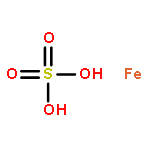
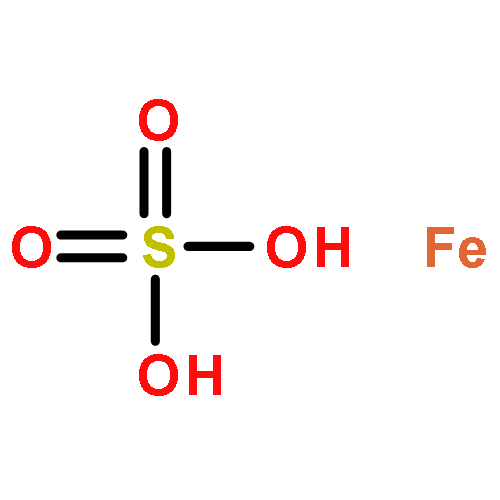
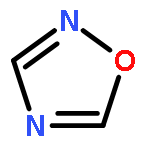
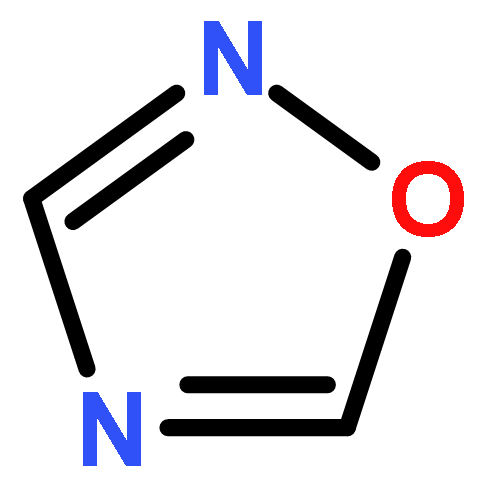
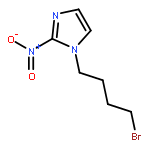
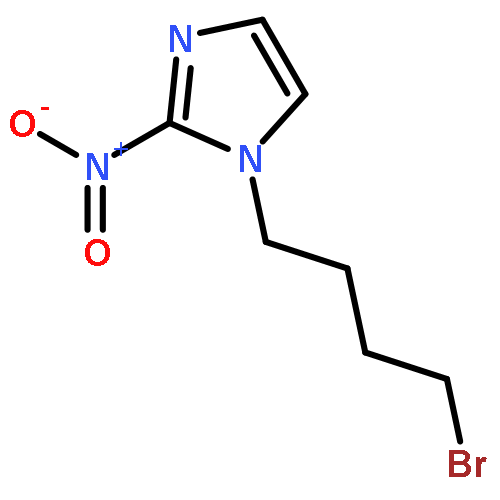
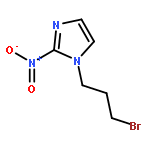
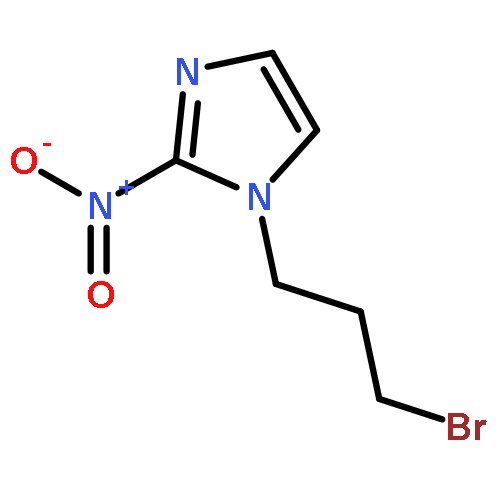
Co-reporter:Lingfei Wang;Jun Wei;Ranran Wu;Gang Cheng;Xinjin Li;Jinbo Hu;Yongzhou Hu
Organic Chemistry Frontiers 2017 vol. 4(Issue 2) pp:214-223
Publication Date(Web):2017/02/01
DOI:10.1039/C6QO00674D
