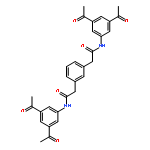
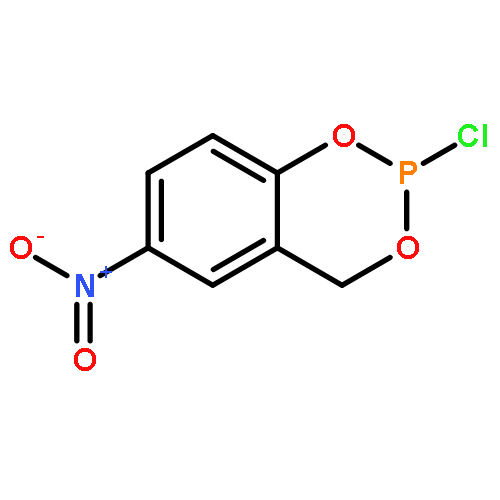
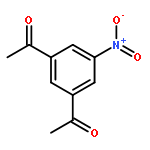
The inhibition of cellular factors that are involved in viral replication may be an important alternative to the commonly used strategy of targeting viral enzymes. The guanylhydrazone CNI-1493, a potent inhibitor of the deoxyhypusine synthase (DHS), prevents the activation of the cellular factor eIF-5A and thereby suppresses HIV replication and a number of other diseases. Here, we report on the design, synthesis and biological evaluation of a series of CNI-1493 analogues. The sebacoyl linker in CNI-1493 was replaced by different alkyl or aryl dicarboxylic acids. Most of the tested derivatives suppress HIV-1 replication efficiently in a dose-dependent manner without showing toxic side effects. The unexpected antiviral activity of the rigid derivatives point to a second binding mode as previously assumed for CNI-1493. Moreover, the chemical stability of CNI-1493 was analysed, showing a successive hydrolysis of the imino bonds. By molecular dynamics simulations, the behaviour of the parent CNI-1493 in solution and its interactions with DHS were investigated.
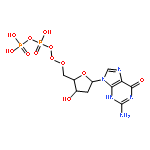
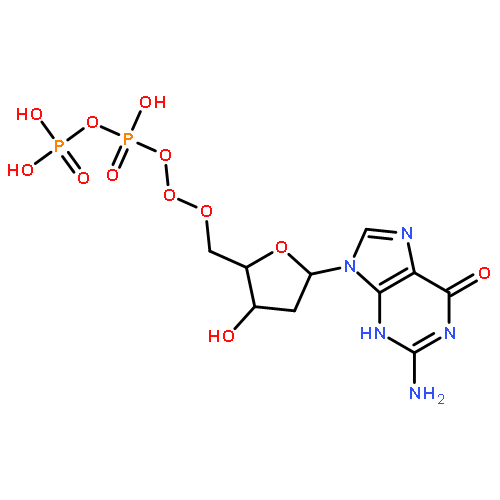
The metabolic conversion of nucleoside analogues into their triphosphates often proceeds insufficiently. Rate-limitations can be at the mono-, but also at the di- and triphosphorylation steps. We developed a nucleoside triphosphate (NTP) delivery system (TriPPPro-approach). In this approach, NTPs are masked by two bioreversible units at the γ-phosphate. Using a procedure involving H-phosphonate chemistry, a series of derivatives bearing approved, as well as potentially antivirally active, nucleoside analogues was synthesized. The enzyme-triggered delivery of NTPs was demonstrated by pig liver esterase, in human T-lymphocyte cell extracts and by a polymerase chain reaction using a prodrug of thymidine triphosphate. The TriPPPro-compounds of some HIV-inactive nucleoside analogues showed marked anti-HIV activity. For cellular uptake studies, a fluorescent TriPPPro-compound was prepared that delivered the triphosphorylated metabolite to intact CEM cells.
The metabolic conversion of nucleoside analogues into their triphosphates often proceeds insufficiently. Rate-limitations can be at the mono-, but also at the di- and triphosphorylation steps. We developed a nucleoside triphosphate (NTP) delivery system (TriPPPro-approach). In this approach, NTPs are masked by two bioreversible units at the γ-phosphate. Using a procedure involving H-phosphonate chemistry, a series of derivatives bearing approved, as well as potentially antivirally active, nucleoside analogues was synthesized. The enzyme-triggered delivery of NTPs was demonstrated by pig liver esterase, in human T-lymphocyte cell extracts and by a polymerase chain reaction using a prodrug of thymidine triphosphate. The TriPPPro-compounds of some HIV-inactive nucleoside analogues showed marked anti-HIV activity. For cellular uptake studies, a fluorescent TriPPPro-compound was prepared that delivered the triphosphorylated metabolite to intact CEM cells.
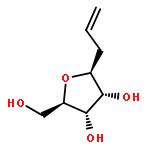
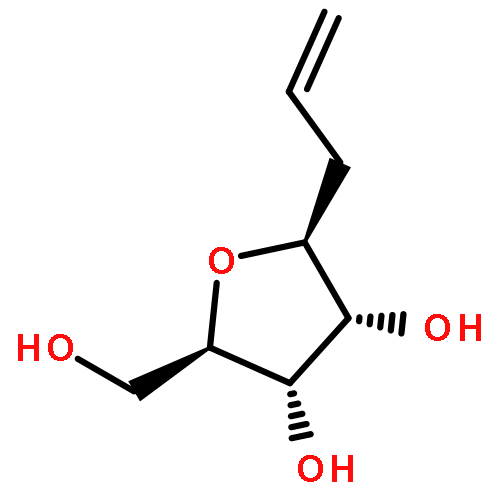
An efficient procedure was developed to prepare homo-C-nucleosides. The β-allyl C-glycoside of D-ribose was transformed into the thienopyrimidine nucleoside and the benzodiazepine nucleoside of 2-deoxy-D-ribose. For incorporation into oligonucleotides by solid-phase synthesis, both derivatives were transformed into the related 3′-phosphoramidite building blocks. These phosphoramidites were then site-specifically incorporated into DNA oligonucleotides. The modified DNA strands were hybridized with different DNA and RNA strands, and their melting temperatures and circular dichroism spectra were studied. Interestingly, although in the case of DNA–DNA hybrids the nonnatural nucleosides caused a marked decrease in melting temperature, to levels even lower than those for mismatched hybrids, in the case of DNA–RNA hybrids very similar melting temperatures were measured for the nonnatural nucleosides and the unmodified oligonucleotides.
