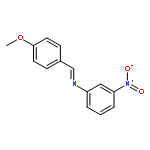
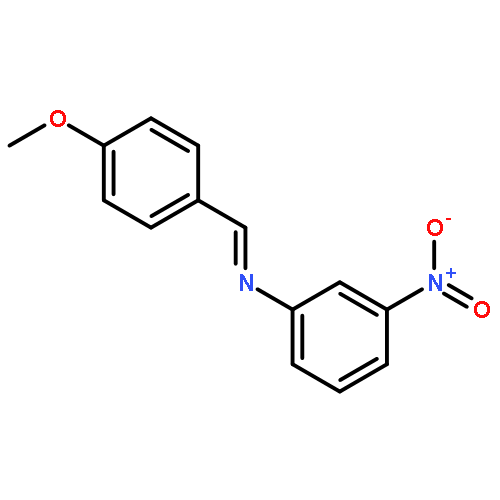
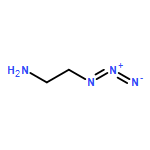
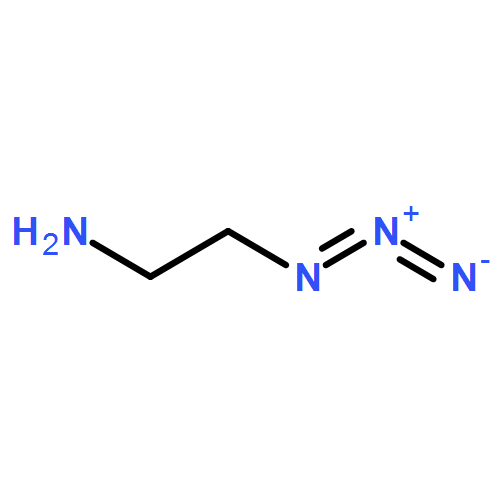
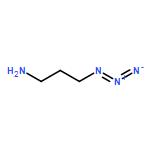
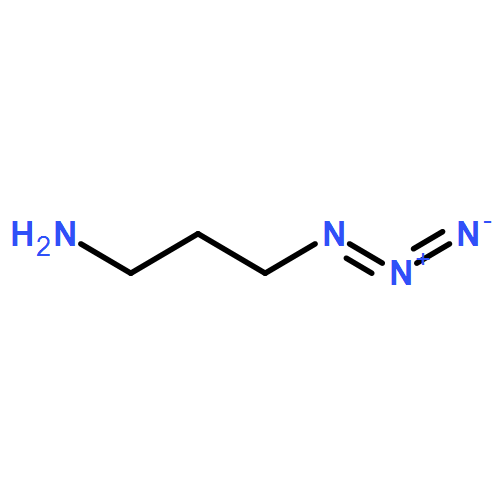
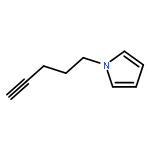
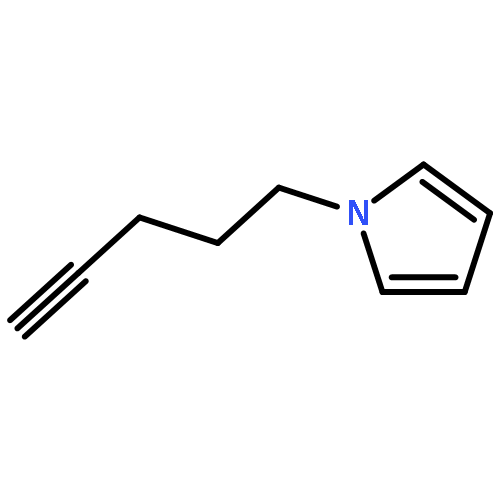
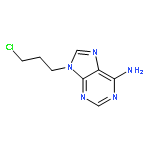
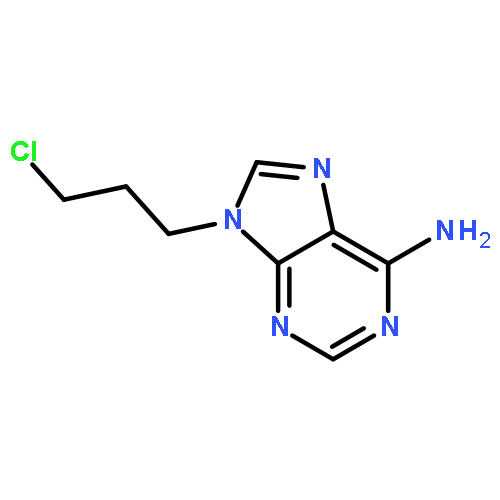
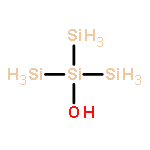
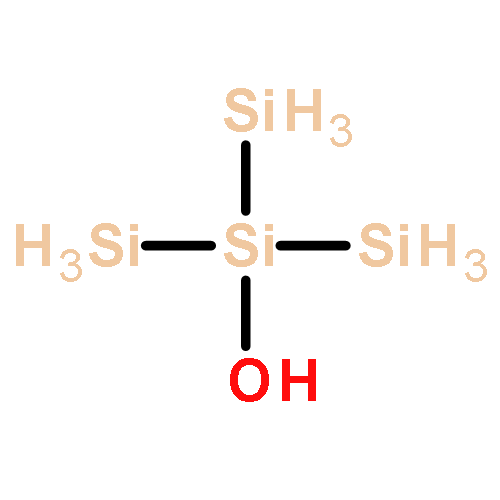
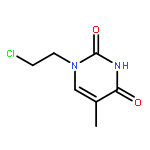
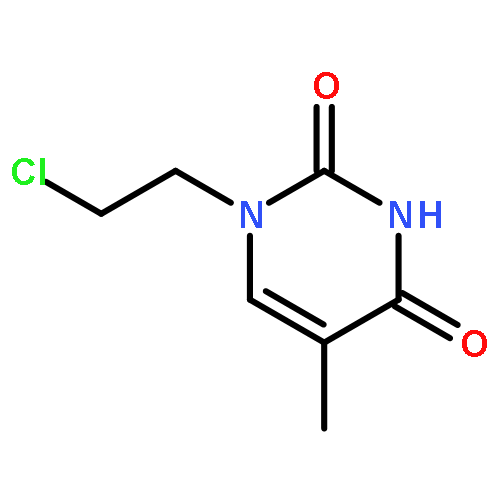
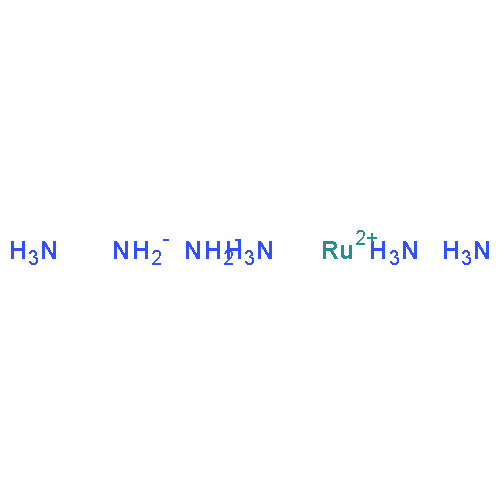
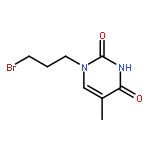
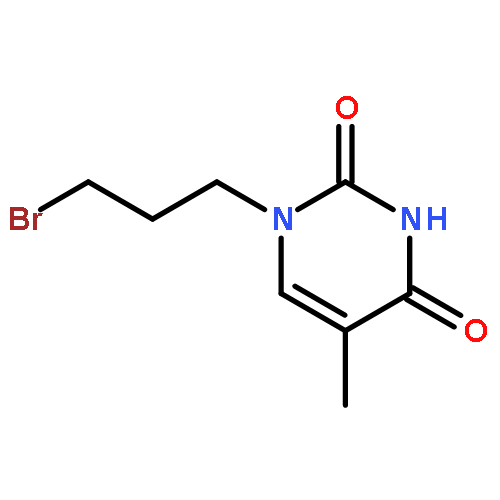
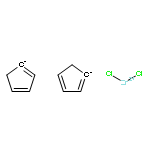
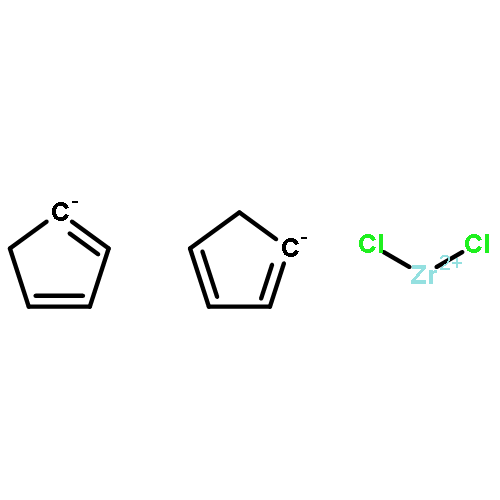
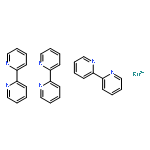
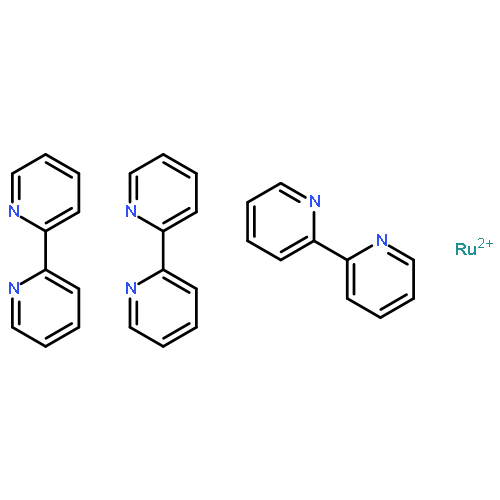
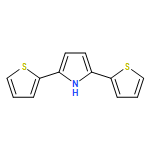
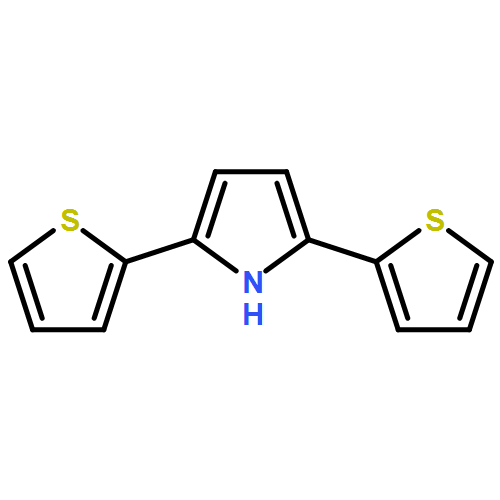
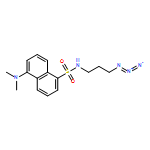
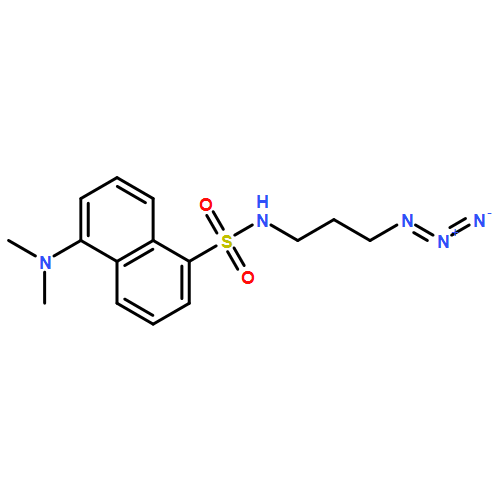
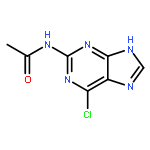
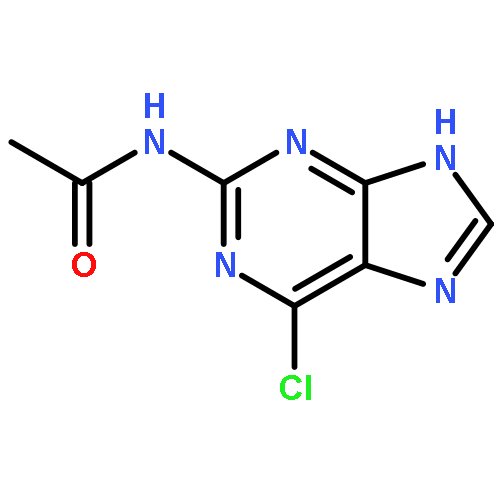
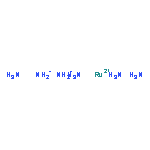Co-reporter:Glenn Lamming, James Kolokotroni, Thomas Harrison, Thomas J. Penfold, William Clegg, Paul G. Waddell, Michael R. Probert, and Andrew Houlton
Crystal Growth & Design November 1, 2017 Volume 17(Issue 11) pp:5753-5753
Publication Date(Web):July 24, 2017
DOI:10.1021/acs.cgd.7b00752
A series of new one-dimensional coordination polymer materials, based on Ag(I)–N bond formation, has been synthesized and structurally characterized by single crystal X-ray diffraction. Reactions between the poly-monodentate ligands based on (1E,1′E′)-N,N′-(-bis(1-pyridin-3-yl)methanimine and Ag(I) salts give products that feature simple coordination chains or metallacyclic- and tape-based structures. For the simple chains these are as either isolated units, or assembled in dimeric and