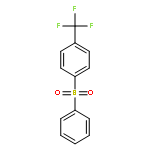
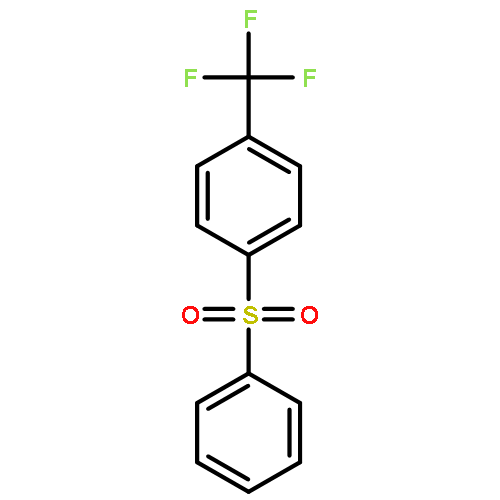
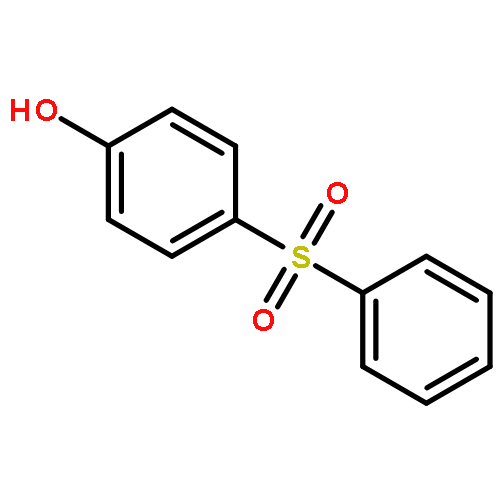
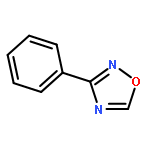
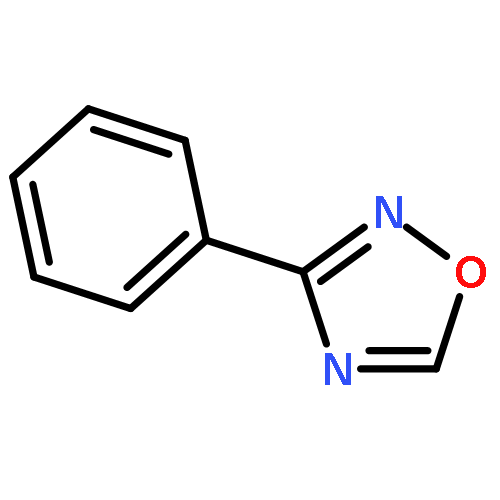
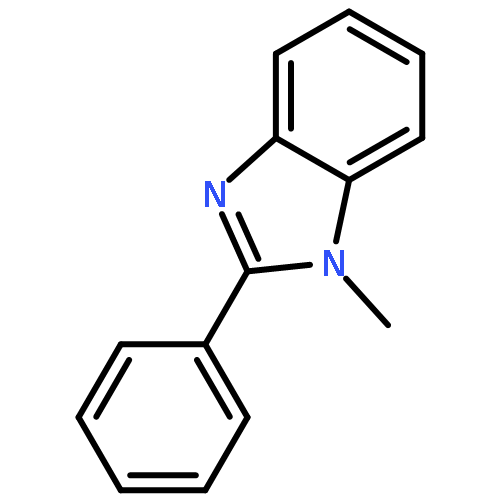
Little attention has been paid to the stability and reactivity of the C–F bond under different conditions. In this study, the stability and reactivity of tri-, di-, and monofluoromethyl/methoxy/methylthio moiety groups on arenes under acidic or basic conditions are carefully investigated.
