Co-reporter:Thitaphat Ngernsutivorakul;Cynthia M. Cipolla
Analytical and Bioanalytical Chemistry 2017 Volume 409( Issue 1) pp:275-285
Publication Date(Web):20 October 2016
DOI:10.1007/s00216-016-9999-5
Fiber optics coupled to components such as lenses and mirrors have seen extensive use as probes for Raman and fluorescence measurements. Probes can be placed directly on or into a sample to allow for simplified and remote application of these optical techniques. The size and complexity of such probes however limits their application. We have used microfabrication in polydimethylsiloxane (PDMS) to create compact probes that are 0.5 mm thick by 1 mm wide. The miniature probes incorporate pre-aligned mirrors, lenses, and two fiber optic guides to allow separate input and output optical paths suitable for Raman and fluorescence spectroscopy measurements. The fabricated probe has 70 % unidirectional optical throughput and generates no spectral artifacts in the wavelength range of 200 to 800 nm. The probe is demonstrated for measurement of fluorescence within microfluidic devices and collection of Raman spectra from a pharmaceutical tablet. The fluorescence limit of detection was 6 nM when using the probe to measure resorufin inside a 150-μm inner diameter glass capillary, 100 nM for resorufin in a 60-μm-deep × 100-μm-wide PDMS channel, and 11 nM for fluorescein in a 25-μm-deep × 80-μm-wide glass channel. It is demonstrated that the same probe can be used on different sample types, e.g., microfluidic chips and tablets. Compared to existing Raman and fluorescence probes, the microfabricated probes enable measurement in smaller spaces and have lower fabrication cost.
Co-reporter:Mohamed Dawod;Natalie E. Arvin
Analyst (1876-Present) 2017 vol. 142(Issue 11) pp:1847-1866
Publication Date(Web):2017/05/30
DOI:10.1039/C7AN00198C
This review article describes the significant recent advances in the analysis of proteins by capillary and microchip electrophoresis during the period from mid-2014 to early 2017. This review highlights the progressions, new methodologies, innovative instrumental modifications, and challenges for efficient protein analysis in human specimens, animal tissues, and plant samples. The protein analysis fields covered in this review include analysis of native, reduced, and denatured proteins in addition to Western blotting, protein therapeutics and proteomics.
Co-reporter:Colleen E. Dugan;James P. Grinias
Analytical and Bioanalytical Chemistry 2017 Volume 409( Issue 1) pp:169-178
Publication Date(Web):2017 January
DOI:10.1007/s00216-016-9983-0
Microfluidics is an enabling technology for both cell biology and chemical analysis. We combine these attributes with a microfluidic device for on-line solid-phase extraction (SPE) and mass spectrometry (MS) analysis of secreted metabolites from living cells in culture on the chip. The device was constructed with polydimethylsiloxane (PDMS) and contains a reversibly sealed chamber for perfusing cells. A multilayer design allowed a series of valves to control an on-chip 7.5 μL injection loop downstream of the cell chamber with operation similar to a six-port valve. The valve collects sample and then diverts it to a packed SPE bed that was connected in-line to treat samples prior to MS analysis. The valve allows samples to be collected and injected onto the SPE bed while preventing exposure of cells to added back pressure from the SPE bed and organic solvents needed to elute collected chemicals. Here, cultured murine 3T3-L1 adipocytes were loaded into the cell chamber and non-esterified fatty acids (NEFAs) that were secreted by the cells were monitored by SPE-MS at 30 min intervals. The limit of detection for a palmitoleic acid standard was 1.4 μM. Due to the multiplexed detection capabilities of MS, a variety of NEFAs were detected. Upon stimulation with isoproterenol and forskolin, secretion of select NEFAs was elevated an average of 1.5-fold compared to basal levels. Despite the 30-min delay between sample injections, this device is a step towards a miniaturized system that allows automated monitoring and identification of a variety of molecules in the extracellular environment.
Co-reporter:Paige A. Malec, Marianna Oteri, Veronica Inferrera, Francesco Cacciola, Luigi Mondello, Robert T. Kennedy
Journal of Chromatography A 2017 Volume 1523(Volume 1523) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.chroma.2017.07.061
•Novel application of benzoyl chloride derivatization in wine.•LC–MS/MS assay developed for 56 amines and phenols in wine.•Metabolites in wine compared by varietal and location of origin.•Differences in 29 of 56 metabolites observed in wine.•Location of origin had larger impact on metabolite profile than varietal.Amine and phenolic metabolites are important contributors to the flavor and health effects of many foods, including wine. Determination of these metabolites often involves UV detection following separation by liquid chromatography. While this is sufficient for some applications, chemical derivatization with LC–MS provides greater sensitivity and selectivity relative to LC-UV.We have developed an assay for 56 amine and phenolic metabolites in wine using benzoyl chloride derivatization and LC–MS. Isotopically labeled benzoyl chloride was used to prepare internal standards for each metabolite. Nanomolar limits of detection were achieved for all metabolites. To demonstrate the application of this assay, we compared metabolite profiles from Merlot and Cabernet Sauvignon wines from California and Australia. We found five metabolites which were significantly different when grouped by varietal, while twenty-four were different when grouped by location of production. This shows that the method can identify differences between various wines.
Co-reporter:Woong Hee Lee, Thitaphat Ngernsutivorakul, Omar S. Mabrouk, Jenny-Marie T. Wong, Colleen E. Dugan, Samuel S. Pappas, Hyeun Joong Yoon, and Robert T. Kennedy
Analytical Chemistry 2016 Volume 88(Issue 2) pp:1230
Publication Date(Web):January 4, 2016
DOI:10.1021/acs.analchem.5b03541
Microdialysis sampling is an essential tool for in vivo neurochemical monitoring. Conventional dialysis probes are over 220 μm in diameter and have limited flexibility in design because they are made by assembly using preformed membranes. The probe size constrains spatial resolution and governs the amount of tissue damaged caused by probe insertion. To overcome these limitations, we have developed a method to microfabricate probes in Si that are 45 μm thick × 180 μm wide. The probes contain a buried, U-shaped channel that is 30 μm deep × 60 μm wide and terminates in ports for external connection. A 4 mm length of the probe is covered with a 5 μm thick nanoporous membrane. The membrane was microfabricated by deep reactive ion etching through a porous aluminum oxide layer. The microfabricated probe has cross-sectional area that is 79% less than that of the smallest conventional microdialysis probes. The probes yield 2–20% relative recovery at 100 nL/min perfusion rate for a variety of small molecules. The probe was successfully tested in vivo by sampling from the striatum of live rats. Fractions were collected at 20 min intervals (2 μL) before and after an intraperitoneal injection of 5 mg/kg amphetamine. Analysis of fractions by liquid chromatography–mass spectrometry revealed reliable detection of 14 neurochemicals, including dopamine and acetylcholine, at basal conditions. Amphetamine evoked a 43-fold rise in dopamine, a result nearly identical to a conventional dialysis probe in the same animal. The microfabricated probes have potential for sampling with higher spatial resolution and less tissue disruption than conventional probes. It may also be possible to add functionality to the probes by integrating other components, such as electrodes, optics, and additional channels.
Co-reporter:Shi Jin, Michael D. Furtaw, Huaxian Chen, Don T. Lamb, Stephen A. Ferguson, Natalie E. Arvin, Mohamed Dawod, and Robert T. Kennedy
Analytical Chemistry 2016 Volume 88(Issue 13) pp:6703
Publication Date(Web):June 7, 2016
DOI:10.1021/acs.analchem.6b00705
Western blotting is a commonly used protein assay that combines the selectivity of electrophoretic separation and immunoassay. The technique is limited by long time, manual operation with mediocre reproducibility, and large sample consumption, typically 10–20 μg per assay. Western blots are also usually used to measure only one protein per assay with an additional housekeeping protein for normalization. Measurement of multiple proteins is possible; however, it requires stripping membranes of antibody and then reprobing with a second antibody. Miniaturized alternatives to Western blot based on microfluidic or capillary electrophoresis have been developed that enable higher-throughput, automation, and greater mass sensitivity. In one approach, proteins are separated by electrophoresis on a microchip that is dragged along a polyvinylidene fluoride membrane so that as proteins exit the chip they are captured on the membrane for immunoassay. In this work, we improve this method to allow multiplexed protein detection. Multiple injections made from the same sample can be deposited in separate tracks so that each is probed with a different antibody. To further enhance multiplexing capability, the electrophoresis channel dimensions were optimized for resolution while keeping separation and blotting times to less than 8 min. Using a 15 μm deep × 50 μm wide × 8.6 cm long channel, it is possible to achieve baseline resolution of proteins that differ by 5% in molecular weight, e.g., ERK1 (44 kDa) from ERK2 (42 kDa). This resolution allows similar proteins detected by cross-reactive antibodies in a single track. We demonstrate detection of 11 proteins from 9 injections from a single Jurkat cell lysate sample consisting of 400 ng of total protein using this procedure. Thus, multiplexed Western blots are possible without cumbersome stripping and reprobing steps.
Co-reporter:Claire M. Ouimet, Hao Shao, Jennifer N. Rauch, Mohamed Dawod, Bryce Nordhues, Chad A. Dickey, Jason E. Gestwicki, and Robert T. Kennedy
Analytical Chemistry 2016 Volume 88(Issue 16) pp:8272
Publication Date(Web):July 19, 2016
DOI:10.1021/acs.analchem.6b02126
Capillary electrophoresis (CE) has been identified as a useful platform for detecting, quantifying, and screening for modulators of protein–protein interactions (PPIs). In this method, one protein binding partner is labeled with a fluorophore, the protein binding partners are mixed, and then, the complex is separated from free protein to allow direct determination of bound to free ratios. Although it possesses many advantages for PPI studies, the method is limited by the need to have separation conditions that both prevent protein adsorption to capillary and maintain protein interactions during the separation. In this work, we use protein cross-linking capillary electrophoresis (PXCE) to overcome this limitation. In PXCE, the proteins are cross-linked under binding conditions and then separated. This approach eliminates the need to maintain noncovalent interactions during electrophoresis and facilitates method development. We report PXCE methods for an antibody–antigen interaction and heterodimer and homodimer heat shock protein complexes. Complexes are cross-linked by short treatments with formaldehyde after reaching binding equilibrium. Cross-linked complexes are separated by electrophoretic mobility using free solution CE or by size using sieving electrophoresis of SDS complexes. The method gives good quantitative results; e.g., a lysozyme–antibody interaction was found to have Kd = 24 ± 3 nM by PXCE and Kd = 17 ± 2 nM using isothermal calorimetry (ITC). Heat shock protein 70 (Hsp70) in complex with bcl2 associated athanogene 3 (Bag3) was found to have Kd = 25 ± 5 nM by PXCE which agrees with Kd values reported without cross-linking. Hsp70–Bag3 binding site mutants and small molecule inhibitors of Hsp70–Bag3 were characterized by PXCE with good agreement to inhibitory constants and IC50 values obtained by a bead-based flow cytometry protein interaction assay (FCPIA). PXCE allows rapid method development for quantitative analysis of PPIs.
Co-reporter:Jenny-Marie T Wong, Paige A Malec, Omar S Mabrouk, Jennifer Ro, Monica Dus, Robert T Kennedy
Journal of Chromatography A 2016 Volume 1446() pp:78-90
Publication Date(Web):13 May 2016
DOI:10.1016/j.chroma.2016.04.006
•Improved reaction conditions for benzoyl chloride labeling for HPLC–MS/MS analysis.•Novel assay of 70 neurologically relevant compounds using benzoyl chloride labeling.•Analysis of rat dialysate, fly tissue homogenate and hemolymph, human CSF and serum.•Stable-isotope labeled internal standard for all analytes for quantification.Widely targeted metabolomic assays are useful because they provide quantitative data on large groups of related compounds. We report a high performance liquid chromatography-tandem mass spectrometry (HPLC–MS/MS) method that utilizes benzoyl chloride labeling for 70 neurologically relevant compounds, including catecholamines, indoleamines, amino acids, polyamines, trace amines, antioxidants, energy compounds, and their metabolites. The method includes neurotransmitters and metabolites found in both vertebrates and insects. This method was applied to analyze microdialysate from rats, human cerebrospinal fluid, human serum, fly tissue homogenate, and fly hemolymph, demonstrating its broad versatility for multiple physiological contexts and model systems. Limits of detection for most assayed compounds were below 10 nM, relative standard deviations were below 10%, and carryover was less than 5% for 70 compounds separated in 20 min, with a total analysis time of 33 min. This broadly applicable method provides robust monitoring of multiple analytes, utilizes small sample sizes, and can be applied to diverse matrices. The assay will be of value for evaluating normal physiological changes in metabolism in neurochemical systems. The results demonstrate the utility of benzoyl chloride labeling with HPLC–MS/MS for widely targeted metabolomics assays.
