Co-reporter:Shiliang Li; Hongling Xu; Shichao Cui; Fangshu Wu; Youli Zhang; Mingbo Su; Yinghui Gong; Shaobing Qiu; Qian Jiao; Chun Qin; Jiwei Shan; Ming Zhang; Jiawei Wang; Qiao Yin; Minghao Xu; Xiaofeng Liu; Rui Wang; Lili Zhu; Jia Li; Yufang Xu; Hualiang Jiang; Zhenjiang Zhao; Jingya Li;Honglin Li
Journal of Medicinal Chemistry 2016 Volume 59(Issue 14) pp:6772-6790
Publication Date(Web):July 9, 2016
DOI:10.1021/acs.jmedchem.6b00505
Starting from the lead isodaphnetin, a natural product inhibitor of DPP-4 discovered through a target fishing docking based approach, a series of novel 2-phenyl-3,4-dihydro-2H-benzo[f]chromen-3-amine derivatives as potent DPP-4 inhibitors are rationally designed utilizing highly efficient 3D molecular similarity based scaffold hopping as well as electrostatic complementary methods. Those ingenious drug design strategies bring us approximate 7400-fold boost in potency. Compounds 22a and 24a are the most potent ones (IC50 ≈ 2.0 nM) with good pharmacokinetic profiles. Compound 22a demonstrated stable pharmacological effect. A 3 mg/kg oral dose provided >80% inhibition of DPP-4 activity within 24 h, which is comparable to the performance of the long-acting control omarigliptin. Moreover, the efficacy of 22a in improving the glucose tolerance is also comparable with omarigliptin. In this study, not only promising DPP-4 inhibitors as long acting antidiabetic that are clinically on demand are identified, but the target fish docking and medicinal chemistry strategies were successfully implemented.
Co-reporter:Wei Zhou, Shiliang Li, Weiqiang Lu, Jun Yuan, Yufang Xu, Honglin Li, Jin Huang and Zhenjiang Zhao
MedChemComm 2016 vol. 7(Issue 2) pp:292-296
Publication Date(Web):25 Nov 2015
DOI:10.1039/C5MD00469A
RSK2 (p90 ribosomal S6 kinase 2) is a serine/threonine kinase expressed in a variety of cancers. Molecular-targeted inhibition of RSK2 as a potential therapeutic strategy for human cancers has been documented. In this work, a series of isoindole-1,3-dione derivatives as novel RSK2 inhibitors were designed and synthesized from a hit discovered in our previous study. Some compounds were confirmed to be moderately potent RSK2 inhibitors with IC50 values of about 0.5 μM. Structure–activity relationship analysis and binding mode studies by molecular docking were performed.
Co-reporter:Liuqing Yang, Wei Liu, Hanbing Mei, Yuan Zhang, Xiaojuan Yu, Yufang Xu, Honglin Li, Jin Huang and Zhenjiang Zhao
MedChemComm 2015 vol. 6(Issue 4) pp:671-676
Publication Date(Web):05 Jan 2015
DOI:10.1039/C4MD00498A
Structure-based virtual screening of a commercial library identified pentanedioic acid derivatives (6 and 13b) as a kind of novel scaffold farnesyltransferase inhibitors (FTIs). Chemical modifications of the lead compounds, biological assays and analysis of the structure–activity relationships (SAR) were conducted to discover more potent FTIs. Some of them displayed excellent inhibition against FTase, and among them, the most active compound 13n with an IC50 value of 0.0029 μM and SAR analysis might be helpful to the discovery of more potent FTIs.
Co-reporter:Rui Gao, Sha Liao, Chen Zhang, Weilong Zhu, Liyan Wang, Jin Huang, Zhenjiang Zhao, Honglin Li, Xuhong Qian, Yufang Xu
European Journal of Medicinal Chemistry 2013 Volume 62() pp:597-604
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2013.01.030
In this study, starting from a lead compound discovered by virtual screening, a series of novel heterocyclic substituted benzenesulfonamides were designed and synthesized as new carbonic anhydrase IX (CA IX) inhibitors. Some compounds exhibited potent inhibitory effects against CA IX (in the low nanomolar range) as well as high selectivity against other carbonic anhydrase isozymes (CA I and CA II). The most potent and selective compound 27 could inhibit CA IX in the subnanomolar level with IC50 of 0.48 nM, which increased the potency by about 40-fold against CA IX compared with the lead compound 26, and presented more than 103 fold selectivity over CA I and CA II. The structure–activity relationship (SAR) based on the docking experiments further elucidated the effects of the compounds on the bioactivity and selectivity.Graphical abstractThrough structural optimization, a more potent and high selectivity CA IX inhibitor 27 was obtained.Highlights► Novel benzenesulfonamides CA IX inhibitors were designed and synthesized. ► Structure optimization was carried out on the basis of the lead compound. ► High activity and selectivity of compound 27 was studied and explained.
Co-reporter:Ye Zhong, Mengzhu Xue, Xue Zhao, Jun Yuan, Xiaofeng Liu, Jin Huang, Zhenjiang Zhao, Honglin Li, Yufang Xu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 7) pp:1724-1734
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmc.2013.01.047
Co-reporter:Qingshan Du, Weiping Zhu, Zhenjiang Zhao, Xuhong Qian, and Yufang Xu
Journal of Agricultural and Food Chemistry 2012 Volume 60(Issue 1) pp:346-353
Publication Date(Web):December 5, 2011
DOI:10.1021/jf203974p
Plant activators are a novel kind of agrochemicals that could induce resistance in many plants against a broad spectrum of diseases. To date, only few plant activators have been commercialized. In order to develop novel plant activators, a series of benzo-1,2,3-thiadiazole-7-carboxylate derivatives were synthesized, and the structures were characterized by 1H NMR, IR, elemental analyses, and HRMS or MS. Their potential systemic acquired resistance as plant activators was evaluated as well. Most of them showed good activity, especially, fluoro-containing compounds 3d and 3e, which displayed excellent SAR-inducing activity against cucumber Erysiphe cichoracearum and Colletotrichum lagenarium in assay screening. Field test results illustrated that compounds 3d and 3e were more potent than the commercial plant activator, S-methyl benzo[1,2,3]thiadiazole-7-carbothioate (BTH) toward these pathogens. Further, the preparation of compound 3d is more facile than BTH with lower cost, which will be helpful for further applications in agricultural plant protection.