Co-reporter:Ping Xu, Minkui Zhang, Rong Sheng, Yongmin Ma
European Journal of Medicinal Chemistry 2017 Volume 127(Volume 127) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.ejmech.2016.12.045
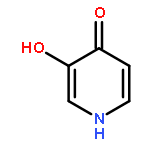
•22 3-Hydroxypyridin-4-one-resveratrol hybrids were designed.•3i and 4f are good antioxidants, metal-chelating agents and Aβ anti aggregation.•SAR between compounds and the Aβ1–42 inhibition activity was outlined.A series of deferiprone-resveratrol hybrids have been designed and synthesized as multitarget-directed ligands (MTDLs) through merging the chelating moiety 3-hydroxypyridin-4-one into the structure of resveratrol, a natural antioxidant agent and β-amyloid peptide (Aβ) aggregation inhibitor. The in vitro biological evaluation revealed that most of these newly synthesized compounds exhibited good inhibitory activity against self-induced Aβ1–42 aggregation, excellent antioxidant activity and potent metal chelating capability. Compounds 3i and 4f were identified as the most promising MTDLs with triple functions, possessing micromolar IC50 values for Aβ1–42 aggregation inhibition, greater 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS•+) scavenging activity than Trolox and similar pFe(III) values to that of deferiprone.Download high-res image (163KB)Download full-size image
Co-reporter:Rong Sheng, Li Tang, Liu Jiang, Lingjuan Hong, Ying Shi, Naiming Zhou, and Yongzhou Hu
ACS Chemical Neuroscience 2016 Volume 7(Issue 1) pp:69
Publication Date(Web):October 19, 2015
DOI:10.1021/acschemneuro.5b00224
A series of novel 1-phenyl-3-hydroxy-4-pyridinone derivatives were designed and synthesized as multifunctional agents for Alzheimer’s disease (AD) therapy through incorporation of 3-hydroxy-4-pyridinone moiety from deferiprone into the scaffold of H3 receptor antagonists. Most of these new compounds displayed designed quadruple functions, H3 receptor antagonism, Aβ aggregation inhibition, metal ion chelation, and radical scavenging. Especially, the most promising compound 5c displayed nanomolar IC50 values in H3 receptor antagonism with high selectivity, efficient capability to interrupt the formation of Aβ1–42 fibrils, good copper and iron chelating properties, and more potent 2,2′-azino-bis(3-ethyl-benzothiazoline-6-sulfonic acid) radical cation (ABTS•+) scavenging activity than Trolox. Further biological evaluation revealed that it did not show obvious cytotoxicity and hERG potassium channel inhibition at micromolar concentration. In addition, compound 5c demonstrated suitable pharmacokinetic properties and acceptable blood–brain barrier (BBB) permeability in vivo. All these results indicate that compound 5c is a potential multifunctional candidate for AD therapy.Keywords: Alzheimer’s disease; Aβ aggregation inhibition; H3R antagonism; metal ion chelation; Multifunctional agents; radical scavenge
Co-reporter:Liu Jiang, Minkui Zhang, Li Tang, Qinjie Weng, Yanhong Shen, Yongzhou Hu and Rong Sheng
RSC Advances 2016 vol. 6(Issue 21) pp:17318-17327
Publication Date(Web):26 Jan 2016
DOI:10.1039/C5RA25788C
A novel series of 2-subsituted benzothiazole derivatives as MTDLs were designed and synthesized for AD therapy using pharmacophore-combine strategy. The benzothiazole moiety from ThT and the HPO moiety from deferiprone were connected with vinyl linker to achieve target compounds. The biological evaluation results revealed that the majority of them demonstrated desirable triple functions by interfering with Aβ aggregation, oxidative stress and metal dyshomeostasis simultaneously. The two most attractive compounds 9c and 9i exhibited excellent self-Aβ1–42 aggregation inhibitory activity, efficient ABTS˙+ scavenging activity, potent biometals chelating properties, as well as disaggregation activity against previous formed Aβ1–42 fibrils. In addition to these advantages, both of them displayed no cytotoxicity to human glioma U251 cells up to 50 μM, thereby meriting further investigation.
Co-reporter:Weiyan Cheng, Shijun Zhu, Xiaodong Ma, Ni Qiu, Peng Peng, Rong Sheng, Yongzhou Hu
European Journal of Medicinal Chemistry 2015 Volume 89() pp:826-834
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.11.010
•18 novel EGFR inhibitors were designed and synthesized.•5 compounds were more potent than gefitinib under normoxia and hypoxia.•15c could be reductive activated to generate reactive radicals under hypoxia.•15c was relatively stable under normoxia and easily activated under hypoxia.A series of novel 6-(nitroimidazole-1H-alkyloxyl)-4-anilinoquinazoline derivatives (15a–15r) were designed, synthesized and evaluated as efficient EGFR inhibitors through introduction of hypoxia activated nitroimidazole moiety into the quinazoline scaffold of EGFR inhibitors. The majority of these newly synthesized compounds exhibited comparable EGFR inhibitory activities to gefitinib and moderate to excellent anti-proliferative activities against HT-29 cells under normoxia and hypoxia. The most promising compound 15c displayed the IC50 value of 0.47 nM against EGFR kinase and excellent cytotoxic effect against HT-29 cells under normoxia and hypoxia with the IC50 values of 2.21 μM and 1.62 μM, respectively. The mimic reductive activation study revealed that compound 15c exerted reductive activation properties under hypoxia, which were consistent with the in vitro metabolic study, wherein 15c was easily reductive activated under hypoxia and much more stable under normoxia. All these results suggested that 15c was a potential cancer therapeutic agent both under normoxia and hypoxia and was worth of further development.