Fucose-containing glycans mediate a variety of biological processes, but there is little information on reaction processes and mechanisms mediated by fucosyltransferases. We recently reported on fluorescently labeled GDP-β-L-fucose-ATTO 550, which enabled monitoring of α1,3-fucosyltransferase activity. Here we present an extension to the previously described results, based on the synthesis of a fluorescein-isothiocyanate (FITC)-labeled and two carboxyfluorescein-labeled (FAM-labeled) NDP-β-L-fucose derivatives, and applied all four compounds in labeling of different glycoproteins with the aid of four different fucosyltransferases. The labeling processes were analyzed by in-gel fluorescence and fluorescence polarization measurements. Comparison with the ATTO-labeled sugar revealed that the FITC-labeled fucose was the best of these substrates, and that the bacterial enzyme HP-FucT tolerated the fluorescent substrates better than human fucosyltransferases.
C8-N-arylamine adducts of 2′-deoxyguanosine (2′-dG) play an important role in the induction of the chemical carcinogenesis caused by aromatic amines. C8-N-acetyl-N-arylamine dG adducts that differ in their substitution pattern in the aniline moiety were converted by cycloSal technology into the corresponding C8-N-acetyl-N-arylamine-2′-deoxyguanosine-5′-triphosphates and C8-NH-arylamine-2′-deoxyguanosine-5′-triphosphates. Their conformation preference has been investigated by NOE spectroscopy and DFT calculations. The substrate properties of the C8-dG adducts were studied in primer-extension assays by using Klenow fragment exo− of Escherichia coli DNA polymerase I and human DNA polymerase β. It was shown that the incorporation was independent of the substitution pattern in the aryl moiety and the N-acetyl group. Although the triphosphates were poor substrates for the human polymerases, they were incorporated twice before the termination of the elongation process occurred; this might demonstrate the importance of C8-N-arylamine-2′-deoxyguanosine-5′-triphosphates in chemical carcinogenesis.
Nucleoside analogues are extensively used as antiviral and anticancer agents. Their efficiency is dependent on their metabolism into the ultimately active nucleoside triphosphates. Often one step or even more in the metabolism of the nucleoside to the triphosphate is inefficient. To overcome this hurdle, prodrugs of the nucleotides are needed. Bis(acyloxybenzyl)nucleoside diphosphates have been reported by us as a first example of an efficient nucleoside diphosphate prodrug (DiPPro nucleotides). Here, the synthesis and the properties of bis(benzoyloxybenzyl)nucleoside diphosphates of the nucleoside analogues d4T and AZT are disclosed. The synthesis was achieved by using a phosphoramidite/oxidation route. In chemical hydrolysis studies, most of the compounds formed a nucleoside diphosphate. This was confirmed in CEM cell extracts, although the prodrug stability in extracts was lower than in phosphate buffer. Furthermore, the stability and the amount of nucleoside diphosphate formed were dependent on the substituent in the benzoyl moiety. Some of the compounds were more active against HIV in thymidine kinase-deficient CEM/TK− cells than were d4T or AZT.
Despite their close structural similarity to nucleoside analogues such as the anti-HIV drugs AZT and d4T, 2′,3′-dideoxyuridine (ddU) and 2′,3′-dideoxy-2′,3′-didehydrouridine (d4U) are entirely inactive against HIV in their nucleoside form. However, it has been shown that the corresponding triphosphates of these two nucleosides can effectively block HIV reverse transcriptase. Herein we report on two types of nucleotide prodrugs (cycloSal and DiPPro nucleotides) of ddU and d4U to investigate their ability to overcome insufficient intracellular phosphorylation, which may be the reason behind their low anti-HIV activity. The release of the corresponding mono- and diphosphates from these compounds was demonstrated by hydrolysis studies in phosphate buffer (pH 7.3) and human CD4+ T-lymphocyte CEM cell extracts. Surprisingly, however, these compounds showed low or no anti-HIV activity in tests with human CD4+ T-lymphocyte CEM cells. Studies of the conversion of ddUDP and d4UDP into their triphosphate metabolites by nucleoside diphosphate kinase (NDPK) showed nearly no conversion of either diphosphate, which may be the reason for low intracellular triphosphate levels that result in low antiviral activity.
A fast, high-yielding and reliable method for the synthesis of DNA- and RNA 5′-triphosphates is reported. After synthesizing DNA or RNA oligonucleotides by automated oligonucleotide synthesis, 5-chloro-saligenyl-N,N-diisopropylphosphoramidite was coupled to the 5′-end. Oxidation of the formed 5′-phosphite using the same oxidizing reagent used in standard oligonucleotide synthesis led to 5′-cycloSal-oligonucleotides. Reaction of the support-bonded 5′-cycloSal-oligonucleotide with pyrophosphate yielded the corresponding 5′-triphosphates. The 5′-triphosphorylated DNA and RNA oligonucleotides were obtained after cleavage from the support in high purity and excellent yields. The whole reaction sequence was adapted to be used on a standard oligonucleotide synthesizer.
