Co-reporter:Cameron L. Brown, Meredith H. Barbee, Jeong Hoon Ko, Heather D. Maynard, and Stephen L. Craig
Journal of Chemical Education November 14, 2017 Volume 94(Issue 11) pp:1752-1752
Publication Date(Web):March 30, 2017
DOI:10.1021/acs.jchemed.6b00806
An easy-to-implement science outreach demonstration featuring a mechanically and photochemically color-changing polymer is described. The active polymeric material is a filled poly(dimethylsiloxane) (PDMS) elastomer that is covalently functionalized with spiropyran (SP), which is both a photochemical and mechanochemical switch. The material can be reversibly changed from colorless to dark purple by exposing it to light from a blue laser pointer or providing a mechanical stimulus such as hitting the polymer with a hammer or dragging a blunt object across the surface. The keynote demonstration is a PDMS chemical-drawing board that allows children literally to “write without ink” using a laser pointer or a blunt stylus. Collectively, these demonstrations are suitable for various student groups and encompass concepts in polymer and materials chemistry, photochemistry, and mechanochemistry. This demonstration has been successfully employed dozens of times in multiple universities across North America.Keywords: Demonstrations; Elementary/Middle School Science; Hands-On-Learning/Manipulatives; Materials Science; Photochemistry; Polymer Chemistry; Public Understanding/Outreach;
Co-reporter:Junpeng Wang, Tatiana B. Kouznetsova, and Stephen L. Craig
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10410-10412
Publication Date(Web):August 8, 2016
DOI:10.1021/jacs.6b06452
The mechanochemical activation of cis-gem-difluorocyclopropane (cis-gDFC) mechanophore in toluene was characterized with single-molecule force spectroscopy. Unlike previously reported behavior in methyl benzoate (MB), two transitions are observed in the force vs extension curves of cis-gDFC polymers in toluene. The first transition occurs at the same force of ∼1300 pN observed previously in MB, but a second transition is observed at forces of ∼1800 pN that reveal the partial formation of the trans-gDFC isomer. The behavior is attributed to competing reactions of the cis-gDFC at the 1300 pN plateau: addition of oxygen to a ring-opened diradicaloid intermediate, and isomerization of cis-gDFC to its trans isomer.
Co-reporter:Gregory R. Gossweiler; Tatiana B. Kouznetsova
Journal of the American Chemical Society 2015 Volume 137(Issue 19) pp:6148-6151
Publication Date(Web):April 28, 2015
DOI:10.1021/jacs.5b02492
The mechanically accelerated ring-opening reaction of spiropyran to a colored merocyanine provides a useful method by which to image the molecular scale stress/strain distribution within a polymer, but the magnitude of the forces necessary for activation has yet to be quantified. Here, we report single molecule force spectroscopy studies of two spiropyran isomers. Ring opening on the time scale of tens of milliseconds is found to require forces of ∼240 pN, well below that of previously characterized covalent mechanophores. The lower threshold force is a combination of a low force-free activation energy and the fact that the change in rate with force (activation length) of each isomer is greater than that inferred in other systems. Finally, regiochemical effects on mechanochemical coupling are characterized, and increasing force reverses the relative ring opening rates of the two isomers.
Co-reporter:Junpeng Wang; Tatiana B. Kouznetsova
Journal of the American Chemical Society 2015 Volume 137(Issue 36) pp:11554-11557
Publication Date(Web):September 3, 2015
DOI:10.1021/jacs.5b06168
Mechanical forces, applied via covalent polymer mechanochemistry, have been used to bias reaction pathways and activate otherwise inaccessible reactions. Here, single-molecule polymer mechanochemistry is used to induce the disrotatory outward ring opening of a cis-dialkyl substituted syn-chloro-gem-chlorofluorocyclopropane, in violation of the Woodward–Hoffmann–DePuy (WHD) rule. The forces required to trigger the anti-WHD pathway on the ∼100 ms time scale of the experiment are about 200 pN greater than those involved in the WHD favored process (1290 vs 1500 pN). The kinetics are complemented by tension trapping experiments that suggest that the reaction proceeds along a reaction pathway that generates substantial diradicaloid character.
Co-reporter:Cameron L. Brown and Stephen L. Craig
Chemical Science 2015 vol. 6(Issue 4) pp:2158-2165
Publication Date(Web):12 Feb 2015
DOI:10.1039/C4SC01945H
Force reactive functional groups, or mechanophores, have emerged as the basis of a potential strategy for sensing and countering stress-induced material failure. The general utility of this strategy is limited, however, because the levels of mechanophore activation in the bulk are typically low and observed only under large, typically irreversible strains. Strategies that enhance activation are therefore quite useful. Molecular-level design principles by which to engineer enhanced mechanophore activity are reviewed, with an emphasis on quantitative structure–activity studies determined for a family of gem-dihalocyclopropane mechanophores.
Co-reporter:Zachary S. Kean, Gregory R. Gossweiler, Tatiana B. Kouznetsova, Gihan B. Hewage and Stephen L. Craig
Chemical Communications 2015 vol. 51(Issue 44) pp:9157-9160
Publication Date(Web):29 Apr 2015
DOI:10.1039/C5CC01836F
Here we present a coumarin dimer (CD) mechanophore that, when embedded near the mid-chain of poly(methyl acrylate) polymers, activates under pulsed ultrasound conditions to yield coumarin chain-end functional polymers. Quantitative photochemical scission of the CD polymers provides a reference against which the activation efficiency of chain-centered mechanophores in polymers synthesized by controlled/living radical polymerization (CRP) can be assessed. Activation efficiency is characterized with respect to the polymer molecular weight (MW), polydispersity index (PDI), and distribution of mechanophores along the backbone.
Co-reporter:Gregory R. Gossweiler, Cameron L. Brown, Gihan B. Hewage, Eitan Sapiro-Gheiler, William J. Trautman, Garrett W. Welshofer, and Stephen L. Craig
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 40) pp:22431
Publication Date(Web):September 21, 2015
DOI:10.1021/acsami.5b06440
The functions of soft robotics are intimately tied to their form—channels and voids defined by an elastomeric superstructure that reversibly stores and releases mechanical energy to change shape, grip objects, and achieve complex motions. Here, we demonstrate that covalent polymer mechanochemistry provides a viable mechanism to convert the same mechanical potential energy used for actuation in soft robots into a mechanochromic, covalent chemical response. A bis-alkene functionalized spiropyran (SP) mechanophore is cured into a molded poly(dimethylsiloxane) (PDMS) soft robot walker and gripper. The stresses and strains necessary for SP activation are compatible with soft robot function. The color change associated with actuation suggests opportunities for not only new color changing or camouflaging strategies, but also the possibility for simultaneous activation of latent chemistry (e.g., release of small molecules, change in mechanical properties, activation of catalysts, etc.) in soft robots. In addition, mechanochromic stress mapping in a functional robotic device might provide a useful design and optimization tool, revealing spatial and temporal force evolution within the robot in a way that might be coupled to autonomous feedback loops that allow the robot to regulate its own activity. The demonstration motivates the simultaneous development of new combinations of mechanophores, materials, and soft, active devices for enhanced functionality.Keywords: mechanochemistry; mechanochromic; PDMS; soft robot; spiropyran; stimuli-responsive
Co-reporter:Junpeng Wang, Ilya Piskun, and Stephen L. Craig
ACS Macro Letters 2015 Volume 4(Issue 8) pp:834
Publication Date(Web):July 20, 2015
DOI:10.1021/acsmacrolett.5b00440
The mechanical stresses that materials experience during use can lead to aging and failure. Recent developments in covalent mechanochemistry have provided a mechanism by which those stresses can be channeled into constructive, rather than destructive, responses, including strengthening in materials. Here, the synthesis and mechanical response of a polymer containing multiple benzocyclobutene (BCB) mechanophores along its backbone are reported. When solutions of the BCB polymer were exposed to the normally destructive elongational flow forces generated by pulsed ultrasonication, the number of intermolecular bond-forming reactions was greater than the number of bond-breaking reactions, leading to a net increase in polymer molecular weight. The molecular weight increase could be turned into gelation by introducing a bismaleimide cross-linker that reacts with the ortho-quinodimethide intermediate generated by mechanically assisted ring opening of the BCB mechanophores and using polymer concentrations in excess of the critical overlap concentration. Unlike a previous mechanically induced gelation of a mechanophore-based polymer, the BCB cross-linking requires no ionic components and represents an attractive, second platform for stress-strengthening materials.