Co-reporter:James P. Grinias, Jenny-Marie T. Wong, Robert T. Kennedy
Journal of Chromatography A 2016 Volume 1461() pp:42-50
Publication Date(Web):26 August 2016
DOI:10.1016/j.chroma.2016.07.043
•Study of thermal effects on gradient UHPLC–MS/MS method in different oven modes.•Temperature increases approach 30 K at viscous friction levels exceeding 25 W/m.•Forced air oven mode reduces magnitude of the axial thermal gradient.•Gradient UHPLC–MS/MS method with derivatization used for neurotransmitter analysis.•Gradient UHPLC–MS/MS method still repeatable with shifting thermal gradients.The impact of viscous friction on eluent temperature and column efficiency in liquid chromatography is of renewed interest as the need for pressures exceeding 1000 bar to use with columns packed with sub-2 μm particles has grown. One way the development of axial and radial temperature gradients that arise due to viscous friction can be affected is by the thermal environment the column is placed in. In this study, a new column oven integrated into an ultrahigh pressure liquid chromatograph that enables both still-air and forced-air operating modes is investigated to find the magnitude of the effect of the axial thermal gradient that forms in 2.1 × 100 mm columns packed with sub-2 μm particles in these modes. Temperature increases of nearly 30 K were observed when the generated power of the column exceeded 25 W/m. The impact of the heating due to viscous friction on the repeatability of peak capacity, elution time, and peak area ratio to an internal standard for a gradient UHPLC-MS/MS method to analyze neurotransmitters was found to be limited. This result indicates that high speed UHPLC-MS/MS gradient methods under conditions of high viscous friction may be possible without the negative effects typically observed with isocratic separations under similar conditions.
Co-reporter:Shuwen Sun, Benjamin C. Buer, E. Neil G. Marsh and Robert T. Kennedy
Analytical Methods 2016 vol. 8(Issue 17) pp:3458-3465
Publication Date(Web):07 Apr 2016
DOI:10.1039/C6AY00698A
Sirtuin 1 (SIRT1) is a NAD+-dependent deacetylase which has been implicated in age-related diseases such as cancer, Alzheimer's disease, type 2 diabetes, and vascular diseases. SIRT1 modulators are of interest for their potential therapeutic use and potential as chemical probes to study the role of SIRT1. Fluorescence-based assays used to identify SIRT1 activators have been shown to have artifacts related to the fluorogenic substrates used in the assays. Such problems highlight the potential utility of a label-free high throughput screening (HTS) strategy. In this work, we describe a label-free SIRT1 assay suitable for HTS based on segmented flow-electrospray ionization-mass spectrometry (ESI-MS). In the assay, 0.5 μM SIRT1 was incubated with 20 μM acetylated 21-amino acid peptide, which acts as substrate for the protein. A stable-isotope labeled product peptide was added to the assay mixture as an internal standard after reaction quenching. The resulting samples are formatted into 100 nL droplets segmented by perfluorodecalin and then infused at 0.8 samples per second into an ESI-MS. To enable direct ESI-MS analysis, 11 μM SIRT1 was dialyzed into a 200 μM ammonium formate (pH 8.0) buffer prior to use in the assay. This buffer was demonstrated to minimally affect enzyme kinetics and yet be compatible with ESI-MS. The assay conditions were optimized through enzyme kinetic study, and tested by screening an 80-compound library. The assay Z-factor was 0.7. Four inhibitors and no activators were detected from the library.
Co-reporter:James P. Grinias; Jason T. Whitfield; Erik D. Guetschow
Journal of Chemical Education 2016 Volume 93(Issue 7) pp:1316-1319
Publication Date(Web):June 22, 2016
DOI:10.1021/acs.jchemed.6b00262
Many research and teaching laboratories rely on USB data acquisition devices to collect voltage signals from instrumentation. However, these devices can be cost-prohibitive (especially when large numbers are needed for teaching laboratories) and require software to be developed for operation. In this article, we describe the development and use of an open-source USB data acquisition device (with 16-bit acquisition resolution) built using simple electronic components and an Arduino Uno that costs under $50. Additionally, open-source software written in Python is included so that data can be acquired using nearly any PC or Mac computer with a simple USB connection. Use of the device was demonstrated for a sophomore-level analytical experiment using gas chromatography and a capillary electrophoresis-UV separation on an instrument used for research purposes.
Co-reporter:James P. Grinias, Robert T. Kennedy
TrAC Trends in Analytical Chemistry 2016 Volume 81() pp:110-117
Publication Date(Web):July–August 2016
DOI:10.1016/j.trac.2015.08.002
•Advances in on-chip microfluidic LC columns are described.•Stationary-phase supports for chip-LC include particles, monoliths, and pillar arrays.•Different substrate materials and selectivities can be used for specific separations.This study focuses on recent advances (from January 2013 through April 2015) in microfluidic liquid chromatography. Articles published during this period are organized by the type of stationary-phase support focusing on device fabrication, column preparation, and specific applications. In addition, a comprehensive table comparing chromatographic figures of merit for this study is provided in Appendix A as a reference for readers.
Co-reporter:Ying Zhou, Jenny-Marie T. Wong, Omar S. Mabrouk, and Robert T. Kennedy
Analytical Chemistry 2015 Volume 87(Issue 19) pp:9802
Publication Date(Web):September 9, 2015
DOI:10.1021/acs.analchem.5b02086
Neuropeptides are an important class of neurochemicals; however, measuring their concentration in vivo by using microdialysis sampling is challenging due to their low concentration and the small samples generated. Capillary liquid chromatography with mass spectrometry (cLC-MS) can yield attomole limits of detection (LOD); however, low recovery and loss of sample to adsorptive surfaces can still hinder detection of neuropeptides. We have evaluated recovery during sampling and transfer to the cLC column for a selection of 10 neuropeptides. Adding acetonitrile to sample eliminated carryover and improved LOD by 1.4- to 60-fold. The amount of acetonitrile required was found to have an optimal value that correlated with peptide molecular weight and retention time on a reversed phase LC column. Treating AN69 dialysis membrane, which bears negative charge due to incorporated sulfonate groups, with polyethylenimine (PEI) improved recovery by 1.2- to 80-fold. The effect appeared to be due to reducing electrostatic interaction between peptides and the microdialysis probe because modification increased recovery only for peptides that carried net positive charge. The combined effects improved LOD of the entire method by 1.3- to 800-fold for the different peptides. We conclude that peptides with both charged and hydrophobic regions require combined strategies to prevent adsorption and yield the best possible detection. The method was demonstrated by determining orexin A, orexin B, and a novel isoform of rat β-endorphin in the arcuate nucleus. Dialysate concentrations were below 10 pM for these peptides. A standard addition study on dialysates revealed that while some peptides can be accurately quantified, some are affected by the matrix.
Co-reporter:Shi Jin, Robert T. Kennedy
Chinese Chemical Letters 2015 Volume 26(Issue 4) pp:416-418
Publication Date(Web):April 2015
DOI:10.1016/j.cclet.2015.01.021
Western blotting is a highly valued method for protein identification and relative quantitation in complex samples. It combines size-based electrophoretic separation with immunoaffinity to identify specific proteins. This technique remains popular and has become a workhorse in biochemical research and clinical laboratories. Despite its utility and popularity, this method has many limitations including slow analysis, incompatibility with limited sample application, low throughput and low information content. Recently there has been significant success in improving different aspects of Western blotting. In this review, we provide an overview of the developments in the area of improving conventional Western blotting methods with a focus on recent developments in microfluidic Western blotting. We overview different separation platforms, and discuss studies on protein transfer methods as well as protein immobilization methods and chemistries. We also describe integrated miniaturized platforms that can perform rapid separations and immunodetections.This photo shows a microchip of the microchip-Western blot apparatus. SDS–protein complexes are separated on chip and captured on a moving membrane as they exit the chip. Direction of flow during separation is indicated by arrows.
Co-reporter:Erik D. Guetschow, Daniel J. Steyer, and Robert T. Kennedy
Analytical Chemistry 2014 Volume 86(Issue 20) pp:10373
Publication Date(Web):September 19, 2014
DOI:10.1021/ac502758h
High-throughput screening (HTS) using multiwell plates and fluorescence plate readers is a powerful tool for drug discovery and evaluation by allowing tens of thousands of assays to be completed in 1 day. Although this method has been successful, electrophoresis-based methods for screening are also of interest to avoid difficulties associated fluorescence assays such as requirements to engineer fluorogenic reactions and false positives. We have developed a method using droplet microfluidics to couple multiwell plate-based assays to microchip electrophoresis (MCE) to screen enzyme modulators. Samples contained in multiwell plates are reformatted in to plugs with a sample volume of 8 nL segmented by an immiscible oil. The segmented flow sample streams are coupled to a hybrid polydimethylsiloxane–glass microfluidic device capable of selectively extracting the aqueous samples from the droplet stream and rapidly analyzing by MCE with laser-induced fluorescence detection. This system was demonstrated by screening a test library of 140 compounds against using protein kinase A. For each sample in the screen, two droplets are generated, allowing approximately 6 MCE injections per sample. Using a 1 s separation at 2000 V/cm, we are able to analyze 96 samples in 12 min. Separation resolution between the internal standard, substrate, and product is 1.2 and average separation efficiency is 16 000 plates/s using real samples. Twenty-five compounds were identified as modulators during primary screening and verified using dose–response curves.
Co-reporter:Shuwen Sun and Robert T. Kennedy
Analytical Chemistry 2014 Volume 86(Issue 18) pp:9309
Publication Date(Web):August 19, 2014
DOI:10.1021/ac502542z
High throughput screening (HTS) is important for identifying molecules with desired properties. Mass spectrometry (MS) is potentially powerful for label-free HTS due to its high sensitivity, speed, and resolution. Segmented flow, where samples are manipulated as droplets separated by an immiscible fluid, is an intriguing format for high throughput MS because it can be used to reliably and precisely manipulate nanoliter volumes and can be directly coupled to electrospray ionization (ESI) MS for rapid analysis. In this study, we describe a “MS Plate Reader” that couples standard multiwell plate HTS workflow to droplet ESI-MS. The MS plate reader can reformat 3072 samples from eight 384-well plates into nanoliter droplets segmented by an immiscible oil at 4.5 samples/s and sequentially analyze them by MS at 2 samples/s. Using the system, a label-free screen for cathepsin B modulators against 1280 chemicals was completed in 45 min with a high Z-factor (>0.72) and no false positives (24 of 24 hits confirmed). The assay revealed 11 structures not previously linked to cathepsin inhibition. For even larger scale screening, reformatting and analysis could be conducted simultaneously, which would enable more than 145 000 samples to be analyzed in 1 day.
Co-reporter:Woong Hee Lee, Thomas R. Slaney, Robert W. Hower, and Robert T. Kennedy
Analytical Chemistry 2013 Volume 85(Issue 8) pp:3828
Publication Date(Web):April 2, 2013
DOI:10.1021/ac400579x
Microfabricated fluidic systems have emerged as a powerful approach for chemical analysis. Relatively unexplored is the use of microfabrication to create sampling probes. We have developed a sampling probe microfabricated in Si by bulk micromachining and lithography. The probe is 70 μm wide by 85 μm thick by 11 mm long and incorporates two buried channels that are 20 μm in diameter. The tip of the probe has two 20 μm holes where fluid is ejected or collected for sampling. Utility of the probe was demonstrated by sampling from the brain of live rats. For sampling, artificial cerebral spinal fluid was infused in through one channel at 50 nL/min while sample was withdrawn at the same flow rate from the other channel. Analysis of resulting fractions collected every 20 min from the striatum of rats by liquid chromatography with mass spectrometry demonstrated reliable detection of 17 neurotransmitters and metabolites. The small probe dimensions suggest it is less perturbing to tissue and can be used to sample smaller brain nuclei than larger sampling devices, such as microdialysis probes. This sampling probe may have other applications such as sampling from cells in culture. The use of microfabrication may also enable incorporation of electrodes for electrochemical or electrophysiological recording and other channels that enable more complex sample preparation on the device.
Co-reporter:Shi Jin, Gwendolyn J. Anderson, and Robert T. Kennedy
Analytical Chemistry 2013 Volume 85(Issue 12) pp:6073
Publication Date(Web):May 15, 2013
DOI:10.1021/ac400940x
