The conversion of axially chiral tetraalkoxyresorcin[4]arenes (cyclochiral resorcinarenes) into the related tetrakis(triflates) in high yields is described; this provides an efficient source of chiral materials for use in palladium-catalyzed transformations, including the reductive removal of triflate groups and the synthesis of compounds that are formally derived from 3-aminophenol, providing axially chiral derivatives that are nitrogen-substituted on the upper rim.
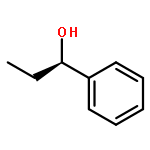
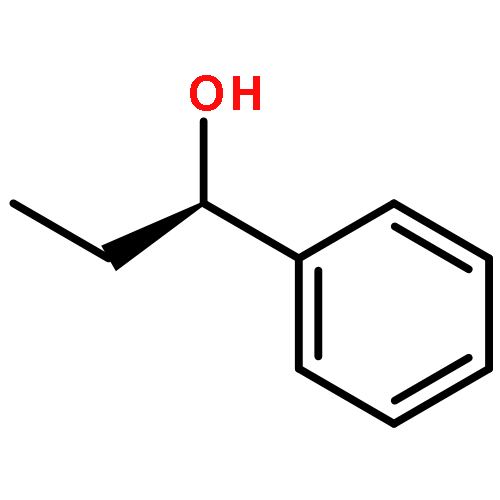
The preparation of optically pure (–)-3-[(2R)-2-methoxy-2-phenylethoxy]phenol from resorcinol monobenzoate and its conversion into diastereoisomeric tetraalkoxyresorcin[4]arenes together with proof of the absolute configurations of the products is reported. The results of the study indicate that diastereoselective ring closure of linear tetrameric intermediates is controlled by the steric demand of the alkyl group in the precursor 3-alkoxyphenol.
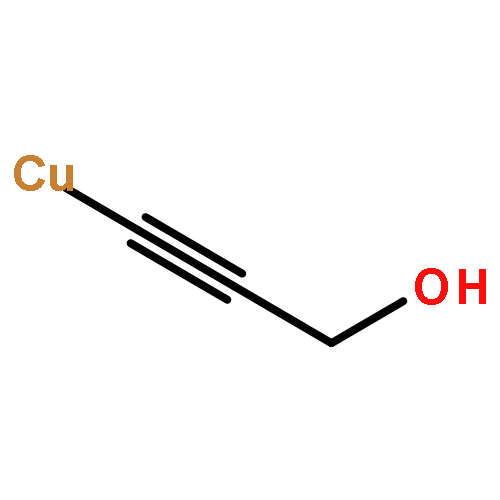
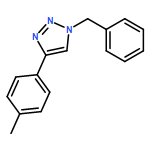
Dinuclear alkynylcopper(I) ladderane complexes are prepared by a robust and simple protocol involving the reduction of Cu2(OH)3OAc or Cu(OAc)2 by easily oxidised alcohols in the presence of terminal alkynes; they function as efficient catalysts in copper-catalysed alkyne–azide cycloaddition reactions as predicted by the Ahlquist–Fokin calculations. The same copper(I) catalysts are formed during reactions by using the Sharpless–Fokin protocol. The experimental results also provide evidence that sodium ascorbate functions as a base to deprotonate terminal alkynes and additionally give a convincing alternative explanation for the fact that the CuI-catalysed reactions of certain 1,3-diazides with phenylacetylene give bis(triazoles) as the major products. The same dinuclear alkynylcopper(I) complexes also function as catalysts in cycloaddition reactions of azides with 1-iodoalkynes.
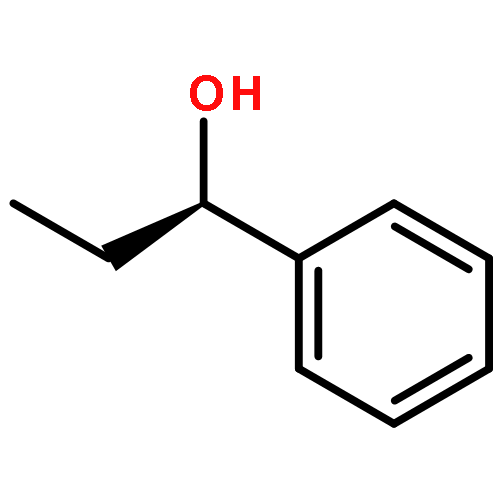
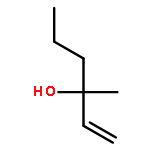
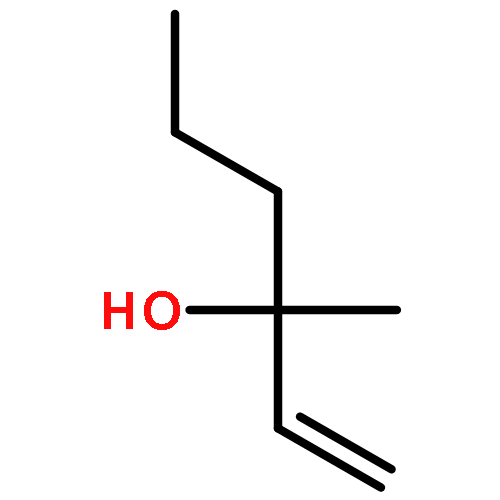
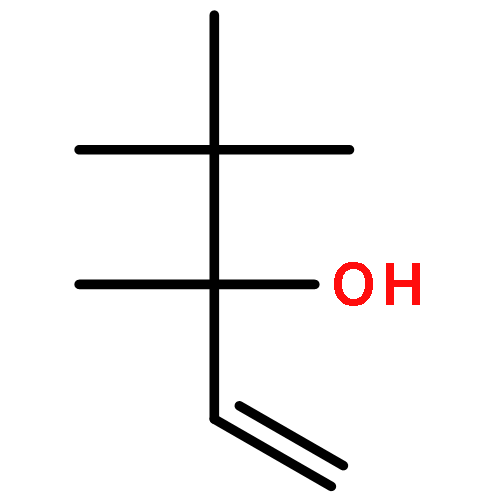
The measurement of the pKa of racemic tetramethoxyresorcin[4]arenes explains the failure to obtain good yields in attempted Mannich reactions of these substrates under classical reaction conditions. The failure is related to the lack of adequate concentrations of the iminium ions that results from the reduced acid strength of tetraalkoxyresorcin[4]arenes compared with that of the parent octahydroxyresorcin[4]arenes. However, the preparation of a series of Mannich bases derived from racemic tetraalkoxyresorcin[4]arenes was accomplished under microwave-assisted aprotic reaction conditions and the use of preformed iminium ion intermediates. When the reactions were carried out with the use of chiral bis(aminol) ethers, mixtures of diastereomers were obtained that could be separated by flash chromatography. The absolute configurations of the enantiomerically pure tetrabenzoxazine derivatives were established in some cases by X-ray crystallographic analysis and by a comparison of the nuclear magnetic resonance spectroscopic data. The alkylation of racemic tetramethoxyresorcin[4]arenes was achieved with the use of an excess of 2-bromo-N-[(R)-(+)-(α-methylbenzyl)]acetamide in acetonitrile containing potassium carbonate. Enantioselective ligand-assisted reactions of aromatic aldehydes are also reported with the use of dialkylzinc reagents both in the absence and in the presence of terminal alkynes.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
The preparation of a series of diastereoisomeric tetracamphorsulfonates derived from racemic tetramethoxyresorcin[4]arenes was achieved by reactions with an excess of (S)-(+)-10-camphorsulfonyl chloride in pyridine followed by isolation using flash chromatography. Tetradeprotonation of a number of tetramethoxyresorcin[4]arenes using n-butyllithium in tetrahydrofuran, followed by reactions using (S)-(+)-10-camphorsulfonyl chloride, gave the same tetracamphorsulfonates. Mono-, di- and tricamphorsulfonates were also prepared following selective deprotonation. In the reactions with tetraisopropyloxy- and tetracyclopentyloxyresorcin[4]arenes, only the mono- and dicamphorsulfonates were formed. X-ray crystallographic analysis established the absolute configurations of three diastereoisomerically pure tetracamphorsulfonates, including a diastereoisomer prepared from 6,12,18,24-tetramethoxy-2,8,14,20-tetrakis(2-methylpropyl)resorcin[4]arene. An additional pair of diastereoisomers was also prepared using (R)-(–)-10-camphorsulfonyl chloride and 6,12,18,24-tetramethoxy-2,8,14,20-tetrakis(2-methylpropyl)resorcin[4]arene, for one of which the structure was confirmed by an additional X-ray structure determination. Hydrolytic removal of the camphorsulfonyl residue(s) from the various diastereoisomers gave enantiomers of known absolute configurations. In some cases, the chiral nonracemic tetraalkoxyresorcin[4]arenes were converted into known tetrabenzoxazine derivatives by using N,N-bis(methoxymethyl)[(S)-(–)-(α-methylbenzyl)]amine in thermal or microwave-assisted reactions. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Methodology is presented for synthesis of the first examples of chiral, bridged resorcinarenes with functionality in the bridge as possible sites for intracavity asymmetric catalysis. A preliminary study comparing unfunctionalised with functionalised lines using the enantioselective addition of diethylzinc to benzaldehyde as a probe reaction, provides compelling evidence for intracavity catalysis. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)












