.
Co-reporter:Tao Liu ; Yu Liu ; Hung-Ying Kao
Journal of Medicinal Chemistry 2010 Volume 53(Issue 6) pp:2494-2501
Publication Date(Web):February 24, 2010
DOI:10.1021/jm901778v
Peptidylprolyl isomerase Pin1 regulates the function and/or stability of phosphoproteins by altering the conformation of specific pSer/pThr-Pro peptide bonds. In this work, a cyclic peptide library was synthesized and screened against the catalytic domain of human Pin1. The selected inhibitors contained a consensus motif of d-pThr-Pip-Nal (where Pip is l-piperidine-2-carboxylic acid and Nal is l-2-naphthylalanine). Representative compounds were tested for binding to Pin1 by isothermal titration calorimetry and inhibition of Pin1 activity, and the most potent inhibitors had KD (and KI) values in the low nanomolar range. Treatment of breast cancer cells with the inhibitors, which were rendered membrane permeable by attachment of an octaarginine sequence, inhibited cell proliferation and increased the protein levels of two previously established Pin1 substrates, PML and SMRT. Finally, a second generation of cell permeable Pin1 inhibitors was designed by replacing the noncritical residues within the cyclic peptide ring with arginine residues and shown to have antiproliferative activity against the cancer cells.
Co-reporter:Qing Xiao, Feiran Zhang, Benjamin A. Nacev, Jun O. Liu and Dehua Pei
Biochemistry 2010 Volume 49(Issue 26) pp:
Publication Date(Web):June 3, 2010
DOI:10.1021/bi1005464
Methionine aminopeptidase (MetAP) catalyzes the hydrolytic cleavage of the N-terminal methionine from newly synthesized polypeptides. The extent of removal of methionyl from a protein is dictated by its N-terminal peptide sequence. Earlier studies revealed that MetAPs require amino acids containing small side chains (e.g., Gly, Ala, Ser, Cys, Pro, Thr, and Val) as the P1′ residue, but their specificity at positions P2′ and beyond remains incompletely defined. In this work, the substrate specificities of Escherichia coli MetAP1, human MetAP1, and human MetAP2 were systematically profiled by screening against a combinatorial peptide library and kinetic analysis of individually synthesized peptide substrates. Our results show that although all three enzymes require small residues at the P1′ position, they have differential tolerance for Val and Thr at this position. The catalytic activity of human MetAP2 toward Met-Val peptides is consistently 2 orders of magnitude higher than that of MetAP1, suggesting that MetAP2 is responsible for processing proteins containing N-terminal Met-Val and Met-Thr sequences in vivo. At positions P2′−P5′, all three MetAPs have broad specificity but are poorly active toward peptides containing a proline at the P2′ position. In addition, the human MetAPs disfavor acidic residues at the P2′−P5′ positions. The specificity data have allowed us to formulate a simple set of rules that can reliably predict the N-terminal processing of E. coli and human proteins.
Co-reporter:Ryan L. Hard, Jiangxin Liu, Juan Shen, Pei Zhou, and Dehua Pei
Biochemistry 2010 Volume 49(Issue 50) pp:
Publication Date(Web):November 19, 2010
DOI:10.1021/bi101014s
The BUZ/Znf-UBP domain is a protein module found in the cytoplasmic deacetylase HDAC6, E3 ubiquitin ligase BRAP2/IMP, and a subfamily of ubiquitin-specific proteases. Although several BUZ domains have been shown to bind ubiquitin with high affinity by recognizing its C-terminal sequence (RLRGG-COOH), it is currently unknown whether the interaction is sequence-specific or whether the BUZ domains are capable of binding to proteins other than ubiquitin. In this work, the BUZ domains of HDAC6 and Ubp-M were subjected to screening against a one-bead-one-compound (OBOC) peptide library that exhibited random peptide sequences with free C-termini. Sequence analysis of the selected binding peptides as well as alanine scanning studies revealed that the BUZ domains require a C-terminal Gly-Gly motif for binding. At the more N-terminal positions, the two BUZ domains have distinct sequence specificities, allowing them to bind to different peptides and/or proteins. A database search of the human proteome on the basis of the BUZ domain specificities identified 11 and 24 potential partner proteins for Ubp-M and HDAC6 BUZ domains, respectively. Peptides corresponding to the C-terminal sequences of four of the predicted binding partners (FBXO11, histone H4, PTOV1, and FAT10) were synthesized and tested for binding to the BUZ domains by fluorescence polarization. All four peptides bound to the HDAC6 BUZ domain with low micromolar KD values and less tightly to the Ubp-M BUZ domain. Finally, in vitro pull-down assays showed that the Ubp-M BUZ domain was capable of binding to the histone H3−histone H4 tetramer protein complex. Our results suggest that BUZ domains are sequence-specific protein-binding modules, with each BUZ domain potentially binding to a different subset of proteins.
Co-reporter:Xianwen Chen, Pauline H. Tan, Yanyan Zhang and Dehua Pei
ACS Combinatorial Science 2009 Volume 11(Issue 4) pp:604
Publication Date(Web):April 28, 2009
DOI:10.1021/cc9000168