Western blotting is a commonly used assay for proteins. Despite the utility of the method, it is also characterized by long analysis times, manual operation, and lack of established miniaturized counterpart. We report a new way to Western blot that addresses these limitations. In the method, sodium dodecyl sulfate (SDS)-protein complexes are separated by sieving electrophoresis in a microfluidic device or chip. The chip is interfaced to a moving membrane so that proteins are captured in discrete zones as they migrate from the chip. Separations of SDS-protein complexes in the molecular weight range of 11–155 kDa were completed in 2 min with 4 × 104 theoretical plates at 460 V/cm. Migration time and peak area relative standard deviations were 3–6% and 0.2%, respectively. Detection limit for actin was 0.7 nM. Assays for actin, AMP-kinase, carbonic anhydrase, and lysozyme are shown to demonstrate versatility of the method. Total analysis time including immunoassay was 22–32 min for a single sample. Because processing membrane for immunoassay is the slow step of the assay, sequential injections from different reservoirs on the chip and capture in different tracks on the same membrane allow increased throughput. As a demonstration, 9 injections were collected on one membrane and analyzed in 43 min (∼5 min/sample). Further improvements in throughput are possible with more reservoirs or parallel channels.
Co-reporter:Jennifer N. Rauch, Jing Nie, Tonia J. Buchholz, Jason E. Gestwicki, and Robert T. Kennedy
Analytical Chemistry 2013 Volume 85(Issue 20) pp:9824
Publication Date(Web):September 2, 2013
DOI:10.1021/ac4023082
Methods for identifying chemical inhibitors of protein–protein interactions (PPIs) are often prone to discovery of false positives, particularly those caused by molecules that induce protein aggregation. Thus, there is interest in developing new platforms that might allow earlier identification of these problematic compounds. Capillary electrophoresis (CE) has been evaluated as a method to screen for PPI inhibitors using the challenging system of Hsp70 interacting with its co-chaperone Bag3. In the method, Hsp70 is labeled with a fluorophore, mixed with Bag3, and the resulting bound and free Hsp70 are separated and detected by CE with laser-induced fluorescence detection. The method used a chemically modified CE capillary to prevent protein adsorption. Inhibitors of the Hsp70–Bag3 interaction were detected by observing a reduction in the bound-to-free ratio. The method was used to screen a library of 3443 compounds, and the results were compared to those from a flow cytometry protein interaction assay. CE was found to produce a lower hit rate with more compounds that were reconfirmed in subsequent testing, suggesting greater specificity. This finding was attributed to the use of electropherograms to detect artifacts such as aggregators and to differences in protein modifications required to perform the different assays. Increases in throughput are required to make the CE method suitable for primary screens, but at the current stage of development it is attractive as a secondary screen to test hits found by higher-throughput methods.
Co-reporter:Thomas R. Slaney, Omar S. Mabrouk, Kirsten A. Porter-Stransky, Brandon J. Aragona, and Robert T. Kennedy
ACS Chemical Neuroscience 2013 Volume 4(Issue 2) pp:321
Publication Date(Web):November 12, 2012
DOI:10.1021/cn300158p
Although populations of neurons are known to vary on the micrometer scale, little is known about whether basal concentrations of neurotransmitters also vary on this scale. We used low-flow push–pull perfusion to test if such chemical gradients exist between several small brain nuclei. A miniaturized polyimide-encased push–pull probe was developed and used to measure basal neurotransmitter spatial gradients within brain of live animals with 0.004 mm3 resolution. We simultaneously measured dopamine (DA), norepinephrine, serotonin (5-HT), glutamate, γ-aminobutyric acid (GABA), aspartate (Asp), glycine (Gly), acetylcholine (ACh), and several neurotransmitter metabolites. Significant differences in basal concentrations between midbrain regions as little as 200 μm apart were observed. For example, dopamine in the ventral tegmental area (VTA) was 4.8 ± 1.5 nM but in the red nucleus was 0.5 ± 0.2 nM. Regions of high glutamate concentration and variability were found within the VTA of some individuals, suggesting hot spots of glutamatergic activity. Measurements were also made within the nucleus accumbens core and shell. Differences were not observed in dopamine and 5-HT in the core and shell; but their metabolites homovanillic acid (460 ± 60 nM and 130 ± 60 nM respectively) and 5-hydroxyindoleacetic acid (720 ± 200 nM and 220 ± 50 nM respectively) did differ significantly, suggesting differences in dopamine and 5-HT activity in these brain regions. Maintenance of these gradients depends upon a variety of mechanisms. Such gradients likely underlie highly localized effects of drugs and control of behavior that have been found using other techniques.Keywords: Dopamine; glutamate; in vivo; microdialysis; push−pull perfusion; spatial resolution
Co-reporter:Neil D. Hershey and Robert T. Kennedy
ACS Chemical Neuroscience 2013 Volume 4(Issue 5) pp:729
Publication Date(Web):January 12, 2013
DOI:10.1021/cn300199m
In vivo calibration of microdialysis probes is required for interpreting measured concentrations. The most popular method of in vivo calibration is no-net-flux (NNF), which requires infusing several concentrations of neurotransmitters to determine in vivo recoveries (extraction fraction or Ed) and extracellular concentrations. A new method for in vivo calibration of microdialysis of neurotransmitters using glutamate (GLU) and dopamine (DA) as model analytes is reported. 13C6-DA and 13C5-GLU were perfused through microdialysis probes as internal calibrators. Using liquid chromatography with mass spectrometry, it was possible to distinguish the 13C-forms from the endogenous forms of each neurotransmitter. Ed was directly calculated by measuring the loss of the 13C-forms during infusion. The measured endogenous 12C forms of the neurotransmitters could be corrected for Ed to give calibrated extracellular concentrations in vivo. Retrodialysis of stable-isotope-labeled (SIL) neurotransmitters gave Ed and extracellular concentrations of 13C5-GLU and 13C6-DA that matched no-net-flux measurements; however, the values were obtained in a fraction of time because no added measurements were required to obtain the calibration. Ed was reduced during uptake inhibition for GLU and DA when measured by SIL retrodialysis. Because Ed is directly measured at each microdialysis fraction, it was possible to monitor changes in Ed under transient conditions created by systemic injection of uptake inhibitors. The results show that DA and GLU concentrations are underestimated by as much as 50% if not corrected for Ed during uptake inhibition. SIL retrodialysis provides equivalent information to NNF at much reduced time and animal use.Keywords: cocaine; dopamine; glutamate; No-net-flux; nucleus accumbens; retrodialysis
Co-reporter:Jing Nie
Journal of Separation Science 2013 Volume 36( Issue 21-22) pp:3471-3477
Publication Date(Web):
DOI:10.1002/jssc.201300725
A challenge for capillary LC (cLC) is fraction collection and the manipulation of fractions from microscale columns. An emerging approach is the use of segmented flow or droplet technology to perform such tasks. In this work, a fraction collection and postcolumn reaction system based on segmented flow was developed for the gradient cLC of proteins. In the system, column effluent and immiscible oil are pumped into separate arms of a tee resulting in regular fractions of effluent segmented by oil. Fractions were generated at 1 Hz corresponding to 5 nL volumes. The fraction collection rate was high enough to generate over 30 fractions per peak and preserve chromatographic resolution achieved for a five-protein test mixture. The resulting fractions could be stored and subsequently derivatized for fluorescence detection by pumping them into a second tee where naphthalene dicarboxyaldehyde, a fluorogenic reagent, was pumped into a second arm and added to each fraction. Proteins were derivatized within the droplets enabling postcolumn fluorescence detection of the proteins. The experiments demonstrate that fraction collection from cLC by segmented flow can be extended to proteins. Further, they illustrate a potential workflow for protein analysis based on postcolumn derivatization for fluorescence detection.
Co-reporter:Ying Zhou;Omar S. Mabrouk
Journal of The American Society for Mass Spectrometry 2013 Volume 24( Issue 11) pp:1700-1709
Publication Date(Web):2013 November
DOI:10.1007/s13361-013-0605-1
Measurement of neuropeptides in the brain through in vivo microdialysis sampling provides direct correlation between neuropeptide concentration and brain function. Capillary liquid chromatography-multistage mass spectrometry (CLC-MSn) has proven to be effective at measuring endogenous neuropeptides in microdialysis samples. In the method, microliter samples are concentrated onto nanoliter volume packed beds before ionization and mass spectrometry analysis. The long times required for extensive preconcentration present a barrier to routine use because of the many samples that must be analyzed and instability of neuropeptides. In this study, we evaluated the capacity of 75 μm inner diameter (i.d.) capillary column packed with 10 μm reversed phase particles for increasing the throughput in CLC-MSn based neuropeptide measurement. Coupling a high injection flow rate for fast sample loading/desalting with a low elution flow rate to maintain detection sensitivity, this column has reduced analysis time from ∼30 min to 3.8 min for 5 μL sample, with 3 pM limit of detection (LOD) for enkephalins and 10 pM LOD for dynorphin A1-8 in 5 μL sample. The use of isotope-labeled internal standard lowered peptide signal variation to less than 5 %. This method was validated for in vivo detection of Leu and Met enkephalin with microdialysate collected from rat globus pallidus. The improvement in speed and stability makes CLC-MSn measurement of neuropeptides in vivo more practical.
Co-reporter:Peng Song, Omar S. Mabrouk, Neil D. Hershey, and Robert T. Kennedy
Analytical Chemistry 2012 Volume 84(Issue 1) pp:412
Publication Date(Web):November 27, 2011
DOI:10.1021/ac202794q
In vivo neurochemical monitoring using microdialysis sampling is important in neuroscience because it allows correlation of neurotransmission with behavior, disease state, and drug concentrations in the intact brain. A significant limitation of current practice is that different assays are utilized for measuring each class of neurotransmitter. We present a high performance liquid chromatography (HPLC)–tandem mass spectrometry method that utilizes benzoyl chloride for determination of the most common low molecular weight neurotransmitters and metabolites. In this method, 17 analytes were separated in 8 min. The limit of detection was 0.03–0.2 nM for monoamine neurotransmitters, 0.05–11 nM for monoamine metabolites, 2–250 nM for amino acids, 0.5 nM for acetylcholine, 2 nM for histamine, and 25 nM for adenosine at sample volume of 5 μL. Relative standard deviation for repeated analysis at concentrations expected in vivo averaged 7% (n = 3). Commercially available 13C benzoyl chloride was used to generate isotope-labeled internal standards for improved quantification. To demonstrate utility of the method for study of small brain regions, the GABAA receptor antagonist bicuculline (50 μM) was infused into a rat ventral tegmental area while recording neurotransmitter concentration locally and in nucleus accumbens, revealing complex GABAergic control over mesolimbic processes. To demonstrate high temporal resolution monitoring, samples were collected every 60 s while neostigmine, an acetylcholine esterase inhibitor, was infused into the medial prefrontal cortex. This experiment revealed selective positive control of acetylcholine over cortical glutamate.
Co-reporter:Peng Song, Neil D. Hershey, Omar S. Mabrouk, Thomas R. Slaney, and Robert T. Kennedy
Analytical Chemistry 2012 Volume 84(Issue 11) pp:4659
Publication Date(Web):May 22, 2012
DOI:10.1021/ac301203m
Developing sensors for in vivo chemical monitoring is a daunting challenge. An alternative approach is to couple sampling methods with online analytical techniques; however, such approaches are generally hampered by lower temporal resolution and slow analysis. In this work, microdialysis sampling was coupled with segmented flow electrospray ionization mass spectrometry (ESI-MS) to perform in vivo chemical monitoring. The use of segmented flow to prevent Taylor dispersion of collected zones and rapid analysis with direct ESI-MS allowed 5 s temporal resolution to be achieved. The MS “sensor” was applied to monitor acetylcholine in the brain of live rats. The detection limit of 5 nM was sufficient to monitor basal acetylcholine as well as dynamic changes elicited by microinjection of neostigmine, an inhibitor of acetycholinesterase, that evoked rapid increases in acetycholine and tetrodotoxin, a blocker of Na+ channels, that lowered the acetylcholine concentration. The versatility of the sensor was demonstrated by simultaneously monitoring metabolites and infused drugs.
Co-reporter:Shuwen Sun, Thomas R. Slaney, and Robert T. Kennedy
Analytical Chemistry 2012 Volume 84(Issue 13) pp:5794
Publication Date(Web):May 31, 2012
DOI:10.1021/ac3011389
Droplet-based microfluidics is an attractive platform for screening and optimizing chemical reactions. Using this approach, it is possible to reliably manipulate nanoliter volume samples and perform operations such as reagent addition with high precision, automation, and throughput. Most studies using droplet microfluidics have relied on optical techniques to detect the reaction; however, this requires engineering color or fluorescence change into the reaction being studied. In this work, we couple electrospray ionization mass spectrometry (ESI-MS) to nanoliter scale segmented flow reactions to enable direct (label-free) analysis of reaction products. The system is applied to a screen of inhibitors for cathepsin B. In this approach, solutions of test compounds (including three known inhibitors) are arranged as an array of nanoliter droplets in a tube segmented by perfluorodecalin. The samples are pumped through a series of tees to add enzyme, substrate (peptides), and quenchant. The resulting reaction mixtures are then infused into a metal-coated, fused silica ESI emitter for MS analysis. The system has potential for high-throughput as reagent addition steps are performed at 0.7 s per sample and ESI-MS at up to 1.2 s per sample. Carryover is inconsequential in the ESI emitter and between 2 and 9% per reagent addition depending on the tee utilized. The assay was reliable with a Z-factor of ∼0.8. The method required 0.8 pmol of test compound, 1.6 pmol of substrate, and 5 fmol of enzyme per reaction. Segmented flow ESI-MS allows direct, label free screening of reactions at good throughput and ultralow sample consumption.