Co-reporter:Junpeng Wang, Tatiana B. Kouznetsova, Zhenbin Niu, Arnold L. Rheingold, and Stephen L. Craig
The Journal of Organic Chemistry 2015 Volume 80(Issue 23) pp:11895-11898
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.joc.5b01465
Mechanical forces have previously been used to drive reactions along pathways that violate the orbital symmetry effects captured in the Woodward–Hoffmann rules. Here, we show that a polymer “lever arm effect” can provide a mechanical advantage in accelerating the symmetry forbidden disrotatory ring opening of benzocyclobutene (BCB). Addition of an α-E-alkene to the BCB mechanophore drops the force required to induce reactions on the ∼0.1 s time scale of single-molecule force spectroscopy experiments from 1370 to 920 pN.
Co-reporter:Junpeng Wang, Mitchell T. Ong, Tatiana B. Kouznetsova, Jeremy M. Lenhardt, Todd J. Martínez, and Stephen L. Craig
The Journal of Organic Chemistry 2015 Volume 80(Issue 23) pp:11773-11778
Publication Date(Web):August 31, 2015
DOI:10.1021/acs.joc.5b01493
The dynamics of reactions at or in the immediate vicinity of transition states are critical to reaction rates and product distributions, but direct experimental probes of those dynamics are rare. Here, s-trans, s-trans 1,3-diradicaloid transition states are trapped by tension along the backbone of purely cis-substituted gem-difluorocyclopropanated polybutadiene using the extensional forces generated by pulsed sonication of dilute polymer solutions. Once released, the branching ratio between symmetry-allowed disrotatory ring closing (of which the trapped diradicaloid structure is the transition state) and symmetry-forbidden conrotatory ring closing (whose transition state is nearby) can be inferred. Net conrotatory ring closing occurred in 5.0 ± 0.5% of the released transition states, in excellent agreement with ab initio molecular dynamics simulations.
Co-reporter:Jeremy M. Lenhardt, Ashley L. Black Ramirez, Bobin Lee, Tatiana B. Kouznetsova, and Stephen L. Craig
Macromolecules 2015 Volume 48(Issue 18) pp:6396-6403
Publication Date(Web):September 11, 2015
DOI:10.1021/acs.macromol.5b01677
Structure–activity relationships in the mechanochemistry of gem-dichlorocyclopropane (gDCC)-based polymer solutions triggered by pulsed ultrasound are reported. Insights into the flow-induced mechanochemical transformations of gDCC mechanophores into the corresponding 2,3-dichloroalkenes are obtained by monitoring the mechanochemistry as a function of initial polymer molecular weight and sonication conditions. The competition between gDCC activation and polymer chain scission is invariant to sonication power, temperature, polymer concentration, and solvent but is sensitive to initial polymer molecular weight. The results have practical implications for the use of polymer sonochemistry as a tool for quantifying the relative mechanical strength of scissile polymers and conceptual implications for thinking about the nature of the force distributions experienced during sonochemical experiments.
Co-reporter:Zachary S. Kean;Jennifer L. Hawk;Shaoting Lin;Xuanhe Zhao;Rint P. Sijbesma
Advanced Materials 2014 Volume 26( Issue 34) pp:6013-6018
Publication Date(Web):
DOI:10.1002/adma.201401570
Co-reporter:Junpeng Wang ; Tatiana B. Kouznetsova ; Zachary S. Kean ; Lin Fan ; Brendan D. Mar ; Todd J. Martínez
Journal of the American Chemical Society 2014 Volume 136(Issue 43) pp:15162-15165
Publication Date(Web):October 16, 2014
DOI:10.1021/ja509585g
Molecular mechanisms by which to increase the activity of a mechanophore might provide access to new chemical reactions and enhanced stress-responsive behavior in mechanochemically active polymeric materials. Here, single-molecule force spectroscopy reveals that the force-induced acceleration of the electrocyclic ring opening of gem-dichlorocyclopropanes (gDCC) is sensitive to the stereochemistry of an α-alkene substituent on the gDCC. On the ∼0.1 s time scale of the experiment, the force required to open the E-alkene-substituted gDCC was found to be 0.4 nN lower than that required in the corresponding Z-alkene isomer, despite the effectively identical force-free reactivities of the two isomers and the distance between the stereochemical permutation and the scissile bond of the mechanophore. Fitting the experimental data with a cusp model provides force-free activation lengths of 1.67 ± 0.05 and 1.20 ± 0.05 Å for the E and Z isomers, respectively, as compared to 1.65 and 1.24 Å derived from computational modeling.
Co-reporter:Gregory R. Gossweiler, Gihan B. Hewage, Gerardo Soriano, Qiming Wang, Garrett W. Welshofer, Xuanhe Zhao, and Stephen L. Craig
ACS Macro Letters 2014 Volume 3(Issue 3) pp:216
Publication Date(Web):February 12, 2014
DOI:10.1021/mz500031q
Covalent mechanochemistry within bulk polymers typically occurs with irreversible deformation of the parent material. Here we show that embedding mechanophores into an elastomeric poly(dimethylsiloxane) (PDMS) network allows for covalent bond activation under macroscopically reversible deformations. Using the colorimetric mechanophore spiropyran, we show that bond activation can be repeated over multiple cycles of tensile elongation with full shape recovery. Further, localized compression can be used to pattern strain-induced chemistry. The platform enables the reversibility of a secondary strain-induced color change to be characterized. We also observe mechanical acceleration of a flex-activated retro-Diels–Alder reaction, allowing a chemical signal to be released in response to a fully reversible deformation.
Co-reporter:Zachary S. Kean;Dr. Sergey Akbulatov;Dr. Yancong Tian; Ross A. Widenhoefer;Dr. Roman Boulatov; Stephen L. Craig
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14508-14511
Publication Date(Web):
DOI:10.1002/anie.201407494
Abstract
A catalyst that couples a photoswitch to the biaryl backbone of a chiral bis(phosphine) ligand, thus allowing photochemical manipulation of ligand geometry without perturbing the electronic structure is reported. The changes in catalyst activity and selectivity upon switching can be attributed to intramolecular mechanical forces, thus laying the foundation for a new class of catalysts whose selectivity can be varied smoothly and in situ over a useful range by controlling molecular stress experienced by the catalyst during turnover. Forces on the order of 100 pN are generated, thus leading to measurable changes in the enantioselectivities of asymmetric Heck arylations and Trost allylic alkylations. The differential coupling between applied force and competing stereochemical pathways is quantified and found to be more efficient for the Heck arylations.
