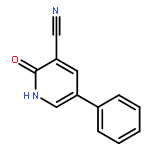
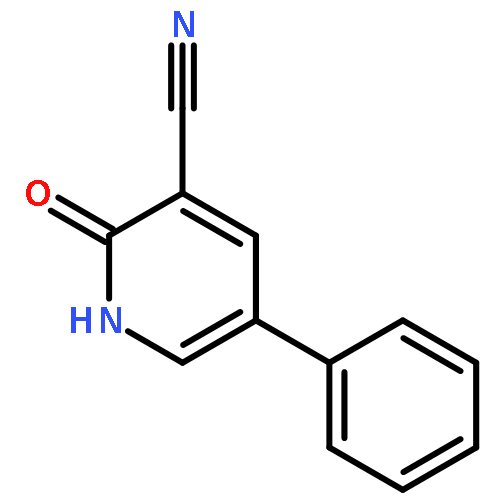
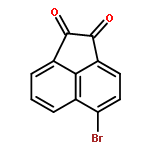
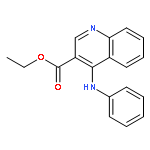
Co-reporter:Ziqian Wang; Ting Song; Yingang Feng; Zongwei Guo; Yudan Fan; Wenjie Xu; Lu Liu; Anhui Wang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:3152-3162
Publication Date(Web):March 16, 2016
DOI:10.1021/acs.jmedchem.5b01913
No α-helical mimetic that exhibits Bcl-2/MDM2 dual inhibition has been rationally designed due to the different helicities of the α-helixes at their binding interfaces. Herein, we extracted a one-turn α-helix-mimicking ortho-triarene unit from o-phenylene foldamers. Linking benzamide substrates with a rotatable C–N bond, we constructed a novel semirigid pyramid-like scaffold that could support its two-turn α-helix mimicry without aromatic stacking interactions and could adopt the different dihedral angles of the key residues of p53 and BH3-only peptides. On the basis of this universal scaffold, a series of substituent groups were installed to capture the key residues of both p53TAD and BimBH3 and balance the differences of the bulks between them. Identified by FP, ITC, and NMR spectroscopy, a compound 6e (zq-1) that directly binds to Mcl-1, Bcl-2, and MDM2 with balanced submicromolar affinities was obtained. Cell-based experiments demonstrated its antitumor ability through Bcl-2/MDM2 dual inhibition simultaneously.
Co-reporter:Ting Song;Gaobo Chai;Yubo Liu;Xiaoyan Yu;Ziqian Wang
British Journal of Pharmacology 2016 Volume 173( Issue 3) pp:471-483
Publication Date(Web):
DOI:10.1111/bph.13370
Background and Purpose
Although the ongoing clinical trials of ABT-263 and ABT-199 in chronic lymphocytic leukaemia (CLL) have indicated that BH3 mimetics hold considerable promise, understanding the mechanism of CLL resistance to BH3 mimetics remains a challenge.
Experimental Approach
The LD50 values of ABT-737, ABT-263 and ABT-199 in a number of primary CLL cells from 40 patients, were determined. The levels of Bcl-2 family proteins, including phosphorylated Bcl-2 (pBcl-2) and their interactions were measured by immunoblotting and co-immunoprecipitation. In vitro binding assays were performed by isothermal titration calorimetry and ELISA. BH3 profiling in isolated mitochondria was analysed.
Key Results
The ratio of (Mcl-1 + pBcl-2) to Bcl-2 expression provided the most significant predictive marker for the cytotoxic potential of ABT-737, ABT-263 and ABT-199 in the panel of CLL samples. Mechanistically, pBcl-2 inhibited the effects of the ABT compounds on the displacement of Bax and Bim from Bcl-2, thereby suppressing mitochondrial apoptosis. The ABT compounds exhibited 100–300-fold lower binding affinity to the glutamic acid, phosphomimetic, mutant of Bcl-2 (T69E, S70E and S87E; EEE-Bcl-2). BH3 peptides exhibited different rank orders of binding affinities to full-length WT-Bcl-2 and full-length EEE-Bcl-2.