Co-reporter:Jonathan G. Shackman;Kendra R. Reid
Analytical and Bioanalytical Chemistry 2012 Volume 402( Issue 9) pp:2797-2803
Publication Date(Web):2012 March
DOI:10.1007/s00216-012-5755-7
A rapid microfluidic based capillary electrophoresis immunoassay (CEIA) was developed for on-line monitoring of glucagon secretion from pancreatic islets of Langerhans. In the device, a cell chamber containing living islets was perfused with buffers containing either high or low glucose concentration. Perfusate was continuously sampled by electroosmosis through a separate channel on the chip. The perfusate was mixed on-line with fluorescein isothiocyanate-labeled glucagon (FITC-glucagon) and monoclonal anti-glucagon antibody. To minimize sample dilution, the on-chip mixing ratio of sampled perfusate to reagents was maximized by allowing reagents to only be added by diffusion. Every 6 s, the reaction mixture was injected onto a 1.5-cm separation channel where free FITC-glucagon and the FITC-glucagon–antibody complex were separated under an electric field of 700 V cm−1. The immunoassay had a detection limit of 1 nM. Groups of islets were quantitatively monitored for changes in glucagon secretion as the glucose concentration was decreased from 15 to 1 mM in the perfusate revealing a pulse of glucagon secretion during a step change. The highly automated system should be enable studies of the regulation of glucagon and its potential role in diabetes and obesity. The method also further demonstrates the potential of rapid CEIA on microfluidic systems for monitoring cellular function.
Co-reporter:Amy L. Payeur, Matthew A. Lorenz, Robert T. Kennedy
Journal of Chromatography B 2012 Volumes 893–894() pp:187-192
Publication Date(Web):15 April 2012
DOI:10.1016/j.jchromb.2012.03.003
A comprehensive two-dimensional gas chromatography (GC × GC) time-of-flight mass spectrometry method was developed for determination of fatty acids (irrespective of origin, i.e., both free fatty acids and fatty acids bound in sources such as triglycerides) in cultured mammalian cells. The method was applied to INS-1 cells, an insulin-secreting cell line commonly used as a model in diabetes studies. In the method, lipids were extracted and transformed to fatty acid methyl esters for analysis. GC × GC analysis revealed the presence of 30 identifiable fatty acids in the extract. This result doubles the number of fatty acids previously identified in these cells. The method yielded linear calibrations and an average relative standard deviation of 8.4% for replicate injections of samples and 12.4% for replicate analysis of different samples. The method was used to demonstrate changes in fatty acid content as a function of glucose concentration on the cells. These results demonstrate the utility of this method for analysis of fatty acids in mammalian cell cultures.Highlights► A GC × GC method for assessing the fatty acid content of a cell culture line was developed. ► The method allowed identification of 15 new fatty acids present in insulin secreting cells, double the number previously known. ► Glucose effects on fatty acid content were determined and revealed decreases at high concentration for many fatty acids that may be related to secretion of fatty acids.
Co-reporter:Gwendolyn J. Anderson, Cynthia M. Cipolla, and Robert T. Kennedy
Analytical Chemistry 2011 Volume 83(Issue 4) pp:1350
Publication Date(Web):January 25, 2011
DOI:10.1021/ac102671n
A microscale Western blotting system based on separating sodium-dodecyl sulfate protein complexes by capillary gel electrophoresis followed by deposition onto a blotting membrane for immunoassay is described. In the system, the separation capillary is grounded through a sheath capillary to a mobile X−Y translation stage which moves a blotting membrane past the capillary outlet for protein deposition. The blotting membrane is moistened with a methanol and buffer mixture to facilitate protein adsorption. Although discrete protein zones could be detected, bands were broadened by ∼1.7-fold by transfer to membrane. A complete Western blot for lysozyme was completed in about one hour with 50 pg mass detection limit from low microgram per milliliter samples. These results demonstrate substantial reduction in time requirements and improvement in mass sensitivity compared to conventional Western blots. Western blotting using capillary electrophoresis shows promise to analyze low volume samples with reduced reagents and time, while retaining the information content of a typical Western blot.
Co-reporter:Matthew A. Lorenz, Charles F. Burant, and Robert T. Kennedy
Analytical Chemistry 2011 Volume 83(Issue 9) pp:3406
Publication Date(Web):April 1, 2011
DOI:10.1021/ac103313x
A simple, fast, and reproducible sample preparation procedure was developed for relative quantification of metabolites in adherent mammalian cells using the clonal β-cell line INS-1 as a model sample. The method was developed by evaluating the effect of different sample preparation procedures on high performance liquid chromatography- mass spectrometry quantification of 27 metabolites involved in glycolysis and the tricarboxylic acid cycle on a directed basis as well as for all detectable chromatographic features on an undirected basis. We demonstrate that a rapid water rinse step prior to quenching of metabolism reduces components that suppress electrospray ionization thereby increasing signal for 26 of 27 targeted metabolites and increasing total number of detected features from 237 to 452 with no detectable change of metabolite content. A novel quenching technique is employed which involves addition of liquid nitrogen directly to the culture dish and allows for samples to be stored at −80 °C for at least 7 d before extraction. Separation of quenching and extraction steps provides the benefit of increased experimental convenience and sample stability while maintaining metabolite content similar to techniques that employ simultaneous quenching and extraction with cold organic solvent. The extraction solvent 9:1 methanol: chloroform was found to provide superior performance over acetonitrile, ethanol, and methanol with respect to metabolite recovery and extract stability. Maximal recovery was achieved using a single rapid (∼1 min) extraction step. The utility of this rapid preparation method (∼5 min) was demonstrated through precise metabolite measurements (11% average relative standard deviation without internal standards) associated with step changes in glucose concentration that evoke insulin secretion in the clonal β-cell line INS-1.
Co-reporter:Thomas R. Slaney, Jing Nie, Neil D. Hershey, Prasanna K. Thwar, Jennifer Linderman, Mark A. Burns, and Robert T. Kennedy
Analytical Chemistry 2011 Volume 83(Issue 13) pp:5207
Publication Date(Web):May 23, 2011
DOI:10.1021/ac2003938
Low-flow push–pull perfusion is a sampling method that yields better spatial resolution than competitive methods like microdialysis. Because of the low flow rates used (50 nL/min), it is challenging to use this technique at high temporal resolution which requires methods of collecting, manipulating, and analyzing nanoliter samples. High temporal resolution also requires control of Taylor dispersion during sampling. To meet these challenges, push–pull perfusion was coupled with segmented flow to achieve in vivo sampling at 7 s temporal resolution at 50 nL/min flow rates. By further miniaturizing the probe inlet, sampling with 200 ms resolution at 30 nL/min (pull only) was demonstrated in vitro. Using this method, l-glutamate was monitored in the striatum of anesthetized rats. Up to 500 samples of 6 nL each were collected at 7 s intervals, segmented by an immiscible oil and stored in a capillary tube. The samples were assayed offline for l-glutamate at a rate of 15 samples/min by pumping them into a reagent addition tee fabricated from Teflon where reagents were added for a fluorescent enzyme assay. Fluorescence of the resulting plugs was monitored downstream. Microinjection of 70 mM potassium in physiological buffered saline evoked l-glutamate concentration transients that had an average maxima of 4.5 ± 1.1 μM (n = 6 animals, 3–4 injections each) and rise times of 22 ± 2 s. These results demonstrate that low-flow push–pull perfusion with segmented flow can be used for high temporal resolution chemical monitoring and in complex biological environments.
Co-reporter:Omar S. Mabrouk;Qiang Li;Peng Song
Journal of Neurochemistry 2011 Volume 118( Issue 1) pp:24-33
Publication Date(Web):
DOI:10.1111/j.1471-4159.2011.07293.x
J. Neurochem. (2011) 10.1111/j.1471-4159.2011.07293.x
Abstract
Pallidal dopamine, GABA and the endogenous opioid peptides enkephalins have independently been shown to be important controllers of sensorimotor processes. Using in vivo microdialysis coupled to liquid chromatography–mass spectrometry and a behavioral assay, we explored the interaction between these three neurotransmitters in the rat globus pallidus. Amphetamine (3 mg/kg i.p.) evoked an increase in dopamine, GABA and methionine/leucine enkephalin. Local perfusion of the dopamine D1 receptor antagonist SCH 23390 (100 μM) fully prevented amphetamine stimulated enkephalin and GABA release in the globus pallidus and greatly suppressed hyperlocomotion. In contrast, the dopamine D2 receptor antagonist raclopride (100 μM) had only minimal effects suggesting a greater role for pallidal D1 over D2 receptors in the regulation of movement. Under basal conditions, opioid receptor blockade by naloxone perfusion (10 μM) in the globus pallidus stimulated GABA and inhibited dopamine release. Amphetamine-stimulated dopamine release and locomotor activation were attenuated by naloxone perfusion with no effect on GABA. These findings demonstrate a functional relationship between pallidal dopamine, GABA and enkephalin systems in the control of locomotor behavior under basal and stimulated conditions. Moreover, these findings demonstrate the usefulness of liquid chromatography–mass spectrometry as an analytical tool when coupled to in vivo microdialysis.
Co-reporter:Qiang Li, Jian Pei, Peng Song and Robert T. Kennedy
Analytical Chemistry 2010 Volume 82(Issue 12) pp:5260
Publication Date(Web):May 21, 2010
DOI:10.1021/ac100669z
Off-line analysis and characterization of samples separated by capillary liquid chromatography (LC) has been problematic using conventional approaches to fraction collection. We demonstrate collection of nanoliter fractions by forming plugs of effluent from a 75 μm inner diameter LC column segmented by an immiscible oil such as perfluorodecalin. The plugs are stored in tubing that can then be used to manipulate the samples. Off-line electrospray ionization mass spectrometry (ESI-MS) was used to characterize the samples. ESI-MS was performed by directly pumping the segmented plugs into a nanospray emitter tip. Critical parameters including the choice of oils, ESI voltage, and flow rates that allow successful direct infusion analysis were investigated. Best signals were obtained under conditions in which the oil did not form an electrospray but was siphoned away from the tip. Off-line analysis showed preservation of the chromatogram with no loss of resolution. The method was demonstrated to allow changes in flow rate during the analysis. Specifically, decreases in flow rate were used to allow extended MS analysis time on selected fractions, similar to “peak parking”.
Co-reporter:Jing Nie and Robert T. Kennedy
Analytical Chemistry 2010 Volume 82(Issue 18) pp:7852
Publication Date(Web):August 25, 2010
DOI:10.1021/ac101723x
A microfluidic device is developed to sample from oil-segmented fluid plugs of nanoliter volume by passive splitting. In the device, plugs are pumped into a loop where they split according to the flow resistance of each arm of the loop. The loop structure prevents changes in flow resistance over time, caused by accumulation of plugs in downstream collector capillaries, from interfering with the split ratio. To prevent plugs from recombining at the downstream junction of the loop, a series of posts is fabricated into the junction. This structure allows oil to cross, enabling pressure equalization, but not the aqueous plugs allowing them to be collected into separate capillaries or channels. The split ratio depended on both dimensions of the loop channels and frequency of plugs entering the loop. Split ratios from 1:1 to 34:1 were achieved for samples from 1.7 to 3.3 nL. Long-term stability was demonstrated by splitting over 7000 plugs with 6.3% relative standard deviation of the daughter plug size. The system will have application to high-throughput chemical analysis on a nanoliter scale by enabling several applications including performing multiple assays on a single sample, preserving sample while some is removed for analysis, and splitting reagents or test compounds for use in multiple assays.
Co-reporter:Jian Pei, Jing Nie, and Robert T. Kennedy
Analytical Chemistry 2010 Volume 82(Issue 22) pp:9261
Publication Date(Web):October 15, 2010
DOI:10.1021/ac101755y