Co-reporter:Zachary S. Kean ; Zhenbin Niu ; Gihan B. Hewage ; Arnold L. Rheingold
Journal of the American Chemical Society 2013 Volume 135(Issue 36) pp:13598-13604
Publication Date(Web):August 13, 2013
DOI:10.1021/ja4075997
A primary goal of covalent mechanochemistry is to develop polymer bound mechanophores that undergo constructive transformations in response to otherwise destructive forces. The [2 + 2] cycloreversion of cyclobutane mechanophores has emerged as a versatile framework to develop a wide range of stress-activated functionality. Herein, we report the development of a class of cyclobutane bearing bicyclo[4.2.0]octane mechanophores. Using carbodiimide polyesterification, these stress-responsive units were incorporated into high molecular weight polymers containing up to 700 mechanophores per polymer chain. Under exposure to the otherwise destructive elongational forces of pulsed ultrasound, these mechanophores unravel by ∼7 Å per monomer unit to form α,β-unsaturated esters that react constructively via thiol–ene conjugate addition to form sulfide functionalized copolymers and cross-linked polymer networks. To probe the dynamics of the mechanochemical ring opening, a series of bicyclo[4.2.0]octane derivatives that varied in stereochemistry, substitution, and symmetry were synthesized and activated. Reactivity and product stereochemistry was analyzed by 1H NMR, which allowed us to interrogate the mechanism of the mechanochemical [2 + 2] cycloreversion. These results support that the ring opening is not concerted but proceeds via a 1,4 diradical intermediate. The bicyclo[4.2.0]octanes hold promise as active functional groups in new classes of stress-responsive polymeric materials.
Co-reporter:Hope M. Klukovich ; Zachary S. Kean ; Ashley L. Black Ramirez ; Jeremy M. Lenhardt ; Jiaxing Lin ; Xiangqian Hu
Journal of the American Chemical Society 2012 Volume 134(Issue 23) pp:9577-9580
Publication Date(Web):May 31, 2012
DOI:10.1021/ja302996n
Epoxidized polybutadiene and epoxidized polynorbornene were subjected to pulsed ultrasound in the presence of small molecules capable of being trapped by carbonyl ylides. When epoxidized polybutadiene was sonicated, there was no observable small molecule addition to the polymer. Concurrently, no appreciable isomerization (cis to trans epoxide) was observed, indicating that the epoxide rings along the backbone are not mechanically active under the experimental conditions employed. In contrast, when epoxidized polynorbornene was subjected to the same conditions, both addition of ylide trapping reagents and net isomerization of cis to trans epoxide were observed. The results demonstrate the mechanical activity of epoxides, show that mechanophore activity is determined not only by the functional group but also the polymer backbone in which it is embedded, and facilitate a characterization of the reactivity of the ring-opened dialkyl epoxide.
Co-reporter:Zachary S. Kean ; Ashley L. Black Ramirez ; Yufan Yan
Journal of the American Chemical Society 2012 Volume 134(Issue 31) pp:12939-12942
Publication Date(Web):July 21, 2012
DOI:10.1021/ja3063666
Force-induced transformations of polymer-bound functionalities have the potential to produce a rich array of stress-responsive behavior. One area of particular interest is the activation of non-scissile mechanophores in which latent reactivity can be unveiled that, under the appropriate conditions, could lead to constructive bond formation in materials exposed to typically destructive stress. Here, the mechanical activation of a bicyclo[3.2.0]heptane (BCH) mechanophore is demonstrated via selective labeling of bis-enone products. BCH ring-opening produces large local elongation (>4 Å) and products that are reactive to conjugate additions under mild conditions. Subsequent photocyclization regenerates the initial BCH functionality, providing switchable structure and reactivity along the polymer backbone in response to stress and visible light.
Co-reporter:Ashley L. Black Ramirez, James W. Ogle, Andrew L. Schmitt, Jeremy M. Lenhardt, Matthew P. Cashion, Mahesh K. Mahanthappa, and Stephen L. Craig
ACS Macro Letters 2012 Volume 1(Issue 1) pp:23
Publication Date(Web):November 9, 2011
DOI:10.1021/mz200005u
The high shear forces generated during the pulsed ultrasound of dilute polymer solutions lead to large tensile forces that are focused near the center of the polymer chain, but quantitative experimental evidence regarding the force distribution is rare. Here, pulsed ultrasound of quantitatively geminal-dihalocyclopropanated (gDHC) polybutadiene provides insights into the distribution. Pulsed ultrasound leads to the mechanochemical ring-opening of the gDHC mechanophore to a 2,3-dihaloalkene. The alkene product is then degraded through ozonolysis to leave behind only those stretches of the polymer that have not experienced large enough forces to be activated. Microstructural and molecular weight analysis reveals that the activated and unactivated regions of the polymer are continuous, indicating a smooth and monotonic force distribution from the midchain peak toward the polymer ends. When coupled to chain scission, the net process constitutes the rapid, specific, and reagentless conversion of a single homopolymer into block copolymers. Despite their compositional polydispersity, the sonicated polymers assemble into ordered lamellar phases that are characterized by small-angle X-ray scattering.
Co-reporter:Zachary S. Kean, Stephen L. Craig
Polymer 2012 Volume 53(Issue 5) pp:1035-1048
Publication Date(Web):28 February 2012
DOI:10.1016/j.polymer.2012.01.018
In the past five years, the field of covalent polymer mechanochemistry has experienced a renaissance. Once limited to the simple scission of polymer chains, mechanical force can now be used to produce a wide array of productive chemistry. These outcomes have both challenged and supported classically held views of chemical reactivity. The impact of these findings has relevance in both synthetic chemistry and material science. Here, we review our efforts to exploit mechanochemical coupling to produce constructive and stress-responsive covalent chemistry in polymer materials.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Donghua Xu, Daisuke Asai, Ashutosh Chilkoti, and Stephen L. Craig
Biomacromolecules 2012 Volume 13(Issue 8) pp:
Publication Date(Web):July 12, 2012
DOI:10.1021/bm300760s
The rheological properties of cysteine-containing elastin-like polypeptide (Cys-ELP) solutions and Cys-ELP hydrogels are reported. The Cys-ELP solutions exhibit a surprisingly high apparent viscosity at low shear rate. The high viscosity is attributed to the formation of an interfacial cross-linked “skin” at the sample surface, rather than the bulk of the Cys-ELP solution. At higher shear rate, the interfacial cross-linked film breaks, and its influence on the viscosity of the Cys-ELP solution can be ignored. Cys-ELP hydrogels are formed by mixing Cys-ELP and hydrogen peroxide (H2O2). At fixed concentration of Cys-ELP, the gelation time can be tuned by the concentration of H2O2. Cys-ELP hydrogels have the typical characteristics of covalent cross-linked networks, as the storage moduli are larger than the loss moduli and are independent of frequency in dynamic oscillatory frequency sweep experiments. The plateau moduli obtained from linear frequency sweep experiments are much lower than those estimated from the number of thiol groups along the Cys-ELP chain, indicating that only a small fraction of thiols form elastically active cross-links. From the small value of the fraction of elastically active cross-links, the Cys-ELP hydrogel is concluded to be an inhomogenous network. Under steady shear, a 2.5 wt % Cys-ELP hydrogel shear thickens at shear rates lower than that necessary for fracture.
Co-reporter:Zachary S. Kean;Ashley L. Black Ramirez
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 17) pp:3481-3484
Publication Date(Web):
DOI:10.1002/pola.26148
Co-reporter:Hope M. Klukovich ; Zachary S. Kean ; Scott T. Iacono
Journal of the American Chemical Society 2011 Volume 133(Issue 44) pp:17882-17888
Publication Date(Web):October 3, 2011
DOI:10.1021/ja2074517
Perfluorocyclobutane (PFCB) polymer solutions were subjected to pulsed ultrasound, leading to mechanically induced chain scission and molecular weight degradation. 19F NMR revealed that the new, mechanically generated end groups are trifluorovinyl ethers formed by cycloreversion of the PFCB groups, a process that differs from thermal degradation pathways. One consequence of the mechanochemical process is that the trifluorovinyl ether end groups can be remended simply by subjecting the polymer solution to the original polymerization conditions, that is, heating to >150 °C. Stereochemical changes in the PFCBs, in combination with radical trapping experiments, indicate that PFCB scission proceeds via a stepwise mechanism with a 1,4-diradical intermediate, offering a potential mechanism for localized functionalization and cross-linking in regions of high stress.