Conclusions and Implications
Our study suggested that a structural alteration in the BH3-binding groove was induced by phosphorylation of Bcl-2. Our data also provided a framework to overcome resistance of CLL cells to the ABT compounds by combining pBcl-2 kinase inhibitors with the ABT compounds.
Co-reporter:Anhui Wang;Ting Song;Ziqian Wang;Yubo Liu;Yudan Fan;Yahui Zhang
Chemical Biology & Drug Design 2016 Volume 87( Issue 4) pp:551-561
Publication Date(Web):
DOI:10.1111/cbdd.12679
Inhibition of interactions between Mcl-1 and proapoptotic proteins is considered to be a therapeutic strategy to induce apoptosis in cancer cells. Here, we adopted molecular dynamics simulation with molecular mechanics–Poisson Boltzmann/surface area method (MM-PB/SA) to study the inhibition mechanism of three Mcl-1 inhibitors, compounds 1, 2 and 3. Analysis of energy components shows that the better binding free energy of compound 3 than compounds 1 and 2 is attributable to the van der Waals energy (ΔEvdw) and non-polar solvation energy (ΔGnp) upon binding. In addition to the excellent agreement with previous experimentally determined affinities, our simulation results further show a bend of helix 4 on Mcl-1 upon compound 3 binding, which is driven by hydrophobic interaction with residue Val253, leading to a narrowed BH3-binding groove to impede PumaBH3 binding. The computational result is consistent with our competitive isothermal titration calorimetry (ITC) assays, which shows that the competitive ability of compound 3 toward Mcl-1/PumaBH3 complex is improved beyond its direct binding affinity toward Mcl-1 itself, and compound 3 exhibits much more efficiency to compete with PumaBH3 than compound 2. Our study provides a new strategy to improve inhibitory activity on Mcl-1 based on the conformational dynamic change.
Co-reporter:Zhichao Zhang;Pengchen Su;Xiangqian Li;Ting Song;Gaobo Chai;Xiaoyan Yu;Keren Zhang
Archiv der Pharmazie 2015 Volume 348( Issue 2) pp:89-99
Publication Date(Web):
DOI:10.1002/ardp.201400296
We have previously reported a small-molecule two-face Bim BH3 mimetic, 2,3-dihydroxy-6-(4-isopropylphenylthio)anthracene-9,10-dione (1). Herein, we linked a polyphenol fragment, which was deconstructed from compound 1, with a drug-derived building block gained from computer-aided molecular design. 2-Phenyl-1H-benzo[d]imidazole as a new scaffold for two-face Bim mimetics was developed; based on this, a series of Mcl-1/Bcl-2 dual inhibitors were obtained. The most potent compound 6d binds to Mcl-1 and Bcl-2 with Ki values of 127 and 607 nM, respectively, and effectively induces apoptosis in a dose-dependent, mechanism-based manner in multiple cancer cell lines.
Co-reporter:Dr. Ting Song;Xiaoyan Yu;Dr. Yubo Liu;Dr. Xiangqian Li;Gaobo Chai;Dr. Zhichao Zhang
ChemBioChem 2015 Volume 16( Issue 5) pp:757-765
Publication Date(Web):
DOI:10.1002/cbic.201402639
Abstract
Although the role of Bcl-2 phosphorylation is still under debate, it has been identified in a resistance mechanism to BH3 mimetics, for example ABT-737 and S1. We identified an S1 analogue, S1-16, as a small-molecule inhibitor of pBcl-2. S1-16 efficiently kills EEE-Bcl-2 (a T69E, S70E, and S87E mutant mimicking phosphorylation)-expressing HL-60 cells and high endogenously expressing pBcl-2 cells, by disrupting EEE-Bcl-2 or native pBcl-2 interactions with Bax and Bak, followed by apoptosis. In vitro binding assays showed that S1-16 binds to the BH3 binding groove of EEE-Bcl-2 (Kd=0.38 μM by ITC; IC50=0.16 μM by ELISA), as well as nonphosphorylated Bcl-2 (npBcl-2; Kd=0.38 μM; IC50=0.12 μM). However, ABT-737 and S1 had much weaker affinities to EEE-Bcl-2 (IC50=1.43 and >10 μM, respectively), compared with npBcl-2 (IC50=0.011 and 0.74 μM, respectively). The allosteric effect on BH3 binding groove by Bcl-2 phosphorylation in the loop region was illustrated for the first time.