tetrameric arrangements through intermetallic, argentophilic interactions. However, crystal packing effects and solvent inclusion are found to readily disrupt this type of bonding. Density functional theory calculations provide an assessment of the bond order and the influence of anion binding on these interactions.
Co-reporter:Glenn Lamming, Osama El-Zubir, James Kolokotroni, Christopher McGurk, Paul G. Waddell, Michael R. Probert, and Andrew Houlton
Inorganic Chemistry 2016 Volume 55(Issue 19) pp:9644-9652
Publication Date(Web):September 15, 2016
DOI:10.1021/acs.inorgchem.6b01365
A series of new two-dimensional coordination framework materials, based on Ag(I)–N bond formation, has been synthesized and structurally characterized by single crystal methods. Reactions between the poly-monodentate bridging ligand N,N′-((1r,4r)-cyclohexane-1,4-diyl)bis(1-(pyridin-3-yl)methanimine), L1, and silver salts yield compounds {[Ag(L1)(MeCN)](CF3SO3–)}n, 1, {[Ag(L1)(PF2O2–)]·H2O}n, 2, and {Ag2(L1)(tosylate)2}n, 3. The frameworks of these materials exhibit two distinct net topologies: 36.46.53 (1 and 2) and 44.62 (3). In all cases, L1 ligands are found to be fully saturated, in terms of metal ion binding, with both sets of pyridyl and imino N atoms involved, though in 1 and 2, crystallographically independent L1 moieties also display pyridyl-only binding. Either solvent (1) or the anion (2 and 3) acts as a terminal ligand to support interlayer interactions in the solid state. For 2 and 3 the molecular sheet orientation lies in the plane of the largest crystal face, indicating that crystal growth is preferentially driven by coordinate bond formation. Despite the relatively labile nature, typical of such Ag(I)–N bonds, solvent-based exfoliation of crystals of 3 was shown to provide dispersions of large, μm2, flakes which readily deposit on oxide surfaces as single-molecule sheets, as revealed by atomic force microscopy.
