Co-reporter:Jie Xu
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 34) pp:7093-7096
Publication Date(Web):2017/08/30
DOI:10.1039/C7OB01654A
A Pd(0)-catalyzed elimination of an allylic acetate generates a π-allyl complex that is postulated to initiate a novel intramolecular Diels–Alder cycloaddition to a tethered furan (IMDAF). Under the reaction conditions, this convergent, microwave-accelerated cascade process provides substituted indoles in moderate to good yields after Pd-hydride elimination, aromatization by dehydration, and in situ N-Boc cleavage.
Co-reporter:John A. Milligan, Carl A. Busacca, Chris H. Senanayake, and Peter Wipf
Organic Letters 2016 Volume 18(Issue 17) pp:4300-4303
Publication Date(Web):August 12, 2016
DOI:10.1021/acs.orglett.6b02051
Hydrophosphination of bicyclo[1.1.0]butyl nitriles with phosphine boranes and phosphites provided novel cyclobutyl-P derivatives. The reaction generally favors the syn-diastereomer, and the nitrile can be reduced and converted to other functional groups, thus enabling the preparation of bidentate ligands that access new conformational space by virtue of their attachment to the torsionally malleable but sterically restrictive cyclobutane scaffold. The enantioselective hydrogenation of dehydrophenylalanine using a bidentate phosphine–phosphite ligand illustrates the synthetic utility of the newly prepared scaffold.
Co-reporter:Joseph M. Salamoun and Peter Wipf
Journal of Medicinal Chemistry 2016 Volume 59(Issue 17) pp:7771-7772
Publication Date(Web):August 19, 2016
DOI:10.1021/acs.jmedchem.6b01210
Despite their extensive involvement in signaling pathways and disease pathologies, targeting protein phosphatases remains an underexplored opportunity in drug discovery. Selective intracellular regulation of phosphatases with small molecule inhibitors has been an unmet challenge. However, recent progress in the development of allosteric modulators encourages renewed efforts to exploit their potential as therapeutic targets.
Co-reporter:Joseph M. Salamoun, Kelley E. McQueeney, Kalyani Patil, Steven J. Geib, Elizabeth R. Sharlow, John S. Lazo and Peter Wipf
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 27) pp:6398-6402
Publication Date(Web):07 Jun 2016
DOI:10.1039/C6OB00946H
The phosphatase PTP4A3 is an attractive anticancer target, but knowledge of its exact role in cells remains incomplete. A potent, structurally novel inhibitor of the PTP4A family was obtained by photooxygenation of a less active, electron-rich thienopyridone (1). Iminothienopyridinedione 13 displays increased solution stability and is readily obtained by two new synthetic routes that converge in the preparation of 1. The late-stage photooxygenation of 1 to give 13 in high yield highlights the potential of this reaction to modify the structure and properties of a biological lead compound and generate value for expanding the scope of an SAR investigation. Analog 13 should become a valuable tool for further exploration of the role of PTP4A3 in tumor progression.
Co-reporter:Celeste Alverez, Stacie L. Bulfer, Ramappa Chakrasali, Michael. S. Chimenti, Raymond J. Deshaies, Neal Green, Mark Kelly, Matthew G. LaPorte, Taber S. Lewis, Mary Liang, William J. Moore, R. Jeffrey Neitz, Vsevolod A. Peshkov, Michael A. Walters, Feng Zhang, Michelle R. Arkin, Peter Wipf, and Donna M. Huryn
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 2) pp:182
Publication Date(Web):December 22, 2015
DOI:10.1021/acsmedchemlett.5b00396
A high-throughput screen to discover inhibitors of p97 ATPase activity identified an indole amide that bound to an allosteric site of the protein. Medicinal chemistry optimization led to improvements in potency and solubility. Indole amide 3 represents a novel uncompetitive inhibitor with excellent physical and pharmaceutical properties that can be used as a starting point for drug discovery efforts.Keywords: AAA ATPase; allosteric inhibitor; indole amide; p97 inhibitor; protein homeostasis; ubiquitin pathway modulator
Co-reporter:James K. Johnson, Erin M. Skoda, Jianhua Zhou, Erica Parrinello, Dan Wang, Katherine O’Malley, Benjamin R. Eyer, Mustafa Kazancioglu, Kurtis Eisermann, Paul A. Johnston, Joel B. Nelson, Zhou Wang, and Peter Wipf
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 8) pp:785
Publication Date(Web):May 27, 2016
DOI:10.1021/acsmedchemlett.6b00186
After a high-throughput screening campaign identified thioether 1 as an antagonist of the nuclear androgen receptor, a zone model was developed for structure–activity relationship (SAR) purposes and analogues were synthesized and evaluated in a cell-based luciferase assay. A novel thioether isostere, cyclopropane (1S,2R)-27, showed the desired increased potency and structural properties (stereospecific SAR response, absence of a readily oxidized sulfur atom, low molecular weight, reduced number of flexible bonds and polar surface area, and drug-likeness score) in the prostate-specific antigen luciferase assay in C4-2-PSA-rl cells to qualify as a new lead structure for prostate cancer drug development.Keywords: advanced prostate cancer; Androgen receptor; CRPC; isoxazoles; luciferase assay; thioether isostere;
Co-reporter:Tanja Krainz, Michael M. Gaschler, Chaemin Lim, Joshua R. Sacher, Brent R. Stockwell, and Peter Wipf
ACS Central Science 2016 Volume 2(Issue 9) pp:653
Publication Date(Web):September 7, 2016
DOI:10.1021/acscentsci.6b00199
Discovering compounds and mechanisms for inhibiting ferroptosis, a form of regulated, nonapoptotic cell death, has been of great interest in recent years. In this study, we demonstrate the ability of XJB-5-131, JP4-039, and other nitroxide-based lipid peroxidation mitigators to prevent ferroptotic cell death in HT-1080, BJeLR, and panc-1 cells. Several analogues of the reactive oxygen species (ROS) scavengers XJB-5-131 and JP4-039 were synthesized to probe structure–activity relationships and the influence of subcellular localization on the potency of these novel ferroptosis suppressors. Their biological activity correlated well over several orders of magnitude with their structure, relative lipophilicity, and respective enrichment in mitochondria, revealing a critical role of intramitochondrial lipid peroxidation in ferroptosis. These results also suggest that preventing mitochondrial lipid oxidation might offer a viable therapeutic opportunity in ischemia/reperfusion-induced tissue injury, acute kidney injury, and other pathologies that involve ferroptotic cell death pathways.
Co-reporter:Raffaele Colombo, Zhiyong Wang, Junyan Han, Raghavan Balachandran, Hikmat N. Daghestani, Daniel P. Camarco, Andreas Vogt, Billy W. Day, David Mendel, and Peter Wipf
The Journal of Organic Chemistry 2016 Volume 81(Issue 21) pp:10302-10320
Publication Date(Web):July 22, 2016
DOI:10.1021/acs.joc.6b01314
We report a second-generation synthesis of the exceedingly potent antimitotic agent N14-desacetoxytubulysin H (1) as well as the preparation of nine analogues of this lead structure. Highlights of our synthetic efforts include an efficient late-stage functionalization that allows for the preparation of new side-chain- and backbone-modified analogues. We also discovered C-terminal modifications that preserve the exquisite biological activity of acid 1 and offer the opportunity for effective conjugation to cell type-targeting moieties. All analogues had antiproliferative activities in the high picomolar to low nanomolar range and caused apoptosis and mitotic arrest as measured in a high content nuclear morphology assay. The ten synthetic agents described herein spanned a range of almost 4 orders of magnitude in biological activity and illustrate the continued potential to discover extraordinarily potent antiproliferative compounds based on natural product leads.
Co-reporter:Maciej A. A. Walczak, Tanja Krainz, and Peter Wipf
Accounts of Chemical Research 2015 Volume 48(Issue 4) pp:1149
Publication Date(Web):March 16, 2015
DOI:10.1021/ar500437h
Mechanistically as well as synthetically, bicyclo[1.1.0]butanes represent one of the most fascinating classes of organic compounds. They offer a unique blend of compact size (four carbon atoms), high reactivity (strain energy of 66 kcal/mol), and mechanistic pathway diversity that can be harvested for the rapid assembly of complex scaffolds. The C(1)–C(3) bond combines the electronic features of both σ and π bonds with facile homolytic and heterolytic bond dissociation properties and thereby readily engages pericyclic, transition-metal-mediated, nucleophilic, and electrophilic pathways as well as radical acceptor and donor substrates.Despite this multifaceted reaction profile and recent advances in the preparation of bicylo[1.1.0]butanes, the current portfolio of synthetic applications is still limited compared with those of cyclopropanes and cyclobutanes. In this Account, we describe our work over the past decade on the exploration of substituent effects on the ring strain and the reactivity of bicyclo[1.1.0]butanes, particularly in the context of metal-mediated processes. We first describe Rh(I)-catalyzed cycloisomerization reactions of N-allyl amines to give pyrrolidine and azepine heterocycles. The regioselectivity of the C,C-bond insertion/ring-opening step in these reactions is controlled by the phosphine ligand. After metal carbene formation, an intramolecular cyclopropanation adds a second fused ring system. A proposed mechanism rationalizes why rhodium(I) complexes with monodentate ligands favor five-membered heterocycles, as opposed to Rh(I)–bidentate ligand catalysts, which rearrange N-allyl amines to seven-membered heterocycles. The scope of Rh(I)-catalyzed cycloisomerization reactions was extended to allyl ethers, which provide a mixture of five- and seven-membered cyclic ethers regardless of the nature of the phosphine additive and Rh(I) precatalyst. The chemical diversity of these cycloisomerization products was further expanded by a consecutive one-pot metathesis reaction.Rh(I)-catalyzed cycloisomerizations of propargyl amides, ethers, and electron-deficient bicyclo[1.1.0]butanes diverged mechanistically and often led to a significant number of decomposition products. In these cases, Pt(II) emerged as a superior, more alkynophilic late transition metal with its own mechanistic peculiarities. While monosubstituted bicyclo[1.1.0]butanes led to the formation of tetrahydropyridines, 1,3-disubstituted and electron-deficient bicyclo[1.1.0]butanes reacted distinctly differently with Pt(II) and ultimately provided a complementary set of nitrogen- and oxygen-containing cyclic scaffolds.The metal-catalyzed ring transformations of bicyclo[1.1.0]butanes presented herein suggest additional strategies for new reaction discoveries that can access a wide variety of novel cyclic frameworks from relatively simple starting materials. In addition, these case studies highlight the considerable potential for future applications in natural products, medicinal, and diversity-oriented synthesis based on the wealth of mechanistic pathways available to these strained small-ring carbocycles.
Co-reporter:Celeste Alverez, Michelle R. Arkin, Stacie L. Bulfer, Raffaele Colombo, Marina Kovaliov, Matthew G. LaPorte, Chaemin Lim, Mary Liang, William J. Moore, R. Jeffrey Neitz, Yongzhao Yan, Zhizhou Yue, Donna M. Huryn, and Peter Wipf
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 12) pp:1225
Publication Date(Web):October 23, 2015
DOI:10.1021/acsmedchemlett.5b00364
Exploratory SAR studies of a new phenyl indole chemotype for p97 inhibition revealed C-5 indole substituent effects in the ADPGlo assay that did not fully correlate with either electronic or steric factors. A focused series of methoxy-, trifluoromethoxy-, methyl-, trifluoromethyl-, pentafluorosulfanyl-, and nitro-analogues was found to exhibit IC50s from low nanomolar to double-digit micromolar. Surprisingly, we found that the trifluoromethoxy-analogue was biochemically a better match of the trifluoromethyl-substituted lead structure than a pentafluorosulfanyl-analogue. Moreover, in spite of their almost equivalent strongly electron-depleting effect on the indole core, pentafluorosulfanyl- and nitro-derivatives were found to exhibit a 430-fold difference in p97 inhibitory activities. Conversely, the electronically divergent C-5 methyl- and nitro-analogues both showed low nanomolar activities.Keywords: AAA ATPase; fluorinated substituent effects; p97 inhibitors; pentafluorosulfanyl-indole; structure−activity relationships; trifluoromethyl-indole
Co-reporter:Kara M. George Rosenker, William D. Paquette, Paul A. Johnston, Elizabeth R. Sharlow, Andreas Vogt, Ahmet Bakan, John S. Lazo, Peter Wipf
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 12) pp:2810-2818
Publication Date(Web):15 June 2015
DOI:10.1016/j.bmc.2015.01.043
The cell division cycle 25B dual specificity phosphatase (Cdc25B) regulates the normal progression of the mammalian cell cycle by dephosphorylating and activating cyclin-dependent kinase (Cdk) complexes, particularly in response to DNA damage. Elevated Cdc25B levels enable a bypass of normal cell cycle checkpoints, and the overexpression of Cdc25B has been linked to a variety of human cancers. Thus, Cdc25B is an attractive target for the development of anticancer therapeutics. Herein we describe the synthesis and biological evaluation of a series of non-quinoid inhibitors of Cdc25B containing the 3-aminoisoquinolin-1(2H)-one pharmacophore. In addition to several strategies that address specific substitution patterns on isoquinolines, we have applied a regioselective Pd-catalyzed cross-coupling methodology to synthesize a new lead structure, 6-(3-aminophenyl)-3-(phenylamino)isoquinolin-1(2H)-one (13), which proved to be a reversible, competitive Cdc25B inhibitor with a Ki of 1.9 μM. Compound 13 prevented human cancer cell growth and blocked Cdc25B-mediated mitotic checkpoint bypass. Molecular docking studies support binding near the catalytic site.
Co-reporter:Wei Qian, Joseph Salamoun, Jingnan Wang, Vera Roginskaya, Bennett Van Houten, Peter Wipf
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 4) pp:856-863
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmcl.2014.12.072
The effective management of tumors resistant to platinum drugs-based anticancer therapies is a critical challenge in current clinical practices. The proapoptotic Bcl-2 family proteins Bax and Bak are essential for cisplatin-induced apoptosis. Unfortunately, Bax and its related upstream endogenous apoptotic signaling pathways are often dysregulated in cancer cells. Strategies that are able to bypass Bax- and Bak-dependent apoptotic pathways will thus provide opportunities to overcome platinum drug resistance. We have identified the thioxodihydroquinazolinone mdivi-1 as a member of a novel class of small molecules that are able to induce Bax- and Bak-independent mitochondrial outer membrane permeabilization when combined with cisplatin, thereby efficiently triggering apoptosis in platinum-resistant tumor cells. In the present structure activity relationship (SAR) study of a computationally selected library of mdivi-1 related small molecules, we established a pharmacophore model that can lead to the enhancement of platinum drug efficacy and Bax/Bak-independent mitochondrial apoptosis. Specifically, we found that a thiourea function is necessary but not sufficient for the synergism of this class of thioxodihydroquinazolinones with cisplatin. We were also able to identify more potent mdivi-1 analogs through this SAR study, which will guide future designs with the goal to develop novel combination regimens for the treatment of platinum- and multidrug-resistant tumors.Structural features of thioxodihydroquinazolinones required for synergism with cisplatin.
