Co-reporter:Jinxing Hu;Bo Kong;Yue Liu;Boxuan Xu; Yanfang Zhao
ChemCatChem 2017 Volume 9(Issue 3) pp:403-406
Publication Date(Web):2017/02/06
DOI:10.1002/cctc.201601345
AbstractA new and efficient method for the synthesis of polysubstituted imidazolidines through the Pd0-catalyzed double addition/cyclization of 2,3-allenylamines with aryl iodides and imines was explored. This catalytic reaction afforded the desired products in good yields with high diastereoselectivities.
Co-reporter:Ming-Ze Qin, Lei Wang, Shuang Yan, Jun-Jie Ma, ... Ping Gong
Chinese Chemical Letters 2017 Volume 28, Issue 5(Volume 28, Issue 5) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.cclet.2016.11.030
Based on our previous work, a series of hydrazone moiety-bearing aminopyrimidines were synthesized. The compounds were evaluated for inhibitory activities against EGFR T790M/L858R and antiproliferative activities against H1975 and A549 NSCLC cell lines harboring different forms of EGFR. Compounds 7f and 7k exhibited potent and selective activity in inhibition of gefitinib-resistant H1975 cancer cells (IC50; 0.45, 0.2 μmol/L) while were much less active on A549 cancer cells (IC50; 52.83, >100 μmol/L). Both compounds could be served as promising lead compounds for further investigation.Download high-res image (69KB)Download full-size imageSome aminopyrimidine analogs were identified as potent agents in inhibition of gefitinib-resistant H1975 NSCLC cells.
Co-reporter:Yunlei Hou;Mingze Qin;Xiuxiu Yang;Qi Shen;Yanfang Zhao;Yajing Liu
RSC Advances (2011-Present) 2017 vol. 7(Issue 12) pp:7401-7405
Publication Date(Web):2017/01/20
DOI:10.1039/C6RA27993G
An efficient three-component tandem cyclization reaction for the synthesis of highly substituted oxazolidines was achieved through the Pd0-catalyzed cyclization of buta-2,3-dien-1-ol with aryl iodides and imines. A range of R1 and R2 functional groups is well-tolerated while affording cyclization products in moderate yields and with moderate to high diastereoselectivities.
Co-reporter:Yunlei Hou;Mingze Qin;Xiuxiu Yang;Qi Shen;Yanfang Zhao;Yajing Liu
RSC Advances (2011-Present) 2017 vol. 7(Issue 12) pp:7401-7405
Publication Date(Web):2017/01/20
DOI:10.1039/C6RA27993G
An efficient three-component tandem cyclization reaction for the synthesis of highly substituted oxazolidines was achieved through the Pd0-catalyzed cyclization of buta-2,3-dien-1-ol with aryl iodides and imines. A range of R1 and R2 functional groups is well-tolerated while affording cyclization products in moderate yields and with moderate to high diastereoselectivities.
Co-reporter:Congjun Xu;Mingze Qin;Jun Yi;Yanjing Wang;Yanfeng Chen;Bingfu Zhang;Yanfang Zhao
RSC Advances (2011-Present) 2017 vol. 7(Issue 40) pp:24643-24646
Publication Date(Web):2017/05/05
DOI:10.1039/C7RA01983A
A protocol for the direct synthesis of 5-aryloxazole-4-carbonitrile from acetophenone was first described with potassium ferricyanide as a cheap and low toxicity cyanide reagent, in which, multiple bond formation was implemented via an oxygen mediated radical mechanism. Potassium ferricyanide played a dual role as a “CN” source and also as a coupling partner for the cyclization of oxazole.
Co-reporter:Yunlei Hou;Qi Shen;Liangyu Zhu;Yufei Han;Yanfang Zhao;Mingze Qin
RSC Advances (2011-Present) 2017 vol. 7(Issue 79) pp:50372-50377
Publication Date(Web):2017/10/26
DOI:10.1039/C7RA08208H
Sulfonyl hydrazones have been identified as an excellent sulfonyl anion surrogate in the base and Pd0-catalyzed three-component tandem reaction with aryl iodides and allenes for the synthesis of functionalized allylic sulfones. By forming a stabilized six-membered palladacycle intermediate, the reaction led to the desired higher substituted allylic sulfones with excellent Z selectivities and high yields.
Co-reporter:Ju Liu, Di Yang, Xiuxiu Yang, Minhua Nie, Guodong Wu, Zhunchao Wang, Wei Li, Yajing Liu, Ping Gong
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmc.2017.06.037
•A series of 4-phenoxyquinoline derivatives bearing an 3-oxo-3,4-dihydroquinoxaline moiety were designed and synthesized.•The target compounds showed potent antitumor activity.•Compound 41 showed more potent against five cell lines than foretinib.•Compound 41 showed an IC50 value of 0.90 nM against c-Met kinase.•Compound 41 showed moderate-to-excellent selectivity against 5 other RTK kinases.A series of novel 4-phenoxyquinoline derivatives containing 3-oxo-3,4-dihydroquinoxaline moiety were synthesized and evaluated for their c-Met kinase inhibitory activity and antiproliferative activity against five cancer cell lines (HT-29, H460, A549, MKN-45 and U87MG) in vitro. Most of the compounds exhibited moderate-to-significant cytotoxicity as compared with foretinib. The most promising compound 41 (with c-Met IC50 value of 0.90 nM) showed remarkable cytotoxicity against HT-29, H460, A549, MKN-45 and U87MG cell lines with IC50 values of 0.06 μM, 0.05 μM, 0.18 μM, 0.023 μM and 0.66 μM, respectively, and thus it was 1.22- to 3.50-fold more potent than foretinib. Their preliminary structure-activity relationships (SARs) studies indicate that electron-withdrawing groups on the terminal phenyl rings are beneficial for improving the antitumor activity.A series of novel 4-(2-fluorophenoxy)quinoline derivatives containing 3-oxo-3,4-dihydroquinoxaline moiety were designed, synthesized and evaluated for their cytotoxicity, enzymatic assays and docking analysis.Download high-res image (153KB)Download full-size image
Co-reporter:Ju Liu, Minhua Nie, Yanjing Wang, Jinxing Hu, Feng Zhang, Yanlin Gao, Yajing Liu, Ping Gong
European Journal of Medicinal Chemistry 2016 Volume 123() pp:431-446
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.07.059
•A series of 4-phenoxyquinoline derivatives containing 1,2,4-triazolone moiety were designed and synthesized.•The target compounds showed potent antitumor activity.•The cytotoxicity of 47 was 2.37-fold higher against HT-29 cells than foretinib.•Compound 47 showed an IC50 value of 1.57 nM against c-Met kinase.•Compound 47 showed good selectivity against 4 other RTK kinases.A series of novel 4-phenoxyquinoline derivatives containing 1,2,4-triazolone moiety were synthesized and evaluated for their in vitro cytotoxic activity against four cancer cell lines (HT-29, H460, A549 and MKN-45). Most of the compounds exhibited moderate-to-significant cytotoxicity. Compounds 33, 37, 39, 44, 46, 47, 53, 55, 61, 64 and 66 were further examined for their inhibitory activity against c-Met kinase. The most promising compound 47 (with c-Met IC50 value of 1.57 nM) showed remarkable cytotoxicity against HT-29, H460, A549 and MKN-45 cell lines with IC50 values of 0.08 μM, 0.14 μM, 0.11 μM and 0.031 μM, respectively, and thus it was 1.1- to 2.3- fold more potent than foretinib. Their preliminary structure-activity relationships (SARs) studies indicate that electron-withdrawing groups on the terminal phenyl rings are beneficial for improving the antitumor activity.A series of novel 4-phenoxyquinoline derivatives containing 1,2,4-triazolone moiety were designed, synthesized and evaluated for their biological activities.
Co-reporter:Yu Wang, Guogang Zhang, Gang Hu, Yanxin Bu, Hongrui Lei, Daiying Zuo, Mengting Han, Xin Zhai, Ping Gong
European Journal of Medicinal Chemistry 2016 Volume 123() pp:80-89
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.06.056
•A series of 4-arylaminopyrimidine derivatives possessing a hydrazone moiety were designed and synthesized.•Most compounds showed high antiproliferative effects toward KARPAS299 and HCC78 cell lines.•The enzymatic assay identified 7b as a potent and selective L1196M ALK and ROS1 dual inhibitor.•Molecular docking of compound 7b was performed in the binding site cavity of ALK.A series of 4-arylaminopyrimidine derivatives possessing a hydrazone moiety were designed, synthesized and evaluated for their biological activity. Most compounds exhibited moderate to excellent cytotoxic activity against ALK-addicted KARPAS299 and ROS1-addicted HCC78, while also showing much less potent activity against A549, H460 and HT-29, whose growth were not dependent on ALK and/or ROS1, as compared with crizotinib and ceritinib. The most promising compound, 7b, showed high antiproliferative effects on ALK-addicted KARPAS299 and ROS1-addicted HCC78 cell lines with IC50 of 20 nM and 28 nM, respectively, but showed no inhibitory activity against A549, H460 and HT-29. The enzymatic assay identified 7b as a potent and selective ALK and ROS1 dual inhibitor with IC50 of 2.5 nM and 2.7 nM, respectively. It also exhibited good inhibitory activity against the L1196M ALK with an IC50 value of 67 nM.
Co-reporter:Mingze Qin, Shuang Yan, Lei Wang, Haotian Zhang, Yanfang Zhao, Shasha Wu, Di Wu, Ping Gong
European Journal of Medicinal Chemistry 2016 Volume 115() pp:1-13
Publication Date(Web):10 June 2016
DOI:10.1016/j.ejmech.2016.02.071
•A series of novel diaryl ureas were identified as potent antitumor agents.•Compound 13i potently suppressed proliferation of cancer cells.•Compound 13i effectively inhibited c-Kit, RET, and FLT3 kinases.•Compounds 13i significantly induced apoptosis of HT-29 cells.Herein, we report a novel series of diaryl urea derivatives bearing a triazole moiety, from which potent antitumor agents have been identified. With a modified triazole, most compounds showed high level activity in both cellular and enzymatic assays, accompanied with a suitable ClogD7.4 value. The most active compound, 13i, effectively suppressed proliferation of HT-29, H460 and MDA-MB-231 cancer cells, with IC50 values of 0.90, 0.85 and 1.54 μM, respectively. Compound 13i also exhibited significant inhibition of tyrosine kinases including c-Kit, RET and FLT3. Furthermore, compound 13i could obviously induce apoptosis of HT-29 cells in a concentration-dependent manner. The study of structure-activity relationships also revealed that a hydrophilic tail at the 4-position of the triazole was crucial for high activity of the compound.
Co-reporter:Kuan Lu, Liancheng Duan, Boxuan Xu, Weile Yin, Di Wu, Yanfang Zhao and Ping Gong
RSC Advances 2016 vol. 6(Issue 59) pp:54277-54280
Publication Date(Web):31 May 2016
DOI:10.1039/C6RA08871F
A mild and efficient method for the synthesis of 3-amino-5-aryl-1,2,4-oxadiazole by intramolecular cyclization using PhI(OAc)2 (PIDA) as an oxidant is developed. Various 3-amino-5-aryl-1,2,4-oxadiazoles are prepared in moderate to good yields, and the PIDA-mediated N–O bond formation mechanism is suggested. In view of the readily available starting materials, operational simplicity, high functionality tolerance, and low toxicity, this protocol provides a novel synthetic strategy for 1,2,4-oxadiazoles.
Co-reporter:Xin Zhai, Guanglong Bao, Limei Wang, Mingke Cheng, Meng Zhao, Sijia Zhao, Hongyang Zhou, Ping Gong
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 6) pp:1331-1345
Publication Date(Web):15 March 2016
DOI:10.1016/j.bmc.2016.02.003
In continuing our efforts to identify small molecules able to inhibit c-Met kinase, three series of novel 6,7-disubstituted-4-phenoxyquinoline derivatives (23a–w, 26a–d and 30a–d) bearing (thio)semicarbazone scaffold were designed, synthesized and evaluated for their cytotoxicity. The biological data revealed that most compounds exhibited moderate-to-excellent activity against HT-29, MKN-45, A549 cancer cell lines and relative poor potency toward MDA-MB-231 cell as well as hardly any cytotoxicity in normal PBL cell. Eleven compounds were further examined for their inhibitory activity against c-Met kinase and three compounds (23h, 23n and 26a) demonstrated good inhibitory activity. This work resulted in the discovery of a potent c-Met inhibitor 23n, bearing 2-hydroxy-3-allylphenyl group at R2 moiety, as a valuable lead molecule, which possessed remarkable cytotoxicity and high selectivity against A549 and HT-29 cell lines with IC50 values of 11 nM and 27 nM. Besides, it displayed excellent c-Met kinase inhibition on a single-digital nanomolar level (IC50 = 1.54 nM). Meanwhile, the results from preliminarily in vivo study reflected that compound 23n showed promising overall PK profiles, consistent with the efficacy in both MKN-45 and HT-29 tumor xenograft mice model. These results clearly indicated that compound 23n is a potent and highly selective c-Met inhibitor and its favorable in vitro and in vivo profiles warrant further investigation.Three series of novel 6,7-disubstituted-4-phenoxyquinoline derivatives bearing semicarbazone scaffold were synthesized and evaluated for their cytotoxicity. Compound 23n was further examined for its c-Met kinase activity in vitro as well as pharmacokinetic profiles and antitumor efficacy in vivo.
Co-reporter:Hongrui Lei;Gang Hu;Yu Wang;Pei Han;Zijian Liu;Yanfang Zhao
Archiv der Pharmazie 2016 Volume 349( Issue 8) pp:651-661
Publication Date(Web):
DOI:10.1002/ardp.201600003
A series of novel 4-phenoxyquinoline derivatives containing the benzo[d]thiazole-2-yl urea moiety were synthesized and evaluated for their cytotoxicity against the HT-29, MKN-45, and H460 cell lines. The structures of the target compounds were confirmed by 1H NMR and MS spectra. Most of them showed moderate to excellent potency against the three tested cell lines. Especially, compound 23 was identified a promising agent (c-Met IC50 = 17.6 nM), showing the most potent anticancer activities with IC50 values of 0.18, 0.06, and 0.01 µM against the HT-29, MKN-45, and H460 cell lines, respectively. The docking results of 23 with the c-Met kinase model 3LQ8 showed a specific binding mode between the ligand and the target protein.
Co-reporter:Fangyang Wang;Yajing Liu;Lihui Wang;Jingyu Yang;Yanfang Zhao;Nannan Wang;Qi Cao;Chunfu Wu
Journal of Cellular and Molecular Medicine 2015 Volume 19( Issue 8) pp:1916-1928
Publication Date(Web):
DOI:10.1111/jcmm.12566
Abstract
Caspase-3 is a critical effector caspase in apoptosis cascade, and is often over-expressed in many cancer tissues. The first synthesized procaspase-3 activator, PAC-1, induces cancer cell apoptosis and exhibits antitumour activity in murine xenograft models. To identify more potent procaspase-3 activators, a series of compounds were designed, synthesized and evaluated for their ability of inducing cancer cell death in culture. Among these compounds, WF-208 stood out by its high cytotoxicity against procaspase-3 overexpressed HL-60 cells. Compared with PAC-1, WF-208 showed higher cytotoxicity in cancer cells and lower toxicity in normal cells. The further investigation described herein showed that WF-208 activated procaspase-3, degraded IAPs (The Inhibitors of apoptosis proteins) and leaded to caspase-3-dependent cell death in tumour cells, which possibly because of the zinc-chelating properties. WF-208 also showed greater antitumour activity than PAC-1 in murine xenograft model. In conclusion, we have discovered WF-208 as a promising procaspase-3 activating compound, with higher activity and higher cell selectivity than PAC-1.
Co-reporter:Mingze Qin, Tingting Wang, Boxuan Xu, Zonghui Ma, Nan Jiang, Hongbo Xie, Ping Gong, Yanfang Zhao
European Journal of Medicinal Chemistry 2015 Volume 104() pp:115-126
Publication Date(Web):2 November 2015
DOI:10.1016/j.ejmech.2015.09.031
•A series of novel aminopyrimidines were identified as selective EGFR inhibitors.•The compounds potently inhibited EGFR expressing T790M mutation.•The compounds effectively suppressed proliferation of H1975 cells.•Compounds 14a, 15g and 15i were promising candidates for further development.The epidermal growth factor receptor (EGFR) T790M mutant is found in approximately 50% of clinically acquired resistance to gefitinib among patients with non-small cell lung cancer (NSCLC). Here, a series of novel aminopyrimidines bearing a hydrazone moiety were identified as potent and selective EGFR inhibitors. Compounds 14a, 15g, and 15i potently inhibited all EGFR mutants including EGFR T790M/L858R, EGFR T790M/delE746_A750, and EGFR T790M while they showed weak effects on the wild type (WT) EGFR. In addition, these compounds effectively suppressed proliferation of gefitinib-resistant H1975 (EGFR T790M/L858R) cells but were less potent against A549 (WT EGFR and k-Ras mutation) and HT-29 (non-special gene type) cells, showing a high safety index. Therefore, 14a, 15g, and 15i might be promising candidates to overcome drug resistance mediated by the EGFR T790M mutant.
Co-reporter:Yanfang Zhao, Mingyan Jiang, Shunguang Zhou, Shasha Wu, Xiaolong Zhang, Longsheng Ma, Kai Zhang, Ping Gong
European Journal of Medicinal Chemistry 2015 Volume 96() pp:369-380
Publication Date(Web):26 May 2015
DOI:10.1016/j.ejmech.2015.04.025
•A series of oxazolidinone derivatives containing pyrrole/indole/thiazole moiety were designed and synthesized.•The target compounds showed potent anticoagulant activity.•Five compounds were further examined for their anti-FXa inhibitory activity.•Compound 7b showed an IC50 value of 2.01 nM against human FXa inhibition.A novel series of potent and efficacious factor Xa inhibitors which possesses pyrrole/indole/thiazole moieties as S4 binding element was identified. Compound 7b showed strong human factor Xa inhibitory activity (IC50 = 2.01 nM) and anticoagulant activities in both human (PTCT2 = 0.15 μM, APPTCT2 = 0.30 μM) and rabbit plasma (PTCT2 = 0.46 μM, APPTCT2 = 0.75 μM). The SARs analyses indicated that the size and water solubility of different alkylamino group at the position of S4 ligand were responsible for the anticoagulant activity.A novel series of potent and efficacious factor Xa inhibitors which possesses pyrrole/indole/thiazole moieties as S4 binding element was identified. Compound 7b showed potent human factor Xa inhibitory activity.
Co-reporter:Junjie Ma, Guanglong Bao, Limei Wang, Wanting Li, Boxuan Xu, Baoquan Du, Jie Lv, Xin Zhai, Ping Gong
European Journal of Medicinal Chemistry 2015 Volume 96() pp:173-186
Publication Date(Web):26 May 2015
DOI:10.1016/j.ejmech.2015.04.018
•31 novel benzothiazole derivatives were designed and synthesized.•The target compounds showed excellent in vitro antitumor potency against four cancer cell lines.•Compound 20d was more cytotoxic than oncrasin-1 and PAC-1.•Enzymatic assays, cell cycle analysis and 3D-QSAR study were performed.Through a structure-based molecular hybridization approach, a series of novel benzothiazole derivatives bearing indole-based moiety were designed, synthesized and screened for in vitro antitumor activity against four cancer cell lines (HT29, H460, A549 and MDA-MB-231). Most of them showed moderate to excellent activity against all the tested cell lines. Among them, compounds 20a–w with substituted benzyl-1H-indole moiety showed better selectivity against HT29 cancer cell line than other compounds. Compound 20d exhibited excellent antitumor activity with IC50 values of 0.024, 0.29, 0.84 and 0.88 μM against HT29, H460, A549 and MDA-MB-231, respectively. Further mechanism studies indicated that the marked pharmacological activity of compound 20d might be ascribed to activation of procaspase-3 (apoptosis-inducing) and cell cycle arrest, which had emerged as a lead for further structural modifications. Furthermore, 3D-QSAR model (training set: q2 = 0.850, r2 = 0.987, test set: r2 = 0.811) was built to provide a comprehensive guide for further structural modification and optimization.A novel series of benzothiazole derivatives bearing indole-based moiety were synthesized and evaluated for their cytotoxicity, enzymatic assays, cell cycle analysis and 3D-QSAR analysis.
Co-reporter:Yajing Liu;Guobing Feng;Zonghui Ma;Chen Xu;Zhuang Guo;Liying Xu
Archiv der Pharmazie 2015 Volume 348( Issue 11) pp:776-785
Publication Date(Web):
DOI:10.1002/ardp.201500238
A series of novel 7-methoxy-3-heterocyclic quinolin-6-ol derivatives were synthesized and evaluated for their anti-hepatitis B virus (HBV) activities and cytotoxicities in the HepG2.2.15 cell line. Five compounds, 14a, 15c, 15e, 16b, and 16f, displayed excellent potency and selectivity toward the HBV, with IC50 values of less than 5.0 µM and selectivity index values of 11.0–71.5. Structure–activity relationship studies indicated that the 1,3,4-thiadiazole and sulfinylmethyl derivatives showed the most potent activities against the HBV.
Co-reporter:Junjie Ma;Gang Hu;Lijun Xie;Lei Chen
Chemical Research in Chinese Universities 2015 Volume 31( Issue 6) pp:958-963
Publication Date(Web):2015 December
DOI:10.1007/s40242-015-5034-1
A series of novel benzothiazole derivatives bearing semicarbazone as a linker was designed and synthesized, and their in vitro antitumor activities were evaluated against four cancer cell lines(HT29, H460, A549 and MDA-MB-231). Most of them showed moderate to excellent activity against all the tested cell lines. Among them, compounds 12a―12i with fluoro-substituted benzyl-1H-indole moiety displayed more potent activity than those with phenyl moiety. The most promising compound 12d exhibited excellent antitumor activity with IC50 values of 0.015, 0.28, 1.53 and 0.68 μmol/L against the four tested cell lines respectively.
Co-reporter:Hao Hu;Mingyan Jiang;Lijun Xie;Gang Hu
Chemical Research in Chinese Universities 2015 Volume 31( Issue 5) pp:746-755
Publication Date(Web):2015 October
DOI:10.1007/s40242-015-5166-3
A series of novel 4-phenoxyquinoline derivatives containing 3-amino-2-cyano-acrylamide framework was designed and synthesized, and the in vitro cytotoxic activities of them against five cancer cell lines(HT-29, H460, A549, MKN-45, and U87MG) were evaluated. Most of the compounds exhibited moderate-to-significant cytotoxicity and high selectivity against one or more cell lines as compared with Foretinib. The studies of their preliminary structure-activity relationships(SARs) indicate that the compounds containing methyl groups, especially methyl groups at 4-position of the phenyl ring(moiety B) are more effective. Among them, compound 36 shows the most potent antitumor activities with IC50 values of 0.04, 0.09, 0.67, 0.39 and 1.10 μmol/L against HT-29, H460, A549, MKN-45 and U87MG cell lines, respectively.
Co-reporter:Weike Liao, Gang Hu, Zhuang Guo, Deyu Sun, Lixia Zhang, Yanxin Bu, Yingxiu Li, Yajing Liu, Ping Gong
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4410-4422
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.026
Co-reporter:Xin Zhai;Limei Wang;Jiyue Shi
Chemical Research in Chinese Universities 2015 Volume 31( Issue 3) pp:372-380
Publication Date(Web):2015 June
DOI:10.1007/s40242-015-4435-5
A novel series of 5H-pyridazino[4,5-b]indoles(L-01—L-32) was synthesized and characterized by means of 1H NMR, MS and elemental analysis. The cytotoxicity of the target compounds against Bel-7402 and HT-1080 cell lines were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT) assay. Most of them exhibited moderate to excellent cytotoxicity, and six compounds(L-04, L-06, L-18, L-20, L-21 and L-23) possessed dramatically increased cytotoxicity superior to Gefitinib. Of these initial hits, compound L-21 displayed remarkable cytotoxicity against the tested cell lines with half maximal inhibitory concentration(IC50) values of 4.6 and 2.1 μmol/L, respectively, which was 13.9- to 25.6-fold more potent than positive control.
Co-reporter:Zijian Liu, Yu Wang, Huafang Lin, Dazhuang Zuo, Lihui Wang, Yanfang Zhao, Ping Gong
European Journal of Medicinal Chemistry 2014 Volume 85() pp:215-227
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.07.099
•Two series of thieno[3,2-d]pyrimidine derivatives containing diaryl urea moiety were designed and synthesized.•The target compounds showed moderate to significant cytotoxic activities.•The cytotoxicity of 29a was 10.7- and 14.8-fold higher than GDC-0941 against H460 and HT-29 cell lines, respectively.•29a could inhibit HT-29 proliferation by inducing apoptosis mechanism.Two series of thieno[3,2-d]pyrimidine derivatives containing diaryl urea moiety were designed, synthesized and evaluated for their biological activity. The preliminary investigation showed that most compounds displayed good to excellent potency against four tested cancer cell lines as compared with GDC-0941 and sorafenib. In particular, the most promising compound 29a showed the most potent antitumor activities with IC50 values of 0.081 μM, 0.058 μM, 0.18 μM, and 0.23 μM against H460, HT-29, MKN-45 and MDA-MB-231 cell lines, respectively. The SAR analyses indicated that compounds with mono-halogen groups at 4-position on the terminal phenyl ring were more active than those with double-halogen groups or methyl groups. In addition, the introduction of chlorine atoms into 6,7-position of thieno[3,2-d]pyrimidine moiety led to a slight decline, but more selective activity against H460 and HT-29 cell lines.Two series of thieno[3,2-d]pyrimidine derivatives containing diaryl urea moiety were designed, synthesized and evaluated for their cytotoxicity, enzymatic assays and apoptosis analysis.
Co-reporter:Shunguang Zhou, Huimin Liao, Chao He, Yanan Dou, Mingyan Jiang, Lixiang Ren, Yanfang Zhao, Ping Gong
European Journal of Medicinal Chemistry 2014 Volume 83() pp:581-593
Publication Date(Web):18 August 2014
DOI:10.1016/j.ejmech.2014.06.068