The high yielding synthesis of pyranonucleoside-6′-triphosphates by using the cycloSal-method is described. Synthesis of the activated cycloSal-pyranonucleoside-6′-phosphate triesters was achieved by applying a synthetic route that had been developed for the synthesis of cycloSal-(glycopyranosyl-6)-phosphates by us. The route involved regioselective 6′-tert-butyldimethylsilyl protection and exchange of the silyl protecting group by the fluorenylmethyloxycarbonyl (Fmoc) group. The 6′-Fmoc-protected derivatives were selectively converted into the cycloSal-triester. These were then very efficiently converted into triphosphates by a “titration-like” reaction with pyrophosphate. Simple purification by first ion exchange followed by reversed phase (RP) column chromatography afforded the triphosphates in very good yields.
The human enzyme deoxyhypusine synthase (DHS) is an important host cell factor that participates in the post-translational hypusine modification of eukaryotic initiation factor 5A (eIF-5A). Hypusine-modified eIF-5A plays a role in a number of diseases, including HIV infection/AIDS. Thus, DHS represents a novel and attractive drug target. So far, four crystal structures are available, and various substances have been tested for inhibition of human DHS. Among these inhibitors, N-1-guanyl-1,7-diaminoheptane (GC7) has been co-crystallized in the active site of DHS. However, despite its potency, GC7 is not selective enough to be used in drug applications. Therefore, new compounds that target DHS are needed. Herein we report the in silico design, chemical synthesis, and biological evaluation of new DHS inhibitors. One of these inhibitors showed dose-dependent inhibition of DHS in vitro, as well as suppression of HIV replication in cell cultures. Furthermore, the compound exhibited no cytotoxic effects at active concentrations. Thus, this designed compound demonstrated proof of principle and represents a promising starting point for the development of new drug candidates to specifically interfere with DHS activity.
Bioreversible protection of the β-phosphate group of nucleoside diphosphates (NDPs) as bis(acyloxybenzyl)phosphate esters is presented. To investigate the structure–activity relationship of these potential NDP prodrugs (DiPPro drugs) a series of DiPPro compounds was synthesized bearing fatty acids of various lengths and d4T as a model nucleoside. For synthesis of the lipophilically modified diphosphate group, preformed phosphoramidites were allowed to react with nucleotides, and the β-PIII moiety was subsequently oxidized. The chemical and enzymatic stability of these prodrugs was studied in different media such as phosphate buffer (pH 7.3) or CEM cell extracts. In all media, the hydrolysis rate was clearly dependent on the acyl moiety and decreased with increasing alkyl chain length. The compounds showed a markedly lower half-life in cell extracts than in pH 7.3 phosphate buffer due to the presence of enzyme-catalyzed cleavage. In all media, the DiPPro compounds released d4T diphosphate (d4TDP) as the main product beside d4TMP. In antiviral assays, the compounds proved to be at least as potent as d4T against HIV-1 and 2 in wild-type CEM/0 cells. As a proof of concept, compounds with longer acyl residues showed very good anti-HIV activities in thymidine-kinase-deficient cells (CEM/TK−), indicating their ability to penetrate cell membranes and the delivery of phosphorylated metabolites.
Beside the predominately found 8-(arylamino)-2′-dG, 8-(acetylarylamino) damages within DNA-strands may also play an important role in the induction of chemical carcinogenesis. A synthesis pathway leading to these 8-(acetylarylamino)-dG adducts using different aromatic amines has been optimized. The 8-modified dGs were converted into the corresponding phosphoramidites and site-specifically incorporated into different oligonucleotides leading to DNA strands. Lesion-bearing hybrids of these damaged DNA-strands with complementary oligonucleotides were used to study their melting properties and their circular dichroism spectra. It was shown that no EcoRI restriction took place with the damage inside the cleavage site. Finally, three different DNA polymerases were used for primer extension studies.
The synthesis of cycloSal-masked glycopyranosyl phosphates demands suitably protected precursors. A highly regioselective strategy for the preparation of cycloSal-(1,2,3,4-tetra-O-acetylglycopyranosyl-6)-phosphates was developed. Intermediate introduction of the Fmoc-group allowed the isolation of the 1,2,3,4-tetra-O-acetyl glycopyranoses to be skipped, thus, no isomerization occurred. Glycopyranoses were first converted into the 6-O-TBDMS-1,2,3,4-tetra-O-acetyl derivatives then, in a one-pot reaction, the silyl ether was cleaved and the resulting 1,2,3,4-tetra-O-acetyl glycopyranoses were trapped with Fmoc-chloride. In this exchange of protecting groups no acetyl group migration occurred. The 6-O-Fmoc-protected intermediates were selectively converted into the cycloSal-masked glycopyranosyl phosphates in a one-pot reaction. Finally, the reactivity of these activated glycopyranosyl-6-phosphates was demonstrated in the synthesis of 1,6-diglycopyranosyl-phosphates.
Fucosylation is often the final process in glycan biosynthesis. The resulting glycans are involved in a variety of biological processes, such as cell adhesion, inflammation, or tumor metastasis. Fucosyltransferases catalyze the transfer of fucose residues from the activated donor molecule GDP-β-L-fucose to various acceptor molecules. However, detailed information about the reaction processes is still lacking for most fucosyltransferases. In this work we have monitored α1,3-fucosyltransferase activity. For both donor and acceptor substrates, the introduction of a fluorescent ATTO dye was the last step in the synthesis. The subsequent conversion of these substrates into fluorescently labeled products by α1,3-fucosyltransferases was examined by high-performance thin-layer chromatography coupled with mass spectrometry as well as dual-color fluorescence cross-correlation spectroscopy, which revealed that both fluorescently labeled donor GDP-β-L-fucose-ATTO 550 and acceptor N-acetyllactosamine-ATTO 647N were accepted by recombinant human fucosyltransferase IX and Helicobacter pylori α1,3-fucosyltransferase, respectively. Analysis by fluorescence cross-correlation spectroscopy allowed a quick and versatile estimation of the progress of the enzymatic reaction and therefore this method can be used as an alternative method for investigating fucosyltransferase reactions.