Co-reporter:Yujie Li, Peng Peng, Li Tang, Yunzhen Hu, Yongzhou Hu, Rong Sheng
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 17) pp:4717-4725
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmc.2014.07.009
A series of novel 2-methoxy-phenyl dimethyl-carbamate derivatives were designed, synthesized and evaluated as site-activated MTDLs based on rivastigmine and curcumin. Most of them exhibited good to excellent AChE and BuChE inhibitory activities with sub-micromolar IC50 values. Among all the compounds, 6a demonstrated the most potent AChE inhibition with IC50 value of 0.097 μM, which is about 20-fold than that of rivastigmine. In addition, the three selected compounds 5a, 6a and 6e demonstrated inhibitory activity against Aβ self-aggregation similar to cucurmin in TEM assay, which is obviously different from the weak activity of rivastigmine. Moreover, the hydrolysate of 6a (compound 7) also showed potent ABTS+ scavenging and moderate copper ion chelating activity in vitro.A series of novel 2-methoxy-phenyl dimethyl-carbamate derivatives were designed, synthesized and evaluated as site-activated MTDLs.
Co-reporter:Weiyan Cheng, Youting Yuan, Ni Qiu, Peng Peng, Rong Sheng, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2014 22(24) pp: 6796-6805
Publication Date(Web):
DOI:10.1016/j.bmc.2014.10.038
Co-reporter:Li Tang, Liying Zhao, Lingjuan Hong, Fenyan Yang, Rong Sheng, Jianzhong Chen, Ying Shi, Naimin Zhou, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 19) pp:5936-5944
Publication Date(Web):1 October 2013
DOI:10.1016/j.bmc.2013.07.051
A series of novel 3-substituted-indole derivatives with a benzyl tertiary amino moiety were designed, synthesized and evaluated as H3 receptor antagonists and free radical scavengers for Alzheimer’s disease therapy. Most of these synthesized compounds exhibited moderate to potent antagonistic activities in CREs driven luciferase assay. In particular, compound 2d demonstrated the most favorable H3 receptor antagonistic activity with the IC50 value of 0.049 μM. Besides, it also displayed high binding affinity to H3 receptor (Ki = 4.26 ± 2.55 nM) and high selectivity over other three histamine receptors. Moreover, 2d and other two 3-substituted indole derivatives 1d and 3d exerted potent ABTS radical cation scavenging capacities similar to melatonin. Above results illustrate that 2d is an interesting lead for extensive optimization to explore new drug candidate for AD therapy.A series of novel 3-substituted-indole derivatives, as exemplified by compound 2d, was designed, synthesized and evaluated as selective H3 receptor antagonists and potent free radical scavengers.
Co-reporter:Lingfei Wang, Jun Wei, Ranran Wu, Gang Cheng, Xinjin Li, Jinbo Hu, Yongzhou Hu and Rong Sheng
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 2) pp:NaN223-223
Publication Date(Web):2016/11/16
DOI:10.1039/C6QO00674D
Little attention has been paid to the stability and reactivity of the C–F bond under different conditions. In this study, the stability and reactivity of tri-, di-, and monofluoromethyl/methoxy/methylthio moiety groups on arenes under acidic or basic conditions are carefully investigated.