Capillary electrophoresis (CE) on microfabricated structures has achieved impressive sample throughput by combining fast separation speed and parallel operations. One obstacle to further increasing throughput has been lack of methods for loading and injecting individual samples at a rate that matches analysis speed. To address this issue, we have developed a microfluidic device in which samples stored as nanoliter volume plugs segmented by a fluorocarbon oil are introduced sequentially to an array of three electrophoresis channels. A microfluidic interface consisting of patterned surface chemistry and geometric restriction was used to extract samples from each segmented flow channel and transfer to the respective electrophoresis channel for separation. Fluorescence detection was achieved by imaging the chip using a fluorescence microscope equipped with a charge-coupled device. Characterization of the system shows that injection volume is controlled by sample plug volume, flow rate during introduction, and voltage applied to the electrophoresis channel. The system was tested for a GTPase assay. Peak area ratios of enzyme product and internal standard had 6% relative standard deviations. Cross-contamination between peaks was 7%. Throughput of 120 samples in 10 min was achieved. Further development of the system may allow application to high-throughput applications such as drug screening.
Co-reporter:Claire N. Chisolm, Charles R. Evans, Colin Jennings, Will A. Black, Frederick J. Antosz, Yangqiao Qiang, Angel R. Diaz, Robert T. Kennedy
Journal of Chromatography A 2010 Volume 1217(Issue 48) pp:7471-7477
Publication Date(Web):26 November 2010
DOI:10.1016/j.chroma.2010.09.066
A push–pull sampling system interfaced on-line to high-performance liquid chromatography (HPLC) was developed for micro-volume real-time monitoring of reaction mixtures. The device consists of concentric tubes wherein sample was continuously withdrawn through the outer tube and reaction quenchant continuously delivered through a recessed inner tube. The device allowed sampling rates of 0.1–6.0 μL/min from a reaction vessel and stopped the reaction by passive mixing with quenchant to preserve the conditions observed in the reaction vessel. A finite element model of the system showed that reaction mixtures could be completely mixed with quenchant within 4.3 s at a flow rate of 1.0 μL/min. The model also showed that an offset distance of 1 mm between the push capillary and sample capillary tips is sufficient to avoid leakage of quenchant/diluent into the bulk sample for push flow rates up to 95% of the pull flow rate. The maximum relative push flow rate was determined to be 90% of the pull flow rate experimentally. Delay between sampling and delivery to the HPLC was from 111 ± 3 s to 317 ± 9 s for pull flow rates from 1.0 to 3.0 μL/min in agreement with expected delays based on tubing volume. Response times were from 27 ± 1 s to 52 ± 6 s over the same flow rate range. The sampler was tested to determine the effects of sample viscosity. The sampler was also used to demonstrate periodic sampling capabilities. As a test of the system, it was used to monitor the base-catalyzed hydrolysis of aspirin for 1.5 h, demonstrating its utility for monitoring an ongoing reaction.
Co-reporter:Maura L. Perry;Gina M. Leinninger;Rong Chen;Kathryn D. Luderman;Hongyan Yang;Margaret E. Gnegy;Martin G. Myers Jr
Journal of Neurochemistry 2010 Volume 114( Issue 3) pp:666-674
Publication Date(Web):
DOI:10.1111/j.1471-4159.2010.06757.x
J. Neurochem. (2010) 114, 666–674.
Abstract
Adipocytes produce the hormone, leptin, in proportion to fat mass to signal the status of body energy stores to the central nervous system, thereby modulating food intake and energy homeostasis. In addition to controlling satiety, leptin suppresses the reward value of food, which is controlled by the mesolimbic dopamine (DA) system. Previous results from leptin-deficient ob/ob animals suggest that chronic leptin deficiency decreases DA content in the mesolimbic DA system, thereby decreasing the response to amphetamine (AMPH). The extent to which these alterations in the mesolimbic DA system of ob/ob animals may mirror the leptin response of normal animals has remained unclear, however. We therefore examined the potential short-term modulation of the mesolimbic DA system by leptin in normal animals. We show that 4 h of systemic leptin treatment enhances AMPH-stimulated DA efflux in the nucleus accumbens (NAc) of Sprague-Dawley rats. While acute leptin treatment increased NAc tyrosine hydroxylase activity, total tyrosine hydroxylase and DA content were unchanged at this early time point. Leptin also increased NAc DA transporter activity in the absence of changes in cell surface or total DA transporter. Thus, leptin modulates the mesolimbic DA system via multiple acute mechanisms, and increases AMPH-mediated DA efflux in normal animals.
Co-reporter:Charles Evans;Katrina L Bogan;Peng Song;Charles F Burant
BMC Chemical Biology 2010 Volume 10( Issue 1) pp:
Publication Date(Web):2010 December
DOI:10.1186/1472-6769-10-2
NAD+ is a coenzyme for hydride transfer enzymes and a substrate for sirtuins and other NAD+-dependent ADPribose transfer enzymes. In wild-type Saccharomyces cerevisiae, calorie restriction accomplished by glucose limitation extends replicative lifespan in a manner that depends on Sir2 and the NAD+ salvage enzymes, nicotinic acid phosphoribosyl transferase and nicotinamidase. Though alterations in the NAD+ to nicotinamide ratio and the NAD+ to NADH ratio are anticipated by models to account for the effects of calorie restriction, the nature of a putative change in NAD+ metabolism requires analytical definition and quantification of the key metabolites.Hydrophilic interaction chromatography followed by tandem electrospray mass spectrometry were used to identify the 12 compounds that constitute the core NAD+ metabolome and 6 related nucleosides and nucleotides. Whereas yeast extract and nicotinic acid increase net NAD+ synthesis in a manner that can account for extended lifespan, glucose restriction does not alter NAD+ or nicotinamide levels in ways that would increase Sir2 activity.The results constrain the possible mechanisms by which calorie restriction may regulate Sir2 and suggest that provision of vitamins and calorie restriction extend lifespan by different mechanisms.
Co-reporter:Jian Pei;Qiang Li
Journal of The American Society for Mass Spectrometry 2010 Volume 21( Issue 7) pp:1107-1113
Publication Date(Web):2010 July
DOI:10.1016/j.jasms.2010.02.013
Electrospray ionization mass spectrometry (ESI-MS) is an attractive analytical tool for high-throughput screening because of its rapid scan time and ability to detect compounds without need for labels. Impediments to the use of ESI-MS for screening have been the relatively large sample consumed and slow sample introduction rates associated with commonly used flow injection analysis. We have previously shown that by segmenting nanoliter plugs of sample with air, an array of discrete samples can be delivered to a platinum-coated emitter tip for ESI-MS analysis with throughput as high as 0.8 Hz and carry-over between samples less than 0.1%. This method was applied to screening for inhibitors of acetylcholinesterase as a demonstration of the potential of segmented flow ESI-MS for such applications. Each enzyme assay consumed 10 nL of sample. At 1 μL/min infusion rate, 102 samples were analyzed, corresponding to a 0.65 Hz sample analysis rate. Linear quantification of choline was achieved from 200 μM to 10 mM using this method and Z′ values were over 0.8 for the assay. Detailed pharmacologic dose-response curves of selected inhibitors were also measured in high-throughput fashion to validate the method.
Co-reporter:Anna M. Clark, Kyle M. Sousa, Colin Jennings, Ormond A. MacDougald and Robert T. Kennedy
Analytical Chemistry 2009 Volume 81(Issue 6) pp:2350
Publication Date(Web):February 20, 2009
DOI:10.1021/ac8026965
A dual-chip microfluidic platform that coupled perfusion of cultured adipocytes with on-line fluorescence-based enzyme assay was developed to monitor glycerol secretions in real time from cultured adipocytes. The perfusion cell chip, which could be reversibly sealed to allow reloading of cells and reuse of the chip, was shown by modeling to generate low shear stress on the cells under study. Effluent from the perfusion chip was pumped into an enzyme assay chip for monitoring of secretion from the cells. The on-line enzyme assay had a limit of detection (LOD) of 4 μM glycerol. The temporal resolution of the combined system for detecting changes in glycerol concentration was 90 s. The microfluidic device was used to continuously monitor glycerol secretion from murine 3T3-L1 adipocytes, grown and differentiated on glass coverslips, for at least 2 h. An average basal glycerol concentration of 28 μM was detected in the effluent. Pharmacological treatment with a β-adrenergic agonist to stimulate lipolysis evoked a 3-fold increase in glycerol secretion followed by sustained release that was 40% higher than basal concentration. The ability to monitor changes in cellular secretion over time may provide insight into adipocyte metabolism and the dysregulation that occurs with obesity-related disorders.
Co-reporter:Qiang Li, Jon-Kar Zubieta and Robert T. Kennedy
Analytical Chemistry 2009 Volume 81(Issue 6) pp:2242
Publication Date(Web):February 5, 2009
DOI:10.1021/ac802391b
A method using capillary liquid chromatography−triple-stage mass spectrometry (LC−MS3) to determine endogenous opioid peptides in microdialysis samples collected in vivo was developed, validated, and applied to measurements in the rat striatum. Peptides in dialysate rapidly degraded when stored at room temperature or −80 °C. Adding acetic acid to a final concentration of 5% stabilized the peptides for 5 days allowing storage of fractions and off-line measurements which proved more convenient and reliable than previously used on-line methods. Study of the effect of dialysis flow rate from 0.2 to 2 μL/min and column inner diameter (i.d.) from 25 to 75 μm on the relative signal obtained for peptides revealed that lowest flow rates and smallest column i.d. gave the highest relative signal. The method was tested for 10 different neuropeptides and limits of detection (LODs) were from 0.5 to 60 pM (4 μL samples) for most. β-Endorphin had an LOD of 5 nM when detected directly, but it could be quantitatively determined by detecting a characteristic peptide produced by tryptic digestion with an LOD of 3 pM. This approach may prove useful for other large neuropeptides as well. The method was used to determine met-enkephalin, leu-enkephalin, dynorphin A1−8, and β-endorphin in vivo. Endomorphin 1 and 2 were below the detection limit of the method in vivo. Quantitative determination of leu-enkephalin using external calibration was verified by standard addition experiments. The improvements over previous approaches using capillary LC−MSn make in vivo neuropeptide monitoring more practical and feasible for a variety of neuropeptides.
Co-reporter:John F. Dishinger, Kendra R. Reid and Robert T. Kennedy
Analytical Chemistry 2009 Volume 81(Issue 8) pp:3119
Publication Date(Web):March 20, 2009
DOI:10.1021/ac900109t
Quantification of insulin release from pancreatic islets of Langerhans is of interest for diabetes research. Typical insulin secretion experiments are performed using offline techniques that are expensive, slow, have low-throughput, and require multiple islets. We have developed a microfluidic device for high-throughput, automated, and online monitoring of insulin secretion from individual islets in parallel. This chip consists of 15 channel networks each capable of superfusing a single islet and mixing superfusate from each islet online with fluorescein isothiocyanate-labeled insulin and anti-insulin antibody for a competitive immunoassay. The resulting continuous reaction streams are periodically injected onto parallel electrophoresis channels where the mixtures are separated. The resulting traces are used to quantify relative insulin released from islets. Serial immunoassays were performed at 10 s intervals on all 15 channels, corresponding to 5400 immunoassays per hour, to create temporally resolved insulin release profiles that captured single islet secretion dynamics. The chip was used to demonstrate that free fatty acid induced lipotoxicity in islets eliminates pulsatile insulin secretion.
Co-reporter:Jian Pei, Qiang Li, Mike S. Lee, Gary A. Valaskovic and Robert T. Kennedy
Analytical Chemistry 2009 Volume 81(Issue 15) pp:6558
Publication Date(Web):June 25, 2009
DOI:10.1021/ac901172a
Droplets or plugs within multiphase microfluidic systems have rapidly gained interest as a way to manipulate samples and chemical reactions on the femtoliter to microliter scale. Chemical analysis of the plugs remains a challenge. We have discovered that nanoliter plugs of sample separated by air or oil can be analyzed by electrospray ionization mass spectrometry when pumped directly into a fused silica nanospray emitter tip. Using leucine-enkephalin in methanol and 1% acetic acid in water (50:50 v:v) as a model sample, we found carry-over between plugs was <0.1% and relative standard deviation of signal for a series of plugs was 3%. Detection limits were 1 nM. Sample analysis rates of 0.8 Hz were achieved by pumping 13 nL samples separated by 3 mm long air gaps in a 75 μm inner diameter tube. Analysis rates were limited by the scan time of the ion trap mass spectrometer. The system provides a robust, rapid, and information-rich method for chemical analysis of sample in segmented flow systems.
Co-reporter:Kendra R. Reid and Robert T. Kennedy
Analytical Chemistry 2009 Volume 81(Issue 16) pp:6837
Publication Date(Web):July 21, 2009
DOI:10.1021/ac901114k