•A series of 4-phenoxyquinoline derivatives containing pyridazinone moiety were designed and synthesized.•The target compounds showed potent antitumor activity.•Six compounds were further examined for their c-Met kinase activity.•The cytotoxicity of 15a was 2.3-fold higher against A549 cells than foretinib.•Compound 15a showed an IC50 value of 2.15 nM against c-Met kinase.A series of novel 4-phenoxyquinoline derivatives containing pyridazinone moiety were synthesized and evaluated for their in vitro cytotoxic activity against five cancer cell lines (HT-29, H460, A549, MKN-45, and U87MG). Most of the compounds exhibited moderate-to-significant cytotoxicity and high selectivity against one or more cell lines. Compounds 15a, 20a, 15b, 15c, 20d, and 16e were further examined for their inhibitory activity against c-Met kinase. The most promising compound 15a (c-Met half-maximal inhibitory concentration [IC50] = 2.15 nM) showed remarkable cytotoxicity against HT-29, H460, and A549 cell lines with IC50 values of 0.10 μM, 0.13 μM, and 0.05 μM, respectively, and thus it was 1.5- to 2.3-fold more potent than foretinib. Their preliminary structure–activity relationships (SARs) studies indicate that electron-withdrawing groups on the terminal phenyl rings are beneficial for improving the antitumor activity.A series of novel 4-phenoxyquinoline derivatives containing pyridazinone moiety were synthesized and evaluated for their cytotoxic activities. Six potent compounds were further examined for their c-Met kinase activity.
Co-reporter:Weike Liao, Chen Xu, Xiaohui Ji, Gang Hu, Lixiang Ren, Yajing Liu, Ruijuan Li, Ping Gong, Tiemin Sun
European Journal of Medicinal Chemistry 2014 Volume 87() pp:508-518
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.095
•A series of 4-(2-fluorophenoxy)quinoline derivatives bearing an acylhydrazone moiety were designed and synthesized.•The target compounds showed potent antitumor activity.•Six compounds were further examined for their c-Met kinase activity.•The cytotoxicity of 40 was 6.8-fold higher against H460 cells than foretinib.•Compound 40 showed an IC50 value of 1.86 nM against c-Met kinase.A series of 4-(2-fluorophenoxy)quinoline derivatives containing an acylhydrazone moiety were designed, synthesized and evaluated for their in vitro biological activities against c-Met kinase and five cancer cell lines (A549, H460, HT-29, MKN-45, and U87MG). Most compounds showed weak to excellent antiproliferative activity. The most promising analog, 40 (c-Met IC50 = 1.86 nM), displayed 1.3-, 6.8-, 1.5-, 3.5-fold increase against HT-29, H460, A549 and U87MG cell lines, respectively, compared with Foretinib. An analysis of structure-activity relationships revealed that an acylhydrazone scaffold with an unsubstituted sp2 hybridized carbon adjacent to the 4-CF3 phenyl ring is favorable for antitumor activity.A series of novel 4-(2-fluorophenoxy)quinoline derivatives containing acylhydrazone moiety were synthesized and evaluated for their cytotoxic activities. Six potent compounds were further examined for their c-Met kinase activity.
Co-reporter:Zijian Liu, Shasha Wu, Yu Wang, Ruijuan Li, Jian Wang, Lihui Wang, Yanfang Zhao, Ping Gong
European Journal of Medicinal Chemistry 2014 Volume 87() pp:782-793
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.10.022
•A series of thieno[3,2-d]pyrimidine derivatives possessing diaryl semicarbazone scaffolds was designed, synthesized.•The enzymatic assays, apoptosis analysis and docking analysis of the most potent compounds 36 were performed.•The cytotoxicity of compound 36 was 15.3- and 22.1-fold higher than GDC-0941 against H460 and HT-29 cell lines, respectively.•Compound 36 showed an IC50 value of 0.027 μM against PI3Kα kinase.A series of novel thieno[3,2-d]pyrimidine derivatives possessing diaryl semicarbazone scaffolds were designed, synthesized and evaluated for their anticancer activity. Most compounds displayed good to excellent potency against four tested cancer cell lines as compared with GDC-0941 and sorafenib. In this study, a promising compound 36 (PI3Kα IC50 = 0.027 μM) was identified, which showed the most potent antitumor activities with IC50 values of 0.057 μM, 0.039 μM, 0.25 μM, and 0.23 μM against H460, HT-29, MKN-45 and MDA-MB-231 cell lines, respectively. In addition, the SAR analyses indicated that compounds with 4-morpholino group at the C-4 position of thieno[3,2-d]pyrimidine moiety exhibited superior activities than compounds bearing chain amino groups. In addition, compounds with mono-methoxy group at the 3-position or dimethyl groups at the 3,5-position on the terminal phenyl ring were more active. The SAR analyses will guide us to further refine the structure of the thieno[3,2-d]pyrimidine derivatives to achieve optimum anticancer activity.A series of thieno[3,2-d]pyrimidine derivatives possessing diaryl semicarbazone scaffolds was designed, synthesized and evaluated for their cytotoxicity, enzymatic assays, apoptosis analysis and docking analysis.
Co-reporter:Junjie Ma, Dong Chen, Kuan Lu, Lihui Wang, Xiaoqi Han, Yanfang Zhao, Ping Gong
European Journal of Medicinal Chemistry 2014 Volume 86() pp:257-269
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.08.058
•36 novel benzothiazole derivatives were designed and synthesized.•Target compounds showed excellent antitumor potency in vitro against 5 cancer cell lines.•The cytotoxic activities of 15g and 16b were 1.8–8.7 times more active than PAC-1.•Enzymatic activities of 15g and 16b were 2.9 times and 16 times better than PAC-1.A series of novel benzothiazole derivatives bearing the ortho-hydroxy N-carbamoylhydrazone moiety were designed and synthesized and their cytotoxic activities against five cancer cell lines (NCI-H226, SK-N-SH, HT29, MKN45, and MDA-MB-231) were screened in vitro. Most of them showed moderate to excellent activity against all the tested cell lines. Among them, compounds 15g (procaspase-3 EC50 = 1.42 μM) and 16b (procaspase-3 EC50 = 0.25 μM) exhibited excellent antitumor activity with IC50 values ranging from 0.14 μM to 0.98 μM against all cancer cell lines, which were 1.8–8.7 times more active than the first procaspase activating compound (PAC-1) (procaspase-3 EC50 = 4.08 μM). The structure–activity relationship (SAR) analyses indicated that the introduction of a lipophilic group (a benzyloxy or heteroaryloxy group) at the 4-position of the 2-hydroxy phenyl ring was beneficial to antitumor activity, and the presence of substituents containing nitrogen that are positively charged at physiological pH could also improve antitumor activity. It was also confirmed that the steric effect of the 4-position substituent of the benzyloxy group had a significant influence on cytotoxic activity.A series of novel benzothiazole derivatives bearing ortho-hydroxy N-carbamoylhydrazone moiety were synthesized and evaluated for their cytotoxic activities. Six potent compounds were further examined for their procaspase-3 kinase activity.
Co-reporter:Mingze Qin, Xin Zhai, Hongbo Xie, Junjie Ma, Kuan Lu, Yu Wang, Lihui Wang, Yucheng Gu, Ping Gong
European Journal of Medicinal Chemistry 2014 Volume 81() pp:47-58
Publication Date(Web):23 June 2014
DOI:10.1016/j.ejmech.2014.04.059
•Some novel antitumor agents were synthesized and evaluated.•The compounds showed excellent antitumor potency and physicochemical profile.•The compounds potently inhibited Raf kinase with a good enzyme selectivity.•The cytotoxic activity of 11f was 1.2–45.5 folds better than sorafenib.New 2-(4-(2-(dimethylamino)ethyl)-4H-1,2,4-triazol-3-yl)pyridine derivatives were synthesized and evaluated for their in vitro cytotoxicity against five cancer cell lines namely MKN-45, H460, HT-29, A549 and U87MG, as well as the normal cell line WI-38. Nearly all the compounds exhibited superior potency to sorafenib with a better selectivity towards the MKN-45, H460 and HT-29 cell lines. In addition, the enzymatic screening result demonstrated that the optimized compounds possessed potent Raf kinase inhibition as well as favorable enzyme selectivity. The most promising compound, 11f, showed high levels of cytotoxicity against MKN-45, H460 and HT-29 cells with IC50 values of 51, 72 and 130 nM, respectively, which are 45.5, 30.4 and 27.8 folds higher than the corresponding IC50 values for sorafenib against these cell lines. Structure–activity relationships revealed that the dimethylaminoethyl group was crucial for high activity.A series of 2-(4-(2-(dimethylamino)ethyl)-4H-1,2,4-triazole-3-yl)pyridine derivatives were synthesized. These analogs demonstrated potent antitumor potency and good enzyme selectivity, many of them could be served as promising candidates for further development.
Co-reporter:Shunguang Zhou, Huimin Liao, Mingmei Liu, Guobing Feng, Baolin Fu, Ruijuan Li, Maosheng Cheng, Yanfang Zhao, Ping Gong
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 22) pp:6438-6452
Publication Date(Web):15 November 2014
DOI:10.1016/j.bmc.2014.09.037
A series of 6,7-disubstituted-4-(2-fluorophenoxy)quinoline derivatives possessing 1,2,3-triazole-4-carboxamide moiety were designed, synthesized and evaluated for their in vitro biological activities against c-Met kinase and five typical cancer cell lines (A549, H460, HT-29, MKN-45 and U87MG). Most compounds showed moderate to excellent antiproliferative activity. In this study, a promising compound 34, with a c-Met IC50 value of 1.04 nM, was identified as a multitargeted receptor tyrosine kinase inhibitor. The SAR analyses indicated that compounds with halogen group, especially fluoro group, at 4-position on the phenyl ring (moiety B) have potent antitumor activity, and methylation on the 5-atom linker played an important role in the c-Met enzymatic activity.A series of 6,7-disubstituted-4-(2-fluorophenoxy)quinoline derivatives possessing 1,2,3-triazole-4-carboxamide moiety were designed, synthesized and evaluated for their enzymatic assays, cytotoxicity and docking analysis. The c-Met active site in complex with compounds 32 (A) and 34 (B) and their hydrophobic surface.
Co-reporter:Junjie Ma;Guangyan Zhang;Xiaoqi Han;Guanglong Bao;Lihui Wang;Xin Zhai
Archiv der Pharmazie 2014 Volume 347( Issue 12) pp:936-949
Publication Date(Web):
DOI:10.1002/ardp.201400230
A novel series of benzothiazole derivatives bearing the ortho-hydroxy-N-acylhydrazone moiety were designed, synthesized, and evaluated for their procaspase-3 kinase activation activities and antiproliferative activities against five cancer cell lines (NCI-H226, SK-N-SH, HT29, MKN-45, and MDA-MB-231). Most target compounds showed moderate to excellent cytotoxic activity against all five tested cancer lines. The most promising compound 18e (procaspase-3 EC50 = 0.31 µM) with IC50 values ranging from 0.24 to 0.92µM against all tested cell lines was 4.24–12.2 times more active than PAC-1 (procaspase-3 EC50 = 0.41 µM). Structure–activity relationship studies indicated that the phenyl group on the 2-hydroxyphenyl ring (moiety A) was critical for pharmacological activity in vitro. In addition, introduction of a benzyloxyl group on moiety A and a mono-electron-withdrawing group at the 4-position of the benzyloxyl group were more favorable for antitumor activity. Moreover, reduction of the electron density in the phenyl ring of the benzyloxy group led to a dramatic decrease in the procaspase-3 kinase activation activity.
Co-reporter:Shunguang Zhou, Jianguo Ren, Mingmei Liu, Lixiang Ren, Yajing Liu, Ping Gong
Bioorganic Chemistry 2014 Volume 57() pp:30-42
Publication Date(Web):December 2014
DOI:10.1016/j.bioorg.2014.07.011
•Two series of 6,7-disubstituted-4-phenoxyquinoline derivatives were designed and synthesized.•Five compounds were further examined for their c-Met kinase activity.•Compound 17 showed an IC50 values of 2.20 nM against c-Met kinase.Two series of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing 2,4-imidazolinedione/pyrazolone scaffold were designed, synthesized and evaluated for their c-Met kinase inhibition and cytotoxicity against HT-29, H460, A549, MKN-45, and U87MG cancer cell lines in vitro. The pharmacological data indicated that most of the tested compounds showed moderate to significant cytotoxicity and high selectivity against HT-29, H460 and A549 cancer cell lines as compared with foretinib. The SAR analyses indicated that compounds with halogen groups, especially trifluoromethyl groups at 2-position on the phenyl ring (moiety B) were more effective. In this study, a promising compound 17 (c-Met IC50 = 2.20 nM, a multi-target tyrosine kinase inhibitor) showed the most potent antitumor activities with IC50 values of 0.14 μM, 0.18 μM, 0.09 μM, 0.03 μM, and 1.06 μM against HT-29, H460, A549, MKN-45, and U87MG cell lines, respectively.Two series of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing 2,4-imidazolinedione/pyrazolone scaffold were synthesized and evaluated for their cytotoxic activities. Five potent compounds were further examined for their c-Met kinase activity.
Co-reporter:Zijian Liu, Rui Wang, Ruiming Guo, Jinxing Hu, Ruijuan Li, Yanfang Zhao, Ping Gong
Bioorganic & Medicinal Chemistry 2014 22(14) pp: 3642-3653
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.013
Co-reporter:Qiang Huang;Qiangqiang Fu;Yajing Liu
Chemical Research in Chinese Universities 2014 Volume 30( Issue 2) pp:257-265
Publication Date(Web):2014 April
DOI:10.1007/s40242-014-3253-5
In an attempt to develop potent and selective anticancer agents, we designed and synthesized a series of novel bis(morpholino-1,3,5-triazine) derivatives bearing aylmethylene hydrazine moiety and evaluated their cytotoxicity, in vitro, against H460(non-small-cell lung cancer), HT-29(human colorectal cancer) and MDA-MB-231(human breast cancer) cell lines. The pharmacological results indicate that all the compounds exhibit enhanced cytotoxicity than BMCL-200908069-1, and six target compounds(7e, 7h, 7j, 9a, 9b, 9c) were superior to PAC-1 against all the tested cancer cell lines. The most active compound 7j, with IC50^(inhibitory concentration 50%) values of 0.75, 0.34 and 0.60 μmol/L against HT-29, H460 and MDA-MB-231 cancer cell lines, was 39-, 28-, and 60-fold more potent than BMCL-200908069-1(29.24, 9.52 and 36.21 μmol/L), respectively.
Co-reporter:Huimin Liao;Lian’e Chong;Li Tan
Chemical Research in Chinese Universities 2014 Volume 30( Issue 5) pp:759-763
Publication Date(Web):2014 October
DOI:10.1007/s40242-014-4081-3
A series of novel 5,7-diphenylimidazo[1,2-a]pyridine derivatives was designed and synthesized. The in vitro cytotoxic activities of all the target compounds against human colorectal cancer(HT-29), human lung cancer(H460), human gastric cancer(MKN45) and human breast cancer(MDA-MB-231) cell lines were evaluated. The pharmacological results indicated that most of the target compounds showed moderate to excellent activities against the tested cell lines. The most promising compound 4h(0.20, 0.006, 0.08, 0.021 μmol/L) was 2.6, 5.1, 3.6 and 21.9 times more active than EPC2407(0.52, 0.031, 0.29, 0.46 μmol/L) against HT-29, H460, MKN45 and MDA-MB-231 cell lines, respectively.
Co-reporter:Baohui Qi, Bin Mi, Xin Zhai, Ziyi Xu, Xiaolong Zhang, Zeru Tian, Ping Gong
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 17) pp:5246-5260
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmc.2013.06.026
A novel series of N1-(3-fluoro-4-(6,7-disubstituted-quinolin-4-yloxy)phenyl)-N4-arylidenesemicarbazide derivatives were synthesized and evaluated for their c-Met kinase inhibition and cytotoxicity against A549, HT-29, MKN-45 and MDA-MB-231 cancer cell lines in vitro. Several potent compounds were further evaluated against three other cancer cell lines (U87MG, NCI-H460 and SMMC7721). Most of compounds tested exhibited moderate to excellent activity. The studies of SARs identified the most promising compound 28 (c-Met IC50 = 1.4 nM) as a c-Met kinase inhibitor. In this study, a promising compound 28 was identified, which displayed 2.1-, 3.3-, 48.4- and 3.6-fold increase against A549, HT-29, U87MG and NCI-H460 cell lines, respectively, compared with that of Foretinib.
Co-reporter:Sai Li, Yanfang Zhao, Kewen Wang, Yali Gao, Jianming Han, Bingbing Cui, Ping Gong
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 11) pp:2843-2855
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmc.2013.04.013
A series of novel 4-(2-fluorophenoxy)quinoline derivatives containing 4-oxo-1,4-dihydrocinnoline-3-carboxamide moiety were designed, synthesized and evaluated for their in vitro biological activities against c-Met kinase and six typical cancer cell lines (A549, H460, HT-29, MKN-45, U87MG and SMMC-7721). All the prepared compounds showed moderate to excellent antiproliferative activity, and the analysis of their structure–activity relationships indicated that 2-chloro or 2-trifluoromethyl substituted phenyl group on the 1-position of cinnoline ring was more favorable for antitumor activity. In this study, a promising compound 33, with a c-Met IC50 value of 0.59 nM, was identified as a multitargeted receptor tyrosine kinase inhibitor.
Co-reporter:Yan-Fang Zhao, Zi-Jian Liu, Xin Zhai, Dan-Dan Ge, Qiang Huang, Ping Gong
Chinese Chemical Letters 2013 Volume 24(Issue 5) pp:386-388
Publication Date(Web):May 2013
DOI:10.1016/j.cclet.2013.02.004
A series of novel diaryl ureas containing 4-[(2-amino-6-trifluromethyl)pyrimidine-4-yl]piperazine-1-yl group were synthesized and evaluated for their cytotoxic activities in a panel of human cancer cell lines. Compared with the reference drug Sorafenib, some compounds showed more potent and a broader spectrum of anti-cancer activities. Among them, compound 2p demonstrated significant inhibitory activities against MDA-MB-231, HT-29 and MCF-7 cell lines with IC50 values of 0.016, 0.63, 0.001 μmol/L, respectively.A series of novel diaryl ureas containing 4-[(2-amino-6-trifluromethyl)pyrimidine-4-yl]piperazine-1-yl group were synthesized and evaluated for their cytotoxic activities in a panel of human cancer cell lines. Among them, compound 2p demonstrated significant inhibitory activities against MDA-MB-231, HT-29 and MCF-7 cell lines with IC50 values of 0.016, 0.63, 0.001 μmol/L, respectively.
Co-reporter:Sai Li;Rui Jiang;Mingze Qin;Haicheng Liu;Guangyan Zhang
Archiv der Pharmazie 2013 Volume 346( Issue 7) pp:521-533
Publication Date(Web):
DOI:10.1002/ardp.201300029
A series of 4-(2-fluorophenoxy)quinoline derivatives bearing the 4-oxo-1,4-dihydroquinoline-3-carboxamide moiety were designed, synthesized, and evaluated for their in vitro antitumor activity against the H460, HT-29, MKN-45, U87MG, and SMMC-7721 cancer cell lines. Most of the tested compounds showed potent activity and high selectivity toward the HT-29 and MKN-45 cell lines. Furthermore, compounds 21b, 21c, and 21i were further examined for their c-Met kinase activity and exhibited strong efficacy with IC50 values in the single-digit nanomolar range, which was comparable with the positive control foretinib. The most promising compound 21c showed excellent cytostatic activity with IC50 values from 0.01 to 0.53 µM against all tested cell lines, thus being 1.7–2.2 times more active than foretinib.
Co-reporter:Baohui Qi;Haiyan Tao;Di Wu;Jinying Bai;Yan Shi
Archiv der Pharmazie 2013 Volume 346( Issue 8) pp:596-609
Publication Date(Web):
DOI:10.1002/ardp.201300087
ABSTRACT
Novel quinoline derivatives bearing acyclic semicarbazones were prepared and their chemical structures as well as the relative stereochemistry were confirmed. All the synthesized compounds were evaluated for their c-Met kinase inhibitory activity and their cytotoxicity against the cell lines HT-29, MKN-45, and MDA-MB-231 in vitro. Several potent compounds were further evaluated against A549 cells. Most compounds displayed moderate to excellent activity, and the structure–activity relationship studies identified the most promising compound 35 as a selective c-Met kinase inhibitor (IC50 = 4.3 nM). Compound 35 showed a 3.5- and 18.8-fold increase in cytotoxicity in vitro against HT-29 and A549 cells, respectively, compared to that of foretinib. Poor off-target effects of compound 35 were further confirmed by the antiproliferative activity against the c-Met inhibition less sensitive MDA-MB-231 cell line (IC50 = 0.77 µM).
Co-reporter:Mingze Qin;Weike Liao;Chen Xu;Baolin Fu;Jianguo Ren;Yucheng Gu
Archiv der Pharmazie 2013 Volume 346( Issue 11) pp:840-850
Publication Date(Web):
DOI:10.1002/ardp.201300188
A series of 4-(2-fluorophenoxy)-2-(1H-tetrazol-1-yl)pyridines bearing semicarbazone moieties were synthesized and evaluated for their in vitro antitumor potency. Some of the compounds (10b, 10c, 10e–10h, 10m–10p, 10r, and 11b) exhibited moderate to excellent antitumor activity as compared to sorafenib and PAC-1, as well as low levels of toxicity toward the human fetal lung fibroblast cell line WI-38. The most promising compound 10p (IC50 = 0.08, 0.36, 0.97 µM) was 45.1-, 6.1-, and 2.4-fold more active than sorafenib (IC50 = 3.61, 2.19, 2.32 µM), and 17, 3.2, and 2.9 times better than PAC-1 (IC50 = 1.36, 1.17, 2.83 µM) against three cancer cell lines (HT-29, H460, and MKN-45), respectively. In addition, further studies examining enzymatic activity suggested that the marked pharmacological activity observed might be ascribed to an inhibitory action against CRAf kinase.
Co-reporter:Xin Zhai;Ying He;Zhen Yang
Chemical Research in Chinese Universities 2013 Volume 29( Issue 1) pp:62-66
Publication Date(Web):2013 February
DOI:10.1007/s40242-013-2136-5
A series of novel N-methylpicolinamide-moiety containing diarylthiosemicarbazide derivatives was prepared and evaluated for their in vitro antiproliferative activity against three cancer cell lines(human alveolar epithelial cell A549, human lung cancer cell H460 and human colorectal cancer cell HT-29) by 3-(4,5-dimethyl)thiazolyl-diphenyltetrazoliumromide(MTT) assay. Six compounds(7b–7g) with halogen substituents exhibited preferable cytotoxicity against one or more cell lines in a low micromolar range. Especially, the most promising compound 7g exhibited remarkable antiproliferative activity with the IC50 values of 2.2, 1.8 and 5.2 μmol/L against A549, H460 and HT-29 cell lines respectively, which is comparable to sorafenib.
Co-reporter:Guogang Zhang;Yajing Liu;Shuobing Wang;Chuan Zhou;Qingchang Huang
Archiv der Pharmazie 2012 Volume 345( Issue 1) pp:49-56
Publication Date(Web):
DOI:10.1002/ardp.201100103
Abstract
In an attempt to develop potent antitumor agents, a series of novel 2-hydrazonylpyrido[2,3-b]pyrazin-3(4H)-one derivatives were designed and synthesized. All the prepared compounds were screened for their cytotoxic activities against A549, MDA-MB-231 and HT-29 cell lines in vitro. Pharmacological data indicated that five of the target compounds showed cytotoxicity against A549 cell line below a concentration of 1 µM. Compound 15g was the most potent one with IC50 values of 0.19, 2.11 and 2.15 µM against A549, MDA-MB-231 and HT29 cell lines, respectively.
Co-reporter:Shuobing Wang;Yanfang Zhao;Wufu Zhu;Ying Liu;Kaixing Guo
Archiv der Pharmazie 2012 Volume 345( Issue 1) pp:73-80
Publication Date(Web):
DOI:10.1002/ardp.201100082
Abstract
A novel series of indolin-2-one derivatives containing the 4-thiazolidinone moiety (5a—5p) was synthesized and the cytotoxicity of these derivatives was evaluated in vitro against three human cancer cell lines (HT-29, H460 and MDA-MB-231) by standard MTT assay. Some prepared compounds exhibited significant cytotoxicity against different human cancer cell lines. Several potent compounds were further evaluated against one normal cell line (WI-38). In particular, the promising compound 5h showed remarkable cytotoxicity and selectivity against the HT-29 and H460 cancer cell lines (IC50 = 0.016 µmol/L, 0.0037 µmol/L, respectively).
Co-reporter:Yajing Liu;Shulan Zhang;Ye Li;Jianqiang Wang;Yu Song
Archiv der Pharmazie 2012 Volume 345( Issue 4) pp:287-293
Publication Date(Web):
DOI:10.1002/ardp.201100250
Abstract
A new series of 1,4-disubstituted phthalazinylpiperazine derivatives 7a–f, 12a–f and 20a–f were designed and synthesized in order to develop potent and selective antitumor agents. The target compounds were screened for their cytotoxic activities against A549, HT-29 and MDA-MB-231 cancer cell lines in vitro. Among them, compounds 7a–f exhibited excellent selectivity for MDA-MB-231 with IC50 values ranging from 0.013 µM to 0.079 µM. The most promising compound, 7e (IC50 = 2.19 µM, 2.19 µM, 0.013 µM), was 9.3, 10, and 4.9 × 103 times more active than vatalanib (IC50 = 20.27 µM, 21.96 µM, 63.90 µM), respectively.
Co-reporter:Xin Zhai;Wei Li;Dong Chen;Ruiwei Lai;Jun Liu
Archiv der Pharmazie 2012 Volume 345( Issue 5) pp:360-367
Publication Date(Web):
DOI:10.1002/ardp.201100064
Abstract
A new series of 2,5-diaryliminothiazolidin-4-ones were designed and synthesized as potent antiproliferative agents. The antiproliferative activities of the 25 target compounds were evaluated against three cancer cell lines (A549, H460 and HT29) by MTT assay. Pharmacological data indicated that most of the compounds possessed moderate activity, some showed remarkable activity against one or more cell lines. As the most promising compound, 8s (with IC50 values of 1.1, 0.01 and 1.3 µM against the A549, H460 and HT29 cell lines) was 1.1- to 270-fold more potent than the reference drug sorafenib. Furthermore, preliminary structure–activity relationships (SARs) were summarized to provide guidance for further design and discovery of 2-iminothiazolidin-4-one-based antiproliferative agents.
Co-reporter:Wufu Zhu;Yajing Liu;Yanfang Zhao;Haiyan Wang;Li Tan;Weijie Fan
Archiv der Pharmazie 2012 Volume 345( Issue 10) pp:812-821
Publication Date(Web):
DOI:10.1002/ardp.201200074
Abstract
A series of 6-hydrazinyl-2,4-bismorpholino pyrimidine and 1,3,5-triazine derivatives (5a–5l and 8a–8o) were synthesized and their chemical structures as well as the relative stereochemistry were confirmed. All the synthesized compounds were evaluated for antiproliferative activity against three cancer cell lines (H460, HT-29, and MDA-MB-231). Several potent compounds were further evaluated against two other cell lines (U87MG, H1975). Most of the prepared compounds, particularly compounds 5c and 5j with IC50 values (0.07 and 0.05 µM, respectively) in the nM range, exhibited moderate to excellent antiproliferative activity and high selectivity against the H460 cancer cell line as compared with compound 1. The most promising compound 5j, possessing a cyano group at the 3-position of the benzene ring, showed strong antiproliferative activity against H460, HT-29, and MDA-MB-231 cell lines with IC50 values of 0.05, 6.31, and 6.50 µM, which were 4.6- to 190.4-fold more active than compound 1 (9.52, 29.24, and 36.21 µM), respectively.