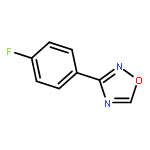
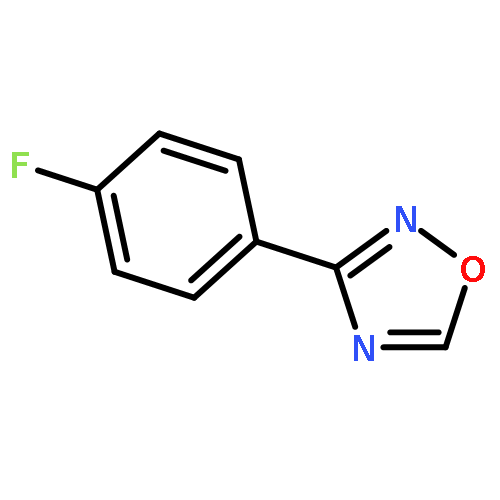



![1,2,4-Oxadiazole, 5-phenyl-3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/425500/425410-72-6.png)
![1,2,4-Oxadiazole, 5-phenyl-3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/425500/425410-72-6_b.png)








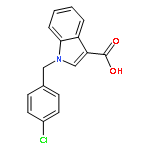
![1,2,4-Oxadiazole, 3-(4-methoxyphenyl)-5-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/89900/89804-67-1.png)
![1,2,4-Oxadiazole, 3-(4-methoxyphenyl)-5-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/89900/89804-67-1_b.png)
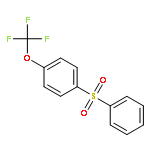
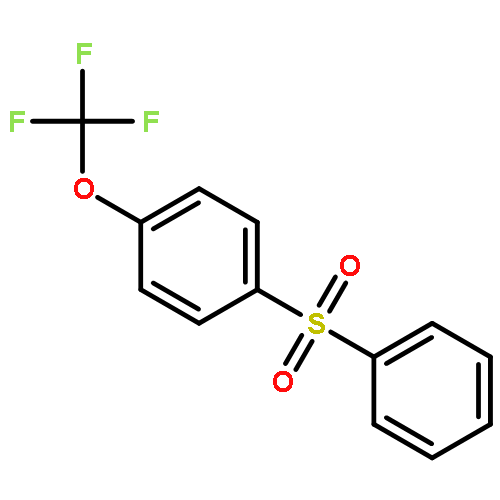

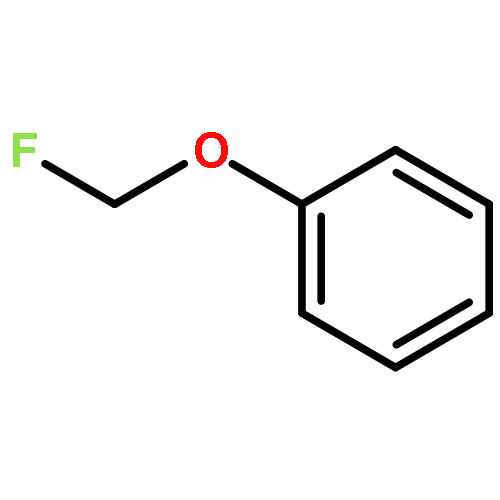
![Benzene, 1-[(difluoromethyl)thio]-4-methoxy-](http://img.cochemist.com/ccimg/82000/81931-98-8.png)
![Benzene, 1-[(difluoromethyl)thio]-4-methoxy-](http://img.cochemist.com/ccimg/82000/81931-98-8_b.png)