Co-reporter:Jeremy M. Lenhardt ; James W. Ogle ; Mitchell T. Ong ; Robert Choe ; Todd J. Martinez
Journal of the American Chemical Society 2011 Volume 133(Issue 10) pp:3222-3225
Publication Date(Web):February 22, 2011
DOI:10.1021/ja107645c
Tension along a polymer chain traps neighboring s-trans/s-trans-1,3-diradicals from the mechanically induced ring opening of gem-difluorocyclopropanes (gDFCs). The diradicals correspond to the transition states of the force-free thermal isomerization reactions of gDFCs, and the tension trapping allows a new disproportionation reaction between two simultaneously trapped diradicals to take place.
Co-reporter:Ashley L. Black, Joshua A. Orlicki and Stephen L. Craig
Journal of Materials Chemistry A 2011 vol. 21(Issue 23) pp:8460-8465
Publication Date(Web):16 Feb 2011
DOI:10.1039/C0JM03875J
Polybutadiene was functionalized with dibromo-, dichloro-, and bromochloro-carbene to give gem-dihalocyclopropanated (gDHC) polymers, in which the gDHCs act as mechanically activated functional groups or mechanophores. The polymers were extruded to determine the mechanophore activity in the solid state. The extent of gDHC ring opening depends on both the polymer composition and the macroscopic shear stress, ranging from 6.0% to over 30% after an hour of extrusion. In addition, the 2,3-dibromoalkene formed from mechanical activation of the gem-dibromocyclopropane was found to undergo subsequent nucleophilic substitution by chloride in the solid state. The number of solid-state substitution reactions far exceeds the number of main-chain bonds broken, a finding with implications for the future use of mechanophores in self-strengthening or self-healing polymers.
Co-reporter:Jeremy M. Lenhardt, Ashley L. Black, Brett A. Beiermann, Brian D. Steinberg, Faiyam Rahman, Tasha Samborski, Joseph Elsakr, Jeffrey S. Moore, Nancy R. Sottos and Stephen L. Craig
Journal of Materials Chemistry A 2011 vol. 21(Issue 23) pp:8454-8459
Publication Date(Web):07 Feb 2011
DOI:10.1039/C0JM04117C
The incorporation of mechanically active functional groups, or mechanophores, along polymer backbones offers opportunities for new stress-responsive material properties and also provides a method by which to probe fundamental questions related to molecular stress distributions in polymeric materials under load. The activation of covalent chemistry in polymers has primarily been demonstrated in solution, but to date little is known regarding activation in the solid state. In the latter regard, recent effort has focused on the use of spectroscopically active mechanophores that directly probe the presence of stress within materials. The distribution of forces within individual polymer chains, however, has yet to be characterized. Herein we report that gem-dihalocyclopropane (gDHC) functionalized polybutadiene is mechanochemically active in the solid state, and that the strain-triggered ring opening of the gDHCs provides quantitative information regarding the number of mechanically active monomers and the size of the mechanically activated domains along individual polymer backbones within bulk materials subjected to compressive and tensile loads. The results show that high mechanical forces are concentrated over lengths of only a few monomers.
Co-reporter:Ashley L. Black, Jeremy M. Lenhardt and Stephen L. Craig
Journal of Materials Chemistry A 2011 vol. 21(Issue 6) pp:1655-1663
Publication Date(Web):01 Nov 2010
DOI:10.1039/C0JM02636K
Current activity in, and future prospects for, the incorporation of mechanochemically active functional groups (“mechanophores”) into polymers is reviewed. This area of research is treated in the context of two categories. The first category is the development of new chemistry in the service of material science, through the design and synthesis of mechanophores to provide stress-sensing and/or stress-responsive elements in materials. The second category is the reverse—the development of new material architectures that efficiently transmit macroscopic forces to targeted molecules in order to generate chemical reactivity that is inaccessible by other means.
Co-reporter:Donghua Xu and Stephen L. Craig
Macromolecules 2011 Volume 44(Issue 18) pp:7478-7488
Publication Date(Web):August 30, 2011
DOI:10.1021/ma201386t
The large amplitude oscillatory shear behavior of metallo-supramolecular polymer networks formed by adding bis-Pd(II) cross-linkers to poly(4-vinylpyridine) (PVP) in dimethyl sulfoxide (DMSO) solution is reported. The influence of scanning frequency, dissociation rate of cross-linkers, concentration of cross-linkers, and concentration of PVP solution on the large amplitude oscillatory shear behavior is explored. In semidilute unentangled PVP solutions, above a critical scanning frequency, strain hardening of both storage moduli and loss moduli is observed. In the semidilute entangled regime of PVP solution, however, strain softening is observed for samples with faster cross-linkers (kd ∼ 1450 s–1), whereas strain hardening is observed for samples with slower cross-linkers (kd ∼ 17 s–1). The mechanism of strain hardening is attributed primarily to a strain-induced increase in the number of elastically active chains, with possible contributions from non-Gaussian stretching of polymer chains at strains approaching network fracture. The divergent strain softening of samples with faster cross-linkers in semidilute entangled PVP solutions, relative to the strain hardening of samples with slower cross-linkers, is consistent with observed shear thinning/shear thickening behavior reported previously and is attributed to the fact that the average time that a cross-linker remains detached is too short to permit the local relaxation of polymer chain segments that is necessary for a net conversion of elastically inactive to elastically active cross-linkers. These and other observations paint a picture in which strain softening and shear thinning arise from the same set of molecular mechanisms, conceptually uniting the two nonlinear responses for this system.
Co-reporter:Donghua Xu, Chen-Yang Liu, and Stephen L. Craig
Macromolecules 2011 Volume 44(Issue 7) pp:2343-2353
Publication Date(Web):March 14, 2011
DOI:10.1021/ma2000916
The steady shear behavior of metallo-supramolecular polymer networks formed by bis-Pd(II) cross-linkers and semidilute entangled solutions of poly(4-vinylpyridine) (PVP) in dimethyl sulfoxide (DMSO) or N,N-dimethylformamide (DMF) is reported. The steady shear behavior of the networks depends on the dissociation rate and association rate of the cross-linkers, the concentration of cross-linkers, and the concentration of the polymer solution. The divergent steady shear behavior—shear thinning versus shear thickening—of samples with identical structure but different cross-linker dynamics [ J. Phys. Chem. Lett. 2010, 1, 1683−1686] is further explored in this paper. The divergent steady shear behavior for networks with different cross-linkers is connected to a competition between different time scales: the average time that a cross-linker remains open (τ1) and the local relaxation time of a segment of polymer chain (τsegment). When τ1 is larger than τsegment, shear thickening is observed. When τ1 is smaller than τsegment, only shear thinning is observed.
Co-reporter:Donghua Xu and Stephen L. Craig
Macromolecules 2011 Volume 44(Issue 13) pp:5465-5472
Publication Date(Web):June 13, 2011
DOI:10.1021/ma200096s
The linear rheological properties of networks formed by adding bis-Pd(II) cross-linkers to poly(4-vinylpyridine) (PVP) solution are examined, and the scaling law relationships between the zero shear viscosity (η0) of the networks versus the concentration of PVP solution (CPVP), the concentration of cross-linkers (CX), and the number density of elastically active chains (νphantom) are experimentally determined. The scaling law relationships are compared to the theoretical expectations of the sticky Rouse and sticky reptation models [ Macromolecules 2001, 34, 1058−1068], and both qualitative and quantitative differences are observed.