![Pyridine, 2-[(2R,4S,5R)-2,3,4-trimethyl-5-phenyl-2-oxazolidinyl]-](http://img.cochemist.com/ccimg/853900/853802-38-7.png)
![Pyridine, 2-[(2R,4S,5R)-2,3,4-trimethyl-5-phenyl-2-oxazolidinyl]-](http://img.cochemist.com/ccimg/853900/853802-38-7_b.png)
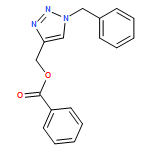
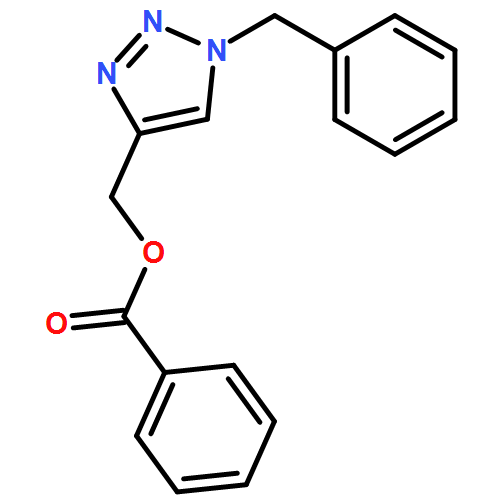
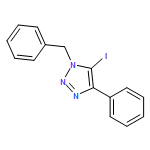
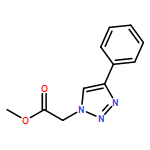
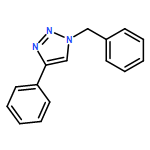
![1H-1,2,3-Triazole, 1-[(4-methylphenyl)methyl]-4-phenyl-](/data/chemimg/3722400/126800-02-0.png)
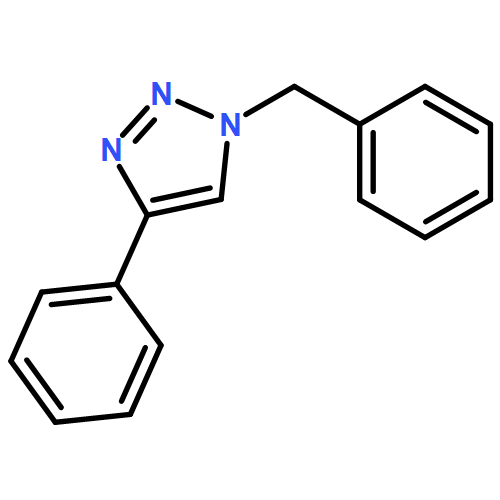
![1H-1,2,3-Triazole, 1-[(4-methylphenyl)methyl]-4-phenyl-](/data/chemimg/3722400/126800-02-0_b.png)