Co-reporter:Yang Xiong Li, Xin Zhai, Wei Ke Liao, Wu Fu Zhu, Ying He, Ping Gong
Chinese Chemical Letters 2012 Volume 23(Issue 4) pp:415-418
Publication Date(Web):April 2012
DOI:10.1016/j.cclet.2012.01.015
In an attempt to develop potent antitumor agents, new rhodacyanine analogues containing the pyridinium ring (5a–5h), the isoquinolinium ring (6a–6c) and the quinolinium ring (7a–7e) linked to the rhodanine ring via N–N covalent bond were designed, synthesized and evaluated for antitumor activity against human lung cancer cell line (H460) by MTT assay in vitro. Most of the tested compounds showed enhanced antitumor activity with IC50 values ranging from 0.006 to 9.2 μmol/L as compared to the lead compound MKT-077. Among them, the most promising compound 7d (IC50 = 0.006 μmol/L) was 216.7 times more active than MKT-077 (IC50 = 1.3 μmol/L). The preliminary structure–activity relationship of the target compounds was discussed.
Co-reporter:Bei Zhang, Yan Fang Zhao, Xin Zhai, Wei Jie Fan, Jun Ling Ren, Chun Fu Wu, Ping Gong
Chinese Chemical Letters 2012 Volume 23(Issue 8) pp:915-918
Publication Date(Web):August 2012
DOI:10.1016/j.cclet.2012.06.009
A new series of diaryl urea derivatives bearing N-acylhydrazone moiety were designed and synthesized. All the target compounds were evaluated for their antiproliferative activities against human leukemia cell line (HL-60), human lung adenocarcinoma epithelial cell line (A549) and human breast cancer cell line (MDA-MB-231) in vitro by standard MTT assay. The pharmacological results indicated that some compounds exhibited promising antitumor activities. Compound 1j showed the most potent antiproliferative activity against the tested three cell lines with IC50 values of 0.13 μmol/L, 0.7 μmol/L and 0.5 μmol/L, respectively.
Co-reporter:DeXiang Guo;YaJing Liu;Ting Li;Nan Wang;Xin Zhai;Chun Hu
Science China Chemistry 2012 Volume 55( Issue 3) pp:347-351
Publication Date(Web):2012 March
DOI:10.1007/s11426-011-4477-6
A new series of 4,5-dihydro-1H-thiochromeno[4,3-d]pyrimidine derivatives have been designed and synthesized. The antitumor activities of the target compounds have been evaluated in vitro against two human cancer cell lines including A549 (human alveolar adenocarcinoma cell) and H460 (human lung cancer) by MTT assay. Most of the target compounds exhibited significant antitumor activities against A549 and H460 cancer cell lines. The most potent compound 4-(benzo[d][1,3]dioxol-5-yl)-8,9-difluoro-2-(4-methylpiperazin-1-yl)-4,5-dihydro-1H-thiochromeno[4,3-d]pyrimidine (CH05) (IC50 = 0.44 μM, 3.07 μM) was 2.0 and 8.4 times more active than gefitinib (IC50 = 0.89 μM, 16.81 μM) against A549 and H460 cell lines, respectively.
Co-reporter:Shuobing Wang, Yanfang Zhao, Guogang Zhang, Yingxiang Lv, Ning Zhang, Ping Gong
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 8) pp:3509-3518
Publication Date(Web):August 2011
DOI:10.1016/j.ejmech.2011.05.017
A series of novel 4-thiazolidinone and indolin-2-one hybrid derivatives 5a–5s and 10a–10s have been designed and synthesized and their cytotoxic activities were evaluated in vitro against three human cancer cell lines including HT-29 (human colon cancer), H460 (human lung cancer), MDA-MB-231 (human breast cancer) by MTT assay. Several potent target compounds (5m, 5p, 5s, 10a, 10c–10g, 10m, 10p) were further evaluated against one cancer cell line SMMC-7721 (human liver cancer) and one normal cell line WI-38 (human fetal lung fibroblasts). Most of the prepared compounds exhibited significant antitumor activities against different human cancer cell lines. Compound 10c (IC50 = 0.025 μM, 0.075 μM, 0.77 μM, 1.95 μM) was 52, 36, 4.8 and 3.3 times more active than Sunitinib (IC50 = 1.3 μM, 2.7 μM, 3.7 μM, 6.47 μM) against HT-29, H460, MDA-MB-231 and SMMC-7721 cancer cell line, respectively.The cytotoxic activity of 4-thiazolidinone and indolin-2-one hybrid compound 10c (IC50 = 0.025 μM, 0.075 μM, 0.77 μM, 1.95 μM) was significant against HT-29, H460, MDA-MB-231 and SMMC-7721 cancer cell line, respectively.Highlights► A series of 4-thiazolidinone and indolin-2-one hybrid derivatives were synthesized. ► The derivatives were evaluated in vitro against HT-29, H460, MDA-MB-231, SMMC-7721 human cancer cell lines. ► Compound 10c showed excellent antitumor activity against four cancer cell lines. ► Combination of tow-privileged structures could lead to potent antitumor agent.
Co-reporter:Fan Zhang, Yanfang Zhao, Li Sun, Lu Ding, Yucheng Gu, Ping Gong
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 7) pp:3149-3157
Publication Date(Web):July 2011
DOI:10.1016/j.ejmech.2011.03.055
A series of novel 2-amino-3-cyano-6-(1H-indol-3-yl)-4-phenylpyridine derivatives were synthesized and their cytotoxic activity against A549, H460, HT-29 and SMMC-7721 cell lines was evaluated in vitro. Among them, ten compounds (10, 11, 14, 16, 17, 26, 27, 29, 30 and 31) displayed excellent anti-tumor activity against different cell lines. The most promising compound 27 showed strong anti-tumor activity against A549, H460, HT-29 and SMMC-7721 cell lines with IC50 values of 22, 0.23, 0.65 and 0.77 nM, which were 2.6-, 83-, 1.1 × 103- and 2.0 × 103- fold more active than MX-58151 (IC50 values of 0.058, 0.019, 0.70 and 1.53 μM), respectively.A series of novel 2-amino-3-cyano-6-(1H-indol-3-yl)-4-phenylpyridine derivatives were synthesized and their cytotoxic activity against A549, H460, HT-29 and SMMC-7721 cell lines was evaluated in vitro by the standard MTT assay.Highlights► Introduction of indole core improved the cytotoxicity of 4,6-diaryl-2-amino-3-cyanopyridines. ► Ten compounds showed potent cytotoxicity against different cell lines with IC50 values in nM range. ► Some compounds exhibited excellent selectivity against H460 or HT-29 cell lines. ► Compound 27 showed the strongest cytotoxicity against four cell lines with IC50 values in nM range.
Co-reporter:Dong Chen;Yajing Liu;Shulan Zhang;Dexiang Guo;Chunhong Liu;Sai Li
Archiv der Pharmazie 2011 Volume 344( Issue 3) pp:158-164
Publication Date(Web):
DOI:10.1002/ardp.201000045
Abstract
A series of ethyl 6-bromo-8-hydroxyimidazo[1,2-a]pyridine-3-carboxylate derivatives were synthesized and evaluated for their anti-hepatitis B virus (HBV) activity and cytotoxicity in HepG2.2.15 cells. Nearly half of the tested compounds were proved to be highly effective in inhibiting the replication of HBV DNA with IC50 values ranging from 1.3 to 9.1 µM. Among them, 10o and 10s were identified as the most promising compounds.
Co-reporter:Lijun Xie;Xin Zhai;Chun Liu;Peng Li;Yangxiong Li;Guoxian Guo
Archiv der Pharmazie 2011 Volume 344( Issue 10) pp:639-647
Publication Date(Web):
DOI:10.1002/ardp.201000391
Abstract
In an attempt to develop potent and selective anti-tumor agents, three new series of artemisinin–chalcone hybrids 10a–10g, 11a–11g and 12a–12h were designed, synthesized and screened for their anti-tumor activity against five cell lines (HT-29, A549, MDA-MB-231, HeLa and H460) in vitro. Among compounds 10a–g and 11a–11g, most of them displayed enhanced activity and good selectivity toward HT-29 and HeLa cell lines with IC50 values ranging from 0.12 to 0.85 µM as compared with DHA (dihydroartemisinin). Compounds 10a and 11a are most active toward HeLa cells with IC50 values of 0.12 and 0.19 µM. The results revealed that the presence of chalcone moiety is beneficial to their activity and selectivity. In addition, compounds 12a-12h containing a ‘reversed chalcone’ moiety showed only slight improvement in activity than those of DHA.
Co-reporter:Lijun Xie;Yanfang Zhao;Xin Zhai;Peng Li;Chun Liu;Yangxiong Li
Archiv der Pharmazie 2011 Volume 344( Issue 10) pp:631-638
Publication Date(Web):
DOI:10.1002/ardp.201000363
Abstract
Three series of novel artemisinin–guanidine hybrids 4a–4f, 8a–8h and 9a–9h have been facilely synthesized via four-component reaction (aza-Wittig reaction) and evaluated for their anti-tumor activities against A549, HT-29 and MDA-MB-231 cell lines in vitro. All of the tested compounds showed enhanced anti-tumor activities with IC50 values ranging from 0.02 µM to 12.0 µM as compared to DHA (dihydroartemisinin). Among them, artemisinin derived dimers, compounds 9b (IC50 = 0.05 µM), 9d (IC50 = 0.06 µM) and 9f (IC50 = 0.02 µM) were found to be most active against HT29 cells.
Co-reporter:Guo Gang Zhang, Ya Jing Liu, Xiao Guang Ma, Hao Dong, Ju Li, Ping Gong
Chinese Chemical Letters 2011 Volume 22(Issue 10) pp:1223-1225
Publication Date(Web):October 2011
DOI:10.1016/j.cclet.2011.05.031
A series of novel 2-hydrazinylpyrido[2,3-b]pyrazin-3(4H)-one derivatives were synthesized and evaluated for their cytotoxic activities against A549, MDA-MB-231 and HT-29 cell lines in vitro. Pharmacological data indicated that compounds 5b, 5c, 10a and 10g possessed marked cytotoxicity, especially 10a (with IC50 values of 0.81, 2.56 and 1.63 μmol/L against A549, MDA-MB-231 and HT29 cell lines, respectively), which had emerged as a lead compound.
Co-reporter:Wei Li;Xin Zhai;Zheng Zhong;Guangyue Li;Yongxiao Pu
Archiv der Pharmazie 2011 Volume 344( Issue 6) pp:349-357
Publication Date(Web):
DOI:10.1002/ardp.201000326
Abstract
A series of rhodanine-containing sorafenib analogs was designed, synthesized and evaluated for their in-vitro antitumor activity against three cancer cell lines (A549, H460 and HT29). Pharmacological data indicated that some of the target compounds possessed marked antiproliferative activity superior to the reference drug sorafenib, especially the most promising compound 7r (with the IC50 value of 0.8, 1.3 and 2.8 µM against A549, H460 and HT29 cell lines, respectively). The activity was found to strongly depend on the substitution pattern of the rhodanine motif at C-5″ position. Results suggested that this series of compounds could serve as the bases for the development of novel antitumor agents.
Co-reporter:Fan Zhang, Xin Zhai, Li Juan Chen, Jian Guo Qi, Bo Cui, Yu Cheng Gu, Ping Gong
Chinese Chemical Letters 2011 Volume 22(Issue 11) pp:1277-1280
Publication Date(Web):November 2011
DOI:10.1016/j.cclet.2011.05.030
A series of 2,5-disubstituted pyrimido[5,4-c]quinoline derivatives were synthesized and their cytotoxic activity against H460, HT-29 and MDA-MB-231 cell lines was evaluated in vitro. It was found that most of the tested compounds especially compound 17, shown stronger activity to the selected three cell lines than ZM447439.
Co-reporter:Shulan Zhang, Yanfang Zhao, Yajing Liu, Dong Chen, Weihuan Lan, Qiaoling Zhao, Chengcheng Dong, Lin Xia, Ping Gong
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 8) pp:3504-3510
Publication Date(Web):August 2010
DOI:10.1016/j.ejmech.2010.05.016
In an attempt to develop potent and selective antitumor agents, a series of novel 1,4-disubstituted phthalazine derivatives was designed and synthesized. All the prepared compounds were screened for their cytotoxic activities against A549, HT-29 and MDA-MB-231 cell lines in vitro. Among them, seven compounds (7a–7e, 7j and 7i) displayed excellent selectivity for MDA-MB-231 cells with IC50 values in the nM range, a desirable range for pharmacological testing. The most promising compound, 7a (IC50 = 3.79 μM, 2.32 μM, 0.84 nM), was 5.6-, 10.8- and 6.9 × 104- times more active than PTK-787 (IC50 = 21.16 μM, 22.11 μM, 57.72 μM), respectively.The most promising compound, 7a (IC50 = 3.79 μM, 2.32 μM, 0.84 nM), showed excellent cytotoxic activities against A549, HT-29 and MDA-MB-231 cell lines in vitro by the MTT method.
Co-reporter:Shu Lan Zhang, Ya Jing Liu, Yan Fang Zhao, Qiu Ting Guo, Ping Gong
Chinese Chemical Letters 2010 Volume 21(Issue 9) pp:1071-1074
Publication Date(Web):September 2010
DOI:10.1016/j.cclet.2010.04.004
A series of 1,4-substituted phthalazine derivatives were designed and synthesized. All the prepared compounds were screened for their cytotoxic activities against A549, HT-29 and MDA-MB-231 cell lines in vitro. Among them, compounds 7a–7h showed excellent selectivity for MDA-MB-231 cell line with IC50 values from 1 nmol/L to 0.92 μmol/L. A preliminary SAR study of these derivatives was performed.
Co-reporter:Dong Chen, Xin Zhai, Qiu Hui Yuan, Jian Luo, Si Chang Xie, Ping Gong
Chinese Chemical Letters 2010 Volume 21(Issue 11) pp:1326-1329
Publication Date(Web):November 2010
DOI:10.1016/j.cclet.2010.05.011
A series of 1H-benzimidazol-5-ol derivatives were synthesized and evaluated for their anti-hepatitis B virus (HBV) activity and cytotoxicity in HepG2.2.15 cells. Half of the tested compounds were found to be potent against HBsAg secretion with IC50 values less than 100 μmol/L. Compounds 14c, 14d, and 14e showed significant inhibitory activity to the viral antigen HBeAg.
Co-reporter:Peng Yao;Xin Zhai;Dong Liu;Bao Hui Qi;Hai Liang Tan;Yong Cai Jin
Archiv der Pharmazie 2010 Volume 343( Issue 1) pp:17-23
Publication Date(Web):
DOI:10.1002/ardp.200900130
Abstract
We herein disclose a series of novel diaryl urea derivatives possessing a 4H-pyrido[1,2-a]pyrimidin-4-one group as novel potent anticancer compounds. The structures were confirmed by IR, 1H-NMR, and MS. All the compounds were screened for their antiprofilerative activity agaist the human breast cancer cell line (MDA-MB-231). The pharmacological results indicated that most of the compounds showed moderate activity. The best of this series is compound 4c (IC50 = 0.7 μmol/L), with a potency 3.6-fold higher than Sorafenib (IC50 = 2.5 μmol/L), which was approved in 2005.
Co-reporter:Shu Lan Zhang, Xin Zhai, Shi Jiao Zhang, Hong Hao Yu, Ping Gong
Chinese Chemical Letters 2010 Volume 21(Issue 8) pp:939-942
Publication Date(Web):August 2010
DOI:10.1016/j.cclet.2010.03.016
A series of quinoline-3-carbonitrile derivatives were designed and synthesized. Their cytotoxicity in vitro against four cancer cell lines (A549, HT-29, MDA-MB-231 and SMMC-7721) were evaluated by standard MTT assay. The pharmacological results showed that most of the prepared compounds displayed excellent selective cytotoxicity toward SMMC-7721 cell line. Among them, compounds 7c, 7e, 11b, 11f and 11g were more active than Gefitinb against SMMC-7721 cell line.
Co-reporter:Yanfang Zhao, Yajing Liu, Dong Chen, Zengquan Wei, Wenzhao Liu, Ping Gong
Bioorganic & Medicinal Chemistry Letters 2010 20(24) pp: 7230-7233
Publication Date(Web):
DOI:10.1016/j.bmcl.2010.10.099
Co-reporter:Wei Jia, Yajing Liu, Wei Li, Yan Liu, Dajun Zhang, Peng Zhang, Ping Gong
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 13) pp:4569-4574
Publication Date(Web):1 July 2009
DOI:10.1016/j.bmc.2009.05.001
A series of novel 6H-[1]benzothiopyrano[4,3-b]quinoline derivatives were prepared and evaluated for their anti-hepatitis B virus (HBV) activity and cytotoxicity in human hepatoblastoma-derived liver Hep-G2 cells. Compounds 10g, 10h, 10j, 10l and 10o were found to be potent anti-HBV compounds with IC50 values less than 50 μM. The most promising compound was 10l, with an IC50 value of 14.7 μM and a SI value of 12.4. This is the first report of the anti-HBV effects of 6H-[1]benzothiopyrano[4,3-b] quinolin-9-ols.
Co-reporter:Wei Jia;Yanfang Zhao;Rongdong Li;Yanjiao Wu;Zebiao Li
Archiv der Pharmazie 2009 Volume 342( Issue 9) pp:507-512
Publication Date(Web):
DOI:10.1002/ardp.200900070
Abstract
A series of 9-methoxy-6H-[1]benzothiopyrano[4,3-b]quinolin-10-ols with a Mannich side chain were synthesized and evaluated for their anti-Hepatitis B virus (HBV) activity in HepG2.2.15 cells. Some compounds showed significant anti-HBV activity with IC50 values less than 41 μM. Among them, compound 9b was the most effective anti-HBV agent (IC50 = 1.7 μM, SI = 60.3).
Co-reporter:Xin Zhai, Nan Jiang, Ke Liang Zhang, Feng Bao, Ping Gong
Chinese Chemical Letters 2009 Volume 20(Issue 10) pp:1179-1182
Publication Date(Web):October 2009
DOI:10.1016/j.cclet.2009.04.027
In our efforts to identify novel potent anticancer agents, we synthesized a series of 2,7-disubstituted triazolo[1,5-a]pyrimidines (6–16). Their antiproliferative activity against Bel-7402, HT-1080 and WI-38 cell lines was tested by MTT assay in vitro. Four of the compounds (9–11 and 16) displayed promising antiproliferative activity superior to gefitinib, especially compound 9. A preliminary SAR study of these derivatives was performed.
Co-reporter:Yajing Liu;Yanfang Zhao;Xin Zhai;Xiuping Liu;Lixue Sun;Yanxia Ren
Archiv der Pharmazie 2008 Volume 341( Issue 7) pp:446-452
Publication Date(Web):
DOI:10.1002/ardp.200800035
Abstract
Some new ethyl 8-imidazolylmethyl-7-hydroxyquinoline-3-carboxylate derivatives have been synthesized and evaluated for their anti-hepatitis B virus (HBV) activities and cytotoxicities in HepG2.2.15 cells stable transfection with HBV. Compounds 13a, 11b, 11c, 12c, 13c, 11g, and 12g inhibited the expression of the viral antigens HBsAg or HBeAg in a low concentration, of which 11c (IC50 = 12.6 μM, SI = 12.4), 12c (IC50 = 3.5 μM, SI = 37.9), and 12g (IC50 = 2.6 μM, SI = 61.6) showed more active abilities to inhibit the replication of HBV DNA than the positive control lamivudine (3TC, IC50 = 343.2 μM, SI = 7.0).
Co-reporter:Xin Zhai, Juan Li, Lei He, Su Zheng, Yao Bin Zhang, Ping Gong
Chinese Chemical Letters 2008 Volume 19(Issue 1) pp:29-32
Publication Date(Web):January 2008
DOI:10.1016/j.cclet.2007.11.018
Eight novel 1,4-disubstituted phthalazines (7–14) were designed and synthesized. The structures of all the synthesized compounds were confirmed by IR, 1H NMR, 13C NMR, MS and elemental analysis. Their in vitro cytotoxicity against cancer cell lines (Bel-7402 and HT-1080) were evaluated by standard MTT assay. Among them, compounds 9 and 11 exhibited more potent cytotoxicity than cisplatin.
Co-reporter:Yan-ling SONG, Xin ZHAI, Zhi-feng SUN, Sang-hee KIM, Ping GONG
Chemical Research in Chinese Universities 2008 Volume 24(Issue 4) pp:445-448
Publication Date(Web):July 2008
DOI:10.1016/S1005-9040(08)60093-8
Abstract
The substituted phenanthrene-9-carboxyaldehydes are very important intermediates for the syntheses of phenanthroindolizidine and phenanthroquinolizidine alkaloids. The novel title compound was prepared from the reaction of 5 steps starting from the condensation of 3-methoxyl-4-methyl-phenylacetic acid and 4-(benzyloxy)-2-iodo-benzaldehyde, followed by esterification, cyclization, reduction, and oxidation. A new method for the preparation of phenanthrene ring via palladium-catalyzed intramolecular Heck reaction was described. The title compound was characterized by IR, 1H NMR, 13C NMR, elemental analysis, and MS.
Co-reporter:Shuchun Guo;Yanfang Zhao;Xianglin Zhao;Sisi Zhang;Lijun Xie;Weiyong Kong
Archiv der Pharmazie 2007 Volume 340(Issue 8) pp:
Publication Date(Web):13 JUL 2007
DOI:10.1002/ardp.200700044
A series of novel methylthio-, sulfinyl-, and sulfonyl-8H-thieno[2,3-b]pyrrolizin-8-oximino derivatives 7A–12P was designed and synthesized as anti-tumor agents. Their structures were confirmed by IR, 1H-NMR, MS, and elemental analysis. The anti-tumor activities of all the target compounds were tested by the MTT method in vitro against Bel-7402 (human liver cancer) and HT-1080 (human fibro sarcoma) cell lines. Among them, compound 11N (IC50 = 18.2 μM, 8.2 μM), was the most promising compound of all synthesized molecules, it was 2.5- and 3.3-times more active than cisplatin (IC50 = 45.2 μM, 26.7 μM), respectively.
Co-reporter:Rong Dong Li;Xin Zhai;Yan Fang Zhao;Shuang Yu
Archiv der Pharmazie 2007 Volume 340(Issue 8) pp:
Publication Date(Web):13 JUL 2007
DOI:10.1002/ardp.200700078
A novel series of 1-anilino-5H-pyridazino[4,5-b]indoles was designed and synthesized in order to find novel potent anti-tumor compounds. Their structures were confirmed by MS, 1H-NMR, and elemental analysis. All compounds were screened for their cytotoxic activity against two human cancer cell lines (Bel-7420, HT-1080). The compounds 8, 9, and 17 showed 50% growth inhibitory activity in low micromolar concentration (IC50 = 7.7∼12.8 μM). Among them, compound 17 displayed the most potent anti-tumor activity with IC50 values of 8.2 μM and 7.9 μM against Bel-7402 and HT-1080, respectively.
Co-reporter:Rong Dong Li, Xin Zhai, Yan Fang Zhao, Ping Gong
Chinese Chemical Letters 2007 Volume 18(Issue 10) pp:1191-1194
Publication Date(Web):October 2007
DOI:10.1016/j.cclet.2007.07.027
A novel series of 5H-pyridazino[4,5-b]indoles were designed and synthesized in order to find novel potent anticancer compounds. The structures were confirmed by 1H NMR and MS. Their antiproliferative activities against two cancer cell lines were tested by the MTT method in vitro. Three of compounds (1e, 1g, and 1h) exhibited potent antiproliferative activities, especially compound 1h (with IC50 values of 5.2 μmol/L and 1.9 μmol/L against Bel-7402 and HT-1080, respectively). The preliminary structure–activity relationships of 5H-pyridazino[4,5-b]indole derivatives were discussed.
Co-reporter:Juan Li;Yan Fang Zhao;Xiang Lin Zhao;Xiao Ye Yuan
Archiv der Pharmazie 2006 Volume 339(Issue 11) pp:
Publication Date(Web):12 OCT 2006
DOI:10.1002/ardp.200600098
A series of novel pyrazolo[1,5-a]pyrimidines were designed and synthesized in order to find novel potent anti-tumor compounds. The structures of all the compounds were confirmed by IR, 1H-NMR, elemental analysis, and MS. Their anti-tumor activities against cancer cell lines were tested by the MTT method in vitro. Compound 19 displayed potent anti-tumor activity.
Co-reporter:Xue-Ying Wu, Yao-Ling Wang, Li Hai, Ping Gong, Yong Wu
Chinese Chemical Letters (February 2017) Volume 28(Issue 2) pp:
Publication Date(Web):February 2017
DOI:10.1016/j.cclet.2016.10.026
Rocuronium bromide has been used as an aminosteroid non-depolarizing neuromuscular blocker and muscle relaxant. In this work, a new and efficient route for preparing a key intermediate 2β-(4-morpholinyl)-16β-(1-pyrrolidinyl)-5α-androstan-3α,17β-diol (6) was developed through a ring-opening of epoxide followed by introducing and pyrrolidine. Compound 6 can easily provide rocuronium bromide and the overall yield of compound 6 in 5 steps increased to 57.8%, which was higher than currently reported methods. Extraordinarily, this method would avoid the generation of disubstituted impurities E and F which are difficult to remove.A new and efficient route for preparing 2β-(4-morpholinyl)-16β-(1-pyrrolidinyl)-5α-androstan-3α,17β-diol (6) as the key intermediate for providing rocuronium bromide (7) was developed. The overall yield of compound 6 in 5 steps increased to 57.8%, which was higher than currently reported methods Extraordinarily, this method could avoid the generation of disubstituted impurities E and F which are difficult to remove.
Co-reporter:Fangyang Wang, Lihui Wang, Yanfang Zhao, Yi Li, ... Chunfu Wu
Molecular Oncology (December 2014) Volume 8(Issue 8) pp:1640-1652
Publication Date(Web):1 December 2014
DOI:10.1016/j.molonc.2014.06.015
•We discovered a novel procaspase-3 activator WF-210.•We demonstrated that WF-210 has higher selection index.•We found that WF-210 could induce tumor cell apoptosis through activate procaspase-3.•WF-210 or PAC-1 induced apoptosis by promoting proteasome-dependent degradation of IAPs.•WF-210 showed greater therapeutic effect in vivo compared with PAC-1.Purpose: Procaspase-3, a proenzyme of apoptotic executioner caspase-3, is overexpressed in numerous tumors. We aimed to characterize a novel procaspase-3 activator, WF-210, which may have potential as an anticancer drug.Experimental design: The procaspase-3 activating ability, antitumor efficacy, mechanisms of action, and toxicity profiles of WF-210 were investigated in vitro and in vivo, using normal cells, cancer cells, and mouse xenograft models. The role of procaspase-3 in WF-210-induced apoptosis was explored by manipulating procaspase-3 expression in cultured cells.Results: WF-210 activated procaspase-3 with an EC50 of 0.95 μM, less than half that of its mother compound PAC-1 (2.08 μM). The mechanism involved the chelation of inhibitory zinc ions, subsequently resulting in an auto-activation of procaspase-3. WF-210 was more cytotoxic than PAC-1 to human cancer cells, but less cytotoxic to normal cells. Cancer cells with high procaspase-3 expression, like HL-60 and U-937, were particularly sensitive. WF-210-induced the apoptosis of HL-60 and U-937 cells by activating procaspases and promoting proteasome-dependent degradation of XIAP and Survivin. The level of WF-210-induced apoptosis in cultured cells was related to the level of procaspase-3 expression. Finally, WF-210 was superior to PAC-1 in retarding the in vivo growth of breast, liver and gallbladder xenograft tumors which overexpress procaspase-3, and induced no substantial weight loss or neurotoxicity. WF-210 and PAC-1 had no effect on the growth of MCF-7 xenograft tumors, which do not express procaspase-3.Conclusion: We identified WF-210 as a potent small-molecule activator of procaspase-3. The favorable antitumor activity and acceptable toxicity profile of WF-210 provide a strong rationale for its clinical evaluation in the treatment of tumors with high procaspase-3 expression.
Co-reporter:Mingze Qin, Shuang Yan, Lei Wang, Haotian Zhang, Ye Tian, Yanfang Zhao, Ping Gong
Bioorganic & Medicinal Chemistry (15 March 2017) Volume 25(Issue 6) pp:1778-1786
Publication Date(Web):15 March 2017
DOI:10.1016/j.bmc.2017.01.039