On-bead screening of one-bead-one-compound (OBOC) libraries provides a powerful method for the rapid identification of active compounds against molecular or cellular targets. However, on-bead screening is susceptible to interference from nonspecific binding, which results in biased screening data and false positives. In this work, we have found that a major source of nonspecific binding is derived from the high ligand loading on the library beads, which permits a macromolecular target (e.g., a protein) to simultaneously interact with multiple ligands on the bead surface. To circumvent this problem, we have synthesized a phosphotyrosyl (pY)-containing peptide library on spatially segregated TentaGel microbeads, which feature a 10-fold reduced peptide loading on the bead surface but a normal peptide loading in the bead interior. The library was screened against a panel of 10 Src homology 2 (SH2) domains including those of Csk and Fyn kinases and adaptor protein SLAP, and the specific recognition motif(s) was successfully identified for each of the domains. In contrast, when the SH2 domains were screened against a control library that contained unaltered (high) ligand loading at the bead surface, six of them exhibited varying degrees of sequence biases, ranging from minor perturbation in the relative abundance of different sequences to the exclusive selection of false positive sequences that have no measurable affinity to the target protein. These results indicate that reduction of the ligand loading on the bead surface represents a simple, effective strategy to largely eliminate the interference from nonspecific binding, while preserving sufficient amounts of materials in the bead interior for compound identification. This finding should further expand the utility of OBOC libraries in biomedical research.
Co-reporter:Ozlem Dogan Ekici, Jinge Zhu, Ivy Yeuk Wah Chung, Mark Paetzel, Ross E. Dalbey and Dehua Pei
Biochemistry 2009 Volume 48(Issue 24) pp:
Publication Date(Web):May 12, 2009
DOI:10.1021/bi900461e
Knowing the substrate specificity of a protease is useful in determining its physiological substrates, developing robust assays, and designing specific inhibitors against the enzyme. In this work, we report the development of a combinatorial peptide library method for systematically profiling the substrate specificity of endopeptidases. A fluorescent donor (Edans) and quencher (Dabcyl) pair was added to the C- and N-termini of a support-bound peptide. Protease cleavage of the peptide removed the N-terminal quencher, resulting in fluorescent beads, which were isolated and individually sequenced by partial Edman degradation and mass spectrometry (PED−MS) to reveal the peptide sequence, as well as the site of proteolytic cleavage. The method was validated with bovine trypsin and Escherichia coli leader peptidase and subsequently applied to determine the substrate specificity of a viral protease, VP4, derived from the blotched snakehead virus (BSNV). The results show that VP4 cleaves peptides with a consensus sequence of (Abu/Ala/Pro)-X-Ala↓X, in agreement with the previously observed cleavage sites in its protein substrates. Resynthesis and a solution-phase assay of several representative sequences against VP4 confirmed the library screening results.
Co-reporter:Yanyan Zhang, Anne-Sophie Wavreille, Andrew R. Kunys and Dehua Pei
Biochemistry 2009 Volume 48(Issue 46) pp:
Publication Date(Web):October 19, 2009
DOI:10.1021/bi9012462
SH2 domain-containing inositol 5-phosphatases 1 (SHIP1) and 2 (SHIP2) are structurally similar proteins that catalyze the degradation of lipid secondary messenger phosphatidylinositol 3,4,5-triphosphate to produce phosphatidylinositol 3,4-diphosphate. Despite their high sequence identity (51%), SHIP1 and SHIP2 share little overlap in their in vivo functions. In this work, the sequence specificity of the SHIP2 SH2 domain was systematically defined through the screening of a combinatorial pY peptide library. Comparison of its specificity profile with that of the SHIP1 SH2 domain showed that the two SH2 domains have similar specificities, both recognizing pY peptides of the consensus sequence pY[S/Y][L/Y/M][L/M/I/V], although there are also subtle differences such as the tolerance of an arginine at the pY + 1 position by the SHIP2 but not SHIP1 SH2 domain. Surface plasmon resonance analysis of their interaction with various pY peptides suggested that the two domains have similar binding affinities but dramatically different binding kinetics, with the SHIP1 SH2 domain having fast association and dissociation rates while the SHIP2 domain showing apparent slow-binding behavior. Site-directed mutagenesis and kinetic studies indicated that the SHIP2 SH2 domain exists as a mixture of two conformational isomers. The major, inactive isomer apparently contains two cis peptidyl−prolyl bonds at positions 88 and 105, whereas the minor, active isomer has both proline residues in their trans configuration. Cis−trans isomerization of the peptidyl−prolyl bonds may provide a potential mechanism for regulating the interaction between SHIP2 and pY proteins. These data suggest that a combination of tissue distribution, specificity, and kinetic differences is likely responsible for their in vivo functional differences.
Co-reporter:Amit Thakkar, Allison S. Cohen, Michael D. Connolly, Ronald N. Zuckermann and Dehua Pei
ACS Combinatorial Science 2009 Volume 11(Issue 2) pp:294
Publication Date(Web):January 20, 2009
DOI:10.1021/cc8001734