A convenient approach to the chemical synthesis of L-altrose (1) and its 6-deoxy derivative 2 has been developed by starting from D-galactose (9) and D-fucose (10), respectively. The 5-epimerization by a Mitsunobu inversion of the open-chain D-hexoses was the key step for these routes. Furthermore, the conversion of 2 into peracetylated TDP-6-deoxy-α-L-altrose (3a) was achieved by the cycloSal approach. However, the final deacetylation led to an unexpected side-reaction resulting in the previously unknown 6-deoxy-α-L-altropyranose 1,3-cyclophosphate (4).
An efficient synthesis of (S)- or (R)-3-(benzyloxy-methyl)-cyclopent-3-enol was developed by appling an enzyme-catalyzed kinetic-resolution approach. This procedure allowed the syntheses of the enantiomeric building blocks (S)- and (R)-cyclopentenol with high optical purity (>98 % ee). In contrast to previous approaches, the key advantage of this procedure is that the resolution is done on the level of enantiomers that only contain one stereogenic center. Owing to this feature, it was possible to chemically convert the enantiomers into each other. By using this route, the starting materials for the syntheses of carbocyclic D- and L-nucleoside analogues were readily accessible. 3′,4′-Unsaturated D- or L-carbocyclic nucleosides were obtained from the condensation of various nucleobases with (S)- or (R)-cyclopentenol. Functionalization of the double bond in 3′-deoxy-3′,4′-didehydro-carba-D-thymidine led to a variety of new nucleoside analogues. By using the cycloSal approach, their corresponding phosphorylated metabolites were readily accessable. Moreover, a new synthetic route to carbocyclic 2′-deoxy-nucleosides was developed, thereby leading to D- and L-carba-dT. D-Carba-dT was tested for antiviral activity against multidrug-resistance HIV-1 strain E2-2 and compared to the known antiviral agent d4T, as well as L-carba-dT. Whilst L-carba-dT was found to be inactive, its D-analogue showed remarkably high activity against the resistant virus and significantly better than that of d4T. However, against the wild-type virus strain NL4/3, d4T was found to be more-active than D-carba-dT.
The cycloSal approach has been used in the past for the synthesis of a range of phosphorylated bioconjugates. In those reports, cycloSal nucleotides were allowed to react with different phosphate nucleophiles. With glycopyranosyl phosphates as nucleophiles, diphosphate-linked sugar nucleotides were formed. Here, cycloSal-nucleotides were used to prepare monophosphate-linked sugar nucleotides successfully in high anomeric purity and high chemical yield. The method was successfully used for the synthesis of three nucleotide glycopyranoses as model compounds. The method was then applied to the syntheses of CMP-N-acetyl-neuraminic acids (CMP-Neu5NAc) and of four derivatives with different modifications at their amino functions (N-propanoyl, N-butanoyl, N-pentanoyl and N-cyclopropylcarbonyl). The compounds were used for initial enzymatic studies with a bacterial polysialyltransferase (polyST). Surprisingly, the enzyme showed marked differences in terms of utilisation of the four derivatives. The N-propanoyl, N-butanoyl, and N-pentanoyl derivatives were efficiently used in a first transfer with a fluorescently labelled trisialo-acceptor. However, elongation of the resulting tetrasialo-acceptors worsened progressively with the size of the N-acyl chain. The N-pentanoyl derivative allowed a single transfer, leading to a capped tetramer. The N-cyclopropylcarbonyl derivative was not transferred.
Adducts of C8-(N-acetyl)-arylamines and 2′-deoxyadenosine were synthesised by palladium-catalysed CN cross-coupling chemistry. These 2′-dA adducts were converted into the corresponding 3′-phosphoramidites and site-specifically incorporated into DNA oligonucleotides, which were characterised by mass spectrometry, UV thermal-stability assays and circular dichroism. These modified oligonucleotides were also used in EcoRI restriction assays and in primer-extension studies with three different DNA polymerases. The incorporation of the 2′-dA lesion close to the EcoRI restriction site dramatically reduced the susceptibility of the DNA strand to cleavage; this indicates a significant local distortion of the DNA double helix. The incorporation of the acetylated C8-2′-dA-phosphoramidites into 20-mer oligonucleotides failed, however, because the N-acetyl group was lost during the deprotection process. Instead the corresponding C8-NH-2′-dA-modified oligonucleotides were obtained. The effect of the C8-NH-arylamine-dA lesion on the replication by DNA polymerases was clearly dependent both on the polymerase used and on the arylamine-dA damage.
The first diastereoselective synthesis of aryloxyphosphoramidate prodrugs of 3′-deoxy-2′,3′-didehydrothymidinemonophosphate (d4TMP) was recently reported. The synthetic approach utilized the chiral auxiliary (S)-4-isopropylthiazolidine-2-thione (2). For this strategy, a stereochemically pure phosphorodiamidate intermediate was needed. The diastereoselective formation of this key compound was investigated by using different phenols and L-alanine methyl or benzyl ester. Generally, the reaction with 3- or 4-substituted phenols led to significantly better diastereoselectivities compared to their 2-substituted counterparts. Moreover, variation of the ester group in the amino acid residue resulted in no significant differences with regard to the obtained diastereoselectivity. From the reported results, a model for the transition state was elaborated. Finally, eight new (SP)-arylphosphoramidates were synthesized with very high diastereoselectivities (up to ≥ 95 % de) and tested for their anti-HIV potency, showing a tendency for higher antiviral activity from the (SP) diastereomers.