Co-reporter:Jing Ma, Chaemin Lim, Joshua R. Sacher, Bennett Van Houten, Wei Qian, Peter Wipf
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 21) pp:4828-4833
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmcl.2015.06.073
Mitochondria play important roles in tumor cell physiology and survival by providing energy and metabolites for proliferation and metastasis. As part of their oncogenic status, cancer cells frequently produce increased levels of mitochondrial-generated reactive oxygen species (ROS). However, extensive stimulation of ROS generation in mitochondria has been shown to be able to induce cancer cell death, and is one of the major mechanisms of action of many anticancer agents. We hypothesized that enhancing mitochondrial ROS generation through direct targeting of a ROS generator into mitochondria will exhibit tumor cell selectivity, as well as high efficacy in inducing cancer cell death. We thus synthesized a mitochondrial targeted version of β-lapachone (XJB-Lapachone) based on our XJB mitochondrial targeting platform. We found that the mitochondrial targeted β-lapachone is more efficient in inducing apoptosis compared to unconjugated β-lapachone, and the tumor cell selectivity is maintained. XJB-Lapachone also induced extensive cellular vacuolization and autophagy at a concentration not observed with unconjugated β-lapachone. Through characterization of mitochondrial function we revealed that XJB-Lapachone is indeed more capable of stimulating ROS generation in mitochondria, which led to a dramatic mitochondrial uncoupling and autophagic degradation of mitochondria. Taken together, we have demonstrated that targeting β-lapachone accomplishes higher efficacy through inducing ROS generation directly in mitochondria, resulting in extensive mitochondrial and cellular damage. XJB-Lapachone will thus help to establish a novel platform for the design of next generation mitochondrial targeted ROS generators for cancer therapy.
Co-reporter:Peter Wipf, Benjamin R. Eyer, Yukihiro Yamaguchi, Feng Zhang, Matthew D. Neal, Chhinder P. Sodhi, Misty Good, Maria Branca, Thomas Prindle Jr., Peng Lu, Jeffrey L. Brodsky, David J. Hackam
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3097-3100
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.11.048
The low-molecular weight isopropyl 2-acetamido-α-glucoside 16 (C34) inhibits toll-like receptor 4 (TLR4) in enterocytes and macrophages in vitro, and reduces systemic inflammation in mouse models of endotoxemia and necrotizing enterocolitis. We used a copper(II)-mediated solvolysis of anomeric oxazolines and an acid-mediated conversion of β-glucosamine and β-galactosamine pentaacetates to generate analogs of 16 at the anomeric carbon and at C-4 of the pyranose ring. These compounds were evaluated for their influence on TLR4-mediated inflammatory signaling in cultured enterocytes and monocytes. Their efficacy was confirmed using a NF-kB-luciferase reporter mouse, thus establishing the first structure–activity relationship (SAR) study in this series and identifying the more efficacious isopropyl 2-acetamido-α-galactoside 17.
Co-reporter:Joseph Salamoun, Shelby Anderson, James C. Burnett, Rick Gussio, and Peter Wipf
Organic Letters 2014 Volume 16(Issue 7) pp:2034-2037
Publication Date(Web):March 18, 2014
DOI:10.1021/ol500620m
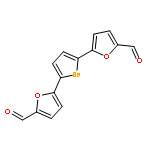
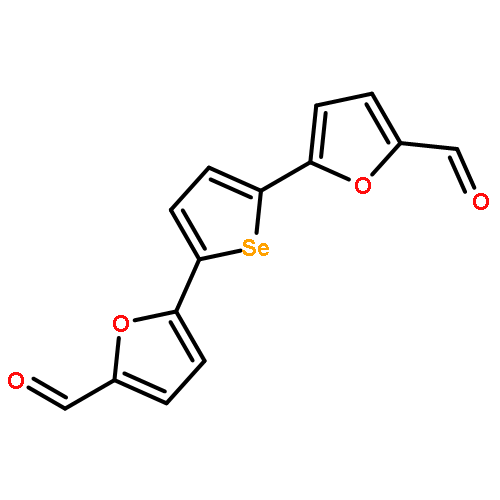
Two complementary approaches for the preparation of linked 5-membered heterocycles were developed. The Pd-catalyzed Suzuki–Miyaura cross-coupling with halogenated furan, thiophene, and selenophene led to higher overall yields, but C,H-bond activation was a more efficient strategy for the coupling at C(2) of oxazoles. Potency and selectivity of the final hydroxymethyl products in renal (A498), lung (NCI-H226), kidney (CAKI-1), and breast (MDA-MB-468, MCF7) carcinoma cell lines were determined.
Co-reporter:Erin M. Skoda, Joshua R. Sacher, Mustafa Z. Kazancioglu, Jaideep Saha, and Peter Wipf
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 8) pp:900
Publication Date(Web):June 27, 2014
DOI:10.1021/ml5001504
Low aqueous solubility is a common challenge in drug discovery and development and can lead to inconclusive biological assay results. Attaching small, polar groups that do not interfere with the bioactivity of the pharmacophore often improves solubility, but there is a dearth of viable neutral moieties available for this purpose. We have modified several poorly soluble drugs or drug candidates with the oxetanyl sulfoxide moiety of the DMSO analog MMS-350 and noted in most cases a moderate to large improvement of aqueous solubility. Furthermore, the membrane permeability of a test sample was enhanced compared to the parent compound.Keywords: Aqueous solubility; JP4-039; MMS-350; oxetane; sulfoxide
Co-reporter:Elaine M. Schuster
Israel Journal of Chemistry 2014 Volume 54( Issue 4) pp:361-370
Publication Date(Web):
DOI:10.1002/ijch.201400045
Abstract
The versatility of photochemical flow processes is rapidly expanding, and we suggest that this technique is capable of broadening the general appeal and impact of photochemistry similar or even to a greater extent to what microwave technology has done to thermal processes. This review highlights the recent developments that support this suggestion as well as the technological challenges that have yet to be resolved. Continuous photoflow reactions allow for better control over heating and mixing, are safer to operate on scale-up, in particular if LEDs are used in place of Hg lamps, they offer a higher surface/volume ratio and more efficient light penetration than batch mode photoreactions, and furthermore allow for the removal of products in-line to avoid secondary decompositions and poor conversions.
Co-reporter:Carl A. Busacca, John A. Milligan, Eakkaphon Rattanangkool, Cyrus Ramavarapu, Anji Chen, Anjan K. Saha, Zhibin Li, Heewon Lee, Steven J. Geib, Guijun Wang, Chris H. Senanayake, and Peter Wipf
The Journal of Organic Chemistry 2014 Volume 79(Issue 20) pp:9878-9887
Publication Date(Web):September 19, 2014
DOI:10.1021/jo501841s
The direct addition of anionic secondary phosphine boranes to carbodiimides yields both chiral and achiral phosphaguanidine boranes under ambient temperature conditions. An analogous preparation of menthol-derived phosphinite boranes is also described. These products can be deborinated to give the corresponding phosphines, and subsequently oxidized to give phosphine oxides. The robustness of this method was further demonstrated in the synthesis of structurally novel cyclic phosphaguanidines.
Co-reporter:Christopher J. Rosenker, Elizabeth H. Krenske, K. N. Houk, and Peter Wipf
Organic Letters 2013 Volume 15(Issue 5) pp:1076-1079
Publication Date(Web):February 13, 2013
DOI:10.1021/ol400094k
The energetics of thiol addition and elimination reactions to bicyclic enones derived from an indole core structure were explored using 1H NMR and density functional theory (DFT) calculations. The agreement between experiment and theory is excellent, and the combined results reveal that even minor changes in the conformation of the enone, substituents on the scaffold, and the use of different bases have a signficant influence on product distribution. A potential application of these principles is in the rational design of new reversible covalent enzyme inhibitors.
Co-reporter:Marie-Céline Frantz, Erin M. Skoda, Joshua R. Sacher, Michael W. Epperly, Julie P. Goff, Joel S. Greenberger and Peter Wipf
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 25) pp:4147-4153
Publication Date(Web):17 May 2013
DOI:10.1039/C3OB40489G
JP4-039 is a lead structure in a series of nitroxide conjugates that are capable of accumulating in mitochondria and scavenging reactive oxygen species (ROS). To explore structure–activity relationships (SAR), new analogs with variable nitroxide moieties were prepared. Furthermore, fluorophore-tagged analogs were synthesized and provided the opportunity for visualization in mitochondria. All analogs were tested for radioprotective and radiomitigative effects in 32Dcl3 cells.
Co-reporter:Matthew LaPorte, Ki Bum Hong, Jie Xu, and Peter Wipf
The Journal of Organic Chemistry 2013 Volume 78(Issue 1) pp:167-174
Publication Date(Web):November 8, 2012
DOI:10.1021/jo3022605
A convergent approach provides a convenient access to synthetically and biologically useful 3,4-disubstituted 5-hydroxyindoles. The one-pot procedure uses microwave heating to initiate an intramolecular [4 + 2]-cycloaddition of an alkynol segment onto a furan followed by a fragmentation, aromatization, and N-Boc deprotection cascade. Yields range from 15 to 74%, with aromatic substituents providing better conversions. 4-Trimethylsilylated analogues undergo a 1,3-silatropic rearrangement to give the O-TMS ethers.
Co-reporter:David M. Arnold, Matthew G. LaPorte, Shelby M. Anderson, Peter Wipf
Tetrahedron 2013 69(36) pp: 7719-7731
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.127
Co-reporter:Liming Cao, John P. Maciejewski, Stephan Elzner, David Amantini and Peter Wipf
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 30) pp:5811-5814
Publication Date(Web):20 Mar 2012
DOI:10.1039/C2OB25353D
The intramolecular Staudinger-aza-Wittig reaction is used for a general synthesis of 1,2,5,6-tetrahydro-1,2,4-triazines, a structural motif reported for the natural product noelaquinone. The DEF moiety of noelaquinone was obtained in 13 steps and 2% overall yield, and the structure of the synthetic product was confirmed by X-ray analysis.
Co-reporter:Erin M. Skoda, Gary C. Davis, and Peter Wipf
Organic Process Research & Development 2012 Volume 16(Issue 1) pp:26-34
Publication Date(Web):January 4, 2012
DOI:10.1021/op2002613
Nucleophilic imine additions with vinyl organometallics have developed into efficient, high-yielding, and robust methodologies to generate structurally diverse allylic amines. We have used the hydrozirconation/transmetalation/imine addition protocol in the synthesis of allylic amine intermediates for peptide bond isosteres, phosphatase inhibitors, and mitochondria-targeted peptide mimetics. The gramicidin S-derived XJB-5-131 and JP4-039 and their analogues have been prepared on up to 160-g scale for preclinical studies. These (E)-alkene peptide isosteres adopt type II′ β-turn secondary structures and display impressive biological properties including selective reactions with reactive oxygen species (ROS) and prevention of apoptosis.
Co-reporter:Mary Liang, Tyler B. Tarr, Karla Bravo-Altamirano, Guillermo Valdomir, Gabriel Rensch, Lauren Swanson, Nicholas R. DeStefino, Cara M. Mazzarisi, Rachel A. Olszewski, Gabriela Mustata Wilson, Stephen D. Meriney, and Peter Wipf
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 12) pp:985
Publication Date(Web):October 1, 2012
DOI:10.1021/ml3002083
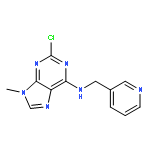
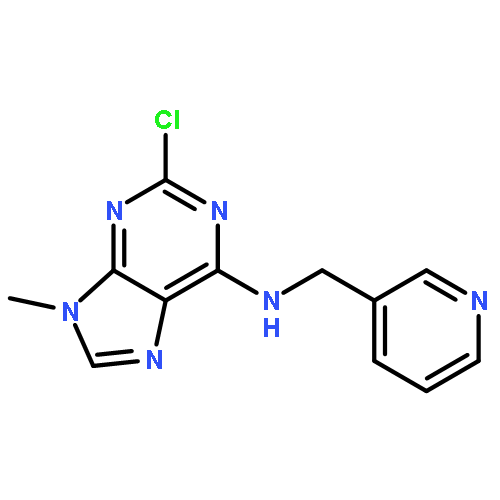
The acute effect of the potent cyclin-dependent kinase (cdk) inhibitor (R)-roscovitine on Ca2+ channels inspired the development of structural analogues as a potential treatment for motor nerve terminal dysfunction. On the basis of a versatile chlorinated purine scaffold, we have synthesized ca. 20 derivatives and characterized their N-type Ca2+ channel agonist action. Agents that showed strong agonist effects were also characterized in a kinase panel for their off-target effects. Among several novel compounds with diminished cdk activity, we identified a new lead structure with a 4-fold improved N-type Ca2+ channel agonist effect and a 22-fold decreased cdk2 activity as compared to (R)-roscovitine. This compound was selective for agonist activity on N- and P/Q-type over L-type calcium channels.Keywords: cdk2; Lambert−Eaton myasthenic syndrome; LEMS; N/P/Q-type calcium channels; neurological autoimmune disorder; roscovitine; selective agonist
Co-reporter:Filip R. Petronijevic
Journal of the American Chemical Society 2011 Volume 133(Issue 20) pp:7704-7707
Publication Date(Web):April 25, 2011
DOI:10.1021/ja2026882
Novel routes to the naturally occurring indole alkaloid cycloclavine and its unnatural C(5)-epimer are described. Key features include the rapid construction of the heterocyclic core segments by two Diels–Alder reactions. An indole annulation was accomplished by a late-stage intramolecular Diels–Alder furan cycloaddition, and a methylenecyclopropane dienophile was used for a stereoselective intramolecular [4 + 2] cycloaddition to give the cyclopropa[c]indoline building block present in cycloclavine.
Co-reporter:Adam T. Hoye and Peter Wipf
Organic Letters 2011 Volume 13(Issue 10) pp:2634-2637
Publication Date(Web):April 21, 2011
DOI:10.1021/ol200743u
A convergent route featuring [3,3]-sigmatropic rearrangements of a linchpin azepinopyrrolidine served to install two of the four contiguous stereocenters present in the tricyclic Stemona alkaloids sessilifoliamide and stemoamide. In addition to the first total synthesis of (–)-sessilifoliamide C, a potential biosynthetic relationship between the sessilifoliamides and previously reported Stemona alkaloids is presented.
Co-reporter:Chad D. Hopkins, John C. Schmitz, Edward Chu, and Peter Wipf
Organic Letters 2011 Volume 13(Issue 15) pp:4088-4091
Publication Date(Web):July 8, 2011
DOI:10.1021/ol2015994
The total synthesis of a bis-cyclopropane analog of the antimitotic natural product (−)-disorazole C1 was accomplished in 23 steps and 1.1% overall yield. A vinyl cyclopropane cross-metathesis reaction generated a key (E)-alkene segment of the target molecule. IC50 determinations of (−)-CP2-disorazole C1 in human colon cancer cell lines indicated low nanomolar cytotoxic properties. Accordingly, this synthetic bioisostere represents the first biologically active disorazole analog not containing a conjugated diene or polyene substructure element.
Co-reporter:Karla Bravo-Altamirano, Kara M. George, Marie-Céline Frantz, Courtney R. LaValle, Manuj Tandon, Stephanie Leimgruber, Elizabeth R. Sharlow, John S. Lazo, Q. Jane Wang, and Peter Wipf
ACS Medicinal Chemistry Letters 2011 Volume 2(Issue 2) pp:154
Publication Date(Web):December 14, 2010
DOI:10.1021/ml100230n