Co-reporter:Dong Wu ; Jeremy M. Lenhardt ; Ashley L. Black ; Boris B. Akhremitchev +
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:15936-15938
Publication Date(Web):October 26, 2010
DOI:10.1021/ja108429h
Single-molecule force spectroscopy is used to observe the irreversible extension of a gem-dibromocyclopropane (gDBC)-functionalized polybutadiene under tension, a process akin to polymer necking at a single-molecule level. The extension of close to 28% in the contour length of the polymer backbone occurs at roughly 1.2 nN (tip velocity of 3 μm/s) and is attributed to the force-induced isomerization of the gDBCs into 2,3-dibromoalkenes. The rearrangement represents a possible new mechanism for localized stress relief in polymers and polymer networks under load, and the quantification of the force dependency provides a benchmark value for further studies of mechanically triggered chemistry in bulk polymers.
Co-reporter:Donghua Xu and Stephen L. Craig
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 11) pp:1683-1686
Publication Date(Web):May 12, 2010
DOI:10.1021/jz1004818
Molecular theories of shear thickening and shear thinning in associative polymer networks are typically united in that they involve a single kinetic parameter that describes the network — a relaxation time that is related to the lifetime of the associative bonds. Here we report the steady-shear behavior of two structurally identical metallo-supramolecular polymer networks, for which single-relaxation parameter models break down in dramatic fashion. The networks are formed by the addition of reversible cross-linkers to semidilute entangled solutions of poly(4-vinylpyridine) (PVP) in dimethylsulfoxide (DMSO), and they differ only in the lifetime of the reversible cross-links. Shear thickening is observed for cross-linkers that have a slower dissociation rate (17 s−1), while shear thinning is observed for samples that have a faster dissociation rate (ca. 1400 s−1). The difference in the steady shear behavior of the unentangled versus entangled regime reveals an unexpected, additional competing relaxation, ascribed to topological disentanglement in the semidilute entangled regime that contributes to the rheological properties.Keywords (keywords): networks; rheology; supramolecular structures;
Co-reporter:Donghua Xu, Jennifer L. Hawk, David M. Loveless, Sung Lan Jeon and Stephen L. Craig
Macromolecules 2010 Volume 43(Issue 7) pp:3556-3565
Publication Date(Web):March 17, 2010
DOI:10.1021/ma100093b
We report here the nonlinear rheological properties of metallo-supramolecular networks formed by the reversible cross-linking of semidilute unentangled solutions of poly(4-vinylpyridine) (PVP) in dimethyl sulfoxide (DMSO). The reversible cross-linkers are bis-Pd(II) or bis-Pt(II) complexes that coordinate to the pyridine functional groups on the PVP. Under steady shear, shear thickening is observed above a critical shear rate, and critical shear rate is experimentally correlated with the lifetime of the metal−ligand bond. The onset and magnitude of the shear thickening depend on the amount of cross-linkers added. In contrast to the behavior observed in most transient networks, the time scale of network relaxation is found to increase during shear thickening. The primary mechanism of shear thickening is ascribed to the shear-induced transformation of intrachain cross-linking to interchain cross-linking, rather than nonlinear high tension along polymer chains that are stretched beyond the Gaussian range.
Co-reporter:Jeremy M. Lenhardt ; Ashley L. Black
Journal of the American Chemical Society 2009 Volume 131(Issue 31) pp:10818-10819
Publication Date(Web):July 15, 2009
DOI:10.1021/ja9036548
When gem-dichlorocyclopropane (gDCC) copolymers derived from polybutadiene are subjected to ultrasonication, the gDCCs undergo ring opening to form 2,3-dichloroalkenes. The reactivity is not observed in low-molecular-weight (6.5 kDa) copolymers or side-chain gDCCs, consistent with mechanically induced reactivity due to the elongational strain of the polymers in the sonication flow fields. The ring openings occur several hundred times more frequently than polymer chain scission, and cis-coupled gDCCs are slightly more likely to react than their trans isomers. The ability to dramatically and specifically alter the structure of the polymer backbone through a coupled restoring force suggests new routes to postsynthetic polymer modification and motivates the design of easily scalable mechanophores for applications in stress-responsive polymers.
Co-reporter:StephenL. Craig
Angewandte Chemie International Edition 2009 Volume 48( Issue 15) pp:2645-2647
Publication Date(Web):
DOI:10.1002/anie.200805603
Co-reporter:Michael J. Serpe, Jason R. Whitehead, Monica Rivera, Robert L. Clark, Stephen L. Craig
Colloids and Surfaces A: Physicochemical and Engineering Aspects 2009 Volume 346(1–3) pp:20-27
Publication Date(Web):20 August 2009
DOI:10.1016/j.colsurfa.2009.05.019
Single-molecule force spectroscopy, as implemented in an atomic force microscope, provides a rarely used method by which to monitor dynamic processes that occur near surfaces. Here, a methodology is presented and characterized that facilitates the study of polymer bridging across nanometer-sized gaps. The model system employed is that of DNA-based reversible polymers, and an automated procedure is introduced that allows the AFM tip–surface contact point to be automatically determined, and the distance d between opposing surfaces to be actively controlled. Using this methodology, the importance of several experimental parameters was systematically studied, e.g. the frequency of repeated tip/surface contacts, the area of the substrate surface sampled by the AFM, and the use of multiple AFM tips and substrates. Experiments revealed the surfaces to be robust throughout pulling experiments, so that multiple touches and pulls could be carried out on a single spot with no measurable affect on the results. Differences in observed bridging probabilities were observed, both on different spots on the same surface and, more dramatically, from one day to another. Data normalization via a reference measurement allows data from multiple days to be directly compared.
Co-reporter:StephenL. Craig
Angewandte Chemie 2009 Volume 121( Issue 15) pp:2683-2685
Publication Date(Web):
DOI:10.1002/ange.200805603
Co-reporter:Hemraj Juwarker;JeremyM. Lenhardt;DavidM. Pham Dr. ;StephenL. Craig
Angewandte Chemie International Edition 2008 Volume 47( Issue 20) pp:3740-3743
Publication Date(Web):
DOI:10.1002/anie.200800548
Co-reporter:Michael J. Serpe, Farrell R. Kersey, Jason R. Whitehead, Scott M. Wilson, Robert L. Clark and Stephen L. Craig
The Journal of Physical Chemistry C 2008 Volume 112(Issue 49) pp:19163-19167
Publication Date(Web):November 14, 2008
DOI:10.1021/jp806649a
A simple, practical, and model-free method is presented, by which kinetic data can be extracted from variable loading-rate single molecule dynamic force spectroscopy experiments. The constant sampling rate of digital acquisition facilitates the sorting of multiple force curves into histograms that reflect the collective force−time history of the bonds of interest. Combining the force−time history with similar histograms of force-induced events, in particular bond rupture, gives a direct measure of rate vs applied force. The utility of the method is demonstrated through its application to simulated data.
Co-reporter:StephenL. Craig
Angewandte Chemie International Edition 2008 Volume 47( Issue 46) pp:8776-8777
Publication Date(Web):
DOI:10.1002/anie.200802334
Co-reporter:Hemraj Juwarker;JeremyM. Lenhardt;DavidM. Pham Dr. ;StephenL. Craig
Angewandte Chemie 2008 Volume 120( Issue 20) pp:3800-3803
Publication Date(Web):
DOI:10.1002/ange.200800548
Co-reporter:StephenL. Craig
Angewandte Chemie 2008 Volume 120( Issue 46) pp:8904-8906
Publication Date(Web):
DOI:10.1002/ange.200802334
Co-reporter:David M. Loveless, Sung Lan Jeon and Stephen L. Craig
Journal of Materials Chemistry A 2007 vol. 17(Issue 1) pp:56-61
Publication Date(Web):03 Nov 2006
DOI:10.1039/B614026B
The precise manipulation of network percolation, combined with the previously reported effects of the kinetics of cross-linking interactions, provide a mechanism by which to optimize the stimulus-responsive mechanical properties of supramolecular gels. Specific metal–ligand coordinative bonds create cross-links between poly(4-vinylpyridine) in DMSO, and an abrupt change in mechanical properties is observed at a critical concentration of cross-linker. The change in mechanical properties is attributed to the onset of percolation within the network, and bulk mechanical properties are shown to be especially sensitive to external stimuli in the vicinity of the percolation threshold. The reversible control of bulk mechanics is demonstrated, and the magnitude of the response (changes of up to five orders of magnitude in modulus) is determined by the concentration and dissociation kinetics of the cross-linkers. Combinations of cross-linkers, individually present at concentrations below the percolation threshold, provide a related mechanism by which complex viscoelastic switching can be programmed at the small-molecule level.