![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1.png)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1_b.png)
![1-[(3,4-DIMETHOXYPHENYL)METHYL]-6,7-DIMETHOXYISOQUINOLINE;SULFURIC ACID](http://img.cochemist.com/ccimg/32900/32808-09-6.png)
![1-[(3,4-DIMETHOXYPHENYL)METHYL]-6,7-DIMETHOXYISOQUINOLINE;SULFURIC ACID](http://img.cochemist.com/ccimg/32900/32808-09-6_b.png)




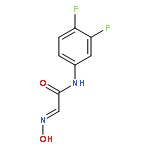
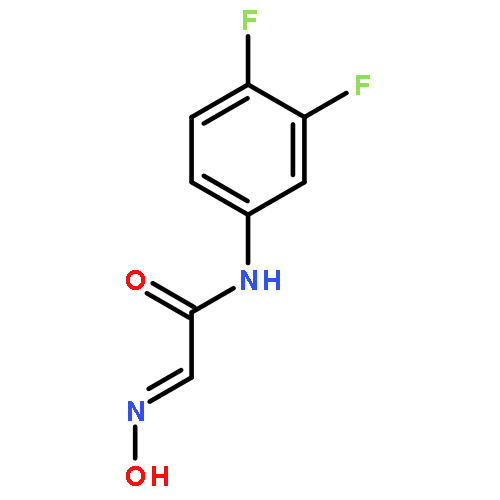