Co-reporter:Xiangqian Li, Xiaomeng Liang, Ting Song, Pengchen Su, Zhichao Zhang
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:5738-5746
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.09.047
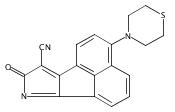
We report herein characteristic studies of Mcl-1 and Bcl-2 dual inhibitors. It was found that a protruding carbonyl group forming hydrogen bond with R263 plays a predominant role compared with the hydrophobic group that occupies the p2 pocket. A series of dual inhibitors representing different parts of the morpholino-1H-phenalene were designed, synthesized and evaluated.
Co-reporter:Ting Song, Xiangqian Li, Ying Yang, Guiye Wu, Shenghui Xie, Pengchen Su, Yingang Feng, Zhichao Zhang
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 1) pp:663-664
Publication Date(Web):1 January 2014
DOI:10.1016/j.bmc.2013.10.043
Co-reporter:Xiangqian Li;Ziqian Wang; Yingang Feng;Ting Song;Pengchen Su;Chengbin Chen;Gaobo Chai;Ying Yang; Zhichao Zhang
ChemBioChem 2014 Volume 15( Issue 9) pp:1280-1285
Publication Date(Web):
DOI:10.1002/cbic.201402040
Abstract
The design of a cross-acridine scaffold mimicking the i, i+3, i+5, and i+7 residues distributed over a two-face, two-turn α-helix is described. Docking studies and 2D 1H,15N HSQC NMR spectroscopy provide compelling evidence that compound 3 d accurately reproduces the arrangement of four hotspots in the Bim BH3 peptide to permit binding to the Mcl-1 and Bcl-2 proteins (Ki 0.079 and 0.056 μM, respectively). Furthermore, the hotspot mutation could also be mimicked by individual or multiple deletions of side chains on the scaffold.
Co-reporter:Ting Song ; Qingbin Chen ; Xiangqian Li ; Gaobo Chai
Journal of Medicinal Chemistry 2013 Volume 56(Issue 22) pp:9366-9367
Publication Date(Web):November 13, 2013
DOI:10.1021/jm401588g
Co-reporter:Yubo Liu;Ting Song;Furong Liang;Mingzhou Xie;Hongkun Sheng
British Journal of Pharmacology 2013 Volume 169( Issue 7) pp:1612-1623
Publication Date(Web):
DOI:10.1111/bph.12243
Background and Purpose
B cell lymphoma 2 (Bcl-2) is a central regulator of cell survival that is overexpressed in the majority of small-cell lung cancers (SCLC) and contributes to both malignant transformation and therapeutic resistance. The purpose of this work was to study the key factors that determine the sensitivity of SCLC cells to Bcl-2 homology domain-3 (BH3) mimetic S1 and the mechanism underlying the resistance of BH3 mimetics.
Experimental Approaches
Western blot was used to evaluate the contribution of Bcl-2 family members to the cellular response of SCLC cell lines to S1. Acquired resistant cells were derived from initially sensitive H1688 cells. Quantitative PCR and gene silencing were performed to investigate Bcl-2 up-regulation.
Key Results
A progressive increase in the relative levels of Bcl-2 and phosphorylated Bcl-2 (pBcl-2) characterized the increased de novo and acquired resistance of SCLC cell lines. Furthermore, acute treatment of S1 induced Bcl-2 expression and phosphorylation. We showed that BH3 mimetics, including S1 and ABT-737, induced endoplasmic reticulum (ER) stress and then activated MAPK/ERK pathway. The dual function of MAPK/ERK pathway in defining BH3 mimetics was illustrated; ERK1/2 activation leaded to Bcl-2 transcriptional up-regulation and sustained phosphorylation in naïve and acquired resistant SCLC cells. pBcl-2 played a key role in creating resistance of S1 and ABT-737 not only by sequestrating pro-apoptotic proteins, but also sequestrating a positive feedback to promote ERK1/2 activation.
Conclusions and Implications
These results provide significant novel insights into the molecular mechanisms for crosstalk between ER stress and endogenously apoptotic pathways in SCLC following BH3 mimetics treatment.