Protein kinase D (PKD) is a member of a novel family of serine/threonine kinases that regulate fundamental cellular processes. PKD is implicated in the pathogenesis of several diseases, including cancer. Progress in understanding the biological functions and therapeutic potential of PKD has been hampered by the lack of specific inhibitors. The benzoxoloazepinolone CID755673 was recently identified as the first potent and selective PKD inhibitor. The study of structure−activity relationships (SAR) of this lead compound led to further improvements in PKD1 potency. We describe herein the synthesis and biological evaluation of novel benzothienothiazepinone analogues. We achieved a 10-fold increase in the in vitro PKD1 inhibitory potency for the second generation lead kb-NB142-70 and accomplished a transition to an almost equally potent novel pyrimidine scaffold, while maintaining excellent target selectivity. These promising results will guide the design of pharmacological tools to dissect PKD function and pave the way for the development of potential anticancer agents.Keywords (keywords): benzothienothiazepinone; CID755673; Protein kinase D; pyrimidines; small molecule inhibitor
Co-reporter:Tingting Mo, Xueling Mi, Erin E. Milner, Geoffrey S. Dow, Peter Wipf
Tetrahedron Letters 2010 Volume 51(Issue 39) pp:5137-5140
Publication Date(Web):29 September 2010
DOI:10.1016/j.tetlet.2010.07.113
The 8-SF5 analog of mefloquine was synthesized in nine steps from commercially available starting materials and in five steps from a novel ortho-SF5–substituted aniline intermediate.
Co-reporter:Jared T. Hammill, Julia Contreras-García, Aaron M. Virshup, David N. Beratan, Weitao Yang, Peter Wipf
Tetrahedron 2010 66(31) pp: 5852-5862
Publication Date(Web):
DOI:10.1016/j.tet.2010.04.112
Co-reporter:Parag Mukhopadhyay, Peter Wipf and David N. Beratan
Accounts of Chemical Research 2009 Volume 42(Issue 6) pp:809
Publication Date(Web):April 20, 2009
DOI:10.1021/ar8002859
Modern chemistry emerged from the quest to describe the three-dimensional structure of molecules: van’t Hoff’s tetravalent carbon placed symmetry and dissymmetry at the heart of chemistry. In this Account, we explore how modern theory, synthesis, and spectroscopy can be used in concert to elucidate the symmetry and dissymmetry of molecules and their assemblies. Chiroptical spectroscopy, including optical rotatory dispersion (ORD), electronic circular dichroism (ECD), vibrational circular dichroism (VCD), and Raman optical activity (ROA), measures the response of dissymmetric structures to electromagnetic radiation. This response can in turn reveal the arrangement of atoms in space, but deciphering the molecular information encoded in chiroptical spectra requires an effective theoretical approach. Although important correlations between ECD and molecular stereochemistry have existed for some time, a battery of accurate new theoretical methods that link a much wider range of chiroptical spectroscopies to structure have emerged over the past decade. The promise of this field is considerable: theory and spectroscopy can assist in assigning the relative and absolute configurations of complex products, revealing the structure of noncovalent aggregates, defining metrics for molecular diversity based on polarization response, and designing chirally imprinted nanomaterials. The physical organic chemistry of chirality is fascinating in its own right: defining atomic and group contributions to optical rotation (OR) is now possible. Although the common expectation is that chiroptical response is determined solely by a chiral solute’s electronic structure in a given environment, chiral imprinting effects on the surrounding medium and molecular assembly can, in fact, dominate the chiroptical signatures. The theoretical interpretation of chiroptical markers is challenging because the optical properties are subtle, resulting from the strong electric dipole and the weaker electric quadrupole and magnetic dipole perturbations by the electromagnetic field. Moreover, OR arises from a combination of nearly canceling contributions to the electronic response. Indeed, the challenge posed by the chiroptical properties delayed the advent of even qualitatively accurate descriptions for some chiroptical signatures until the past decade when, for example, prediction of the observed sign of experimental OR became accessible to theory. The computation of chiroptical signatures, in close coordination with synthesis and spectroscopy, provides a powerful framework to diagnose and interpret the dissymmetry of chemical structures and molecular assemblies. Chiroptical theory now produces new schemes to elucidate structure, to describe the specific molecular sources of chiroptical signatures, and to assist in our understanding of how dissymmetry is templated and propagated in the condensed phase.
Co-reporter:Peter Wipf, Tingting Mo, Steven J. Geib, Diana Caridha, Geoffrey S. Dow, Lucia Gerena, Norma Roncal and Erin E. Milner
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 20) pp:4163-4165
Publication Date(Web):07 Aug 2009
DOI:10.1039/B911483A
Two novel SF5 analogs of the antimalarial agent mefloquine were synthesized in 5 steps and 10–23% overall yields and found to have improved activity and selectivity against malaria parasites. This work also represents the first report of SF5-substituted quinolines.
Co-reporter:Amir H. Faraji, Peter Wipf
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:2950-2962
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.02.043
This review highlights the properties of nanoparticles used in targeted drug delivery, including delivery to cells as well as organelle targets, some of the known pharmacokinetic properties of nanoparticles, and their typical modifications to allow for therapeutic delivery. Nanoparticles exploit biological pathways to achieve payload delivery to cellular and intracellular targets, including transport past the blood-brain barrier. As illustrative examples of their utility, the evaluation of targeted nanoparticles in the treatment of cancers and diseases of the central nervous system, such as glioblastoma multiforme, neurovascular disorders, and neurodegenerative diseases, is discussed.Nanoparticles exploit biological pathways to achieve payload delivery of small molecules to cellular and intracellular targets. Synthetic strategies, including surface, porosity, stealthing, and size modifications, can be utilized to refine the pharmacokinetic properties of nanoparticles and allow for efficient delivery.
Co-reporter:J.C. Burnett, C. Wang, J.E. Nuss, T.L. Nguyen, A.R. Hermone, J.J. Schmidt, R. Gussio, P. Wipf, S. Bavari
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 19) pp:5811-5813
Publication Date(Web):1 October 2009
DOI:10.1016/j.bmcl.2009.01.111
Botulinum neurotoxins, responsible for the neuroparalytic syndrome botulism, are the deadliest of known biological toxins. The work described in this study was based on a three-zone pharmacophore model for botulinum neurotoxin serotype A light chain inhibition. Specifically, the pharmacophore defined a separation between the overlaps of several different, non-zinc(II)-coordinating small molecule chemotypes, enabling the design and synthesis of a new structural hybrid possessing a Ki = 600 nM (±100 nM).The 3-zone pharmacophore for botulinum neurotoxin serotype A metalloprotease inhibition guided the design of a potent regioisomeric pair possessing a Ki = 600 (±100) nM.
Co-reporter:Mary Liang, Cecilia Saiz, Chiara Pizzo, Peter Wipf
Tetrahedron Letters 2009 50(49) pp: 6810-6813
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.09.107
Co-reporter:Adam T. Hoye, Jennifer E. Davoren and Peter Wipf, Mitchell P. Fink, Valerian E. Kagan
Accounts of Chemical Research 2008 Volume 41(Issue 1) pp:87
Publication Date(Web):January 15, 2008
DOI:10.1021/ar700135m
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are closely linked to degenerative diseases such as Alzheimer’s disease, Parkinson’s, neuronal death including ischemic and hemorrhagic stroke, acute and chronic degenerative cardiac myocyte death, and cancer. As a byproduct of oxidative phosphorylation, a steady stream of reactive species emerge from our cellular energy plants, the mitochondria. ROS and RNS potentially cause damage to all cellular components. Structure alteration, biomolecule fragmentation, and oxidation of side chains are trade-offs of cellular energy production. ROS and RNS escape results in the activation of cytosolic stress pathways, DNA damage, and the upregulation of JNK, p38, and p53. Incomplete scavenging of ROS and RNS particularly affects the mitochondrial lipid cardiolipin (CL), triggers the release of mitochondrial cytochrome c, and activates the intrinsic death pathway. Due to the active redox environment and the excess of NADH and ATP at the inner mitochondrial membrane, a broad range of agents including electron acceptors, electron donors, and hydride acceptors can be used to influence the biochemical pathways. The key to therapeutic value is to enrich selective redox modulators at the target sites. Our approach is based on conjugating nitroxides to segments of natural products with relatively high affinity for mitochondrial membranes. For example, a modified gramicidin S segment was successfully used for this purpose and proven to be effective in preventing superoxide production in cells and CL oxidation in mitochondria and in protecting cells against a range of pro-apoptotic triggers such as actinomycin D, radiation, and staurosporine. More importantly, these mitochondria-targeted nitroxide/gramicidin conjugates were able to protect against apoptosis in vivo by preventing CL oxidation induced by intestinal hemorrhagic shock. Optimization of nitroxide carriers could lead to a new generation of effective antiapoptotic agents acting at an early mitochondrial stage. Alternative chemistry-based approaches to targeting mitochondria include the use of proteins and peptides, as well as the attachment of payloads to lipophilic cationic compounds, sulfonylureas, anthracyclines, and other agents with proven or hypothetical affinities for mitochondria. Manganese superoxide dismutase (MnSOD), SS tetrapeptides with 2′,6′-dimethyltyrosine (Dmt) residues, rhodamine, triphenylphosphonium salts, nonopioid analgesics, adriamycin, and diverse electron-rich aromatics and stilbenes were used to influence mitochondrial biochemistry and the biology of aging. Some general structural principles for effective therapeutic agents are now emerging. Among these are the presence of basic or positively charged functional groups, hydrophobic substructures, and, most promising for future selective strategies, classes of compounds that are actively shuttled into mitochondria, bind to mitochondria-specific proteins, or show preferential affinity to mitochondria-specific lipids.
Co-reporter:Filip Petronijevic, Cody Timmons, Anthony Cuzzupe and Peter Wipf
Chemical Communications 2008 (Issue 1) pp:2008
Publication Date(Web):11 Nov 2008
DOI:10.1039/B816989F
The key steps of a versatile new protocol for the convergent synthesis of 3,4-disubstituted indoles are the addition of an α-lithiated alkylaminofuran to a carbonyl compound, a microwave-accelerated intramolecular Diels–Alder cycloaddition and an in situ double aromatization reaction.
Co-reporter:Peter Wipf;Nilukshi Jayasuriya
Chirality 2008 Volume 20( Issue 3-4) pp:425-430
Publication Date(Web):
DOI:10.1002/chir.20468
Abstract
The in situ hydrozirconation–transmetalation-aldehyde addition process is a convenient method for the generation of allylic alcohols. Ongoing research has focused on enhancing the enantioselectivity and substrate scope of this process. A chiral β-amino thiol scaffold was evaluated in the addition reaction. Amino thiols tend to provide the highest ee's, in part due to the higher affinity of sulfur for zinc over zirconium. A class of valine-based thiol ligands was identified to be effective for the formation of enantiomerically enriched allylic alcohols in terms of low ligand loading and high % ee. Chirality, 2008. © 2007 Wiley-Liss, Inc.
Co-reporter:David L. Waller, Corey R. J. Stephenson and Peter Wipf
Organic & Biomolecular Chemistry 2007 vol. 5(Issue 1) pp:58-60
Publication Date(Web):14 Nov 2006
DOI:10.1039/B612992G
Oxidative rearrangement of cyclic enol ethers leads to α-alkoxyesters. In the presence of a neighboring spiroether, this approach provides a stereoselective access to spiroketals. A modified proposal for the biosynthesis of acutumine is presented.
Co-reporter:Peter Wipf;Stefan Werner;Leslie A. Twining;Christopher Kendall
Chirality 2007 Volume 19(Issue 1) pp:5-9
Publication Date(Web):6 NOV 2006
DOI:10.1002/chir.20331
Evaporative light-scattering detection (ELSD) is widely applied in the HPLC analysis of organic compounds lacking a UV chromophore. However, this detection method is generally unsuitable for determination of enantiomeric ratios (er). The er calculated from a UV trace and an ELS trace of the same compound differs significantly. Because of the nonlinear concentration response of the ELS detector, a compound with an er of 95:5 appears to be enantiomerically pure by ELS detection. It is possible to obtain a calibration curve and to calculate a correction factor, but this procedure is time consuming and therefore not very practical for routine analyses. In contrast, a charged aerosol detector allows a very accurate determination of the enantiomeric ratios. Like the ELS detection, the CA detection is independent of the chromophore properties of the substrate. Therefore, we recommend the use of CA instead of ELS detection for determination of the enantiomeric ratios of non-UV active compounds. Chirality, 2006. © 2006 Wiley-Liss, Inc.
Co-reporter:Peter Wipf;Thomas H. Graham;Andreas Vogt;Rachel P. Sikorski;Alexer P. Ducruet and;John S. Lazo
Chemical Biology & Drug Design 2006 Volume 67(Issue 1) pp:
Publication Date(Web):8 DEC 2005
DOI:10.1111/j.1747-0285.2005.00313.x
Structure–activity analyses of synthetic disorazole C1 and eight of its analogs indicate that the presence of a vinyl oxirane moiety or a tetraene sequence is not necessary for potent cytotoxic and antimitotic properties. Using an automated multiparameter fluorescence-based cellular assay to simultaneously probe the effects of disorazole analogs on cellular microtubules, mitotic arrest, and cytotoxicity, we found that disorazole C1 enhanced the mitotic index and chromatin condensation and arrested cells in the G2/M phase of the cell cycle. All structural analogs and synthesis precursors of disorazole C1 were at least two orders of magnitude less potent than the parent compound, thus indicating that both the functional group array and the three-dimensional conformation of the parent compound are critical for interaction with the biological target. We conclude that disorazole C1 is a potent inducer of mitotic arrest and hypothesize that this biological activity may be mediated by microtubule perturbation.
Co-reporter:Peter Wipf ;Maciej A. A. Walczak
Angewandte Chemie 2006 Volume 118(Issue 25) pp:
Publication Date(Web):19 MAY 2006
DOI:10.1002/ange.200600723
Direkter Zugang: Phasentransfer-N-Allylierung oder -N-Propargylierung von (Bicyclo[1.1.0]butylmethyl)aminen löst diastereoselektive pericyclische Kaskadenreaktionen aus, die letztlich auf formalen En- oder [2+2]-Wegen neuartige spirocyclische und tricyclische Pyrrolidinheterocyclen liefern.
Co-reporter:Peter Wipf ;Maciej A. A. Walczak
Angewandte Chemie International Edition 2006 Volume 45(Issue 25) pp:
Publication Date(Web):19 MAY 2006
DOI:10.1002/anie.200600723