Microchip electrophoresis is an emerging analytical technology with several useful attributes including rapid separation time, small sample requirements, and automation. In numerous potential applications, such as chemical monitoring or high-throughput screening, it may be desirable to use a system for many analyses without operator intervention; however, long-term operation of microchip electrophoresis systems has received little attention. We have developed a microchip electrophoresis system that can automatically inject samples at 6 s intervals for 24 h resulting in collection of 14 400 assays in one session. Continuous operation time of a prototype of the device was limited to 2 h due to degradation of reagents and electrophoresis buffers on the chip; however, modification so that all reagents were continuously perfused into reservoirs on the device ensured fresh reagents were always used for analysis and enabled extended operating sessions. The electrophoresis chip incorporated a cell perfusion chamber and reagent addition channels to allow chemical monitoring of fluid around cells cultured on the chip by serial electrophoretic immunoassays. The immunoassay had detection limits of 0.4 nM for insulin and generated ∼4% relative standard deviation over an entire 24 h period with no evidence of signal drift. The combined system was used to monitor insulin secretion from single islets of Langerhans for 6−39 h. The monitoring experiments revealed that islets have secretion dynamics that include spontaneous oscillations after extended nonoscillating periods and possible ultradian rhythms.
Co-reporter:Maura Perry, Qiang Li, Robert T. Kennedy
Analytica Chimica Acta 2009 Volume 653(Issue 1) pp:1-22
Publication Date(Web):19 October 2009
DOI:10.1016/j.aca.2009.08.038
Methods and advances for monitoring neurotransmitters in vivo or for tissue analysis of neurotransmitters over the last five years are reviewed. The review is organized primarily by neurotransmitter type. Transmitter and related compounds may be monitored by either in vivo sampling coupled to analytical methods or implanted sensors. Sampling is primarily performed using microdialysis, but low-flow push–pull perfusion may offer advantages of spatial resolution while minimizing the tissue disruption associated with higher flow rates. Analytical techniques coupled to these sampling methods include liquid chromatography, capillary electrophoresis, enzyme assays, sensors, and mass spectrometry. Methods for the detection of amino acid, monoamine, neuropeptide, acetylcholine, nucleoside, and soluble gas neurotransmitters have been developed and improved upon. Advances in the speed and sensitivity of these methods have enabled improvements in temporal resolution and increased the number of compounds detectable. Similar advances have enabled improved detection at tissue samples, with a substantial emphasis on single cell and other small samples. Sensors provide excellent temporal and spatial resolution for in vivo monitoring. Advances in application to catecholamines, indoleamines, and amino acids have been prominent. Improvements in stability, sensitivity, and selectivity of the sensors have been of paramount interest.
Co-reporter:Qihui Ni, Kendra R. Reid and Charles F. Burant, Robert T. Kennedy
Analytical Chemistry 2008 Volume 80(Issue 10) pp:3539
Publication Date(Web):April 10, 2008
DOI:10.1021/ac800406f
Reversed-phase, packed capillary liquid chromatography interfaced by electrospray ionization to mass spectrometry was explored as an analytical method for determination of metabolites in microscale tissue samples using single islets of Langerhans as a model system. With the use of a 75 µm inner diameter column coupled to a quadrupole ion trap mass spectrometer in full scan mode, detection limits of 0.1−33 fmol were achieved for glycoloytic and tricarboxylic acid cycle metabolites. Reproducible processing of islets for analysis with little loss of metabolites was performed by rapid freezing followed by methanol−water extraction. The method yielded 20 µL of extract of which just 15 nL was injected suggesting the potential for performing multiple assays on the same islet. Approximately 200 presumed metabolites could be detected, of which 22 were identified by matching retention times and MS/MS spectra to standards. Relative standard deviations for peak detection was from 7 to 18% and was unaffected by storage for up to 11 days. The method was used to detect changes in metabolism associated with increasing extracellular islet glucose concentration from 3 to 20 mM yielding results largely consistent with known metabolism of islets. Because most previous studies of islet metabolism have only observed a few compounds at once and require far more tissue, this measurement method represents a significant advance for studies of metabolism of islets and other microscale samples.
Co-reporter:Jian Pei, John F. Dishinger, David L. Roman, Chetwana Rungwanitcha, Richard R. Neubig and Robert T. Kennedy
Analytical Chemistry 2008 Volume 80(Issue 13) pp:5225
Publication Date(Web):May 9, 2008
DOI:10.1021/ac800553g
A microfluidic chip consisting of parallel channels designed for rapid electrophoretic enzyme assays was developed. Radial arrangement of channels and a common waste channel allowed chips with 16 and 36 electrophoresis units to be fabricated on a 7.62 × 7.62 cm2 glass substrate. Fluorescence detection was achieved using a Xe arc lamp source and commercial charge-coupled device (CCD) camera to image migrating analyte zones in individual channels. Chip performance was evaluated by performing electrophoretic assays for G protein GTPase activity on chip using BODIPY−GTP as enzyme substrate. A 16-channel design proved to be useful in extracting kinetic information by allowing serial electrophoretic assays from 16 different enzyme reaction mixtures at 20 s intervals in parallel. This system was used to rapidly determine enzyme concentrations, optimal enzymatic reaction conditions, and Michaelis−Menten constants. A chip with 36 channels was used for screening for modulators of the G protein−RGS protein interaction by assaying the amount of product formed in enzyme reaction mixtures that contained test compounds. Thirty-six electrophoretic assays were performed in 30 s suggesting the potential throughput up to 4320 assays/h with appropriate sample handling procedures. Both designs showed excellent reproducibility of peak migration time and peak area. Relative standard deviations of normalized peak area of enzymatic product BODIPY−GDP were 5% and 11%, respectively, in the 16- and 36-channel designs.
Co-reporter:Meng Wang, Gregory T. Roman, Kristin Schultz, Colin Jennings and Robert T. Kennedy
Analytical Chemistry 2008 Volume 80(Issue 14) pp:5607
Publication Date(Web):June 12, 2008
DOI:10.1021/ac800622s
Microdialysis sampling probes were interfaced to a segmented flow system to improve temporal resolution for monitoring concentration dynamics. Aqueous dialysate was segmented into nanoliter plugs by pumping sample stream into the base of a tee channel structure microfabricated on a PDMS chip that had an immiscible carrier phase (perfluorodecalin) pumped into the cross arm of the tee. Varying the oil flow rate from 0.22 to 6.3 μL/min and sample flow rate from 42 to 328 nL/min allowed control of plug volume, interval between plugs, and frequency of plug generation between 6 and 28 nL, 0.6 and 10 s, and 0.1 and 1.7 Hz, respectively. Temporal resolution of the system, determined by measuring fluorescence in individual sample plugs following step changes of fluorescein concentration at the sampling probe surface, was as good as 15 s. Temporal resolution was independent of both sampling flow rate and distance that samples were pumped from the sampling probe. This effect is due to the prevention of Taylor dispersion of the sample as it was transported by segmented flow. In contrast, without flow segmentation, temporal resolution was worsened from 25 to 160 s as the detection point was moved from the sampling probe to 40 cm downstream. Glucose was detected by modifying the chip to allow enzyme assay reagents to be mixed with dialysate as sample plugs formed. The resulting assay had a detection limit of 50 μM and a linear range of 0.2−2 mM. This system was used to measure glucose in the brain of anesthetized rats. Basal concentration was 1.5 ± 0.1 mM (n = 3) and was decreased 60% by infusion of high-K+ solution through the probe. These results demonstrate the potential of microdialysis with segmented flow to be used for in vivo monitoring experiments with high temporal resolution.
Co-reporter:Gregory T. Roman, Meng Wang, Kristin N. Shultz, Colin Jennings and Robert T. Kennedy
Analytical Chemistry 2008 Volume 80(Issue 21) pp:8231
Publication Date(Web):October 3, 2008
DOI:10.1021/ac801317t
A method for sampling and electrophoretic analysis of aqueous plugs segmented in a stream of immiscible oil is described. In the method, an aqueous buffer and oil stream flow parallel to each other to form a stable virtual wall in a microfabricated K-shaped fluidic element. As aqueous sample plugs in the oil stream make contact with the virtual wall, coalescence occurs and sample is electrokinetically transferred to the aqueous stream. Using this virtual wall, two methods of injection for channel electrophoresis were developed. In the first, discrete sample zones flow past the inlet of an electrophoresis channel and a portion is injected by electroosmotic flow, termed the “discrete injector”. With this approach at least 800 plugs could be injected without interruption from a continuous segmented stream with 5.1% RSD in peak area. This method generated up to 1,050 theoretical plates, although analysis of the injector suggested that improvements may be possible. In a second method, aqueous plugs are sampled in a way that allows them to form a continuous stream that is directed to a microfluidic cross-style injector, termed the “desegmenting injector”. This method does not analyze each individual plug but instead allows periodic sampling of a high-frequency stream of plugs. Using this system at least 1000 injections could be performed sequentially with 5.8% RSD in peak area and 53,500 theoretical plates. This method was demonstrated to be useful for monitoring concentration changes from a sampling device with 10 s temporal resolution. Aqueous plugs in segmented flows have been applied to many different chemical manipulations including synthesis, assays, sampling processing and sampling. Nearly all such studies have used optical methods to analyze plug contents. This method offers a new way to analyze such samples and should enable new applications of segmented flow systems.
Co-reporter:Jennifer R. W. Furchak, Peilin Yang, Colin Jennings, Nils G. Walter and Robert T. Kennedy
Analytical Chemistry 2008 Volume 80(Issue 21) pp:8195
Publication Date(Web):October 9, 2008
DOI:10.1021/ac801410k
A naturally occurring aptazyme, the glmS ribozyme, is adapted to an assay for glucosamine 6-phosphate, an effector molecule for the aptazyme. In the assay, binding of analyte allosterically activates aptazyme to cleave a fluorescently labeled oligonucleotide substrate. The extent of reaction, and hence analyte concentration, is detected by either fluorescence resonance energy transfer (FRET) or capillary electrophoresis with laser-induced fluorescence (CE-LIF). With FRET, assay signal is the rate of increase in FRET in presence of analyte. With CE-LIF, the assay signal is the peak height of cleavage product formed after a fixed incubation time. The assay has a linear response up to 100 (CE-LIF) or 500 μM (FRET) and detection limit of ∼500 nM for glucosamine 6-phosphate under single-turnover conditions. When substrate is present in excess of the aptazyme, it is possible to amplify the signal by multiple turnovers to achieve a 13-fold improvement in sensitivity and detection limit of 50 nM. Successful signal amplification requires a temperature cycle to alternately dissociate cleaved substrate and allow fresh substrate to bind aptazyme. The results show that aptazymes have potential utility as analytical reagents for quantification of effector molecules.
Co-reporter:Peilin Yang, Robert T. Kennedy
Journal of Chromatography A 2008 Volume 1194(Issue 2) pp:225-230
Publication Date(Web):20 June 2008
DOI:10.1016/j.chroma.2008.04.072
Reversed-phase HPLC was coupled on-line to a rapid, competitive affinity probe capillary electrophoresis (APCE) assay to screen mixtures for compounds that inhibit protein–ligand interactions. The Fyn Src homology 2 (SH2) domain and its phosphopeptide binding partner were used as a model interaction for demonstration of this technique. In the method, mixtures containing possible inhibitors of binding were separated by HPLC at a flow rate of 0.3 mL/min. A small portion of effluent was directed to a fluidic tee where it was mixed on-line with Fyn SH2 domain and a fluorescent phosphopeptide (“affinity probe”) known to bind selectively to Fyn SH2 domain. Electropherograms of the reaction mixture were collected on-line at ∼6 s intervals using a flow-gated interface to control injections onto the capillary electrophoresis with laser-induced fluorescence system. The resulting electropherograms contained two peaks, one corresponding to the free affinity probe and the other a complex of the affinity probe and Fyn SH2 domain. Compounds that bound the protein were detected as a decrease in the peak height of the complex and an increase in the peak height of affinity probe with relative standard deviations of <5%. The assay was shown to resolve multiple peptidergic inhibitors and selectively detect them within a complex mixture of peptides. Signals were dependent upon both concentration of active peptide and its potency in binding inhibition. Detection limits were in the range of 2–11 μM depending upon the peptide. Common organic solvents used in HPLC were shown to have minimal effect in the on-line measurement up to ∼60% in the mobile phase.
Co-reporter:James L. Edwards, Rachel L. Edwards, Kendra R. Reid, Robert T. Kennedy
Journal of Chromatography A 2007 Volume 1172(Issue 2) pp:127-134
Publication Date(Web):23 November 2007
DOI:10.1016/j.chroma.2007.09.075