Co-reporter:Hasan Daw A. Mohamed, Scott M. D. Watson, Benjamin R. Horrocks and Andrew Houlton
Journal of Materials Chemistry A 2015 vol. 3(Issue 2) pp:438-446
Publication Date(Web):19 Nov 2014
DOI:10.1039/C4TC02307B
Two methods for the preparation of rhodium nanowires are described: (i) electroless metal deposition at duplex DNA ‘template’ molecules in bulk solution and (ii) electrochemical reduction in DNA-containing solution at a modified electrode. Both methods render essentially similar 1D nanostructures with a Rh/Rh-oxide core–shell structure. AFM studies revealed the resulting nanostructures are typically <10 nm in diameter with continuous and smooth metal coatings. However, the latter method was less effective with samples containing an ∼3-fold increase in the bare template DNA remaining. A combination of SPM methods demonstrated the structures to be electrically conducting, hence confirming their nanowire nature. The conductivity was, however, several orders of magnitude lower than that of bulk Rh; a fact attributed to the presence of resistance-increasing mechanisms, such as grain boundaries present in the Rh coatings and electron surface scattering.
Co-reporter:Scott M. D. Watson ; Miguel A. Galindo ; Benjamin R. Horrocks
Journal of the American Chemical Society 2014 Volume 136(Issue 18) pp:6649-6655
Publication Date(Web):April 8, 2014
DOI:10.1021/ja500439v
Details of the mechanism of formation of supramolecular polymer nanowires by templating on DNA are revealed for the first time using AFM. Overall these data reveal that the smooth, regular, structures produced are rendered by highly dynamic supramolecular transformations occurring over the micrometre scale. In the initial stages of the process a low density of conducting polymer (CP) binds to the DNA as, essentially, spherical particles. Further reaction time produces DNA strands which are more densely packed with particles giving a beads-on-a-string appearance. The particles subsequently undergo dynamic reconfiguration so as to elongate along the template axis and merge to yield the highly regular, smooth morphology of the final nanowire. MD simulations illustrate the early stages of the process showing the binding of globular CP to duplex DNA, while the latter stages can be modeled effectively by a linear thermodynamic description based on the balance between the line energy, which accounts for adhesion of the material to the template, and its surface tension. This model accounts for the phenomena observed in the AFM studies: the relative success of DNA templating of polymers compared to metals; the slow approach to equilibrium; and the observed thinning and ‘necking’ phenomena as the structures transform from beads-on-a-string to smooth nanowire.
Co-reporter:Jonathan Pate, Felix Zamora, Scott M. D. Watson, Nicholas G. Wright, Benjamin R. Horrocks and Andrew Houlton
Journal of Materials Chemistry A 2014 vol. 2(Issue 43) pp:9265-9273
Publication Date(Web):25 Sep 2014
DOI:10.1039/C4TC01632G
Templating the electroless reduction of metal ions on DNA is now an established route to the preparation of nanowires and can be particularly useful for the formation of nanowires in the desirable <10 nm size range. However, different preparation conditions produce nanowires of widely different morphologies and conductivities. We describe a method for the synthesis of Cu nanowires in which electroless metal deposition is carried out on DNA ‘template’ molecules in bulk solution. Though analogous to previous surface-based routes, importantly this now produces conductive material. AFM was used to evaluate the size and morphology of the resulting nanowires; a mean nanowire diameter of 7.1 nm (standard deviation = 4.7 nm) was determined from a statistical analysis of 100 nanowires and the Cu coatings were continuous and smooth. These findings represent a notable improvement in nanowire morphology in comparison to the previous surface-based routes. UV-vis spectroscopy, X-ray diffraction, and X-ray photoelectron spectroscopy were used to confirm formation of Cu(0) metal takes place during nanowire synthesis, and additional scanning probe microscopy techniques were employed to probe the electrical properties of the nanowires. The nanowires are less conductive [resistivity ∼ 2 Ω cm] than bulk Cu, but much more conductive than nanowires prepared by the analogous method on surface-bound DNA. Using an extension of our thermodynamic model for DNA-templating, we show that the templating process in bulk solution favours the formation of continuous nanowires compared to templating on surface-bound DNA.
Co-reporter:Scott M. D. Watson, Hasan Daw A. Mohamed, Benjamin R. Horrocks and Andrew Houlton
Nanoscale 2013 vol. 5(Issue 12) pp:5349-5359
Publication Date(Web):24 Apr 2013
DOI:10.1039/C3NR00716B
The fabrication of electrically conducting magnetic nanowires has been achieved using electrochemical DNA-templating of iron. In this approach, binding of the Fe2+ cations to the DNA “template” molecules has been utilised to promote growth along the molecular axis. Formation of Fe within the product material was verified by XRD and XPS studies, which confirmed an iron/oxide “core–shell” structure. The effectiveness of the DNA duplex to direct the metal growth in one dimension was highlighted by AFM which reveals the product material to comprise high aspect ratio nanostructured architectures. These “nanowires” were observed to have morphologies consisting of densely packed linear arrangements of metal particles along the template, with wire diameters up to 26 nm. The structures were confirmed to be electrically conductive, as expected for such Fe-based materials, and to display superparamagnetic behaviour, consistent with the small size and particulate nature of the nanowires.
Co-reporter:Pilar Amo-Ochoa, Oscar Castillo, Ross W. Harrington, Félix Zamora, and Andrew Houlton
Inorganic Chemistry 2013 Volume 52(Issue 4) pp:2174-2181
Publication Date(Web):February 1, 2013
DOI:10.1021/ic302602c
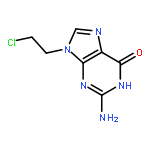
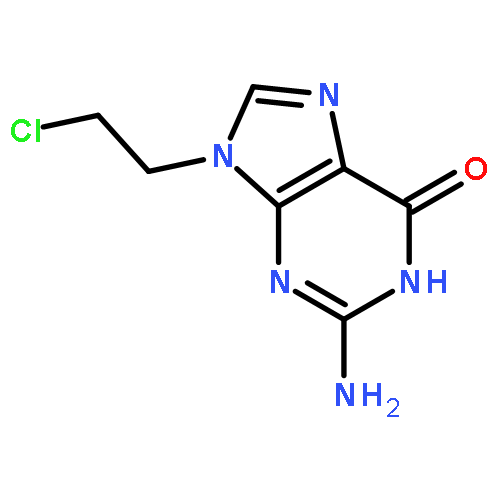
Reactions between [Rh2(CH3COO)4] with 2,6-diaminopurine (HDap) or 6-chloro-2-aminopurine (HClap) and [Rh2((CH3)3CCOO)4] with HClap produce, three new dirhodium(II) carboxylate complexes of the general form, [Rh2(RCOO)4(Purine)2] (R= CH3, (CH3)3C). Single crystal X-ray diffraction studies confirm that in all cases the purine coordinates to the axial position of the dirhodium(II)tetracarboxylate unit. However, while the complex obtained with HDap features the typical purine binding mode via N(7), complexes containing HClap show unusual N3 coordination. This is an extremely rare instance of an unrestricted purine binding via N3. Some rationalization of these data is offered based on a series of DFT calculations.
Co-reporter:Hasan Daw A. Mohamed, Scott M. D. Watson, Benjamin R. Horrocks and Andrew Houlton
Nanoscale 2012 vol. 4(Issue 19) pp:5936-5945
Publication Date(Web):02 Aug 2012
DOI:10.1039/C2NR31559A
The synthesis of nanowires made of magnetite (Fe3O4) phase iron oxide was achieved using DNA as a template to direct formation of the metal oxide and confine its growth in two dimensions. This simple solution-based approach involves initial association of Fe2+ and Fe3+ to the DNA “template” molecules, and subsequent co-precipitation of the Fe3O4 material, upon increasing the solution pH, to give the final metal oxide nanowires. Analysis of the DNA-templated material, using a combination of FTIR, XRD, XPS, and Raman spectroscopy, confirmed the iron oxide formed to be the Fe3O4 crystal phase. Investigation of the structural character of the nanowires, carried out by AFM, revealed the metal oxide to form regular coatings of nanometre-scale thickness around the DNA templates. Statistical analysis showed the size distribution of the nanowires to follow a trimodal model, with the modal diameter values identified as 5–6 nm, 14–15 nm, and 23–24 nm. Additional scanning probe microscopy techniques (SCM, MFM) were also used to verify that the nanowire structures are electrically conducting and exhibit magnetic behaviour. Such properties, coupled with the small dimensions of these materials, make them potentially good candidates for application in a host of future nanoscale device technologies.