A method for the rapid sequence determination of peptoids [oligo(N-substituted glycines)] and peptide−peptoid hybrids selected from one-bead-one-compound combinatorial libraries has been developed. In this method, beads carrying unique peptoid (or peptide−peptoid) sequences were subjected to multiple cycles of partial Edman degradation (PED) by treatment with a 1:3 (mol/mol) mixture of phenyl isothiocyanate (PITC) and 9-fluorenylmethyl chloroformate (Fmoc-Cl) to generate a series of N-terminal truncation products for each resin-bound peptoid. After PED, the Fmoc group was removed from the N-terminus and any reacted side chains via piperidine treatment. The resulting mixture of the full-length peptoid and its truncation products was analyzed by matrix-assisted laser desorption ionization (MALDI) mass spectrometry, to reveal the sequence of the full-length peptoid. With a slight modification, the method was also effective in the sequence determination of peptide−peptoid hybrids. This rapid, high-throughput, sensitive, and inexpensive sequencing method should greatly expand the utility of combinatorial peptoid libraries in biomedical and materials research.
Co-reporter:Tao Liu, Sang Hoon Joo, Jeffrey L. Voorhees, Charles L. Brooks, Dehua Pei
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 3) pp:1026-1033
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmc.2008.01.015
Prolactin receptor is involved in normal lactation and reproduction; however, excessive prolactin levels can cause various reproductive disorders such as prolactinomas. Small-molecule antagonists against the human prolactin receptor (hPRLr) thus have potential clinical applications and may serve as useful molecular probes in biomedical research. In this work, we synthesized a large, support-bound cyclic peptide library (theoretical diversity of 1.2 × 107) on 90-μm TentaGel beads and screened it against the extracellular domain of hPRLr. To facilitate hit identification, each TentaGel bead was spatially segregated into outer and inner layers, with a cyclic peptide displayed on the bead surface while the bead interior contained the corresponding linear peptide. The identity of a positive bead was revealed by sequencing the linear encoding peptide within the bead by partial Edman degradation/mass spectrometry. Screening of the library resulted in 20 hits, two of which were selected for further analysis and shown to bind to hPRLr with dissociation constants of 2–3 μM.A cyclic peptide library was screened against the extracellular domain of human prolactin receptor and ligands with low-micromolar dissociation constants were discovered.
Co-reporter:Jinge Zhu and Dehua Pei
ACS Chemical Biology 2008 Volume 3(Issue 2) pp:110
Publication Date(Web):January 24, 2008
DOI:10.1021/cb7002048
Autoinducer 2 (AI-2), which enables different bacterial species to engage in interspecies communication, has been difficult to detect quantitatively. Currently, the most commonly used method for AI-2 detection employs an engineered Vibrio harveyi reporter strain, which produces bioluminescence in response to AI-2. However, the bioassay is not quantitative and is sensitive to assay conditions. In this work, we have developed two protein sensors for AI-2 by modifying AI-2 receptor proteins LuxP and LsrB with environmentally sensitive fluorescent dyes. The protein sensors bind specifically to AI-2 and produce dose-dependent changes in their fluorescence yield. The new assay method has been applied to monitor the enzymatic synthesis of AI-2 in real time and determine the extracellular and intracellular AI-2 concentrations in several bacterial culture fluids.
Co-reporter:Yanyan Zhang, Shanggen Zhou, Anne-Sophie Wavreille, James DeWille and Dehua Pei
ACS Combinatorial Science 2008 Volume 10(Issue 2) pp:247
Publication Date(Web):February 8, 2008
DOI:10.1021/cc700185g
Cyclic peptides provide attractive lead compounds for drug discovery and excellent molecular probes in biomedical research. In this work, a novel method has been developed for the high-throughput synthesis, screening, and identification of cyclic peptidyl ligands against macromolecular targets. Support-bound cyclic phosphotyrosyl peptide libraries containing randomized amino acid sequences and different ring sizes (theoretical diversity of 3.2 × 106) were synthesized and screened against the SH2 domains of Grb2 and tensin. Potent, selective inhibitors were identified from the libraries and were generally more effective than the corresponding linear peptides. One of the inhibitors selected against the Grb2 SH2 domain inhibited human breast cancer cell growth and disrupted actin filaments. This method should be applicable to the development of cyclic peptidyl inhibitors against other protein domains, enzymes, and receptors.
Co-reporter:
Biochemistry 2008 Volume 47(Issue 9) pp:3061-3072
Publication Date(Web):January 31, 2008
DOI:10.1021/bi7023628
Preparation of support-bound combinatorial peptide libraries with free C-termini has been challenging in the past because solid-phase peptide synthesis usually starts from the C-terminus, which must be covalently attached to the solid support. In this work, we have developed a general methodology to synthesize and screen one-bead-one-compound peptide libraries containing free C-termini. TentaGel microbeads (90 μm) were spatially segregated into outer and inner layers, and peptides were synthesized on the beads in the conventional C → N manner, with their C-termini attached to the support through an ester linkage on the bead surface but through an amide bond in the bead interior. The surface peptides were cyclized between their N-terminal amine and a carboxyl group installed at a C-terminal linker sequence, while the internal peptides were kept in the linear form. Base hydrolysis of the ester linkage in the cyclic peptides regenerated linear peptides that contained a free α-carboxyl group at their C-termini but remained covalently attached to the resin via the N-termini (“inverted” peptides). An inverted peptide library containing five random residues (theoretical diversity of 3.2 × 106) was synthesized and screened for binding to four postsynaptic density-95/discs large/zona occluden-1 (PDZ) domains of sodium-hydrogen exchanger regulatory factor-1 (NHERF1) and channel-interacting PDZ domain protein (CIPP). The identity of the binding peptides was determined by sequencing the linear encoding peptides inside the bead by partial Edman degradation/mass spectrometry. Consensus recognition motifs were identified for the PDZ domains, and representative peptides were resynthesized and confirmed for binding to their cognate PDZ domains. This method should be generally applicable to all PDZ domains as well as other protein domains and enzymes that recognize the C-terminus of their target proteins.