Co-reporter:David M. Loveless;Nehal I. Abu-Lail Dr.;Marian Kaholek Dr.;Stefan Zauscher
Angewandte Chemie International Edition 2006 Volume 45(Issue 46) pp:
Publication Date(Web):25 OCT 2006
DOI:10.1002/anie.200602508
Detailed brushwork: Structurally and thermodynamically similar cross-linkers have opposite effects on the lateral resistance of grafted thin films of poly(4-vinylpyridine) brushes. The chemically reversible changes in mechanics are influenced by the dissimilar kinetics of the cross-linking interaction.
Co-reporter:David M. Loveless;Nehal I. Abu-Lail Dr.;Marian Kaholek Dr.;Stefan Zauscher
Angewandte Chemie 2006 Volume 118(Issue 46) pp:
Publication Date(Web):25 OCT 2006
DOI:10.1002/ange.200602508
Gründlich gebürstet: Strukturell und thermodynamisch ähnliche Vernetzer wirken entgegengesetzt auf den lateralen Widerstand gepfropfter dünner Filme aus Poly(4-vinylpyridin)-Bürsten. Die chemisch reversiblen Änderungen in der Mechanik werden durch die unähnliche Kinetik der vernetzenden Wechselwirkung beeinflusst.
Co-reporter:J. Kim;Y. Liu;S. J. Ahn;S. Zauscher;J. M. Karty;Y. Yamanaka;S. L. Craig
Advanced Materials 2005 Volume 17(Issue 14) pp:
Publication Date(Web):19 MAY 2005
DOI:10.1002/adma.200401355
Supramolecular polymer films can be formed by DNA-based reversible polymers on a surface with grafted anchor points (see Figure). The adhesive mechanical properties of the self-assembled polymer brushes have been investigated by atomic force microscopy. Reversible polymer-mediated adhesion is found to be sensitive to surface anchor density and the thermodynamics and conformational flexibility of the monomer in this system.
Co-reporter:Wayne C. Yount;David M. Loveless
Angewandte Chemie 2005 Volume 117(Issue 18) pp:
Publication Date(Web):4 APR 2005
DOI:10.1002/ange.200500026
Festhalten: Die Dynamik und nicht die Thermodynamik reversibler Verknüpfungen bestimmt vor allem die makroskopische Viskosität in einer Familie supramolekularer Netzwerke. Dies folgt aus einer Untersuchung, in der eine Reihe strukturell ähnlicher Pinzettenkomplexe (siehe Bild, OTf=CF3SO3−) mit unterschiedlicher Ligandenaustausch-Dynamik verwendet wurde, um Poly(4-vinylpyridin) zu vernetzen.
Co-reporter:Wayne C. Yount;David M. Loveless
Angewandte Chemie International Edition 2005 Volume 44(Issue 18) pp:
Publication Date(Web):1 APR 2005
DOI:10.1002/anie.200500026
Hold on tight: The dynamics, rather than thermodynamics, of reversible cross-links are the primary determinants of bulk viscosities in a family of supramolecular networks. This result comes from a study in which a series of structurally similar pincer complexes (see picture, OTf=CF3SO3−) that have different ligand-exchange dynamics is used to cross-link poly(4-vinylpyridine).
Co-reporter:Yan Liu, Jun Xu and Stephen L. Craig
Chemical Communications 2004 (Issue 16) pp:1864-1865
Publication Date(Web):30 Jun 2004
DOI:10.1039/B405982D
Amphiphiles defined by noncovalent inclusion complexes between an alkylated β-cyclodextrin and PEG-conjugated guests assemble into higher-ordered structures whose thermodynamic stability reflects that of the defining intermolecular interactions.
Co-reporter:Elizabeth A. Fogleman;Wayne C. Yount;Jun Xu
Angewandte Chemie 2002 Volume 114(Issue 21) pp:
Publication Date(Web):31 OCT 2002
DOI:10.1002/1521-3757(20021104)114:21<4198::AID-ANGE4198>3.0.CO;2-Z
Grundsätzliches über reversible Polymerisationen lässt sich aus Experimenten mit Monomeren auf Oligonucleotid-Basis ableiten; diese bestehen aus Oligonucleotid-Sequenzen, die direkt oder über einen Spacer kovalent verknüpft sind. Thermodynamik und Kinetik der Assoziation werden durch die Variabilität des jeweiligen Systems bestimmt. Die Zahl reversibler Wechselwirkungen und die konformative Flexibilität entlang der Polymerhauptkette hängen vom Spacer ab.
Co-reporter:Elizabeth A. Fogleman;Wayne C. Yount;Jun Xu
Angewandte Chemie International Edition 2002 Volume 41(Issue 21) pp:
Publication Date(Web):31 OCT 2002
DOI:10.1002/1521-3773(20021104)41:21<4026::AID-ANIE4026>3.0.CO;2-E
Fundamental studies of reversible polymerizations can be achieved with intrinsically modular oligonucleotide-based monomers (OMs) comprised of oligonucleotide sequences that are covalently linked directly or through a synthetic spacer. The thermodynamics and kinetics of association are determined by the variable OM base system. The number of reversible interactions and the conformational flexibility along the polymer backbone are dependent on the spacer.
Co-reporter:Bobin Lee; Zhenbin Niu; Junpeng Wang; Carla Slebodnick
Journal of the American Chemical Society () pp:
Publication Date(Web):August 6, 2015
DOI:10.1021/jacs.5b06937
The mechanical strength of scissile chemical bonds plays a role in material failure and in the mechanical activation of latent reactivity, but quantitative measures of mechanical strength are rare. Here, we report the relative mechanical strength of polymers bearing three putatively “weak” scissile bonds: the carbon–nitrogen bond of an azobisdialkylnitrile (<30 kcal mol–1), the carbon–sulfur bond of a thioether (71–74 kcal mol–1), and the carbon–oxygen bond of a benzylphenyl ether (52–54 kcal mol–1). The mechanical strengths are assessed in the context of chain scission triggered by pulsed sonication of polymer solutions, by using two complementary techniques: (i) the competition within a single polymer chain between the bond scission of interest and the nonscissile mechanochemical ring opening of gem-dichlorocyclopropane mechanophores and (ii) the molecular weights at long (4 h) sonication times of multimechanophore polymers. The two methods produce a consistent story: in contrast to their thermodynamic strengths, the relative mechanical strengths of the three weak bonds are azobisdialkylnitrile (weakest) < thioether < benzylphenyl ether. The greater mechanical strength of the benzylphenyl ether relative to the thermodynamically stronger carbon–sulfur bond is ascribed to poor mechanochemical coupling, at least in part as a result of the rehybridization that accompanies carbon–oxygen bond scission.
Co-reporter:David M. Loveless, Sung Lan Jeon and Stephen L. Craig
Journal of Materials Chemistry A 2007 - vol. 17(Issue 1) pp:NaN61-61
Publication Date(Web):2006/11/03
DOI:10.1039/B614026B
The precise manipulation of network percolation, combined with the previously reported effects of the kinetics of cross-linking interactions, provide a mechanism by which to optimize the stimulus-responsive mechanical properties of supramolecular gels. Specific metal–ligand coordinative bonds create cross-links between poly(4-vinylpyridine) in DMSO, and an abrupt change in mechanical properties is observed at a critical concentration of cross-linker. The change in mechanical properties is attributed to the onset of percolation within the network, and bulk mechanical properties are shown to be especially sensitive to external stimuli in the vicinity of the percolation threshold. The reversible control of bulk mechanics is demonstrated, and the magnitude of the response (changes of up to five orders of magnitude in modulus) is determined by the concentration and dissociation kinetics of the cross-linkers. Combinations of cross-linkers, individually present at concentrations below the percolation threshold, provide a related mechanism by which complex viscoelastic switching can be programmed at the small-molecule level.