![Benzenamine, 4-[2-(dimethylamino)ethoxy]-2-methoxy-](http://img.cochemist.com/ccimg/927700/927672-74-0.png)
![Benzenamine, 4-[2-(dimethylamino)ethoxy]-2-methoxy-](http://img.cochemist.com/ccimg/927700/927672-74-0_b.png)


![2-METHOXY-4-[4-(4-METHYLPIPERAZIN-1-YL)PIPERIDIN-1-YL]ANILINE](http://img.cochemist.com/ccimg/761500/761440-75-9.png)
![2-METHOXY-4-[4-(4-METHYLPIPERAZIN-1-YL)PIPERIDIN-1-YL]ANILINE](http://img.cochemist.com/ccimg/761500/761440-75-9_b.png)






![Benzoic acid, 3-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/61500/61471-37-2.png)
![Benzoic acid, 3-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/61500/61471-37-2_b.png)








![Benzoic acid, 4-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/200/122-92-9.png)
![Benzoic acid, 4-[[(aminothioxomethyl)hydrazono]methyl]-](http://img.cochemist.com/ccimg/200/122-92-9_b.png)




![Benzene, 1-chloro-2-[(2-chloro-4-nitrophenoxy)methyl]-3-fluoro-](http://img.cochemist.com/ccimg/867300/867288-06-0.png)
![Benzene, 1-chloro-2-[(2-chloro-4-nitrophenoxy)methyl]-3-fluoro-](http://img.cochemist.com/ccimg/867300/867288-06-0_b.png)
![Benzenamine, 3-chloro-4-[(2-chloro-6-fluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/867300/867288-04-8.png)
![Benzenamine, 3-chloro-4-[(2-chloro-6-fluorophenyl)methoxy]-](http://img.cochemist.com/ccimg/867300/867288-04-8_b.png)
![[1,1'-Biphenyl]-4-amine, 3,5-difluoro-3'-methoxy-](http://img.cochemist.com/ccimg/867300/867288-00-4.png)
![[1,1'-Biphenyl]-4-amine, 3,5-difluoro-3'-methoxy-](http://img.cochemist.com/ccimg/867300/867288-00-4_b.png)

![Methanesulfonamide, N,N-bis[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/852400/852358-71-5.png)
![Methanesulfonamide, N,N-bis[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/852400/852358-71-5_b.png)