![Phenol, 4-methyl-2-[[methyl(phenylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/258900/258827-68-8.png)
![Phenol, 4-methyl-2-[[methyl(phenylmethyl)amino]methyl]-](http://img.cochemist.com/ccimg/258900/258827-68-8_b.png)




![Phenol, 2-[(diethylamino)methyl]-4,6-dimethyl-](http://img.cochemist.com/ccimg/69100/69084-00-0.png)
![Phenol, 2-[(diethylamino)methyl]-4,6-dimethyl-](http://img.cochemist.com/ccimg/69100/69084-00-0_b.png)










![COPPER, [3-(4-METHOXYPHENOXY)-1-PROPYNYL]-](http://img.cochemist.com/ccimg/66400/66397-28-2.png)
![COPPER, [3-(4-METHOXYPHENOXY)-1-PROPYNYL]-](http://img.cochemist.com/ccimg/66400/66397-28-2_b.png)
![Phenol, 2,4-bis[(diethylamino)methyl]-3,6-dimethyl-](http://img.cochemist.com/ccimg/121000/120983-08-6.png)
![Phenol, 2,4-bis[(diethylamino)methyl]-3,6-dimethyl-](http://img.cochemist.com/ccimg/121000/120983-08-6_b.png)
![2H-1,3-Benzoxazine, 3,4-dihydro-6-methyl-3-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/237500/237431-64-0.png)
![2H-1,3-Benzoxazine, 3,4-dihydro-6-methyl-3-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/237500/237431-64-0_b.png)