Co-reporter:Ashley L. Black, Joshua A. Orlicki and Stephen L. Craig
Journal of Materials Chemistry A 2011 - vol. 21(Issue 23) pp:NaN8465-8465
Publication Date(Web):2011/02/16
DOI:10.1039/C0JM03875J
Polybutadiene was functionalized with dibromo-, dichloro-, and bromochloro-carbene to give gem-dihalocyclopropanated (gDHC) polymers, in which the gDHCs act as mechanically activated functional groups or mechanophores. The polymers were extruded to determine the mechanophore activity in the solid state. The extent of gDHC ring opening depends on both the polymer composition and the macroscopic shear stress, ranging from 6.0% to over 30% after an hour of extrusion. In addition, the 2,3-dibromoalkene formed from mechanical activation of the gem-dibromocyclopropane was found to undergo subsequent nucleophilic substitution by chloride in the solid state. The number of solid-state substitution reactions far exceeds the number of main-chain bonds broken, a finding with implications for the future use of mechanophores in self-strengthening or self-healing polymers.
Co-reporter:Jeremy M. Lenhardt, Ashley L. Black, Brett A. Beiermann, Brian D. Steinberg, Faiyam Rahman, Tasha Samborski, Joseph Elsakr, Jeffrey S. Moore, Nancy R. Sottos and Stephen L. Craig
Journal of Materials Chemistry A 2011 - vol. 21(Issue 23) pp:NaN8459-8459
Publication Date(Web):2011/02/07
DOI:10.1039/C0JM04117C
The incorporation of mechanically active functional groups, or mechanophores, along polymer backbones offers opportunities for new stress-responsive material properties and also provides a method by which to probe fundamental questions related to molecular stress distributions in polymeric materials under load. The activation of covalent chemistry in polymers has primarily been demonstrated in solution, but to date little is known regarding activation in the solid state. In the latter regard, recent effort has focused on the use of spectroscopically active mechanophores that directly probe the presence of stress within materials. The distribution of forces within individual polymer chains, however, has yet to be characterized. Herein we report that gem-dihalocyclopropane (gDHC) functionalized polybutadiene is mechanochemically active in the solid state, and that the strain-triggered ring opening of the gDHCs provides quantitative information regarding the number of mechanically active monomers and the size of the mechanically activated domains along individual polymer backbones within bulk materials subjected to compressive and tensile loads. The results show that high mechanical forces are concentrated over lengths of only a few monomers.
Co-reporter:Ashley L. Black, Jeremy M. Lenhardt and Stephen L. Craig
Journal of Materials Chemistry A 2011 - vol. 21(Issue 6) pp:NaN1663-1663
Publication Date(Web):2010/11/01
DOI:10.1039/C0JM02636K
Current activity in, and future prospects for, the incorporation of mechanochemically active functional groups (“mechanophores”) into polymers is reviewed. This area of research is treated in the context of two categories. The first category is the development of new chemistry in the service of material science, through the design and synthesis of mechanophores to provide stress-sensing and/or stress-responsive elements in materials. The second category is the reverse—the development of new material architectures that efficiently transmit macroscopic forces to targeted molecules in order to generate chemical reactivity that is inaccessible by other means.
Co-reporter:Cameron L. Brown and Stephen L. Craig
Chemical Science (2010-Present) 2015 - vol. 6(Issue 4) pp:NaN2165-2165
Publication Date(Web):2015/02/12
DOI:10.1039/C4SC01945H
Force reactive functional groups, or mechanophores, have emerged as the basis of a potential strategy for sensing and countering stress-induced material failure. The general utility of this strategy is limited, however, because the levels of mechanophore activation in the bulk are typically low and observed only under large, typically irreversible strains. Strategies that enhance activation are therefore quite useful. Molecular-level design principles by which to engineer enhanced mechanophore activity are reviewed, with an emphasis on quantitative structure–activity studies determined for a family of gem-dihalocyclopropane mechanophores.
Co-reporter:Zachary S. Kean, Gregory R. Gossweiler, Tatiana B. Kouznetsova, Gihan B. Hewage and Stephen L. Craig
Chemical Communications 2015 - vol. 51(Issue 44) pp:NaN9160-9160
Publication Date(Web):2015/04/29
DOI:10.1039/C5CC01836F