Co-reporter:Anne-Sophie Wavreille and Dehua Pei
ACS Chemical Biology 2007 Volume 2(Issue 2) pp:109
Publication Date(Web):January 26, 2007
DOI:10.1021/cb600433g
Many protein–protein interactions are mediated by small modular domains, which recognize short peptide motifs in their partner proteins. However, for the great majority of these domains, the identity of their partner proteins remains unknown. In this work, a chemical/bioinformatics approach has been developed to identify phosphotyrosyl (pY) proteins that bind to tensin, a protein involved in the formation of actin cytoskeleton and signal transduction. A pY peptide library was chemically synthesized and screened against the Src homology 2 (SH2) domain of tensin to identify the peptide motifs that bind to the SH2 domain. Next, protein databases were searched for proteins containing the SH2 domain-binding peptide motifs. Finally, the potential tensin-binding proteins were confirmed (or disproved) by in vitro pull-down and coimmunoprecipitation assays. This procedure identified phosphoinositide-dependent kinase-1 and downstream of tyrosine kinase 2 as novel tensin-binding proteins. In addition, a cell-permeable pY peptide was designed as tensin SH2 domain inhibitor, which caused the disruption of actin filaments in NIH 3T3 cells. This method should be generally applicable to other modular domains that recognize small peptide motifs.
Co-reporter:Dehua Pei and Anne-Sophie Wavreille
Molecular BioSystems 2007 vol. 3(Issue 8) pp:536-541
Publication Date(Web):18 Jun 2007
DOI:10.1039/B706041F
Identification of binding partners is the crucial first step towards understanding the biological function of a protein. Many protein–protein interactions occur via modular domains that recognize short peptide motifs in their target proteins. Here we describe a chemical/bioinformatics approach for predicting the binding partners of modular domains. The optimal binding motif(s) of a protein domain is identified by screening a combinatorial peptide library. The resulting consensus sequence is used to search protein and genomic databases for potential bindingproteins, which are subsequently confirmed (or disproved) by conventional protein binding assays (e.g. pull-down and co-immunoprecipitation).
Co-reporter:Kiet T. Nguyen, Jen-Chieh Wu, Julie A. Boylan, Frank C. Gherardini, Dehua Pei
Archives of Biochemistry and Biophysics (15 December 2007) Volume 468(Issue 2) pp:
Publication Date(Web):15 December 2007
DOI:10.1016/j.abb.2007.09.023
Peptide deformylase (PDF, E.C. 3.5.1.88) catalyzes the removal of N-terminal formyl groups from nascent ribosome-synthesized polypeptides. PDF contains a catalytically essential divalent metal ion, which is tetrahedrally coordinated by three protein ligands (His, His, and Cys) and a water molecule. Previous studies revealed that the metal cofactor is a Fe2+ ion in Escherichia coli and many other bacterial PDFs. In this work, we found that PDFs from two iron-deficient bacteria, Borrelia burgdorferi and Lactobacillus plantarum, are stable and highly active under aerobic conditions. The native B. burgdorferi PDF (BbPDF) was purified 1200-fold and metal analysis revealed that it contains ∼1.1 Zn2+ ion/polypeptide but no iron. Our studies suggest that PDF utilizes different metal ions in different organisms. These data have important implications in designing PDF inhibitors and should help address some of the unresolved issues regarding PDF structure and catalytic function.
Co-reporter:Ziqing Qian, Patrick G. Dougherty and Dehua Pei
Chemical Communications 2015 - vol. 51(Issue 11) pp:NaN2165-2165
Publication Date(Web):2014/12/17
DOI:10.1039/C4CC09441G
We report a simple, effective method to assess the cytosolic delivery efficiency and kinetics of cell-penetrating peptides using a pH-sensitive fluorescent probe, naphthofluorescein.
Co-reporter:Walaa Bedewy, Hui Liao, Nageh A. Abou-Taleb, Sherif F. Hammad, Tamer Nasr and Dehua Pei
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 21) pp:NaN4543-4543
Publication Date(Web):2017/05/18
DOI:10.1039/C7OB00430C
Cyclic peptides are capable of binding and modulating challenging drug targets including protein–protein interactions. However, their lack of membrane permeability prevents their application against intracellular targets. In this study, we show that it is possible to design a cell-permeable and biologically active cycloheptapeptide inhibitor against the intracellular enzyme peptidyl–prolyl isomerase Pin1 by integrating cell-penetrating and target-binding sequences.
.jpg)
![2-Oxazolidinone,3-[(2R)-2-(hydroxymethyl)-1-oxo-6-heptenyl]-4-(phenylmethyl)-, (4S)-](http://img.cochemist.com/ccimg/596200/596111-55-6.png)
![2-Oxazolidinone,3-[(2R)-2-(hydroxymethyl)-1-oxo-6-heptenyl]-4-(phenylmethyl)-, (4S)-](http://img.cochemist.com/ccimg/596200/596111-55-6_b.png)