Direct access: Phase-transfer N-allylation and N-propargylation of (bicyclo[1.1.0]butylmethyl)amines initiate diastereoselective pericyclic cascade reactions that culminate in novel spirocyclic and tricyclic pyrrolidine heterocycles through formal ene or [2+2] pathways.
Co-reporter:Parag Mukhopadhyay;Gérard Zuber Dr.;Michael-Rock Goldsmith Dr. ;David N. Beratan
ChemPhysChem 2006 Volume 7(Issue 12) pp:2483-2486
Publication Date(Web):30 OCT 2006
DOI:10.1002/cphc.200600477
Explicit solute–solvent interactions: MD simulations and TD-DFT are used to determine the solvent dependence of the optical rotatory dispersion (ORD) for methyloxirane in water (see figure). The inclusion of explicit solute–solvent interactions is essential to describe the influence of the solvent on the OR of the system and the MD simulations provide a suitable means to analyze and predict chiroptical solvation effects.
Co-reporter:Peter Wipf and Tamara D. Hopkins
Chemical Communications 2005 (Issue 27) pp:3421-3423
Publication Date(Web):01 Jun 2005
DOI:10.1039/B505100B
The convergent total synthesis of the marine natural product (+)-bistramide C confirms the a priori assignments of its relative and absolute configurations, which were originally based on the combined application of [α]D analysis, NMR, and synthesis.
Co-reporter:Peter Wipf;Jingbo Xiao;Steven J. Geib
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 11-13) pp:
Publication Date(Web):19 OCT 2005
DOI:10.1002/adsc.200505171
Divergent multi-component reactions (DMCR) involving CC bond formations can provide large increases in structural diversity and allow the rapid assembly of complex products from readily available starting materials. Cascade hydrozirconation-Zr/Zn transmetalation-imine addition of alkynes represents a versatile methodology for the synthesis of (E)-alkene and cyclopropane dipeptide isosteres. Appropriate substitutions at the sp2-carbon of (E)-alkene peptide isosteres allow a range of Pd-catalyzed cross-coupling reactions, which can be used for the fine-tuning of the conformational and electronic properties of the parent peptide bond mimic. CC bond formation by microwave-accelerated Stille coupling of stannylalkenes represents a fast, convergent synthetic approach toward trisubstituted (E)-alkene dipeptide isosteres.
Co-reporter:Peter Wipf, Stephen M. Lynch, Garth Powis, Anne Birmingham and Erikah E. Englund
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 21) pp:3880-3882
Publication Date(Web):28 Sep 2005
DOI:10.1039/B510718K
A series of palmarumycin prodrugs and water-soluble analogs has been synthesized and assayed for inhibition of the thioredoxin–thioredoxin reductase system. Increased aqueous solubility led to an improved in vivo activity profile.
Co-reporter:Peter Wipf and Robert J. Halter
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 11) pp:2053-2061
Publication Date(Web):20 Apr 2005
DOI:10.1039/B504418A
Recent synthetic and biological studies of the viridin class of steroidal furans have revealed multiple opportunities for fundamental discoveries as well as advanced drug design. Wortmannin is a potent enzyme inhibitor that binds to the ATP site of important regulatory kinases such as PI-3 kinase and Polo-like kinase. The natural product shares a unique mechanism-based biological activation pathway with other viridins. Furthermore, while there have been several encouraging approaches toward the total synthesis of these compounds, there is still ample room for improvements in synthetic strategies and tactics, and the development of structurally simplified analogs that exert more specific biological effects and are devoid of toxicity issues that have thwarted the clinical development of the parent compounds.
Co-reporter:Peter Wipf and Thomas H. Graham
Organic & Biomolecular Chemistry 2005 vol. 3(Issue 1) pp:31-35
Publication Date(Web):25 Nov 2004
DOI:10.1039/B413604G
Stereoselective conjugate additions of alcohols, amines, thiols, and halides to C(2)-alkynyl oxazoles and oxazolines provide a versatile entry to heterocyclic building blocks.
Co-reporter:Gérard Zuber;Michael-Rock Goldsmith;David N. Beratan
Chirality 2005 Volume 17(Issue 8) pp:507-510
Publication Date(Web):24 AUG 2005
DOI:10.1002/chir.20190
In this study, we report theoretical specific rotation values for a series of cis-/trans-alkylated-[5]-ladderanes and cis-/trans-methylated-[n]-ladderanes. Using time-dependent density functional response theory optical rotation calculations, we can assign (+) and (−) optical rotation signs to trans-(S)-alkyl-[5]-ladderane and trans-(R)-alkyl-[5]-ladderane configurations, respectively. In order to qualitatively validate our absolute configuration predictions, we computed optical rotation values at three different levels of theory—B3LYP, RI-BP86, and Hartree–Fock—using the aug-cc-pVDZ basis set. We observe a novel rung–parity-controlled oscillatory optical rotatory phenomenon in our computations, which, to the best of our knowledge, has never been reported or observed before. Furthermore, this preliminary analysis of optical rotation properties in this class of compounds should facilitate the correct absolute stereochemical assignment of natural and synthetic ladderanes, such as the trans-isomer of pentacyclic C20-fatty acid methyl ester (pentacycloanammoxic methyl ester), without the need for derivatization, in particular for cases where NMR or X-ray crystal structures are not readily available. © 2005 Wiley-Liss, Inc. Chirality 17:507–510, 2005.
Co-reporter:Peter Wipf, Stephen M. Lynch, Anne Birmingham, Giselle Tamayo, Allan Jiménez, Nefertiti Campos and Garth Powis
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 11) pp:1651-1658
Publication Date(Web):11 May 2004
DOI:10.1039/B402431A
Spiroketal naphthodecalins are readily assembled by Barton's base mediated Ullmann binaphthyl ether coupling, Dakin reactions and hypervalent iodine spirocyclization. The core structures can be further diversified by enone addition and Stille coupling reactions. Nanomolar inhibitors for the Trx/TrxR redox control system were prepared by this approach and compared to series of natural product isolates. Cytotoxicity in MCF-7 cell assays ranged from an IC50 of 1.6 to >100 µM.
Co-reporter:Peter Wipf, Beomjun Joo, Theresa Nguyen and John S. Lazo
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 15) pp:2173-2174
Publication Date(Web):05 Jul 2004
DOI:10.1039/B408184F
The marine bryozoan metabolites caulibugulone A–E were prepared from a readily available isoquinoline dione. These natural products were found to be potent and selective inhibitors of the dual specificity phosphatase Cdc25B.
Co-reporter:Peter Wipf, Joshua G. Pierce, Xiaodong Wang
Tetrahedron: Asymmetry 2003 Volume 14(Issue 22) pp:3605-3611
Publication Date(Web):14 November 2003
DOI:10.1016/j.tetasy.2003.07.022
The recently developed (cyclohexylsulfonylamino)oxazoline (CHAOx) ligand was found to provide high ee's and consistent reaction rates in the asymmetric diethylzinc addition to benzaldehyde over a remarkably large loading range of 0.05–10 mol%. Turnover numbers of 1000–2000 can be explained by the absence of a nonlinear effect and the formation of a catalytically active monomer complex. Substituents at the nitrogen donor atoms of the bidentate ligand prevent zinc-complex dimerization.Graphic
Co-reporter:Peter Wipf;Nilukshi Jayasuriya;Seth Ribe
Chirality 2003 Volume 15(Issue 3) pp:208-212
Publication Date(Web):7 FEB 2003
DOI:10.1002/chir.10176
Unusual nonlinear asymmetric amplification and chiral ligand loading effects were discovered for the use of catalytic quantities of chiral aminoalcohols in the in situ hydrozirconation—transmetalation—aldehyde addition processes. While the stereochemically most efficient aminothiol ligands demonstrated mechanistically conventional reaction parameters in excellent agreement with Kagan's ML2 system, the asymmetric induction in the presence of a chiral aminoalcohol was found to vary greatly with loading and %ee of the ligand. Aminothiols remain the ligands of choice for the highly enantioselective formation of allylic alcohols and provide experimentally more predictable reaction variables. However, new, optimized conditions lead to a synthetically useful product %ee using the readily available and scalable aminoalcohol 2a. Chirality 15:208–212, 2003. © 2003 Wiley-Liss, Inc.
Co-reporter:Michael A. Lyon,
Alexander P. Ducruet,
Peter Wipf
&
John S. Lazo
Nature Reviews Drug Discovery 2002 1(12) pp:961
Publication Date(Web):
DOI:10.1038/nrd963
Dual-specificity protein phosphatases are a subclass of protein tyrosine phosphatases that are uniquely able to hydrolyse the phosphate ester bond on both a tyrosine and a threonine or serine residue on the same protein. Dual-specificity phosphatases have a central role in the complex regulation of signalling pathways that are involved in cell stress responses, proliferation and death. Although this enzyme family is increasingly the target of drug discovery efforts in pharmaceutical companies, a summary of the salient developments in the biology and medicinal chemistry of dual-specificity phosphatases has been lacking. We hope that this comprehensive overview will stimulate further progress in the development of small-molecule inhibitors that could form the basis for a new class of target-directed therapeutic agents.
Co-reporter:Peter Wipf and Jonathan T. Reeves
Chemical Communications 2002 (Issue 18) pp:2066-2067
Publication Date(Web):16 Aug 2002
DOI:10.1039/B207383H
A convergent synthesis of the macrocyclic core of the marine macrolide leucascandrolide A has been accomplished.
Co-reporter:Peter Wipf, Tamara D. Hopkins, Jae-Kyu Jung, Sonia Rodriguez, Anne Birmingham, Eileen C. Southwick, John S. Lazo, Garth Powis
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 19) pp:2637-2641
Publication Date(Web):8 October 2001
DOI:10.1016/S0960-894X(01)00525-X
Natural products of the naphthoquinone spiroketal structural type served as lead structures for the development of novel inhibitors of the thioredoxin–thioredoxin reductase redox system. The most potent compound in this series inhibited thioredoxin with an IC50 of 350 nM, and many derivatives showed low micromolar activities for growth inhibition against two breast cancer cell lines.Synthetic analogues of palmarumycin CP1 were evaluated as new nanomolar to low-micromolar inhibitors of the thioredoxin–thioredoxin reductase redox system. Many derivatives showed significant growth inhibition against two breast cancer cell lines.
Co-reporter:Peter Wipf, Diana C. Aslan, Eileen C. Southwick, John S. Lazo
Bioorganic & Medicinal Chemistry Letters 2001 Volume 11(Issue 3) pp:313-317
Publication Date(Web):2 February 2001
DOI:10.1016/S0960-894X(00)00658-2
Based on a previously identified lead structure, SC-ααδ9, we have developed a versatile new chemical scaffold that can be readily modified to generate libraries of both Tyr and dual specificity phosphatase inhibitors with reduced molecular weight and lipophilicity. The most potent analogue identified to date, aminothiazole 8z, inhibits the dual specificity phosphatase Cdc25B with a Ki of 4.6 ± 0.4 μM and a Hill coefficient of 2.Sulfonylated aminothiazoles represent a versatile new chemical scaffold that can be readily modified to generate libraries of both Tyr and dual specificity phosphatase inhibitors with reduced molecular weight and lipophilicity. The most potent analogue in this series inhibits the dual specificity phosphatase Cdc25B with a Ki of 4.6 ± 0.4 μM.
Co-reporter:David L. Waller, Corey R. J. Stephenson and Peter Wipf
Organic & Biomolecular Chemistry 2007 - vol. 5(Issue 1) pp:NaN60-60
Publication Date(Web):2006/11/14
DOI:10.1039/B612992G
Oxidative rearrangement of cyclic enol ethers leads to α-alkoxyesters. In the presence of a neighboring spiroether, this approach provides a stereoselective access to spiroketals. A modified proposal for the biosynthesis of acutumine is presented.
Co-reporter:Peter Wipf, Tingting Mo, Steven J. Geib, Diana Caridha, Geoffrey S. Dow, Lucia Gerena, Norma Roncal and Erin E. Milner
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 20) pp:NaN4165-4165
Publication Date(Web):2009/08/07
DOI:10.1039/B911483A
Two novel SF5 analogs of the antimalarial agent mefloquine were synthesized in 5 steps and 10–23% overall yields and found to have improved activity and selectivity against malaria parasites. This work also represents the first report of SF5-substituted quinolines.
Co-reporter:Marie-Céline Frantz, Erin M. Skoda, Joshua R. Sacher, Michael W. Epperly, Julie P. Goff, Joel S. Greenberger and Peter Wipf
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 25) pp:NaN4153-4153
Publication Date(Web):2013/05/17
DOI:10.1039/C3OB40489G
JP4-039 is a lead structure in a series of nitroxide conjugates that are capable of accumulating in mitochondria and scavenging reactive oxygen species (ROS). To explore structure–activity relationships (SAR), new analogs with variable nitroxide moieties were prepared. Furthermore, fluorophore-tagged analogs were synthesized and provided the opportunity for visualization in mitochondria. All analogs were tested for radioprotective and radiomitigative effects in 32Dcl3 cells.
Co-reporter:Liming Cao, John P. Maciejewski, Stephan Elzner, David Amantini and Peter Wipf
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 30) pp:NaN5814-5814
Publication Date(Web):2012/03/20
DOI:10.1039/C2OB25353D
The intramolecular Staudinger-aza-Wittig reaction is used for a general synthesis of 1,2,5,6-tetrahydro-1,2,4-triazines, a structural motif reported for the natural product noelaquinone. The DEF moiety of noelaquinone was obtained in 13 steps and 2% overall yield, and the structure of the synthetic product was confirmed by X-ray analysis.
Co-reporter:Joseph M. Salamoun, Kelley E. McQueeney, Kalyani Patil, Steven J. Geib, Elizabeth R. Sharlow, John S. Lazo and Peter Wipf
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 27) pp:NaN6402-6402
Publication Date(Web):2016/06/07
DOI:10.1039/C6OB00946H
The phosphatase PTP4A3 is an attractive anticancer target, but knowledge of its exact role in cells remains incomplete. A potent, structurally novel inhibitor of the PTP4A family was obtained by photooxygenation of a less active, electron-rich thienopyridone (1). Iminothienopyridinedione 13 displays increased solution stability and is readily obtained by two new synthetic routes that converge in the preparation of 1. The late-stage photooxygenation of 1 to give 13 in high yield highlights the potential of this reaction to modify the structure and properties of a biological lead compound and generate value for expanding the scope of an SAR investigation. Analog 13 should become a valuable tool for further exploration of the role of PTP4A3 in tumor progression.
Co-reporter:Filip Petronijevic, Cody Timmons, Anthony Cuzzupe and Peter Wipf
Chemical Communications 2008(Issue 1) pp:-106
Publication Date(Web):2008/11/11
DOI:10.1039/B816989F
The key steps of a versatile new protocol for the convergent synthesis of 3,4-disubstituted indoles are the addition of an α-lithiated alkylaminofuran to a carbonyl compound, a microwave-accelerated intramolecular Diels–Alder cycloaddition and an in situ double aromatization reaction.
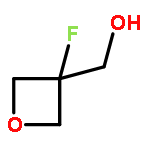
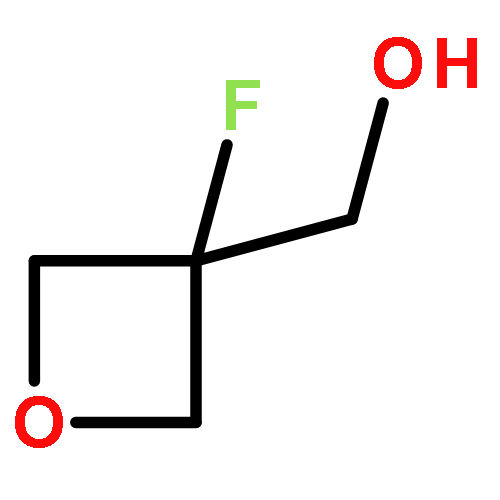
.jpg)