Co-reporter:Dr. Scott M. D. Watson;Joseph H. Hedley;Dr. Miguel A. Galindo;Dr. Said A. F. Al-Said; Nick G. Wright; Bernard A. Connolly;Dr. Benjamin R. Horrocks; Andrew Houlton
Chemistry - A European Journal 2012 Volume 18( Issue 38) pp:12008-12019
Publication Date(Web):
DOI:10.1002/chem.201201495
Abstract
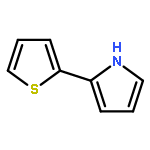
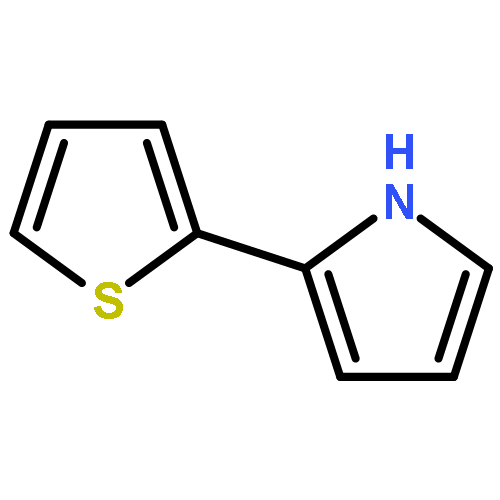
Supramolecular polymer nanowires have been prepared by using DNA-templating of 2,5-(bis-2-thienyl)-pyrrole (TPT) by oxidation with FeCl3 in a mixed aqueous/organic solvent system. Despite the reduced capacity for strong hydrogen bonding in polyTPT compared to other systems, such as polypyrrole, the templating proceeds well. FTIR spectroscopic studies confirm that the resulting material is not a simple mixture and that the two types of polymer interact. This is indicated by shifts in bands associated with both the phosphodiester backbone and the nucleobases. XPS studies further confirm the presence of DNA and TPT, as well as dopant Cl− ions. Molecular dynamics simulations on a [{dA24:dT24}/{TPT}4] model support these findings and indicate a non-coplanar conformation for oligoTPT over much of the trajectory. AFM studies show that the resulting nanowires typically lie in the 7–8 nm diameter range and exhibit a smooth, continuous, morphology. Studies on the electrical properties of the prepared nanowires by using a combination of scanned conductance microscopy, conductive AFM and variable temperature two-terminal I–V measurements show, that in contrast to similar DNA/polymer systems, the conductivity is markedly reduced compared to bulk material. The temperature dependence of the conductivity shows a simple Arrhenius behaviour consistent with the hopping models developed for redox polymers.
Co-reporter:Miguel A. Galindo, Jennifer Hannant, Ross W. Harrington, William Clegg, Benjamin R. Horrocks, Andrew R. Pike and Andrew Houlton
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 5) pp:1555-1564
Publication Date(Web):12 Nov 2010
DOI:10.1039/C0OB00466A
A series of modified nucleosides based on thymidine have been prepared by Pd-catalysed cross-coupling between N-alkyl-alkynyl functionalised pyrrolyl- (py), 2-(2-thienyl)pyrrolyl- (tp) or 2,5-bis(2-thienyl)pyrrolyl (tpt) groups with 5-iodo-2′-deoxyuridine. The length of the alkyl chain linking the nucleoside and pyrrolyl-containing unit, N(CH2)nCC-nucleoside (where n = 1–3) was also varied. The compounds have been characterised by 1H NMR, ES-MS, UV–vis, cyclic voltammetry (CV) and, in some cases, single-crystal X-ray diffraction. Cyclic voltammetry studies demonstrated that all the py-, tp- and tpt-alkynyl derivatives 1–7 can be electrochemically polymerised to form conductive materials. It was found that increasing the N-alkyl chain length in these cases resulted in only minor changes in the oxidation potential. The same behaviour was observed for the tp- and tpt-modified nucleosides 9–12; however, the py-derivative, 8, produced a poorly conducting material. DFT calculations on the one-electron oxidised cation of the modified nucleosides bearing tp or tpt showed that spin density is located on the pyrrolyl and thienyl units in all cases and that the coplanarity of adjacent rings increases upon oxidation. In contrast, in the corresponding pyrrolyl cases the spin density is distributed over the whole molecule, suggesting that polymerisation does not occur solely at the pyrrolyl-Cα position and the conjugation is interrupted.
Co-reporter:Scott M. D. Watson, Nicholas G. Wright, Benjamin R. Horrocks and Andrew Houlton
Langmuir 2010 Volume 26(Issue 3) pp:2068-2075
Publication Date(Web):September 16, 2009
DOI:10.1021/la902583j
The synthesis of one-dimensional metal nanostructures can be achieved through the use of DNA molecules as templates to control and direct metal deposition. Copper nanostructures have been fabricated using this strategy, through association of Cu2+ ions to DNA templates and reduced with ascorbic acid. Due to the possibility that the reduction of the Cu2+ can result in the preferential formation of Cu2O over metallic Cu0, X-ray photoelectron spectroscopy and X-ray diffraction have been carried out to establish the chemical identity of the nanostructures. Conclusive evidence is found that reduction of the Cu2+ ions does result in the formation of the desired metallic Cu0 structures. The morphology of the nanostructured Cu0 material has also been observed by atomic force microscopy, showing the structures to have a “beads-on-a-string” appearance and being 3.0−5.5 nm in height. The electrical properties of the structures have been investigated by scanned conductance microscopy, showing the Cu0 structures exhibit much larger electrical resistance than expected for a metallic nanowire. This is thought to be a consequence of their “beads-on-a-string” morphology and small lateral dimensions (sub-10 nm); both these factors would be expected to increase the electron scattering rate, and, further, there are likely to be significant tunneling barriers at the Cu0 particle−particle junctions.
Co-reporter:Andrew Houlton, Andrew R. Pike, Miguel Angel Galindo and Benjamin R. Horrocks
Chemical Communications 2009 (Issue 14) pp:1797-1806
Publication Date(Web):18 Feb 2009
DOI:10.1039/B818456A
The controlled preparation and assembly of opto-electronic nanoscale materials is being tackled by top-down and bottom-up approaches. The latter draws inspiration from biology, where complex hierarchical systems are assembled from simpler building blocks. One of these, DNA, is proving especially useful: its size, stability, topology; the assorted chemical functional groups; plus its capacity for self-assembly provide a powerful nanoscale toolbox for materials preparation. Here we review recent research that shows the roles DNA can play in the preparation and organisation of semiconductor nanomaterials. Studies show that both hard inorganic and soft polymer materials can be directed to grow at nanoscale lengths using DNA and its constituents. In some cases the resulting materials have been used as components in simple electrical devices and the methodology has been extended to analytical tools. Intriguingly, these DNA–semiconductor hybrid materials have been found to self-assemble themselves, forming highly regular rope-like assemblies and conducting network structures.
Co-reporter:Miguel A. Galindo, David Amantia, William Clegg, Ross W. Harrington, Richard J. Eyre, Jonathon P. Goss, Patrick R. Briddon, William McFarlane and Andrew Houlton
Chemical Communications 2009 (Issue 20) pp:2833-2835
Publication Date(Web):02 Mar 2009
DOI:10.1039/B817329J
The tetrahedral bis(adeninyl)–Cu(I) complex, 1, self-associates in polar solvent through complementary hydrogen-bonding interactions and appears to mimic the natural assembly of duplex DNA.