![6-Heptenoic acid, 2-[[(phenylmethoxy)amino]methyl]-, (2R)-](http://img.cochemist.com/ccimg/596200/596111-58-9.png)
![6-Heptenoic acid, 2-[[(phenylmethoxy)amino]methyl]-, (2R)-](http://img.cochemist.com/ccimg/596200/596111-58-9_b.png)







![L-ARGININE, N-[[4-(BROMOACETYL)PHENOXY]ACETYL]GLYCYL-L-ARGINYL-](http://img.cochemist.com/ccimg/524800/524711-90-8.png)
![L-ARGININE, N-[[4-(BROMOACETYL)PHENOXY]ACETYL]GLYCYL-L-ARGINYL-](http://img.cochemist.com/ccimg/524800/524711-90-8_b.png)
![[1,1'-BIPHENYL]-2-CARBOXYLIC ACID, 4'-(BROMOACETYL)-](http://img.cochemist.com/ccimg/524800/524711-88-4.png)
![[1,1'-BIPHENYL]-2-CARBOXYLIC ACID, 4'-(BROMOACETYL)-](http://img.cochemist.com/ccimg/524800/524711-88-4_b.png)


![Benzoic acid, 4-[(1E)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/464000/463961-90-2.png)
![Benzoic acid, 4-[(1E)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/464000/463961-90-2_b.png)
![BENZOIC ACID, 4-[2-(1,3-DIOXOLAN-2-YL)ETHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/464000/463961-88-8.png)
![BENZOIC ACID, 4-[2-(1,3-DIOXOLAN-2-YL)ETHENYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/464000/463961-88-8_b.png)
