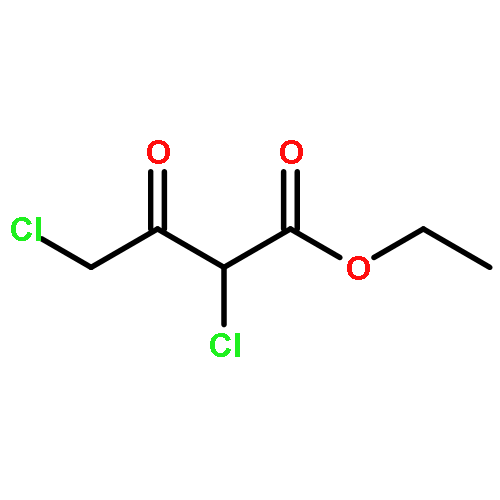
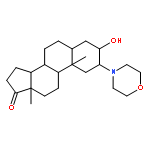
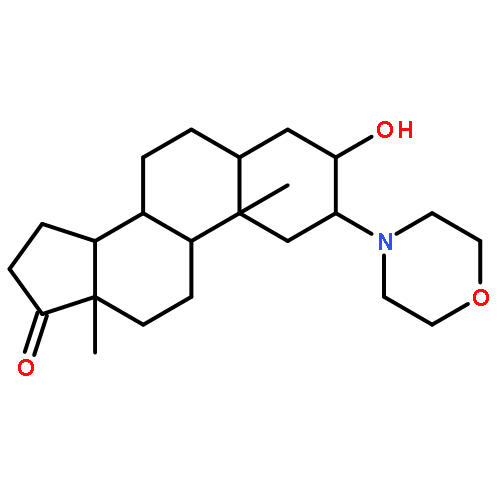