CycloSal-nucleosyl-phosphate triesters are a known class of highly effective nucleotide prodrugs (pronucleotides) of antivirally active nucleoside analogues. Until recently, the synthesis of these compounds always gave diastereoisomeric mixtures. Then, a convergent route for the stereospecific synthesis of cycloSal-triesters was described to give isomerically pure cycloSal-prodrugs for the treatment of viral diseases. Here, the development of a stereoselective synthesis of these pronucleotides using various chiral auxiliaries is described. In contrast to pyrrolidine- or pyrrolidinone derivatives it was found that a thiazolidine derived from valinol fulfilled all three requirements to act as a suitable chiral moiety, allowing: (i) strong chirality transfer, (ii) the formation of separable diastereoisomeric intermediates, and (iii) a suitable leaving group that allows the introduction of the nucleoside analogue (e.g., d4T) in the final step under mild reaction conditions. The title compounds were obtained with very high diastereoisomeric excesses of more than 95 %.
Recently, we reported an efficient chemical method for the synthesis of a variety of naturally occurring nucleoside diphosphate (NDP) sugars. This method, which is based on the cycloSal approach, can also be used, in principle, for the preparation of rare or even nonnatural NDP sugars. Herein, the syntheses of sulfoquinovose-, glucose-6-sulfate-, L-galactose-, and 2-fluoroglycopyranoside-containing NDP sugars are presented, as well as the synthesis of NDP sugars with non-natural nucleosides. The reactions described gavestereoisomerically defined NDP sugars in high yields and short reaction times.
New divergent approaches to 2′,3′-modified carbocyclic L-nucleoside analogues starting from enantiomerically pure (1R,2S)- or (1S,2R)-2-(benzyloxymethyl)cyclopent-3-enol are described. In the key step, stereochemically pure cyclopentanols were condensed with N3-protected thymine through a modified Mitsunobu protocol. Moreover, several routes to different cyclopentanol derivatives, to prepare carbocyclic L-2′,3′-didehydro-2′,3′-dideoxynucleosides (L-d4N), L-2′,3′-dideoxynucleosides (L-ddN), and L-ribonucleosides are reported.
Succinyl-cycloSal-phosphate triesters of ribo- and 2′-deoxyribonucleosides were attached to aminomethyl polystyrene as an insoluble solid support and reacted with phosphate-containing nucleophiles yielding nucleoside di- and triphosphates, nucleoside diphosphate sugars, and dinucleoside polyphosphates in high purity after cleavage from the solid support. Here, reactive cycloSal-phosphate triesters were used as immobilized reagents that led to a generally applicable method for the efficient synthesis of phosphorylated biomolecules and phosphate-bridged bioconjugates.
A diastereoselective synthesis of cycloSal-phosphotriesters (cycloSal=cycloSaligenyl) based on chiral auxiliaries has been developed that allows the synthesis of single diastereomers of the cycloSal-pronucleotides. In previously described synthesis routes, the cycloSal-compounds were always obtained as 1:1 diastereomeric mixtures that could be separated in only rare cases. However, it was shown that the diastereomers have different antiviral activity, toxicity, and hydrolysis stabilities. Here, first a chiral thiazoline derivative was used to prepare nonsubstituted and 5-methyl-cycloSal-phosphotriesters in 48 and ≥95 % de (de=diastereomeric excess). However, this approach failed to give the important group of 3-substituted cycloSal-nucleotides. Therefore, two other chiral groups were discovered that allowed the synthesis of (RP)- and (SP)-3-methyl-cycloSal-phosphotriesters as well. The antiviral activity was found to be five- to 20-fold different between the two individual diastereomers, which proved the importance of this approach.
A reliable and high yielding synthetic pathway for the synthesis of the biologically highly important class of nucleoside diphosphate sugars (NDP-sugars) was developed by using various cycloSal-nucleotides 1 and 9 as active ester building blocks. The reaction with anomerically pure pyranosyl-1-phosphates 2 led to the target NDP-sugars 20–45 in a nucleophilic displacement reaction, which cleaves the cycloSal moiety in anomerically pure forms. As nucleosides cytidine, uridine, thymidine, adenosine, 2′-deoxy-guanosine and 2′,3′-dideoxy-2′,3′-didehydrothymidine were used while the phosphates of D-glucose, D-galactose, D-mannose, D-NAc-glucosamine, D-NAc-galactosamine, D-fucose, L-fucose as well as 6-deoxy-D-gulose were introduced.
C8-Arylamine-dG and C8-arylamine-dA adducts have been prepared using palladium cross-coupling chemistry. These adducts were subsequently converted into the corresponding 5′-O-DMTr-C8-arylamine-3′-O-phosphoramidites and then used for the automated synthesis of different site-specifically modified oligonucleotides. These “damaged” oligonucleotides have been characterized by ESI-MS, UV thermal stability assays, and circular dichroism, and they have been used in EcoRI assays as well as in primer extension studies using various DNA polymerases.