Co-reporter:Miguel A. Galindo, David Amantia, Alberto Martinez Martinez, William Clegg, Ross W. Harrington, Virtudes Moreno Martinez and Andrew Houlton
Inorganic Chemistry 2009 Volume 48(Issue 21) pp:10295-10303
Publication Date(Web):October 2, 2009
DOI:10.1021/ic9013448
The effect of the 2-amino group on metal ion binding at the N3-position of a purine base has been investigated using chelate-tethered derivatives. Reactions of diamine-tethered 2,6-diaminopurine (DAP) with divalent d-block metal ions Cu(II) and Cd(II) confirm that binding can occur, but this is much less prevalent than with adenine. In this regard DAP is similar to guanine where we have previously observed a general lack of N3-binding by divalent metal ions compared to adenine (e.g., Houlton et al., Angew. Chem., Int. Ed. 2000, 39, 2360; Chem.—Eur. J. 2000, 6, 4371). For the univalent d-block metals ions, Cu(I) and Ag(I), binding to adenine N3 is not observed in the solid state, as shown by reactions with dithioether-tethered adenine derivatives. Instead, depending on stoichiometry of the reaction, discrete (with metal/ligand ratio 1:2) or polymeric (with metal/ligand ratio 1:1) complexes were isolated and characterized by single crystal X-ray methods. In the former the nucleobases are pendant and involved in base-pair interactions, with both Watson−Crick···Watson−Crick and Hoogsteen···Hoogsteen type pairings present. For the coordination polymers a rather unexpected influence of the tether length on the site of nucleobase binding is found for bridging ligand binding modes involving the chelating diamine and the adeninyl group. Polymer chains derived with the shorter ethyl tether show binding at the N7 site of adeninyl, while binding at N1 is found in the longer propyl chain length.
Co-reporter:Miguel A. Galindo ; David Amantia ; Alberto Martinez-Martinez ; William Clegg ; Ross W. Harrington ; Virtudes Moreno Martinez
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11085-11091
Publication Date(Web):October 26, 2009
DOI:10.1021/ic901475y
Alkyldiamine-tethered derivatives of 2,6-diaminopurine, ethylenediamine-N9-propyl-2,6-diaminopurine, L1, and ethylenediamine-N9-ethyl-2,6-diaminopurine, L2, react with Pd(II) to give N3-coordinated complexes. However, the exact nature of the resulting complex is dependent on the reaction conditions. With PdCl2(MeCN)2 in MeCN/H2O the expected [PdCl(N3-2,6-DAP-alkyl-en)]+ complex, 1, is formed with L1 chelating the metal center in a tridentate manner through the diamine function and N3 of the purine base. However, under the same conditions the shorter, ethyl-tethered, L2 gives a complex dication, 2, containing a tetradentate ligand forming simultaneously 5-, 6-, and 7-membered chelate rings. This resulting acetamidine, derived by addition to coordinated MeCN, appears to be the first such case involving the 2-amino group of a purine. The ethyl-analogue of 1, [PdCl(N3-2,6-DAP-Et-en)]+ 3, was prepared by reaction of L2 with K2PdCl4 in aqueous media.
Co-reporter:Miguel A. Galindo, Andrew Houlton
Inorganica Chimica Acta 2009 Volume 362(Issue 3) pp:625-633
Publication Date(Web):20 February 2009
DOI:10.1016/j.ica.2008.04.049
Over the last decade chelate-tethered nucleobases have been explored as models for metal ion–DNA interactions as well as to extend the molecular architecture of metal complexes capable of base-pair hydrogen bonding. Here we present an overview of the area.An overview of the coordination chemistry of chelate-tethered nucleobases is presented detailing uses as models for metal ion–DNA interactions as well as to examples which extend the molecular architecture of metal–nucleobase complexes capable of base-pair hydrogen bonding.
Co-reporter:Miguel A. Galindo, Andrew Houlton, William Clegg, Ross W. Harrington, José Dobado, Francisco Santoyo-Gonzalez, Fatima Linares, M. Angustias Romero and Jorge A. R. Navarro
Chemical Communications 2008 (Issue 32) pp:3735-3737
Publication Date(Web):18 Jun 2008
DOI:10.1039/B805705B
The cyclic trinuclear system, [(en)3Pd3(4,7-phen)3]6+, undergoes a ligand exchange reaction with 5-R-2-hydroxypyrimidine derivatives (HRpymo; R = ethynylferrocene, 5-(dimethylamino)-N-(2-propynyl)-1-naphthalene sulfonamide) to give [(en)3Pd3(4,7-phen)2(Rpymo)]5+, functional supramolecular receptors of mononucleotides.
Co-reporter:T. Hollis;L. Dong;B. A. Connolly;N. G. Wright;B. R. Horrocks;A. Houlton
Advanced Materials 2007 Volume 19(Issue 13) pp:1748-1751
Publication Date(Web):6 JUN 2007
DOI:10.1002/adma.200602543
Highly organized CdS growth by DNA templating is used to produce either linear arrangements of Q-particles along the DNA strands or continuous conducting nanowires (see figure). By optimizing the reaction conditions, the potential of biopolymers in general and DNA in particular for producing nanometer-scale electronics is exemplified.
Co-reporter:Liqin Dong Dr.;Tom Hollis;Steven Fishwick;Bernard A. Connolly ;Nicholas G. Wright ;Benjamin R. Horrocks Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 3) pp:
Publication Date(Web):11 DEC 2006
DOI:10.1002/chem.200601320
The synthesis of supramolecular conducting nanowires can be achieved by using DNA and pyrrole. Oxidation of pyrrole in DNA-containing solutions yields a material that contains both the cationic polypyrrole (PPy) and the anionic DNA polymers. Intimate interaction of the two polymer chains in the self-assembled nanowires is indicated by FTIR spectroscopy. AFM imaging shows individual nanowires to be continuous, ≈5 nm high and conformationally flexible. This feature allows them to be aligned by molecular combing in a similar manner to bare DNA and provides a convenient method for fabricating a simple electrical device by stretching DNA/PPy strands across an electrode gap. Current–voltage measurements confirm that the nanowires are conducting, with values typical for a polypyrrole-based material. In contrast to polymerisation of pyrrole on a DNA template in bulk solution, attempts to form similar wires by polymerisation at surface-immobilised DNA do not give a continuous coverage; instead, a beads-on-a-string appearance is observed suggesting that immobilisation inhibits the assembly process.
Co-reporter:Marta Morell Cerdà, David Amantia, Burkhard Costisella, Andrew Houlton and Bernhard Lippert
Dalton Transactions 2006 (Issue 32) pp:3894-3899
Publication Date(Web):26 Jun 2006
DOI:10.1039/B603650C
Simultaneous metal coordination to N7 (PtII) and N3 (PdII) of N9-blocked guanine leads to a 104 fold acidification of the guanine-N(1)H position and hence to a virtual complete deprotonation of the N(1)H position at neutral pH. The chelate-tethered nucleobase ethylenediamine-N9-ethylguanine was employed and relevant acid–base equilibria were studied by pD dependent 1H NMR spectroscopy. CH2 resonances of the tether were assigned on the basis of NOESY and COSY experiments. Our findings suggest a plausible method of formation of a previously reported trinuclear PtII complex of 9-ethylguanine with metals coordinated to N1, N3 and N7. According to this, a sequence with the first metal binding to N7, the second one binding to N3, and only the third one binding to N1 with deprotonation of this site is proposed.