Here we present a coumarin dimer (CD) mechanophore that, when embedded near the mid-chain of poly(methyl acrylate) polymers, activates under pulsed ultrasound conditions to yield coumarin chain-end functional polymers. Quantitative photochemical scission of the CD polymers provides a reference against which the activation efficiency of chain-centered mechanophores in polymers synthesized by controlled/living radical polymerization (CRP) can be assessed. Activation efficiency is characterized with respect to the polymer molecular weight (MW), polydispersity index (PDI), and distribution of mechanophores along the backbone.
.jpg)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-1'(3'H)-ethanol, 5'-hydroxy-3',3'-dimethyl-6-nitro-](/data/chemimg/171100/1253909-23-7.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-1'(3'H)-ethanol, 5'-hydroxy-3',3'-dimethyl-6-nitro-](/data/chemimg/171100/1253909-23-7_b.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diol, 1',3'-dihydro-1',3',3'-trimethyl-6-nitro-](/data/chemimg/171100/1253909-21-5.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diol, 1',3'-dihydro-1',3',3'-trimethyl-6-nitro-](/data/chemimg/171100/1253909-21-5_b.png)
![Chloro(1,3-dimesityl-2-imidazolidinylidene)(2-{(E)-[(4-methylphenyl)imino]methyl}-4-nitrophenolato-κO)(3-phenyl-1H-inden-1-ylidene)ruthenium](http://img.cochemist.com/ccimg/934600/934538-04-2.png)
![Chloro(1,3-dimesityl-2-imidazolidinylidene)(2-{(E)-[(4-methylphenyl)imino]methyl}-4-nitrophenolato-κO)(3-phenyl-1H-inden-1-ylidene)ruthenium](http://img.cochemist.com/ccimg/934600/934538-04-2_b.png)





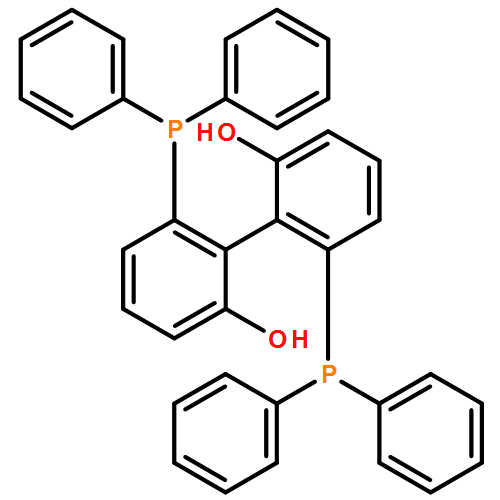
![[1,1'-Biphenyl]-2,2'-diol, 6,6'-bis(diphenylphosphino)-](/data/chemimg/3726200/270253-34-4.png)
![[1,1'-Biphenyl]-2,2'-diol, 6,6'-bis(diphenylphosphino)-](/data/chemimg/3726200/270253-34-4_b.png)



![Bicyclo[6.1.0]non-4-ene, 9,9-difluoro- (9CI)](http://img.cochemist.com/ccimg/154500/154413-30-6.png)
![Bicyclo[6.1.0]non-4-ene, 9,9-difluoro- (9CI)](http://img.cochemist.com/ccimg/154500/154413-30-6_b.png)


![1-[1,1,1,3,3,3-hexafluoro-2-[4-(1,2,2-trifluoroethenoxy)phenyl]propan-2-yl]-4-(1,2,2-trifluoroethenoxy)benzene](http://img.cochemist.com/ccimg/134200/134174-11-1.png)
![1-[1,1,1,3,3,3-hexafluoro-2-[4-(1,2,2-trifluoroethenoxy)phenyl]propan-2-yl]-4-(1,2,2-trifluoroethenoxy)benzene](http://img.cochemist.com/ccimg/134200/134174-11-1_b.png)
![1,2,3,4,5-CYCLOPENTANEPENTAYL, COMPD. WITH 1-[(1S)-1-(DIBICYCLO[2.2.1]HEPT-2-YLPHOSPHINO)ETHYL]-2-[2-(DIPHENYLPHOSPHINO)PHENYL]-1,2,3,4,5-CYCLOPENTANEPENTAYL, IRON SALT (1:1:1)](http://img.cochemist.com/ccimg/134000/133978-15-1.png)
![1,2,3,4,5-CYCLOPENTANEPENTAYL, COMPD. WITH 1-[(1S)-1-(DIBICYCLO[2.2.1]HEPT-2-YLPHOSPHINO)ETHYL]-2-[2-(DIPHENYLPHOSPHINO)PHENYL]-1,2,3,4,5-CYCLOPENTANEPENTAYL, IRON SALT (1:1:1)](http://img.cochemist.com/ccimg/134000/133978-15-1_b.png)








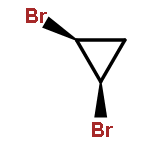
![(2Z)-9,9-dichlorobicyclo[6.1.0]non-2-ene](http://img.cochemist.com/ccimg/58200/58189-58-5.png)
![(2Z)-9,9-dichlorobicyclo[6.1.0]non-2-ene](http://img.cochemist.com/ccimg/58200/58189-58-5_b.png)




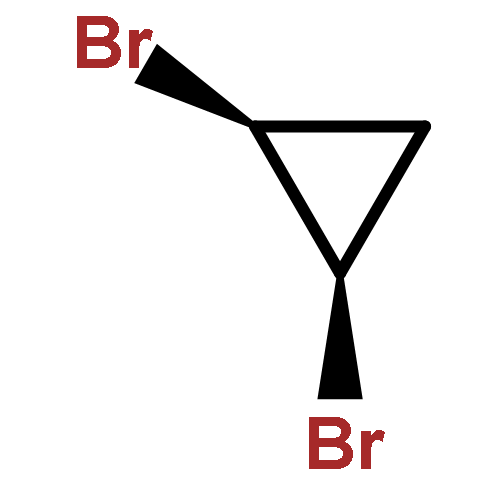
![(4Z)-9,9-dibromobicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/24500/24449-05-6.png)
![(4Z)-9,9-dibromobicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/24500/24449-05-6_b.png)
![2,4-Cyclohexadien-1-one, 6-[2-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)ethylidene]-4-nitro-](/data/chemimg/3778900/18457-95-9.png)
![2,4-Cyclohexadien-1-one, 6-[2-(1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)ethylidene]-4-nitro-](/data/chemimg/3778900/18457-95-9_b.png)


![9-oxabicyclo[6.1.0]non-2-ene](http://img.cochemist.com/ccimg/6700/6690-12-6.png)
![9-oxabicyclo[6.1.0]non-2-ene](http://img.cochemist.com/ccimg/6700/6690-12-6_b.png)
![1,4,5,6-tetrachloro-7,7-dimethoxybicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/5300/5259-71-2.png)
![1,4,5,6-tetrachloro-7,7-dimethoxybicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/5300/5259-71-2_b.png)







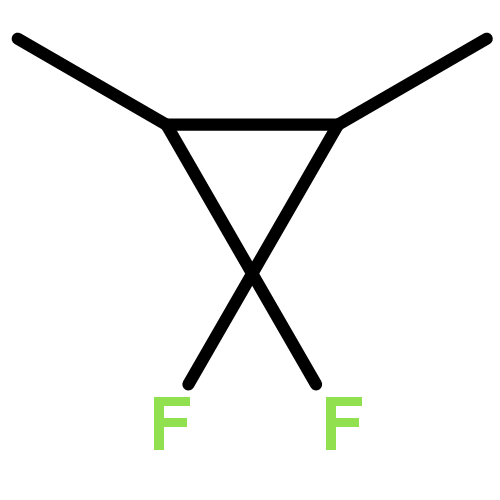


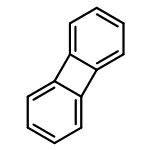
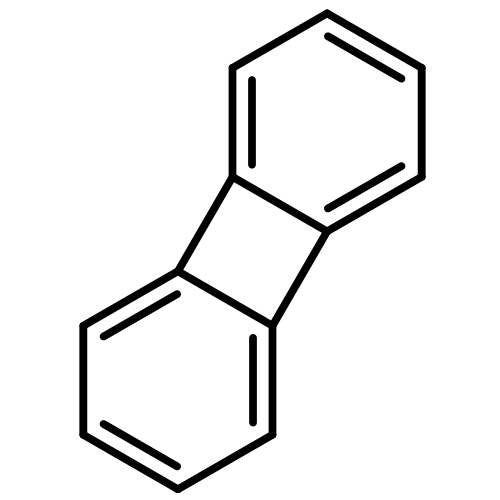




![4-Pentenoic acid, 1',3'-dihydro-3',3'-dimethyl-6-nitro-1'-[2-[(1-oxo-4-penten-1-yl)oxy]ethyl]spiro[2H-1-benzopyran-2,2'-[2H]indol]-8-yl ester](/data/chemimg/3778900/1554286-40-6.png)
![4-Pentenoic acid, 1',3'-dihydro-3',3'-dimethyl-6-nitro-1'-[2-[(1-oxo-4-penten-1-yl)oxy]ethyl]spiro[2H-1-benzopyran-2,2'-[2H]indol]-8-yl ester](/data/chemimg/3778900/1554286-40-6_b.png)


![Poly[1,3-cyclopentanediyl-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/65600/65556-06-1.png)
![Poly[1,3-cyclopentanediyl-(1Z)-1,2-ethenediyl]](http://img.cochemist.com/ccimg/65600/65556-06-1_b.png)


![(Z)-cis-9-oxabicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/19800/19740-90-0.png)
![(Z)-cis-9-oxabicyclo[6.1.0]non-4-ene](http://img.cochemist.com/ccimg/19800/19740-90-0_b.png)

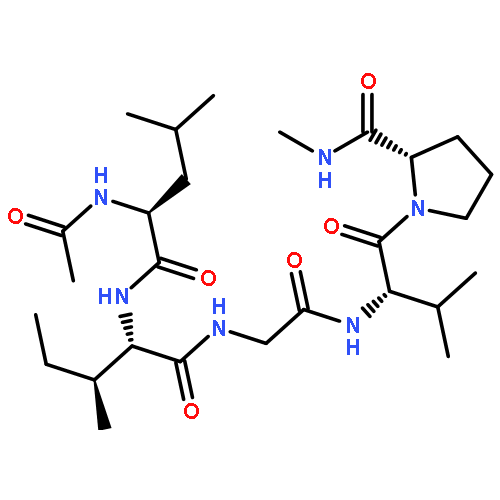




![Bicyclo[6.1.0]non-4-ene, 9,9-dichloro-](http://img.cochemist.com/ccimg/4800/4729-12-8.png)
![Bicyclo[6.1.0]non-4-ene, 9,9-dichloro-](http://img.cochemist.com/ccimg/4800/4729-12-8_b.png)