![Butanoic acid,3-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-4-[(4-methyl-2-oxo-2H-1-benzopyran-7-yl)amino]-4-oxo-,(3S)-](http://img.cochemist.com/ccimg/238100/238084-15-6.png)
![Butanoic acid,3-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-4-[(4-methyl-2-oxo-2H-1-benzopyran-7-yl)amino]-4-oxo-,(3S)-](http://img.cochemist.com/ccimg/238100/238084-15-6_b.png)
![Ethanone,2-bromo-1-[6-(dimethylamino)-2-naphthalenyl]-](http://img.cochemist.com/ccimg/210900/210832-86-3.png)
![Ethanone,2-bromo-1-[6-(dimethylamino)-2-naphthalenyl]-](http://img.cochemist.com/ccimg/210900/210832-86-3_b.png)


























![L-Leucine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-alanyl-](http://img.cochemist.com/ccimg/177700/177609-08-4.png)
![L-Leucine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-alanyl-](http://img.cochemist.com/ccimg/177700/177609-08-4_b.png)
![L-Phenylalanine, N-[N-[N2-[N-(N-L-seryl-L-seryl)-L-leucyl]-L-arginyl]glycyl]-](http://img.cochemist.com/ccimg/176000/175992-46-8.png)
![L-Phenylalanine, N-[N-[N2-[N-(N-L-seryl-L-seryl)-L-leucyl]-L-arginyl]glycyl]-](http://img.cochemist.com/ccimg/176000/175992-46-8_b.png)













![L-Lysine,N6-[4-[2-[4-(dimethylamino)phenyl]diazenyl]benzoyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/147000/146998-27-8.png)
![L-Lysine,N6-[4-[2-[4-(dimethylamino)phenyl]diazenyl]benzoyl]-N2-[(9H-fluoren-9-ylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/147000/146998-27-8_b.png)