![Piperidine, 1,1'-[(1-methyl-1H-pyrrole-2,5-diyl)bis(methylene)]bis-](http://img.cochemist.com/ccimg/117400/117326-58-6.png)
![Piperidine, 1,1'-[(1-methyl-1H-pyrrole-2,5-diyl)bis(methylene)]bis-](http://img.cochemist.com/ccimg/117400/117326-58-6_b.png)
![PYRIDINE, 2-[(2S,4S,5S)-2,3,4-TRIMETHYL-5-PHENYL-2-OXAZOLIDINYL]-](http://img.cochemist.com/ccimg/853900/853802-39-8.png)
![PYRIDINE, 2-[(2S,4S,5S)-2,3,4-TRIMETHYL-5-PHENYL-2-OXAZOLIDINYL]-](http://img.cochemist.com/ccimg/853900/853802-39-8_b.png)
![Phenol, 2-[[(2-hydroxyethyl)methylamino]methyl]-5-methoxy-](http://img.cochemist.com/ccimg/123400/123341-20-8.png)
![Phenol, 2-[[(2-hydroxyethyl)methylamino]methyl]-5-methoxy-](http://img.cochemist.com/ccimg/123400/123341-20-8_b.png)
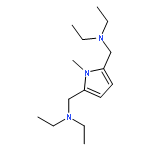





![Ethanol, 2-[methyl[(5-methyl-2-furanyl)methyl]amino]-](http://img.cochemist.com/ccimg/123400/123387-82-6.png)
![Ethanol, 2-[methyl[(5-methyl-2-furanyl)methyl]amino]-](http://img.cochemist.com/ccimg/123400/123387-82-6_b.png)




![PYRAZINO[1,2-A]INDOLE, 4-ETHENYL-1,2,3,4-TETRAHYDRO-2-(PHENYLMETHYL)-](http://img.cochemist.com/ccimg/770000/769929-36-4.png)
![PYRAZINO[1,2-A]INDOLE, 4-ETHENYL-1,2,3,4-TETRAHYDRO-2-(PHENYLMETHYL)-](http://img.cochemist.com/ccimg/770000/769929-36-4_b.png)


![Stannane, trimethyl[4-[(tetrahydro-2H-pyran-2-yl)oxy]phenyl]-](http://img.cochemist.com/ccimg/108700/108613-04-3.png)
![Stannane, trimethyl[4-[(tetrahydro-2H-pyran-2-yl)oxy]phenyl]-](http://img.cochemist.com/ccimg/108700/108613-04-3_b.png)




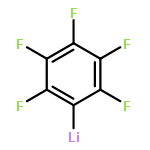
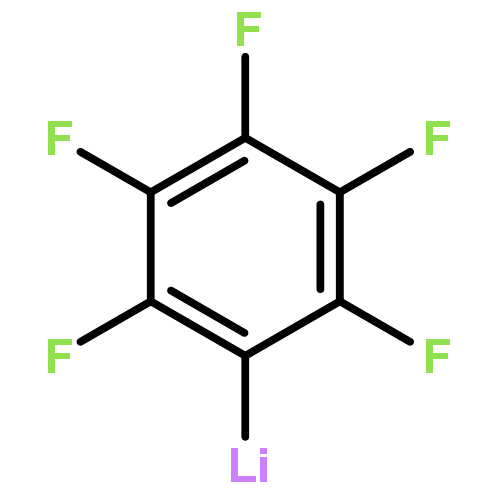
![Benzo[h]quinoline, 2-phenyl-4-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/193400/193337-18-7.png)
![Benzo[h]quinoline, 2-phenyl-4-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/193400/193337-18-7_b.png)
![1-NAPHTHALENAMINE, 2,4-BIS[(1,1-DIMETHYLETHYL)THIO]-](http://img.cochemist.com/ccimg/525000/524948-58-1.png)
![1-NAPHTHALENAMINE, 2,4-BIS[(1,1-DIMETHYLETHYL)THIO]-](http://img.cochemist.com/ccimg/525000/524948-58-1_b.png)


![1,3(2H,4H)-ISOQUINOLINEDIONE, 2-[2-(3,4-DIMETHOXYPHENYL)ETHYL]-](http://img.cochemist.com/ccimg/59200/59168-22-8.png)
![1,3(2H,4H)-ISOQUINOLINEDIONE, 2-[2-(3,4-DIMETHOXYPHENYL)ETHYL]-](http://img.cochemist.com/ccimg/59200/59168-22-8_b.png)






![4-NITROPHENOL [1-14C]](http://img.cochemist.com/ccimg/58500/58402-66-7.png)
![4-NITROPHENOL [1-14C]](http://img.cochemist.com/ccimg/58500/58402-66-7_b.png)










![1H-1,2,3-Triazole, 1-[(4-fluorophenyl)methyl]-4-phenyl-](/data/chemimg/2342000/848950-93-6.png)
![1H-1,2,3-Triazole, 1-[(4-fluorophenyl)methyl]-4-phenyl-](/data/chemimg/2342000/848950-93-6_b.png)
![butyl[(5-methylfuran-2-yl)methyl]amine](http://img.cochemist.com/ccimg/130600/130539-97-8.png)
![butyl[(5-methylfuran-2-yl)methyl]amine](http://img.cochemist.com/ccimg/130600/130539-97-8_b.png)