Co-reporter:David Amantia, Michelle A. Shipman, Clayton Price, Mark R.J. Elsegood, William Clegg, Andrew Houlton
Inorganica Chimica Acta 2006 Volume 359(Issue 11) pp:3515-3520
Publication Date(Web):1 August 2006
DOI:10.1016/j.ica.2006.01.009
The single crystal X-ray structures of two different polynuclear metal–nucleobase complexes are reported. Crystals isolated from the reaction of CdBr2 with ethylenediamine-N9-ethyladenine, A-Et-en, were characterised by X-ray structural analysis as a coordination polymer [μ-{CdBr(N3,N7-A-Et-en)}2CdBr4] (2). Reaction of the Pd(II)–adenine complex, [PdCl(N3-A-Et-en)]+, with sodium uracilate yielded [(Pd(N3-A-Et-en)2(N1,N3-U)][ClO4]2 (3). Despite the considerable differences in the nature of the compounds the crystal structures feature essentially similar supramolecular architectures. A building block analysis demonstrates how this arises and reveals an approach, based on bond-type substitution and molecular replacement, to conserving such features. This may be a useful approach that could be more widely adopted in crystal engineering strategies.A building block analysis of the X-ray crystal structures of quite different polymetallic nucleobase complexes illustrates that molecular replacement and bond-type substitution allow essential features of the supramolecular structures to be retained.
Co-reporter:Andrew R. Pike Dr.;Lyndsey C. Ryder;Benjamin R. Horrocks Dr.;William Clegg ;Bernard A. Connolly
Chemistry - A European Journal 2005 Volume 11(Issue 1) pp:
Publication Date(Web):18 NOV 2004
DOI:10.1002/chem.200400632
The ferrocenyl-nucleoside, 5-ethynylferrocenyl-2′-deoxycytidine (1) has been prepared by Pd-catalyzed cross-coupling between ethynylferrocene and 5-iodo-2′-deoxycytidine and incorporated into oligonucleotides by using automated solid-phase synthesis at both silica supports (CPG) and modified single-crystal silicon electrodes. Analysis of DNA oligonucleotides prepared and cleaved from conventional solid supports confirms that the ferrocenyl-nucleoside remains intact during synthesis and deprotection and that the resulting strands may be oxidised and reduced in a chemically reversible manner. Melting curve data show that the ferrocenyl-modified oligonucleotides form duplex structures with native complementary strands. The redox potential of fully solvated ferrocenyl 12-mers, 350 mV versus SCE, was shifted by +40 mV to a more positive potential upon treatment with the complement contrary to the anticipated negative shift based on a simple electrostatic basis. Automated solid-phase methods were also used to synthesise 12-mer ferrocenyl-containing oligonucleotides directly at chemically modified silicon <111> electrodes. Hybridisation to the surface-bound ferrocenyl-DNA caused a shift in the reduction potential of +34 mV to more positive values, indicating that, even when a ferrocenyl nucleoside is contained in a film, the increased density of anions from the phosphate backbone of the complement is still dominated by other factors, for example, the hydrophobic environment of the ferrocene moiety in the duplex or changes in the ferrocene–phosphate distances. The reduction potential is shifted >100 mV after hybridisation when the aqueous electrolyte is replaced by THF/LiClO4, a solvent of much lower dielectric constant; this is consistent with an explanation based on conformation-induced changes in ferrocene–phosphate distances.
Co-reporter:A.R. Pike;S.N. Patole;N.C. Murray;T. Ilyas;B.A. Connolly;B.R. Horrocks;A. Houlton
Advanced Materials 2003 Volume 15(Issue 3) pp:
Publication Date(Web):13 FEB 2003
DOI:10.1002/adma.200390060
Co-reporter:Ashleigh E. Gibson, Clayton Price, William Clegg and Andrew Houlton
Dalton Transactions 2002 (Issue 2) pp:131-133
Publication Date(Web):17 Dec 2001
DOI:10.1039/B108427P
Na+ and K+ ions exhibit different modes of interaction with the N3 site of the DNA base adenine in a model system.
Co-reporter:Andrew R. Pike;Lars H. Lie;Robert A. Eagling;Lyndsey C. Ryder;Samson N. Patole;Bernard A. Connolly ;Benjamin R. Horrocks Dr.
Angewandte Chemie 2002 Volume 114(Issue 4) pp:
Publication Date(Web):14 FEB 2002
DOI:10.1002/1521-3757(20020215)114:4<637::AID-ANGE637>3.0.CO;2-2
Komplementäre Stränge und Redox-Intercalatoren können sich an Siliciumhalbleiteroberflächen, die durch kovalente Anknüpfung von DNA modifiziert sind, zu nanometergroßen Gebilden anordnen (siehe schematische Darstellung); an der Elektrodenoberfläche kann Ladungstransfer erfolgen.
Co-reporter:Andrew R. Pike;Lars H. Lie;Robert A. Eagling;Lyndsey C. Ryder;Samson N. Patole;Bernard A. Connolly ;Benjamin R. Horrocks Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 4) pp:
Publication Date(Web):14 FEB 2002
DOI:10.1002/1521-3773(20020215)41:4<615::AID-ANIE615>3.0.CO;2-Y
Complementary strands and redox intercalators can be self-assembled into nanoscale structures capable of charge transfer at the electrode surface from DNA oligomers synthesized directly at covalently modified semiconductor silicon surfaces (see schematic representation).
Co-reporter:Clayton Price;Benjamin R. Horrocks;Annabelle Mayeux;Mark R. J. Elsegood;William Clegg
Angewandte Chemie International Edition 2002 Volume 41(Issue 6) pp:
Publication Date(Web):15 MAR 2002
DOI:10.1002/1521-3773(20020315)41:6<1047::AID-ANIE1047>3.0.CO;2-J
Hoogsteen base pairing between coordinated adenine (A) and pendant thymine (T) residues of the Pt complex of a dithioether ligand bearing A and T groups results in the formation of infinite chains in the solid state (see picture). The chains are further involved in π–π stacking interactions.
Co-reporter:Clayton Price;Benjamin R. Horrocks;Annabelle Mayeux;Mark R. J. Elsegood;William Clegg
Angewandte Chemie 2002 Volume 114(Issue 6) pp:
Publication Date(Web):15 MAR 2002
DOI:10.1002/1521-3757(20020315)114:6<1089::AID-ANGE1089>3.0.CO;2-I
Hoogsteen-Paarbildung zwischen den Thymin(T)- und koordinierten Adenin(A)-Resten des Pt-Komplexes eines Dithioether-Liganden, an den diese Reste gebunden sind, führt im festen Zustand zur Bildung unendlich ausgedehnter Ketten (siehe Bild). Bei den Ketten wurden auch π-π-Stapel-Wechselwirkungen festgestellt.
Co-reporter:Andrew R. Pike;Lyndsey C. Ryder;Benjamin R. Horrocks Dr.;William Clegg ;Mark R. J. Elsegood Dr.;Bernard A. Connolly Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 13) pp:
Publication Date(Web):24 JUN 2002
DOI:10.1002/1521-3765(20020703)8:13<2891::AID-CHEM2891>3.0.CO;2-B
Ferrocenylthymidine derivatives have been prepared by Pd-catalysed cross-coupling between ethynylferrocene or vinylferrocene and 5-iodo-2′-deoxyuridine. In the latter case a mixture of trans (2 a) and gem (2 b) isomers was obtained. The cis-vinylferrocenyl (2 c), and ethylferrocenyl (3) derivatives were obtained by catalytic hydrogenation of ethynylferrocenyl-dT (1 a), and 2 c respectively. Single-crystal X-ray data for 1 a, the ferrocenyl-2′furano-pyrimidone 1 b, and 2 a show that the nucleobase is essentially co-planar with the substituted Cp ring of the metallocene. The selective reduction of the linkage between the ferrocenyl and thymidine moieties, from –CC- to -CH2CH2-, causes a shift in the reduction potential of −124 mV. DFT calculations for the one-electron oxidised species indicate that the diminished conjugation reduces the spin transfer onto the bridging C2 group, but has less effect on the extent transferred to the nucleobase from the ferrocenyl group. Compound 1 a was incorporated site-specifically into DNA oligonucleotides by using automated solid-phase methods. However, some interconversion of 1 a1 b occurs, even under rapid mild conditions of deprotection.