![2-Bromo-6,7-dihydrothieno[3,2-c]pyridin-4(5H)-one](http://img.cochemist.com/ccimg/1078200/1078150-17-0.png)
![2-Bromo-6,7-dihydrothieno[3,2-c]pyridin-4(5H)-one](http://img.cochemist.com/ccimg/1078200/1078150-17-0_b.png)


![L-Ornithine, L-prolyl-L-valyl-N5-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/852200/852156-57-1.png)
![L-Ornithine, L-prolyl-L-valyl-N5-[(phenylmethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/852200/852156-57-1_b.png)


![2-phenyl-5h-thieno[3,2-c]pyridin-4-one](http://img.cochemist.com/ccimg/690700/690636-03-4.png)
![2-phenyl-5h-thieno[3,2-c]pyridin-4-one](http://img.cochemist.com/ccimg/690700/690636-03-4_b.png)




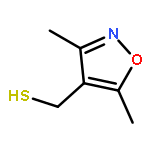
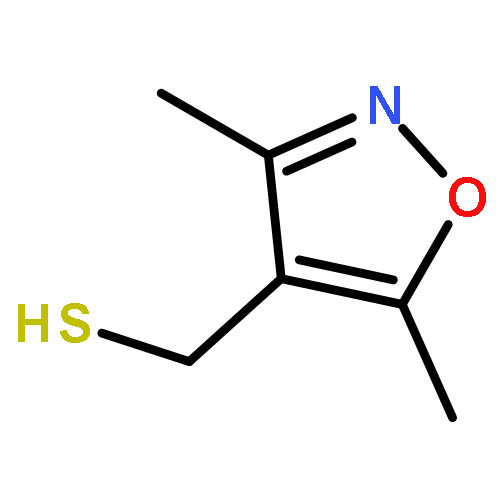
![2-[(3,5-dimethyl-1,2-oxazol-4-yl)methylsulfanyl]acetic Acid](http://img.cochemist.com/ccimg/446900/446830-00-8.png)
![2-[(3,5-dimethyl-1,2-oxazol-4-yl)methylsulfanyl]acetic Acid](http://img.cochemist.com/ccimg/446900/446830-00-8_b.png)


![Thieno[2,3-d]pyrimidin-4(1H)-one, 2,3-dihydro-6-phenyl-2-thioxo-](http://img.cochemist.com/ccimg/185300/185202-49-7.png)
![Thieno[2,3-d]pyrimidin-4(1H)-one, 2,3-dihydro-6-phenyl-2-thioxo-](http://img.cochemist.com/ccimg/185300/185202-49-7_b.png)
![8-Boc-3-(trifluoromethylsulfonyloxy)-8-azabicyclo[3.2.1]oct-3-ene](http://img.cochemist.com/ccimg/185100/185099-68-7.png)
![8-Boc-3-(trifluoromethylsulfonyloxy)-8-azabicyclo[3.2.1]oct-3-ene](http://img.cochemist.com/ccimg/185100/185099-68-7_b.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)
![2-Thioxo-2,3-dihydrothieno[2,3-d]pyrimidin-4(1H)-one](http://img.cochemist.com/ccimg/117600/117516-97-9.png)
![2-Thioxo-2,3-dihydrothieno[2,3-d]pyrimidin-4(1H)-one](http://img.cochemist.com/ccimg/117600/117516-97-9_b.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthene]-ar-carboxylicacid, 2',7'-dichloro-3',6'-dihydroxy-3-oxo-](http://img.cochemist.com/ccimg/111900/111843-78-8.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthene]-ar-carboxylicacid, 2',7'-dichloro-3',6'-dihydroxy-3-oxo-](http://img.cochemist.com/ccimg/111900/111843-78-8_b.png)


![1-Propanol, 2,3-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/108700/108670-86-6.png)
![1-Propanol, 2,3-bis[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/108700/108670-86-6_b.png)
![Hexanoic acid,6-[(aminothioxomethyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/99000/98998-60-8.png)
![Hexanoic acid,6-[(aminothioxomethyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/99000/98998-60-8_b.png)




![L-Valine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](http://img.cochemist.com/ccimg/75000/74912-90-6.png)
![L-Valine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](http://img.cochemist.com/ccimg/75000/74912-90-6_b.png)
![6,7-dihydro-5H-thieno[3,2-c]pyridin-4-one](http://img.cochemist.com/ccimg/68600/68559-60-4.png)
![6,7-dihydro-5H-thieno[3,2-c]pyridin-4-one](http://img.cochemist.com/ccimg/68600/68559-60-4_b.png)
![2-CHLORO-1-[4-(2-METHYLPHENYL)PIPERAZIN-1-YL]ETHANONE](http://img.cochemist.com/ccimg/60200/60121-79-1.png)
![2-CHLORO-1-[4-(2-METHYLPHENYL)PIPERAZIN-1-YL]ETHANONE](http://img.cochemist.com/ccimg/60200/60121-79-1_b.png)

![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9.png)
![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9_b.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2_b.png)













![Benzene, [1-(chloromethyl)ethenyl]-](/data/chemimg/554200/3360-52-9.png)
![Benzene, [1-(chloromethyl)ethenyl]-](/data/chemimg/554200/3360-52-9_b.png)





![Bicyclo[1.1.0]butane-1-carbonitrile, 3-methyl-](http://img.cochemist.com/ccimg/700/694-25-7.png)
![Bicyclo[1.1.0]butane-1-carbonitrile, 3-methyl-](http://img.cochemist.com/ccimg/700/694-25-7_b.png)
![7,8,9,11a-tetrahydro-5H-Azepino[2,1-a]isoindol-5-one](http://img.cochemist.com/ccimg/956800/956736-71-3.png)
![7,8,9,11a-tetrahydro-5H-Azepino[2,1-a]isoindol-5-one](http://img.cochemist.com/ccimg/956800/956736-71-3_b.png)
![Benzene,1-chloro-4-[isocyano[(4-methylphenyl)sulfonyl]methyl]-](http://img.cochemist.com/ccimg/918900/918892-30-5.png)
![Benzene,1-chloro-4-[isocyano[(4-methylphenyl)sulfonyl]methyl]-](http://img.cochemist.com/ccimg/918900/918892-30-5_b.png)


![Bicyclo[3.3.1]nonan-3-one, 9-amino-7-methylene-](http://img.cochemist.com/ccimg/918900/918830-96-3.png)
![Bicyclo[3.3.1]nonan-3-one, 9-amino-7-methylene-](http://img.cochemist.com/ccimg/918900/918830-96-3_b.png)
![1H-Pyrazolo[3,4-d]pyrimidine, 4,6-dichloro-1-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/864300/864292-49-9.png)
![1H-Pyrazolo[3,4-d]pyrimidine, 4,6-dichloro-1-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/864300/864292-49-9_b.png)

![2H-Indol-2-one, 1,3-dihydro-3-[(3-methoxy-1H-pyrrol-2-yl)methylene]-](http://img.cochemist.com/ccimg/856500/856436-10-7.png)
![2H-Indol-2-one, 1,3-dihydro-3-[(3-methoxy-1H-pyrrol-2-yl)methylene]-](http://img.cochemist.com/ccimg/856500/856436-10-7_b.png)
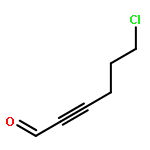
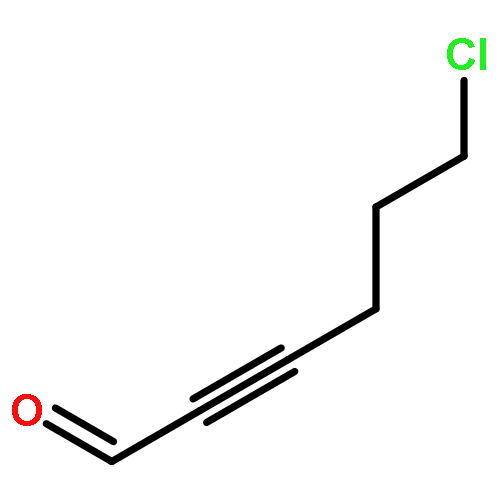
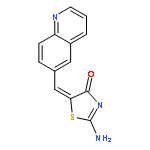
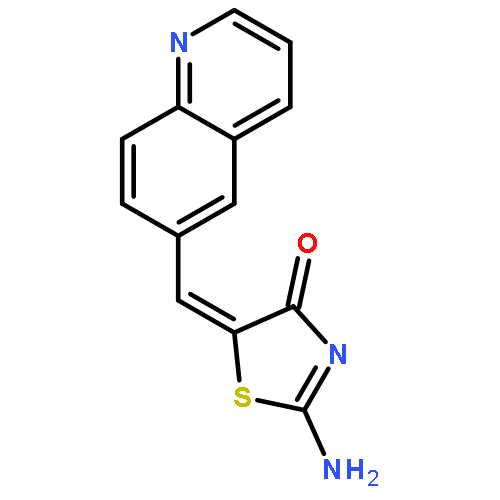
![4(5H)-Thiazolone, 2-[(phenylmethyl)amino]-5-(6-quinolinylmethylene)-](http://img.cochemist.com/ccimg/843700/843641-28-1.png)
![4(5H)-Thiazolone, 2-[(phenylmethyl)amino]-5-(6-quinolinylmethylene)-](http://img.cochemist.com/ccimg/843700/843641-28-1_b.png)




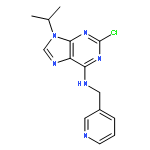
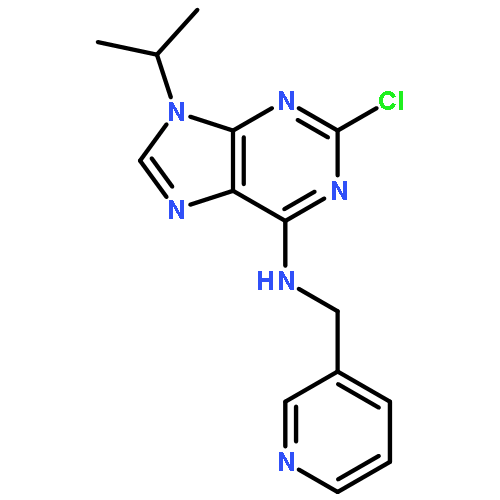