![(1R,2S)-2-[benzyl(methyl)amino]-1-phenylpropan-1-ol](http://img.cochemist.com/ccimg/52300/52253-64-2.png)
![(1R,2S)-2-[benzyl(methyl)amino]-1-phenylpropan-1-ol](http://img.cochemist.com/ccimg/52300/52253-64-2_b.png)
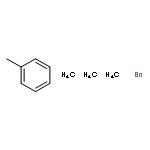
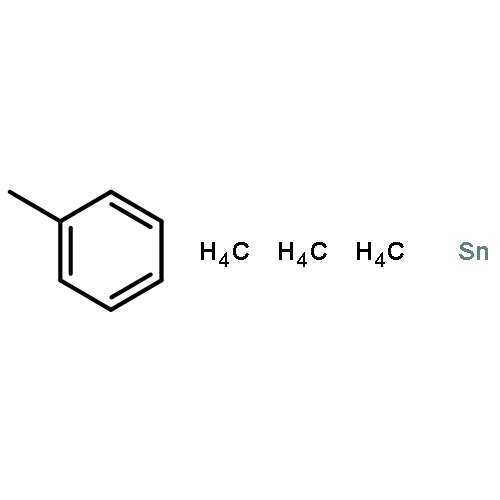




![Thiophene, 2-[[(4-methylphenyl)sulfonyl]methyl]-](http://img.cochemist.com/ccimg/20900/20895-79-8.png)
![Thiophene, 2-[[(4-methylphenyl)sulfonyl]methyl]-](http://img.cochemist.com/ccimg/20900/20895-79-8_b.png)
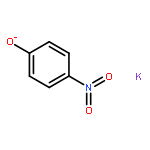



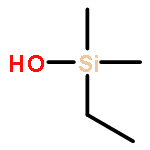
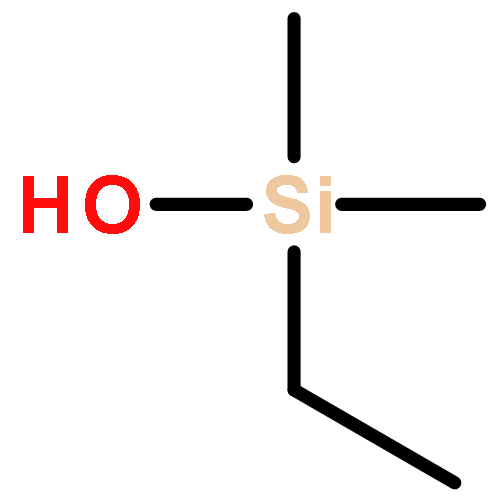







![Benzene, [(S)-methylsulfinyl]-](http://img.cochemist.com/ccimg/18500/18453-46-8.png)
![Benzene, [(S)-methylsulfinyl]-](http://img.cochemist.com/ccimg/18500/18453-46-8_b.png)






![2-[(diethylamino)methyl]-3,6-dimethylphenol](http://img.cochemist.com/ccimg/6700/6630-03-1.png)
![2-[(diethylamino)methyl]-3,6-dimethylphenol](http://img.cochemist.com/ccimg/6700/6630-03-1_b.png)


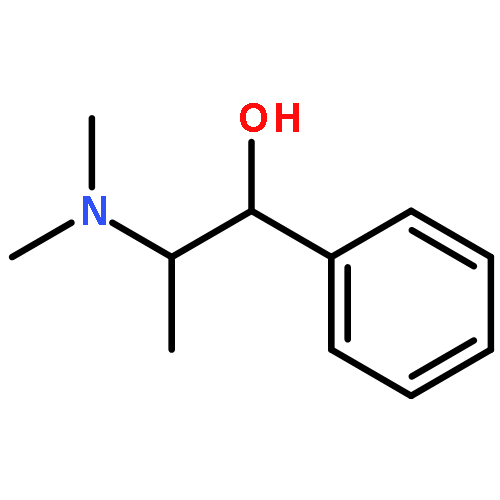
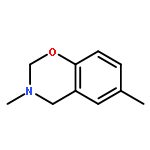
![3-Benzyl-6-methyl-3,4-dihydro-2H-benzo[e][1,3]oxazine](http://img.cochemist.com/ccimg/52100/52055-73-9.png)
![3-Benzyl-6-methyl-3,4-dihydro-2H-benzo[e][1,3]oxazine](http://img.cochemist.com/ccimg/52100/52055-73-9_b.png)


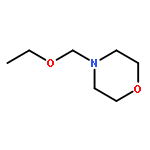



![Benzene, [[(1-phenyl-3-butenyl)oxy]methyl]-](http://img.cochemist.com/ccimg/137500/137438-51-8.png)
![Benzene, [[(1-phenyl-3-butenyl)oxy]methyl]-](http://img.cochemist.com/ccimg/137500/137438-51-8_b.png)