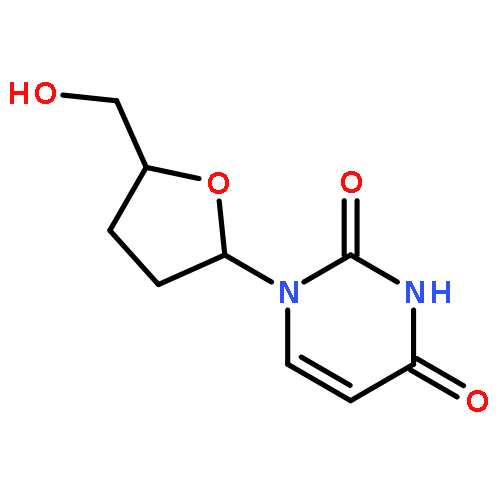
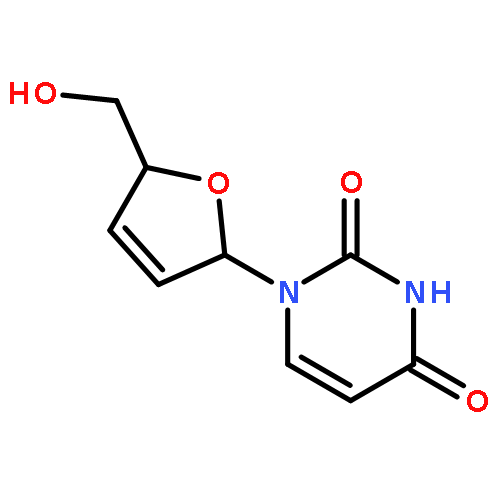
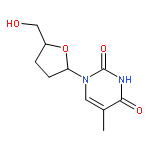
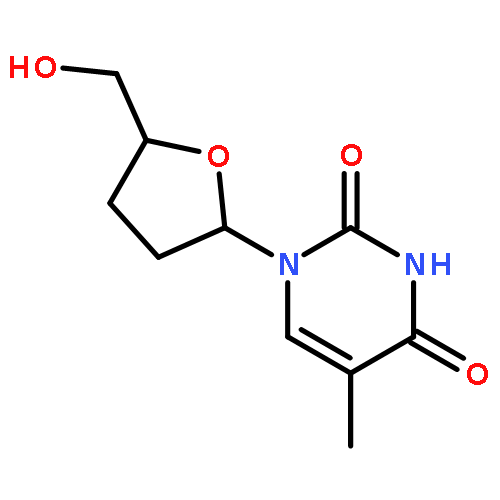
The synthesis of the fluorescent thymidine nucleoside analog 5-methyl-pyrimidin-2-one nucleoside 1 (m5K) and that of its 2′,3′-dideoxy derivative 2 (dm5K) are described. Moreover, the conversion of 1 and 2 into the corresponding 3-methyl-cycloSal-phosphate triesters 11 and 12 or an enzyme-cleavable prodrug 5-acetoxymethylpropionate-cycloSal-phosphate triester 13, and the fluorescence and hydrolysis properties of these new lipophilic triesters are reported. Finally, the suitability of these cycloSal pronucleotides as probes is demonstrated in a cell-extract hydrolysis experiment and a model study for cell uptake. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
The influence of the N3-protection group of thymine on the regioselectivity of the N1- vs. O2-alkylation under Mitsunobu conditions is described. A series of N3-protected thymine derivatives 8a–f was prepared and coupled to cyclopentanol as model compound for carbocyclic nucleoside precursors. Finally, the N3-BOM group was selected to improve our previously reported synthetic strategy to carbocyclic thymidine (carba-dT). Moreover, the 2,6-dimethyl-Bz group led exclusively to the O2-analogue of carba-dT. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Carbocyclic analogues of the anti-HIV dideoxynucleoside 3′-azido-3′-deoxythymidine AZT (1) were synthesized. Starting from the enantiomerically pure carbocyclic 2′-deoxythymidine 2, four different carbocyclic AZT analogues 2,4–6 were prepared. Moreover, the nucleoside analogues were converted into their membrane-permeable cycloSal-phosphate triesters. All compounds were tested in vitro for their anti-HIV activity in human T-lymphocytes (CEM/0). (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Pronucleotides represent a promising alternative to improve the biological activity of nucleoside analogs in antiviral and cancer chemotherapy. In addition, pronucleotides are valuable tools for studies regarding the