Co-reporter:Clayton Price;Michelle A. Shipman;Nicholas H. Rees;Mark R. J. Elsegood Dr.;Andrew J. Edwards;William Clegg Dr.
Chemistry - A European Journal 2001 Volume 7(Issue 6) pp:
Publication Date(Web):8 MAR 2001
DOI:10.1002/1521-3765(20010316)7:6<1194::AID-CHEM1194>3.0.CO;2-3
The reactions of PdII ions with a series of chelate-tethered derivatives of adenine and guanine have been studied and reveal a difference in the reactivity of the purine bases. Reactions of [PdCl2(MeCN)2] and A-alkyl-enH⋅Cl (alkyl=propyl or ethyl, A=adenine, en=ethylenediamine) yield the monocationic species [PdCl(A-N3-Et-en)]+ (1) and [PdCl(A-N3-Pr-en)]+ (2). Both involve co-ordination at the minor groove site N3 of the nucleobase as confirmed by single-crystal X-ray analysis. Reactions with the analogous G-alkyl-enH⋅Cl derivatives (G=guanine, alkyl=ethyl or propyl) were more complex with a mixture of species being observed. For G-Et-en⋅HCl a product was isolated which was identified as [PdCl(G-C8-Et-en)]+ (3). This compound contains a biomolecular metal–carbon bond involving C8 of the purine base. Crystallography of a product obtained from reaction of G-Pr-enH⋅Cl and [Pd(MeCN)4][NO3]2 reveals an octacationic tetrameric complex (4), in which each ligand acts to bridge two metal ions through a combination of a tridentate binding mode involving the diamine and N3 and monodentate coordination at N7.
Co-reporter:Michelle A. Shipman;Clayton Price Dr.;Ashleigh E. Gibson Dr.;Mark R. J. Elsegood Dr.;William Clegg Dr.
Chemistry - A European Journal 2000 Volume 6(Issue 23) pp:
Publication Date(Web):10 NOV 2000
DOI:10.1002/(SICI)1521-3765(20001201)6:23<4239::AID-CHEM4239>3.0.CO;2-#
The cover picture shows an inverted G-tetrad (top left) formed through metal–ligand bond formation between chelate-tethered guanine nucleobases and CdSO4. Individual tetramer units assembled into columnar aggregates as a result of sulfate anions bridging adjacent Cd centres (bottom). Further association of the cylindrical polymers occurs through stacking interactions involving the six-membered rings of the nucleobase (right). This work is described in more detail by A. Houlton et al. on pp. 4371 ff.
Co-reporter:Michelle A. Shipman;Clayton Price Dr.;Ashleigh E. Gibson Dr.;Mark R. J. Elsegood Dr.;William Clegg Dr.
Chemistry - A European Journal 2000 Volume 6(Issue 23) pp:
Publication Date(Web):10 NOV 2000
DOI:10.1002/1521-3765(20001201)6:23<4371::AID-CHEM4371>3.0.CO;2-X
A series of ZnII and CdII complexes of adenine and guanine derivatives containing a diamine tether have been isolated from aqueous solutions and characterised by single crystal X-ray analysis. These studies reveal a wide range of structural types including monomeric, dimeric, tetrameric and polymeric architectures. The extended structures arise from the ability of the ligands to bridge metal ions using the chelating tether in conjunction with N7 of the nucleobase. Additional metal–nucleobase co-ordination is generally observed at the N3-site of the adenine derivatives. With CdII, ethylenediamine-N9-ethylguanine forms an inverted G-tetrad type structure.
Co-reporter:Michelle A. Shipman;Clayton Price Dr.;Mark R. J. Elsegood Dr.;William Clegg Dr.
Angewandte Chemie 2000 Volume 112(Issue 13) pp:
Publication Date(Web):4 JUL 2000
DOI:10.1002/1521-3757(20000703)112:13<2450::AID-ANGE2450>3.0.CO;2-2
Co-reporter:James E. Bateman;Robert D. Eagling;David R. Worrall;Benjamin R. Horrocks
Angewandte Chemie International Edition 1998 Volume 37(Issue 19) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19981016)37:19<2683::AID-ANIE2683>3.0.CO;2-Y
The hydrogen-terminated surface of porous silicon (PS) is sufficiently reactive for the uncatalyzed hydrosilation of alkenes and alkynes. These modifications produce dramatic changes to both the physical and chemical properties of the PS.
Co-reporter:James E. Bateman;Robert D. Eagling;David R. Worrall;Benjamin R. Horrocks
Angewandte Chemie 1998 Volume 110(Issue 19) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19981002)110:19<2829::AID-ANGE2829>3.0.CO;2-5
Wasserstoff-funktionalisiertes poröses Silicium (PS) ist gegenüber Alkenen und Alkinen für eine unkatalysierte Hydrosilylierung ausreichend reaktiv. Die dabei erhaltenen alkylmodifizierten PS-Oberflächen weisen erheblich veränderte physikalische wie auch chemische Eigenschaften auf.
Co-reporter:Miguel A. Galindo;William Clegg;Ross W. Harrington;José Dobado;Francisco Santoyo-Gonzalez;Fatima Linares;M. Angustias Romero;Jorge A. R. Navarro
Chemical Communications 2008(Issue 32) pp:NaN3737-3737
Publication Date(Web):2008/08/07
DOI:10.1039/B805705B
The cyclic trinuclear system, [(en)3Pd3(4,7-phen)3]6+, undergoes a ligand exchange reaction with 5-R-2-hydroxypyrimidine derivatives (HRpymo; R = ethynylferrocene, 5-(dimethylamino)-N-(2-propynyl)-1-naphthalene sulfonamide) to give [(en)3Pd3(4,7-phen)2(Rpymo)]5+, functional supramolecular receptors of mononucleotides.
Co-reporter:Andrew Houlton, Andrew R. Pike, Miguel Angel Galindo and Benjamin R. Horrocks
Chemical Communications 2009(Issue 14) pp:NaN1806-1806
Publication Date(Web):2009/02/18
DOI:10.1039/B818456A
The controlled preparation and assembly of opto-electronic nanoscale materials is being tackled by top-down and bottom-up approaches. The latter draws inspiration from biology, where complex hierarchical systems are assembled from simpler building blocks. One of these, DNA, is proving especially useful: its size, stability, topology; the assorted chemical functional groups; plus its capacity for self-assembly provide a powerful nanoscale toolbox for materials preparation. Here we review recent research that shows the roles DNA can play in the preparation and organisation of semiconductor nanomaterials. Studies show that both hard inorganic and soft polymer materials can be directed to grow at nanoscale lengths using DNA and its constituents. In some cases the resulting materials have been used as components in simple electrical devices and the methodology has been extended to analytical tools. Intriguingly, these DNA–semiconductor hybrid materials have been found to self-assemble themselves, forming highly regular rope-like assemblies and conducting network structures.
Co-reporter:Miguel A. Galindo, David Amantia, William Clegg, Ross W. Harrington, Richard J. Eyre, Jonathon P. Goss, Patrick R. Briddon, William McFarlane and Andrew Houlton
Chemical Communications 2009(Issue 20) pp:NaN2835-2835
Publication Date(Web):2009/03/02
DOI:10.1039/B817329J
The tetrahedral bis(adeninyl)–Cu(I) complex, 1, self-associates in polar solvent through complementary hydrogen-bonding interactions and appears to mimic the natural assembly of duplex DNA.