![9-hydroxy-4-phenyl-6h-pyrrolo[3,4-c]carbazole-1,3-dione](http://img.cochemist.com/ccimg/622900/622864-54-4.png)
![9-hydroxy-4-phenyl-6h-pyrrolo[3,4-c]carbazole-1,3-dione](http://img.cochemist.com/ccimg/622900/622864-54-4_b.png)
![2-Methyl-2-propanyl (3-exo)-9-azabicyclo[3.3.1]non-3-ylcarbamate](http://img.cochemist.com/ccimg/599200/599165-35-2.png)
![2-Methyl-2-propanyl (3-exo)-9-azabicyclo[3.3.1]non-3-ylcarbamate](http://img.cochemist.com/ccimg/599200/599165-35-2_b.png)
![Acetamide, N-(2,3,4,9-tetrahydro-1-oxo-1H-pyrido[3,4-b]indol-6-yl)-](http://img.cochemist.com/ccimg/583900/583827-57-0.png)
![Acetamide, N-(2,3,4,9-tetrahydro-1-oxo-1H-pyrido[3,4-b]indol-6-yl)-](http://img.cochemist.com/ccimg/583900/583827-57-0_b.png)
![Benzofuro[2,3-c]pyridin-1(2H)-one, 3,4-dihydro-6-hydroxy-](http://img.cochemist.com/ccimg/522000/521937-03-1.png)
![Benzofuro[2,3-c]pyridin-1(2H)-one, 3,4-dihydro-6-hydroxy-](http://img.cochemist.com/ccimg/522000/521937-03-1_b.png)


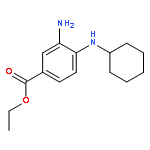
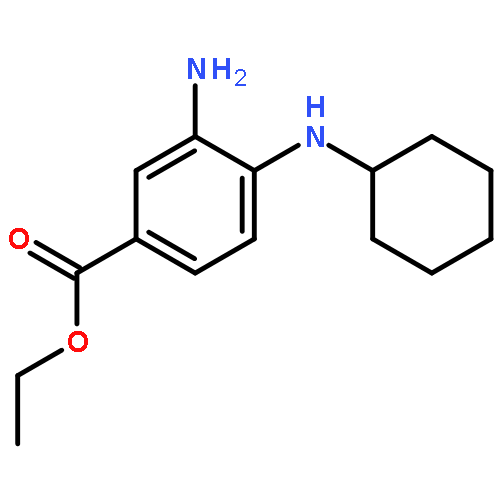
![Benzoic acid, 4-[(1,2-dihydro-1-oxo-3-isoquinolinyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/500200/500198-65-2.png)
![Benzoic acid, 4-[(1,2-dihydro-1-oxo-3-isoquinolinyl)amino]-, ethyl ester](http://img.cochemist.com/ccimg/500200/500198-65-2_b.png)
![1(2H)-Isoquinolinone, 3-[(4-methylphenyl)amino]-](http://img.cochemist.com/ccimg/500200/500198-64-1.png)
![1(2H)-Isoquinolinone, 3-[(4-methylphenyl)amino]-](http://img.cochemist.com/ccimg/500200/500198-64-1_b.png)














![9H-PURIN-6-AMINE, N-([1,1'-BIPHENYL]-4-YLMETHYL)-2-CHLORO-9-PROPYL-](http://img.cochemist.com/ccimg/441100/441055-14-7.png)
![9H-PURIN-6-AMINE, N-([1,1'-BIPHENYL]-4-YLMETHYL)-2-CHLORO-9-PROPYL-](http://img.cochemist.com/ccimg/441100/441055-14-7_b.png)
![9H-Purin-6-amine, N-([1,1'-biphenyl]-4-ylmethyl)-2-chloro-9-methyl-](http://img.cochemist.com/ccimg/441100/441055-11-4.png)
![9H-Purin-6-amine, N-([1,1'-biphenyl]-4-ylmethyl)-2-chloro-9-methyl-](http://img.cochemist.com/ccimg/441100/441055-11-4_b.png)






![NSC 663284;6-CHLORO-7-[[2-(4-MORPHOLINYL)ETHYL]AMINO]-5,8-QUINOLINEDIONE](http://img.cochemist.com/ccimg/384000/383907-43-5.png)
![NSC 663284;6-CHLORO-7-[[2-(4-MORPHOLINYL)ETHYL]AMINO]-5,8-QUINOLINEDIONE](http://img.cochemist.com/ccimg/384000/383907-43-5_b.png)
![Phenol, 2-[4-[(3-aminopropyl)amino]-6-methyl-2-pyrimidinyl]-](/data/chemimg/124700/378217-35-7.png)
![Phenol, 2-[4-[(3-aminopropyl)amino]-6-methyl-2-pyrimidinyl]-](/data/chemimg/124700/378217-35-7_b.png)
![Benzenesulfonamide, N-[1-butyl-3-(phenylsulfonyl)-1H-pyrrolo[2,3-b]quinoxalin-2-yl]-4-methyl-](/data/chemimg/109500/377064-07-8.png)
![Benzenesulfonamide, N-[1-butyl-3-(phenylsulfonyl)-1H-pyrrolo[2,3-b]quinoxalin-2-yl]-4-methyl-](/data/chemimg/109500/377064-07-8_b.png)




![<br>8-Methyl-3a,4,5,9b-tetrahydro-3 H -cyclopenta[ c ]quinoline-4-carboxylic ac id](http://img.cochemist.com/ccimg/354900/354815-91-1.png)
![<br>8-Methyl-3a,4,5,9b-tetrahydro-3 H -cyclopenta[ c ]quinoline-4-carboxylic ac id](http://img.cochemist.com/ccimg/354900/354815-91-1_b.png)
![3a,4,5,9b-Tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylic acid](http://img.cochemist.com/ccimg/354900/354815-90-0.png)
![3a,4,5,9b-Tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylic acid](http://img.cochemist.com/ccimg/354900/354815-90-0_b.png)
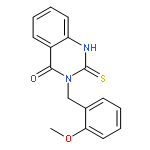
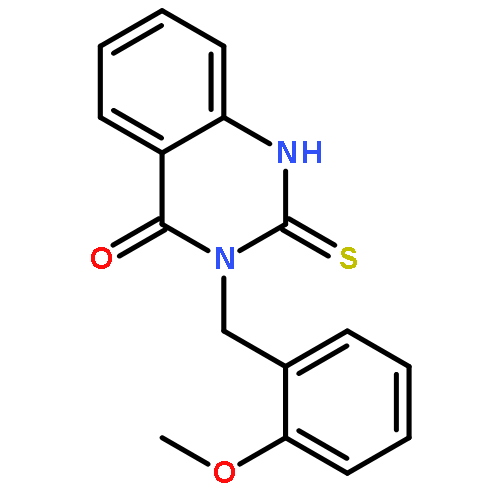




![(ne)-4-methyl-n-[4-oxo-3-(1h-1,2,4-triazol-5-ylsulfanyl)naphthalen-1-ylidene]benzenesulfonamide](http://img.cochemist.com/ccimg/315700/315698-91-0.png)
![(ne)-4-methyl-n-[4-oxo-3-(1h-1,2,4-triazol-5-ylsulfanyl)naphthalen-1-ylidene]benzenesulfonamide](http://img.cochemist.com/ccimg/315700/315698-91-0_b.png)
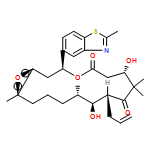
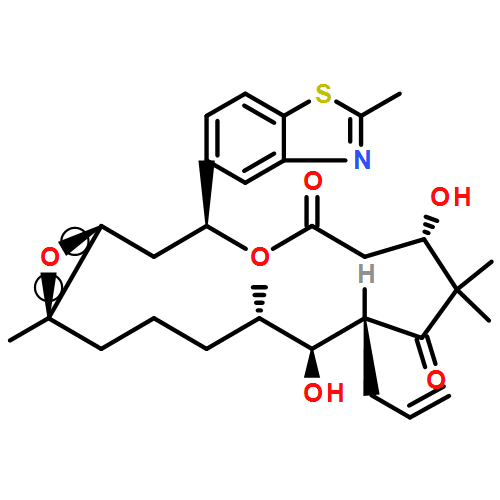
![Benzamide,4-[4-(2,3-dihydro-1,4-benzodioxin-6-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]-](/data/chemimg/25000/301836-43-1.png)
![Benzamide,4-[4-(2,3-dihydro-1,4-benzodioxin-6-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]-](/data/chemimg/25000/301836-43-1_b.png)


![<br>3-Benzyl-7-(tert-butyl)-2-mercapto-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d ]pyrimidin-4(3H)-one](http://img.cochemist.com/ccimg/300000/299921-58-7.png)
![<br>3-Benzyl-7-(tert-butyl)-2-mercapto-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d ]pyrimidin-4(3H)-one](http://img.cochemist.com/ccimg/300000/299921-58-7_b.png)
![9H-Purin-6-amine,N-([1,1'-biphenyl]-4-ylmethyl)-2-chloro-9-(1-methylethyl)-](http://img.cochemist.com/ccimg/294700/294648-35-4.png)
![9H-Purin-6-amine,N-([1,1'-biphenyl]-4-ylmethyl)-2-chloro-9-(1-methylethyl)-](http://img.cochemist.com/ccimg/294700/294648-35-4_b.png)








![1H-Pyrrolo[3,2-b]pyridine, 3-(4-fluorophenyl)-2-(4-pyridinyl)-](http://img.cochemist.com/ccimg/223800/223738-94-1.png)
![1H-Pyrrolo[3,2-b]pyridine, 3-(4-fluorophenyl)-2-(4-pyridinyl)-](http://img.cochemist.com/ccimg/223800/223738-94-1_b.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine,1-(1,1-dimethylethyl)-3-(1-naphthalenyl)-](/data/chemimg/3000/221243-82-9.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine,1-(1,1-dimethylethyl)-3-(1-naphthalenyl)-](/data/chemimg/3000/221243-82-9_b.png)
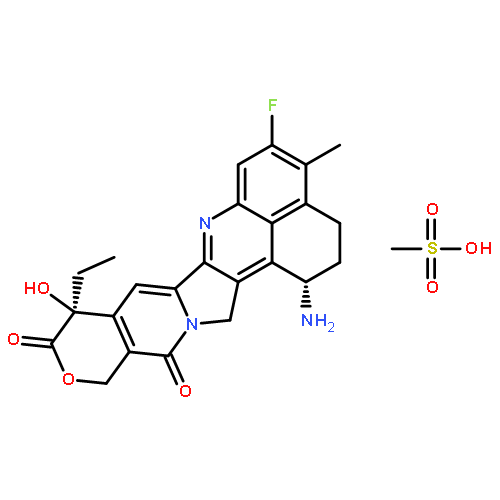
![3H,15H-Oxepino[3',4':6,7]indolizino[1,2-b]quinoline-3,15-dione,5-ethyl-9,10-difluoro-1,4,5,13-tetrahydro-5-hydroxy-, (5R)-](http://img.cochemist.com/ccimg/221000/220997-97-7.png)
![3H,15H-Oxepino[3',4':6,7]indolizino[1,2-b]quinoline-3,15-dione,5-ethyl-9,10-difluoro-1,4,5,13-tetrahydro-5-hydroxy-, (5R)-](http://img.cochemist.com/ccimg/221000/220997-97-7_b.png)
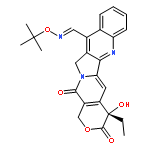
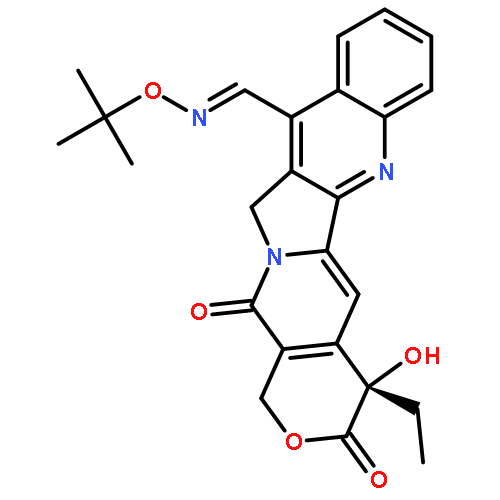
![1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione,11-[(1,1-dimethylethyl)dimethylsilyl]-4-ethyl-4,9-dihydroxy-, (4S)-](http://img.cochemist.com/ccimg/221000/220913-32-6.png)
![1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione,11-[(1,1-dimethylethyl)dimethylsilyl]-4-ethyl-4,9-dihydroxy-, (4S)-](http://img.cochemist.com/ccimg/221000/220913-32-6_b.png)


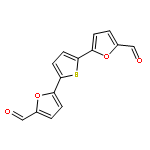
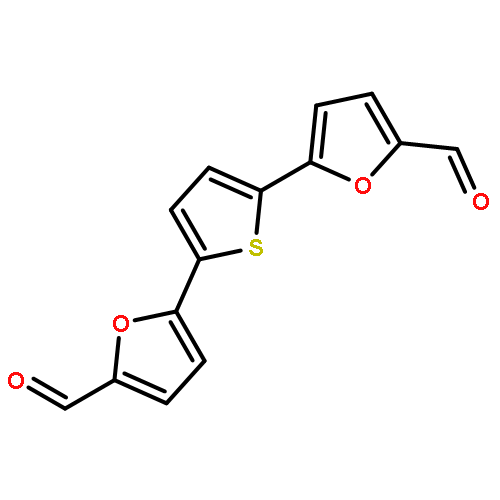
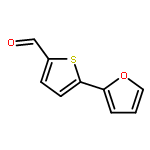
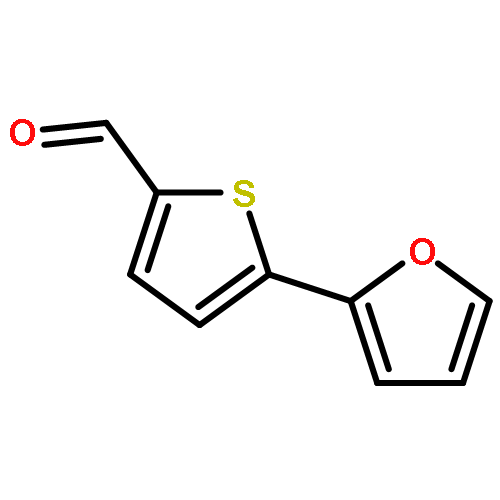
![[5-[5-[5-(hydroxymethyl)thiophen-2-yl]furan-2-yl]thiophen-2-yl]methanol](http://img.cochemist.com/ccimg/213300/213261-59-7.png)
![[5-[5-[5-(hydroxymethyl)thiophen-2-yl]furan-2-yl]thiophen-2-yl]methanol](http://img.cochemist.com/ccimg/213300/213261-59-7_b.png)