nucleoside/nucleotide metabolism. The aim is to achieve nucleotide delivery into cells and thereby bypass limitations during intracellular formation of nucleotides from their nucleoside precursors. The cycloSal approach is one of several conceptually different pronucleotide systems known but is the only approach in which a pronucleotide is cleaved successfully by simple but selective chemical hydrolysis. The basic concept, chemistry, different structural modifications, and their effects on the antiviral potency of the cycloSal d4TMP triesters are briefly discussed first. Then, the application of the approach to various biologically active nucleoside analogs against different targets is summarized. In the second part, the results of a conceptual extension of the cycloSal approach are presented: once cycloSal pronucleotides have passed the membrane, they should be trapped inside the cells after an enzyme-catalyzed process and then release the nucleotide. Finally, results are summarized that demonstrate that the cycloSal approach is not restricted to the delivery of bioactive nucleotides but is also applicable to the intracellular delivery of hexose-1-phosphates. Chemical synthesis, biophysical studies, and biological evaluation will be discussed in combination throughout this paper to demonstrate the strength of the cycloSal approach. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
![1-((2R,4S,5S)-4-azido-5-(((6-chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)methyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/195900/195885-79-1.png)
![1-((2R,4S,5S)-4-azido-5-(((6-chloro-2-oxido-4H-benzo[d][1,3,2]dioxaphosphinin-2-yl)oxy)methyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/195900/195885-79-1_b.png)
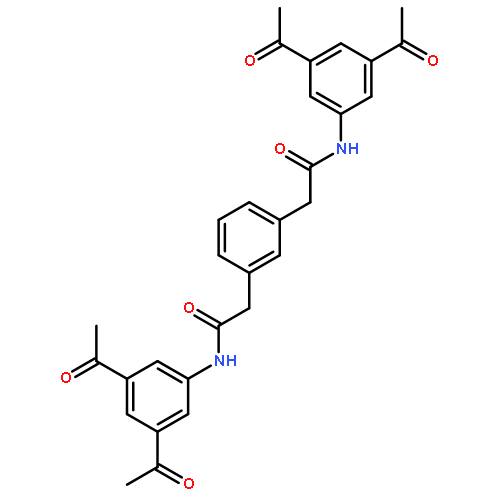
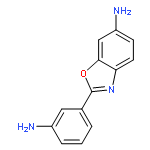
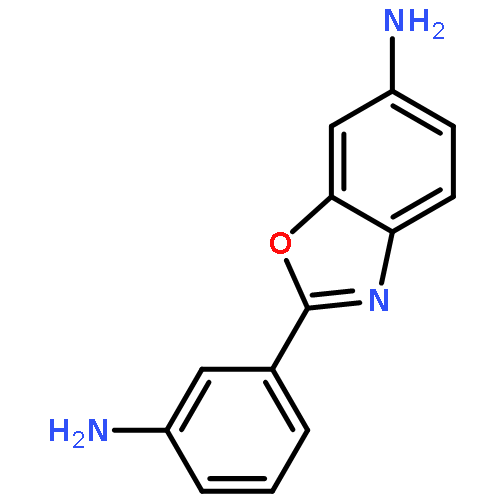
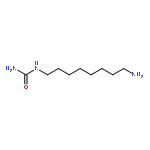
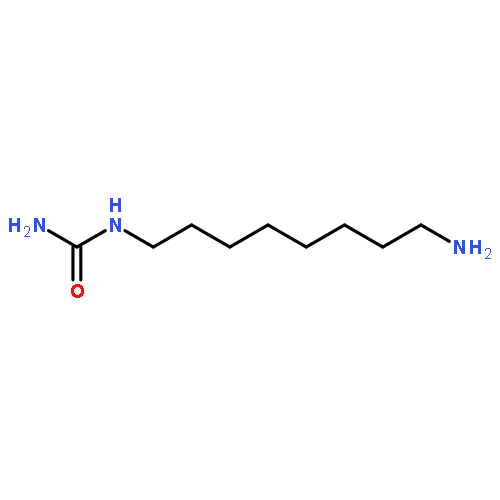
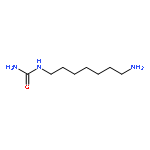
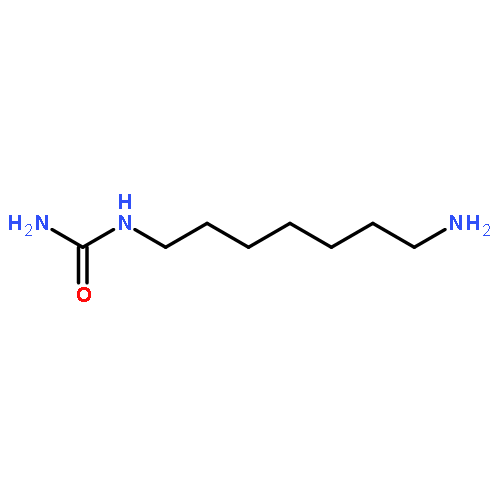
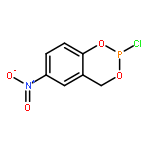
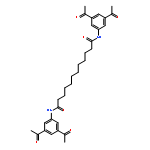