Co-reporter:Zhichao Zhang, Ting Song, Xiangqian Li, Zhiyong Wu, Yingang Feng, Feibo Xie, Chengwu Liu, Jianquan Qin, Hongbo Chen
European Journal of Medicinal Chemistry 2013 Volume 59() pp:141-149
Publication Date(Web):January 2013
DOI:10.1016/j.ejmech.2012.10.050
Based on a known nanomolar Bcl-2 homology domain 3 (BH3) mimetic 3-thiomorpholin-8-oxo-8H-acenaphtho[1,2-b] pyrrole-9-carbonitrile (S1, MW: 331), we applied a fragment-based approach to obtain BH3 mimetics with improved affinity and improved solubility in a water–ethanol (9:1) cosolvent. After the deconstruction of 1 (S1), we obtained fragment cyanoacetamide (4), which was determined to be a ligand efficiency (LE) hot part. After a rational optimization through fragment evolution beginning with fragment 4, a smaller Mcl-1 inhibitor (E,E)-2-(benzylaminocarbonyl)-3-styrylacrylonitrile (4g, MW: 288) with a 6-fold increase in affinity compared to 1 was obtained, as predicted by our optimization curve and identified by Mcl-1 protein nuclear magnetic resonance (NMR).Graphical abstractA series of new Mcl-1 inhibitors were synthesized. 4g binds Mcl-1 with a Ki value of 0.16 μM, and selectively induces apoptosis in Mcl-1-dependent NCI-H23 cells (IC50 = 0.38 μM).Highlights► A novel Mcl-1 inhibitor 4g was synthesized using a fragment-based approach. ► Mcl-1 protein NMR was employed to identify the binding mode of 4g with Mcl-1. ► 4g selectively induced apoptosis in Mcl-1-dependent cancer cells. ► A prediction map for Mcl-1 inhibitors was constructed.
Co-reporter:Zhichao Zhang, Xiaomeng Liang, Xiangqian Li, Ting Song, Qingbin Chen, Hongkun Sheng
European Journal of Medicinal Chemistry 2013 Volume 69() pp:711-718
Publication Date(Web):November 2013
DOI:10.1016/j.ejmech.2013.09.030
•We analysed a quinazolone scaffold which could mimic two faces of Bim α-helix.•A rigid bicyclic ring was necessary for mimicking peptide amphipathic feature.•6c interacted with R263 and occupied two hydrophobic pockets of Mcl-1.•Compound 6c exhibited nanomolar affinities toward Mcl-1/Bcl-2 protein.Based on our previous discovery of an anthraquinone scaffold mimicking two faces of Bim α-helix, we derived a quinazolone scaffold through structure simplification and optimization. It was inferred that a rigid bicyclic ring was necessary and efficient to maintain the two-faced binding mode. A novel dual inhibitor 6c [6,7,8-trihydroxy-3-(2-hydroxy-5-methylbenzyl)-2-phenylquinazolin-4(3H)-one] was obtained based on this scaffold. 6c exhibited dual binding activity with Ki values of 0.123 μM for Mcl-1 and 0.179 μM for Bcl-2.
Co-reporter:Ting Song, Xiangqian Li, Xilong Chang, Xiaomeng Liang, Yan Zhao, Guiye Wu, Shenghui Xie, Pengchen Su, Zhiyong Wu, Yingang Feng, Zhichao Zhang
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 1) pp:11-20
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmc.2012.11.008
Based on the binding mode of our previously discovered dual inhibitor of Bcl-2 and Mcl-1, 3-thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (3, S1), a library of 9-substituted 3 derivatives was synthesized to further probe the p4 pocket of the two targets. By NMR, structure–activity relationship study, and site-directed mutation, compound 6d (3-(4-aminophenylthio)-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-3-phenyl)propylamine) was identified to span p2–p4 pockets of Mcl-1, Bcl-2 and Bcl-xL, and then exhibited 9- to 35-fold better affinity to the three targets than 3 (IC50 = 10, 20 and 18 nM, respectively), which led to greater activity in induction of apoptosis in multiple cancer cell lines. Different contribution of p4 pocket to binding Bcl-2 and Mcl-1 was also investigated by plotting the potency and the HAC of the derivatives.S1 derivatives spanning p2–p4 pockets of Mcl-1, Bcl-2 and Bcl-xL were synthesized. Compound 6d exhibited 9- to 35-fold improved affinity toward the three targets than S1 (IC50 = 10, 20 and 18 nM, respectively). Different contribution of p4 to binding Bcl-2 and Mcl-1 was investigated by plotting the potency and the HAC of the derivatives.