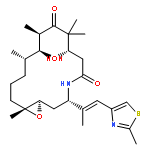
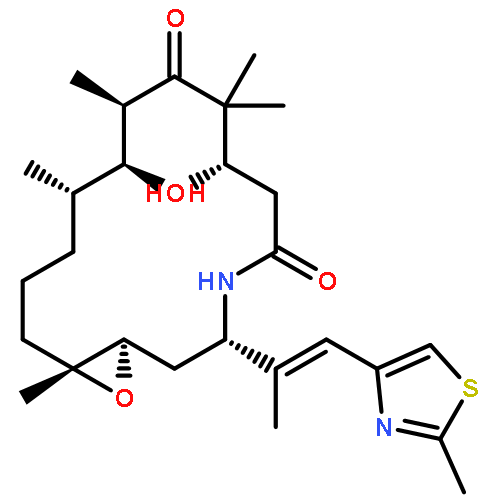
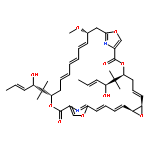
![Oxacyclohexadec-13-ene-2,6-dione,4,8-dihydroxy-5,5,7,9-tetramethyl-16-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-,(4S,7R,8S,9S,13Z,16S)-](http://img.cochemist.com/ccimg/186700/186692-73-9.png)
![Oxacyclohexadec-13-ene-2,6-dione,4,8-dihydroxy-5,5,7,9-tetramethyl-16-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-,(4S,7R,8S,9S,13Z,16S)-](http://img.cochemist.com/ccimg/186700/186692-73-9_b.png)
![Butanoic acid, 2-[(4-bromophenyl)methylene]-3-oxo-, ethyl ester](/data/chemimg/130800/186682-26-8.png)
![Butanoic acid, 2-[(4-bromophenyl)methylene]-3-oxo-, ethyl ester](/data/chemimg/130800/186682-26-8_b.png)

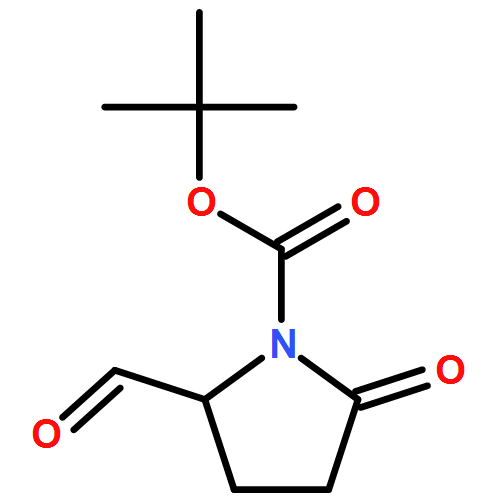
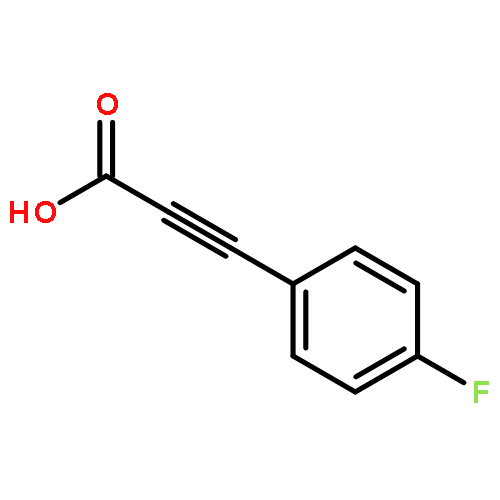
![2-Propynal, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/183900/183801-13-0.png)
![2-Propynal, 3-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/183900/183801-13-0_b.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine,1-(1,1-dimethylethyl)-3-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/181000/180903-17-7.png)
![1H-Pyrazolo[3,4-d]pyrimidin-4-amine,1-(1,1-dimethylethyl)-3-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/181000/180903-17-7_b.png)




![Benzaldehyde,4-[(2-furanylmethyl)thio]-3-nitro-](http://img.cochemist.com/ccimg/175300/175278-53-2.png)
![Benzaldehyde,4-[(2-furanylmethyl)thio]-3-nitro-](http://img.cochemist.com/ccimg/175300/175278-53-2_b.png)


![2H-Pyran, tetrahydro-2-[(1-methyl-2-methylenecyclopropyl)methoxy]-](http://img.cochemist.com/ccimg/164700/164665-65-0.png)
![2H-Pyran, tetrahydro-2-[(1-methyl-2-methylenecyclopropyl)methoxy]-](http://img.cochemist.com/ccimg/164700/164665-65-0_b.png)
![L-Valinamide, N,b,b,1-tetramethyl-L-tryptophyl-N-[(1S,2E)-3-carboxy-1-(1-methylethyl)-2-buten-1-yl]-N,3-dimethyl-](http://img.cochemist.com/ccimg/157300/157207-90-4.png)
![L-Valinamide, N,b,b,1-tetramethyl-L-tryptophyl-N-[(1S,2E)-3-carboxy-1-(1-methylethyl)-2-buten-1-yl]-N,3-dimethyl-](http://img.cochemist.com/ccimg/157300/157207-90-4_b.png)


![endo-3-(Boc-amino)-9-azabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/155600/155560-04-6.png)
![endo-3-(Boc-amino)-9-azabicyclo[3.3.1]nonane](http://img.cochemist.com/ccimg/155600/155560-04-6_b.png)






![4,17-Dioxabicyclo[14.1.0]heptadecane-5,9-dione,7,11-dihydroxy-8,8,10,12-tetramethyl-3-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-,(1S,3S,7S,10R,11S,12S,16R)-](http://img.cochemist.com/ccimg/152100/152044-53-6.png)
![4,17-Dioxabicyclo[14.1.0]heptadecane-5,9-dione,7,11-dihydroxy-8,8,10,12-tetramethyl-3-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]-,(1S,3S,7S,10R,11S,12S,16R)-](http://img.cochemist.com/ccimg/152100/152044-53-6_b.png)
























![Pyrano[3,2-e]indole, 3,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/140500/140427-33-4.png)
![Pyrano[3,2-e]indole, 3,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/140500/140427-33-4_b.png)











![5-Isoquinolinesulfonamide,N-[2-[[3-(4-bromophenyl)-2-propen-1-yl]amino]ethyl]-, hydrochloride (1:2)](http://img.cochemist.com/ccimg/131000/130964-39-5.png)
![5-Isoquinolinesulfonamide,N-[2-[[3-(4-bromophenyl)-2-propen-1-yl]amino]ethyl]-, hydrochloride (1:2)](http://img.cochemist.com/ccimg/131000/130964-39-5_b.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)




![3-[1-(3-Hydroxypropyl)-1H-indol-3-yl]-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione](http://img.cochemist.com/ccimg/125400/125313-60-2.png)
![3-[1-(3-Hydroxypropyl)-1H-indol-3-yl]-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione](http://img.cochemist.com/ccimg/125400/125313-60-2_b.png)


![Benzo[b]thiophene-2-carbonyl chloride, 3-chloro-5-(phenylmethoxy)-](http://img.cochemist.com/ccimg/122100/122024-71-9.png)
![Benzo[b]thiophene-2-carbonyl chloride, 3-chloro-5-(phenylmethoxy)-](http://img.cochemist.com/ccimg/122100/122024-71-9_b.png)
![Benzenamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/122000/121942-75-4.png)
![Benzenamine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/122000/121942-75-4_b.png)
![5H-Indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione,12,13-dihydro-](http://img.cochemist.com/ccimg/118500/118458-54-1.png)
![5H-Indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione,12,13-dihydro-](http://img.cochemist.com/ccimg/118500/118458-54-1_b.png)


![9,13-Epoxy-1H,9H-diindolo[1,2,3-gh:3',2',1'-lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one,2,3,10,11,12,13-hexahydro-3-hydroxy-10-methoxy-9-methyl-11-(methylamino)-,(3R,9S,10R,11R,13R)-](http://img.cochemist.com/ccimg/113000/112953-11-4.png)
![9,13-Epoxy-1H,9H-diindolo[1,2,3-gh:3',2',1'-lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one,2,3,10,11,12,13-hexahydro-3-hydroxy-10-methoxy-9-methyl-11-(methylamino)-,(3R,9S,10R,11R,13R)-](http://img.cochemist.com/ccimg/113000/112953-11-4_b.png)




![Ethanol,2-[[9-methyl-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/101700/101622-51-9.png)
![Ethanol,2-[[9-methyl-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/101700/101622-51-9_b.png)




![3H-Benz[e]indole-1-carboxylic acid, 4,5-dihydro-2-methyl-4,5-dioxo-, ethyl ester](/data/chemimg/2048700/88418-58-0.png)
![3H-Benz[e]indole-1-carboxylic acid, 4,5-dihydro-2-methyl-4,5-dioxo-, ethyl ester](/data/chemimg/2048700/88418-58-0_b.png)
![Benzene,1,1'-[(3-butyn-1-yloxy)(1,1-dimethylethyl)silylene]bis-](http://img.cochemist.com/ccimg/88200/88158-68-3.png)
![Benzene,1,1'-[(3-butyn-1-yloxy)(1,1-dimethylethyl)silylene]bis-](http://img.cochemist.com/ccimg/88200/88158-68-3_b.png)




![2-Furancarboxamide, 5-bromo-N-[(phenylamino)thioxomethyl]-](http://img.cochemist.com/ccimg/82400/82366-65-2.png)
![2-Furancarboxamide, 5-bromo-N-[(phenylamino)thioxomethyl]-](http://img.cochemist.com/ccimg/82400/82366-65-2_b.png)






![1H-ISOINDOLE-1,3(2H)-DIONE, 2,2'-[CARBONYLBIS(OXY)]BIS-](http://img.cochemist.com/ccimg/78900/78816-91-8.png)
![1H-ISOINDOLE-1,3(2H)-DIONE, 2,2'-[CARBONYLBIS(OXY)]BIS-](http://img.cochemist.com/ccimg/78900/78816-91-8_b.png)




![9-Azabicyclo[3.3.1]nonan-3-amine, 9-(phenylmethyl)-, (3-endo)-](/data/chemimg/1968700/76272-58-7.png)
![9-Azabicyclo[3.3.1]nonan-3-amine, 9-(phenylmethyl)-, (3-endo)-](/data/chemimg/1968700/76272-58-7_b.png)








![LYSINE, N6-ACETYL-N2-[(PHENYLMETHOXY)CARBONYL]-](http://img.cochemist.com/ccimg/68300/68223-08-5.png)
![LYSINE, N6-ACETYL-N2-[(PHENYLMETHOXY)CARBONYL]-](http://img.cochemist.com/ccimg/68300/68223-08-5_b.png)







![8-BUTYL-10-[(Z)-[(5Z)-3-METHOXY-5-PYRROL-2-YLIDENEPYRROL-2-YLIDENE]METHYL]-11-AZABICYCLO[7.2.1]DODECA-1(12),9-DIENE](http://img.cochemist.com/ccimg/56300/56208-07-2.png)
![8-BUTYL-10-[(Z)-[(5Z)-3-METHOXY-5-PYRROL-2-YLIDENEPYRROL-2-YLIDENE]METHYL]-11-AZABICYCLO[7.2.1]DODECA-1(12),9-DIENE](http://img.cochemist.com/ccimg/56300/56208-07-2_b.png)
![1(2H)-Isoquinolinone, 3-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/56200/56100-51-7.png)
![1(2H)-Isoquinolinone, 3-[(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/56200/56100-51-7_b.png)
![2H-Pyran, tetrahydro-2-[(2-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/53300/53250-10-5.png)
![2H-Pyran, tetrahydro-2-[(2-methyl-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/53300/53250-10-5_b.png)


![1H-Pyrido[3,4-b]indol-1-one, 2,3,4,9-tetrahydro-6-(phenylmethoxy)-](http://img.cochemist.com/ccimg/51100/51086-22-7.png)
![1H-Pyrido[3,4-b]indol-1-one, 2,3,4,9-tetrahydro-6-(phenylmethoxy)-](http://img.cochemist.com/ccimg/51100/51086-22-7_b.png)
![1H-Pyrido[3,4-b]indol-1-one,2,3,4,9-tetrahydro-6-hydroxy-](http://img.cochemist.com/ccimg/51100/51085-95-1.png)
![1H-Pyrido[3,4-b]indol-1-one,2,3,4,9-tetrahydro-6-hydroxy-](http://img.cochemist.com/ccimg/51100/51085-95-1_b.png)
![2-[(4-methoxyphenyl)methyl]guanidine](http://img.cochemist.com/ccimg/46300/46234-16-6.png)
![2-[(4-methoxyphenyl)methyl]guanidine](http://img.cochemist.com/ccimg/46300/46234-16-6_b.png)
![5,7-dibromo-Tricyclo[3.3.1.13,7]decane-2-carboxylic acid](http://img.cochemist.com/ccimg/35900/35856-02-1.png)
![5,7-dibromo-Tricyclo[3.3.1.13,7]decane-2-carboxylic acid](http://img.cochemist.com/ccimg/35900/35856-02-1_b.png)
![Tricyclo[3.3.1.13,7]decane-2-carbonitrile](http://img.cochemist.com/ccimg/35900/35856-00-9.png)
![Tricyclo[3.3.1.13,7]decane-2-carbonitrile](http://img.cochemist.com/ccimg/35900/35856-00-9_b.png)
![Butanoic acid, 2-[[4-(dimethylamino)phenyl]methylene]-3-oxo-, ethylester](http://img.cochemist.com/ccimg/35400/35388-37-5.png)
![Butanoic acid, 2-[[4-(dimethylamino)phenyl]methylene]-3-oxo-, ethylester](http://img.cochemist.com/ccimg/35400/35388-37-5_b.png)


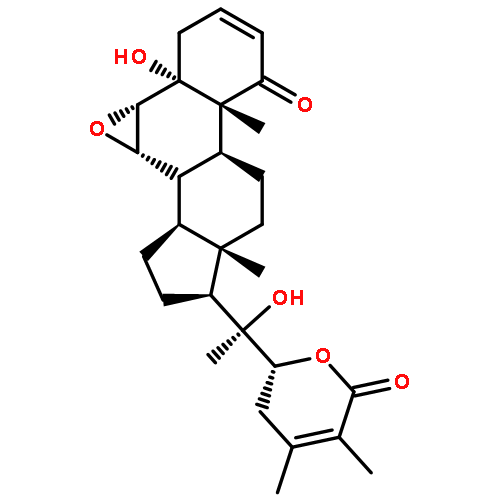
![Pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione, 1,3,6-trimethyl-](http://img.cochemist.com/ccimg/32600/32502-62-8.png)
![Pyrimido[5,4-e]-1,2,4-triazine-5,7(1H,6H)-dione, 1,3,6-trimethyl-](http://img.cochemist.com/ccimg/32600/32502-62-8_b.png)
![9-Azabicyclo[3.3.1]non-9-yloxy](/data/chemimg/1187500/31785-68-9.png)
![9-Azabicyclo[3.3.1]non-9-yloxy](/data/chemimg/1187500/31785-68-9_b.png)