![Benzaldehyde, 4-[3-(4-methoxyphenyl)-1,2,4-oxadiazol-5-yl]-](http://img.cochemist.com/ccimg/72100/72094-41-8.png)
![Benzaldehyde, 4-[3-(4-methoxyphenyl)-1,2,4-oxadiazol-5-yl]-](http://img.cochemist.com/ccimg/72100/72094-41-8_b.png)
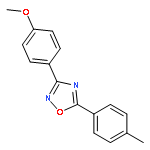
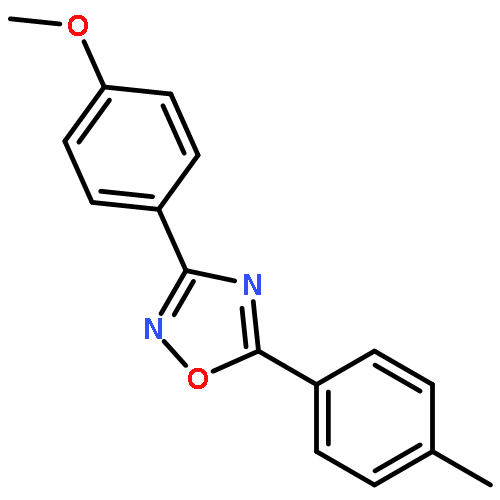
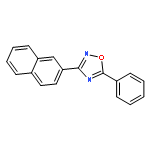
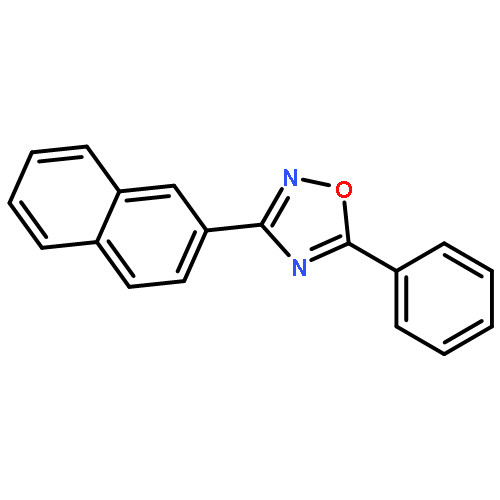
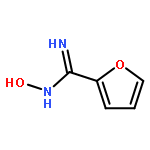
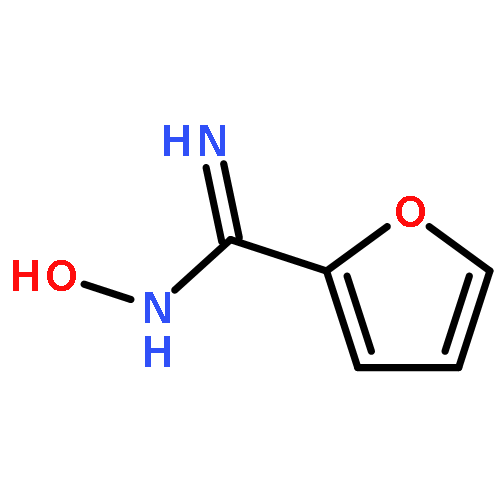
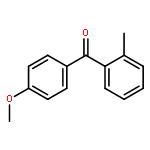
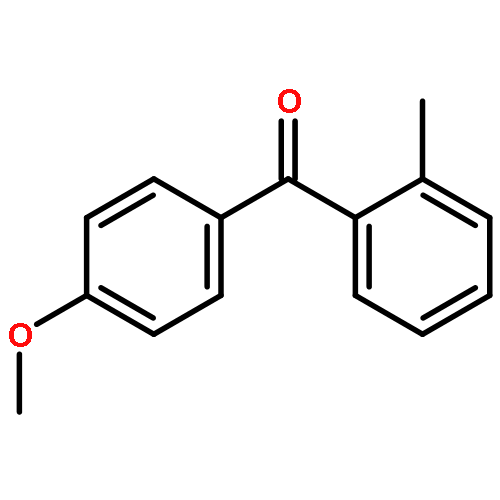
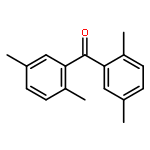
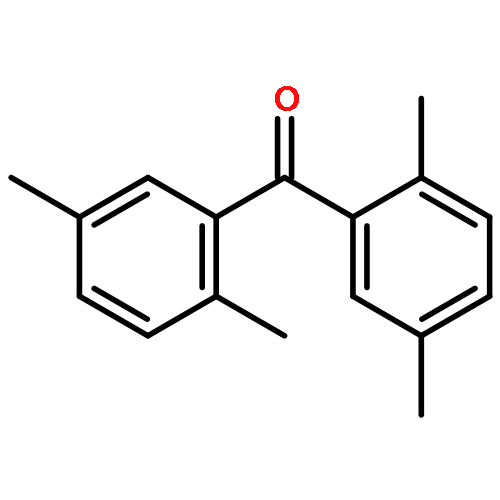
![Benzene,2-[(2,5-dimethylphenyl)methyl]-1,4-dimethyl-](http://img.cochemist.com/ccimg/29100/29050-59-7.png)
![Benzene,2-[(2,5-dimethylphenyl)methyl]-1,4-dimethyl-](http://img.cochemist.com/ccimg/29100/29050-59-7_b.png)
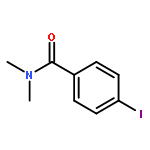

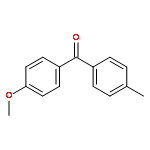
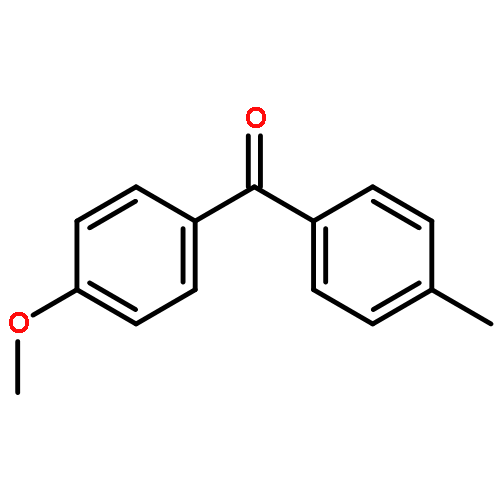