Co-reporter:Hasan Daw A. Mohamed, Scott M. D. Watson, Benjamin R. Horrocks and Andrew Houlton
Journal of Materials Chemistry A 2015 - vol. 3(Issue 2) pp:NaN446-446
Publication Date(Web):2014/11/19
DOI:10.1039/C4TC02307B
Two methods for the preparation of rhodium nanowires are described: (i) electroless metal deposition at duplex DNA ‘template’ molecules in bulk solution and (ii) electrochemical reduction in DNA-containing solution at a modified electrode. Both methods render essentially similar 1D nanostructures with a Rh/Rh-oxide core–shell structure. AFM studies revealed the resulting nanostructures are typically <10 nm in diameter with continuous and smooth metal coatings. However, the latter method was less effective with samples containing an ∼3-fold increase in the bare template DNA remaining. A combination of SPM methods demonstrated the structures to be electrically conducting, hence confirming their nanowire nature. The conductivity was, however, several orders of magnitude lower than that of bulk Rh; a fact attributed to the presence of resistance-increasing mechanisms, such as grain boundaries present in the Rh coatings and electron surface scattering.
Co-reporter:Miguel A. Galindo, Jennifer Hannant, Ross W. Harrington, William Clegg, Benjamin R. Horrocks, Andrew R. Pike and Andrew Houlton
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 5) pp:NaN1564-1564
Publication Date(Web):2010/11/12
DOI:10.1039/C0OB00466A
A series of modified nucleosides based on thymidine have been prepared by Pd-catalysed cross-coupling between N-alkyl-alkynyl functionalised pyrrolyl- (py), 2-(2-thienyl)pyrrolyl- (tp) or 2,5-bis(2-thienyl)pyrrolyl (tpt) groups with 5-iodo-2′-deoxyuridine. The length of the alkyl chain linking the nucleoside and pyrrolyl-containing unit, N(CH2)nCC-nucleoside (where n = 1–3) was also varied. The compounds have been characterised by 1H NMR, ES-MS, UV–vis, cyclic voltammetry (CV) and, in some cases, single-crystal X-ray diffraction. Cyclic voltammetry studies demonstrated that all the py-, tp- and tpt-alkynyl derivatives 1–7 can be electrochemically polymerised to form conductive materials. It was found that increasing the N-alkyl chain length in these cases resulted in only minor changes in the oxidation potential. The same behaviour was observed for the tp- and tpt-modified nucleosides 9–12; however, the py-derivative, 8, produced a poorly conducting material. DFT calculations on the one-electron oxidised cation of the modified nucleosides bearing tp or tpt showed that spin density is located on the pyrrolyl and thienyl units in all cases and that the coplanarity of adjacent rings increases upon oxidation. In contrast, in the corresponding pyrrolyl cases the spin density is distributed over the whole molecule, suggesting that polymerisation does not occur solely at the pyrrolyl-Cα position and the conjugation is interrupted.
Co-reporter:Jonathan Pate, Felix Zamora, Scott M. D. Watson, Nicholas G. Wright, Benjamin R. Horrocks and Andrew Houlton
Journal of Materials Chemistry A 2014 - vol. 2(Issue 43) pp:NaN9273-9273
Publication Date(Web):2014/09/25
DOI:10.1039/C4TC01632G
Templating the electroless reduction of metal ions on DNA is now an established route to the preparation of nanowires and can be particularly useful for the formation of nanowires in the desirable <10 nm size range. However, different preparation conditions produce nanowires of widely different morphologies and conductivities. We describe a method for the synthesis of Cu nanowires in which electroless metal deposition is carried out on DNA ‘template’ molecules in bulk solution. Though analogous to previous surface-based routes, importantly this now produces conductive material. AFM was used to evaluate the size and morphology of the resulting nanowires; a mean nanowire diameter of 7.1 nm (standard deviation = 4.7 nm) was determined from a statistical analysis of 100 nanowires and the Cu coatings were continuous and smooth. These findings represent a notable improvement in nanowire morphology in comparison to the previous surface-based routes. UV-vis spectroscopy, X-ray diffraction, and X-ray photoelectron spectroscopy were used to confirm formation of Cu(0) metal takes place during nanowire synthesis, and additional scanning probe microscopy techniques were employed to probe the electrical properties of the nanowires. The nanowires are less conductive [resistivity ∼ 2 Ω cm] than bulk Cu, but much more conductive than nanowires prepared by the analogous method on surface-bound DNA. Using an extension of our thermodynamic model for DNA-templating, we show that the templating process in bulk solution favours the formation of continuous nanowires compared to templating on surface-bound DNA.

![9H-Purin-6-amine, 9-[2-[[2-(methylthio)ethyl]thio]ethyl]-](http://img.cochemist.com/ccimg/530100/530087-74-2.png)
![9H-Purin-6-amine, 9-[2-[[2-(methylthio)ethyl]thio]ethyl]-](http://img.cochemist.com/ccimg/530100/530087-74-2_b.png)
![CYTIDINE, 5'-O-[BIS(4-METHOXYPHENYL)PHENYLMETHYL]-2'-DEOXY-5-IODO-](http://img.cochemist.com/ccimg/845700/845620-48-6.png)
![CYTIDINE, 5'-O-[BIS(4-METHOXYPHENYL)PHENYLMETHYL]-2'-DEOXY-5-IODO-](http://img.cochemist.com/ccimg/845700/845620-48-6_b.png)
![9H-PURIN-6-AMINE, 9-[2-[[2-(METHYLTHIO)ETHYL]THIO]ETHYL]-, MONONITRATE](http://img.cochemist.com/ccimg/530100/530087-75-3.png)
![9H-PURIN-6-AMINE, 9-[2-[[2-(METHYLTHIO)ETHYL]THIO]ETHYL]-, MONONITRATE](http://img.cochemist.com/ccimg/530100/530087-75-3_b.png)
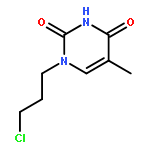
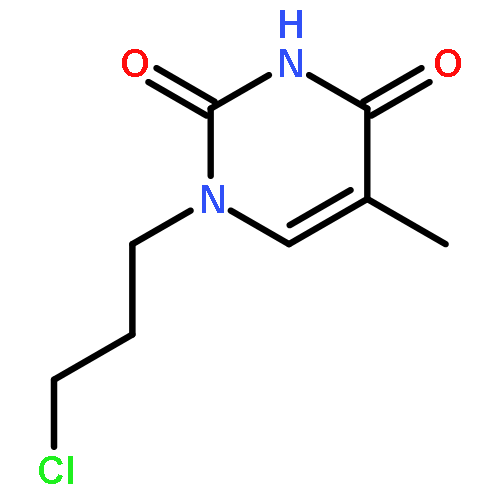
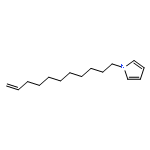
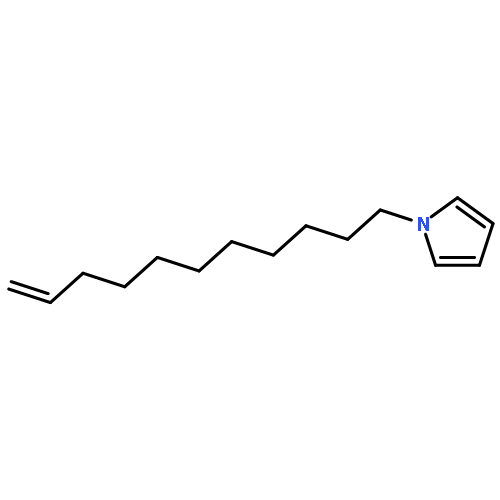
![9H-PURIN-6-AMINE, 9-[3-[[2-(METHYLTHIO)ETHYL]THIO]PROPYL]-](http://img.cochemist.com/ccimg/530100/530087-81-1.png)
![9H-PURIN-6-AMINE, 9-[3-[[2-(METHYLTHIO)ETHYL]THIO]PROPYL]-](http://img.cochemist.com/ccimg/530100/530087-81-1_b.png)