Capillary liquid chromatography coupled with electrospray ionization to a quadrupole ion trap mass spectrometer was explored as a method for the analysis of polar anionic compounds in complex metabolome mixtures. A ternary mobile phase gradient, consisting of aqueous acidic, aqueous neutral and organic phases in combination with an aqueous compatible reversed-phase stationary phase allowed metabolites with a wide range of polarities to be resolved and detected. Detection limits in the full scan mode for glycolysis and tricarboxylic acid cycle intermediates were from 0.9 to 36 fmol. Using this system, 111 ± 9 (n = 3) metabolites were detected in Escherichia coli lysate samples. Reducing column I.D. from 50 to 25 μm increased the number of metabolites detected to 156 ± 17 (n = 3). The improvement in number of metabolites detected was attributed to an increase in separation efficiency, an increase in sensitivity, and a decrease in adduct formation. Implementation of a second separation mode, strong anion exchange, to fractionate the sample prior to capillary RPLC increased the number of metabolites detected to 244 ± 21 (n = 3). This improvement was attributed to the increased peak capacity which decreased co-elution of molecules enabling more sensitive detection by mass spectrometry. This system was also applied to islets of Langerhans where more significant improvements in metabolite detection were observed. In islets, 391 ± 33 small molecules were detected using the two-dimensional separation. The results demonstrate that column miniaturization and use of two-dimensional separations can yield a significant improvement in the coverage of the metabolome.
Co-reporter:Gregory T. Roman, Robert T. Kennedy
Journal of Chromatography A 2007 Volume 1168(1–2) pp:170-188
Publication Date(Web):19 October 2007
DOI:10.1016/j.chroma.2007.06.010
Over the past decade a tremendous amount of research has been performed using microfluidic analytical devices to detect over 200 different chemical species. Most of this work has involved substantial integration of fluid manipulation components such as separation channels, valves, and filters. This level of integration has enabled complex sample processing on miniscule sample volumes. Such devices have also demonstrated high throughput, sensitivity, and separation performance. Although the miniaturization of fluidics has been highly valuable, these devices typically rely on conventional ancillary equipment such as power supplies, detection systems, and pumps for operation. This auxiliary equipment prevents the full realization of a “lab-on-a-chip” device with complete portability, autonomous operation, and low cost. Integration and/or miniaturization of ancillary components would dramatically increase the capability and impact of microfluidic separations systems. This review describes recent efforts to incorporate auxiliary equipment either as miniaturized plug-in modules or directly fabricated into the microfluidic device.
Co-reporter:Nicholas A. Cellar and Robert T. Kennedy
Lab on a Chip 2006 vol. 6(Issue 9) pp:1205-1212
Publication Date(Web):20 Jul 2006
DOI:10.1039/B603561B
A chip fabricated by multilayer soft lithography of poly(dimethylsiloxane) was created for separations-based sensing of neurotransmitters in vivo. The chip incorporated a pneumatically actuated peristaltic pump and valving system to combine low-flow push–pull perfusion sampling, on-line derivatization, and flow-gated injection onto an embedded fused-silica capillary for high speed separation of amine neurotransmitters from the brain of living animals. Six 160 µm wide by 10 µm high control channels, actuated with an overlapping 60° pulse sequence, simultaneously drove sample and buffers through fluidic channels of the same dimensions. Tunable sampling flow rates of 40 to 130 nL min−1 and separation buffer flow rates of 380 to 850 nL min−1 were achieved with actuation frequencies between 3 and 10 Hz. On-line sampling of amine neurotransmitters with separation efficiencies in excess of 250000 plates, detection limits of ∼40 nM, and relative standard deviations of 4% for glutamate and aspartate were achieved in vitro. Electropherograms with resolution of γ-aminobutyric acid, glutamine, taurine, serine, glycine, o-phosphorylethanolamine, glutamate, and aspartate could be collected every 30 s for over 4 h in vivo. It was also shown that pharmacological agents could be delivered and subsequent changes in neurotransmitter profile could be measured when delivering either 70 mM K+ artificial cerebrospinal fluid or 200 µM L-trans-pyrrolidine-2,4-dicarboxilic acid with the chip. These results demonstrate the ability of this chip to sample and monitor chemicals in the complex environment of the central nervous system with high selectivity and sensitivity over extended periods.
Co-reporter:James L. Edwards, Claire N. Chisolm, Jonathan G. Shackman, Robert T. Kennedy
Journal of Chromatography A 2006 Volume 1106(1–2) pp:80-88
Publication Date(Web):17 February 2006
DOI:10.1016/j.chroma.2005.08.082
Capillary electrophoresis (CE) was coupled to negative mode electrospray ionisation-mass spectrometry (MS) for separation and detection of phosphorylated and acidic metabolites in extracts of prokaryotes. Unlike previous CE–MS systems for metabolite analysis, a sheathless interface was used to improve sensitivity. To accomplish this, the separation capillary was modified by creating a porous junction near the outlet where the electrospray voltage and cathodic voltage for CE were applied. The outlet of the capillary was pulled to a 5 μm inner diameter to form an electrospray emitter and had a frit fabricated near the exit to prevent clogging. During analysis pressure was applied at the inlet of the separation column to create sufficient flow towards the detector. Limits of detection for 19 metabolites in full scan mode ranged from 20 nM for ADP ribose to 2.5 μM for α-ketoglutarate for 40 nL injections. Extracts of Escherichia coli, strain DH5-α, were analyzed using this system. In full scan mode, 118 different metabolites were detected. Tandem mass spectrometry was also employed to attempt identification. Reproducible fragmentation of 19 parent peaks was found and 10 of these produced spectra that were consistent with identification obtained from matching to compounds in the MetaCyc database. These results demonstrate the utility of a sensitive CE–MS system for large scale metabolite detection in biological samples.
Co-reporter:Robert T Kennedy, Christopher J Watson, William E Haskins, David H Powell, Robert E Strecker
Current Opinion in Chemical Biology 2002 Volume 6(Issue 5) pp:659-665
Publication Date(Web):1 October 2002
DOI:10.1016/S1367-5931(02)00373-3
Microdialysis is valuable for studying the neurochemical changes underlying behavior. Recent advances include the application of the high-sensitivity methods of capillary electrophoresis and capillary liquid chromatography with mass spectrometry to dialysate analysis. These methods have improved temporal resolution, spatial resolution, multi-analyte capability and potential for compound discovery.
Co-reporter:Meng Wang, Thomas Slaney, Omar Mabrouk, Robert T. Kennedy
Journal of Neuroscience Methods (30 June 2010) Volume 190(Issue 1) pp:39-48
Publication Date(Web):30 June 2010
DOI:10.1016/j.jneumeth.2010.04.023
An off-line in vivo neurochemical monitoring approach was developed based on collecting nanoliter microdialysate fractions as an array of “plugs” segmented by immiscible oil in a piece of Teflon tubing. The dialysis probe was integrated with the plug generator in a polydimethlysiloxane microfluidic device that could be mounted on the subject. The microfluidic device also allowed derivatization reagents to be added to the plugs for fluorescence detection of analytes. Using the device, 2 nL fractions corresponding to 1–20 ms sampling times depending upon dialysis flow rate, were collected. Because axial dispersion was prevented between them, each plug acted as a discrete sample collection vial and temporal resolution was not lost by mixing or diffusion during transport. In vitro tests of the system revealed that the temporal resolution of the system was as good as 2 s and was limited by mass transport effects within the dialysis probe. After collection of dialysate fractions, they were pumped into a glass microfluidic chip that automatically analyzed the plugs by capillary electrophoresis with laser-induced fluorescence at 50 s intervals. By using a relatively low flow rate during transfer to the chip, the temporal resolution of the samples could be preserved despite the relatively slow analysis time. The system was used to detect rapid dynamics in neuroactive amino acids evoked by microinjecting the glutamate uptake inhibitor l-trans-pyrrolidine-2,4-dicarboxylic acid (PDC) or K+ into the striatum of anesthetized rats. The resulted showed increases in neurotransmitter efflux that reached a peak in 20 s for PDC and 13 s for K+.
Co-reporter:Omar S. Mabrouk, Robert T. Kennedy
Journal of Neuroscience Methods (30 July 2012) Volume 209(Issue 1) pp:127-133
Publication Date(Web):30 July 2012
DOI:10.1016/j.jneumeth.2012.06.006
Oxytocin (OXT) and arg-vasopressin (AVP) are nonapeptides with many important functions both peripherally and centrally. Intracerebral microdialysis has helped characterize their importance in regulating complex social and emotional processes. Radioiummunoassay is the most commonly used analytical method used for OXT and AVP measurements in microdialysates. These measurements have several well-known issues including single peptide per assay limit, possible cross-reactivity between structurally related peptides, and laborious sample preparation with radioactive materials. Here we demonstrate the use of capillary LC–MS3 for measuring OXT and AVP simultaneously in dialysates at a 10 min sampling frequency. Microdialysate samples required no preparation and instrumentation was commercially available. Microdialysis probes made with polyacrylonitrile membranes were suitable for high level recovery of the peptides in vitro and in vivo. Responses were linear from 1 to 100 pM. Matrix effect was assessed by standard addition experiments and by comparing signal intensities of OXT and AVP standards made in aCSF or dialysate. It was determined that the online washing step used on this setup was adequate for removing contaminants which interfere with electrospray ionization efficiency. In vivo, both peptides were stimulated by high K+ (75 mM) aCSF perfusion in the paraventricular nucleus (PVN). Also, a systemic injection of high Na+ (2 M) caused a rapid and transient increase in PVN OXT while AVP increased only after 1.5 h. Our findings suggest that capillary LC–MS3 is a straightforward method for monitoring OXT and AVP simultaneously from complex samples such as dialysates.
Co-reporter:David E. Cepeda, Leah Hains, David Li, Joseph Bull, Stephen I. Lentz, Robert T. Kennedy
Journal of Neuroscience Methods (15 March 2015) Volume 242() pp:97-105
Publication Date(Web):15 March 2015
DOI:10.1016/j.jneumeth.2015.01.019
•Push–pull perfusion at 50 nL/min was evaluated for tissue damage by infusing stains.•Push–pull damaged 24% of cells, less than the 33% observed with microdialysis.•Modeling and data reveal that flow did not contribute to damage.•Low-flow Push–pull perfusion provides high spatial resolution for in vivo sampling.BackgroundNeurochemical monitoring via sampling probes is valuable for deciphering neurotransmission in vivo. Microdialysis is commonly used; however, the spatial resolution is poor.New MethodRecently push–pull perfusion at low flow rates (50 nL/min) has been proposed as a method for in vivo sampling from the central nervous system. Tissue damage from such probes has not been investigated in detail. In this work, we evaluated acute tissue response to low-flow push–pull perfusion by infusing the nuclear stains Sytox Orange and Hoechst 33342 through probes implanted in the striatum for 200 min, to label damaged and total cells, respectively, in situ.ResultsUsing the damaged/total labeled cell ratio as a measure of tissue damage, we found that 33 ± 8% were damaged within the dye region around a microdialysis probe. We found that low-flow push–pull perfusion probes damaged 24 ± 4% of cells in the sampling area. Flow had no effect on the number of damaged cells for low-flow push–pull perfusion. Modeling revealed that shear stress and pressure gradients generated by the flow were lower than thresholds expected to cause damage.Comparison with existing methods.Push–pull perfusion caused less tissue damage but yielded 1500-fold better spatial resolution.ConclusionsPush–pull perfusion at low flow rates is a viable method for sampling from the brain with potential for high temporal and spatial resolution. Tissue damage is mostly caused by probe insertion. Smaller probes may yield even lower damage.
Co-reporter:Holly M. Shackman, Minshan Shou, Nicholas A. Cellar, Christopher J. Watson, Robert T. Kennedy
Journal of Neuroscience Methods (15 January 2007) Volume 159(Issue 1) pp:86-92
Publication Date(Web):15 January 2007
DOI:10.1016/j.jneumeth.2006.06.020
Capillary liquid chromatography–mass spectrometry (cLC–MS) was coupled on-line to microdialysis sampling to monitor endogenous acetylcholine (ACh) from the rodent brain. In vivo microdialysate sampled at 0.6 μL/min from the striatum of ketamine or chloral hydrate anesthetized rats was loaded onto a sample loop and then injected onto a ∼5 cm long strong cation exchange (SCX) capillary column. A step gradient was used to separate the analyte from ionization suppressing salts contained in dialysate in 2.4 min. Sampling coupled on-line with cLC–MS allowed for high temporal resolution (data points at 2.4 min intervals), good reproducibility (10–15% relative standard deviation, R.S.D.), and sensitive limits of detection (0.04 nM or 8 amol injected). The method successfully monitored basal and stimulated levels (induced by increased K+ concentrations) of ACh from the anesthetized rat without necessitating perfusion of an acetylcholinesterase (AChE) inhibitor. Absolute and percent basal levels of ACh from rats receiving different anesthetics were also compared.
.jpg)
![2-(4-ethoxyphenyl)-6-[6-(4-methylpiperazin-1-yl)-1H-benzimidazol-2-yl]-1H-benzimidazole,trihydrochloride](http://img.cochemist.com/ccimg/875800/875756-97-1.png)
![2-(4-ethoxyphenyl)-6-[6-(4-methylpiperazin-1-yl)-1H-benzimidazol-2-yl]-1H-benzimidazole,trihydrochloride](http://img.cochemist.com/ccimg/875800/875756-97-1_b.png)