![2-Naphthalenesulfonicacid, 6-[[4-[(2-iodoacetyl)amino]phenyl]amino]-, sodium salt (1:1)](http://img.cochemist.com/ccimg/143800/143756-46-1.png)
![2-Naphthalenesulfonicacid, 6-[[4-[(2-iodoacetyl)amino]phenyl]amino]-, sodium salt (1:1)](http://img.cochemist.com/ccimg/143800/143756-46-1_b.png)












![2-Methyl-2-propanyl N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-tyrosinate](http://img.cochemist.com/ccimg/133900/133852-23-0.png)
![2-Methyl-2-propanyl N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-tyrosinate](http://img.cochemist.com/ccimg/133900/133852-23-0_b.png)







![(3aR,5R,6S,6aR)-5-((tert-butyldiphenylsilyloxy)methyl)-2,2-dimethyl-dihydro-5H-furo[3,2-d][1,3]dioxol-6-ol](http://img.cochemist.com/ccimg/114900/114861-14-2.png)
![(3aR,5R,6S,6aR)-5-((tert-butyldiphenylsilyloxy)methyl)-2,2-dimethyl-dihydro-5H-furo[3,2-d][1,3]dioxol-6-ol](http://img.cochemist.com/ccimg/114900/114861-14-2_b.png)




















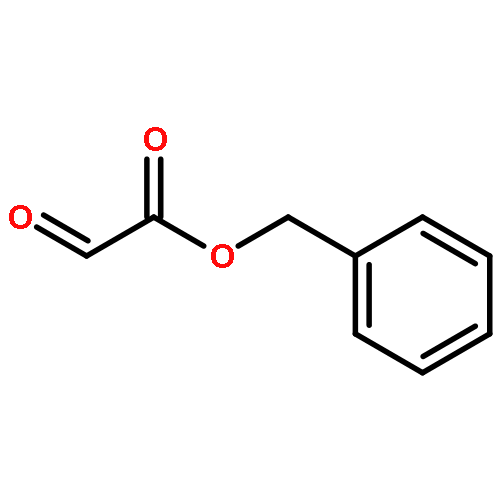
![Benzoic acid, 4-[(1E)-3-oxo-1-propen-1-yl]-, methyl ester](/data/chemimg/709300/78024-60-9.png)
![Benzoic acid, 4-[(1E)-3-oxo-1-propen-1-yl]-, methyl ester](/data/chemimg/709300/78024-60-9_b.png)
![Pentanoic acid, 2-[(acetylthio)methyl]-4-methyl-](http://img.cochemist.com/ccimg/76800/76789-49-6.png)
![Pentanoic acid, 2-[(acetylthio)methyl]-4-methyl-](http://img.cochemist.com/ccimg/76800/76789-49-6_b.png)




![Butanoic acid, 2-[(acetylthio)methyl]-](http://img.cochemist.com/ccimg/69900/69810-55-5.png)
![Butanoic acid, 2-[(acetylthio)methyl]-](http://img.cochemist.com/ccimg/69900/69810-55-5_b.png)




![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1.png)
![N-[9-(2-carboxyphenyl)-6-(dimethylamino)-3H-xanthen-3-ylidene]-N-methylmethanaminium perchlorate](http://img.cochemist.com/ccimg/62700/62669-72-1_b.png)




![L-Arginine, N2-[N-(N-formyl-L-methionyl)-L-leucyl]-](http://img.cochemist.com/ccimg/59900/59880-99-8.png)
![L-Arginine, N2-[N-(N-formyl-L-methionyl)-L-leucyl]-](http://img.cochemist.com/ccimg/59900/59880-99-8_b.png)


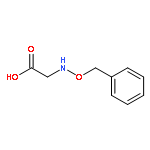

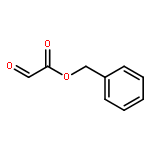

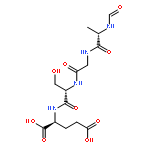
![Propanedinitrile, 2-[2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2138200/51325-91-8.png)
![Propanedinitrile, 2-[2-[2-[4-(dimethylamino)phenyl]ethenyl]-6-methyl-4H-pyran-4-ylidene]-](/data/chemimg/2138200/51325-91-8_b.png)