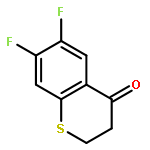

![5-Chloro-N-[[[4-[6-fluoro-1,4-dihydro-7-(methylamino)-2,4-dioxo-3(2H)-quinazolinyl]phenyl]amino]carbonyl]-2-thiophenesulfonamide](http://img.cochemist.com/ccimg/936600/936500-94-6.png)
![5-Chloro-N-[[[4-[6-fluoro-1,4-dihydro-7-(methylamino)-2,4-dioxo-3(2H)-quinazolinyl]phenyl]amino]carbonyl]-2-thiophenesulfonamide](http://img.cochemist.com/ccimg/936600/936500-94-6_b.png)

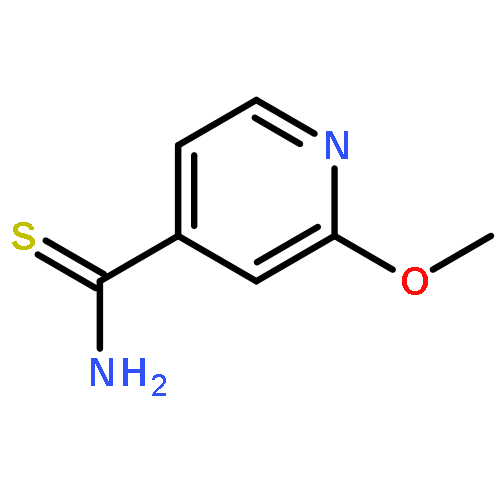
![Phthalazine, 1-chloro-4-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/929700/929613-64-9.png)
![Phthalazine, 1-chloro-4-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/929700/929613-64-9_b.png)
![1(2H)-Phthalazinone, 4-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/929700/929613-62-7.png)
![1(2H)-Phthalazinone, 4-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/929700/929613-62-7_b.png)
![Benzoic acid, 2-[2-(phenylthio)acetyl]-, methyl ester](http://img.cochemist.com/ccimg/929700/929613-60-5.png)
![Benzoic acid, 2-[2-(phenylthio)acetyl]-, methyl ester](http://img.cochemist.com/ccimg/929700/929613-60-5_b.png)
![2-BUTENOIC ACID, 4-[(3-METHOXYPHENYL)THIO]-3-(METHYLAMINO)-, ETHYL ESTER](http://img.cochemist.com/ccimg/886100/886059-37-6.png)
![2-BUTENOIC ACID, 4-[(3-METHOXYPHENYL)THIO]-3-(METHYLAMINO)-, ETHYL ESTER](http://img.cochemist.com/ccimg/886100/886059-37-6_b.png)
![2-Butenoic acid, 3-(methylamino)-4-[(4-methylphenyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/886100/886059-34-3.png)
![2-Butenoic acid, 3-(methylamino)-4-[(4-methylphenyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/886100/886059-34-3_b.png)
![Thieno[3,2-d]pyrimidine-6-methanol, 2-chloro-4-(4-morpholinyl)-](/data/chemimg/1610400/885698-97-5.png)
![Thieno[3,2-d]pyrimidine-6-methanol, 2-chloro-4-(4-morpholinyl)-](/data/chemimg/1610400/885698-97-5_b.png)
![4-(2-Chloro-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine](http://img.cochemist.com/ccimg/885700/885675-66-1.png)
![4-(2-Chloro-6-((4-(methylsulfonyl)piperazin-1-yl)methyl)thieno[3,2-d]pyrimidin-4-yl)morpholine](http://img.cochemist.com/ccimg/885700/885675-66-1_b.png)
![Thieno[3,2-d]pyrimidine-6-carboxaldehyde, 2-chloro-4-(4-morpholinyl)-](/data/chemimg/975500/885618-31-5.png)
![Thieno[3,2-d]pyrimidine-6-carboxaldehyde, 2-chloro-4-(4-morpholinyl)-](/data/chemimg/975500/885618-31-5_b.png)
![1H-Pyrrole-2-carboxaldehyde, 1-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/883600/883541-16-0.png)
![1H-Pyrrole-2-carboxaldehyde, 1-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/883600/883541-16-0_b.png)
![1H-Indole-3-carboxylic acid,6-bromo-1-cyclopropyl-5-hydroxy-2-[(phenylthio)methyl]-, ethyl ester](http://img.cochemist.com/ccimg/874600/874595-65-0.png)
![1H-Indole-3-carboxylic acid,6-bromo-1-cyclopropyl-5-hydroxy-2-[(phenylthio)methyl]-, ethyl ester](http://img.cochemist.com/ccimg/874600/874595-65-0_b.png)