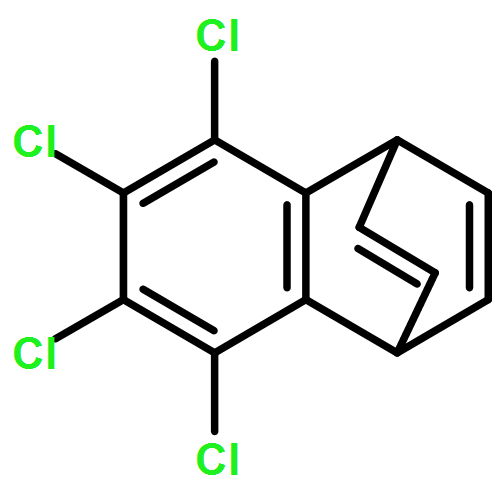




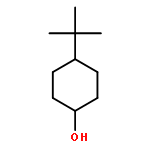



![7,8,13,13b-tetrahydro-5H-benzo[1,2]indolizino[8,7-b]indol-5-one](http://img.cochemist.com/ccimg/65100/65020-20-4.png)
![7,8,13,13b-tetrahydro-5H-benzo[1,2]indolizino[8,7-b]indol-5-one](http://img.cochemist.com/ccimg/65100/65020-20-4_b.png)


![Methanone, phenyl[(2R,3S)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-92-4.png)
![Methanone, phenyl[(2R,3S)-3-phenyloxiranyl]-](http://img.cochemist.com/ccimg/61900/61840-92-4_b.png)




![Thieno[3,2-b][1,5]naphthyridine, 7-methoxy-9-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/785800/785778-20-3.png)
![Thieno[3,2-b][1,5]naphthyridine, 7-methoxy-9-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/785800/785778-20-3_b.png)



![4-[(1-methylpyrrol-2-yl)methyl]morpholine](http://img.cochemist.com/ccimg/7000/6974-95-4.png)
![4-[(1-methylpyrrol-2-yl)methyl]morpholine](http://img.cochemist.com/ccimg/7000/6974-95-4_b.png)


![Silane, trimethyl[(1-phenoxy-1-propenyl)oxy]-](http://img.cochemist.com/ccimg/66000/65946-50-1.png)
![Silane, trimethyl[(1-phenoxy-1-propenyl)oxy]-](http://img.cochemist.com/ccimg/66000/65946-50-1_b.png)


![(1as,6ar)-6,6a-dihydro-1ah-indeno[1,2-b]oxirene](http://img.cochemist.com/ccimg/67600/67528-26-1.png)
![(1as,6ar)-6,6a-dihydro-1ah-indeno[1,2-b]oxirene](http://img.cochemist.com/ccimg/67600/67528-26-1_b.png)
![Benzenemethanol, α-[1-(ethylmethylamino)ethyl]-, [R-(R*,S*)]-](http://img.cochemist.com/ccimg/48200/48141-64-6.png)
![Benzenemethanol, α-[1-(ethylmethylamino)ethyl]-, [R-(R*,S*)]-](http://img.cochemist.com/ccimg/48200/48141-64-6_b.png)




![Piperidine, 1-[(1-methyl-1H-pyrrol-2-yl)methyl]-](http://img.cochemist.com/ccimg/117400/117326-56-4.png)
![Piperidine, 1-[(1-methyl-1H-pyrrol-2-yl)methyl]-](http://img.cochemist.com/ccimg/117400/117326-56-4_b.png)
![Acetamide, 2-bromo-N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/166800/166771-57-9.png)
![Acetamide, 2-bromo-N-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/166800/166771-57-9_b.png)




![Isoxazolo[3,4-f]quinoline, 5-methoxy-3-(2-thienyl)-](http://img.cochemist.com/ccimg/785800/785778-25-8.png)
![Isoxazolo[3,4-f]quinoline, 5-methoxy-3-(2-thienyl)-](http://img.cochemist.com/ccimg/785800/785778-25-8_b.png)



![[1,1'-Biphenyl]-2-amine, 2'-methoxy-](http://img.cochemist.com/ccimg/1300/1206-76-4.png)
![[1,1'-Biphenyl]-2-amine, 2'-methoxy-](http://img.cochemist.com/ccimg/1300/1206-76-4_b.png)



















![2-[(DIMETHYLAMINO)METHYL]-4-METHYLPHENOL](http://img.cochemist.com/ccimg/7300/7204-35-5.png)
![2-[(DIMETHYLAMINO)METHYL]-4-METHYLPHENOL](http://img.cochemist.com/ccimg/7300/7204-35-5_b.png)