Co-reporter:Ting Song;Zuguang Xue;Xiaoyun Shen;Xiangqian Li
Basic & Clinical Pharmacology & Toxicology 2013 Volume 113( Issue 3) pp:145-151
Publication Date(Web):
DOI:10.1111/bcpt.12074
Abstract
Small molecule S1 is a pan-BH3 mimetic that can bind antiapoptotic Bcl-2, Bcl-xL and Mcl-1 proteins. Herein, different Bcl-2 member expression cancer cell lines (NCI-H345, MCF-7, SMMC-7721 and Hela) and cells deficient in Bax and/or Bak by shRNA were used to unravel the cascade of events by which S1 promotes apoptosis compared with Bcl-2/Bcl-xL inhibitor ABT-737. We identified that S1 exhibited broader antitumour spectrum than ABT-737 through disruption of more Bcl-2 interactions including Mcl-1/Bak interaction. Moreover, the individual and combined roles of Bax and Bak in S1-induced apoptosis were revealed. Our results showed that S1 induced a Bak-mediated apoptosis. Bak played a predominant role in either S1 or ABT-737-induced apoptosis through the cooperation with Bax on the formation of large oligomers on mitochondrial membrane.
Co-reporter:Zhichao Zhang;Yan Zhao;Ting Song;Yubo Liu;Xiangqian Li;Pengchen Su;Shenghui Xie
Chemical Biology & Drug Design 2013 Volume 82( Issue 4) pp:394-400
Publication Date(Web):
DOI:10.1111/cbdd.12161
The use of small molecule B-cell lymphoma 2 homology domain 3 mimetics to neutralize the B-cell lymphoma 2 protein is an attractive strategy for cancer treatment due to its ability to cause targeted cell apoptosis. We have previously reported the design and optimization of a series of B-cell lymphoma 2 homology domain 3-mimetics, called compounds 1–6. In this study, we evaluated the optimization of B-cell lymphoma 2 homology domain 3-mimetics from a thermodynamic perspective. Understanding the thermodynamic parameters of B-cell lymphoma 2 homology domain 3-mimetics plays a critical role in the development of B-cell lymphoma 2 small-molecule inhibitors. The thermodynamic parameters for the interactions of these compounds with the myeloid cell leukemia sequence 1 protein were obtained using isothermal titration calorimetry. Owing to compounds 1–6 overcoming enthalpy–entropy compensation, the affinities of them improved gradually. Toward binding to the myeloid cell leukemia sequence 1 protein, compound 6 was deemed optimal with an obtained Kd value of 238 nm, which is a 104-fold improvement compared with 1. Analysis of the enthalpy and −TΔS efficiencies showed that ligand efficiencies with respect to molecular size are correlated with the enthalpic efficiencies. Notably, an enthalpy gain of 4.65 kcal/mol identified that an additional hydrogen bond is formed by 2 with myeloid cell leukemia sequence 1 compared with compound 1. For the first time, hydrogen bonding between a small-molecule inhibitor of B-cell lymphoma 2 was demonstrated experimentally.
Co-reporter:Zhichao Zhang ; Xiangqian Li ; Ting Song ; Yan Zhao ;Yingang Feng
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10735-10741
Publication Date(Web):November 20, 2012
DOI:10.1021/jm301504b
Bim BH3 peptide features an α-helix with hotspot residues on multiple faces. Compound 5 (6-bromo-2,3-dihydroxyanthracene-9,10-dione), which adopts a rigid-plan amphipathic conformation, was designed and evaluated as a scaffold to mimic two faces of Bim α-helix. It reproduced the functionalities of both D67 and I65 on two opposing helical sides. Moreover, it maintained the two-faced binding mode during further evolution. A putative BH3 α-helix mimic and nanomolar Bcl-2/Mcl-1 dual inhibitor, 6, was obtained based on the structure of 5.