![3-[(3,4-difluorophenyl)thio]propanoic acid](http://img.cochemist.com/ccimg/863700/863667-96-3.png)
![3-[(3,4-difluorophenyl)thio]propanoic acid](http://img.cochemist.com/ccimg/863700/863667-96-3_b.png)





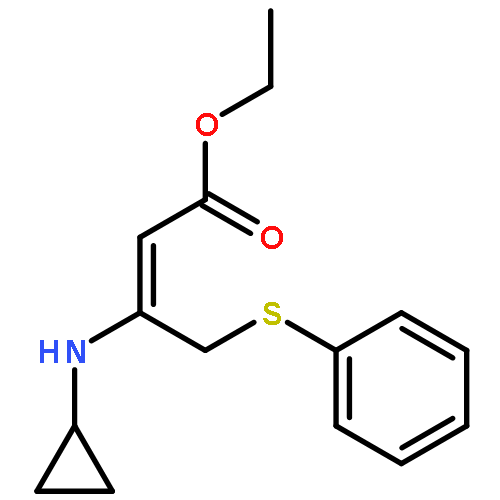

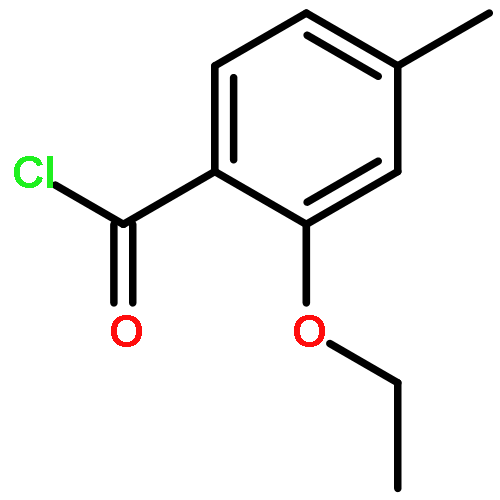


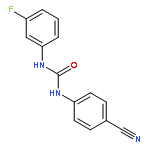
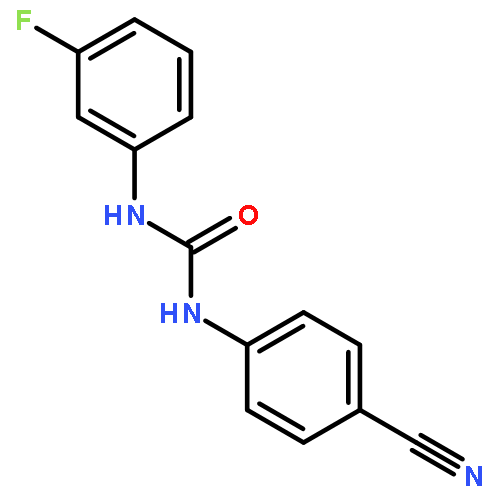




![2-Thiophenecarboxylic acid, 3-[(4-nitrobenzoyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/183500/183495-96-7.png)
![2-Thiophenecarboxylic acid, 3-[(4-nitrobenzoyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/183500/183495-96-7_b.png)







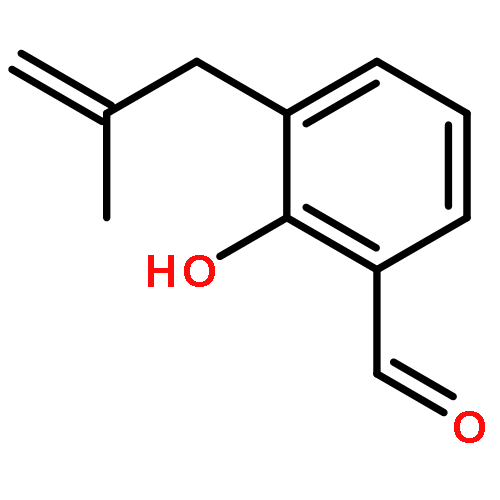

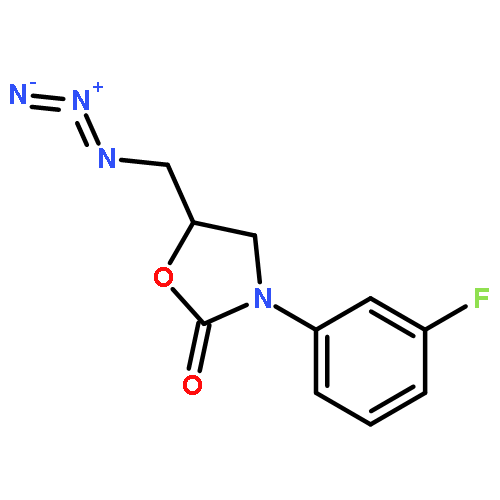






![(S)-N-[[3-(3-Fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]acetamide](http://img.cochemist.com/ccimg/139100/139071-79-7.png)
![(S)-N-[[3-(3-Fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]acetamide](http://img.cochemist.com/ccimg/139100/139071-79-7_b.png)


![2-Propenoic acid, 3-[(4-methylphenyl)sulfonyl]-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/117700/117659-25-3.png)
![2-Propenoic acid, 3-[(4-methylphenyl)sulfonyl]-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/117700/117659-25-3_b.png)




![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, 10-(2-bromoethoxy)decahydro-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,10S,12R,12aR)-](http://img.cochemist.com/ccimg/101900/101834-30-4.png)
![3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, 10-(2-bromoethoxy)decahydro-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,10S,12R,12aR)-](http://img.cochemist.com/ccimg/101900/101834-30-4_b.png)


![Butanoic acid, 4-[(3-methylphenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/83900/83821-46-9.png)
![Butanoic acid, 4-[(3-methylphenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/83900/83821-46-9_b.png)


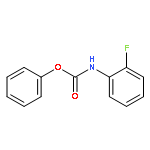

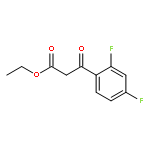
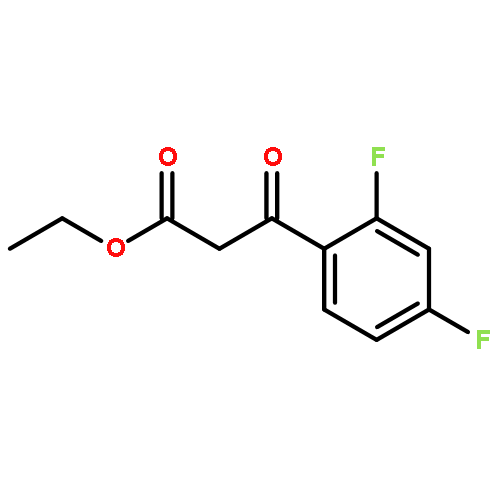
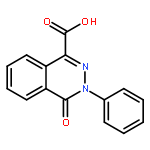


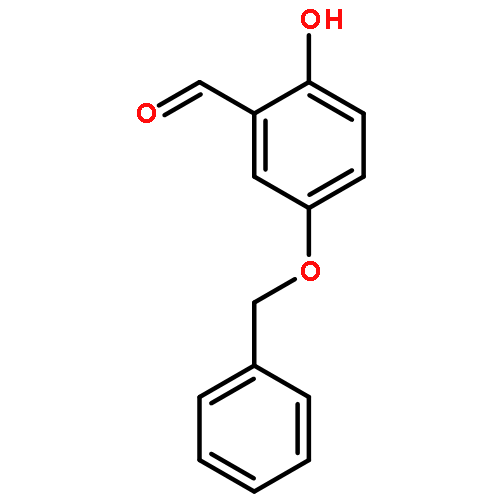










![2-[(3S,4R)-3-HYDROXY-2,2-DIMETHYL-6-(TRIFLUOROMETHOXY)-3,4-DIHYDR<WBR />O-2H-CHROMEN-4-YL]-1-ISOINDOLINONE](http://img.cochemist.com/ccimg/37600/37522-19-3.png)
![2-[(3S,4R)-3-HYDROXY-2,2-DIMETHYL-6-(TRIFLUOROMETHOXY)-3,4-DIHYDR<WBR />O-2H-CHROMEN-4-YL]-1-ISOINDOLINONE](http://img.cochemist.com/ccimg/37600/37522-19-3_b.png)





![Butanoic acid, 4-[(4-methylphenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/18200/18168-82-6.png)
![Butanoic acid, 4-[(4-methylphenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/18200/18168-82-6_b.png)


![Urea, [2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/13200/13114-85-7.png)
![Urea, [2-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/13200/13114-85-7_b.png)





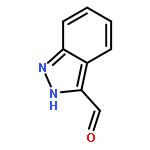
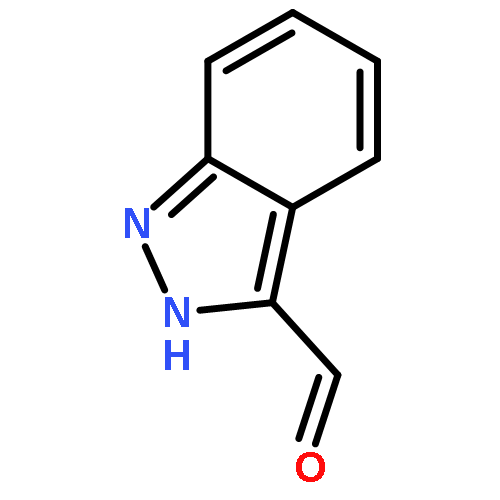
![N-{2-[(4-FLUOROBENZYL)AMINO]-2-OXOETHYL}-N-(2-METHOXYBENZYL)-1,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/5000/4981-92-4.png)
![N-{2-[(4-FLUOROBENZYL)AMINO]-2-OXOETHYL}-N-(2-METHOXYBENZYL)-1,2,3-THIADIAZOLE-4-CARBOXAMIDE](http://img.cochemist.com/ccimg/5000/4981-92-4_b.png)
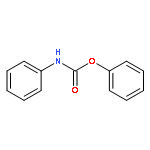
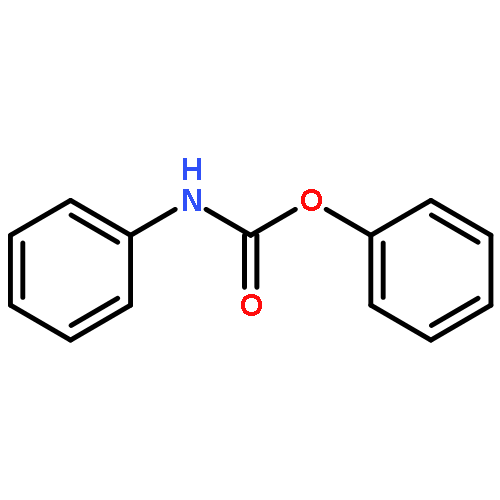
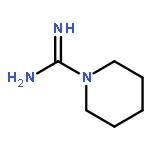

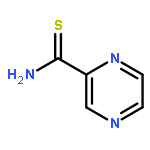

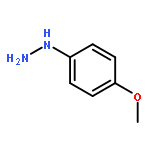
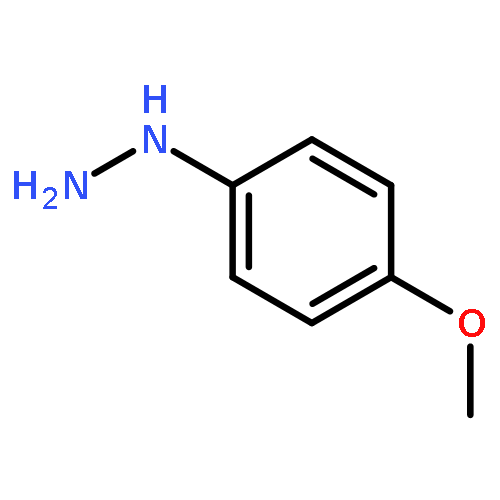


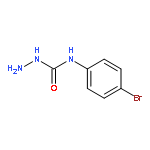
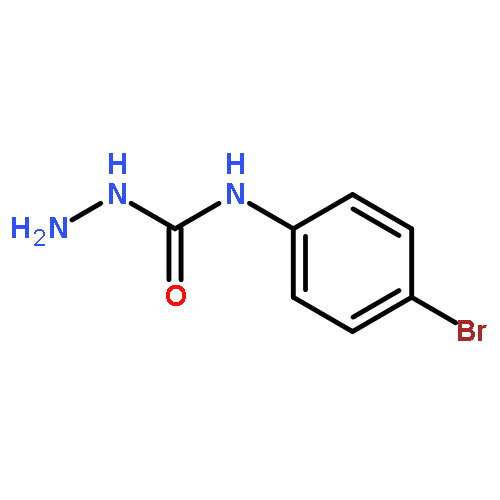





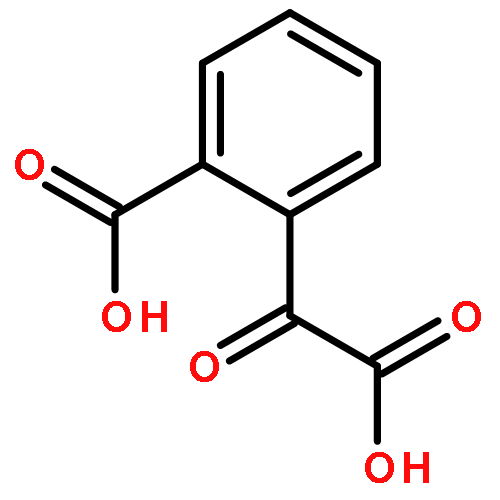
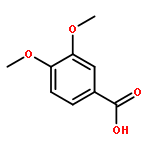
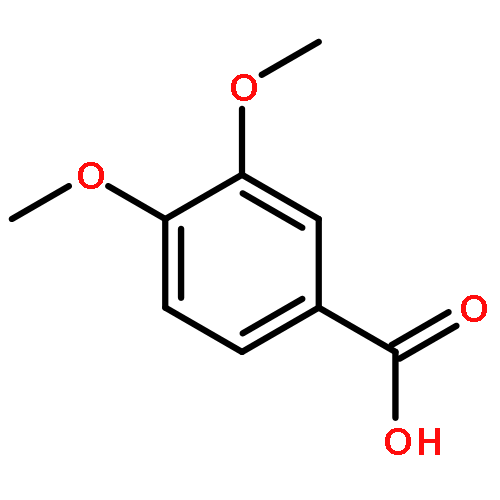