![1H-Isoindole-1,3(2H)-dione, 2-[[(4-hydroxyphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/313500/313493-98-0.png)
![1H-Isoindole-1,3(2H)-dione, 2-[[(4-hydroxyphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/313500/313493-98-0_b.png)


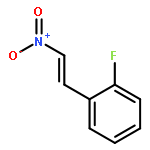
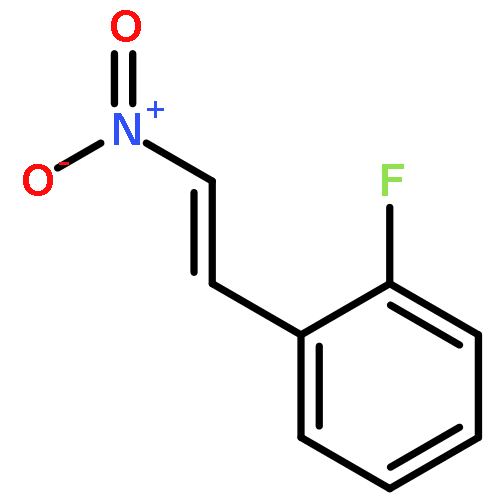
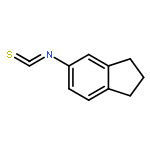



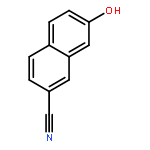



![[1,1'-Biphenyl]-4-amine, 3-fluoro-](http://img.cochemist.com/ccimg/76400/76302-56-2.png)
![[1,1'-Biphenyl]-4-amine, 3-fluoro-](http://img.cochemist.com/ccimg/76400/76302-56-2_b.png)


















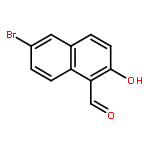
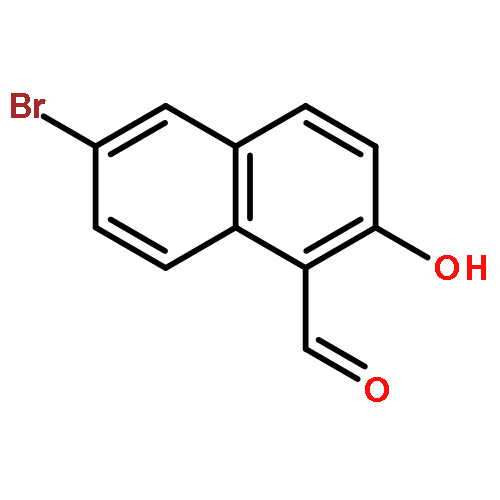


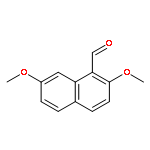
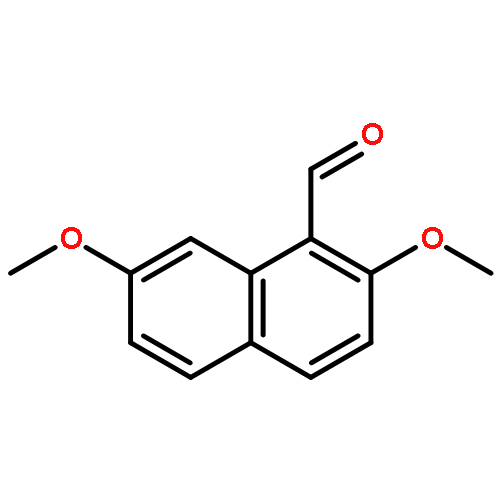
![[(8S)-5,5,8-TRIMETHYLDECALIN-2-YL] ACETATE](http://img.cochemist.com/ccimg/34800/34762-56-6.png)
![[(8S)-5,5,8-TRIMETHYLDECALIN-2-YL] ACETATE](http://img.cochemist.com/ccimg/34800/34762-56-6_b.png)

![1H-Isoindole-1,3(2H)-dione, 2-[[(4-methylphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/30900/30818-66-7.png)
![1H-Isoindole-1,3(2H)-dione, 2-[[(4-methylphenyl)amino]methyl]-](http://img.cochemist.com/ccimg/30900/30818-66-7_b.png)


![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6.png)
![1-chloro-2-[(e)-2-nitrovinyl]benzene](http://img.cochemist.com/ccimg/22600/22568-07-6_b.png)

![Thiourea,N-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/19300/19250-03-4.png)
![Thiourea,N-[1,1'-biphenyl]-4-yl-](http://img.cochemist.com/ccimg/19300/19250-03-4_b.png)