![1-Naphthalenesulfonicacid, 5-[[2-[(2-iodoacetyl)amino]ethyl]amino]-](http://img.cochemist.com/ccimg/37000/36930-63-9.png)
![1-Naphthalenesulfonicacid, 5-[[2-[(2-iodoacetyl)amino]ethyl]amino]-](http://img.cochemist.com/ccimg/37000/36930-63-9_b.png)








![Benzoic acid, 4-[[4-(dimethylamino)phenyl]azo]-, ethyl ester](http://img.cochemist.com/ccimg/27000/26962-53-8.png)
![Benzoic acid, 4-[[4-(dimethylamino)phenyl]azo]-, ethyl ester](http://img.cochemist.com/ccimg/27000/26962-53-8_b.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2_b.png)






![L-Cystine,N,N'-bis[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/18500/18468-46-7.png)
![L-Cystine,N,N'-bis[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]-](http://img.cochemist.com/ccimg/18500/18468-46-7_b.png)













![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-, 2,5-dioxo-1-pyrrolidinyl ester](http://img.cochemist.com/ccimg/3700/3674-06-4.png)
![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-, 2,5-dioxo-1-pyrrolidinyl ester](http://img.cochemist.com/ccimg/3700/3674-06-4_b.png)




![2-[(2-amino-4-methylpentanoyl)amino]-3-phenylpropanoic Acid](http://img.cochemist.com/ccimg/3100/3063-05-6.png)
![2-[(2-amino-4-methylpentanoyl)amino]-3-phenylpropanoic Acid](http://img.cochemist.com/ccimg/3100/3063-05-6_b.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)


![2-Hydrazono-3-methyl-2,3-dihydrobenzo[d]thiazole](http://img.cochemist.com/ccimg/1200/1128-67-2.png)
![2-Hydrazono-3-methyl-2,3-dihydrobenzo[d]thiazole](http://img.cochemist.com/ccimg/1200/1128-67-2_b.png)










![Formamide,N-[[(3S,6R)-3-(1,1-dimethylethyl)-2,5-dioxo-1,4-diazacyclopentadec-10-en-6-yl]methyl]-N-(phenylmethoxy)-](http://img.cochemist.com/ccimg/596200/596111-59-0.png)
![Formamide,N-[[(3S,6R)-3-(1,1-dimethylethyl)-2,5-dioxo-1,4-diazacyclopentadec-10-en-6-yl]methyl]-N-(phenylmethoxy)-](http://img.cochemist.com/ccimg/596200/596111-59-0_b.png)


![6-Heptenoic acid, 2-[[formyl(phenylmethoxy)amino]methyl]-, (2R)-](http://img.cochemist.com/ccimg/596200/596111-53-4.png)
![6-Heptenoic acid, 2-[[formyl(phenylmethoxy)amino]methyl]-, (2R)-](http://img.cochemist.com/ccimg/596200/596111-53-4_b.png)






![L-Serine, N-[N-(N-acetyl-L-methionyl)-L-alanyl]-](http://img.cochemist.com/ccimg/149200/149151-19-9.png)
![L-Serine, N-[N-(N-acetyl-L-methionyl)-L-alanyl]-](http://img.cochemist.com/ccimg/149200/149151-19-9_b.png)















![Acetic acid, [4-(bromoacetyl)phenoxy]-](http://img.cochemist.com/ccimg/30000/29936-81-0.png)
![Acetic acid, [4-(bromoacetyl)phenoxy]-](http://img.cochemist.com/ccimg/30000/29936-81-0_b.png)
![Acetic acid, [4-(chloroacetyl)phenoxy]-](http://img.cochemist.com/ccimg/24500/24483-61-2.png)
![Acetic acid, [4-(chloroacetyl)phenoxy]-](http://img.cochemist.com/ccimg/24500/24483-61-2_b.png)










![Tert-butyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-sulfanylbutanoate](http://img.cochemist.com/ccimg/630200/630108-94-0.png)
![Tert-butyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-sulfanylbutanoate](http://img.cochemist.com/ccimg/630200/630108-94-0_b.png)