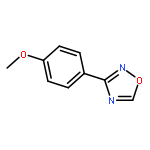
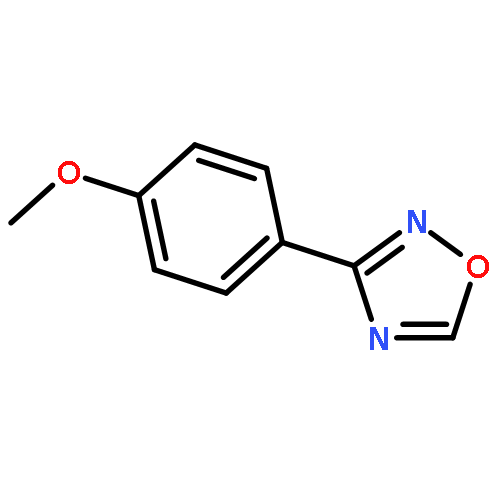
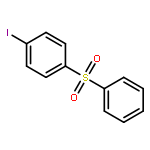
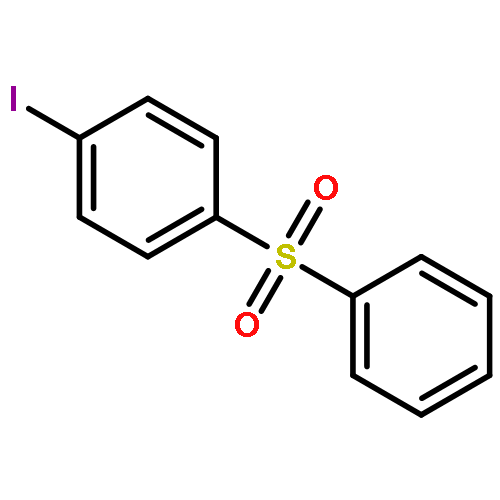


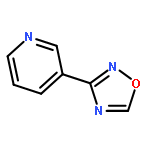
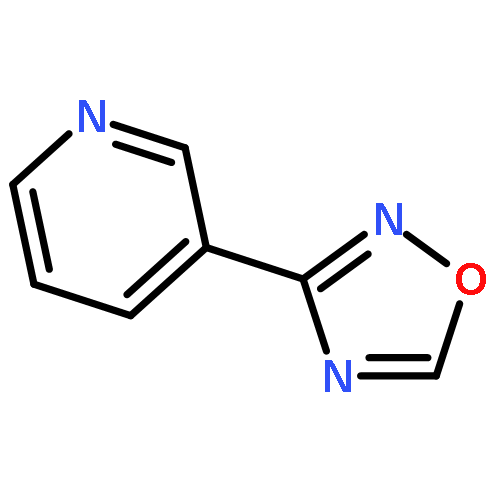
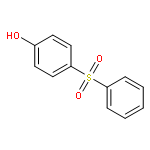
![1-iodo-4-[(4-iodophenyl)disulfanyl]benzene](http://img.cochemist.com/ccimg/6400/6345-64-8.png)
![1-iodo-4-[(4-iodophenyl)disulfanyl]benzene](http://img.cochemist.com/ccimg/6400/6345-64-8_b.png)

![Benzene,[(difluoromethyl)thio]-](http://img.cochemist.com/ccimg/1600/1535-67-7.png)
![Benzene,[(difluoromethyl)thio]-](http://img.cochemist.com/ccimg/1600/1535-67-7_b.png)









![7-QUINAZOLINOL, 4-[(3-CHLORO-4-FLUOROPHENYL)AMINO]-6-METHOXY-](http://img.cochemist.com/ccimg/451500/451494-91-0.png)
![7-QUINAZOLINOL, 4-[(3-CHLORO-4-FLUOROPHENYL)AMINO]-6-METHOXY-](http://img.cochemist.com/ccimg/451500/451494-91-0_b.png)
![6-Quinazolinol, 4-[(3-bromophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/295400/295330-61-9.png)