Co-reporter:Zhichao Zhang ; Guiye Wu ; Feibo Xie ; Ting Song ;Xilong Chang
Journal of Medicinal Chemistry 2011 Volume 54(Issue 4) pp:1101-1105
Publication Date(Web):January 14, 2011
DOI:10.1021/jm101181u
We recently described the discovery of a dual inhibitor of Bcl-2 and Mcl-1, 3-thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (3, S1). Here we report a structure-guided design in combination with structure−activity relationship studies to exploit the difference in the p2 binding pocket of Bcl-2 and Mcl-1, from which a novel dual inhibitor 3-(4-aminophenylthio)-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (6h) was obtained, which showed significant enhanced IC50 value against Mcl-1 (5 nM), greater Mcl-1/Bak disruption potential, and accordingly a 10-fold increased cytotoxicity over 3.
Co-reporter:Zhichao Zhang, Hongna Yang, Guiye Wu, Zhiqiang Li, Ting Song, Xiang qian Li
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 9) pp:3909-3916
Publication Date(Web):September 2011
DOI:10.1016/j.ejmech.2011.05.062
Based on our previous discovery of a dual inhibitor of Mcl-1 and Bcl-2, 3-thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (1, S1), and guided by structure insight of 1 complex with Mcl-1 and Bcl-2, we exploited the spatial orientation of BH3 groove of the two proteins by a series of analogues of 1. These analogues contain substitutes with various steric hindrance designed to explore the width and length of the p2 pocket. The structure–activity relationships (SARs) studies together with docking studies and cell-based assays proved that the p2 pocket of Mcl-1 is relatively wider and shorter than that of Bcl-2. A novel dual inhibitor 6 was obtained based on these new findings that it exhibited nanomalar affinities toward Mcl-1 and Bcl-2, as well as nanomalar cytotoxicity activity against multiple cancer cell lines.Predicted binding models of compound 3c (a) in complex with Mcl-1 (b) and Bcl-2 (c). The p1 and p2 pockets of Mcl-1 and Bcl-2 are labeled.Highlights► We have synthesized two series dual inhibitors of Mcl-1 and Bcl-2. ► The spatial orientation of BH3 groove of the two proteins was exploited. ► The p2 pocket of Mcl-1 is relatively wider and shorter than that of Bcl-2. ► Compound 6 exhibits nanomalar affinities and nanomalar cytotoxicity activity.
Co-reporter:Zhichao Zhang, Guiye Wu, Jin Gao and Ting Song
Molecular Pharmaceutics 2010 Volume 7(Issue 4) pp:1348-1354
Publication Date(Web):June 15, 2010
DOI:10.1021/mp100081x
Small molecule inhibitors always exhibit poor water solubility due to the inherent hydrophobic property. It is an important challenge when they are developed as a real drug. S1, a structure-specific Bcl-2 inhibitor encountered this issue when moved forward in preclinical development. Herein, we prepared a 1:1 type of S1−γ-cyclodextrin (S1−γ-CD) inclusion complex to enhance the solubility. Bioevaluation of this new formulation was carried out totally in water solution. The cell internalization and cellular accumulation of S1−γ-CD was illustrated by its fluorescence analogue S2. Disruption of Bcl-2/Bax heterodimerization in MCF-7 cells further revealed S1−γ-CD could reach the subcellular function site. Moreover, the even stronger disruption by S1−γ-CD than free S1 was found due to the higher local concentrations. Furthermore, the in vivo antitumor activity of S1−γ-CD was evaluated in the H22 xenograft model. Results showed it exhibited significant antitumor activity with a decrease of tumor size (average tumor volume = 234 ± 76 mm3 vs control group, 398 ± 121 mm3, P < 0.01, and S1 group, 296 ± 65 mm3, P < 0.05), and a much longer survival time (the median time to the end point = 39.9 days vs control group, 29.2 days, P < 0.01). More importantly, the similar disruption of Bcl-2/Bax was found in S1−γ-CD treated mice and free S1 treated ones. It demonstrated that S1−γ-CD not only obtains a pharmaceutical level in vivo but also maintains the mechanism-based antitumor ability of S1 itself. It has been identified that cyclodextrin is appropriate to deliver a structure-specific molecule to its subcellular function site without any adverse effects on its mechanism-based potency, in either cultured cells or animals.Keywords: antitumor; cyclodextrins; in vivo; Small molecule inhibitors;