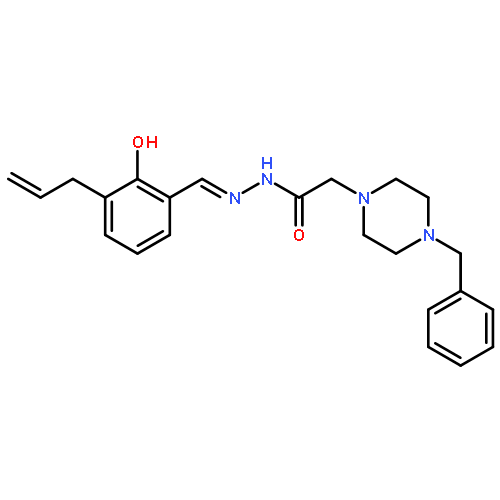
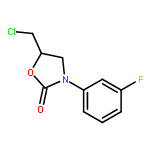
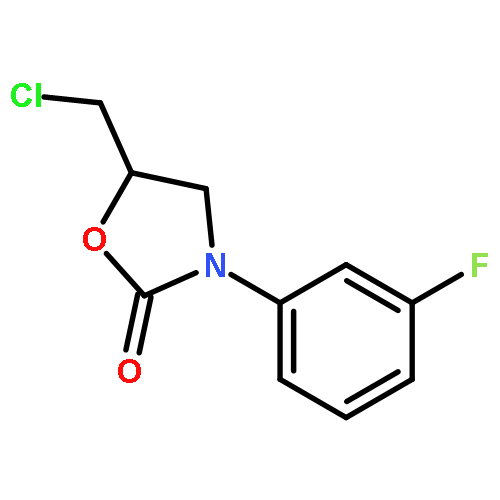
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole-2-carbaldehyde](http://img.cochemist.com/ccimg/537100/537010-34-7.png)
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole-2-carbaldehyde](http://img.cochemist.com/ccimg/537100/537010-34-7_b.png)
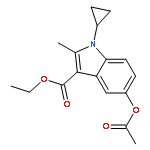


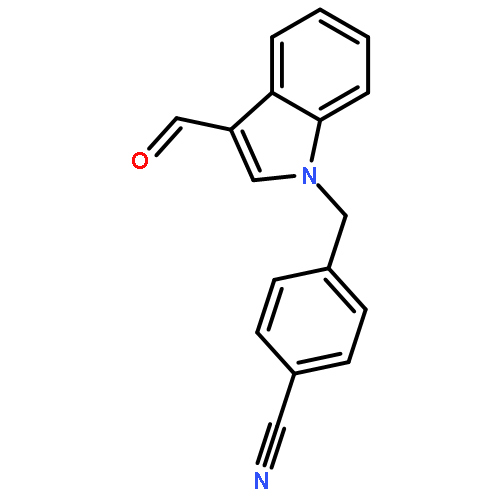



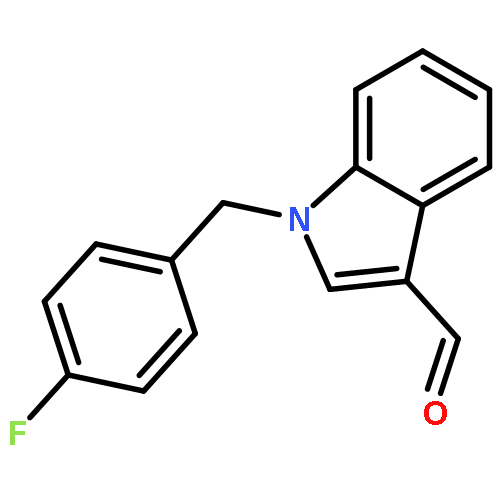




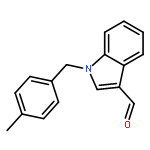



![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/107500/107456-42-8.png)
![1-(4-Chlorobenzyl)-1H-benzo[d]imidazole](http://img.cochemist.com/ccimg/107500/107456-42-8_b.png)

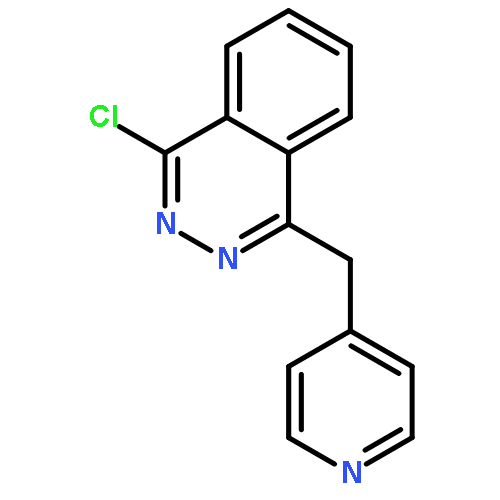

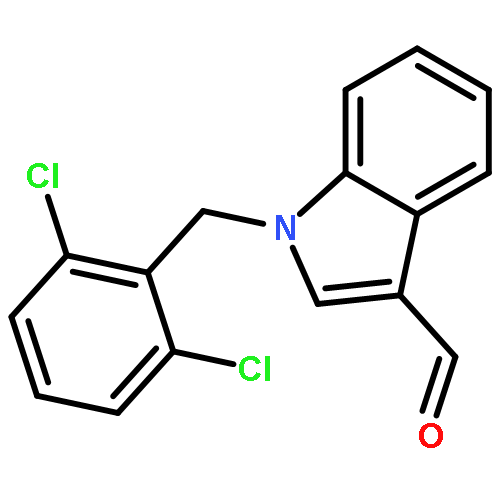
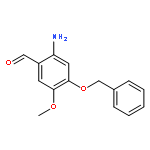
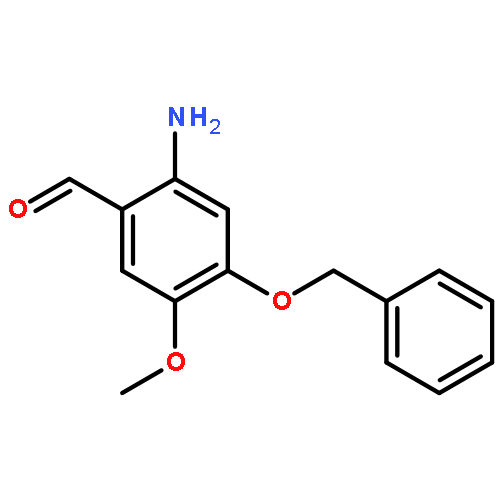




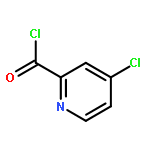





![3-[(4-fluorophenyl)thio]propanoic acid](http://img.cochemist.com/ccimg/19600/19543-85-2.png)
![3-[(4-fluorophenyl)thio]propanoic acid](http://img.cochemist.com/ccimg/19600/19543-85-2_b.png)
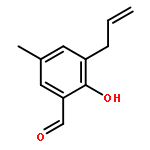
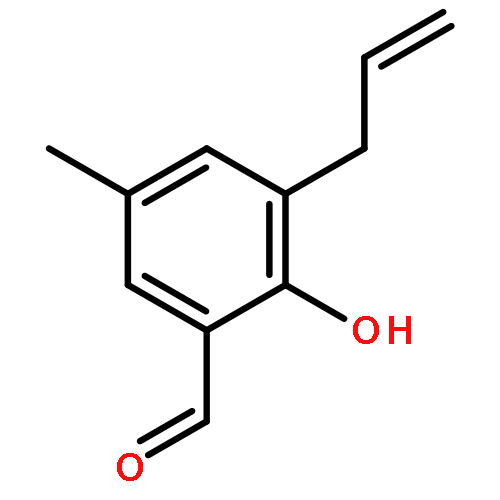
![Butanoic acid, 4-[(3-methoxyphenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/16800/16768-98-2.png)
![Butanoic acid, 4-[(3-methoxyphenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/16800/16768-98-2_b.png)


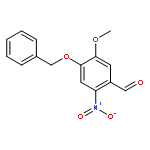





![Pyrazolo[1,5-a]pyrimidine-3-carbonitrile, 7-chloro-5-(chloromethyl)-](http://img.cochemist.com/ccimg/925700/925675-86-1.png)
![Pyrazolo[1,5-a]pyrimidine-3-carbonitrile, 7-chloro-5-(chloromethyl)-](http://img.cochemist.com/ccimg/925700/925675-86-1_b.png)
![Butanoic acid, 4-[(3,4-difluorophenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/886100/886059-29-6.png)
![Butanoic acid, 4-[(3,4-difluorophenyl)thio]-3-oxo-, ethyl ester](http://img.cochemist.com/ccimg/886100/886059-29-6_b.png)


![1-[(4-TERT-BUTYLPHENYL)METHYL]INDOLE-3-CARBALDEHYDE](http://img.cochemist.com/ccimg/405300/405274-91-1.png)
![1-[(4-TERT-BUTYLPHENYL)METHYL]INDOLE-3-CARBALDEHYDE](http://img.cochemist.com/ccimg/405300/405274-91-1_b.png)
![4-[(4-methylpiperidin-1-yl)methyl]aniline](http://img.cochemist.com/ccimg/262400/262368-64-9.png)
![4-[(4-methylpiperidin-1-yl)methyl]aniline](http://img.cochemist.com/ccimg/262400/262368-64-9_b.png)


![1H-Indole-3-carboxaldehyde,1-[(2-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/193000/192997-17-4.png)
![1H-Indole-3-carboxaldehyde,1-[(2-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/193000/192997-17-4_b.png)


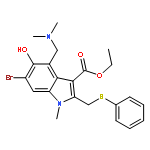
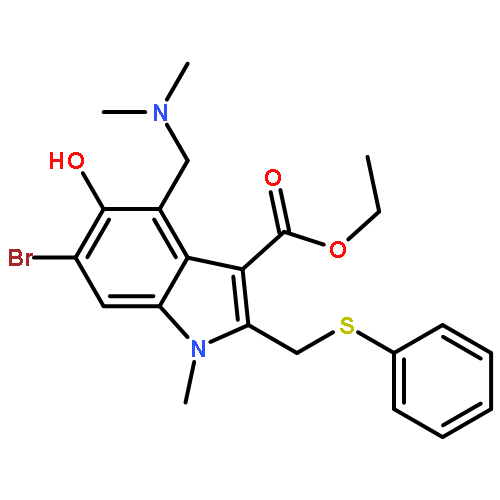

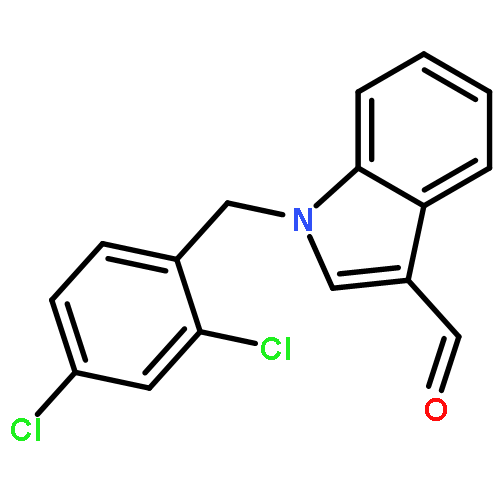
![1-[(3,4-dichlorophenyl)methyl]indole-3-carbaldehyde](http://img.cochemist.com/ccimg/90900/90815-02-4.png)
![1-[(3,4-dichlorophenyl)methyl]indole-3-carbaldehyde](http://img.cochemist.com/ccimg/90900/90815-02-4_b.png)
![ONCRASIN 1;1-[(4-CHLOROPHENYL)METHYL]-1H-INDOLE-3-CARBOXALDEHYDE](http://img.cochemist.com/ccimg/75700/75629-57-1.png)
![ONCRASIN 1;1-[(4-CHLOROPHENYL)METHYL]-1H-INDOLE-3-CARBOXALDEHYDE](http://img.cochemist.com/ccimg/75700/75629-57-1_b.png)


![Thieno[3,2-d]pyrimidine, 2-chloro-4-(4-morpholinyl)-](/data/chemimg/1041500/16234-15-4.png)
![Thieno[3,2-d]pyrimidine, 2-chloro-4-(4-morpholinyl)-](/data/chemimg/1041500/16234-15-4_b.png)
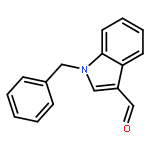



![Benzaldehyde, 4-[(4-chlorophenyl)methoxy]-2-hydroxy-](http://img.cochemist.com/ccimg/929300/929291-04-3.png)
![Benzaldehyde, 4-[(4-chlorophenyl)methoxy]-2-hydroxy-](http://img.cochemist.com/ccimg/929300/929291-04-3_b.png)

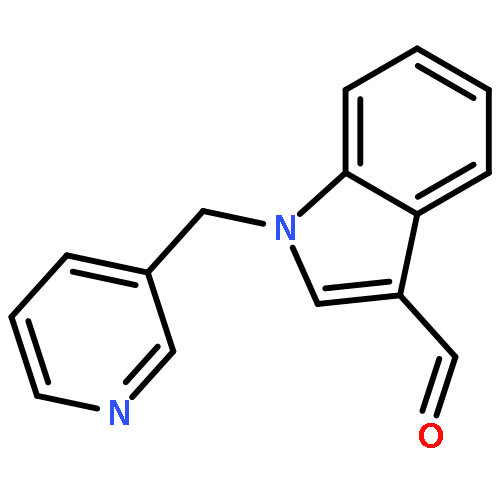
![BUTANOIC ACID, 4-[(4-FLUOROPHENYL)THIO]-3-OXO-, ETHYL ESTER](http://img.cochemist.com/ccimg/886100/886059-31-0.png)
![BUTANOIC ACID, 4-[(4-FLUOROPHENYL)THIO]-3-OXO-, ETHYL ESTER](http://img.cochemist.com/ccimg/886100/886059-31-0_b.png)










![QUINOLINE, 4-CHLORO-6-METHOXY-7-[3-(1-PIPERIDINYL)PROPOXY]-](http://img.cochemist.com/ccimg/861900/861881-05-2.png)
![QUINOLINE, 4-CHLORO-6-METHOXY-7-[3-(1-PIPERIDINYL)PROPOXY]-](http://img.cochemist.com/ccimg/861900/861881-05-2_b.png)
![Quinoline, 4-chloro-6-methoxy-7-[3-(4-morpholinyl)propoxy]-](http://img.cochemist.com/ccimg/205500/205448-32-4.png)
![Quinoline, 4-chloro-6-methoxy-7-[3-(4-morpholinyl)propoxy]-](http://img.cochemist.com/ccimg/205500/205448-32-4_b.png)