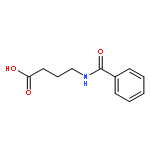


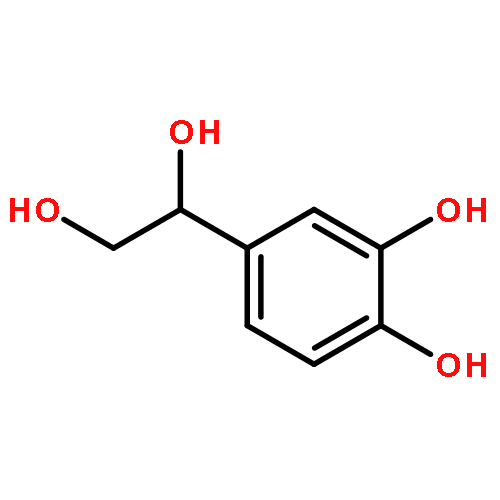
](http://img.cochemist.com/ccimg/27000/26913-06-4.png)
](http://img.cochemist.com/ccimg/27000/26913-06-4_b.png)












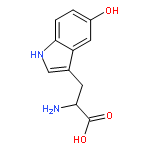

![2-aminoethoxy-[[5-(4-amino-2-oxo-pyrimidin-1-yl)-3,4-dihydroxy-oxolan-2-yl]methoxy-hydroxy-phosphoryl]oxy-phosphinic Acid](http://img.cochemist.com/ccimg/3100/3036-18-8.png)
![2-aminoethoxy-[[5-(4-amino-2-oxo-pyrimidin-1-yl)-3,4-dihydroxy-oxolan-2-yl]methoxy-hydroxy-phosphoryl]oxy-phosphinic Acid](http://img.cochemist.com/ccimg/3100/3036-18-8_b.png)
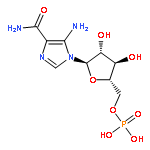
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)
![Acetamide,N-[2-(5-hydroxy-1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/1300/1210-83-9.png)
![Acetamide,N-[2-(5-hydroxy-1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/1300/1210-83-9_b.png)









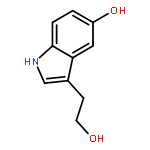
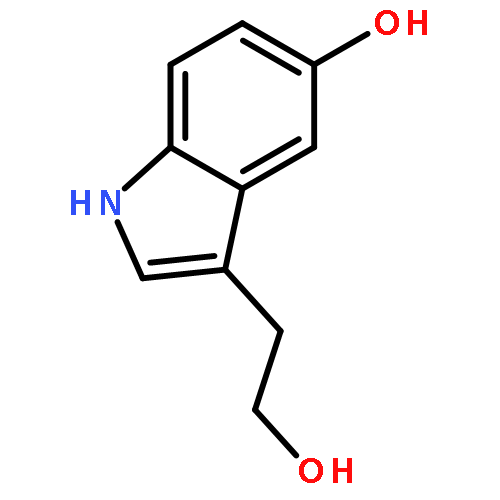
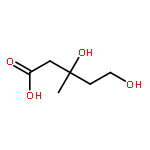


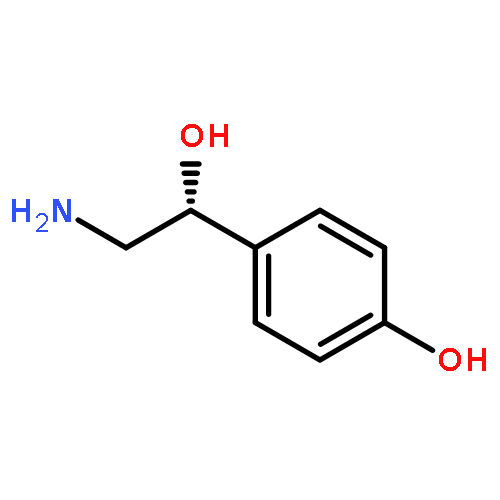




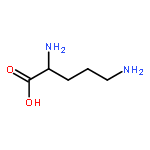

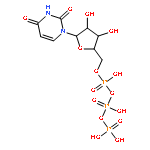
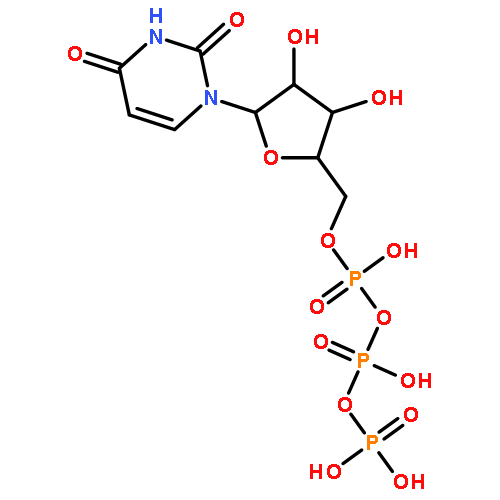
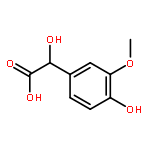



![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)






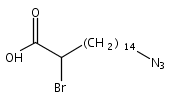
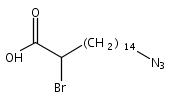






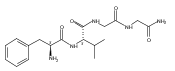

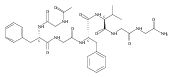










![Benzoic acid,4-[(2-formyl-3-quinolinyl)carbonyl]-](http://img.cochemist.com/ccimg/131200/131124-59-9.png)
![Benzoic acid,4-[(2-formyl-3-quinolinyl)carbonyl]-](http://img.cochemist.com/ccimg/131200/131124-59-9_b.png)




![acetic acid; (2S)-2-amino-N-[(1R)-1-[[[(1S)-1-(2-hydroxyethylcarbamoyl)-2-phenyl-ethyl]-methyl-carbamoyl]methylcarbamoyl]ethyl]-3-(4-hydroxyphenyl)propanamide](http://img.cochemist.com/ccimg/101000/100929-53-1.png)
![acetic acid; (2S)-2-amino-N-[(1R)-1-[[[(1S)-1-(2-hydroxyethylcarbamoyl)-2-phenyl-ethyl]-methyl-carbamoyl]methylcarbamoyl]ethyl]-3-(4-hydroxyphenyl)propanamide](http://img.cochemist.com/ccimg/101000/100929-53-1_b.png)


![L-Argininamide,N2-[(phenylmethoxy)carbonyl]-L-arginyl-N-(4-methyl-2-oxo-2H-1-benzopyran-7-yl)-](http://img.cochemist.com/ccimg/89000/88937-61-5.png)
![L-Argininamide,N2-[(phenylmethoxy)carbonyl]-L-arginyl-N-(4-methyl-2-oxo-2H-1-benzopyran-7-yl)-](http://img.cochemist.com/ccimg/89000/88937-61-5_b.png)






![DAMGO;[D-ALA2,NME-PHE4,GLY-OL5]-ENKEPHALIN](http://img.cochemist.com/ccimg/78200/78123-71-4.png)
![DAMGO;[D-ALA2,NME-PHE4,GLY-OL5]-ENKEPHALIN](http://img.cochemist.com/ccimg/78200/78123-71-4_b.png)





![L-Leucinamide,N-acetyl-L-leucyl-N-[(1S)-4-[(aminoiminomethyl)amino]-1-formylbutyl]-](http://img.cochemist.com/ccimg/55200/55123-66-5.png)
![L-Leucinamide,N-acetyl-L-leucyl-N-[(1S)-4-[(aminoiminomethyl)amino]-1-formylbutyl]-](http://img.cochemist.com/ccimg/55200/55123-66-5_b.png)















![1-[5-[[[[5-(6-Aminopurin-9-yl)-3,4-dihydroxy-oxolan-2-yl]methoxy-hydroxy-phosphoryl]oxy-hydroxy-phosphoryl]oxymethyl]-3,4-dihydroxy-oxolan-2-yl]pyridine-5-carboxylate](http://img.cochemist.com/ccimg/6500/6450-77-7.png)
![1-[5-[[[[5-(6-Aminopurin-9-yl)-3,4-dihydroxy-oxolan-2-yl]methoxy-hydroxy-phosphoryl]oxy-hydroxy-phosphoryl]oxymethyl]-3,4-dihydroxy-oxolan-2-yl]pyridine-5-carboxylate](http://img.cochemist.com/ccimg/6500/6450-77-7_b.png)





















![(2R)-2-AMINO-1-PHENYL-PROPAN-1-OL; (E)-4-(4-BROMOPHENOXY)-4-OXO-B<WBR />UT-2-ENOIC ACID; N,N-DIMETHYL-3-(2-PYRIDYL)PROPAN-1-AMINE; 3-[(1R<WBR />)-1-HYDROXY-2-METHYLAMINO-ETHYL]PHENOL; DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/400/320-77-4.png)
![(2R)-2-AMINO-1-PHENYL-PROPAN-1-OL; (E)-4-(4-BROMOPHENOXY)-4-OXO-B<WBR />UT-2-ENOIC ACID; N,N-DIMETHYL-3-(2-PYRIDYL)PROPAN-1-AMINE; 3-[(1R<WBR />)-1-HYDROXY-2-METHYLAMINO-ETHYL]PHENOL; DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/400/320-77-4_b.png)














![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)









![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6.png)
![(3aS,8aR)-1,3a,8-Trimethyl-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indol-5-yl methylcarbamate](http://img.cochemist.com/ccimg/100/57-47-6_b.png)