Co-reporter:Zhichao Zhang, Yuanyuan Wang, Ting Song, Jin Gao, Guiye Wu, Jing Zhang and Xuhong Qian
Chemical Research in Toxicology 2009 Volume 22(Issue 3) pp:483
Publication Date(Web):January 30, 2009
DOI:10.1021/tx800288v
We have previously shown the binding modes of two DNA interacting analogues 1a {3-(4-methyl-piperazin)-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile} and 3a {3-(3-dimethylamino-propylamino)-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile} with the DNA double helix. In this study, we have determined the notably different DNA damage signal pathway elicited by 1a and 3a due to the different extents to which they unwind the DNA double helix. First, we have identified that ataxia-telangiectasia-mutated (ATM) protein kinase can respond to DNA double helix unwinding caused by both 1a and 3a. In addition, the amount of ATM activation is consistent with the degree to which the DNA double helix was unwound. Consequently, we used 1a and 3a to semiquantitatively probe the response of RNA polymerase II (RNAPII) and p53 toward DNA double helix unwinding in vivo. By means of flow cytometry, immunocytochemistry, ChIP, quantitative real-time polymerase chain reaction, and Western blot analyses, we measured the level of p53 and RNAPII phosphorylation, in addition to the dynamics of the RNAPII distribution along the c-Myc gene. These results provided novel evidence for the impact of subtle DNA structural changes on the activity of RNAPII and p53. Moreover, DNA double helix conformational damage-dependent apoptosis was studied for the first time. These results indicated that 1a can induce transcriptional blockage following a shift of the unphosphorylated IIa form of RNAPII to the phosphorylated IIo form, while 3a is unable to induce the same effect. Subsequently, p53 accumulation and phosphorylation events occur that lead to apoptosis in the case of 1a exposure. This suggests that the transcriptional blockage is also correlated to the degree of double helix unwinding. Furthermore, we found that the degree of DNA conformational damage determines whether or not apoptosis occurs through transcriptional blockage. Under our experimental conditions, ATM does not participate in the downstream events even when it has been activated. Thus, p53-mediated apoptosis may be independently triggered by transcriptional blockage.
Co-reporter:Liji Jin;Yuanyuan Wang
Science China Life Sciences 2007 Volume 50( Issue 5) pp:624-629
Publication Date(Web):2007 October
DOI:10.1007/s11427-007-0079-0
Recently, the heterocyclic compound 8-oxo-3-thiomorpholino-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (S1) was synthesized and shown to induce apoptosis in both (H22) hematoma and (MCF-7) adenocarcinoma cells. The IC50 values of S1 against the two cell lines were 0.17 and 0.09 μmol/L, respectively. Furthermore, the apoptosis-inducing activity of this compound was highlighted both in vivo and in vitro. Subsequent experiments identified Bcl-2 as the primary target of S1, as a significant reduction in Bcl-2 protein levels was observed in H22 cells following a two-hour treatment with 10 μmol/L S1. While rapid depolarization of mitochondrial membranes led immediately to caspase 9 activation, no changes were identified in either caspase 8 levels or levels in Bcl-2 mRNA. These data were consistent with the results of circular dichroism (CD) spectra analysis, revealing that S1 inactivated the Bcl-2 protein by destroying its critical alpha helices. Taken together, these results suggest the potential of S1 in the development of new therapeutic agents.