![6-Quinazolinol, 4-[(3-bromophenyl)amino]-7-methoxy-](http://img.cochemist.com/ccimg/295400/295330-61-9_b.png)
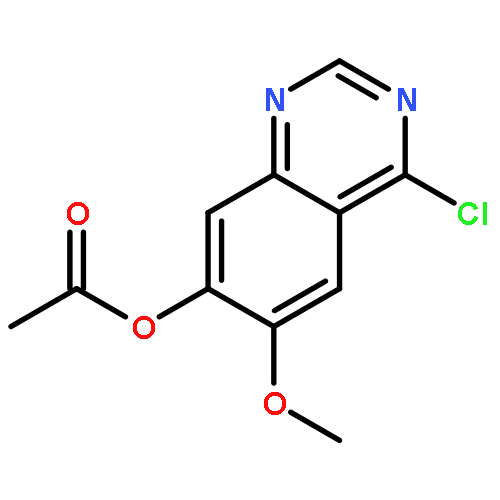
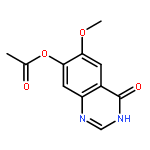
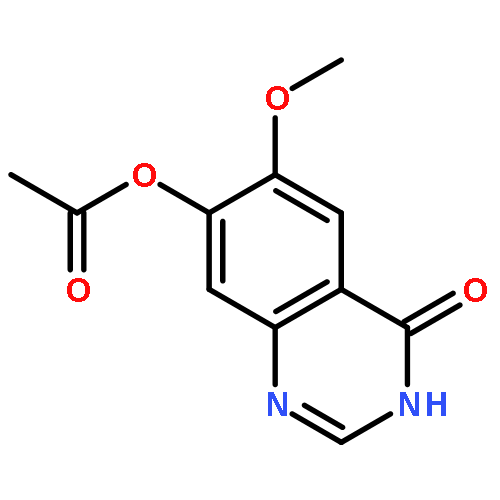








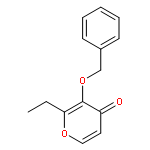


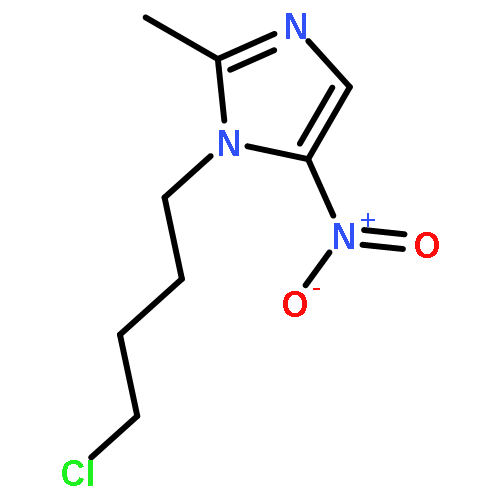
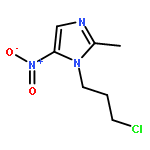
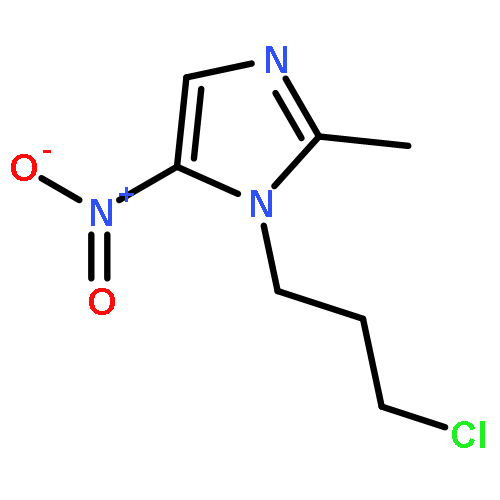
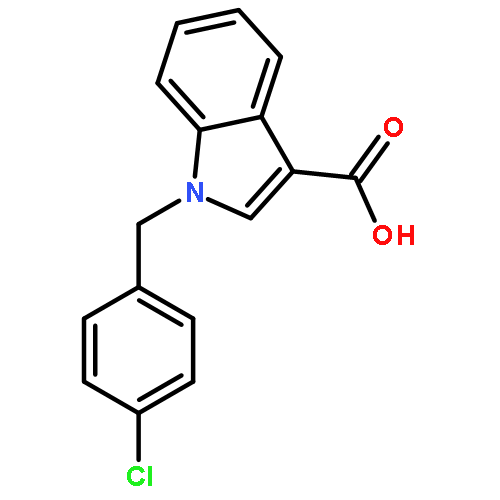
![1H-Indazole-3-carboxylicacid, 1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/50300/50264-86-3.png)
![1H-Indazole-3-carboxylicacid, 1-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/50300/50264-86-3_b.png)
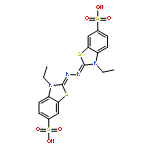
![3-Aminobenzo[e][1,2,4]triazine 1,4-dioxide](http://img.cochemist.com/ccimg/27400/27314-97-2.png)
![3-Aminobenzo[e][1,2,4]triazine 1,4-dioxide](http://img.cochemist.com/ccimg/27400/27314-97-2_b.png)