![Phenol,4-methyl-2-[(methylamino)methyl]-](http://img.cochemist.com/ccimg/237500/237431-66-2.png)
![Phenol,4-methyl-2-[(methylamino)methyl]-](http://img.cochemist.com/ccimg/237500/237431-66-2_b.png)




![Pyrrolidine, 1-[(5-methyl-2-furanyl)methyl]-](http://img.cochemist.com/ccimg/61500/61480-99-7.png)
![Pyrrolidine, 1-[(5-methyl-2-furanyl)methyl]-](http://img.cochemist.com/ccimg/61500/61480-99-7_b.png)















![D-erythro-Pent-1-enitol,1,4-anhydro-2-deoxy-3,5-bis-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/173400/173327-56-5.png)
![D-erythro-Pent-1-enitol,1,4-anhydro-2-deoxy-3,5-bis-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/173400/173327-56-5_b.png)

![Ethanol, 2-[(2-furanylmethyl)methylamino]-](http://img.cochemist.com/ccimg/17300/17254-98-7.png)
![Ethanol, 2-[(2-furanylmethyl)methylamino]-](http://img.cochemist.com/ccimg/17300/17254-98-7_b.png)







![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3.png)
![Propanedioic acid, [(1S,2E)-1,3-diphenyl-2-propenyl]-, dimethyl ester](http://img.cochemist.com/ccimg/96500/96482-64-3_b.png)








![1,4-Dioxaspiro[4.5]decane, 8-methyl-](http://img.cochemist.com/ccimg/1000/935-51-3.png)
![1,4-Dioxaspiro[4.5]decane, 8-methyl-](http://img.cochemist.com/ccimg/1000/935-51-3_b.png)
![Benzenecarbothioamide, N-[(2-iodophenyl)methyl]-](http://img.cochemist.com/ccimg/95600/95517-42-3.png)
![Benzenecarbothioamide, N-[(2-iodophenyl)methyl]-](http://img.cochemist.com/ccimg/95600/95517-42-3_b.png)



![Benzene,[(methylsulfinyl)methyl]-](http://img.cochemist.com/ccimg/900/824-86-2.png)
![Benzene,[(methylsulfinyl)methyl]-](http://img.cochemist.com/ccimg/900/824-86-2_b.png)


![2,3-DIMETHOXY-6,12B-DIHYDRO-5H-ISOINDOLO[1,2-A]ISOQUINOLIN-8-ONE](http://img.cochemist.com/ccimg/26500/26477-10-1.png)
![2,3-DIMETHOXY-6,12B-DIHYDRO-5H-ISOINDOLO[1,2-A]ISOQUINOLIN-8-ONE](http://img.cochemist.com/ccimg/26500/26477-10-1_b.png)























![Piperidine,1-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/29200/29175-53-9.png)
![Piperidine,1-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/29200/29175-53-9_b.png)










![2,4-bis[(dimethylamino)methyl]phenol](http://img.cochemist.com/ccimg/5500/5424-54-4.png)
![2,4-bis[(dimethylamino)methyl]phenol](http://img.cochemist.com/ccimg/5500/5424-54-4_b.png)

![2-[(2-FURYLMETHYL)AMINO]-4-PHENOXY-5-SULFAMOYLBENZENESULFONIC ACI<WBR />D](http://img.cochemist.com/ccimg/33200/33143-28-1.png)
![2-[(2-FURYLMETHYL)AMINO]-4-PHENOXY-5-SULFAMOYLBENZENESULFONIC ACI<WBR />D](http://img.cochemist.com/ccimg/33200/33143-28-1_b.png)
![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(3-METHOXYPHENYL)-1,2,3-THI<WBR />ADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/30200/30186-48-2.png)
![N-[2-(CYCLOPENTYLAMINO)-2-OXOETHYL]-N-(3-METHOXYPHENYL)-1,2,3-THI<WBR />ADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/30200/30186-48-2_b.png)