Co-reporter:Ting Song, Gaobo Chai, Yubo Liu, Mingzhou Xie, Qingbin Chen, Xiaoyan Yu, Hongkun Sheng, Zhichao Zhang
European Journal of Pharmaceutical Sciences (5 April 2015) Volume 70() pp:64-71
Publication Date(Web):5 April 2015
DOI:10.1016/j.ejps.2015.01.003
Paclitaxel is an alternative chemotherapeutic agent for chronic myelogenous leukemia (CML) when primary or secondary resistance of tyrosine kinase inhibitors (TKI) is emerging, because paclitaxel could bypass the apoptotic deficiencies linked to p53 and fas ligand pathways in CML. However, high levels of Bcl-2 family proteins in CML could resist paclitaxel-induced apoptosis. Herein, we utilized two BH3 mimetics ABT-737 and S1 to study the potential of BH3 mimetics in combination with paclitaxel in treatment of CML cells and illustrated the mechanism by which BH3 mimetics synergize with paclitaxel. As a single agent, S1 could induce apoptosis in CML-derived cell line K562, whereas ABT-737 was largely ineffective. However, both of the two agents could efficiently synergize with paclitaxel through intrinsic apoptosis pathway. By using Bcl-2 siRNA, Bcl-XL siRNA or Mcl-1 siRNA, we found although each of the three members exhibited activities to block paclitaxel-induced apoptosis, Mcl-1 was the determinant for the synergistic effect between paclitaxel and ABT-737 or S1. Furthermore, paclitaxel/ABT737 synergized to drastically upregulate Bim to displace Bak from Mcl-1, whereas S1 directly binds Mcl-1 to release both Bim and Bak. As such, ABT-737 and S1 sensitized CML to paclitaxel by Mcl-1 inhibition, indirect inhibition through Bim antagonizing Mcl-1, or direct inhibition through binding to Mcl-1 itself. Finally, activation of JNK/Bim pathway was identified as the apical mechanism for ABT-737/paclitaxel synergism. Together, our results demonstrated potent synergy between BH3 mimetics and paclitaxel in the killing of CML cells and revealed an important role for Mcl-1 in mediating synergism by these agents.Download high-res image (58KB)Download full-size image

![BENZENESULFONYL CHLORIDE, 4-[(3,4-DIFLUOROPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/625900/625814-23-5.png)
![BENZENESULFONYL CHLORIDE, 4-[(3,4-DIFLUOROPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/625900/625814-23-5_b.png)
![Benzenesulfonyl chloride, 4-[(3-methylphenyl)methoxy]-](http://img.cochemist.com/ccimg/625900/625814-22-4.png)
![Benzenesulfonyl chloride, 4-[(3-methylphenyl)methoxy]-](http://img.cochemist.com/ccimg/625900/625814-22-4_b.png)
![BENZENESULFONYL CHLORIDE, 4-[(3-CHLOROPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/625900/625814-21-3.png)
![BENZENESULFONYL CHLORIDE, 4-[(3-CHLOROPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/625900/625814-21-3_b.png)
![BENZENESULFONYL CHLORIDE, 4-[(2-METHYLPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/609900/609817-17-6.png)
![BENZENESULFONYL CHLORIDE, 4-[(2-METHYLPHENYL)METHOXY]-](http://img.cochemist.com/ccimg/609900/609817-17-6_b.png)















![1H-Benzimidazole,1-[(4-methylphenyl)methyl]-2-(3,4,5-trimethoxyphenyl)-](http://img.cochemist.com/ccimg/175800/175713-06-1.png)
![1H-Benzimidazole,1-[(4-methylphenyl)methyl]-2-(3,4,5-trimethoxyphenyl)-](http://img.cochemist.com/ccimg/175800/175713-06-1_b.png)
![1H-Benzimidazole,1-[(3-methylphenyl)methyl]-2-(3,4,5-trimethoxyphenyl)-](http://img.cochemist.com/ccimg/175800/175712-95-5.png)
![1H-Benzimidazole,1-[(3-methylphenyl)methyl]-2-(3,4,5-trimethoxyphenyl)-](http://img.cochemist.com/ccimg/175800/175712-95-5_b.png)
![1H-Benzimidazole,1-[(2-methylphenyl)methyl]-2-(3,4,5-trimethoxyphenyl)-](http://img.cochemist.com/ccimg/175800/175712-94-4.png)
![1H-Benzimidazole,1-[(2-methylphenyl)methyl]-2-(3,4,5-trimethoxyphenyl)-](http://img.cochemist.com/ccimg/175800/175712-94-4_b.png)












![1H-Isoindole-1,3(2H)-dione, 5-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/64200/64146-72-1.png)
![1H-Isoindole-1,3(2H)-dione, 5-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/64200/64146-72-1_b.png)