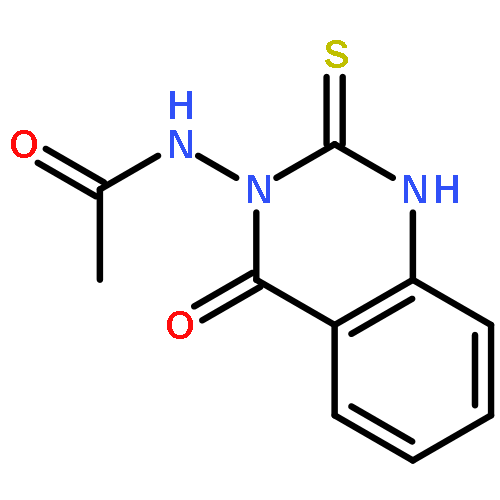
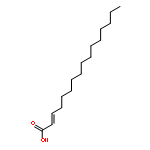

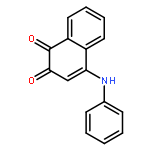
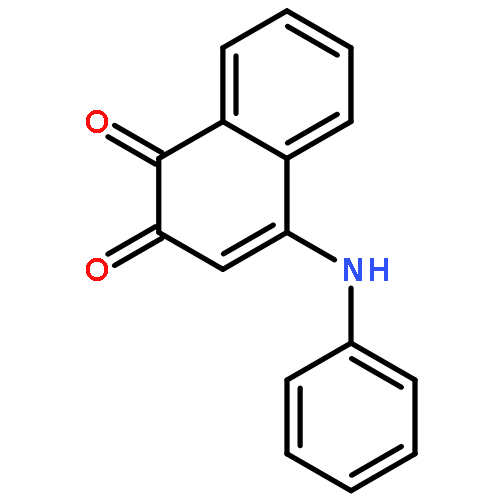


![6-amino-2,3,4,9-tetrahydro-1H-Pyrido[3,4-b]indol-1-one](http://img.cochemist.com/ccimg/26500/26405-64-1.png)
![6-amino-2,3,4,9-tetrahydro-1H-Pyrido[3,4-b]indol-1-one](http://img.cochemist.com/ccimg/26500/26405-64-1_b.png)




![Butanoic acid, 2-[(4-chlorophenyl)methylene]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/19500/19411-80-4.png)
![Butanoic acid, 2-[(4-chlorophenyl)methylene]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/19500/19411-80-4_b.png)


![Thieno[3,2-d]pyrimidine, 2,4-dimethoxy-](http://img.cochemist.com/ccimg/16300/16229-02-0.png)
![Thieno[3,2-d]pyrimidine, 2,4-dimethoxy-](http://img.cochemist.com/ccimg/16300/16229-02-0_b.png)





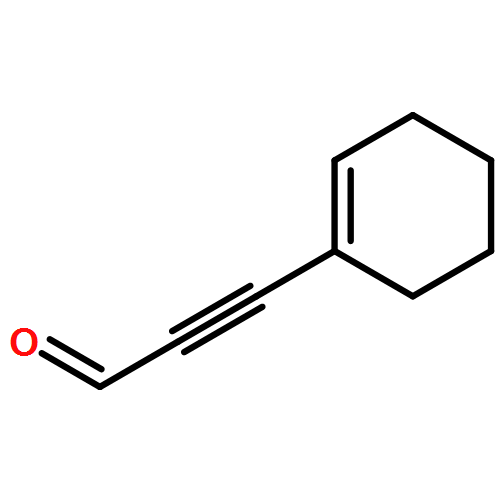
![2H-Naphtho[1,2-b]pyran-5,6-dione,3,4-dihydro-3-hydroxy-2,2-dimethyl-](http://img.cochemist.com/ccimg/15300/15297-98-0.png)
![2H-Naphtho[1,2-b]pyran-5,6-dione,3,4-dihydro-3-hydroxy-2,2-dimethyl-](http://img.cochemist.com/ccimg/15300/15297-98-0_b.png)
![5H-Pyrano[3,2-d]oxazole-5-methanol,6,7-bis(acetyloxy)-3a,6,7,7a-tetrahydro-2-methyl-, acetate (ester)](/data/chemimg/1034800/10378-06-0.png)
![5H-Pyrano[3,2-d]oxazole-5-methanol,6,7-bis(acetyloxy)-3a,6,7,7a-tetrahydro-2-methyl-, acetate (ester)](/data/chemimg/1034800/10378-06-0_b.png)


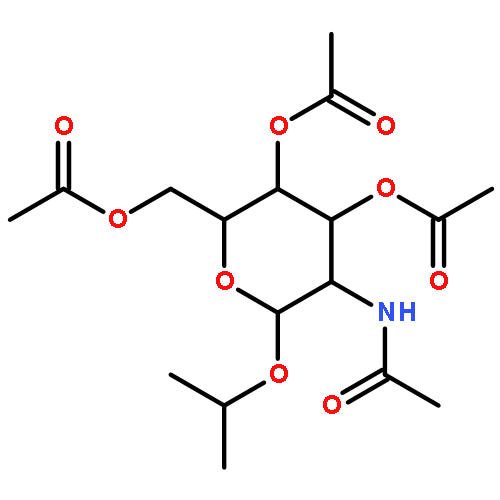


![N~2~-acetyl-N~6~-[(benzyloxy)carbonyl]lysine](http://img.cochemist.com/ccimg/6400/6367-08-4.png)
![N~2~-acetyl-N~6~-[(benzyloxy)carbonyl]lysine](http://img.cochemist.com/ccimg/6400/6367-08-4_b.png)



![6-chloro-1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5500/5413-96-7.png)
![6-chloro-1-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5500/5413-96-7_b.png)
![6-chloro-N,1-dimethyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5400/5326-77-2.png)
![6-chloro-N,1-dimethyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/5400/5326-77-2_b.png)


![Benzamide,N-[(2-pyridinylamino)thioxomethyl]-](http://img.cochemist.com/ccimg/5000/4921-86-2.png)
![Benzamide,N-[(2-pyridinylamino)thioxomethyl]-](http://img.cochemist.com/ccimg/5000/4921-86-2_b.png)
![2,2-Dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione](/data/chemimg/41800/4707-32-8.png)
![2,2-Dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione](/data/chemimg/41800/4707-32-8_b.png)
![2,4(1H,3H)-Quinazolinedione, 3-[3-(4-morpholinyl)propyl]-](http://img.cochemist.com/ccimg/4300/4266-54-0.png)
![2,4(1H,3H)-Quinazolinedione, 3-[3-(4-morpholinyl)propyl]-](http://img.cochemist.com/ccimg/4300/4266-54-0_b.png)
![5-{[(2-METHOXYPHENYL)AMINO]METHYL}-2-THIOXO-2,3-DIHYDRO-4(1H)-PYR<WBR />IMIDINONE](http://img.cochemist.com/ccimg/3200/3161-15-7.png)
![5-{[(2-METHOXYPHENYL)AMINO]METHYL}-2-THIOXO-2,3-DIHYDRO-4(1H)-PYR<WBR />IMIDINONE](http://img.cochemist.com/ccimg/3200/3161-15-7_b.png)














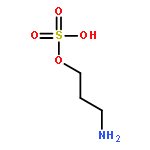
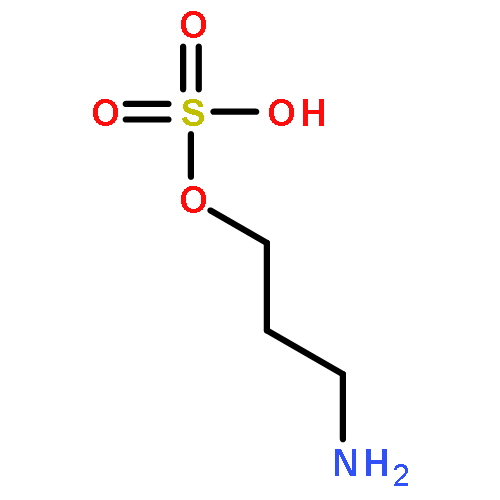



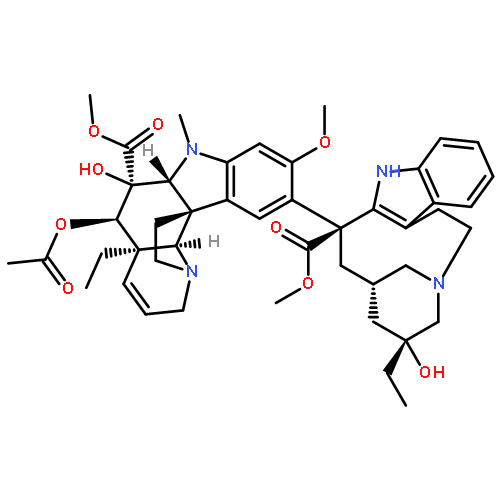
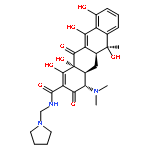



![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)






![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3.png)
![2,2'-Bi-1H-pyrrole,4-methoxy-5-[(5-methyl-4-pentyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/100/82-89-3_b.png)




![7-BROMO-2-METHOXYTHIENO[3,2-D]PYRIMIDINE](/data/chemimg/429700/1259978-35-2.png)
![7-BROMO-2-METHOXYTHIENO[3,2-D]PYRIMIDINE](/data/chemimg/429700/1259978-35-2_b.png)
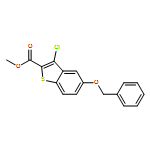
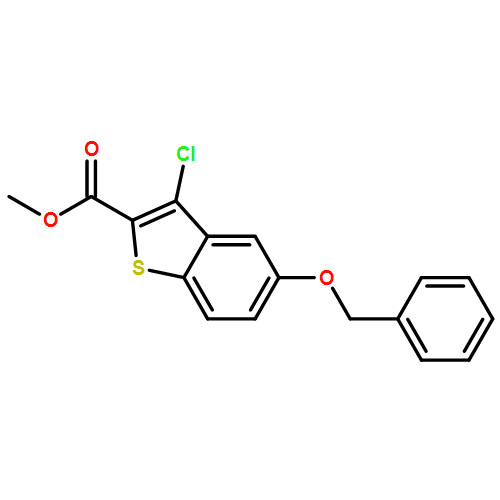


















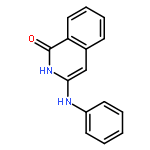
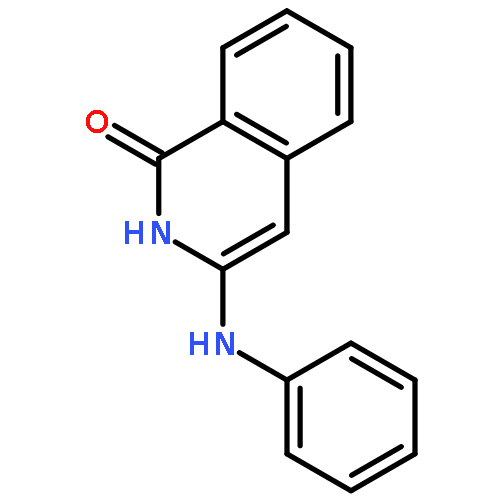
![(3S,4S,5S,6S)-6-[4-(2-aminoethyl)-2-hydroxy-phenoxy]-3,4,5-trihydroxy-tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/36000/35954-65-5.png)
![(3S,4S,5S,6S)-6-[4-(2-aminoethyl)-2-hydroxy-phenoxy]-3,4,5-trihydroxy-tetrahydropyran-2-carboxylic acid](http://img.cochemist.com/ccimg/36000/35954-65-5_b.png)

![Benzo[b]thiophene-2-carboxylicacid, 3-hydroxy-5-nitro-, methyl ester](http://img.cochemist.com/ccimg/26800/26759-52-4.png)
![Benzo[b]thiophene-2-carboxylicacid, 3-hydroxy-5-nitro-, methyl ester](http://img.cochemist.com/ccimg/26800/26759-52-4_b.png)






![Hexanoic acid,6-[(aminocarbonyl)amino]-](http://img.cochemist.com/ccimg/1500/1468-42-4.png)
![Hexanoic acid,6-[(aminocarbonyl)amino]-](http://img.cochemist.com/ccimg/1500/1468-42-4_b.png)
![Bicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/200/157-33-5.png)
![Bicyclo[1.1.0]butane](http://img.cochemist.com/ccimg/200/157-33-5_b.png)


![9-HYDROXY-1,2,3,4-TETRAHYDROCHROMENO[3,4-B]PYRIDIN-5-ONE](http://img.cochemist.com/ccimg/370600/370586-05-3.png)
![9-HYDROXY-1,2,3,4-TETRAHYDROCHROMENO[3,4-B]PYRIDIN-5-ONE](http://img.cochemist.com/ccimg/370600/370586-05-3_b.png)






![3-Hydroxy-5,6,7,8-tetrahydro-10-oxa-8-aza-benzo[a]azulen-9-one](http://img.cochemist.com/ccimg/522000/521937-07-5.png)
![3-Hydroxy-5,6,7,8-tetrahydro-10-oxa-8-aza-benzo[a]azulen-9-one](http://img.cochemist.com/ccimg/522000/521937-07-5_b.png)


![9-Methoxy-2,3,4,5-tetrahydro[1]benzothieno[2,3-f]-1,4-thiazepin-5-one](http://img.cochemist.com/ccimg/172900/172832-10-9.png)
![9-Methoxy-2,3,4,5-tetrahydro[1]benzothieno[2,3-f]-1,4-thiazepin-5-one](http://img.cochemist.com/ccimg/172900/172832-10-9_b.png)






![3,4-dihydro-9-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one](http://img.cochemist.com/ccimg/1233600/1233533-04-4.png)
![3,4-dihydro-9-hydroxy-[1]benzothieno[2,3-f]-1,4-thiazepin-5(2H)-one](http://img.cochemist.com/ccimg/1233600/1233533-04-4_b.png)