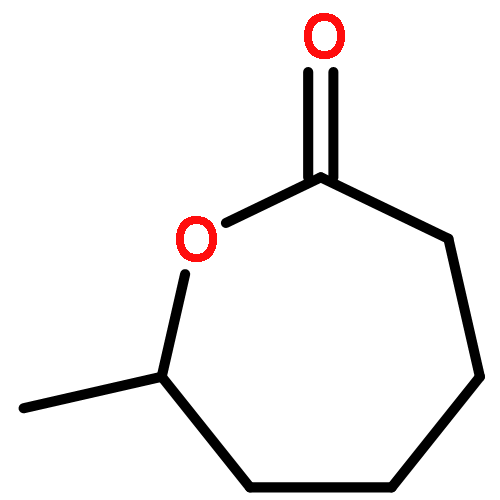


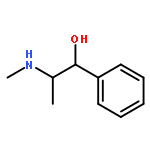
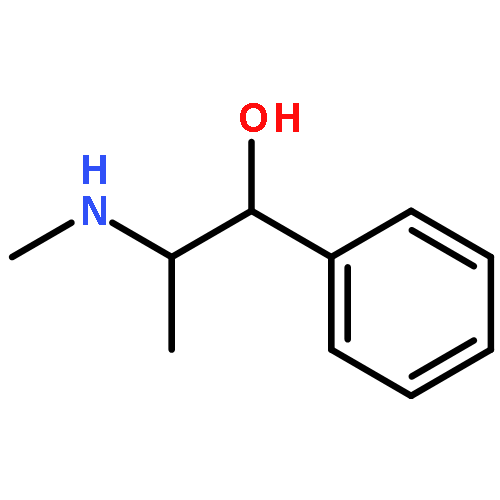


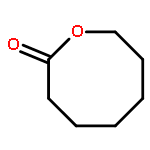
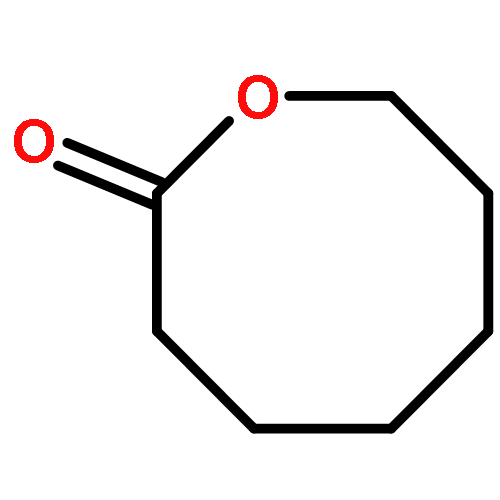




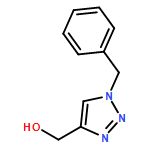



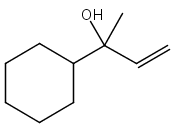
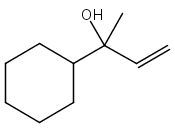














![2-[[2-amino-3-[(2-methylpropan-2-yl)oxy]-3-oxopropyl]-[2-(5-methyl-2,4-dioxopyrimidin-1-yl)acetyl]amino]acetic Acid](http://img.cochemist.com/ccimg/139200/139166-80-6.png)
![2-[[2-amino-3-[(2-methylpropan-2-yl)oxy]-3-oxopropyl]-[2-(5-methyl-2,4-dioxopyrimidin-1-yl)acetyl]amino]acetic Acid](http://img.cochemist.com/ccimg/139200/139166-80-6_b.png)






![1-[(5-methylfuran-2-yl)methyl]piperidine](http://img.cochemist.com/ccimg/46300/46209-55-6.png)
![1-[(5-methylfuran-2-yl)methyl]piperidine](http://img.cochemist.com/ccimg/46300/46209-55-6_b.png)
![Phenol,4-[(dimethylamino)methyl]-](http://img.cochemist.com/ccimg/200/103-87-7.png)
![Phenol,4-[(dimethylamino)methyl]-](http://img.cochemist.com/ccimg/200/103-87-7_b.png)

![Silane, trimethyl[(1-phenoxyethenyl)oxy]-](http://img.cochemist.com/ccimg/111100/111061-31-5.png)
![Silane, trimethyl[(1-phenoxyethenyl)oxy]-](http://img.cochemist.com/ccimg/111100/111061-31-5_b.png)
![METHYL (1S,3AS,3BS,5AR,9AR,9BS,11AS)-9A,11A-DIMETHYL-7-OXO-1,2,3,3A,3B,4,5,5A,6,9B,10,11-DODECAHYDROINDENO[5,4-F]QUINOLINE-1-CARBOXYLATE](http://img.cochemist.com/ccimg/103400/103335-41-7.png)
![METHYL (1S,3AS,3BS,5AR,9AR,9BS,11AS)-9A,11A-DIMETHYL-7-OXO-1,2,3,3A,3B,4,5,5A,6,9B,10,11-DODECAHYDROINDENO[5,4-F]QUINOLINE-1-CARBOXYLATE](http://img.cochemist.com/ccimg/103400/103335-41-7_b.png)





![Glycine,N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[[6-[[(phenylmethoxy)carbonyl]amino]-9H-purin-9-yl]acetyl]-](http://img.cochemist.com/ccimg/149400/149376-69-2.png)
![Glycine,N-[2-[[(1,1-dimethylethoxy)carbonyl]amino]ethyl]-N-[[6-[[(phenylmethoxy)carbonyl]amino]-9H-purin-9-yl]acetyl]-](http://img.cochemist.com/ccimg/149400/149376-69-2_b.png)