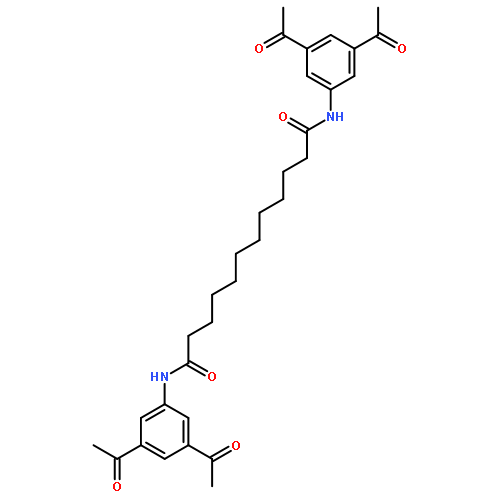
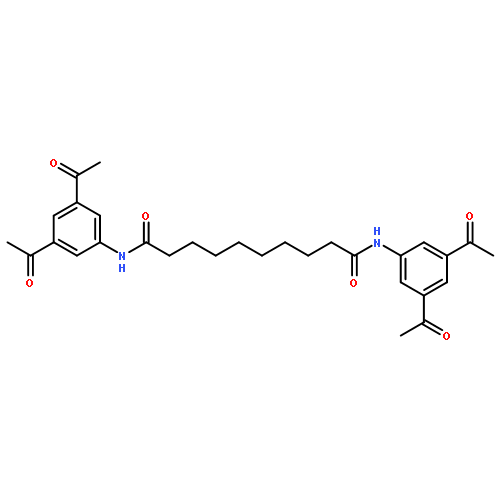
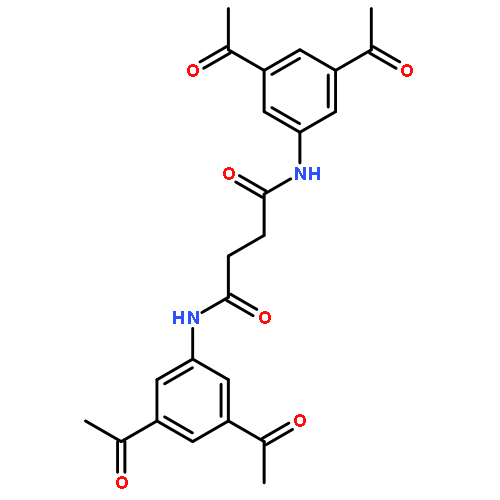
![N,N'-Bis[3,5-bis[1-(2-amidinohydrazono)ethyl]phenyl]decanediamide tetrahydrochloride](http://img.cochemist.com/ccimg/164400/164301-51-3.png)
![N,N'-Bis[3,5-bis[1-(2-amidinohydrazono)ethyl]phenyl]decanediamide tetrahydrochloride](http://img.cochemist.com/ccimg/164400/164301-51-3_b.png)
![Phenol, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126100/126070-20-0.png)
![Phenol, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-](http://img.cochemist.com/ccimg/126100/126070-20-0_b.png)
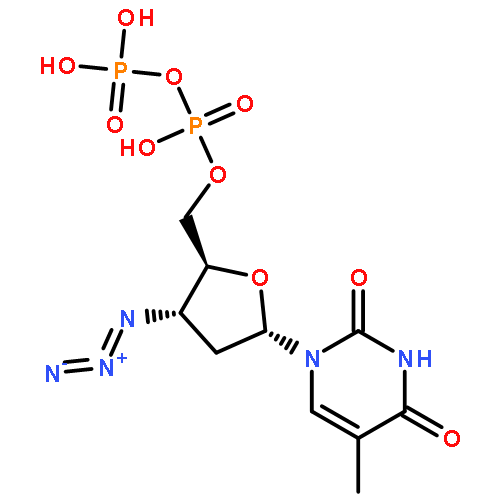
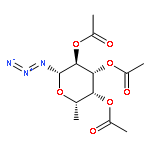
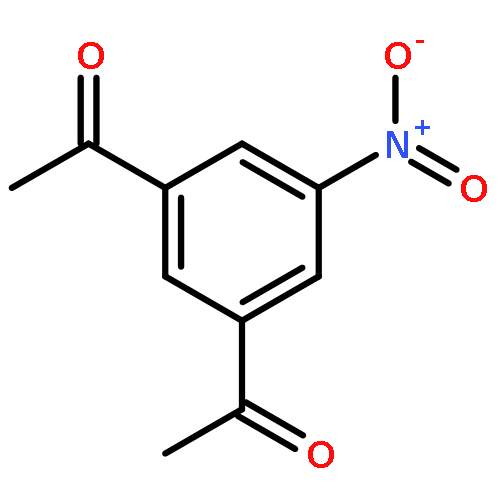
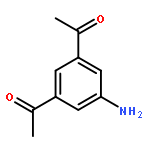
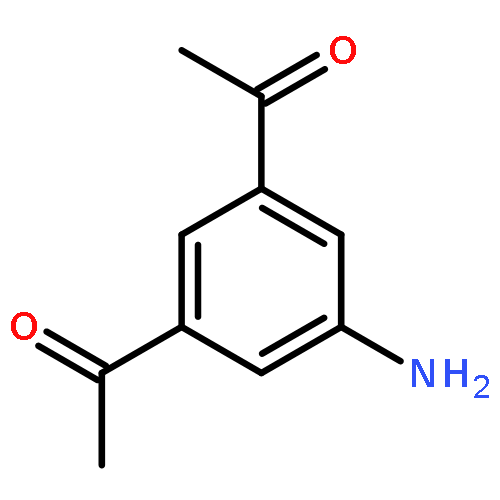

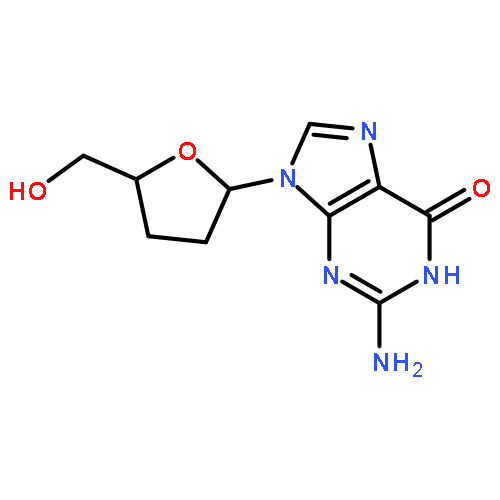
![Uridine, 5-[(1E)-2-bromoethenyl]-2'-deoxy-, 3'-acetate](http://img.cochemist.com/ccimg/84300/84218-88-2.png)
![Uridine, 5-[(1E)-2-bromoethenyl]-2'-deoxy-, 3'-acetate](http://img.cochemist.com/ccimg/84300/84218-88-2_b.png)


![Guanosine, N-acetyl-5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-deoxy-](http://img.cochemist.com/ccimg/67300/67219-54-9.png)
![Guanosine, N-acetyl-5'-O-[bis(4-methoxyphenyl)phenylmethyl]-2'-deoxy-](http://img.cochemist.com/ccimg/67300/67219-54-9_b.png)

![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8.png)
![2-[4-(aminoiminomethyl)phenyl]-1H-Indole-6-carboximidamide](http://img.cochemist.com/ccimg/47200/47165-04-8_b.png)
![[(2r,3r,4s,5r,6r)-4,5-diacetyloxy-6-azido-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/30900/30854-62-7.png)
![[(2r,3r,4s,5r,6r)-4,5-diacetyloxy-6-azido-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/30900/30854-62-7_b.png)





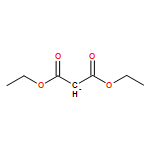





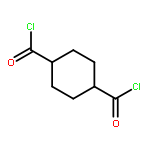
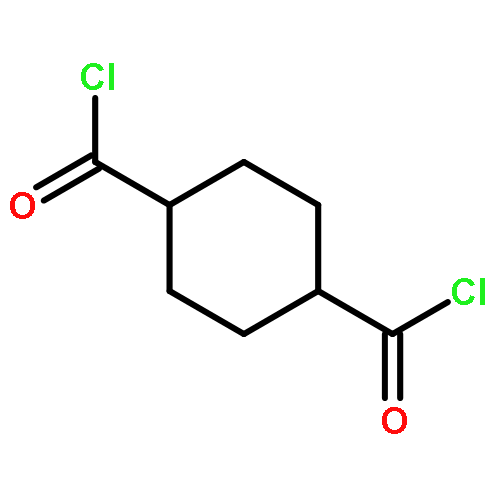
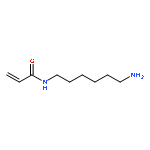












![[4-(hydroxymethyl)phenyl] Acetate](http://img.cochemist.com/ccimg/6400/6309-46-2.png)
![[4-(hydroxymethyl)phenyl] Acetate](http://img.cochemist.com/ccimg/6400/6309-46-2_b.png)